
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of unsymmetrical dialkoxydiarylsilanes and diarylsilanediols from tetraalkoxysilane having a dioxasilepane unit†
Kenshiro
Hitoshio
 ,
Hiroki
Maeda
,
Kento
Teranishi
,
Jun
Shimokawa
* and
Hideki
Yorimitsu
*
,
Hiroki
Maeda
,
Kento
Teranishi
,
Jun
Shimokawa
* and
Hideki
Yorimitsu
*
Department of Chemistry Graduate School of Science, Kyoto University Sakyo-ku, Kyoto 606-8502, Japan. E-mail: shimokawa@kuchem.kyoto-u.ac.jp; yori@kuchem.kyoto-u.ac.jp
First published on 19th June 2024
Abstract
The tetraalkoxysilane carrying a stable seven-membered dioxasilepane moiety and two trifluoroethoxy groups undergoes reliable iterative substitution of the two trifluoroethoxy groups by sequential treatment with different aryl Grignard reagents while keeping the seven-membered structure intact. The process results in the synthesis of unsymmetrical dialkoxydiarylsilanes and eventually diarylsilanediols after proper hydrolysis.
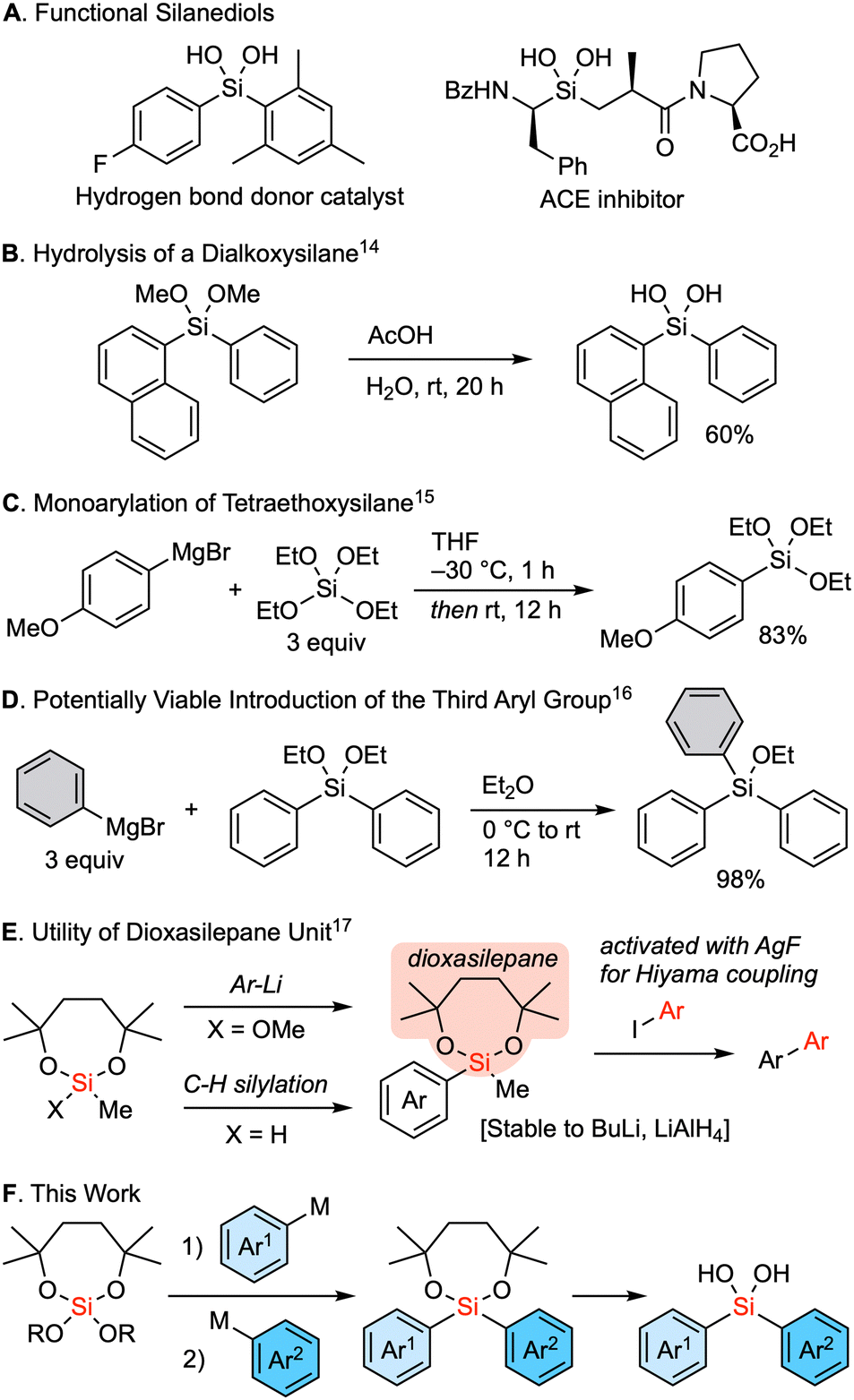
Silanediols constitute a class of compounds characterized by two hydroxy groups attached to a silicon atom. These compounds are known for their unique ability to donate hydrogen bonds and have therefore attracted interest as a core structure of organocatalysts1–7 as well as a substructure of bioactive compounds that is bioisosteric to a hydrated carbonyl group (Fig. 1A).8–13 The hydrolysis of dialkoxysilanes represents a well-established method for the synthesis of these silanediols (Fig. 1B).14 In an ideal scenario, such dialkoxysilanes could be synthesized via sequential reactions from a tetraalkoxysilane and organometallic reagents, wherein two substituents are sequentially introduced to the silicon center. Nevertheless, a significant challenge persists in controlling the sequential substitutions. An earlier solution for the selective synthesis of alkyl or aryl trialkoxysilanes involved the reaction of an excess amount of tetraalkoxysilane with Grignard reagent at low temperature (Fig. 1C).15 The necessity of the excess amount of an alkoxysilane underscores the similar reactivity for the first nucleophilic substitution of an alkoxy group compared to the second introduction of an aryl group to form a dialkoxy diarylsilane. Moreover, dialkoxysilanes readily react with organometallic reagents to introduce the third substituent (Fig. 1D).16 As a result, the controlled stepwise introduction of two different carbon substituents into a tetraalkoxysilane remains a major challenge. Thus, we considered the use of a seven-membered dialkoxysilyl moiety, the dioxasilepanyl group to circumvent this drawback.17 This functional moiety was designed to provide both kinetic and thermodynamic stability to the alkoxysilyl moiety, allowing it to survive the reaction even when using butyllithium or LiAlH4 (Fig. 1E). The seven-membered dialkoxy moiety also imparts unique reactivity to this silyl group. The methoxy group on the silicon atom can be cleanly substituted with an aryl group upon treatment with aryllithiums, and a hydrosilane can be used for C–H silylation of aryls. The silyl group can then be converted via the Hiyama cross-coupling reaction upon activation with AgF. In this context, here we report a reliable strategy for the sequential introduction of different aryl groups by carefully selecting a leaving alkoxy group onto the silicon center of a dioxasilepane moiety. Our method has successfully demonstrated the synthesis of a series of unsymmetrical dialkoxy diarylsilanes, which are practical precursors of diarylsilanediols (Fig. 1F).
 | ||
| Fig. 1 Overview of the current study. | ||
The nucleophilic substitution reaction involving a tetraalkoxysilane 1, bearing a dioxasilepane unit, was initially explored (Scheme 1A). Using a 4-tBu phenyl Grignard reagent, the reaction involving dimethoxy dioxasilepane 1-OMe proceeded, with the methoxy group serving as the leaving group to give 2a-OMe. At the same time, the overreaction yielded diarylsilane 3aa in 25% yield (entry 1). This tendency to give a mixture showed a partial improvement with the ethoxy-substituted 1-OEt as a substrate, decreasing the yield of the diarylsilane 3aa due to the slower introduction of the second aryl group (entry 2). Isopropoxy group as a leaving group was not examined because the substrate 1-OiPr could not be synthesized. Interestingly, the use of the tetraalkoxysilane 1-OTFE, substituted with two trifluoroethoxy groups, showed satisfactory selectivity. Our mechanistic rationale for this phenomenon is that the first arylation of the silicon center is enhanced due to the electron withdrawing nature of the two trifluoroethoxy groups. In addition, the introduction of the second aryl group is slowed by the steric hindrance of the first aryl group and the remaining trifluoroethoxy group, which kinetically delays further nucleophilic attack. Thus, a single substitution product 2a-OTFE was obtained in 93% NMR yield along with a trace amount of 3aa (entry 3). In essence, the combination of the 7-membered ring structure and the trifluoroethoxy group successfully controlled the single substitution reaction. Of note, this seven-membered moiety helps to improve the stability of alkoxysilanes,17 which are normally labile during chromatography and on contact with air and water. It is interesting to note that the substrate with the cyclic 1,3-propanediol diether (1-PDO) only led to the selective formation of disubstituted diarylsilane 3aa. The reaction would probably proceed through the first substitution and the formation of a cyclic biscoordinating magnesium complex, which would further facilitate the leaving of a propanediolate through the second arylation.
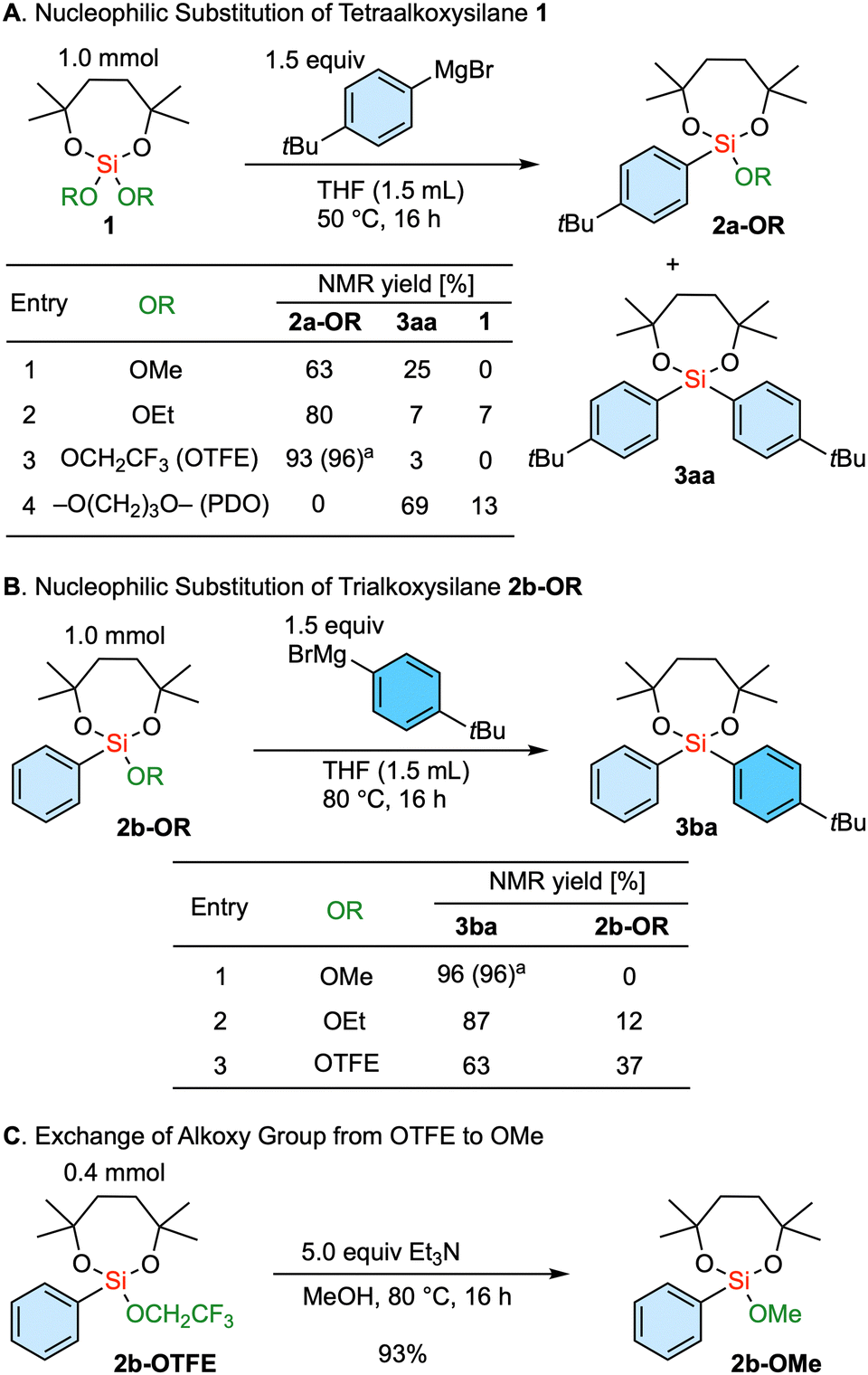 | ||
Scheme 1 Exploration of alkoxy substituents on silicon center for the reaction with Grignard reagents. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Isolated yield. OTFE = 2,2,2-trifluoroethoxy, PDO = 1,3-propanediol. Isolated yield. OTFE = 2,2,2-trifluoroethoxy, PDO = 1,3-propanediol. | ||
Next, we embarked on the exploration of the introduction of the second aryl group (Scheme 1B). In this case, the methoxy-substituted substrate 2b-OMe gave the product 3ba in very high yield (entry 1). In contrast, the ethoxy substrate 2b-OEt resulted in a diminished reactivity as demonstrated above (entry 2), and the trifluoroethoxy-substituted 2b-OTFE underwent the even more sluggish introduction of the second aryl group (entry 3). Thus, the methoxy group serves as the optimal leaving group for the introduction of the second aryl group.
In an attempt to combine these two findings for selective sequential diarylation, the exchange of the remaining alkoxy group from trifluoroethoxy to methoxy was investigated. This transformation was achieved upon treatment with triethylamine in methanol (Scheme 1C).18,19 Therefore, the established protocol involves the use of a trifluoroethoxy group as a leaving group for the first aryl introduction. Subsequently, the remaining alkoxy group is converted to a methoxy group, which is then followed by the introduction of the second aryl group through the substitution of the methoxy group. It is of note that the dioxasilepane moiety remained unaffected during the introductions of the two aryl groups as well as the transalkoxylation. This underlines the fact that the introduction of the third aryl group is completely suppressed in the current process.
Based on the established method, we investigated the introduction of an array of aryl groups to the structure of diaryl silanes. Scheme 2A delineates the reaction of bis(trifluoroethoxy) dioxasilepane 1-OTFE with a variety of aryl Grignard reagents. In this instance, a trace amount of diarylsilane 3 is formed, which complicates the purification of the product 2-OTFE. Consequently, the crude 2-OTFE was directly subjected to transalkoxylation to yield its methoxy form 2-OMe, with the 2-step yields indicated in Scheme 2A. The reaction proceeds with a series of aryl Grignard reagents that are substituted with either electron-donating or electron-withdrawing substituents. The reaction can be scaled up to 10 mmol, providing 2a-OMe in 81% yield. In the case of 4-dimethylaminophenylmagnesium bromide, a decrease in the yield was observed. This phenomenon can be attributed to the decreased efficiency of transalkoxylation due to the increased electron density on the silicon atom, culminating in the sluggish formation of 2g-OMe. In the case of the bulky Grignard reagents, the above protocol resulted in a very slow conversion. Therefore, the more reactive 1-OMe was used for those bulky nucleophiles (Scheme 2B). Thus, single substitution of 1-OMe was observed in the case of 1-naphthyl and o-tolyl Grignard reagents as nucleophiles.
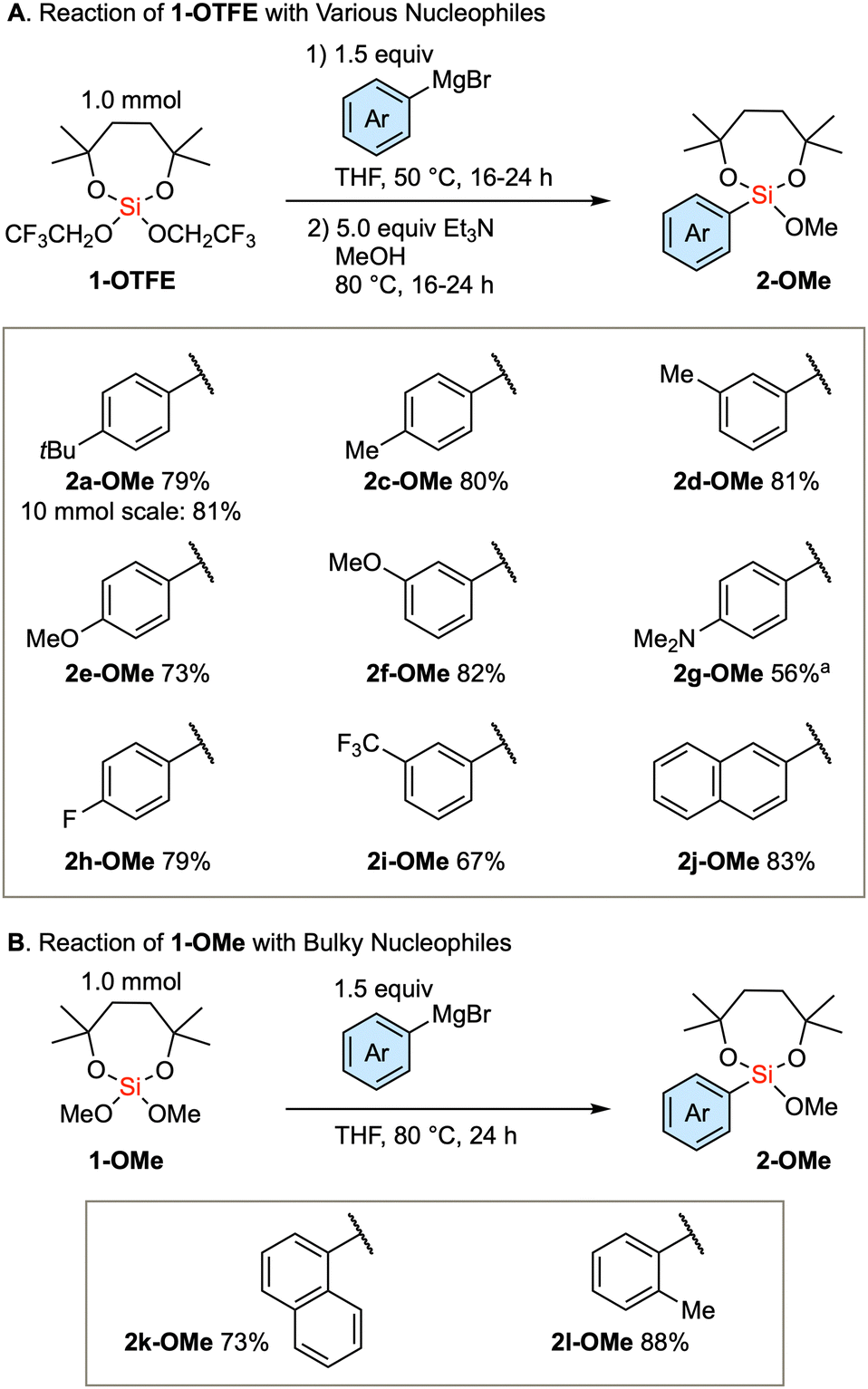 | ||
| Scheme 2 Scope of the first arylation with respect to the substituent on the aryl groups. aSecond step: 72 h. | ||
We subsequently scrutinized the scope of the nucleophilic substitution reaction concerning the structural variation of the initially introduced aryl group, denoted as Ar1, and the second aryl group, Ar2. As the substrate, we employed 2a-OMe (Ar1 = 4-tBu-phenyl), 2b-OMe (Ar1 = phenyl), 2e-OMe (Ar1 = 4-OMe-phenyl), 2h-OMe (Ar1 = 4-F-phenyl), and sterically demanding 2l-OMe (Ar1 = 2-Me-phenyl). In any of these substrates, aryl Grignard reagents (Ar2MgBr) with electron-donating or electron-withdrawing substituents successfully gave the product diarylsilanes (Scheme 3).
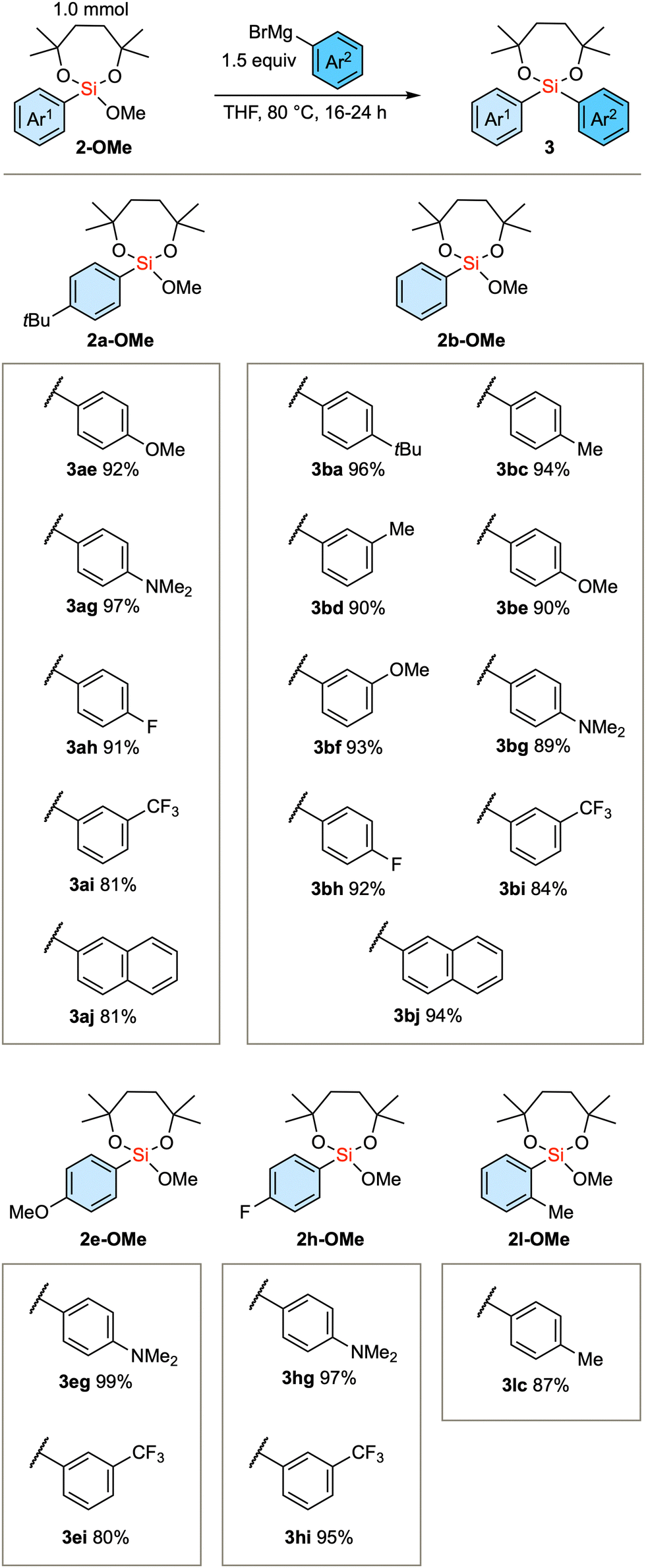 | ||
| Scheme 3 Substrate scope of the reaction with respect to Ar1 and Ar2 groups. | ||
The diarylsilanes synthesized via the aforementioned methodology were subjected to conversion into silanediols (Scheme 4). Due to the confirmed stability of diaryl dioxasilepanes, the removal of the seven-membered structure was initially not successful using the conventional acidic conditions (TsOH, TFA, AcOH, or PPTS in CH2Cl2). Therefore, the removal of the cyclic structure was achieved by first converting the compound to bis-trimethylsilyl ether under conditions using a combination of chlorotrimethylsilane and NaI in acetonitrile to generate Me3SiI in situ.20 The cyclic structure was swiftly removed to afford the bis(trimethylsiloxy)silane intermediate 4. The TMS group can be readily hydrolyzed with aqueous NaOH in acetonitrile under mild basic conditions,21 resulting in the conversion to diarylsilanediols 5. Not only the parent diphenylsilanediol 5bb but also diarylsilanediols such as 5bh, 5aj, 5ah, and 5lc can be synthesized from the corresponding diarylsilane 3.
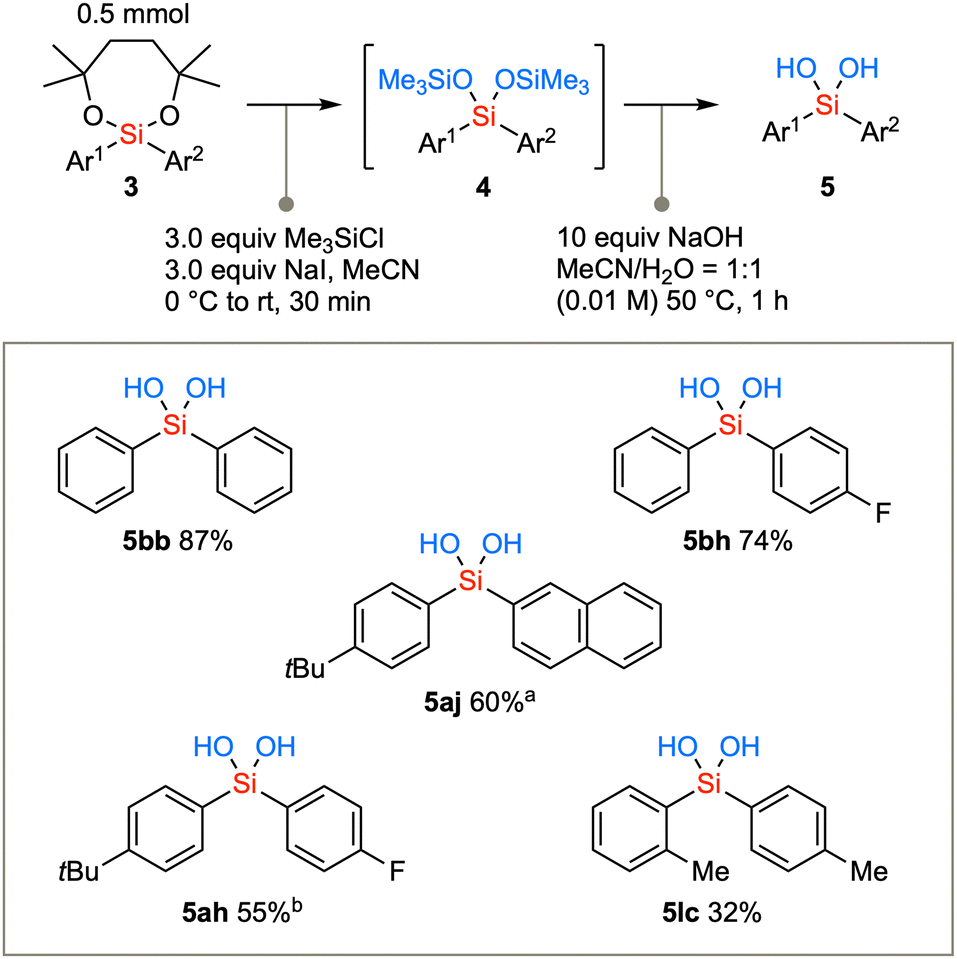 | ||
| Scheme 4 Hydrolysis of diaryl dioxasilepanes via the transetherification generates diarylsilanediols. aReaction time was 6 h for the second step. bReaction time was 2 h for the second step. | ||
In conclusion, we have discovered a method for the controlled nucleophilic substitution of two alkoxy groups on a tetraalkoxysilane having a dioxasilepane moiety. Even two different aryl groups can be introduced sequentially to the central silicon atom. We have also established a method for the cleanly removing the seven-membered ring structure of the product diarylsilane using TMS iodide and a mild conversion method for deprotecting diarylsilanediols. Thus, a general method for the synthesis of a variety of unsymmetrical diarylsilanediols has been established.
This work was supported by JSPS KAKENHI Grant Numbers JP21H01934 and partly by JST CREST Grant Number JPMJCR19R4, Japan. KH acknowledges JST SPRING, Grant Number JPMJSP2110 for financial support.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
-
A. M. Hardman-Baldwin and A. E. Mattson, Science of Synthesis Knowledge Updates, Thieme, Stuttgart, 2017, vol. 2017/1, pp. 213–245 Search PubMed
.
-
A. Leveille and A. Mattson, in Anion-Binding Catalysis, ed. O. G. Mancheño, Wiley-VCH, Weinheim, 2022, 6, pp. 201–220 Search PubMed
- N. T. Tran, T. Min and A. K. Franz, Chem. – Eur. J., 2011, 17, 9897–9900 CrossRef CAS
- A. G. Schafer, J. M. Wieting and A. E. Mattson, Org. Lett., 2011, 13, 5228–5231 CrossRef CAS
- N. T. Tran, S. O. Wilson and A. K. Franz, Org. Lett., 2012, 14, 186–189 CrossRef CAS PubMed
- J. M. Wieting, T. J. Fisher, A. G. Schafer, M. D. Visco, J. C. Gallucci and A. E. Mattson, Eur. J. Org. Chem., 2015, 525–533 CrossRef CAS
- A. M. Hardman-Baldwin, M. D. Visco, J. M. Wieting, C. Stern, S.-I. Kondo and A. E. Mattson, Org. Lett., 2016, 18, 3766–3769 CrossRef CAS PubMed
- C.-A. Chen, S. M. Sieburth, A. Glekas, G. W. Hewitt, G. L. Trainor, S. Erickson-Viitanen, S. S. Garber, B. Cordova, S. Jeffry and R. M. Klabe, Chem. Biol., 2001, 8, 1161–1166 CrossRef CAS PubMed
- M. w Mutahi, T. Nittoli, L. Guo and S. M. Sieburth, J. Am. Chem. Soc., 2002, 124, 7363–7375 CrossRef CAS PubMed
- J. Kim and S. M. Sieburth, J. Org. Chem., 2004, 69, 3008–3014 CrossRef CAS PubMed
- S. M. Sieburth and C. A. Chen, Eur. J. Org. Chem., 2006, 311–322 CrossRef CAS
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed
- R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779–3798 CrossRef CAS PubMed
- J. F. Kannengießer, M. Briesenick, D. Meier, V. Huch, B. Morgenstern and G. Kickelbick, Chem. – Eur. J., 2021, 27, 16461–16476 CrossRef PubMed
- A. S. Manoso, C. Ahn, A. Soheili, C. J. Handy, R. Correia, W. M. Seganish and P. DeShong, J. Org. Chem., 2004, 69, 8305–8314 CrossRef CAS PubMed
- Y. Jorapur and T. Shimada, Synlett, 2012, 1633–1638 CAS
- H. Saito, J. Shimokawa and H. Yorimitsu, Chem. Sci., 2021, 12, 9546–9555 RSC
- N. Li, D.-F. Chen, P.-S. Wang, Z.-Y. Han and L.-Z. Gong, Synthesis, 2014, 1355–1361 Search PubMed
- Detailed screening of the reaction conditions is described in the ESI† (Table S3).
- T. Morita, Y. Okamoto and H. Sakurai, J. Chem. Soc., Chem. Commun., 1978, 874–875 RSC
- A. S. Madsen, H. M. E. Kristensen, G. Lanz and C. A. Olsen, ChemMedChem, 2014, 9, 614–626 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02051k |
| This journal is © The Royal Society of Chemistry 2024 |