
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of protein–protein and protein–membrane interactions by isotope-edited infrared spectroscopy
Suren A.
Tatulian

Department of Physics, University of Central Florida, Orlando, FL 32816, USA. E-mail: statulia@ucf.edu
First published on 23rd July 2024
Abstract
The objective of this work is to highlight the power of isotope-edited Fourier transform infrared (FTIR) spectroscopy in resolving important problems encountered in biochemistry, biophysics, and biomedical research, focusing on protein–protein and protein membrane interactions that play key roles in practically all life processes. An overview of the effects of isotope substitutions in (bio)molecules on spectral frequencies and intensities is given. Data are presented demonstrating how isotope-labeled proteins and/or lipids can be used to elucidate enzymatic mechanisms, the mode of membrane binding of peripheral proteins, regulation of membrane protein function, protein aggregation, and local and global structural changes in proteins during functional transitions. The use of polarized attenuated total reflection FTIR spectroscopy to identify the spatial orientation and the secondary structure of a membrane-bound interfacial enzyme and the mode of lipid hydrolysis is described. Methods of production of site-directed, segmental, and domain-specific labeling of proteins by the synthetic, semisynthetic, and recombinant strategies, including advanced protein engineering technologies such as nonsense suppression and frameshift quadruplet codons are overviewed.
1. Introduction
Protein–protein interactions play a crucial role in nearly all aspects of cell physiology, such as cell signaling, reception, immunology, metabolism, muscle contraction, cell adhesion etc.1–3 Moreover, defects in protein–protein interactions constitute the core cause of many diseases.4–6 On the other hand, interactions of proteins with cellular membranes are central to many vital processes such as transport and homeostasis of ions and molecules, endo- and exocytosis, interfacial enzymology, etc.7–9 Therefore, development of capable methods of analysis and characterization of such interactions is of pivotal importance for understanding the intricate molecular machinery driving life processes. Apart from the atomic- or near-atomic-resolution structural techniques like X-ray crystallography, nuclear magnetic resonance, cryo-electron microscopy and tomography,10–12 single molecule imaging,13,14 and advanced versions of atomic force microscopy,13–16 other methods that are conventionally considered low-resolution comprise incompletely appreciated structure-resolving power. One of such techniques is Fourier transform infrared (FTIR) spectroscopy, including its various versions such as polarized attenuated total reflection (ATR) and isotope-edited FTIR.FTIR is vibrational spectroscopy that can be described with rather high fidelity using the harmonic oscillator (HO) approach (see the next section), according to which the vibrational frequency (ν) of a diatomic molecule of atomic masses of m1 and m2 is:17
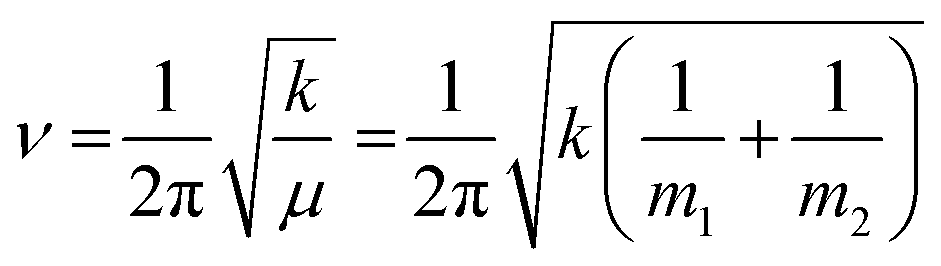 | (1) |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O covalent bond and thereby reduces the vibrational frequency, allowing identification of the presence and the strength of H-bonding that stabilizes protein structure. Furthermore, replacement of one or both atoms with a heavier isotope will generate a downshifted vibrational frequency, permitting analysis of a protein's local structure and dynamics, as described in the forthcoming sections.
O covalent bond and thereby reduces the vibrational frequency, allowing identification of the presence and the strength of H-bonding that stabilizes protein structure. Furthermore, replacement of one or both atoms with a heavier isotope will generate a downshifted vibrational frequency, permitting analysis of a protein's local structure and dynamics, as described in the forthcoming sections.
The multiple vibrational modes of the amide group of proteins, i.e., amide I through amide VII as well as the amide A and B modes, generate absorbance bands at specific mid-infrared frequencies that are sensitive to the protein's secondary and tertiary structure.18 The amide I mode (1700–1600 cm−1) is mostly due to the CO stretching vibration, with small contributions from the C–N, C–C–N, and N–H vibrations. Its frequency depends on the local geometry of the protein backbone and interactions between chemical groups such as H-bonding between the main-chain CO and N–H groups and through space vibrational coupling effects. Hence, the amide I band is exquisitely sensitive to the protein secondary structure. Due to the spectral overlap between the H2O bending mode and the protein amide I mode, a D2O-based buffer is usually used in FTIR experiments. The use of D2O results in a slight red shift of amide I frequencies caused by amide hydrogen/deuterium exchange (HX). The main secondary structures of a protein, such as various β- and γ-turns, α-helix, β-sheet, irregular structure, can be pinpointed based on their characteristic amide I wavenumbers (W = 1/λ = ν/c, where λ is the wavelength and c is the speed of light). The approximate wavenumber ranges for proteins in H2O (and D2O, shown in parentheses) are as follows: 1700–1660 cm−1 (1690–1650 cm−1) for γ-turns, 1685–1655 cm−1 (1675–1640 cm−1) for β-turns, 1660–1648 cm−1 (1655–1638 cm−1) for α-helix, 1660–1652 cm−1 (1648–1640 cm−1) for irregular structure, and 1638–1630 cm−1 (1635–1625 cm−1) for β-sheet. Moreover, various subtypes of same secondary structure can be distinguished. For example, the αII-helix, which has identical geometric parameters with the regular α-helix (rise per amino acid along the helical axis, number of amino acids per turn) but slightly tilted amide plane and hence weaker CO⋯H–N H-bonding and stronger CO and H–N covalent bonds, generates amide I signal at higher wavenumbers, ∼1665 cm−1, making FTIR a unique tool for detection of this structure. FTIR is also uniquely suited to distinguish between parallel and antiparallel and between intramolecular and intermolecular β-sheets as antiparallel β-sheets generate an additional (albeit weak) component at higher wavenumbers (around 1680 cm−1) and the wavenumbers of intermolecular β-sheets, e.g. in aggregated proteins or amyloid peptides, are shifted towards lower wavenumbers (1625–1615 cm−1).18
The amide II mode occurs around 1550 cm−1 in H2O and is mostly due to the amide N–H in-plane bending mode. In D2O, the amide hydrogens undergo exchange with deuterium of the solvent, if exposed and not involved in strong intramolecular H-bonding, resulting in a ∼90 cm−1 downshift of the amide II mode. Thus, the extent and the kinetics of the amide HX, i.e. the rate of reduction of the ∼1550 cm−1 signal, is diagnostic for the extent of solvent exposure (tertiary structure) and the degree of involvement in H-bonding (secondary structure). Other amide modes are less sensitive to protein structure or dynamics, yet have been used for protein structural studies, as described in more detail elsewhere.18
The ATR-FTIR spectroscopy is a surface-sensitive version of FTIR that is based on creation of an evanescent field at the surface of an infrared-transparent internal reflection element (IRE), such as a germanium plate, during internal reflection of the infrared light within the IRE. As the evanescent wave decays with a characteristic length of 300–400 nm, this technique detects the sample, such as a lipid membrane with reconstituted protein(s) deposited on the surface of the IRE. Plane-polarized infrared light is usually used to study not only the structural features of membrane proteins and lipids but also their spatial orientation.
The isotope-edited FTIR, which is the main topic of this article, is based on the spectral shift of the infrared signal due to replacement of atoms in biomolecules, such as H, C, N, by nonradioactive isotopes, such as 2H (D), 13C, 15N. For example, a lipid molecule with one unlabeled and one deuterated acyl chain generates infrared signals separated by ∼730 cm−1, which allows identification of selective cleavage of the sn-1 and sn-2 chains by phospholipase A2 (PLA2), reflecting the enzyme's mechanism.19 Combination of a 13C-labeled protein with an unlabeled protein results in amide I band splitting, allowing analysis of dynamic structural changes in each protein during intermolecular interactions.20 Site directed or segmental 13C (or 13C,15N or 13C18O) labeling of a protein allows determination of the local conformational dynamics during functional transitions.21–23 The production of isotope-labeled proteins ranges from easy (e.g., uniform labeling by expression in bacteria grown a minimal medium containing a sole labeled nutrient) to moderate difficulty (e.g., chemical ligation of an unlabeled fragment of a protein with a labeled peptide or incorporation of a single labeled amino acid by the nonsense suppression method).
Apart from the advanced capabilities of FTIR spectroscopy, other features such as the relative simplicity of the experimental setup, the ease of the measurements and data analysis, the small amount of material per experiment (typically, around 50 μg of protein or lipid), and the wealth of the information on the structure of both protein and lipid (if present) from just one spectrum measured in 5 minutes, make the method attractive for broad applications, e.g. protein–protein and protein–lipid interactions and beyond.
2. Isotope effects on absorbance frequency and intensity
2.1. Model compounds
Replacement of an atom in a chemical group –X–Y by an isotope of different nuclear mass will shift the X–Y vibrational frequency. If Y has a much smaller mass than X, the X–Y stretching vibration can be considered as harmonic oscillation since Y will be essentially involved in the oscillation (like a ping pong ball attached to a bowling ball by a spring) and the frequency will be determined mainly by its mass (μ ≈ mY). Then, the frequency shift resulting from replacement of Y by an isotope (e.g., replacement of C–H with C–D or N–H with N–D) can be predicted using eqn (1). If the mass of Y is not very small, the HO approach is still valid when the X–Y bond is much stronger than the other bonds in a polyatomic molecule, such as the CO double bond.17 Thus, the C–H to C–D, N–H to N–D, and 12C16O to 13C16O or to 13C18O isotopic replacements, often used in protein and lipid FTIR spectroscopy, are expected to obey the HO rules, in particular the eqn (1), with acceptable accuracy.
The HO theory predicts that the C–H to C–D isotopic change will decrease the vibrational frequency by a factor of νi/ν = 0.7338 (νi and ν are the frequencies of the heavier isotopic and normal forms, respectively). This prediction is valid so long as the molecule is free from any kind of intermolecular interactions, ideally, when it is in gas phase.17 Vibrational coupling effects or interactions with the solvent or other solute molecules, e.g. via H-bonding, may drastically affect the resulting frequencies and intensities (see below). For example, the methylene asymmetric stretching wavenumbers of the CH4 and CD4 isotopomers of methane were 3019 cm−1 and 2259 cm−1, respectively.17 This corresponds to a factor of 0.7483, which agrees with the HO prediction with 2% accuracy. The respective wavenumber of 13CH4 was 3010 cm−1, as expected for a diatomic HO. The 13CO stretching vibration of 3-13C-2,4-dimethyl-3-pentanone, 1-13C-2-methylpropionoc acid, and C6H513COOH (benzoic acid) dissolved in organic solvents was downshifted by 35–37 cm−1 as compared to the wavenumbers of their normal (unlabeled) counterparts, in good agreement with the HO theory.17
Linear hydrocarbons in condensed phase produced strong infrared peaks in spectral regions 2926–2915 cm−1 and 2855–2845 cm−1 attributed to the antisymmetric and symmetric methylene stretching vibrations, and the respective wavenumbers of their perdeuterated (i.e., totally deuterated) counterparts were red shifted by >700 cm−1.24,25 For n-hexadecane, the spectral shifts were from 2926 cm−1 to 2198 cm−1 and from 2854 cm−1 to 2095 cm−1.25 These data indicate that the spectral shift of the symmetric stretching vibration agrees with the diatomic HO prediction (νi/ν = 0.7341 as compared to the predicted value of 0.7338) but is less for the antisymmetric vibration (νi/ν = 0.7512), resulting in a wider separation between the perdeuterated peaks. The symmetric mode has been shown to be affected by intra- and intermolecular Fermi resonance interactions with the methylene bending mode, which results in secondary bands that contribute to the apparent intensity increase in the antisymmetric stretching region.26 Such interactions are symmetry-forbidden for the antisymmetric stretching mode of an extended chain.
For molecules in gas phase, the change in the integrated molar absorption intensity (A) is predicted to be proportional to the square of the change in frequency:17
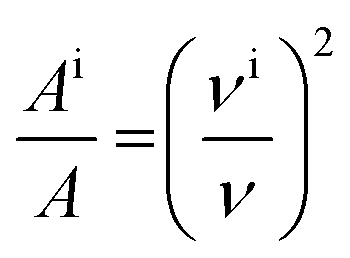 | (2) |
2.2. Peptides and proteins
For proteins or lipid bilayers, which are in the condensed phase and are involved in a multitude of trough-space and trough-bond intermolecular interactions, the HO predictions are not expected to strictly hold. In addition, the spectral shifts of the amide I signal due to the most frequently utilized 13CO labeling strategy cannot be described by a simple diatomic HO modeling as the amide I mode is not a pure CO stretching vibration. Nonetheless, incorporation of just one or up to 5 consecutive backbone 13CO-labeled amino acid residues in synthetic peptides produced a new amide I peak red shifted from that of the unsubstituted peptide by 37 ± 4 cm−1, in agreement with the HO theory applied to an isolated CO bond.27–34 Incorporation of 13C-labeled amino acids located consecutively or separated by one or more 12C units in a α-helical 25-mer peptide revealed a complex behavior of both the vibrational frequency and intensity.33,34 The frequency of a single 13C amide unit was downshifted by 36–37 cm−1 (from 1632–1630 cm−1 to 1596–1593 cm−1) and increasing number of 13C units resulted in increase in both 12C and 13C frequencies, increase in 13C intensity, and decrease in 12C intensity.34 Variably spaced 13C units produced a non-monotonous (“damped sinusoidal”) dependence of the downshifted frequency on the number of intervening 12C units. Strikingly, the presence of one or two 12C units between two 13C units produced amide I signal of reduced intensity downshifted by additional ∼10 cm−1.33,34 These effects were interpreted in terms of distinct distance dependencies of the through-covalent-bonding mechanical coupling and through-space electrostatic and transition dipole couplings as well as relative dipolar orientations of the amide oscillators at defined positions along the spiraling helical chain.21,34 The increased frequency and reduced intensity of the 12C signal upon incorporation of more 13C unis is thought to be caused by vibrational decoupling between the 12C amide oscillators.34
The amide I intensity depends on the secondary structure and its continuity and is different for 12CO and 13CO peptide segments. For example, the molar absorption coefficient at the maximum of the amide I band (εmax) was 640 M−1 cm−1 at 1 °C (mostly helical structure) and 392 M−1 cm−1 at 91 °C (mostly unordered) for the unlabeled (12CO) peptide30 (here, M−1 means per M of amino acid units). The respective values of εmax were smaller for the 13C-labeled segments by a factor of 0.7–0.8. This difference in molar absorptivity was explained by higher helicity and longer chains of the unlabeled segments rather than by intrinsic, isotope-specific differences30 echoing with the concept of disruption of the 12CO/12CO coupling by intervening 13CO units mentioned above.34 This is reminiscent of the up to 2-fold stronger amide I signal from the 13C-labeled 4-residue segment located centrally compared to that when located at the N- or C-termini of a 20-mer helical peptide.28,35
The agreement between the observed and diatomic HO-predicted frequency shift of ∼37 cm−1 upon a ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) 12CO → 13CO substitution may be fortuitous due to compensation of diverse effects on the resulting vibrational frequency. When other amide I components such as the C–N stretching mode are included in the computation, the 13C-induced spectral shift is around 43 cm−1 rather than 37 cm−1.36 Uniform 13C-labeling of 40–42 amino acid residue peptides reduced the amide I frequency from 1658 cm−1 to 1617 cm−1 in α-helical state and from 1633 cm−1 to 1590 cm−1 in β-sheet state, i.e. a 41–43 cm−1 downshift as predicted by the more accurate simulation.37 Likewise, other studies on chemically synthesized or recombinantly produced proteins reported 40–44 cm−1 downshift of the amide I peak due to singly, segmentally, or uniform 13CO labeling,38–41 while the spectral shift due to uniform 13C,15N labeling of recombinant proteins was 45–55 cm−1.42,43
12CO → 13CO substitution may be fortuitous due to compensation of diverse effects on the resulting vibrational frequency. When other amide I components such as the C–N stretching mode are included in the computation, the 13C-induced spectral shift is around 43 cm−1 rather than 37 cm−1.36 Uniform 13C-labeling of 40–42 amino acid residue peptides reduced the amide I frequency from 1658 cm−1 to 1617 cm−1 in α-helical state and from 1633 cm−1 to 1590 cm−1 in β-sheet state, i.e. a 41–43 cm−1 downshift as predicted by the more accurate simulation.37 Likewise, other studies on chemically synthesized or recombinantly produced proteins reported 40–44 cm−1 downshift of the amide I peak due to singly, segmentally, or uniform 13CO labeling,38–41 while the spectral shift due to uniform 13C,15N labeling of recombinant proteins was 45–55 cm−1.42,43
Apart from intramolecular vibrational couplings, the amide I frequency also depends on H-bonding with the solvent. Thus, for totally hydrated α-helical peptides that form H-bonding with water, the frequency is reduced by as much as 17 cm−1, e.g., from ∼1650 cm−1 down to ∼1633 cm−1.21 Peptides in dry state (dried from hexafluoroisopropanol solution)37,44 or embedded in lipid bilayers40,45 show amide I peaks around 1661–1657 cm−1 as compared to 1633 ± 4 cm−1 displayed by peptides totally hydrated in a D2O buffer.28,30,33,34 A similar effect has been reported for a β-sheet peptide.46
Studies on peptides in β-sheet conformation revealed position-dependent 13C isotopic shifts in spectral frequencies and absorbance intensities.29,46–49 Incorporation of one or two 13CO amino acids in a synthetic, antiparallel β-sheet forming tetradecapeptide produced a spectrally downshifted (from 1634–1628 cm−1 to 1611–1606 cm−1) amide I component of anomalously high intensity.29 The integrated molar absorptivity of the downshifted amide I component of a single 13CO label was more than doubled relative to that of the main 12CO part, i.e. constituted around 17% of the total amide I contour as opposed to the expected 1/14 ≈ 7%. The peptide with two consecutive labels generated a similar amide I band. However, when the two 13CO labels were separated by one 12CO unit, the absorptivity increased nearly 2-fold compared to the species with two consecutive 13CO labels. These anomalous intensities were interpreted by through-space transition dipole coupling and through covalent bonding and through H-bonding interactions between the 13CO and 12CO amide oscillators. In case of single or sequential double labeled peptides, the 13C-substituted oscillators borrow intensity from the unlabeled oscillators of both opposite (through H-bonding) and same (through covalent bonding) strands whereas the separated oscillators do the same but pick up more intensity from neighboring 12CO units through covalent bonding. Interestingly, in case of an α-helical peptide, a single 13CO substitution produced an amide I component downshifted by ∼38 cm−1 as expected for an isolated oscillator, suggesting the absence of (or weaker) through-H-bond interactions in α-helix compared to β-sheet, consistent with a shorter N⋯O distance by 0.034 Å in the latter case.29 In addition to this difference, Huang et al.33 reported that in α-helical structure the isotopically substituted groups are coupled essentially to each other but not to the unlabeled units, in contrast to β-sheet structure.
An important feature observed in in β-sheet peptides, which has helped distinguish between parallel and antiparallel β-sheets, is that the spectral downshift of the 13CO substituted units that are involved in coupling with each other is larger by ∼10 cm−1 compared to that caused by 13CO/12CO coupling (1594–1591 cm−1 and 1604–1601 cm−1, respectively).46,47,50,51 The effects of vibrational couplings between same or different isotope units on vibrational frequencies and intensities has been demonstrated in studies on a dodecamer that forms a β-hairpin structure with amide I peak at 1633 cm−1.48 Incorporation of two 13CO units in opposing strands produced a new component that was downshifted more when the labeled 13CO oscillators were spatially (not along the chain) closer. In addition, the more downshifted signal (closer spacing) was less intense than the less downshifted signal generated by more remotely spaced units. These features were interpreted by 13CO/13CO and 12CO/13CO vibrational coupling effects: closely spaced 13CO oscillators couple stronger resulting in a larger downshift but do not mix with the 12CO modes whereas those spaced more distantly (more 12CO units in between) experience less 13CO/13CO coupling (higher frequency) and more 12CO/13CO coupling (higher intensity through mixing). These data underscore the dependence of spectral frequencies and intensities caused by 13C-labeling on the number and location of labeled residues. Upon insertion of 3 backbone 13CO units in a 3-stranded antiparallel β-sheet peptide (one per strand), the 12CO frequency of the main β-sheet rose to 1642 cm−1, resulting in a 54 cm−1 spectral gap between labeled and unlabeled signals.49 This effect is explained by reduction of the strand length and subsequent disruption of coupling between 12CO oscillators,49 like in case of α-helix discussed above.34
Double isotopic labeling of the amide carbonyl with 13C and 18O provides a better resolution of the local structure of proteins or peptides.40 Labeling one out of 25 residues of an α-helical peptide with 13C18O resulted in an amide I component shifted to 1595 cm−1 from the main (unlabeled) component centered at 1659 cm−1.40 This 64 cm−1 shift is less than the 78 cm−1 predicted by diatomic HO calculations but is consistent with a more rigorous Hartree–Fock theory. The robust spectral resolution permitted determination of the local structure and orientation of the transmembrane domains of membrane proteins by ATR-52,53 or transmission FTIR54 on oriented peptide/lipid films. 2D infrared studies on another helical peptide 13C18O labeled at 1 position at a time spanning the central 11 residues, mixed with the unlabeled peptide at various proportions in lipid vesicles, revealed excitonic through-space vibrational coupling (delocalization) between the labeled oscillators, or the absence of such coupling at higher dilutions, which allowed construction of the structure of the transmembrane helical dimer.55
For peptides forming cross-β-sheet amyloid fibrils, such as the amyloid β(1–40) peptide, a main amide I peak was detected at 1625 cm−1 and a downshifted component in the 1585–1575 cm−1 range depending on the position of the 13C18O substituted residue.56 The spectral shift was stronger for the sample composed of the labeled peptide due to vibrational coupling along the fibril axis and was blue shifted by ∼20 cm−1 (from ∼1575 cm−1 to ∼1595 cm−1) when combined with a large molar excess of the unlabeled peptide. In contrast to α-helix, where a single label may not produce a diagnostic signal because of the absence of coupling with another labeled residue, the isotopic substitution of a single amino acid in parallel in-register fibril forming peptides produces a strongly delocalized and red shifted normal mode indicative of interstrand coupling along the fibril axis.57,58 A heptadecapeptide with 13C18O substituted alanine at position 5 or 15 displayed amide I spectra with a major peak at 1632 or 1629 cm−1 and a minor (isotope-induced) peak at 1587 or 1591 cm−1, signifying a 45 cm−1 or 38 cm−1 red shift for the N- and C-terminal segments that were deduced to be in ordered cross-β and unordered conformations, respectively.59 Thus, the 13C18O isotope-induced frequency shift in β-sheet peptides was significantly less (∼45–50 cm−1)56,59 than that in α-helices (∼65 cm−1).40,52–54
The human islet amyloid polypeptide in both α-helical and β-sheet conformations, incorporating 1 or 2 adjacent 13C18O labeled residues, was studied by 2D infrared spectroscopy.58 Incorporation of a single label into the α-helical or β-sheet structure caused a ∼55 cm−1 and 32–42 cm−1 red shift, respectively, in qualitative agreement with the above assessment. The respective red shifts caused by two adjacent labels were ∼45 cm−1 and ∼52 cm−1 however, reversing the disparity between α-helical and β-sheet structures. This effect was consistent with normal mode analysis which highlighted differences between spectral shifts due to single vs. double (adjacent) labels in the two structures: two adjacent isotope labels cause a red shift from the uncoupled local mode frequency of the isotope substituted group for a β-sheet structure (through intra- and interstrand couplings) and a blue shift for α-helix (through covalent bond coupling).58
The above discussions underscore the complexity of the behavior of vibrational frequencies and intensities and the involved isotopic effects. The frequencies and intensities of labeled vibrations depend on the secondary structure, the number of the labels and their sequentiality. The isotope labeling strategy should be selected taking into account the targeted secondary structure. For example, spectral features of a single isotope in an α-helix will produce only a local mode bearing little structural information but in a parallel in-register β-sheet structure or a cross-β-sheet structure a single label will be strongly coupled along and across the sheet plane producing the diagnostic amide I signal.49,58
Interpretation of the shifted frequency in terms of secondary structure is not straightforward. For example, the ∼1633 cm−1 signal can be generated by β-sheet48,49 or by hydrated and totally amide deuterated α-helix.21,30–32,34 Additional methods such as circular dichroism or NMR are required to identify whether the spectral shift, e.g. from ∼1655 cm−1 to ∼1635 cm−1, reflects an α → β transition or hydration of the protein's backbone carbonyls plus amide HX.29,30,49 The amide I signal in the spectrum of a 13CO or 13C18O labeled peptide or protein in the 1600–1565 cm−1 region should be treated cautiously as it may be produced by either the labeled amino acid or the side chains of certain amino acids such as arginine or glutamic acid.30,48,56 Finally, possible artifacts stemming from the admixture of 13C due to natural abundance (∼1.1%) should be kept in mind to avoid misinterpretation of the intensity of the signal generated by the labeled site.
2.3. Lipids
Artificial membranes composed of isotopically labeled lipids have been used to study thermal phase transitions, lateral segregation, lipid transfer, lipid–protein interactions, and the influence thereof on the function of membrane proteins.60–64 Mostly, the acyl chain deuterated or carbonyl 13CO substituted lipids have been used to gain structural information on the membrane core or the polar headgroup region. The acyl chain methylene groups generate antisymmetric and symmetric stretching vibrations centered around 2920 cm−1 and 2850 cm−1 in protiated (CH2) form while chain perdeuteration shifts these wavenumbers down to ∼2192 cm−1 and ∼2090 cm−1, respectively.25,60,65,66 The methylene stretching modes of perdeuterated chains are usually less intense in terms of peak height and the spectral spacing between the symmetric and antisymmetric peaks is larger (∼100 cm−1) compared to the unlabeled lipids (∼70 cm−1), similar to the features detected for n-alkanes discussed above. Analysis of ATR-FTIR spectra of unlabeled and acyl chain perdeuterated phospholipids produced nearly equal integrated molar absorption intensities: Ai/A ≈ 1.0, indicating no isotope-induced changes in intrinsic absorptivities.60
Temperature-induced gel to liquid crystal phase transitions of bilayer membranes composed of unlabeled and acyl chain perdeuterated lipids cause increase in the stretching wavenumbers by 2–5 cm−1.63,65–68 This spectral shift is interpreted in terms of decreased vibrational interactions between adjacent methylene units due to increasing ratio of gauche/trans conformational isomers.62,65,67 Similar methylene stretching vibrational shifts have been detected upon isotopic dilution of n-alkanes as well as phospholipids without involvement of conformational changes, explained by alterations in intermolecular Fermi resonance between like isotopic species.25 Use of lipids with perdeuterated acyl chains has permitted analysis of thermal phase transitions of individual membrane components composed of binary or ternary lipid mixtures.62,68,69 Other vibrational modes of lipids such as the methylene rocking and scissoring modes and their spectral shifts upon deuteration have been analyzed to reveal membrane depth-dependent fractions of gauche rotamers and to detect lipid microdomain formation.70,71
The carbonyl stretching mode of glycerophospholipids in aqueous suspensions generates a band with a peak wavenumber in the 1740–1730 cm−1 region which, upon resolution enhancement, reveals two components around 1742 cm−1 and 1727 cm−1.67,72,73 These two components have been assigned to the carbonyls at sn-1 and sn-2 ester groups, respectively, and the splitting was ascribed to their distinct conformations.67 Further studies on lipids selectively 13CO labeled at either sn-1 or sn-2 carbonyls resulted in two bands, the one generated by the labeled carbonyl red shifted by 40–43 cm−1.72–75 Strikingly, both the unlabeled and the 13CO labeled (and downshifted) bands were in turn composed of at least two components of their own separated by the same gap, i.e. 11–17 cm−1.72,74,75 It was concluded that the higher and lower frequency components of the carbonyl stretching vibrations are generated by dehydrated and hydrated fractions of the carbonyl groups rather than structural difference that could account for a smaller (3–4 cm−1) spectral difference.72,74 In general, an increasing polarity and H-bonding capability of the solvent results in lower frequency and wider vibrational bands.72,75 The red shift upon hydration can be explained by weakening of the CO double bond (parameter k in eqn (1)) due to strong CO⋯H–O–H hydrogen bonding.
FTIR studies on phosphatidylcholine selectively 13CO labeled at one of the two ester carbonyls suggested that cholesterol forms H-bonding with the sn-2 but not sn-1 carbonyl group.76 Polarized ATR-FTIR studies on substrate-supported multilayers of selectively 13CO labeled phosphatidylcholines reported a ∼40 cm−1 red shift of the CO double bond and a ∼20 cm−1 red shift of the ester CO–O single bond stretching vibrations, allowing determination of similar orientations of the CO bonds of both chains (≥60°) and radically different orientations of the CO–O bonds relative to the membrane normal.73
Both the methylene and the carbonyl stretching modes of lipids have been used to assess protein–lipid interactions and activities of membrane proteins. FTIR data on acyl chain perdeuterated phospholipids combined with unlabeled diacylglycerol identified domains of phosphatidylserine/diacylglycerol complexes that facilitated activation of protein kinase C.62 Membrane binding of human 5-lipoxygenase caused a shift of the carbonyl stretching band to higher wavenumbers, suggesting membrane dehydration.77 Fluorescence data on lipid vesicles containing a surface polarity-sensitive probe implied membrane dehydration upon PLA2 binding, however the lower and higher frequency components of the lipid carbonyl stretching bands (1728 cm−1 and 1742 cm−1) became stronger and weaker, respectively, indicating H-bonding between the protein and membrane lipids.78 The mode of membrane binding and selective hydrolysis of membrane lipids by PLA2 are described in detail in the forthcoming sections.
3. Protein–membrane interactions
3.1. Hierarchy of membrane proteins and modes of interactions with membranes
Around one-third of all gene products are membrane proteins,79,80 clearly implying that protein–membrane interactions constitute one of the major pillars of the temple of life. These proteins employ distinct mechanisms of membrane binding. While the integral proteins possess well-defined nonpolar stretches that interact with membrane lipids via hydrophobic contacts, the lipid-tethered proteins utilize a covalently attached fatty acid (most frequently, palmitic or myristic), glycosylphosphatidylinositol, or isoprenoid (farnesyl or geranylgeranyl) moiety for membrane binding.81–91 Polytopic proteins constitute the most abundant subclass of membrane proteins (>25%)8 and may contain 2 to 16 transmembrane helices.81 A smaller group of integral proteins utilizes an antiparallel transmembrane β-sheet structure arranged in a β-barrel-like architecture to serve as a transport passageway for relatively large molecules or as receptors, cell adhesion and/or signaling molecules in mitochondria of animal cells, chloroplasts of plant cells and plasma membranes of prokaryotes such as the bacterial toxin α-hemolysin.8,82–84The peripheral proteins have been estimated to constitute ∼9% of the human proteome and perform their function through transient association with plasma or organellar membranes.7–9 Some of these proteins are enzymes targeting membrane lipids for phosphorylation (e.g., diacylglycerol kinase, phosphatidylinositol 3-kinase), oxygenation (lipoxygenases), or hydrolysis (e.g., sphingomyelinase or phospholipases A1, A2, B, C, D). Typically, the peripheral proteins bind to membranes using an interfacial binding site (IBS).9 As the IBS is not always a clearly defined motif or domain, identification of the mode of membrane interaction of peripheral proteins remains a challenge. Analysis of 1328 experimentally determined and 1194 simulated (AlphaFold) peripheral protein structures identified some common features of the IBS, i.e., protrusions rich in nonpolar amino acids (protruding hydrophobes)92 and lysine, which support membrane binding through hydrophobic and ionic contacts, respectively.9 In addition, there are many membrane binding modules specifically recognizing individual lipids or their assemblies. For example, most isoforms of protein kinase C become activated through binding to membrane-residing diacylglycerol using the C1 domain. Around 150 human proteins, including protein kinase C, contain one or more C2 domains, which mediate membrane binding, often in a calcium-dependent manner.93 A range of signaling proteins bind to phosphatidylinositol phosphorylated at certain sites through the pleckstrin homology or FYVE domain, which recruit the respective proteins to distinct membranes where their function is most physiologically relevant.7 Other specialized lipid binding domain families are described elsewhere.9,93
Peripheral proteins, including enzymes such as lipoxygenases, annexins, lipid kinases, phosphatases, and hydrolases, rely on binding to membranes with certain lipid composition, at a defined mode, including the depth of membrane insertion and the spatial orientation. The mode of membrane binding is crucial for the function of these proteins,94 yet the “characterization of these fundamentally important lipid–protein interactions with experimental techniques is…prohibitively challenging”.95 The forthcoming text will illustrate an example of how isotope-edited FTIR spectroscopy can determine the precise mode of membrane binding and thereby provide insight into the mechanism of action of a typical interfacial enzyme, a secreted PLA2.
3.2. Selective hydrolysis of lipids by PLA2
Secreted PLA2s are medium-sized (13–16 kDa) extracellular enzymes that hydrolyze the sn-2 acyl chain of glycerophospholipids of cell membranes and liberate unsaturated fatty acids, in particular arachidonic acid, and lyso-phospholipid. These products serve as precursors for the biosynthesis of various eicosanoids or platelet-activating factor, respectively, that possess multiple biological activities, including cell signaling, apoptosis, inflammation, allergy, and tumorigenesis. Secreted PLA2s do not have special membrane binding or activation domains like many other interfacial enzymes, although an interfacial adsorption surface has been identified for each isoform. They bind to the cellular plasma membrane in a unique, “productive” mode that results in enzyme activation.94 One of the factors that facilitate this process of interfacial activation of cationic PLA2s is the presence of anionic lipids in the membrane.96 To understand whether the elevated enzyme activity results from more efficient binding of the cationic protein to the negatively charged membrane surface or from higher intrinsic activity of the enzyme towards the anionic lipid, ATR-FTIR experiments have been conducted on lipid bilayers composed of an equimolar mixture of a zwitterionic lipid dipalmitoylphosphatidylcholine (DPPC) with uniformly perdeuterated acyl chains, DP(d62)PC, and an unlabeled anionic lipid dipalmitoylphosphatidylglycerol (DPPG). In an ATR-FTIR experiment, the sample, in this case a lipid bilayer, is deposited on a germanium plate (Fig. 1). The incident light is directed into the germanium IRE at one edge with the aid of a set of mirrors and travels to the other edge by means of a series of internal reflections. At each internal reflection, an evanescent wave is created above the surface of the IRE that is absorbed by the sample deposited on it.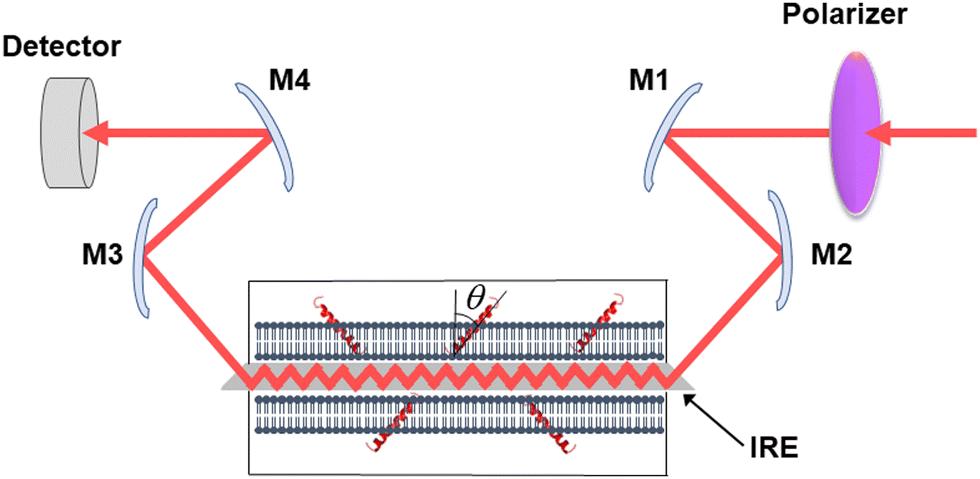 | ||
| Fig. 1 Schematic presentation of the ATR-FTIR setting. The infrared light is plane-polarized via a polarizer and directed into the internal reflection element by mirrors M1 and M2. The incident light performs a series of internal reflections, exits the IRE and is directed to the detector by mirrors M3 and M4. The lipid membrane with embedded protein, shown as a single helix, is deposited at both surfaces of the IRE. Reproduced from ref. 94, with permission. | ||
As described in Section 2.3, deuteration of the lipid acyl chains of DP(d62)PC results in a >700 cm−1 downshift of the methylene stretching vibrational frequency thereby producing FTIR spectra with well-resolved signals from the acyl chains of both unlabeled and deuterated lipids (Fig. 2a). The CD2 absorbance band displays lower intensity and larger width compared to the CH2 band, consistent with earlier observations.60 Addition of PLA2 results in hydrolysis of both lipids and removal of a fraction of the reaction products (free fatty acid and lyso-phospholipid) from the membrane, as evidenced by the reduction of the methylene stretching absorbance intensities. This is consistent with removal of >30% of the bilayer material from membranes caused by lipid hydrolysis by snake venom PLA2s.97–99 A plot of the normalized intensities of the methylene stretching bands versus PLA2 concentration indicates that both the zwitterionic and the acidic lipids are hydrolyzed to similar extents at a given PLA2 concentration (Fig. 2b). Thus, the result of this isotope-edited ATR-FTIR experiment implies that the higher enzymatic activity of PLA2 towards membranes containing negatively charged lipid(s) stems from a stronger electrostatic attraction between the enzyme and the membrane rather than from the intrinsic preference of the enzyme for the anionic lipid.19
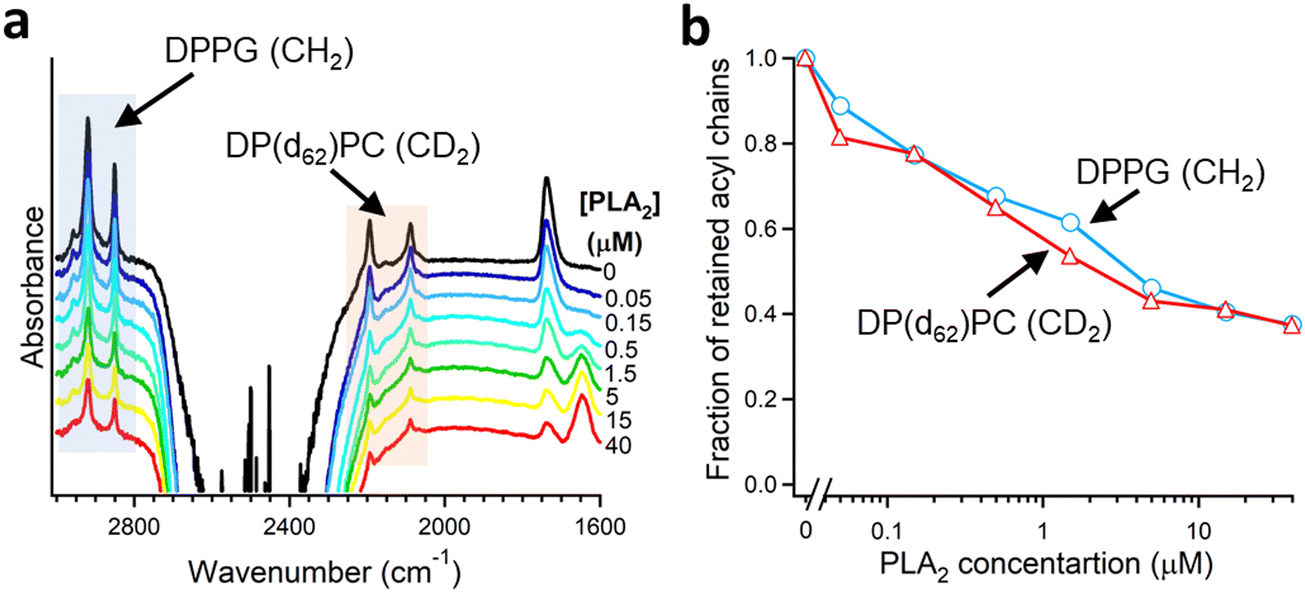 | ||
| Fig. 2 Hydrolysis of the two components of a binary lipid bilayer by PLA2. (a) ATR-FTIR spectra of a lipid bilayer composed of an equimolar combination of DPPG and chain-perdeuterated DP(d62)PC at different concentrations of PLA2, as indicated. (b) Dependence of the integrated methylene absorbance intensities of both lipids on PLA2 concentration. Adapted from ref. 19, with permission. | ||
PLA2 activity depends on the presence of the reaction products, i.e. the free fatty acid and the lysolipid, in the membrane. The membrane binding affinity as well as the burst activation of PLA2 following the initial lag period positively correlated with the presence of lipid hydrolysis products.100–103 Some studies suggested retention of the reaction products in the membrane100,101 while others demonstrated removal of a significant fraction of both products from the membrane upon hydrolysis by PLA2.97–99 Isotope-edited FTIR has been used to determine whether both products of lipid hydrolysis dissociate from the membrane after lipid hydrolysis to equal extents or one of them preferentially accumulates in the membrane and plays a prevalent role in PLA2 activity. PLA2 was applied to supported membranes of DPPC with perdeuterated sn-1 chain and unlabeled sn-2 chain (Fig. 3a). In this case, the >700 cm−1 separation of the methylene stretching vibrational signals provided spectral resolution of both acyl chains and thereby allowed monitoring the removal of each chain from the membrane upon lipid hydrolysis (Fig. 3b and c). As shown in Fig. 3d, while both CH2 and CD2 signals monotonously decrease with increasing PLA2 concentration, the reduction in the CD2 signal, which represents the lyso-phospholipid containing the sn-1 chain, prevails that of the CH2 signals, which represents the free fatty acid. This result implies that the free fatty acid preferentially accumulates in the membrane following hydrolysis by PLA2 and may modulate the enzyme's function by affecting the membranes surface charge and the structure.
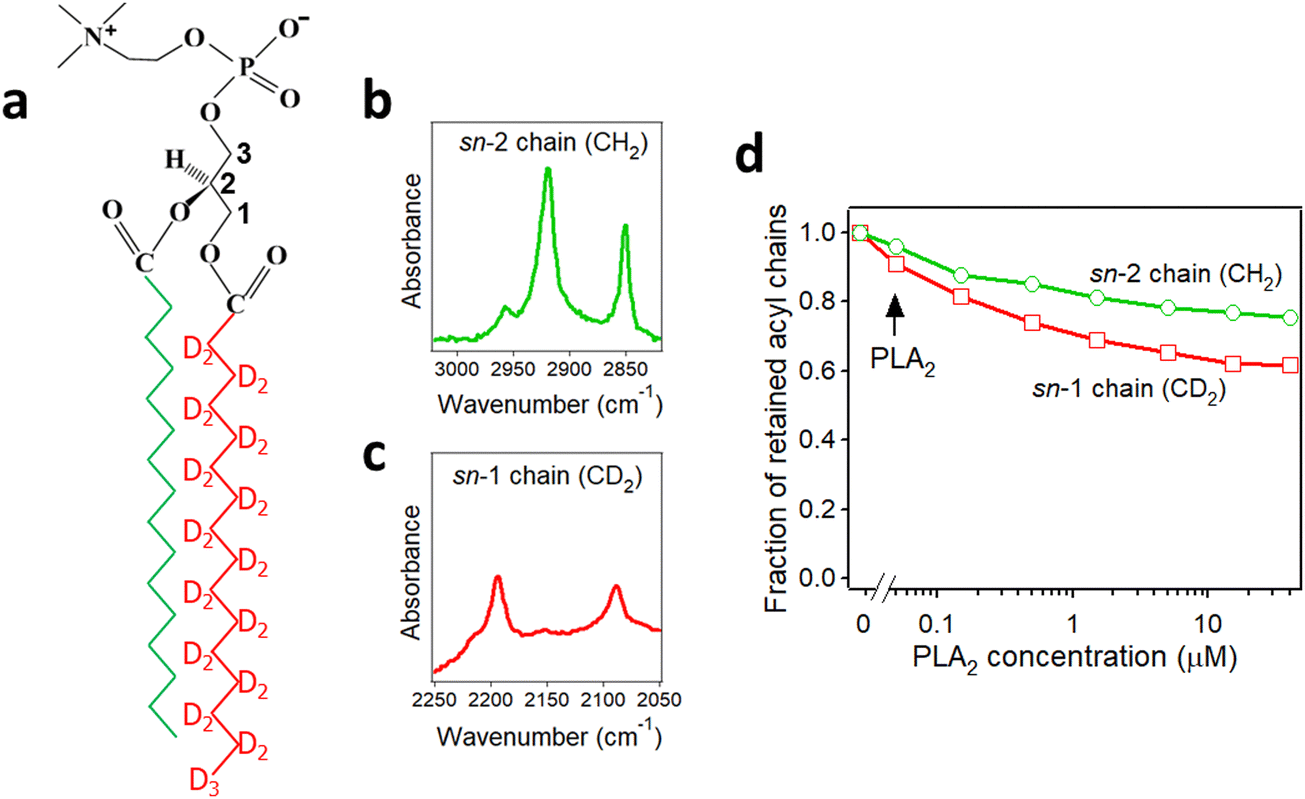 | ||
| Fig. 3 Hydrolysis of the acyl chains of DPPC with deuterated sn-1 chain and unlabeled sn-2 chain by PLA2. (a) Structure of DPPC with deuterated sn-1 chain (red) and unlabeled sn-2 chain (green). (b) and (c) The methylene stretching bands of the unlabeled (b) and deuterated (c) acyl chains of the lipid. (d) Dependence of the integrated methylene absorbance intensities of the unlabeled (green) and deuterated (red) acyl chains on PLA2 concentration. Adapted from ref. 19, with permission. | ||
3.3. Positioning of PLA2 with respect to the membrane
As mentioned above, interfacial enzymes are activated by binding to biomembranes and forming a protein–membrane complex of highly specific configuration, including the depth of membrane insertion and the geometric orientation of the protein with respect to the membrane. For enzymes that use a membrane lipid as substrate, such as various PLA2 isoforms, this specific configuration is a prerequisite for efficient enzyme function as it determines the strength of membrane binding and the mode of substrate accession and product release, i.e. the most important steps of the whole catalytic cycle.104 Determination of the positioning of a secreted PLA2 on a lipid membrane has been achieved by combination of protein engineering, isotope-edited FTIR, and fluorescence spectroscopy, as described below.As shown in Fig. 1, the orientation of a molecule that has a rotational axis of symmetry, such as a helix, can be determined by polarized ATR-FTIR spectroscopy. Briefly, the spectra obtained with infrared light polarized coplanar and orthogonal to the plane of incidence are used to determine the dichroic ratio, i.e. the ratio of absorbances at two polarizations. The measured dichroic ratio, together with known parameters such as the angle of the helical amide I transition dipole with respect to the helical axis (39° ± 1°), the Cartesian electric vector components of the evanescent wave, and the refractive indices of the IRE, the membrane, and the bathing solution, allow determination of the helical order parameter which directly provides the average orientation of the helix relative to the membrane (angle θ in Fig. 1).18,105 Determination of the orientation of a protein of irregular shape is not trivial, however, as the protein may have multiple helices of arbitrary orientations. It has been shown that determination of the orientation of the whole protein requires the orientations of at least two different helices.106 Human pancreatic PLA2 has been chosen to solve the problem because it is a relatively simple protein with three α-helices, an N-terminal helix (residues 1–10) and two internal helices. A semisynthetic, segmentally 13C-labeled PLA2 was produced where an unlabeled synthetic peptide corresponding to the N-terminal helix, with a thioester functional group at the C-terminus, was ligated to a uniformly 13C-labeled construct corresponding to the rest of the protein.106 The residue 11 (the first residue of the 13C-labeled construct) is a cysteine and is ideally located for thioester-to-cysteine peptide ligation (Fig. 4). Analysis of the protein structure showed that the two internal helices were nearly antiparallel, so, in terms of orientation, the protein could be treated as a two-helix system (in ATR-FTIR, parallel and antiparallel orientations are equivalent). ATR-FTIR spectra with coplanar and orthogonal polarizations of the incident light produced multicomponent amide I bands, with the components generated by the unlabeled N-terminal helix and the two 13C-labeled internal helices located at 1656 and 1616 cm−1, respectively (Fig. 5a and b). These data, along with specially developed analytic geometry algorithms, allowed determination of the orientations of the helices and hence of the whole protein relative to the membrane. The depth of membrane insertion was determined by experiments of quenching of the fluorescence of the single tryptophan-3 of pancreatic PLA2 by lipids brominated at various positions along the acyl chains. Isotope-edited polarized ATR-FTIR and fluorescence data thus produced the structure of the protein–membrane complex with a unique positioning of the protein on the membrane in terms of geometric configuration and depth of membrane insertion (Fig. 5c).106
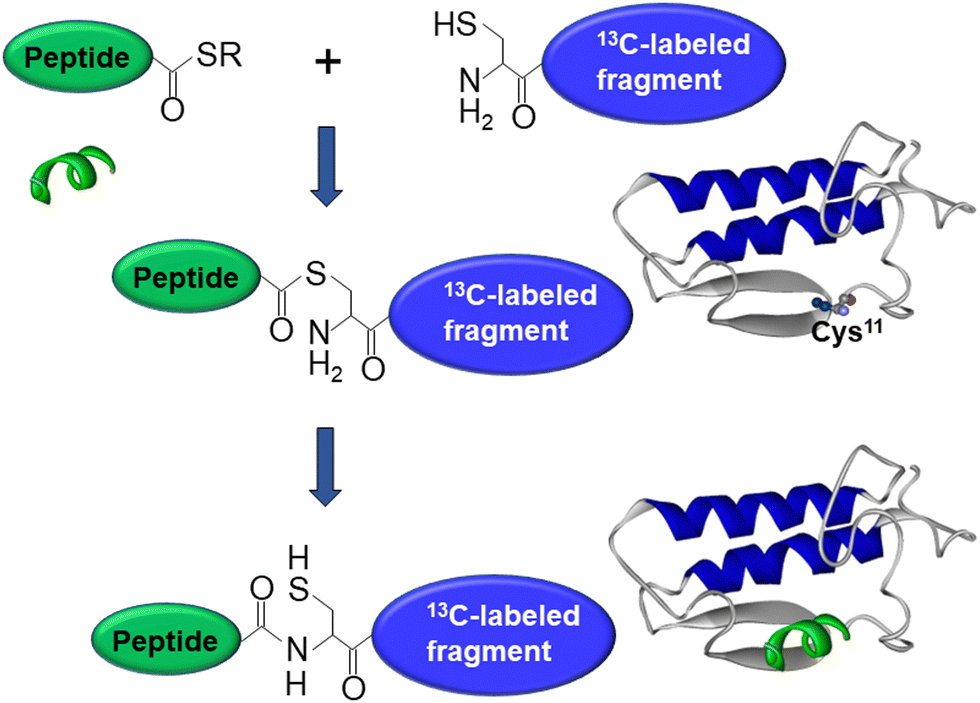 | ||
| Fig. 4 Graphical description of the thioester–cysteine chemical ligation. The synthetic unlabeled peptide (green) with a C-terminal thioester group reacts with the thiol of the N-terminal cysteine of a recombinant, uniformly 13C-labeled protein fragment (blue) followed by S-to-N acyl transfer and formation of a natural peptide bond, resulting in a semisynthetic, segmentally isotope-labeled protein. Adapted from ref. 94, with permission. | ||
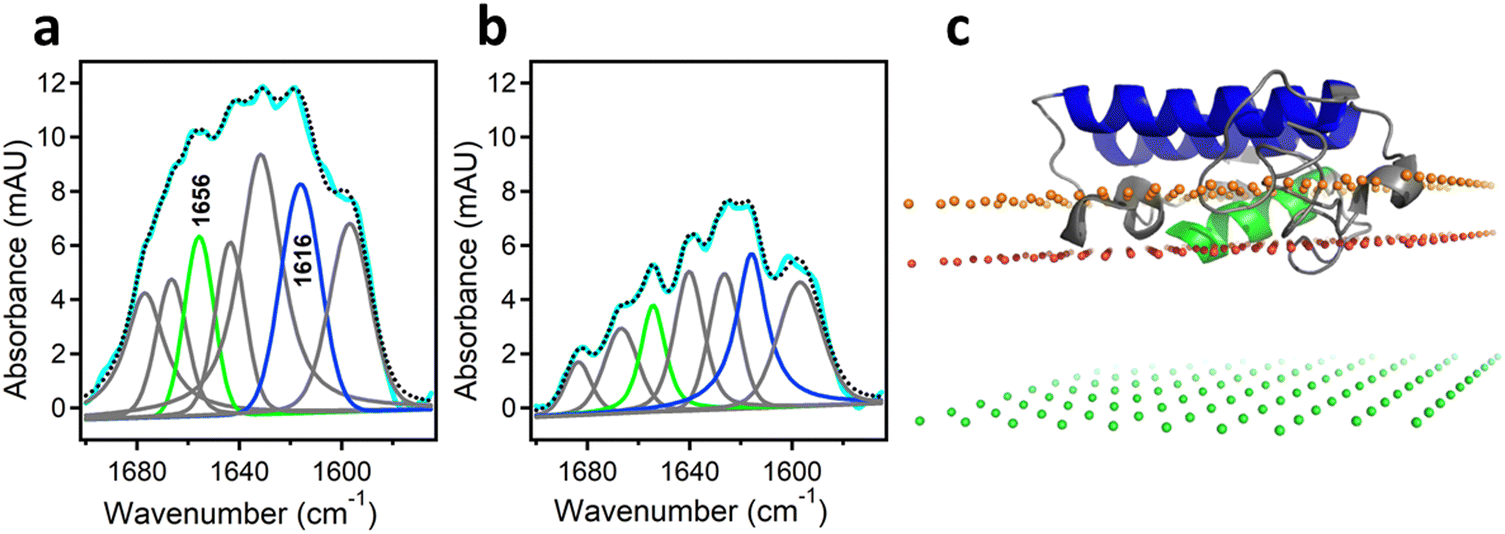 | ||
| Fig. 5 Determination of the orientation of a membrane-bound protein by polarized ATR-FTIR spectroscopy. (a) and (b) ATR-FTIR spectra of membrane-bound human pancreatic PLA2 with unlabeled N-terminal helix and 13C-labeled fragment at coplanar (a) and orthogonal (b) polarizations of the incident light with respect to the plane of incidence. The measured spectrum is shown in cyan and the sum of all amide I components is shown as dotted black line. The amide I components generated by the unlabeled and 13C-labeled helices are shown in green and blue, respectively. (c) The structure of the protein bound to the membrane with its N-terminal and internal helices shown in green and blue, respectively. The three planes indicate the locations of the membrane center (green), the sn-1 carbonyl oxygens (red) and the phosphate atoms (orange) of the lipid. Adapted from ref. 106, with permission. | ||
In this structure, the z coordinate of each atom of the protein is its distance from the membrane center so the location of each amino acid can be easily pinpointed. The protein was found to significantly penetrate into the membrane hydrocarbon core, with tryptophan-3 located at 9 ± 1 Å from the membrane center and providing a hydrophobic anchor for membrane docking of PLA2 together with phenylalanine-19 and leucine-20. Penetration of a cobra venom PLA2 into phosphatidylcholine bilayers 5–7 Å below the lipid headgroup plane has been reported,99 in good agreement with this finding. In addition to hydrophobic interactions, several cationic amino acid residues were involved in H-bonding with lipid carbonyl oxygens or in ionic interactions with lipid phosphate groups. Inspection of the structure shown in Fig. 5c indicates that the opening of the substrate-binding cleft faces the membrane surface so that the catalytic histidine-48 is located at ∼7 Å from the sn-2 ester group of the glycerophospholipid allowing the lipid to travel a shorter distance to reach the catalytic site compared to ∼15 Å proposed earlier.107,108 Thus, this technique, where isotope-edited FTIR plays a central role, provides an efficient tool to elucidate the mode of membrane binding of a peripheral protein and the mechanism of its action.
4. Protein–protein interactions
4.1. Overview and importance of protein–protein interactions
Protein–protein interactions play crucial roles in cell function by supporting vitally important biomolecular structures or processes. In fact, from 300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 to 650000 promiscuous protein–protein interactions are involved in human physiology.2 Protein assemblies can be divided into two classes, i.e., stable multiprotein structures that serve specific functions and transient complexes that are mostly involved in the dynamic events of cell signaling.1,3 Interactions between protein subunits are facilitated by a combination of nonspecific (hydrophobic, ionic, H-bonding) and specific (e.g., shape-complementary, hand-in-glove- or lock-and-key-type) contacts between domains and motifs that often contain critically important “hot spot” amino acids.1,3,81,109–111 As an antithesis of the lock-and-key interactions between (relatively) rigid domains, the “induced fit” or “folding-upon-binding” interactions between initially flexible protein modules such as antibodies or receptors or their ligands that acquire defined shape upon interaction result in promiscuity and polyreactivity effects of profound biological importance.3,112–116
000 to 650000 promiscuous protein–protein interactions are involved in human physiology.2 Protein assemblies can be divided into two classes, i.e., stable multiprotein structures that serve specific functions and transient complexes that are mostly involved in the dynamic events of cell signaling.1,3 Interactions between protein subunits are facilitated by a combination of nonspecific (hydrophobic, ionic, H-bonding) and specific (e.g., shape-complementary, hand-in-glove- or lock-and-key-type) contacts between domains and motifs that often contain critically important “hot spot” amino acids.1,3,81,109–111 As an antithesis of the lock-and-key interactions between (relatively) rigid domains, the “induced fit” or “folding-upon-binding” interactions between initially flexible protein modules such as antibodies or receptors or their ligands that acquire defined shape upon interaction result in promiscuity and polyreactivity effects of profound biological importance.3,112–116
The transient complexes between proteins are formed through highly specific interactions of moderate affinity (typically, nanomolar to micromolar dissociation constants).117–119 Important examples of transient protein–protein complexes are the enzyme–substrate,117–120 enzyme-inhibitor,121,122 ligand–receptor,123–125 and antigen–antibody interactions.3,111,126 More than 100 signaling proteins in human genome contain one or more Src homology 2 domain(s) that mediate binding to phosphorylated tyrosine involved in signal transduction.127 Other protein modules such as the PDZ domain, breast cancer susceptibility protein 1, forkhead-associated domain, mad homology domain 2, interferon regulatory factor 3, polo-box domain unique to the polo-like kinases, the FF, WW, and WD40 motifs containing the respective dipeptides mediate the transient binding of signaling molecules to phosphoserine or phosphothreonine.2,128 The 14-3-3 family proteins bind to and regulate around 1200 partners involved in a plethora of cellular functions and are regulated themselves by caspase-mediated proteolysis, lysine acetylation, or serine/threonine phosphorylation.2,129 The Src homology 3 (SH3) domain, on the other hand, binds proline-rich protein motifs that fit into a groove on the surface of the SH3 domain.130 Finally, regulatory (effector) proteins up- or down-regulate the activity of enzymes or switch from one to another function of multidomain modular proteins via interacting with an allosteric site.131
A special type of protein–protein interactions is aggregation of misfolded proteins and formation of amyloid fibrils that are involved in many pathologies. Some prominent examples are involvement of the amyloid β (Aβ) and tau proteins in Alzheimer's disease, α-synuclein in Parkinson's disease, prion protein in Creutzfeldt–Jakob disease, insulin in iatrogenic amyloidosis, immunoglobulin light chain and transthyretin in cardiac amyloidosis and polyneuropathy, islet amyloid polypeptide (amylin) in type II diabetes, hemoglobin in sickle cell anemia etc.132–141 Amyloidogenesis occurs with or without genetic, inheritable mutations in the respective protein, and the fibrils display remarkable structural similarities, such as formation of the cross-β-sheet structure, despite the absence of sequence similarities.137,140
The importance of protein–protein interactions in cell physiology has led the researchers to develop multiple methods to analyze protein complex formation and the structure and function of the complexes. An incomplete list of these methods includes the yeast two-hybrid system, the protein–fragment complementation assay, fluorescence resonance energy transfer, co-immunoprecipitation, tandem affinity purification coupled to mass spectrometry, (in-cell) cross-linking mass spectrometry, (in-cell) single-molecule tracking, stable isotope labeling by amino acids in cell culture, and computational (in silico) methods.110,142–149 Described in the following sections are the use of isotope-edited FTIR to analyze the structural aspects of Aβ fibrillogenesis and the regulation of sarcoplasmic reticulum (SR) Ca2+-ATPase by phospholamban (PLB).
4.2. Protein aggregation
The Aβ peptide aggregates constitute a histopathological hallmark of the Alzheimer's disease (AD).150 Aβ is derived from the amyloid precursor protein, a bitopic protein in neuronal membranes, and represents mostly its α-helical transmembrane part. Upon enzymatic cleavage and exposure to aqueous medium, the peptide undergoes transition to β-sheet structure followed by fibrillogenesis. Aβ occurs in various forms in human brain in terms of the number of amino acids and enzymatic modifications; in addition to the prevalent 40- and 42-residue species (Aβ1–40, Aβ1–42), N-terminally truncated and pyroglutamylated forms (AβpE3–40, AβpE3–42) are present as well and are hypertoxic.37 Since in human brain these peptides are intermixed and undergo aggregation into hetero-complexes, it is of interest to understand the structural changes in each species during aggregation and the mutual structural effects. Isotope-edited FTIR has been used to achieve this goal.The synthetic, lyophilized, uniformly 13C-labeled Aβ1–42 and unlabeled AβpE3–42 peptides were dissolved in hexafluoroisopropanol to disperse possible aggregates and were dried on a CaF2 FTIR window. These samples generated amide I spectra with peaks around 1617 cm−1 and 1658 cm−1 and amide II peaks around 1533 cm−1 and 1544 cm−1, respectively (Fig. 6a and c, black lines). In view of the ∼40 cm−1 downshift due to 13C-labeling, these data indicate α-helical structure for both peptides before hydration. To observe the kinetics of α-helix to β-sheet transition upon hydration as well as the intermediate oligomeric forms that are the main toxic species, the structural change was slowed down by pumping D2O-saturated nitrogen gas into the chamber of the spectrometer allowing measurements at distinct stages of the transition. Hydration with D2O vapor resulted in gradual shift of the α-helical peaks to 1588 cm−1 for 13C-Aβ1–42 and to 1632 cm−1 for AβpE3–42, indicating α-helix to β-sheet transition of both proteins (Fig. 6a and c). Concomitantly, the intensity in the initial (non-deuterated) amide II band declined because of the downshift of the deuterated amide II mode by ∼90 cm−1 (see Introduction). The kinetics of the HX, i.e. the time-dependence of amide II intensity, and of the α-helix-β-sheet transition, i.e. the ratio of absorbance intensities of the β-sheet and α-helical amide I signals, are shown in Fig. 6b and d. 13C-Aβ1–42 readily underwent amide HX and α-helix to β-sheet transition once exposed to D2O vapor (Fig. 6b) but the peptide was not entirely amide-deuterated after 5 hours of exposure to D2O vapor as indicated by the residual amide II intensity (red line in Fig. 6a and blue line in Fig. 6b), indicating partial solvent protection possibly due to a compact tertiary structure. AβpE3–42 behaved differently. Despite the significant decrease in the amide II intensity during the first 1.0–1.5 hours of deuteration, indicative of extensive HX, the peptide initially experienced little structural changes (Fig. 6d). Following the lag period, the peptide underwent α–β transition in a sigmoidal manner, yet a significant fraction of the initial α-helix as well as β-turn structure persisted after the transition leveled off (shoulder around 1677–1658 cm−1 of the red spectrum of Fig. 6c). In contrast to 13C-Aβ1–42, nearly all amide protons of AβpE3–42 exchanged for deuterium, as indicated by the reduction of the amide II signal to zero (Fig. 6c), suggesting a more open, solvent-exposed tertiary fold. This result is consistent with recently published fluorescence data showing solvent-inaccessibility of tyrosine-10 of Aβ1–42 and solvent exposure of same residue of AβpE3–42.151
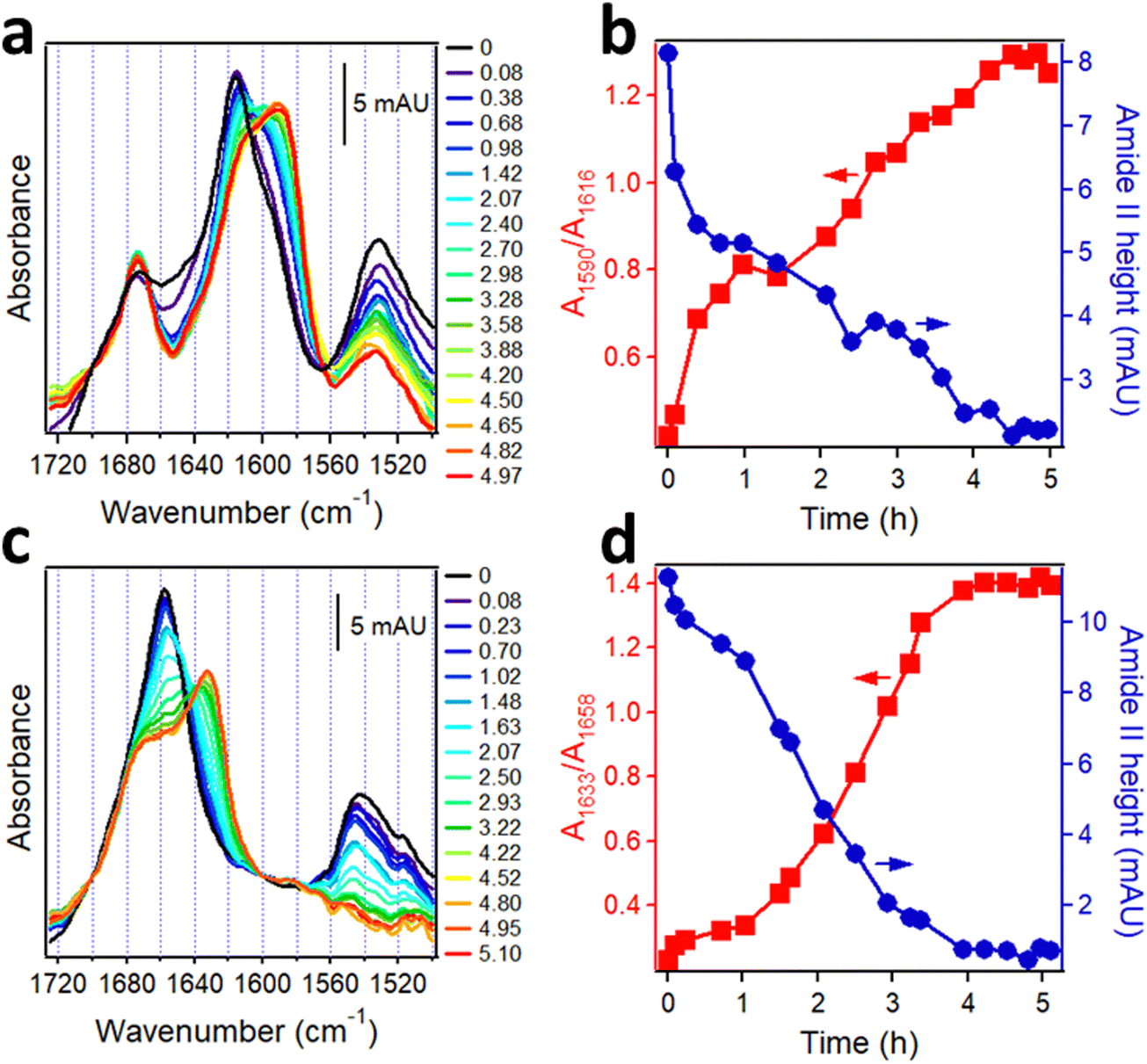 | ||
| Fig. 6 Structural changes in Aβ peptides upon hydration with D2O-saturated nitrogen gas. FTIR spectra of 13C-Aβ1–42 (a) and AβpE3–42 (c) dried on a CaF2 window (black lines) and upon injection of D2O-saturated nitrogen for ∼5 h (blue to red). Panels a and c are appended with columns indicating time of exposure to D2O vapor (in hours). Panels (b) and (d) show the kinetics of β-sheet structure formation in 13C-Aβ1–42 (b) and AβpE3–42 (d) (red) and change in amide II intensities (blue) as a function of time of exposure to D2O vapor. Adapted from ref. 37, with permission. | ||
Above data uncover distinct structural pathways of α-helix to β-sheet structural transitions of the unmodified and pyroglutamylated Aβ peptides, i.e., resistance of the latter to transition to β-sheet structure and a less compact tertiary fold. However, in brain tissue and in the extracellular plaques various forms of Aβ are blended and may affect each other's structural behavior. Such mutual structural effects have been analyzed as follows. AβpE3–42 and 13C-Aβ1–42 have been dissolved in hexafluoroisopropanol and deposited separately on the opposite sides of a CaF2 FTIR window as well as mixed at 1:1 molar ratio and deposited on the same side of another CaF2 window. These two samples were dried by desiccation and mounted on two mutually orthogonal vertical faces of a rotatable stage and placed in the spectrometer. Consecutive FTIR spectra of both separate and combined samples were measured alternately by turning the stage back and forth by 90 degrees about a 4-fold vertical axis while the chamber was purged with D2O-saturated nitrogen. The difference between these spectra at a given time point, i.e. at a given level of amide deuteration, then revealed the mutual structural effects.37
These samples generated complex spectra incorporating the features of both unlabeled and 13C-labeled peptides. In the isolated system, both 13C-Aβ1–42 and AβpE3–42 were α-helical in dry state indicated by the amide I peaks around 1618 cm−1 and 1658 cm−1, respectively (black line in Fig. 7a). The weaker amide I intensity of 13C-Aβ1–42 is consistent with observations of 20–30% lower amide I peak heights of 13C-labeled helical peptide segments compared to the unlabeled segments.30 Hydration by D2O vapor transformed the spectra and produced downshifted peaks at 1586 cm−1 and 1628 cm−1 with higher frequency shoulders located at 1597 cm−1 and 1639 cm−1 (red line in Fig. 7a). The strong signal at 1586 cm−1 and 1628 cm−1 is readily assigned to intermolecular β-sheet structure in 13C-Aβ1–42 and AβpE3–42, respectively, while the upshifted weaker signal at 1597 cm−1 and 1639 cm−1 indicates a smaller fraction of β-sheet with intramolecular H-bonding.18,152 An additional peak around 1673 cm−1 can be assigned to β-turn structure in AβpE3–42.
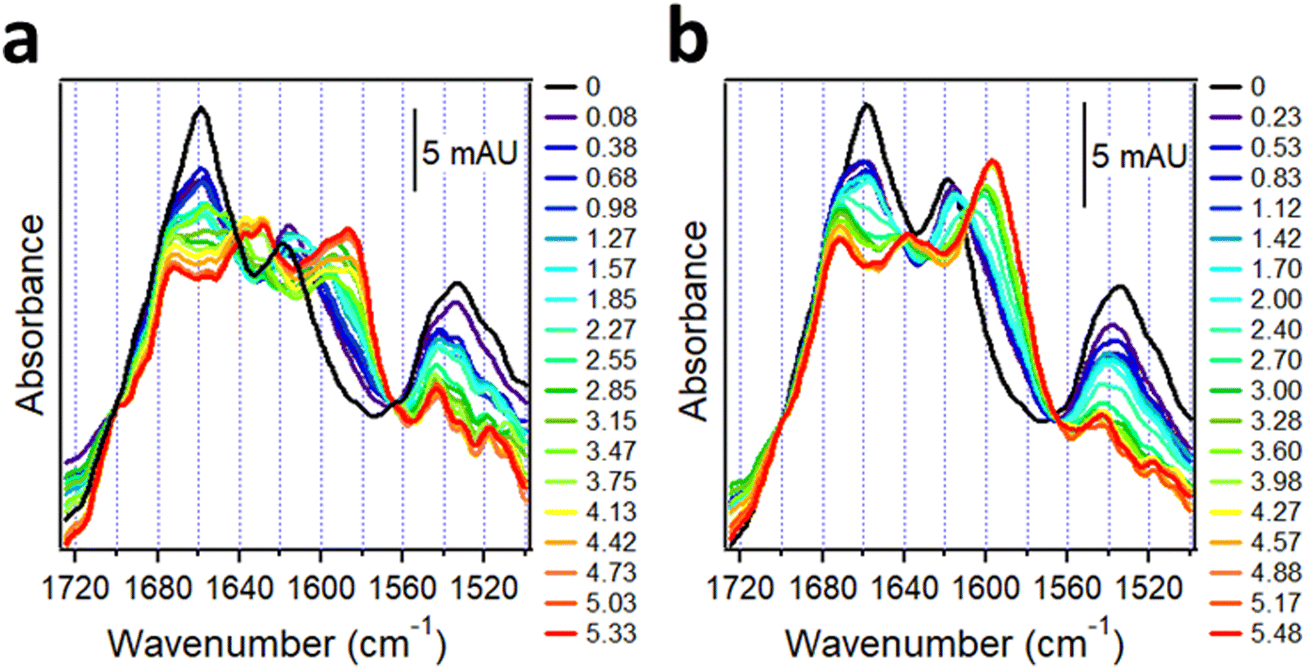 | ||
| Fig. 7 Structural changes in 13C-Aβ1–42 and AβpE3–42 upon hydration with D2O-saturated nitrogen gas when physically separated (a) and combined at 1:1 molar ratio (b). Both panels are appended with columns indicating time of exposure to D2O vapor (in hours). Reproduced from ref. 37, with permission. | ||
Spectra of the combined 13C-Aβ1–42/AβpE3–42 sample hydrated by D2O vapor displayed a stronger higher frequency signal assigned to intramolecular H-bonded β-sheet and a weaker lower frequency, intermolecular H-bonded β-sheet signal (red line in Fig. 7b). In fact, the signal at 1586 cm−1 assigned to the intermolecular β-sheet of 13C-Aβ1–42 was not present in the combined peptide sample at all. These spectral effects can be amplified by resolution enhancement techniques. The second derivative spectra shown in Fig. 8 identify strong signals of the isolated sample located at 1584 cm−1 and 1627 cm−1 and weaker signal around 1597 cm−1 and 1537 cm−1 (blue line of Fig. 8) signifying larger fractions of aggregates with intermolecular H-bonding for both 13C-Aβ1–42 and AβpE3–42. In the spectrum of the combined sample, the relative intensities of these signals were reversed (red line of Fig. 8), indicating conversion of the intermolecular H-bonded aggregates to β-sheet structures stabilized by intramolecular H-bonding, as schematically shown above the respective spectral features in Fig. 8. These data identify mutual structural effects of the unmodified and pyroglutamylated Aβ peptides, i.e., diversion of the aggregation process from fibril formation stabilized by intermolecular H-bonding to oligomeric structures of hairpin-like β-sheets that may exert a stronger cytotoxic effect as the oligomers are more toxic than the fibrils.37,141,153–155 This is an example of how isotope-edited FTIR spectroscopy can provide insight into the molecular mechanism of a disease such as AD.
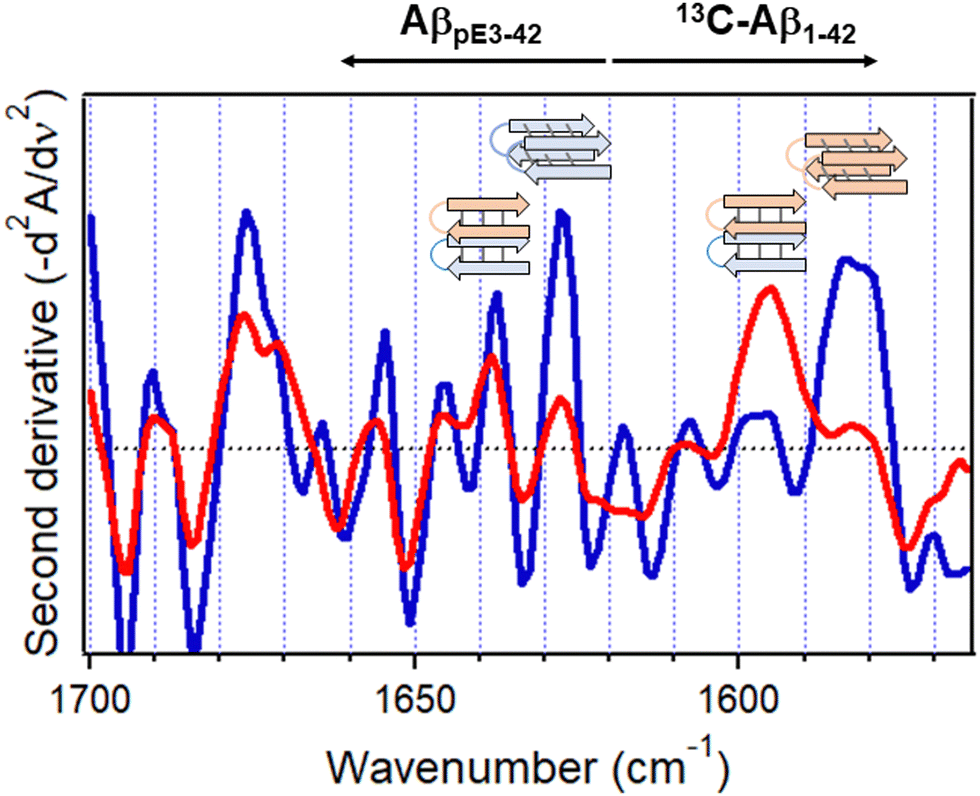 | ||
| Fig. 8 Inverted second derivative amide I spectra of 13C-Aβ1–42 and AβpE3–42 after 5 h hydration with D2O-saturated nitrogen gas when physically separated (blue) and combined at 1:1 molar ratio (red). Cartoons for homo- or hetero-oligomers stabilized by intermolecular and intramolecular H-bonding, respectively, are shown above respective spectral features (13C-Aβ1–42, rose, and AβpE3–42, blue). Reproduced from ref. 37, with permission. | ||
4.3. In-membrane protein–protein interactions
Described in this section is the use of isotope-edited FTIR spectroscopy to analyze the regulatory interaction between two membrane proteins, i.e., regulation of the activity of SR Ca2+-ATPase by PLB that plays a central role in the contraction–relaxation cycle of heart muscle.156 Excitation of the cardiomyocyte triggers Ca2+ influx through the voltage-sensitive Ca2+ channels, which in turn activates the type-2 ryanodine receptor Ca2+ channels in the SR membrane resulting in additional increase in the cytosolic Ca2+ level and contraction by the Ca2+-sensitive contractile proteins. This is followed by relaxation that takes place mainly by pumping the cytosolic Ca2+ back into the SR lumen by the SR Ca2+-ATPase. The activity of the Ca2+-ATPase is regulated by PLB, a relatively small bitopic protein in the SR membrane, as schematically shown in Fig. 9a. Specifically, PLB moderates the enzymatic activity of the Ca2+-ATPase and phosphorylation of PLB by cAMP- and/or Ca2+/calmodulin-dependent protein kinases ceases the inhibitory effect.157–159 Various forms of cardiomyopathy in humans are associated with reduction of the levels of both the SR Ca2+-ATPase and PLB at mRNA and protein levels.156 The regulatory effect of PLB has been shown to involve direct molecular contact between the ATPase and PLB and several intermolecular interaction sites have been identified but the mechanism of regulation remains unclear.158–160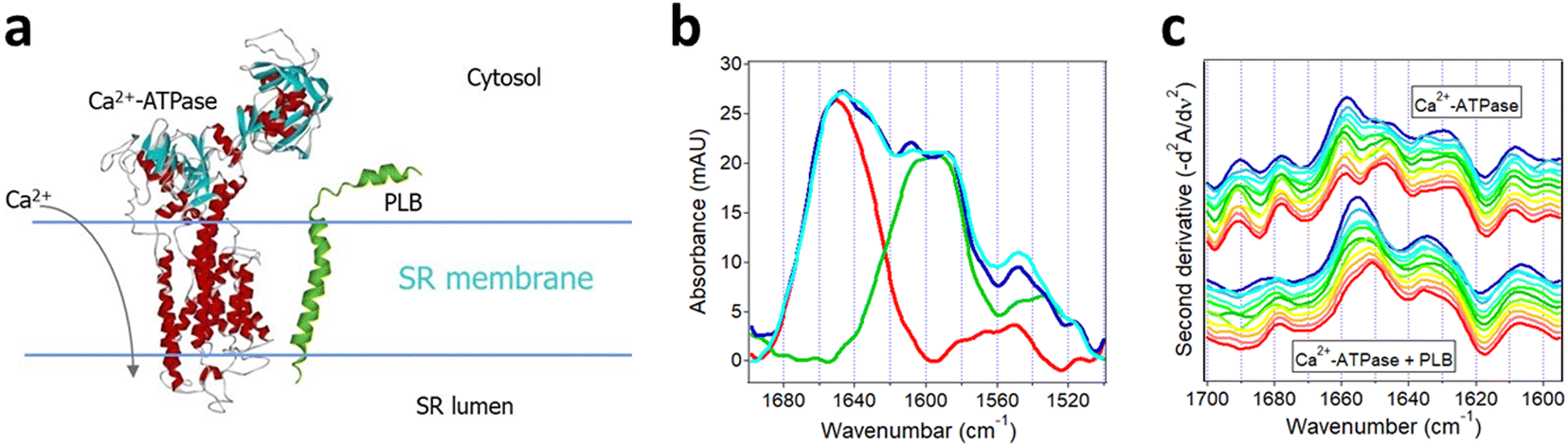 | ||
| Fig. 9 Structural effects resulting from interaction of SR Ca2+-ATPase with uniformly 13C-labeled PLB. (a) Cartoon for the Ca2+-ATPase and PLB in the SR membrane. (b) ATR-FTIR spectra of membrane-reconstituted Ca2+-ATPase (red), uniformly 13C-labeled PLB (green), the sum of the spectra of Ca2+-ATPase and 13C-PLB measured separately (blue), and the spectrum of the 1:11 molar combination of Ca2+-ATPase and 13C-PLB (cyan). (c) Inverted second derivative spectra of Ca2+-ATPase alone (upper group of curves) and with 13C-PLB at 11-fold molar excess (lower group of curves). Time of exposure to D2O-based buffer was 2 min, 4 min, 9 min, 19 min, 36 min, 1 h, 1.4 h, 3 h, 4.4 h, 6 h, and 8 h (blue to red). Partially adopted from ref. 157, with permission. | ||
Since PLB exerts similar regulatory effects on both the cardiac and skeletal muscle SR Ca2+-ATPases and the latter maintains higher functional stability following purification, ATR-FTIR studies have been carried out on recombinant, uniformly 13C-labeled PLB and skeletal muscle SR Ca2+-ATPases co-reconstituted in supported membranes composed of SR-extracted lipids.157 First, spectra of each membrane-embedded protein were measured separately. The Ca2+-ATPase generated amide I and amide II bands centered at 1658 cm−1 and 1546 cm−1, respectively (red line in Fig. 9b). The strong signal at 1658 cm−1 is consistent with the mostly α-helical secondary structure of the Ca2+-ATPase (Fig. 9a). 13C-PLB produced a wide amide I band composed of two components at 1608 cm−1 and 1585 cm−1 indicating α-helical and β-sheet structures, in accord with earlier FTIR studies,161 and an amide II band at 1525 cm−1 (green line in Fig. 9b). The 50 cm−1 spectral shift between the α-helical amide I modes of the two proteins is consistent with the expected isotopic effect.
The spectrum of the Ca2+-ATPase and 13C-PLB co-reconstituted at 11:1 PLB:ATPase molar ratio displayed the structural features of both proteins (cyan line in Fig. 9b). A resolution enhancement procedure, i.e. second derivative spectral analysis in the amide I region of Ca2+-ATPase, was conducted to discern structural effects exerted by PLB. Time-dependent changes in the second derivative spectra over an 8 hour period unveiled interesting effects. In the initial stages of exposure of the proteins to D2O, a strong component at 1658 cm−1 and weaker one at 1647 cm−1 were present in the spectrum of the Ca2+-ATPase (Fig. 9c, upper family of curves), which can be readily assigned to non-deuterated and deuterated α-helices.18 By 8 hours of exposure to D2O, the relative intensities of these components switched toward a larger fraction of the deuterated helices. The spectral features in the 1637–1625 cm−1 and 1700–1670 cm−1 regions represent β-sheet and turn structures while those below 1610 cm−1 are assigned to side chains. In the presence of PLB, the α-helical signal at 1658 cm−1 was downshifted by 3–4 cm−1 and its further downshift due to deuteration occurred much slower (Fig. 9c, lower family of curves). Both effects suggest stabilization of the α-helices of Ca2+-ATPase upon interaction with PLB. More stable helices involve stronger intramolecular H-bonding and hence weaker CO covalent bonds, corresponding to lower vibrational frequency. The stronger CO⋯H–N H-bonding, on the other hand, will slow down the amide deuteration, as detected. In addition, the relative intensities of non-deuterated vs. deuterated β-sheets of the Ca2+-ATPase (signals around 1637 cm−1 and 1625 cm−1) was shifted toward the former in the presence of PLB, in agreement with overall slower HX. Finally, the more stable structure and less effective HX of the Ca2+-ATPase in the presence of PLB was confirmed by a stronger amide II band of the Ca2+-ATPase around 1546 cm−1 as compared to that in the spectral sum of the two proteins measured separately (cf. cyan and blue lines of Fig. 9b). The data thus indicate stabilization of the secondary structure of the Ca2+-ATPase by PLB with no significant structural changes in PLB itself. These findings offer a mechanism for the inhibitory action of PLB on Ca2+-ATPase through increased rigidity of the ATPase structure by PLB and hindrance of the conformational changes involved in the catalytic turnover.
5. Site-specific structure determination
5.1. Isotope labeling schemes
Data described above indicate the remarkable sensitivity of FTIR spectroscopy to the structural dynamics of proteins. However, FTIR usually provides the global rather than local structural information and hence is considered a “low-resolution” technique. Local, site-specific structure of proteins can be determined by isotope-edited FTIR spectroscopy using proteins that are isotopically labeled at selected amino acid residues. Since the conformation-sensitive amide I mode is produced mostly by the main chain CO stretching vibration (76%) as well as by the C–N stretching, C–C–N deformation, and N–H in-pane bending modes,18 the most common labeling schemes include carbonyl 13C labeling, double 13C,15N-labeling, or double 13C18O-labeling of main chain atoms, which result in significant spectral shifts and resolution of the targeted site(s), as described in Section 2.2.
The incorporation of the labeled amino acids can be achieved by various methods, such as chemical synthesis162–165 or recombinant expression in an E. coli strain that is auxotrophic for a certain amino acid which is added into the growth medium in isotope-labeled form.166–168 For example, functionally active insulin has been produced by total chemical synthesis 13C18O-labeled at a selected residue.164 The synthetic method works so long as the synthesis of the whole sequence is practicable, and with the advent of the automated fast-flow peptide synthesis technology the limit of the number of amino acids in the synthetic protein has been pushed beyond 200.169,170 The use of E. coli auxotrophs is a facile strategy as currently around 220 BL21(DE3)-based strains are available allowing production of proteins incorporating various combinations of isotopically labeled amino acids.168 Native chemical ligation of synthetic or recombinantly expressed proteins is another strategy to generate relatively long protein chains incorporating isotopically labeled segments or individual amino acids; an example of a semisynthetic, segmentally isotope-labeled fully functional PLA2 obtained by this method is described above (Section 3.3). FTIR analysis of the lens protein γD-crystallin that was produced by thioester/cysteine chemical ligation of the unlabeled N-terminal domain with the 13C-labeled C-terminal domain allowed elucidation of the molecular mechanism of fibril formation.171 Transmembrane proteins, such as the cation-specific ion channel p7 of hepatitis C virus (63 amino acid residues), have been produced by thioester/cysteine or hydrazide-based chemical ligation of the p7(1–26) and p7(27–63) chains that were selectively labeled with 13C or 15N for FTIR and NMR studies.172,173 Other methods of incorporation of labeled amino acids in single or multiple sites, such as the use of modified tRNA to achieve nonsense suppression, are described below (Section 6.2).
5.2. Local and global structure of Aβ by segmental isotope labeling
The structure of the oligomers formed by the Aβ1–42 peptide and its truncated/pyroglutamylated counterpart, AβpE3–42, has been studied using synthetic peptides harboring uniformly 13C,15N-labeled amino acids (Cambridge Isotope Laboratories). The labeled amino acids should constitute at least ∼5–10% of the total number of residues to (a) produce a readily distinguishable amide I signal and (b) generate a normal mode vibration via inter-residue couplings that can be used for site-specific structure assessment. Aβ1–42 and AβpE3–42 were labeled at three or four consecutive amino acid residues, i.e. the K16L17V18 or V36G37G38V39 segments that play an important role in initiation of Aβ aggregation174,175 and were studied separately as well as combined at an equimolar ratio to gain insight into the structural pathways of fibrillogenesis. All peptide samples, incubated in aqueous buffer for 1 hour, featured a major amide I peak located between 1635 cm−1 and 1630 cm−1 that constituted 50 ± 5% of the total amide I band area (Fig. 10). These amide I wavenumbers indicate formation of oligomers of peptides in β-sheet structure with intramolecular H-bonding as the cross-β sheet fibrils generate amide I signal below 1630 cm−1.50,56–58,176 Smaller fractions of turn structure (1677–1672 cm−1), α-helix (1657–1652 cm−1), and in some cases irregular (unordered) structure (∼1644 cm−1) were present as well.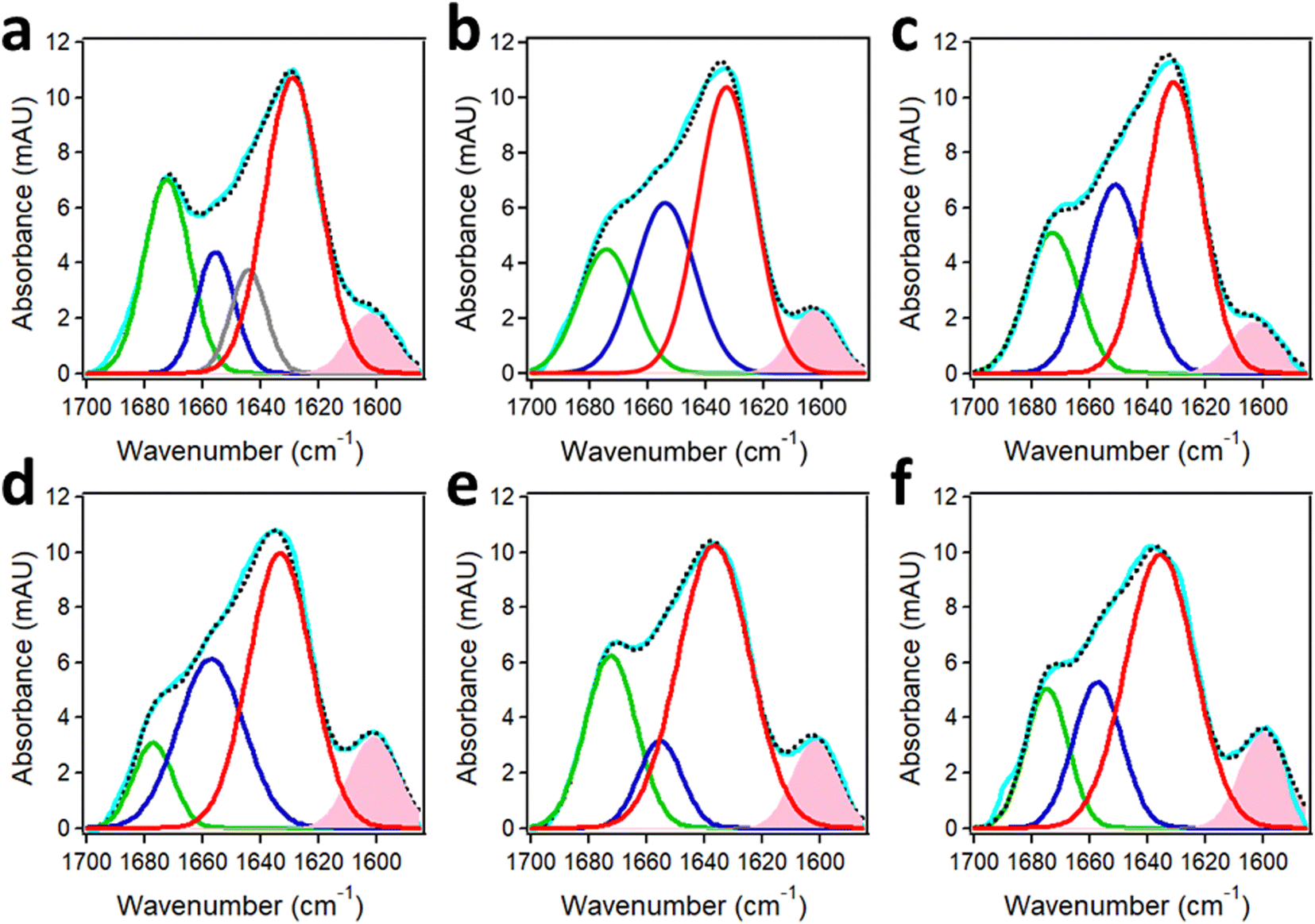 | ||
| Fig. 10 Structures of Aβ1–42 and AβpE3–42 from FTIR spectra of segmentally 13C,15N-labeled peptides. Peak-fitted FTIR spectra of K16L17V18-labeled Aβ1–42 (a), K16L17V18-labeled AβpE3–42 (b), their 1:1 combination (c), V36G37G38V39-labeled Aβ1–42 (d), V36G37G38V39-labeled AβpE3–42 (e), and their 1:1 combination (f) incubated in 50 mM NaCl + 50 mM Na,K-phosphate buffer (pD 7.2) for 1 hour. Color code: measured spectrum is cyan; the turn, α-helix, irregular, and β-sheet components are shown in green, blue, gray, and red lines, respectively. The component generated by the 13C,15N-labeled segment is shaded pink. The sum of all components is shown in black dotted line. Total peptide concentration was 50 μM. Adapted from ref. 44, with permission. | ||
The spectral component generated by the 13C,15N-labeled stretches K16L17V18 and V36G37G38V39 were clearly manifested at 1603–1602 cm−1 and 1601–1600 cm−1 and constituted around 7% and 10% of the amide I area, respectively. These spectral features can be used to determine the mode of peptide aggregation. As indicated in Section 2.2, 13CO/13CO vibrational dipolar coupling, which would occur in case of in-register cross-β-sheet formation, generates lower frequency signals (1594–1591 cm−1) whereas the 13CO/12CO coupling produces signal in the 1604–1601 cm−1 range.46,47,50,51 This implies that the labeled stretches are involved in β-sheet structure as well, thus increasing to total β-sheet to around 60%. More importantly, none of the isotopically labeled segments, i.e. the 16–18 or the 36–39 stretches, are involved in coupling with labeled segments, excluding the parallel in-register β-sheet formation. Instead, both peptides form β-hairpin-like structures upstream of residue 16 stabilized by intramolecular H-bonding. The α-helix is most likely located at the N-terminus and the turn is most likely between the two β-strands.44 The aggregation occurs via non-H-bonding interactions between these hairpin-like structures, which can exert the cytotoxic effect through various mechanisms including membrane insertion and permeabilization.151,177
6. Conclusions and outlook
6.1. Strengths and limitations
Isotope-edited FTIR spectroscopy offers a variety of options to analyze protein–protein and protein–membrane interactions. One of the strengths of this approach is that isotope labeling, i.e. addition of one or two neutrons in the nucleus of an atom, preserves the chemical (and most of physical) properties of the amino acid as opposed to labeling with relatively bulky exogenous molecular probes such as fluorophores or paramagnetic agents often used in fluorescence, EPR, or NMR spectroscopy. Interactions between two proteins and the accompanying conformational changes in each protein can be probed by studies on binary samples composed of an unlabeled and a uniformly 13C or 13C,15N-labeled protein.20,43,157,178 Domain-specific, segmental, or single amino acid labeling of proteins with 13C, 13C,15N, or 13C18O allows for amide I spectral resolution and structural characterization of the labeled site or stretch.21–23,41,162–164,166,171,179 Spectral shifts in the amide II band provide an opportunity to assess the rates of the amide HX and to gain information about the solvent accessibility and the structural dynamics of the labeled and unlabeled parts of the protein. In addition, vibrational coupling effects between amide oscillators of distinct isotope content provide information on the packing of the labeled and unlabeled segments relative to each other and hence on the overall tertiary fold of the protein.44,48,49,57,58 The sensitivity of the vibrational frequency to the strength of the involved covalent bonds, and consequently to the strength of H-bonding, permits distinguishing between very similar secondary structures, such as αI- and αII-helices, which is difficult to accomplish by other methods.180 Polarized ATR-FTIR studies on segmentally labeled proteins reconstituted in lipid bilayers or multilayers allow determination of the orientation of the labeled segment relative to the membrane, thereby providing clues to the mode of its interaction with the membrane.106
Studies on isotope-labeled membrane lipids produce useful information on membrane structure. FTIR studies on glycerophospholipids 13C-labeled at sn-1 or sn-2 ester carbons yielded information on the hydration properties of the membrane surface and the orientations of the carbonyl and other chemical groups of the lipid headgroup, as well as changes of these parameters during thermal phase transitions and interdigitated structure formation.72–74 Data on membranes composed of selectively deuterated lipids at the sn-1 or sn-2 hydrocarbon chains can be used to determine the details of the arrangement and packing of lipids in a bilayer structure.74 As shown in Section 3.2, ATR-FTIR studies on supported membranes composed of an unlabeled and a chain-deuterated lipid can identify which component is preferentially hydrolyzed by PLA2.19 On the other hand, selective deuteration of one of the chains allows deciphering which reaction product (fatty acid or lyso-phospholipid) is mostly removed from the membrane upon hydrolysis by PLA2.19
Despite these advantages, isotope-edited FTIR spectroscopy has its limitations. One inherent issue is the spectral overlap between the amide I signals generated by the unlabeled and 13C or 13C,15N-labeled chains. 13C18O labeling results in additional downshift of the amide I signal and thereby may offer a better spectral resolution (see Section 2.2). Single amino acid labeling may be problematic for several reasons: (a) technical difficulties of incorporation of a labeled amino acid in large proteins, (b) too weak signal compared to the whole amide I band, and (c) uncertainty regarding the secondary structure based on a local amide I mode generated by one amino acid. Solutions to (a) are described in the next section that describes nonsense suppression strategies. Even when the amide I signal of a single amino acid is reliably resolved, its spectral shift and its relation to the local secondary structure is not straightforward because the secondary structure is determined not by the frequency of an isolated amide oscillator (local mode) but by vibrational coupling with neighboring oscillators (normal mode).21,57,58 The 12C/13C vibrational coupling occurs in β-sheet but not in α-helix conformation. Therefore, the signal generated by a single 13C-labeled amino acid will be diagnostic for β-sheet structure but cannot be used to characterize α-helix structure or orientation.46,181 Production of proteins isotopically labeled at single or multiple sites by the synthetic method or via nonsense suppression may present useful solutions to such problems, as discussed in the next section.
6.2. Future directions
Coupling of FTIR spectroscopy with atomic force microscopy (AFM-IR) is a powerful technique that provides spatial resolution of ≤10 nm and thereby allows secondary structural analysis of protein samples or segments of distinct morphologies as opposed to the averaged FTIR spectra. Formation of prefibrillar oligomers and mature fibrils of the Aβ peptide,182,183 α-synuclein,184 or transthyretin185 during the process of aggregation have been resolved by this method. Through improvements of the stability, sensitivity, and accuracy of AFM-IR, single protein molecule resolution has been achieved.186 This technique has been used to analyze the response of cancer cells to drugs at single cell level and at nanoscale resolution.187 Further developments in this area may involve the combination of isotope-edited protein samples with AFM-IR. For example, the molecular compositions of protein aggregates or the structural transitions in individual proteins during functional interactions can be resolved at single molecule spatial resolution.Isotopic labeling of selected domains of proteins using techniques such as native chemical ligation represents an attractive strategy to discern domain-specific structural changes during functional turnover.171 For proteins composed of disulfide bridged domains, such as the insulin receptor family proteins, the protein can be produced in unlabeled and uniformly 13C-labeled forms followed by disulfide reduction and re-association of the unlabeled hormone-binding α-subunit with the 13C-labeled tyrosine-kinase β-subunit.188 Various cell-based or cell-free expression systems for functional mammalian proteins are available that are suitable for post-translational modifications such as glycosylation. These include HEK293 cells, insect cells, wheat germ cells and cell-free systems such as rabbit reticulocyte of HeLa cell extracts,189,190 which can be supplemented with isotopically labeled amino acids or labeled nutrients such as 13C6-D-glucose. ATR-FTIR studies on the hormone-activation of a domain-specific isotope labeled membrane-reconstituted receptor will reveal the dynamic conformational changes in each domain reflecting the transmission of the hormone signal across the membrane. The peptide hormone itself may be produced in 13C18O isotopic form to move its amide I signal further away towards lower frequencies.164
Isotope labeling of one or several amino acids in a protein is crucial for obtaining site-specific structural information. A rapidly developing protein engineering strategy is the use of a genetically modified cell or cell-free expression system containing a stop (nonsense, termination) codon in a given gene, such as TAG (amber), at the site of interest and a nonsense suppressor tRNA aminoacylated with a labeled amino acid and harboring an anticodon complementary to the stop codon (CUA, in this case).191–194 The selected stop codon as well as the suppressor tRNA/amino acyl tRNA synthetase (aaRS) pair should be orthogonal (heterologous) to the host organism or the cell-free system to prevent cross-reaction. In the semisynthetic method, the tRNACUA lacking the last two nucleotides is prepared by in vitro transcription with T7 RNA polymerase and the synthetic, labeled amino acid dinucleotide is appended to the 3′ end with the T4 RNA ligase.192,195 A similar method uses flexible tRNA acylation ribozymes coupled with a translation system containing the activated, labeled or otherwise modified amino acid.196,197 More than one labeled amino acids can be incorporated in a protein by using other stop codons such as TAA (ochre) or TGA (opal) coupled with respective suppressor tRNAs, i.e. tRNAUUA or tRNAUCA provided the expression system is using a third stop codon.198,199 Artificial, such as quadruplet codons can be introduced alone or in tandem with an orthogonal stop codon resulting in double nonsense/frameshift suppression and incorporation of two labeled or unnatural amino acids allowing for simultaneous targeting of two sites in a protein.191,193,200 Three or four noncanonical amino acids have been incorporated in one protein chain by using all three natural stop codons194 or by engineering four suppressor tRNA/aaRS pairs targeting four frameshift quadruplet codons.201 Future efforts will likely expand the scope of protein engineering allowing facile production of proteins labeled at multiple sites through orthogonal tRNA/aaRS pairs or chemically aminoacylated tRNAs that work efficiently in mammalian expression systems, which will enhance the ability of isotope-edited FTIR spectroscopy to gain higher resolution structural insight into the function of important proteins.
Abbreviations
| aaRS | Amino acyl tRNA synthetase |
| Aβ | Amyloid β |
| AD | Alzheimer's disease |
| AFM | Atomic force microscopy |
| ATR | Attenuated total reflection |
| FTIR | Fourier transform infrared |
| DPPC | Dipalmitoylphosphatidylcholine |
| DPPG | Dipalmitoylphosphatidylglycerol |
| HO | Harmonic oscillator |
| HX | Hydrogen–deuterium exchange |
| IBS | Interfacial binding site |
| IR | Infrared |
| IRE | Internal reflection element |
| PLA2 | Phospholipase A2 |
| PLB | Phospholamban |
| SR | Sarcoplasmic reticulum |
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The author declares no conflict of interest.Acknowledgements
This work has been supported, in part, by Florida Department of Health, Ed and Ethel Moore Alzheimer's Disease Research Program (grant 21A06).References
- C. Corbi-Verge and P. M. Kim, Motif mediated protein–protein interactions as drug targets, Cell Commun. Signaling, 2016, 14, 8 CrossRef PubMed.
- N. N. Sluchanko and D. M. Bustos, Intrinsic disorder associated with 14-3-3 proteins and their partners, Prog. Mol. Biol. Transl. Sci., 2019, 166, 19–61 CrossRef CAS PubMed.
- A. B. Kapingidza, K. Kowal and M. Chruszcz, Antigen-Antibody Complexes, Subcell. Biochem., 2020, 94, 465–497 CrossRef CAS PubMed.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Small molecules, big targets: drug discovery faces the protein–protein interaction challenge, Nat. Rev. Drug Discovery, 2016, 15(8), 533–550 CrossRef CAS PubMed.
- D. Wu, Y. Li, L. Zheng, H. Xiao, L. Ouyang, G. Wang and Q. Sun, Small molecules targeting protein–protein interactions for cancer therapy, Acta Pharm. Sin. B, 2023, 13(10), 4060–4088 CrossRef CAS PubMed.
- A. Murabito, J. Bhatt and A. Ghigo, It Takes Two to Tango! Protein–Protein Interactions behind cAMP-Mediated CFTR Regulation, Int. J. Mol. Sci., 2023, 24(13), 10538 CrossRef CAS PubMed.
- M. Overduin and T. A. Kervin, The phosphoinositide code is read by a plethora of protein domains, Expert Rev. Proteomics, 2021, 18(7), 483–502 CrossRef CAS PubMed.
- M. Overduin, T. A. Kervin, Z. Klarenbach, T. R. C. Adra and R. K. Bhat, Comprehensive classification of proteins based on structures that engage lipids by COMPOSEL, Biophys. Chem., 2023, 295, 106971 CrossRef CAS PubMed.
- T. Tubiana, I. Sillitoe, C. Orengo and N. Reuter, Dissecting peripheral protein-membrane interfaces, PLoS Comput. Biol., 2022, 18(12), e1010346 CrossRef CAS PubMed.
- M. Guaita, S. C. Watters and S. Loerch, Recent advances and current trends in cryo-electron microscopy, Curr. Opin. Struct. Biol., 2022, 77, 102484 CrossRef CAS PubMed.
- C. Berger, N. Premaraj, R. B. G. Ravelli, K. Knoops, C. López-Iglesias and P. J. Peters, Cryo-electron tomography on focused ion beam lamellae transforms structural cell biology, Nat. Methods, 2023, 20(4), 499–511 CrossRef CAS PubMed.
- J. H. Viles, Imaging amyloid-β membrane interactions: Ion-channel pores and lipid-bilayer permeability in Alzheimer's disease, Angew. Chem., Int. Ed., 2023, 62(25), e202215785 CrossRef CAS PubMed.
- E. M. Irvin and H. Wang, Single-molecule imaging of genome maintenance proteins encountering specific DNA sequences and structures, DNA Repair, 2023, 128, 103528 CrossRef CAS PubMed.
- S. P. S. Deopa and S. Patil, Viscoelasticity of single folded proteins using dynamic atomic force microscopy, Soft Matter, 2023, 19(23), 4188–4203 RSC.
- D. J. Müller, A. C. Dumitru, C. Lo Giudice, H. E. Gaub, P. Hinterdorfer, G. Hummer, J. J. De Yoreo, Y. F. Dufrêne and D. Alsteens, Atomic Force Microscopy-Based Force Spectroscopy and Multiparametric Imaging of Biomolecular and Cellular Systems, Chem. Rev., 2021, 121(19), 11701–11725 CrossRef PubMed.
- H. Flechsig and T. Ando, Protein dynamics by the combination of high-speed AFM and computational modeling, Curr. Opin. Struct. Biol., 2023, 80, 102591 CrossRef CAS PubMed.
- S. Pinchas and I. Laulicht, Infrared Spectra of Labeled Compounds, Academic Press, London and New York, 1971 Search PubMed.
- S. A. Tatulian, FTIR Analysis of Proteins and Protein-Membrane Interactions, Methods Mol. Biol., 2019, 2003, 281–325 CrossRef CAS PubMed.
- S. A. Tatulian, Toward understanding interfacial activation of secretory phospholipase A2 (PLA2): membrane surface properties and membrane-induced structural changes in the enzyme contribute synergistically to PLA2 activation, Biophys. J., 2001, 80(2), 789–800 CrossRef CAS PubMed.
- P. I. Haris, Probing protein–protein interaction in biomembranes using Fourier transform infrared spectroscopy, Biochim. Biophys. Acta, 2013, 1828(10), 2265–2271 CrossRef CAS PubMed.
- S. M. Decatur, Elucidation of residue-level structure and dynamics of polypeptides via isotope-edited infrared spectroscopy, Acc. Chem. Res., 2006, 39(3), 169–175 CrossRef CAS PubMed.
- I. T. Arkin, Isotope-edited IR spectroscopy for the study of membrane proteins, Curr. Opin. Chem. Biol., 2006, 10(5), 394–401 CrossRef CAS PubMed.
- J. Manor and I. T. Arkin, Gaining insight into membrane protein structure using isotope-edited FTIR, Biochim. Biophys. Acta, 2013, 1828(10), 2256–2264 CrossRef CAS PubMed.
- R. A. MacPhail, H. L. Strauss, R. G. Snyder and C. A. Elliger, C-H Stretching Modes and the Structure of n-Alkyl Chains. 2. Long, All-Trans Chains, J. Phys. Chem., 1984, 88, 334–341 CrossRef CAS.
- V. R. Kodati, R. El-Jastimi and M. Lafleur, Contribution of the Intermolecular Coupling and Librotorsional Mobility in the Methylene Stretching Modes in the Infrared Spectra of Acyl Chains, J. Phys. Chem., 1994, 98, 12191–12197 CrossRef CAS.
- R. G. Snyder, S. L. Hsu and S. Krimm, Vibrational spectra in the C–H stretching region and the structure of the polymethylene chain, Spectrochim. Acta, Part A, 1978, 34, 395–406 CrossRef.
- L. Tadesse, R. Nazarbaghi and L. Walters, Isotopically Enhanced Infrared Spectroscopy: A Novel Method for Examining Secondary Structure at Specific Sites in Conformationally Heterogeneous Peptides, J. Am. Chem. Soc., 1991, 113, 7036–7037 CrossRef CAS.
- R. A. Silva, J. Kubelka, P. Bour, S. M. Decatur and T. A. Keiderling, Site-specific conformational determination in thermal unfolding studies of helical peptides using vibrational circular dichroism with isotopic substitution, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(15), 8318–8323 CrossRef CAS PubMed.
- J. W. Brauner, C. Dugan and R. Mendelsohn, 13C Isotope Labeling of Hydrophobic Peptides. Origin of the Anomalous Intensity Distribution in the Infrared Amide I Spectral Region of β-Sheet Structures, J. Am. Chem. Soc., 2000, 122, 677–683 CrossRef CAS.
- S. Y. Venyaminov, J. F. Hedstrom and F. G. Prendergast, Analysis of the segmental stability of helical peptides by isotope-edited infrared spectroscopy, Proteins, 2001, 45(1), 81–89 CrossRef CAS PubMed.
- B. Pastrana-Rios, Mechanism of unfolding of a model helical peptide, Biochemistry, 2001, 40(31), 9074–9081 CrossRef CAS PubMed.
- C. Y. Huang, Z. Getahun, Y. Zhu, J. W. Klemke, W. F. DeGrado and F. Gai, Helix formation via conformation diffusion search, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(5), 2788–2793 CrossRef CAS PubMed.
- R. Huang, J. Kubelka, W. Barber-Armstrong, R. A. Silva, S. M. Decatur and T. A. Keiderling, Nature of vibrational coupling in helical peptides: an isotopic labeling study, J. Am. Chem. Soc., 2004, 126(8), 2346–2354 CrossRef CAS PubMed.
- W. Barber-Armstrong, T. Donaldson, H. Wijesooriya, R. A. Silva and S. M. Decatur, Empirical relationships between isotope-edited IR spectra and helix geometry in model peptides, J. Am. Chem. Soc., 2004, 126(8), 2339–2345 CrossRef CAS PubMed.
- S. M. Decatur, IR spectroscopy of isotope-labeled helical peptides: probing the effect of N-acetylation on helix stability, Biopolymers, 2000, 54(3), 180–185 CrossRef CAS PubMed.
- P. Bouř and T. A. Keiderling, Ab initio modeling of amide I coupling in antiparallel beta-sheets and the effect of 13C isotopic labeling on infrared spectra, J. Phys. Chem. B, 2005, 109(11), 5348–5357 CrossRef PubMed.
- G. Goldblatt, L. Cilenti, J. O. Matos, B. Lee, N. Ciaffone, Q. X. Wang, L. Tetard, K. Teter and S. A. Tatulian, Unmodified and pyroglutamylated amyloid β peptides form hypertoxic hetero-oligomers of unique secondary structure, FEBS J., 2017, 284(9), 1355–1369 CrossRef CAS PubMed.
- I. T. Arkin, K. R. MacKenzie and A. T. Brünger, Site-directed dichroism as a method for obtaining rotational and orientational constraints for oriented polymers, J. Am. Chem. Soc., 1997, 119(38), 8973–8980 CrossRef CAS.
- K. P. Das, L. P. Choo-Smith, J. M. Petrash and W. K. Surewicz, Insight into the secondary structure of non-native proteins bound to a molecular chaperone alpha-crystallin. An isotope-edited infrared spectroscopic study, J. Biol. Chem., 1999, 274(47), 33209–33212 CrossRef CAS PubMed.
- J. Torres, A. Kukol, J. M. Goodman and I. T. Arkin, Site-specific examination of secondary structure and orientation determination in membrane proteins: the peptidic (13)C(18)O group as a novel infrared probe, Biopolymers, 2001, 59(6), 396–401 CrossRef CAS PubMed.
- C. M. Baronio, M. Baldassarre and A. Barth, Insight into the internal structure of amyloid-β oligomers by isotope-edited Fourier transform infrared spectroscopy, Phys. Chem. Chem. Phys., 2019, 21(16), 8587–8597 RSC.
- P. I. Haris, G. T. Robillard, A. A. van Dijk and D. Chapman, Potential of 13C and 15N labeling for studying protein–protein interactions using Fourier transform infrared spectroscopy, Biochemistry, 1992, 31(27), 6279–6284 CrossRef CAS PubMed.
- M. Zhang, H. Fabian, H. H. Mantsch and H. J. Vogel, Isotope-edited Fourier transform infrared spectroscopy studies of calmodulin's interaction with its target peptides, Biochemistry, 1994, 33(36), 10883–10888 CrossRef CAS PubMed.
- G. Goldblatt, J. O. Matos, J. Gornto and S. A. Tatulian, Isotope-edited FTIR reveals distinct aggregation and structural behaviors of unmodified and pyroglutamylated amyloid β peptides, Phys. Chem. Chem. Phys., 2015, 17(48), 32149–32160 RSC.
- S. A. Tatulian and L. K. Tamm, Secondary structure, orientation, oligomerization, and lipid interactions of the transmembrane domain of influenza hemagglutinin, Biochemistry, 2000, 39(3), 496–507 CrossRef CAS PubMed.
- C. Paul, J. Wang, W. C. Wimley, R. M. Hochstrasser and P. H. Axelsen, Vibrational coupling, isotopic editing, and beta-sheet structure in a membrane-bound polypeptide, J. Am. Chem. Soc., 2004, 126(18), 5843–5850 CrossRef CAS PubMed.
- S. A. Petty and S. M. Decatur, Experimental evidence for the reorganization of beta-strands within aggregates of the Abeta(16–22) peptide, J. Am. Chem. Soc., 2005, 127(39), 13488–13489 CrossRef CAS PubMed.
- R. Huang, V. Setnička, M. A. Etienne, J. Kim, J. Kubelka, R. P. Hammer and T. A. Keiderling, Cross-Strand Coupling of a β-Hairpin Peptide Stabilized with an Aib-Gly Turn Studied Using Isotope-Edited IR Spectroscopy, J. Am. Chem. Soc., 2007, 129(44), 13592–13603 CrossRef CAS PubMed.
- D. Scheerer, H. Chi, D. McElheny, T. A. Keiderling and K. Hauser, Enhanced Sensitivity to Local Dynamics in Peptides by Use of Temperature-Jump IR Spectroscopy and Isotope Labeling, Chemistry, 2020, 26(16), 3524–3534 CrossRef CAS PubMed.
- C. Paul and P. H. Axelsen, beta Sheet structure in amyloid beta fibrils and vibrational dipolar coupling, J. Am. Chem. Soc., 2005, 127(16), 5754–5755 CrossRef CAS PubMed.
- S. A. Petty and S. M. Decatur, Intersheet rearrangement of polypeptides during nucleation of {beta}-sheet aggregates, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(40), 14272–14277 CrossRef CAS PubMed.
- A. Kukol, J. Torres and I. T. Arkin, A structure for the trimeric MHC class II-associated invariant chain transmembrane domain, J. Mol. Biol., 2002, 320(5), 1109–1117 CrossRef CAS PubMed.
- A. J. Beevers and A. Kukol, The transmembrane domain of the oncogenic mutant ErbB-2 receptor: a structure obtained from site-specific infrared dichroism and molecular dynamics, J. Mol. Biol., 2006, 361(5), 945–953 CrossRef CAS PubMed.
- E. Arbely, I. Kass and I. T. Arkin, Site-specific dichroism analysis utilizing transmission FTIR, Biophys. J., 2003, 85(4), 2476–2483 CrossRef CAS PubMed.
- A. Remorino, I. V. Korendovych, Y. Wu, W. F. DeGrado and R. M. Hochstrasser, Residue-specific vibrational echoes yield 3D structures of a transmembrane helix dimer, Science, 2011, 332(6034), 1206–1209 CrossRef CAS PubMed.
- Y. S. Kim, L. Liu, P. H. Axelsen and R. M. Hochstrasser, Two-dimensional infrared spectra of isotopically diluted amyloid fibrils from Abeta40, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(22), 7720–7725 CrossRef CAS PubMed.
- S. D. Moran and M. T. Zanni, How to Get Insight into Amyloid Structure and Formation from Infrared Spectroscopy, J. Phys. Chem. Lett., 2014, 5(11), 1984–1993 CrossRef CAS PubMed.
- M. Maj, J. P. Lomont, K. L. Rich, A. M. Alperstein and M. T. Zanni, Site-specific detection of protein secondary structure using 2D IR dihedral indexing: a proposed assembly mechanism of oligomeric hIAPP, Chem. Sci., 2018, 9(2), 463–474 RSC.
- J. C. Rodríguez-Pérez, I. W. Hamley, S. L. Gras and A. M. Squires, Local orientational disorder in peptide fibrils probed by a combination of residue-specific 13C–18O labelling, polarised infrared spectroscopy and molecular combing, Chem. Commun., 2012, 48(97), 11835–11837 RSC.
- F. R. Rana, A. J. Mautone and R. A. Dluhy, Combined Infrared and 31P NMR Spectroscopic Method for Determining the Fractional Composition in Langmuir-Blodgett Films of Binary Phospholipid Mixtures, Appl. Spectrosc., 1993, 47(7), 1015–1023 CrossRef CAS.
- H. M. Reinl and T. M. Bayerl, Lipid transfer between small unilamellar vesicles and single bilayers on a solid support: self-assembly of supported bilayers with asymmetric lipid distribution, Biochemistry, 1994, 33(47), 14091–14099 CrossRef CAS PubMed.
- A. R. Dibble, A. K. Hinderliter, J. J. Sando and R. L. Biltonen, Lipid lateral heterogeneity in phosphatidylcholine/phosphatidylserine/diacylglycerol vesicles and its influence on protein kinase C activation, Biophys. J., 1996, 71(4), 1877–1890 CrossRef CAS PubMed.
- P. Lasch, C. P. Schultz and D. Naumann, The influence of poly-(L-lysine) and porin on the domain structure of mixed vesicles composed of lipopolysaccharide and phospholipid: an infrared spectroscopic study, Biophys. J., 1998, 75(2), 840–852 CrossRef CAS PubMed.
- J. M. Brockman, Z. Wang, R. H. Notter and R. A. Dluhy, Effect of hydrophobic surfactant proteins SP-B and SP-C on binary phospholipid monolayers: II. Infrared external reflectance-absorption spectroscopy, Biophys. J., 2003, 84(1), 326–340 CrossRef CAS PubMed.
- R. A. Dluhy, D. Moffatt, D. G. Cameron, R. Mendelsohn and H. H. Mantsch, Characterization of cooperative conformational transitions by Fourier transform infrared spectroscopy: application to phospholipid binary mixtures, Can. J. Chem., 1985, 63, 1925–1932 CrossRef CAS.
- D. J. Moore, R. H. Sills, N. Patel and R. Mendelsohn, Conformational order of phospholipids incorporated into human erythrocytes: an FTIR spectroscopy study, Biochemistry, 1996, 35(1), 229–235 CrossRef CAS PubMed.
- H. L. Casal and H. H. Mantsch, Polymorphic phase behaviour of phospholipid membranes studied by infrared spectroscopy, Biochim. Biophys. Acta, 1984, 779(4), 381–401 CrossRef CAS PubMed.
- J. Villalaín and J. C. Gómez-Fernández, Fourier transform infrared spectroscopic study of mixtures of palmitic acid with dipalmitoylphosphatidylcholine using isotopic substitution, Chem. Phys. Lipids, 1992, 62, 19–29 CrossRef.
- F. López-García, J. Villalaín, J. C. Gómez-Fernández and P. J. Quinn, The phase behavior of mixed aqueous dispersions of dipalmitoyl derivatives of phosphatidylcholine and diacylglycerol, Biophys. J., 1994, 66(6), 1991–2004 CrossRef PubMed.
- R. Mendelsohn, M. A. Davies, J. W. Brauner, H. F. Schuster and R. A. Dluhy, Quantitative determination of conformational disorder in the acyl chains of phospholipid bilayers by infrared spectroscopy, Biochemistry, 1989, 28(22), 8934–8939 CrossRef CAS PubMed.
- R. Mendelsohn, G. L. Liang, H. L. Strauss and R. G. Snyder, IR spectroscopic determination of gel state miscibility in long-chain phosphatidylcholine mixtures, Biophys. J., 1995, 69(5), 1987–1998 CrossRef CAS PubMed.
- A. Blume, W. Hübner and G. Messner, Fourier transform infrared spectroscopy of 13CO-labeled phospholipids hydrogen bonding to carbonyl groups, Biochemistry, 1988, 27(21), 8239–8249 CrossRef CAS PubMed.
- W. Hübner and H. H. Mantsch, Orientation of specifically 13CO labeled phosphatidylcholine multilayers from polarized attenuated total reflection FT-IR spectroscopy, Biophys. J., 1991, 59(6), 1261–1272 CrossRef PubMed.
- R. N. Lewis and R. N. McElhaney, Studies of mixed-chain diacyl phosphatidylcholines with highly asymmetric acyl chains: a Fourier transform infrared spectroscopic study of interfacial hydration and hydrocarbon chain packing in the mixed interdigitated gel phase, Biophys. J., 1993, 65(5), 1866–1877 CrossRef CAS PubMed.
- R. N. Lewis, R. N. McElhaney, W. Pohle and H. H. Mantsch, Components of the carbonyl stretching band in the infrared spectra of hydrated 1,2-diacylglycerolipid bilayers: a reevaluation, Biophys. J., 1994, 67(6), 2367–2375 CrossRef CAS PubMed.
- J. Villalain and J. C. Gomez-Fernandez, FT-IR study of the hydrogen bonding interaction between cholesterol and DPPC, Biochem. Soc. Trans., 1992, 20(2), 122S CrossRef CAS PubMed.
- A. H. Pande, D. Moe, K. N. Nemec, S. Qin, S. Tan and S. A. Tatulian, Modulation of human 5-lipoxygenase activity by membrane lipids, Biochemistry, 2004, 43(46), 14653–14666 CrossRef CAS PubMed.
- S. A. Tatulian, Structural effects of covalent inhibition of phospholipase A2 suggest allosteric coupling between membrane binding and catalytic sites, Biophys. J., 2003, 84(3), 1773–1783 CrossRef CAS PubMed.
- K. N. Allen, S. Entova, L. C. Ray and B. Imperiali, Monotopic Membrane Proteins Join the Fold, Trends Biochem. Sci., 2019, 44(1), 7–20 CrossRef CAS PubMed.
- I. Boulos, J. Jabbour, S. Khoury, N. Mikhael, V. Tishkova, N. Candoni, H. E. Ghadieh, S. Veesler, Y. Bassim, S. Azar and F. Harb, Exploring the World of Membrane Proteins: Techniques and Methods for Understanding Structure, Function, and Dynamics, Molecules, 2023, 28(20), 7176 CrossRef CAS PubMed.
- B. Li, J. Mendenhall and J. Meiler, Interfaces Between Alpha-helical Integral Membrane Proteins: Characterization, Prediction, and Docking, Comput. Struct. Biotechnol. J., 2019, 17, 699–711 CrossRef CAS PubMed.
- J. T. Marinko, H. Huang, W. D. Penn, J. A. Capra, J. P. Schlebach and C. R. Sanders, Folding and Misfolding of Human Membrane Proteins in Health and Disease: From Single Molecules to Cellular Proteostasis, Chem. Rev., 2019, 119(9), 5537–5606 CrossRef CAS PubMed.
- L. A. Mayse and L. Movileanu, Gating of β-Barrel Protein Pores, Porins, and Channels: An Old Problem with New Facets, Int. J. Mol. Sci., 2023, 24(15), 12095 CrossRef CAS PubMed.
- Y. C. Chiu, M. C. Yeh, C. H. Wang, Y. A. Chen, H. Chang, H. Y. Lin, M. C. Ho and S. M. Lin, Structural basis for calcium-stimulating pore formation of Vibrio α-hemolysin, Nat. Commun., 2023, 14(1), 5946 CrossRef CAS PubMed.
- C. W. Fhu and A. Ali, Protein Lipidation by Palmitoylation and Myristoylation in Cancer, Front. Cell Dev. Biol., 2021, 9, 673647 CrossRef PubMed.
- S. Shang, J. Liu and F. Hua, Protein acylation: mechanisms, biological functions and therapeutic targets, Signal Transduction Targeted Ther., 2022, 7(1), 396 CrossRef CAS PubMed.
- K. M. J. H. Dennis and L. C. Heather, Post-translational palmitoylation of metabolic proteins, Front. Physiol., 2023, 14, 1122895 CrossRef PubMed.
- G. A. Müller and T. D. Müller, (Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins I: Localization at Plasma Membranes and Extracellular Compartments, Biomolecules, 2023, 13(5), 855 CrossRef PubMed.
- G. A. Müller and T. D. Müller, Patho)Physiology of Glycosylphosphatidylinositol-Anchored Proteins II: Intercellular Transfer of Matter (Inheritance?) That Matters, Biomolecules, 2023, 13(6), 994 CrossRef PubMed.
- D. Jung and H. S. Bachmann, Regulation of protein prenylation, Biomed. Pharmacother., 2023, 164, 114915 CrossRef CAS PubMed.
- C. E. Villanueva and B. Hagenbuch, Palmitoylation of solute carriers, Biochem. Pharmacol., 2023, 215, 115695 CrossRef CAS PubMed.
- E. Fuglebakk and N. Reuter, A model for hydrophobic protrusions on peripheral membrane proteins, PLoS Comput. Biol., 2018, 14(7), e1006325 CrossRef PubMed.
- J. G. Pemberton and T. Balla, Polyphosphoinositide-Binding Domains: Insights from Peripheral Membrane and Lipid-Transfer Proteins, Adv. Exp. Med. Biol., 2019, 1111, 77–137 CrossRef CAS PubMed.
- S. A. Tatulian, Interfacial Enzymes: Membrane Binding, Orientation, Membrane Insertion, and Activity, Methods Enzymol., 2017, 583, 197–230 CAS.
- M. P. Muller, T. Jiang, C. Sun, M. Lihan, S. Pant, P. Mahinthichaichan, A. Trifan and E. Tajkhorshid, Characterization of Lipid-Protein Interactions and Lipid-Mediated Modulation of Membrane Protein Function through Molecular Simulation, Chem. Rev., 2019, 119(9), 6086–6161 CrossRef CAS PubMed.
- S. Ray, J. L. Scott and S. A. Tatulian, Effects of lipid phase transition and membrane surface charge on the interfacial activation of phospholipase A2, Biochemistry, 2007, 46(45), 13089–13100 CrossRef CAS PubMed.
- H. Speijer, P. L. Giesen, R. F. Zwaal, C. E. Hack and W. T. Hermens, Critical micelle concentrations and stirring are rate limiting in the loss of lipid mass during membrane degradation by phospholipase A2, Biophys. J., 1996, 70(5), 2239–2247 CrossRef CAS PubMed.
- T. H. Callisen and Y. Talmon, Direct imaging by cryo-TEM shows membrane break-up by phospholipase A2 enzymatic activity, Biochemistry, 1998, 37(31), 10987–10993 CrossRef CAS PubMed.
- H. P. Vacklin, F. Tiberg, G. Fragneto and R. K. Thomas, Phospholipase A2 hydrolysis of supported phospholipid bilayers: a neutron reflectivity and ellipsometry study, Biochemistry, 2005, 44(8), 2811–2821 CrossRef CAS PubMed.
- M. K. Jain, J. Rogers, D. V. Jahagirdar, J. F. Marecek and F. Ramirez, Kinetics of interfacial catalysis by phospholipase A2 in intravesicle scooting mode, and heterofusion of anionic and zwitterionic vesicles, Biochim. Biophys. Acta, 1986, 860(3), 435–447 CrossRef CAS PubMed.
- T. Bayburt, B. Z. Yu, H. K. Lin, J. Browning, M. K. Jain and M. H. Gelb, Human nonpancreatic secreted phospholipase A2: interfacial parameters, substrate specificities, and competitive inhibitors, Biochemistry, 1993, 32(2), 573–582 CrossRef CAS PubMed.
- W. R. Burack and R. L. Biltonen, Lipid bilayer heterogeneities and modulation of phospholipase A2 activity, Chem. Phys. Lipids, 1994, 73(1–2), 209–222 CrossRef CAS PubMed.
- W. R. Burack, A. R. Dibble, M. M. Allietta and R. L. Biltonen, Changes in vesicle morphology induced by lateral phase separation modulate phospholipase A2 activity, Biochemistry, 1997, 36(34), 10551–10557 CrossRef CAS PubMed.
- A. H. Pande, S. Qin, K. N. Nemec, X. He and S. A. Tatulian, Isoform-specific membrane insertion of secretory phospholipase A2 and functional implications, Biochemistry, 2006, 45(41), 12436–12447 CrossRef CAS PubMed.
- S. A. Tatulian, Attenuated total reflection Fourier transform infrared spectroscopy: a method of choice for studying membrane proteins and lipids, Biochemistry, 2003, 42(41), 11898–11907 CrossRef CAS PubMed.
- S. A. Tatulian, S. Qin, A. H. Pande and X. He, Positioning membrane proteins by novel protein engineering and biophysical approaches, J. Mol. Biol., 2005, 351(5), 939–947 CrossRef CAS PubMed.
- B. Z. Yu, M. J. W. Janssen, H. M. Verheij and M. K. Jain, Control of the chemical step by leucine-31 of pancreatic phospholipase A2, Biochemistry, 2000, 39, 5702–5711 CrossRef CAS PubMed.
- O. G. Berg, M. H. Gelb, M. D. Tsai and M. K. Jain, Interfacial enzymology: the secreted phospholipase A2-paradigm, Chem. Rev., 2001, 101, 2613–2654 CrossRef CAS PubMed.
- O. Keskin, A. Gursoy, B. Ma and R. Nussinov, Principles of protein–protein interactions: what are the preferred ways for proteins to interact?, Chem. Rev., 2008, 108(4), 1225–1244 CrossRef CAS PubMed.
- J. R. Perkins, I. Diboun, B. H. Dessailly, J. G. Lees and C. Orengo, Transient protein–protein interactions: structural, functional, and network properties, Structure, 2010, 18(10), 1233–1243 CrossRef CAS PubMed.
- H. Akiba and K. Tsumoto, Thermodynamics of antibody-antigen interaction revealed by mutation analysis of antibody variable regions, J. Biochem., 2015, 158(1), 1–13 CrossRef CAS PubMed.
- S. A. Tatulian, INSR, in Encyclopedia of Signaling Molecules, ed. S. Choi, Springer, 2nd edn, 2018, pp. 2608–2619 Search PubMed.
- C. J. Oldfield, J. Meng, J. Y. Yang, M. Q. Yang, V. N. Uversky and A. K. Dunker, Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners, BMC Genomics, 2008, 9(Suppl 1), S1 CrossRef PubMed.
- Y. Xu, G. K. Kong, J. G. Menting, M. B. Margetts, C. A. Delaine, L. M. Jenkin, V. V. Kiselyov, P. De Meyts, B. E. Forbes and M. C. Lawrence, How ligand binds to the type 1 insulin-like growth factor receptor, Nat. Commun., 2018, 9(1), 821 CrossRef PubMed.
- H. Kaur and D. M. Salunke, Antibody promiscuity: Understanding the paradigm shift in antigen recognition, IUBMB Life, 2015, 67(7), 498–505 CrossRef CAS PubMed.
- M. Arai, S. Suetaka and K. Ooka, Dynamics and interactions of intrinsically disordered proteins, Curr. Opin. Struct. Biol., 2023, 84, 102734 CrossRef PubMed.
- O. Hennig, S. Philipp, S. Bonin, K. Rollet, T. Kolberg, T. Jühling, H. Betat, C. Sauter and M. Mörl, Adaptation of the Romanomermis culicivorax CCA-Adding Enzyme to Miniaturized Armless tRNA Substrates, Int. J. Mol. Sci., 2020, 21(23), 9047 CrossRef CAS PubMed.
- E. Gattkowski, T. J. Rutherford, F. Möckl, A. Bauche, S. Sander, R. Fliegert and H. Tidow, Analysis of ligand binding and resulting conformational changes in pyrophosphatase NUDT9, FEBS J., 2021, 288(23), 6769–6782 CrossRef CAS PubMed.
- D. Su, T. Kosciuk, M. Yang, I. R. Price and H. Lin, Binding Affinity Determines Substrate Specificity and Enables Discovery of Substrates for N-Myristoyltransferases, ACS Catal., 2021, 11(24), 14877–14883 CrossRef CAS PubMed.
- M. Hager, M. T. Pöhler, F. Reinhardt, K. Wellner, J. Hübner, H. Betat, S. Prohaska and M. Mörl, Substrate Affinity Versus Catalytic Efficiency: Ancestral Sequence Reconstruction of tRNA Nucleotidyltransferases Solves an Enzyme Puzzle, Mol. Biol. Evol., 2022, 39(12), msac250 CrossRef CAS PubMed.
- E. N. Moreno-Córdova, A. A. Arvizu-Flores, E. M. Valenzuela-Soto, K. D. García-Orozco, A. Wall-Medrano, E. Alvarez-Parrilla, J. F. Ayala-Zavala, J. A. Domínguez-Avila and G. A. González-Aguilar, Gallotannins are uncompetitive inhibitors of pancreatic lipase activity, Biophys. Chem., 2020, 264, 106409 CrossRef PubMed.
- E. C. Dacol, S. Wang, Y. Chen and A. P. Lepique, The interaction of SET and protein phosphatase 2A as target for cancer therapy, Biochim. Biophys. Acta, Rev. Cancer, 2021, 1876(1), 188578 CrossRef CAS PubMed.
- R. A. Mitchell, R. B. Luwor and A. W. Burgess, Epidermal growth factor receptor: Structure-function informing the design of anticancer therapeutics, Exp. Cell Res., 2018, 371(1), 1–19 CrossRef CAS PubMed.
- M. M. Hoffmann and J. E. Slansky, T-cell receptor affinity in the age of cancer immunotherapy, Mol. Carcinog., 2020, 59(7), 862–870 CrossRef CAS PubMed.
- S. Zheng, M. Zou, Y. Shao, H. Wu, H. Wu and X. Wang, Two-dimensional measurements of receptor-ligand interactions, Front. Mol. Biosci., 2023, 10, 1154074 CrossRef CAS PubMed.
- K. S. Midelfort and K. D. Wittrup, Context-dependent mutations predominate in an engineered high-affinity single chain antibody fragment, Protein Sci., 2006, 15(2), 324–334 CrossRef CAS PubMed.
- A. Diop, D. Santorelli, F. Malagrinò, C. Nardella, V. Pennacchietti, L. Pagano, L. Marcocci, P. Pietrangeli, S. Gianni and A. Toto, SH2 Domains: Folding, Binding and Therapeutical Approaches, Int. J. Mol. Sci., 2022, 23(24), 15944 CrossRef CAS PubMed.
- J. F. Amacher, L. Brooks, T. H. Hampton and D. R. Madden, Specificity in PDZ-peptide interaction networks: computational analysis and review, J. Struct. Biol.: X, 2020, 4, 100022 CAS.
- B. Thurairajah, A. J. Hudson and R. G. Doveston, Contemporary biophysical approaches for studying 14-3-3 protein–protein interactions, Front. Mol. Biosci., 2022, 9, 1043673 CrossRef CAS PubMed.
- N. H. Shah, J. F. Amacher, L. M. Nocka and J. Kuriyan, The Src module: an ancient scaffold in the evolution of cytoplasmic tyrosine kinases, Crit. Rev. Biochem. Mol. Biol., 2018, 53(5), 535–563 CrossRef CAS PubMed.
- S. Abhishek, W. Deeksha, K. R. Nethravathi, M. D. Davari and E. Rajakumara, Allosteric crosstalk in modular proteins: Function fine-tuning and drug design, Comput. Struct. Biotechnol. J., 2023, 21, 5003–5015 CrossRef CAS PubMed.
- R. Akter, P. Cao, H. Noor, Z. Ridgway, L. H. Tu, H. Wang, A. G. Wong, X. Zhang, A. Abedini, A. M. Schmidt and D. P. Raleigh, Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology, J. Diabetes Res., 2016, 2016, 2798269 Search PubMed.
- K. Yuzu, M. Lindgren, S. Nyström, J. Zhang, W. Mori, R. Kunitomi, T. Nagase, K. Iwaya, P. Hammarström and T. Zako, Insulin amyloid polymorphs: implications for iatrogenic cytotoxicity, RSC Adv., 2020, 10(62), 37721–37727 RSC.
- M. A. Liz, T. Coelho, V. Bellotti, M. I. Fernandez-Arias, P. Mallaina and L. Obici, A Narrative Review of the Role of Transthyretin in Health and Disease, Neurol. Ther., 2020, 9(2), 395–402 CrossRef PubMed.
- D. Maity and D. Pal, Molecular Dynamics of Hemoglobin Reveals Structural Alterations and Explains the Interactions Driving Sickle Cell Fibrillation, J. Phys. Chem. B, 2021, 125(35), 9921–9933 CrossRef CAS PubMed.
- P. H. Nguyen, A. Ramamoorthy, B. R. Sahoo, J. Zheng, P. Faller, J. E. Straub, L. Dominguez, J. E. Shea, N. V. Dokholyan, A. De Simone, B. Ma, R. Nussinov, S. Najafi, S. T. Ngo, A. Loquet, M. Chiricotto, P. Ganguly, J. McCarty, M. S. Li, C. Hall, Y. Wang, Y. Miller, S. Melchionna, B. Habenstein, S. Timr, J. Chen, B. Hnath, B. Strodel, R. Kayed, S. Lesné, G. Wei, F. Sterpone, A. J. Doig and P. Derreumaux, Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer's Disease, Parkinson's Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis, Chem. Rev., 2021, 121(4), 2545–2647 CrossRef CAS PubMed.
- J. N. Buxbaum, A. Dispenzieri, D. S. Eisenberg, M. Fändrich, G. Merlini, M. J. M. Saraiva, Y. Sekijima and P. Westermark, Amyloid nomenclature 2022: update, novel proteins, and recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee, Amyloid, 2022, 29(4), 213–219 CrossRef PubMed.
- K. Y. Kulichikhin, O. A. Malikova, A. E. Zobnina, N. M. Zalutskaya and A. A. Rubel, Interaction of Proteins Involved in Neuronal Proteinopathies, Life, 2023, 13(10), 1954 CrossRef CAS PubMed.
- N. Starr, A. Ioannou and A. Martinez-Naharro, Monitoring cardiac amyloidosis with multimodality imaging, Rev. Esp. Cardiol., 2023, S1885–5857(23), 00237-2 Search PubMed.
- N. Louros, J. Schymkowitz and F. Rousseau, Mechanisms and pathology of protein misfolding and aggregation, Nat. Rev. Mol. Cell Biol., 2023, 24(12), 912–933 CrossRef CAS PubMed.
- K. Al Adem and S. Lee, Structural polymorphism and cytotoxicity of brain-derived β-amyloid extracts, Protein Sci., 2023, 32(5), e4639 CrossRef CAS PubMed.
- K. B. S. Swamy, S. C. Schuyler and J. Y. Leu, Protein Complexes Form a Basis for Complex Hybrid Incompatibility, Front. Genet., 2021, 12, 609766 CrossRef CAS PubMed.
- T. R. Kanitkar, N. Sen, S. Nair, N. Soni, K. Amritkar, Y. Ramtirtha and M. S. Madhusudhan, Methods for Molecular Modelling of Protein Complexes, Methods Mol. Biol., 2021, 2305, 53–80 CrossRef CAS PubMed.
- L. Piersimoni, P. L. Kastritis, C. Arlt and A. Sinz, Cross-Linking Mass Spectrometry for Investigating Protein Conformations and Protein–Protein Interactions—A Method for All Seasons, Chem. Rev., 2022, 122(8), 7500–7531 CrossRef CAS PubMed.
- I. Couée and G. Gouesbet, Protein–Protein Interactions in Abiotic Stress Signaling: An Overview of Biochemical and Biophysical Methods of Characterization, Methods Mol. Biol., 2023, 2642, 319–330 CrossRef PubMed.
- F. J. O'Reilly, A. Graziadei, C. Forbrig, R. Bremenkamp, K. Charles, S. Lenz, C. Elfmann, L. Fischer, J. Stülke and J. Rappsilber, Protein complexes in cells by AI-assisted structural proteomics, Mol. Syst. Biol., 2023, 19(4), e11544 CrossRef PubMed.
- J. R. Prindle, O. I. C. de Cuba and A. Gahlmann, Single-molecule tracking to determine the abundances and stoichiometries of freely-diffusing protein complexes in living cells: past applications and future prospects, J. Chem. Phys., 2023, 159(7), 071002 CrossRef CAS PubMed.
- I. Manipur, M. Giordano, M. Piccirillo, S. Parashuraman and L. Maddalena, Community Detection in Protein–Protein Interaction Networks and Applications, IEEE/ACM Trans. Comput. Biol. Bioinf., 2023, 20(1), 217–237 CAS.
- Y. Y. Wang, W. Li, B. C. Ye and X. B. Bi, Chemical and Biological Strategies for Profiling Protein–Protein Interactions in Living Cells, Chem. – Asian J., 2023, 18(14), e202300226 CrossRef CAS PubMed.
- S. A. Tatulian, Challenges and hopes for Alzheimer's disease, Drug Discovery Today, 2022, 27(4), 1027–1043 CrossRef PubMed.
- A. G. Karkisaval, R. Hassan, A. Nguyen, B. Balster, F. Abedin, R. Lal and S. A. Tatulian, The structure of tyrosine-10 favors ionic conductance of Alzheimer's disease-associated full-length amyloid-β channels, Nat. Commun., 2024, 15(1), 1296 CrossRef CAS PubMed.
- M. Jackson and H. H. Mantsch, The use and misuse of FTIR spectroscopy in the determination of protein structure, Crit. Rev. Biochem. Mol. Biol., 1995, 30(2), 95–120 CrossRef CAS PubMed.
- E. N. Cline, M. A. Bicca, K. L. Viola and W. L. Klein, The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade, J. Alzheimer's Dis., 2018, 64(s1), S567–S610 CAS.
- S. Li and D. J. Selkoe, A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Aβ oligomers from Alzheimer's brain, J. Neurochem., 2020, 154(6), 583–597 CrossRef CAS PubMed.
- M. Khaled, I. Rönnbäck, L. L. Ilag, A. Gräslund, B. Strodel and N. Österlund, A Hairpin Motif in the Amyloid-β Peptide Is Important for Formation of Disease-Related Oligomers, J. Am. Chem. Soc., 2023, 145(33), 18340–18354 CrossRef CAS PubMed.
- U. Ravens and D. Dobrev, Regulation of sarcoplasmic reticulum Ca(2+)-ATPase and phospholamban in the failing and nonfailing heart, Cardiovasc. Res., 2000, 45(1), 245–252 CrossRef CAS PubMed.
- S. A. Tatulian, B. Chen, J. Li, S. Negash, C. R. Middaugh, D. J. Bigelow and T. C. Squier, The inhibitory action of phospholamban involves stabilization of alpha-helices within the Ca-ATPase, Biochemistry, 2002, 41(3), 741–751 CrossRef CAS PubMed.
- M. Gustavsson, R. Verardi, D. G. Mullen, K. R. Mote, N. J. Traaseth, T. Gopinath and G. Veglia, Allosteric regulation of SERCA by phosphorylation-mediated conformational shift of phospholamban, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17338–17343 CrossRef CAS PubMed.
- M. E. Arnold, W. R. Dostmann, J. Martin, M. J. Previs, B. Palmer, M. LeWinter and M. Meyer, SERCA2a-phospholamban interaction monitored by an interposed circularly permutated green fluorescent protein, Am. J. Physiol.: Heart Circ. Physiol., 2021, 320(6), H2188–H2200 CrossRef CAS PubMed.
- T. Morita, D. Hussain, M. Asahi, T. Tsuda, K. Kurzydlowski, C. Toyoshima and D. H. Maclennan, Interaction sites among phospholamban, sarcolipin, and the sarco(endo)plasmic reticulum Ca(2+)-ATPase, Biochem. Biophys. Res. Commun., 2008, 369(1), 188–194 CrossRef CAS PubMed.
- S. A. Tatulian, L. R. Jones, L. G. Reddy, D. L. Stokes and L. K. Tamm, Secondary structure and orientation of phospholamban reconstituted in supported bilayers from polarized attenuated total reflection FTIR spectroscopy, Biochemistry, 1995, 34(13), 4448–4456 CrossRef CAS PubMed.
- J. A. Ihalainen, B. Paoli, S. Muff, E. H. Backus, J. Bredenbeck, G. A. Woolley, A. Caflisch and P. Hamm, Alpha-Helix folding in the presence of structural constraints, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(28), 9588–9593 CrossRef CAS PubMed.
- J. Manor, E. Arbely, A. Beerlink, M. Akkawi and I. T. Arkin, Use of Isotope-Edited FTIR to Derive a Backbone Structure of a Transmembrane Protein, J. Phys. Chem. Lett., 2014, 5(15), 2573–2579 CrossRef CAS PubMed.
- B. Dhayalan, A. Fitzpatrick, K. Mandal, J. Whittaker, M. A. Weiss, A. Tokmakoff and S. B. Kent, Efficient Total Chemical Synthesis of (13)C(18)O Isotopomers of Human Insulin for Isotope-Edited FTIR, ChemBioChem, 2016, 17(5), 415–420 CrossRef CAS PubMed.
- S. Tang, C. Zuo, D. L. Huang, X. Y. Cai, L. H. Zhang, C. L. Tian, J. S. Zheng and L. Liu, Chemical synthesis of membrane proteins by the removable backbone modification method, Nat. Protoc., 2017, 12(12), 2554–2569 CrossRef CAS PubMed.
- C. M. Davis, A. K. Cooper and R. B. Dyer, Fast helix formation in the B domain of protein A revealed by site-specific infrared probes, Biochemistry, 2015, 54(9), 1758–1766 CrossRef CAS PubMed.
- C. Aisenbrey, M. Cusan, S. Lambotte, P. Jasperse, J. Georgescu, U. Harzer and B. Bechinger, Specific isotope labeling of colicin E1 and B channel domains for membrane topological analysis by oriented solid-state NMR spectroscopy, ChemBioChem, 2008, 9(6), 944–951 CrossRef CAS PubMed.
- T. Iwasaki, Y. Miyajima-Nakano, R. Fukazawa, M. T. Lin, S. I. Matsushita, E. Hagiuda, A. T. Taguchi, S. A. Dikanov, Y. Oishi and R. B. Gennis, Escherichia coli amino acid auxotrophic expression host strains for investigating protein structure-function relationships, J. Biochem., 2021, 169(4), 387–394 CrossRef CAS PubMed.
- A. Saebi, J. S. Brown, V. M. Marando, N. Hartrampf, N. M. Chumbler, S. Hanna, M. Poskus, A. Loas, L. L. Kiessling, D. T. Hung and B. L. Pentelute, Rapid Single-Shot Synthesis of the 214 Amino Acid-Long N-Terminal Domain of Pyocin S2, ACS Chem. Biol., 2023, 18(3), 518–527 CrossRef CAS PubMed.
- K. Sato, C. E. Farquhar, J. Rodriguez and B. L. Pentelute, Automated Fast-Flow Synthesis of Chromosome 9 Open Reading Frame 72 Dipeptide Repeat Proteins, J. Am. Chem. Soc., 2023, 145(24), 12992–12997 CrossRef CAS PubMed.
- T. O. Zhang, M. Grechko, S. D. Moran and M. T. Zanni, Isotope-Labeled Amyloids via Synthesis, Expression, and Chemical Ligation for Use in FTIR, 2D IR, and NMR Studies, Methods Mol. Biol., 2016, 1345, 21–41 CrossRef CAS PubMed.
- J. S. Zheng, Y. He, C. Zuo, X. Y. Cai, S. Tang, Z. A. Wang, L. H. Zhang, C. L. Tian and L. Liu, Robust Chemical Synthesis of Membrane Proteins through a General Method of Removable Backbone Modification, J. Am. Chem. Soc., 2016, 138(10), 3553–3561 CrossRef CAS PubMed.
- S. W. Gan, W. Surya, A. Vararattanavech and J. Torres, Two different conformations in hepatitis C virus p7 protein account for proton transport and dye release, PLoS One, 2014, 9(1), e78494 CrossRef PubMed.
- A. Vandersteen, M. F. Masman, G. De Baets, W. Jonckheere, K. van der Werf, S. J. Marrink, J. Rozenski, I. Benilova, B. De Strooper, V. Subramaniam, J. Schymkowitz, F. Rousseau and K. Broersen, Molecular plasticity regulates oligomerization and cytotoxicity of the multipeptide-length amyloid-β peptide pool, J. Biol. Chem., 2012, 287(44), 36732–36743 CrossRef CAS PubMed.
- F. Abedin, N. Kandel and S. A. Tatulian, Effects of Aβ-derived peptide fragments on fibrillogenesis of Aβ, Sci. Rep., 2021, 11(1), 19262 CrossRef CAS PubMed.
- J. O. Matos, G. Goldblatt, J. Jeon, B. Chen and S. A. Tatulian, Pyroglutamylated amyloid-β peptide reverses cross β-sheets by a prion-like mechanism, J. Phys. Chem. B, 2014, 118(21), 5637–5643 CrossRef CAS PubMed.
- Y. Xu, C. T. Filice and Z. Leonenko, Protective effect of trehalose sugar on amyloid-membrane interactions using BLM electrophysiology, Biophys. J., 2024, S0006–3495(24), 00326 Search PubMed.
- F. Abedin and S. A. Tatulian, Mutual structural effects of unmodified and pyroglutamylated amyloid β peptides during aggregation, J. Pept. Sci., 2021, 27(6), e3312 CrossRef CAS PubMed.
- Y. Kimura, M. Imanishi, Y. Li, Y. Yura, T. Ohno, Y. Saga, M. T. Madigan and Z. Y. Wang-Otomo, Identification of metal-sensitive structural changes in the Ca2+-binding photocomplex from Thermochromatium tepidum by isotope-edited vibrational spectroscopy, J. Chem. Phys., 2022, 156(10), 105101 CrossRef CAS PubMed.
- S. A. Tatulian, R. L. Biltonen and L. K. Tamm, Structural changes in a secretory phospholipase A2 induced by membrane binding: a clue to interfacial activation?, J. Mol. Biol., 1997, 268(5), 809–815 CrossRef CAS PubMed.
- C. R. Flach, P. Cai, D. Dieudonné, J. W. Brauner, K. M. Keough, J. Stewart and R. Mendelsohn, Location of structural transitions in an isotopically labeled lung surfactant SP-B peptide by IRRAS, Biophys. J., 2003, 85(1), 340–349 CrossRef CAS PubMed.
- J. Waeytens, V. Van Hemelryck, A. Deniset-Besseau, J. M. Ruysschaert, A. Dazzi and V. Raussens, Characterization by Nano-Infrared Spectroscopy of Individual Aggregated Species of Amyloid Proteins, Molecules, 2020, 25(12), 2899 CrossRef CAS PubMed.
- S. Banerjee, T. Naik, D. Baghel and A. Ghosh, Intermediate Antiparallel Fibrils in Aβ40 Dutch Mutant Aggregation: Insights from Nanoscale Infrared Spectroscopy, J. Phys. Chem. B, 2023, 127(26), 5799–5807 CrossRef CAS PubMed.
- T. Dou, L. Zhou and D. Kurouski, Unravelling the Structural Organization of Individual α-Synuclein Oligomers Grown in the Presence of Phospholipids, J. Phys. Chem. Lett., 2021, 12(18), 4407–4414 CrossRef CAS PubMed.
- A. Rodriguez, A. Ali, A. P. Holman, T. Dou, K. Zhaliazka and D. Kurouski, Nanoscale structural characterization of transthyretin aggregates formed at different time points of protein aggregation using atomic force microscopy-infrared spectroscopy, Protein Sci., 2023, 32(12), e4838 CrossRef CAS PubMed.
- F. S. Ruggeri, B. Mannini, R. Schmid, M. Vendruscolo and T. P. J. Knowles, Single molecule secondary structure determination of proteins through infrared absorption nanospectroscopy, Nat. Commun., 2020, 11(1), 2945 CrossRef CAS PubMed.
- E. Pięta, Nanoscale insight into biochemical changes in cervical cancer cells exposed to adaptogenic drug, Micron, 2023, 170, 103462 CrossRef PubMed.
- S. A. Tatulian, Structural Dynamics of Insulin Receptor and Transmembrane Signaling, Biochemistry, 2015, 54(36), 5523–5532 CrossRef CAS PubMed.
- E. Kinoshita-Kikuta, E. Kinoshita, M. Suga, M. Higashida, Y. Yamane, T. Nakamura and T. Koike, Characterization of Phosphorylation Status and Kinase Activity of Src Family Kinases Expressed in Cell-Based and Cell-Free Protein Expression Systems, Biomolecules, 2021, 11(10), 1448 CrossRef CAS PubMed.
- L. A. Gurzeler, J. Ziegelmüller, O. Mühlemann and E. D. Karousis, Production of human translation-competent lysates using dual centrifugation, RNA Biol., 2022, 19(1), 78–88 CrossRef CAS PubMed.
- J. C. Anderson, N. Wu, S. W. Santoro, V. Lakshman, D. S. King and P. G. Schultz, An expanded genetic code with a functional quadruplet codon, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(20), 7566–7571 CrossRef CAS PubMed.
- R. Gao, Y. Zhang, A. K. Choudhury, L. M. Dedkova and S. M. Hecht, Analogues of vaccinia virus DNA topoisomerase I modified at the active site tyrosine, J. Am. Chem. Soc., 2005, 127(10), 3321–3331 CrossRef CAS PubMed.
- H. Xiao, A. Chatterjee, S. H. Choi, K. M. Bajjuri, S. C. Sinha and P. G. Schultz, Genetic incorporation of multiple unnatural amino acids into proteins in mammalian cells, Angew. Chem., Int. Ed., 2013, 52(52), 14080–14083 CrossRef CAS PubMed.
- A. O. Osgood, Y. Zheng, S. J. S. Roy, N. Biris, M. Hussain, C. Loynd, D. Jewel, J. S. Italia and A. Chatterjee, An Efficient Opal-Suppressor Tryptophanyl Pair Creates New Routes for Simultaneously Incorporating up to Three Distinct Noncanonical Amino Acids into Proteins in Mammalian Cells, Angew. Chem., Int. Ed., 2023, 62(19), e202219269 CrossRef CAS PubMed.
- S. Peuker, H. Andersson, E. Gustavsson, K. S. Maiti, R. Kania, A. Karim, S. Niebling, A. Pedersen, M. Erdelyi and S. Westenhoff, Efficient Isotope Editing of Proteins for Site-Directed Vibrational Spectroscopy, J. Am. Chem. Soc., 2016, 138(7), 2312–2318 CrossRef CAS PubMed.
- Y. Goto, T. Katoh and H. Suga, Flexizymes for genetic code reprogramming, Nat. Protoc., 2011, 6(6), 779–790 CrossRef CAS PubMed.
- J. N. Coronado, P. Ngo, E. V. Anslyn and A. D. Ellington, Chemical insights into flexizyme-mediated tRNA acylation. Cell, Chem. Biol., 2022, 29(7), 1071–1112 CAS.
- Y. Zheng, P. S. Addy, R. Mukherjee and A. Chatterjee, Defining the current scope and limitations of dual noncanonical amino acid mutagenesis in mammalian cells, Chem. Sci., 2017, 8(10), 7211–7217 RSC.
- B. Meineke, J. Heimgärtner, R. Caridha, M. F. Block, K. J. Kimler, M. F. Pires, M. Landreh and S. J. Elsässer, Dual stop codon suppression in mammalian cells with genomically integrated genetic code expansion machinery, Cell Rep. Methods, 2023, 3(11), 100626 CrossRef CAS PubMed.
- M. Sisido, K. Ninomiya, T. Ohtsuki and T. Hohsaka, Four-base codon/anticodon strategy and non-enzymatic aminoacylation for protein engineering with non-natural amino acids, Methods, 2005, 36(3), 270–278 CrossRef CAS PubMed.
- D. L. Dunkelmann, S. B. Oehm, A. T. Beattie and J. W. Chin, A 68-codon genetic code to incorporate four distinct non-canonical amino acids enabled by automated orthogonal mRNA design, Nat. Chem., 2021, 13(11), 1110–1117 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2024 |