
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Reversible oxidative dimerization of 4-thiouridines in tRNA isolates†
Larissa
Bessler
a,
Jonathan
Groß
b,
Christopher J.
Kampf
 b,
Till
Opatz
b and
Mark
Helm
*a
b,
Till
Opatz
b and
Mark
Helm
*a
aInstitute of Pharmaceutical and Biomedical Sciences, Johannes Gutenberg University Mainz, Staudingerweg 5, 55128 Mainz, Germany. E-mail: mhelm@uni-mainz.de
bDepartment of Chemistry, Johannes Gutenberg University Mainz, Duesbergweg 10–14, 55128 Mainz, Germany
First published on 1st February 2024
Abstract
The occurrence of non-canonical nucleoside structures in RNA of biological or synthetic origin has encountered several recent boosts in attention, namely in the context of RNA modifications, and with an eye to RNA vaccines. New nucleoside structures introduce added functionality and function into biopolymers that are otherwise rather homogenous in their chemical structure. Here, we report the discovery of a presumed RNA modification that was identified by combination of liquid chromatography–tandem mass spectrometry (LC–MS/MS) with stable isotope labelling as a dimer of the known RNA modification 4-thiouridine (s4U). The disulfide-linked structure, which had previously been synthetically introduced into RNA, was here formed spontaneously in isolates of E. coli tRNA. Judicious application of stable isotope labelling suggested that this presumed new RNA modification was rather generated ex vivo by oxidation with ambient oxygen. These findings do not only underscore the need for caution in the discovery of new RNA modifications with respect to artifacts, but also raise awareness of an RNA vulnerability, especially to oxidative damage, during its transport or storage.
Introduction
Non-canonical nucleoside structures in native RNA comprise some 170 modified ribonucleoside structures as part of so-called epitranscriptomes, and their chemical diversity is thought to enable control and fine-tuning of a plethora of cellular processes.1–3 Recent developments have shown the tremendous importance even of otherwise rare modifications such as 1-methylpseudouridine (m1Ψ) in biomedical applications.4–7 New biological structures are continuously being discovered, especially in transfer RNA (tRNA) which harbours not only the greatest number of modifications per molecule but also the greatest structural variety of modifications among different RNA species.8–12 Beyond RNA modifications that are enzymatically installed on purpose, the structure of ribonucleosides can also be altered in vivo in the context of RNA damage upon exposure to endogenous and exogenous agents, resulting in alkylated or oxidized nucleosides which hinder RNA in its proper functioning and are associated with a variety of pathophysiological conditions.13,14 Yet further, in addition to structural alterations resulting in both, RNA modifications and damaged nucleosides that are introduced in vivo, the storage and handling of RNA can also lead to structural aberrations ex vivo. For example, new non-canonical nucleosides were found to be formed in vitro during RNA isolation, storage or sample preparation.15–17 Improved instrumentation and lowered detection limits have enabled the detection of ever more nucleoside species. Elucidation of such structures is important to sort genuine artifacts from biological or chemical damage. A very recent example illustrates the importance of the respective analytical chemistry of new structures in quality control of medicinal RNA.18 Many artifacts resulting from deamination, Dimroth rearrangement, oxidation or hydrolysis may also occur as genuine modifications in native RNA whereas a minor fraction consists of non-natural structures, which were shown to be generated in vitro.16,19,20 The occurrence of such structures may not only lead to wrong appreciation of the abundance of known modifications but can also confuse and impede the LC–MS-based identification of new RNA modifications.16,21 More important to the wider community, certain structures can also impair functionality of the sample in downstream experiments or applications.22 One example scrutinised in this context is 8-oxo-guanine (8-oxo-G), the flagship among oxidative lesions in both DNA and RNA, which originates from guanine both in vivo e.g. resulting from reaction with reactive oxygen species (ROS) and in vitro, either by addition of oxidizing agents or as a consequence of sample preparation and storage.17,23–25 As a case in point, 8-oxo-G sites may serve as an example of the effects of RNA lesions on their functionality. Thus, in mRNAs used in in vitro translation systems or being administered to living cells, 8-oxo-G residues were demonstrated to not only reduce translational efficiency and induce ribosome stalling,22,26,27 but also lead to rapid RNA degradation by surveillance mechanisms.28–32 Especially in view of the high therapeutic potential of mRNA as showcased in the form of vaccines during the SARS-CoV-2 pandemic, it is important to consider the effects of manufacturing, long-term storage, and contact with atmospheric oxygen on the integrity of an RNA sample. A pertinent example is the recent discovery of lipid adducts of vaccine mRNA upon extended storage accompanied by significant reduction of translation efficacy.18 The above examples underline the necessity to improve detection and structure elucidation techniques for new nucleoside species, and to aim for a comprehensive understanding of all processes contributing to the chemical space of nucleoside species encountered in vivo and in vitro, be their origin of biological or synthetic nature.We have scrutinised the nucleoside content of tRNA isolates from E. coli for novel structures using a specifically designed LC–MS regimen termed neutral loss scan (NLS, vide infra), and previously reported on two newly identified modification types, along with three dozen candidates of yet unidentified structures.11,33,34 Here we report on the pursuit of a group of four unusual LC–MS signals, which we found to originate from what was initially a putatively new RNA modification. Its structure turned out to be an oxidatively damaged dimeric form of s4U, linked via a disulfide. Isotope labelling experiments argue for formation of this nucleoside structure in vitro, namely upon storage in the presence of ambient oxygen. This constitutes first experimental evidence hinting at possible storage artifacts in RNA samples containing sulphur, particularly s4U, e.g. in correspondingly labelled samples intended for bioconjugation, RNA enrichment, photo-crosslinking or RNA lifetime analysis.35–40
Results and discussion
In previous studies we established an NLS method for the highly sensitive identification of potentially new ribonucleoside structures, using a triple quadrupole (QQQ) mass spectrometer coupled to an LC system. Accordingly, we exploited the predominant fragmentation of the vast majority of nucleoside compounds at the N-glycosidic bond which leads to the (neutral) loss of the ribose moiety and is accompanied by a mass shift of −132 Da.11,34 In order to account for bulky hypermodifications, we set the upper mass limit to an m/z value of 620 Da. After several filtering steps, the most promising candidates, named according to their mass-to-charge ratios, were subjected to further characterisation, which included several types of MS experiments. While the QQQ-NLS method was used to screen for nucleoside species (vide supra) and to determine mass shifts in labelling experiments, we also used the QQQ in a multistage fragmentation approach, a so-called pseudo–MS3 scan, by combining a product ion scan with increased fragmentor voltages, in order to induce in-source fragmentation at the most labile bond. The fragmentation patterns obtained from such experiments can provide important information on certain functional groups within an unknown structure. Another powerful tool in the course of the characterisation of candidate structures was high-resolution mass spectrometry (HRMS), which provided exact masses and therefore allowed the calculation of potential ion formulas which can be transferred into potential structure proposals. Of note, due to sensitivity issues, both the QQQ-pseudo MS3 scan and the HRMS experiments require higher input of starting material than the initial NLS. A practical solution consisted in collecting fractions of candidates and pooling them after evaporation of the eluate to obtain enriched samples.11 At the onset of the present study, within the data set of potentially new ribonucleoside structures, we noticed an early eluting peak at 14.6 min and a later eluting peak at 25.7 min in our reversed-phase chromatography which both were characterised by the simultaneous detection of the same two ions with a mass-to-charge ratio of 541 and 557, respectively (Fig. 1a and d). The exact retention times varied according to the type of experiment, given the use of differentially configured instrumentation (e.g. QQQ and HRMS), reversed-phase columns and associated gradients. Hence, for the sake of simplicity, these are referred to as the early and the late peak in the following; methodological details on each experiment are provided in the experimental section. Interestingly, further characterisation of the candidates 541 and 557 by stable isotope labelling revealed identical composition, i.e. 18 carbon atoms, four nitrogen atoms and two sulphur atoms, not only for the late and the early peak of each candidate individually but also for both candidates (Fig. 1b, e and Fig. S1a, d, ESI†). On one hand this indicated that the different elution times might be related to different conformations of the same structure, but on the other hand this also drew our attention to a possible relation between the candidates 541 and 557. Typically, the use of NH4OAc for precipitation of nucleic acids during sample preparation efficiently minimises adduct formation of nucleoside species and alkali ions during the MS analysis. However, in this case the mass difference of 16 between candidates 541 and 557 demanded to consider the possibility of an adduct formation with the alkaline metal ions Na+ and K+. Going back to the NLS dataset, a closer inspection did indeed reveal a minute peak of m/z 519, corresponding to the related H+ species at both retention times (Fig. S2, ESI†). Further characteristics shared among the four species were found in stable isotope labelling, specifically during an inverse feeding approach. Here, supplementation of a 15N-labelled E. coli culture with 14N-uridine, identified a pyrimidine species as the underlying nucleoside for all candidates (Fig. 1c, f and Fig. S1b, e, ESI†). Of note, due to the intertwined metabolic pathways yielding cytidine and uridine, it is not possible to distinguish between the two pyrimidine species within the used approach. In a next step, we collected fractions of the eluate around the early retention time of the candidates for enrichment and reinjected this sample for a pseudo–MS3 scan. For both candidates, the fragmentation spectra pointed to the loss of a second ribose moiety after the initial loss of the ribose during in-source fragmentation (Fig. 1g–i), further substantiating the assumed relation between candidates 541 and 557. In combination with their comparatively high number of carbon atoms, these findings indicated a potentially dimeric character (Fig. 1h) for both candidates which, due to the limited number of nitrogen atoms, increased the plausibility of a uridine-based dimer. Another intriguing observation during the pseudo-MS3 experiment was the fact that reinjection of material collected from either early or late peak resulted in renewed occurrence of two peaks, between which the later was predominant (Fig. S1c and f, ESI†). Given the previously described evidence for a relation of both species with the same m/z, this finding strongly implied that these presumably different isomers of the same structure might be interconvertible conformers. These findings were recapitulated in different LC–MS settings and corresponding experiments (Fig. 2a (541) and Fig. S3, ESI† (557)), and thus demonstrated that the early and the late eluting compounds are indeed related interconvertible species, presumably conformers. Given that the late peak was consistently predominant in all reinjection experiments, it represents the more stable form under the pertinent experimental conditions. Collected early fractions were also submitted to analysis by high-resolution mass spectrometry (HRMS), and the ensuing calculation of complete sum formulas and thereby unravelled candidates 541 and 557 to be sodium and potassium adducts of a compound with m/z 519 (Fig. 2b). Interestingly, there was a very pronounced difference in the distribution of protonated versus adduct species when comparing analyses from the HRMS instrument (Fig. 2b) and the QQQ instrument (Fig. S2, ESI†) which is most likely a system specific phenomenon but might also be traced to smallest changes in different batches of the mobile phases. Since QQQ measurements were used for further MS analyses and both candidates were shown to be adducts of the same compound with the sodium adduct being the most abundant species during QQQ analysis, further characterisation experiments are representatively shown for this candidate.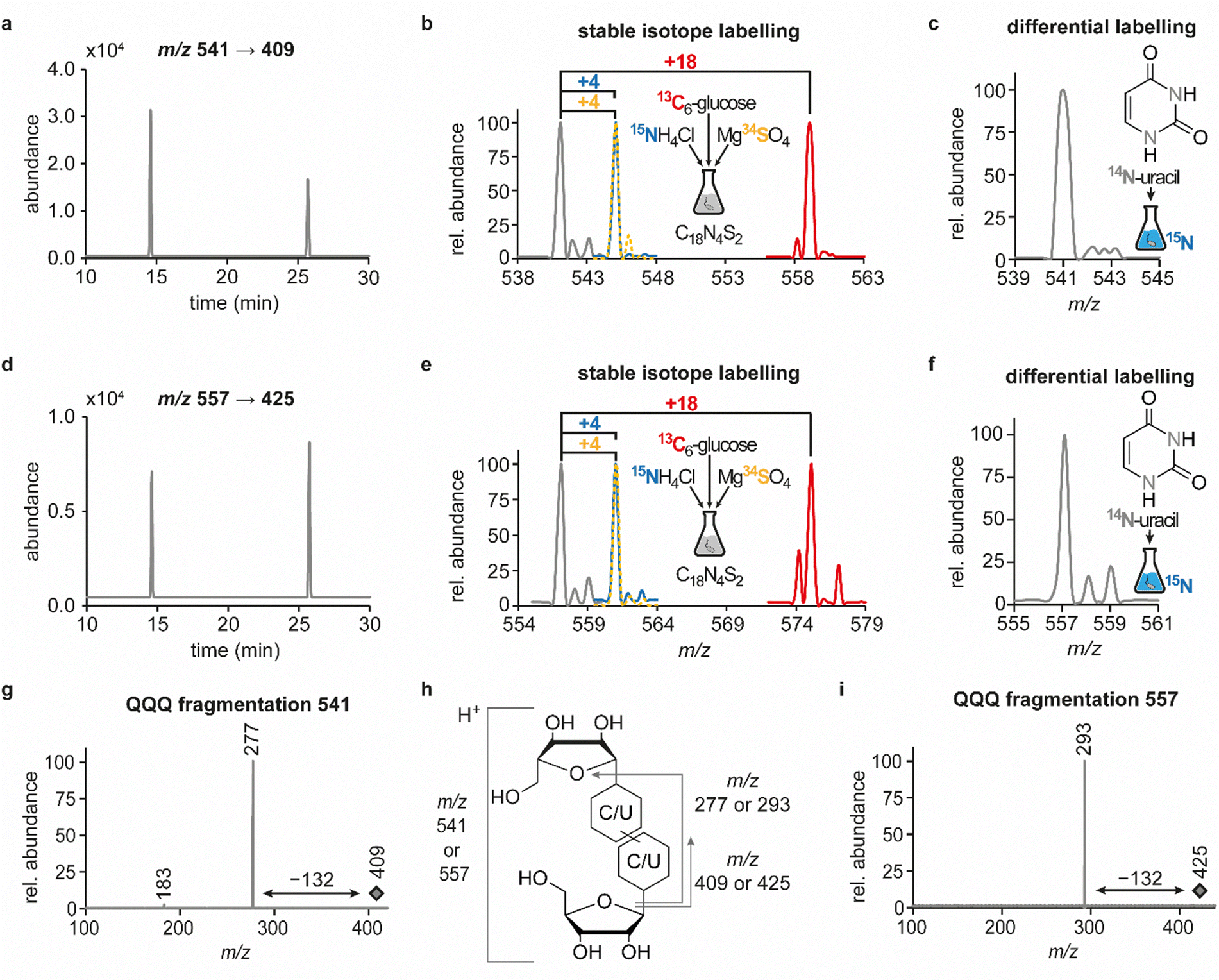 | ||
| Fig. 1 Structural characterisation of candidates 541 and 557 by LC–MS/MS. Abundances were set in relation to the highest peak or signal in the respective extracted ion chromatogram (EIC) or mass spectrum (relative abundance). (a) EIC at m/z 541 from a neutral loss (−132) measurement of an E. coli tRNA hydrolysate. (b) Overlay of QQQ mass spectra at 25.7 min corresponding to the late peak of candidate 541, recorded from an NLS of a hydrolysate of unlabelled (grey) or labelled (13C = red, 15N = blue, 34S = yellow) E. coli tRNA. (c) QQQ NLS mass spectrum for the peak at 25.7 min, recorded from hydrolysed E. coli tRNA isolated from a 15N-labelled culture that was supplemented with 14N-uridine and displayed for candidate 541. (d) EIC at m/z 557 from a neutral loss (−132) measurement of an E. coli tRNA hydrolysate. (e) Overlay of QQQ mass spectra at 25.7 min corresponding to the late peak of candidate 557, recorded and displayed as described in (b). (f) QQQ NLS mass spectrum for the peak at 25.7 min for the sample described in c, displayed for candidate 557. (g) QQQ fragmentation mass spectrum for the precursor m/z 409 at 25.7 min, resulting from the compound with m/z 541 after in-source fragmentation (pseudo-MS3 scan) and illustrating the potential loss of another ribosyl moiety (h) Scheme to illustrate the different product ions occurring in QQQ fragmentation spectra of candidates 541 and 557. (i) QQQ fragmentation mass spectrum for the precursor m/z 425 at 25.7 min, resulting from the compound with m/z 557 after in-source fragmentation, similarly to (g), illustrating the potential loss of another ribosyl moiety. | ||
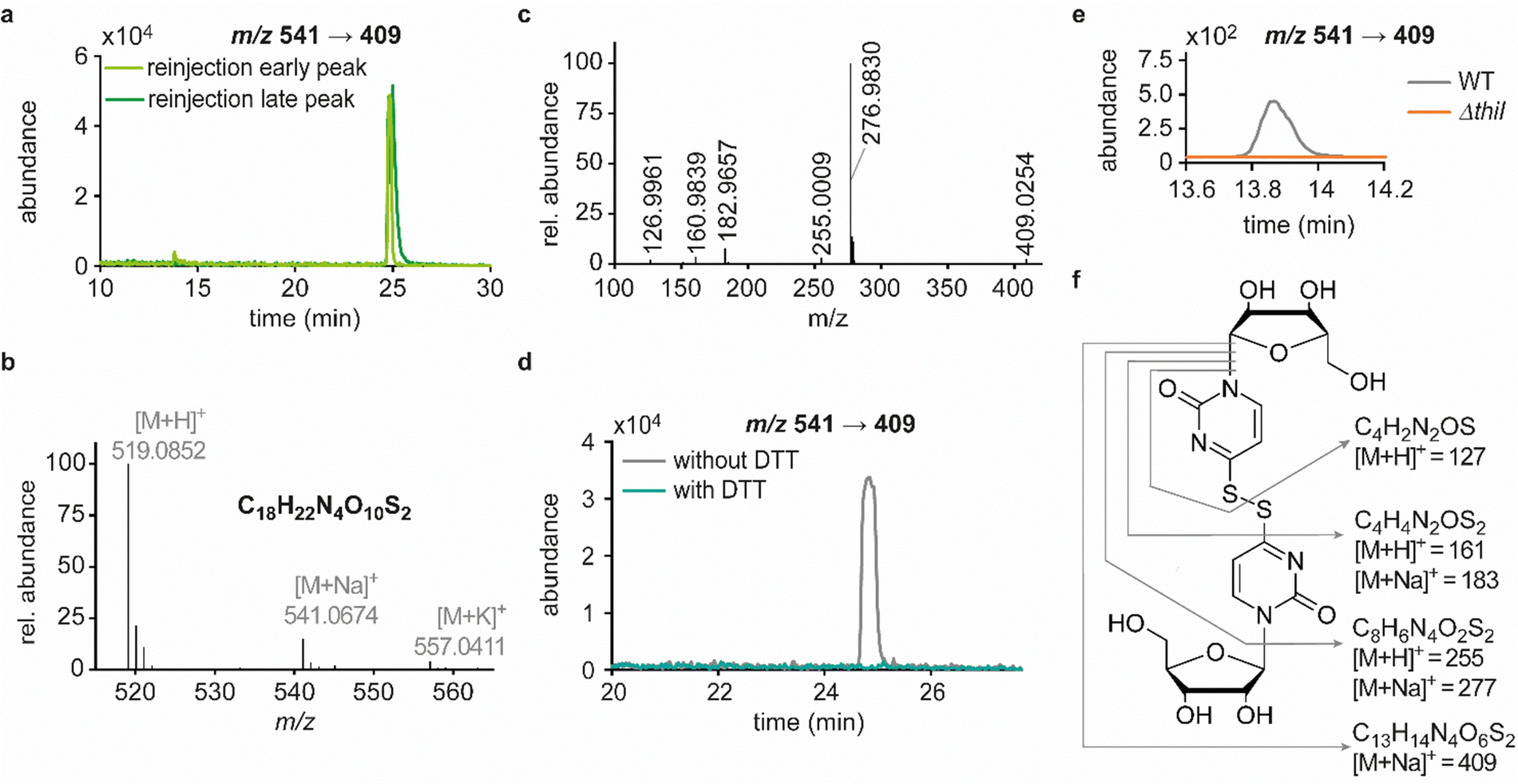 | ||
| Fig. 2 Elucidation of structural details of candidate 541. Abundances were set in relation to the highest peak or signal in the respective extracted ion chromatogram (EIC) or mass spectrum (relative abundance). (a) Merged extracted ion chromatograms (m/z 541 → 409) for reinjection of the separately collected early (light green) and the late (dark green) peak. (b) Mass spectrum and sum formula calculated for the species of interest from the exact mass detected by HRMS analysis of digested tRNA enriched for candidates 541 and 557. (c) HRMS fragmentation pattern of candidate 541. Sum formulas calculated from exact masses are displayed in panel f of this figure. (d) Merged EICs for the mass transition m/z 541 → 409 in a QQQ measurement of hydrolysed E. coli tRNA as an untreated reference sample (grey) and a sample of collected candidate 541 after dithiothreitol (DTT) treatment (teal). (e) Occurrence of candidate 541 (early RT) in a QQQ measurement of tRNA isolated from the Keio wild type (WT) strain and the thiouridine-related knockout strain ΔthiI. (f) Structure proposal for candidate 541 with product ions and their sum formulas being assigned within this structure. | ||
In line with the earlier observation of two consecutive ribose losses in the QQQ fragmentation spectrum for candidate 541, its behaviour during HRMS fragmentation (Fig. 2c) further substantiated the presumed dimeric character since the sum formula calculated for the main fragment (m/z = 276.9830, C13H7N4O2S2+) corresponded to the loss of a second ribosyl moiety. Moreover, the calculated sum formulas of other fragments, which mostly occurred as both the sodium adduct and the protonated species, indicated that even with progressing fragmentation the two sulphur atoms remained within the structure. Consequently, we considered a uridine-derived dimer, linked via a disulfide, a plausible structure proposal. In keeping with this presumed structure, collected fractions, upon treatment with dithiothreitol (DTT) did not show any more signals for either of the four initial candidates (Fig. 2d). Thus, in the course of further structural elucidation, the two sulphur atoms came to the fore and their position within the molecule had to be determined. In combination with the elemental composition of the potentially new structure, the modifications 2-thiouridine (s2U) and s4U were of particular interest and theoretically plausible structures included their homodimers as well as a heterodimer, both linked via a disulfide bond. Since s2U very rarely occurs as such in physiological conditions but rather appears in the context of complex hypermodifications,41 we initially focused on the involvement of s4U and investigated the effect of a single gene knockout (KO) of its modifying enzyme ThiI on the levels of candidate 541 in tRNA. Importantly, in tRNA isolated from an E. coli ThiI knockout the candidate was completely absent (Fig. 2e) which suggested that the potentially dimeric structure arises from s4U as a homodimer consisting of two s4U monomers whose respective 4-positions are linked to each other via a disulfide. The exact masses of product ions obtained during HRMS fragmentation (Fig. 2c) together with the ion formulas calculated from them were consistent with this structure proposal (Fig. 2f). For validation, we synthesized the s4U-dimer by treatment of s4U with an I2-KI-solution (Fig. 3a), following a protocol by Fox et al.,42 and spiked a hydrolysate of 13C-labelled E. coli tRNA with this synthetic (12C) dinucleoside, to collate their chromatographic behaviour. Here, we observed perfectly matching retention times of the synthetic dimer and the compound occurring in the E. coli tRNA at 25.8 min (Fig. 3b), providing ultimate proof that the structure of candidate 541 is a s4U-homodimer linked via a disulfide bridge. While in earlier experiments we usually observed the joint occurrence of the early and the late eluting substance with m/z 541 (Fig. 1a), interestingly, the extracted ion chromatogram for the synthetic compound exclusively showed the late peak. Of note, the absence of the early peak suggests that the molecular mechanism of the I2-KI oxidation specifically yields a single conformer, which did not equilibrate before analysis. In general, the appearance of two different conformations of the supposed structure was plausible with regard to an earlier study by Irie et al. who reported an asymmetric nature of this disulfide and thus two different conformers.43 With regard to the biological importance of these findings it is important to mention that already in the 1960s and 1970s the formation of such a structure in tRNA as a result of its exposure to an I2-KI-solution was reported.44 In order to test if the disulfide dimers detected back then coincided with the detection of candidate 541, we treated E. coli tRNA analogously with an I2-KI-solution and analysed a hydrolysate of accordingly treated tRNA by LC–MS. This analysis showed significantly increased levels of candidate 541 (Fig. 3c), again with an exclusive occurrence in the late eluting form. Importantly, although the incubation with I2-KI-solution stimulated an increased formation of disulfide dimers, in contrast to the above mentioned earlier studies, we also observed the occurrence of this dimeric nucleoside without further treatment of the tRNA in the present case.
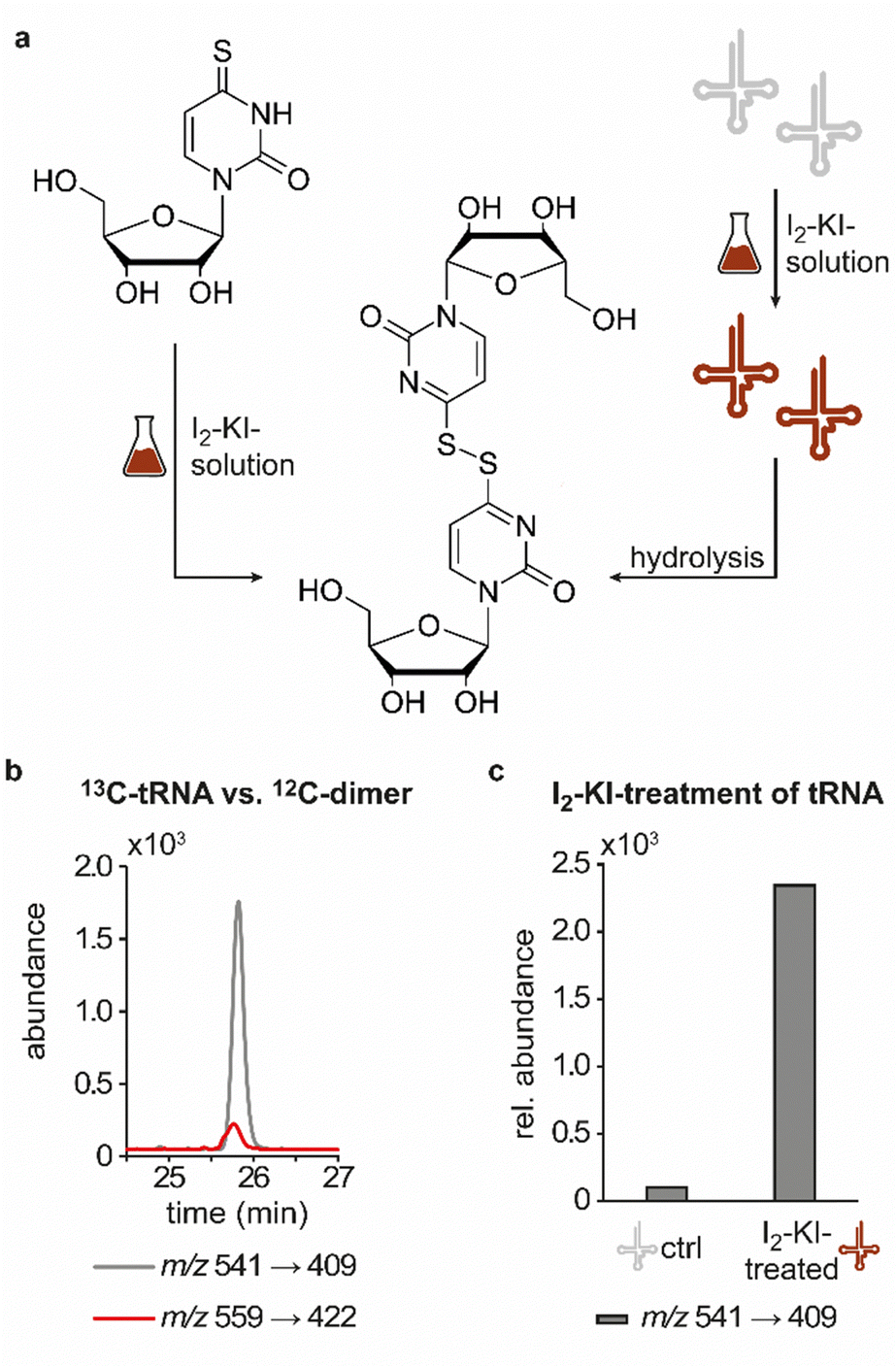 | ||
| Fig. 3 Structural confirmation of the disulfide dimer. (a) Scheme displaying the synthesis of the disulfide dimer via treatment of 4-thiouridine with I2-KI-solution (left) and induced formation of the disulfide dimer in tRNA via its treatment with I2-KI-solution with release of the dinucleoside after hydrolysis (right). (b) QQQ LC–MS/MS analysis of a digested 13C-labelled tRNA sample (red) spiked with the synthetic 12C-dimer (grey). An overlay of the extracted ion chromatograms at a mass transition of m/z 541 → 409 and m/z 559 → 422 is shown. (c) Occurrence of candidate 541 in a QQQ measurement of commercial tRNA as an untreated control (ctrl) sample and commercial tRNA treated with I2-KI-solution. The abundance of candidate 541 was normalised to the UV-signal of adenosine in the respective sample and the control sample set to 100%. | ||
In light of previous reports on diverse circumstances affecting the integrity of RNA modifications,15,16 we saw a need to examine whether the disulfide dimer was generated in vivo or in vitro. Therefore, we separately grew an unlabelled and a 15N-labelled E. coli culture and mixed these samples for the isolation and hydrolysis procedure (Fig. 4). A dimer formed in vivo in a biological context, would yield signals corresponding only to compounds containing only a single of the two nitrogen isotopes, i.e. m/z 541 (unlabelled) and m/z 545 (15N-labelled). In contrast, artificial formation in vitro would be expected to yield, in addition to these pure dimers, a signal of an m/z of 543 mixed dimer, containing equal amounts of 14N and 15N. Indeed, the occurrence of such a mixed dimer was observed within the described experiment, and at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 (Fig. 4) that is compatible with what would be expected for exclusive formation of dimers in vitro. This confers the character of an artifact rather than a genuine RNA modification to this non-canonical nucleoside species, described and structurally elucidated within this study. Consequently, the absence of the candidate in the ThiI KO is merely due to the fact that the artificial formation of the dimer requires the presence of s4U. Importantly, when we probed for dimer formation after incubation of s4U nucleoside under the conditions of hydrolysis, we did not observe any formation of the disulfide (data not shown), indicating that the oxidation occurs on the intact tRNA.
:2:1 (Fig. 4) that is compatible with what would be expected for exclusive formation of dimers in vitro. This confers the character of an artifact rather than a genuine RNA modification to this non-canonical nucleoside species, described and structurally elucidated within this study. Consequently, the absence of the candidate in the ThiI KO is merely due to the fact that the artificial formation of the dimer requires the presence of s4U. Importantly, when we probed for dimer formation after incubation of s4U nucleoside under the conditions of hydrolysis, we did not observe any formation of the disulfide (data not shown), indicating that the oxidation occurs on the intact tRNA.
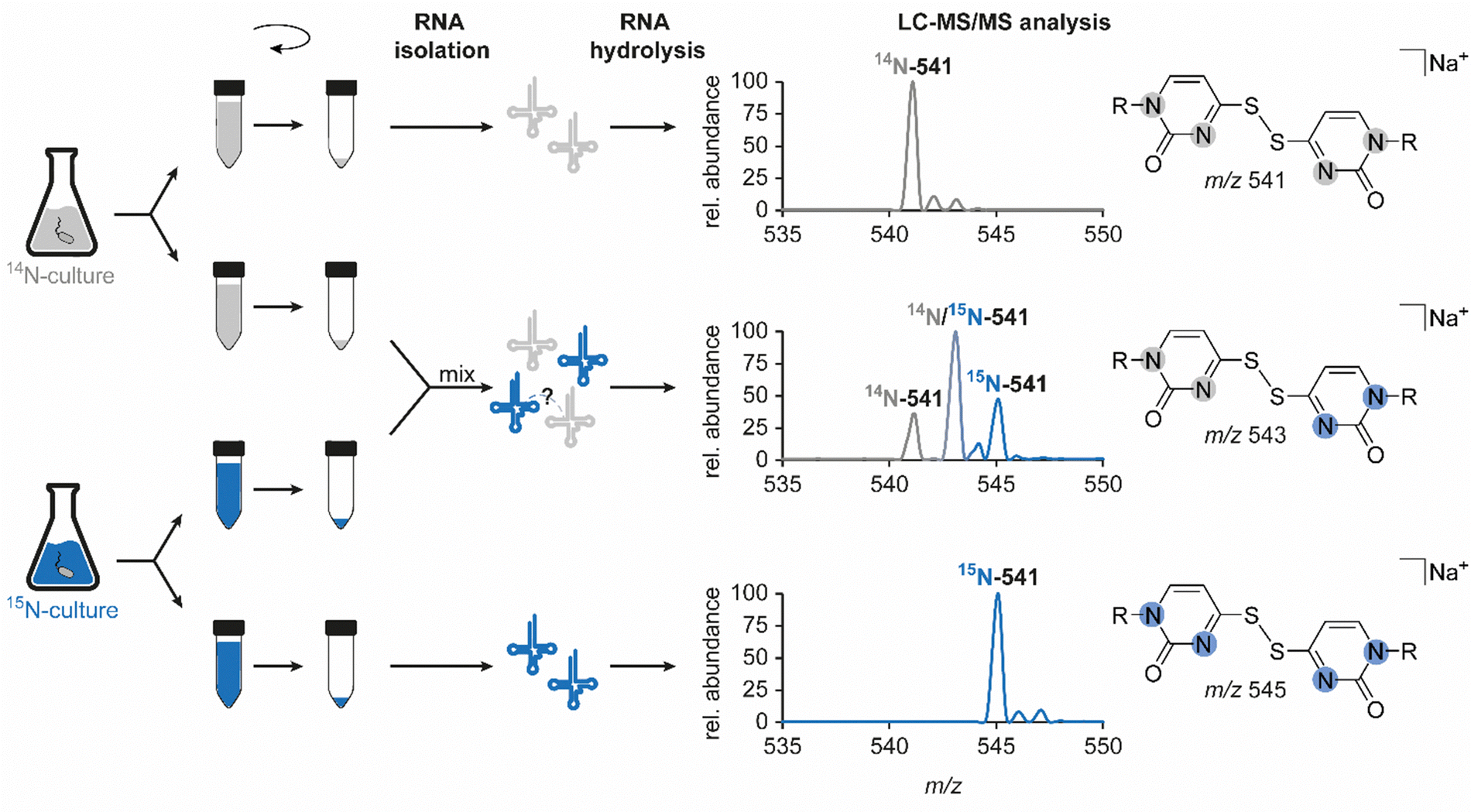 | ||
| Fig. 4 Examination of the authenticity of candidate 541 as a genuine RNA modification. Left: Schematic workflow to examine a potential artificial formation of the dimer during RNA isolation or sample preparation. Right: Results of the LC–MS/MS analysis for respective samples, illustrated by the mass spectrum at the corresponding retention time which was extracted from the neutral loss scan data (−132 Da). 14N-541 describes the mass transition of unlabelled candidate 541 (m/z 541 → 409), 15N-541 corresponds to the mass transition of the uniformly 15N-labelled compound (m/z 545 → 413) and the mixed dimer, half unlabelled and half labelled, is illustrated as 14N/15N-541 (m/z 543 → 411). Abundances were set in relation to the highest signal in the respective mass spectrum (relative abundance). | ||
Conclusions
In search of new RNA nucleoside structures, we detected and structurally elucidated what turned out to be a disulfide-linked dimer of 4-thiouridine in bacterial tRNA. While this structure was not covered in the portfolio of (native) RNA modifications known to date,3 early experiments on the oxidation of tRNA concluded the formation of a thiouridine-dimer upon oxidation of tRNA with I2-KI-solution. Reported biological consequences of such oxidative tRNA dimerization included loss of acceptor activity.44–47 While those early studies attached a biological significance to the dimeric structure, hypothesizing that it might constitute a reversibly deactivated tRNA with regulatory function,46 our data (Fig. 4) is fully consistent with oxidative dimerization occurring after RNA isolation. Specifically, any dimerization having occurred in vivo would have been expected to shift the ratio of the observed dimers away from the 14N–15N hybrid species to the homogeneously labelled 14N–14N and 15N–15N isotopologues. Given that in vivo conditions are typically reductive, and that we have recapitulated reductive cleavage of the dimer by thiols,44,47 we conclude that the dimerization took place in vitro, presumably during work-up and storage, with the oxidative dimerization likely caused by ambient oxygen. Our data suggests that we are looking at two conformers of the same compound, whose interconversion barrier is high enough that they elute in separate peaks, i.e. they do not interconvert during chromatography. Interestingly, oxidation by I2-KI exclusively yields the late peak, while air oxidation also produces some of the early peak, presumably because mechanistic details of both oxidation reactions differ. Oxidation in air has been considered a potential problem in protocols exploiting the chemical biology of thiouridines, but no experimental data nor structures were put forward.36,38,40 Our present work fills this gap and provides a characterisation of this process that is relevant for, among others, medicinal applications of thio-containing RNAs. Indeed, s2U containing mRNA was found to suppress TLR-mediated cytokine emission in dendritic cells.48 Given that, in the case at hand, formation of the disulfide linkage occurs ex vivo, storage time and conditions of RNA must be considered in general. Even though the consequences in the present case may be limited to a minor falsification of s4U levels, oxidative damage to thio-containing RNA during its transport or storage may have dramatic effects on its functionality and will thus have to be subject of thorough studies in the future.Experimental section
RNA preparation
In order to prepare RNA samples, E. coli cells (strain MC4100) were cultured in standard M9-medium which contained 6.8 g L−1 Na2HPO4 (Carl Roth, Germany), 3 g L−1 KH2PO4 (Carl Roth, Germany), 0.5 g L−1 NaCl (Carl Roth, Germany), 1 g L−1 NH4Cl (Merck-Millipore, Germany), 2 mM MgSO4 (Carl Roth, Germany), 0.1 mM CaCl2 (Grüssing GmbH, Germany) and 0.4% glucose (Carl Roth, Germany). For unlabelled RNA, the standard M9-medium was used, whereas single ingredients, namely glucose, NH4Cl or MgSO4 were replaced with their 13C-, 15N- or 34S-labelled counterparts (all Sigma-Aldrich, Germany), to obtain 13C-, 15N- or 34S-labelled RNA. For differential nucleobase labelling, 15N-M9 medium was prepared and the E. coli culture was supplemented with either uracil (Carl Roth, Germany), guanine (Carl Roth, Germany) or adenine (Sigma-Aldrich, Germany) in a final concentration of 300 μM.Respective E. coli cultures were grown to an OD600 of 1.8, the cells harvested (10 min, 8000 g, 4 °C) and subjected to RNA extraction using TRI reagent® (Sigma-Aldrich, Germany) according to the manufacturer's protocol.
For E. coli single gene knockout experiments, we made use of the E. coli Keio knockout collection (GE Healthcare (Dharmacon™), United Kingdom) and used the parent strain (BW25113) as well as the ΔthiI strain (b0423). The E. coli cells were cultured in standard M9-medium and RNA was isolated.
Oxidation of tRNA in vitro
Following the protocol published by Carbon et al., commercial tRNA (Sigma Aldrich, Germany) was dissolved to a concentration of 5 mg mL−1 in 0.01 M Tris–HCl (pH 7).46 After addition of an equal volume of 1 mM iodine in 0.5% KI-0.01 M Tris–HCl (pH 7), the mixture was incubated on ice for 20 min. Subsequently, the tRNA was precipitated twice by addition of 1/10 volume of 5 mM NH4OAc and 2.5 volumes of ice-cold ethanol, respectively.Synthesis of the s4U-dimer
The reaction previously described by Fox et al. was carried out in a sub-milligram scale using 102.5 μL of an aqueous solution of s4U (10 mM), 25 μL Sörensen phosphate buffer (pH 6.8) and 0.5 μL of 1 N iodine solution.42 A colourless solid precipitated, the supernatant was discarded after centrifugation and the product was lyophilized. In order to obtain an amount sufficient for NMR analysis, the synthesis and purification procedure were repeated several times and pooled products were dissolved in DMSO-d6.NMR spectra were recorded on a Bruker Avance III spectrometer (Bruker Corporation, USA). Chemical shifts were reported in parts per million (ppm) relative to tetramethylsilane and internally referenced to DMSO-d6 (2.50 ppm for 1H-NMR and 39.5 ppm for 13C-NMR). 1H–1H COSY, 1H–13C HSQC and 1H–13C HMBC spectra provided the basis for signal assignment. Coupling constants J are given in Hz and multiplicities were abbreviated as follows: d = doublet, dd = doublet of doublets, dt = doublet of triplets.
1H-NMR (600 MHz, DMSO-d6): δ = 8.53 (d, J = 7.1 Hz, 2H, H-6,6′), 6.79 (d, J = 7.1 Hz, 2H, H-5,5′), 5.70 (d, J = 2.6 Hz, 2H, H-7,7′), 3.98 (dd, J = 4.5, 2.6 Hz, 2H, H-8,8′), 3.94 (dd, J = 6.9, 4.5 Hz, 2H, H-9,9′), 3.91 (dt, J = 6.9, 2.6 Hz, 2H, H-10,10′), 3.74 (dd, J = 12.4, 2.6 Hz, 2H, H-11a,11′a), 3.59 (dd, J = 12.4, 2.7 Hz, 2H, H-11b,11′b) ppm.
13C-NMR (151 MHz, DMSO-d6): δ = 174.0 (2C, C-4,4′), 152.4 (2C, C-2,2′), 144.3 (2C, C-6,6′), 99.8 (2C, C-5,5′), 90.7 (2C, C-7,7′), 84.1 (2C, C-10,10′), 74.4 (2C, C-8,8′), 68.1 (2C, C-9,9′), 59.4 (2C, C-11,11′) ppm.
RNA hydrolysis to nucleoside level
Prior to its LC–MS/MS analysis, total tRNA (up to 10 μg) was digested to nucleoside level by incubation with the following enzymes and deaminase inhibitors in 25 mM ammonium acetate (pH 7.5; Sigma-Aldrich, Germany) overnight at 37 °C: 0.6 U nuclease P1 from P. citrinum (Sigma-Aldrich, Germany), 0.2 U snake venom phosphodiesterase from C. adamanteus (Worthington, USA), 2 U FastAP (Thermo Fisher Scientific, Germany), 10 U benzonase (Sigma-Aldrich, Germany), 200 ng Pentostatin (Sigma-Aldrich, Germany) and 500 ng Tetrahydrouridine (Merck-Millipore, Germany).LC–MS experiments
For all LC–MS/MS measurements an Agilent 1260 Infinity (II) series HPLC in combination with either an Agilent 6460A or 6470B triple quadrupole (QQQ) mass spectrometer or a high-resolution Agilent 6545 quadrupole time-of-flight (Q-ToF) mass spectrometer, each of them equipped with an Agilent Jet Stream electrospray ionisation source (ESI), was used (all devices: Agilent Technologies, Germany). Initially and mostly, a YMC-Triart C8 column (150 × 3 mm, 3 μm, 120 Å; YMC Europe GmbH, Germany) at a flow of 0.35 mL min−1 and a temperature of 35 °C was used for separation, which was later on replaced by a Synergi Fusion RP-C18 column (250 × 2.0 mm, 4 μM, 80 Å; Phenomenex, Germany) that was employed under the same conditions and whose use is stated accordingly. The mobile phase consisted of 5 mM ammonium acetate buffer (pH 5.3; solvent A) and LC–MS grade acetonitrile (solvent B; Honeywell). Starting at 100% solvent A, in a first step of the gradient, solvent B was linearly increased to 10% at 20 min. After another linear increase of solvent B to 100% at 50 min, which was held for 3 min, the initial conditions were restored within 7 min and the column re-equilibrated at 100% solvent A for 5 min. With regard to the use of different instrumentation or columns, it is important to mention, that the identity of the candidates was ensured by comparing the results of stable isotope labelled samples and the retention time relative to known modifications. The LC systems contained a diode array detector (DAD) which allowed to monitor the main nucleosides at a wavelength of 254 nm (UV trace). All mass spectrometers were operated in the positive ion mode and details on respective source parameters are provided in Table 1.| Parameters | 6460A (QQQ) | 6470B (QQQ) | 6545 (Q-ToF) |
|---|---|---|---|
| Gas temperature | 350 | 300 | 325 |
| Gas flow (L min−1) | 5 | 7 | 10 |
| Nebulizer pressure (psi) | 35 | 60 | 33 |
| Sheath gas temperature (°C) | 350 | 400 | 325 |
| Sheath gas flow (L min−1) | 12 | 12 | 10 |
| Capillary voltage (V) | 3500 | 3000 | 3500 |
In order to compare the chromatographic behaviour of the candidate occurring in RNA and the synthetic s4U-dimer, 5 μg of hydrolysed 13C-labelled RNA and 1 pmol of synthetic s4U-dimer were co-injected and separated on the C18 column. During this analysis the 6460A QQQ was used with the Agilent MassHunter software (version B.05.00) being employed in the dynamic multiple reaction monitoring (dMRM) mode. The mass transitions for the predefined nucleosides were as follows: 541-12C (m/z 541 → 409) and 541-13C (m/z 559 → 422). Further dMRM settings were a fragmentor voltage of 80 V, a collision energy of 15 eV and a cell accelerator voltage of 2 V.
For analysis of the oxidized RNA samples obtained after I2-KI-treatment of tRNA, 5 μg of hydrolysed RNA sample were injected into the LC–MS system. Here, the C18 column was used for separation and the 6470B QQQ (Agilent MassHunter software, version 10.0) was used in the dMRM mode, detecting the mass transition of candidate 541 (m/z 541 → 409). Using the Agilent MassHunter Qualitative Analysis software (version 10.0), the peaks of interest were integrated and the total area under the curve (abundance) resulting from addition of the respective area under the curve of the early and the late peak was normalised to the peak area of adenosine in this sample, to account for different amounts of RNA.
Fragmentation patterns of candidates 541 and 557 were recorded in so-called pseudo-MS3 scans by using the 6460A QQQ and the Agilent MassHunter software (version B.05.00) in the product ion scan mode with an increased fragmentor voltage of 200 V. The latter leads to an in-source fragmentation of the nucleosides yielding nucleobases in the MS1 detector which filters for the programmed precursor ions m/z 409 and m/z 425. In the collision cell the nucleobase was subjected to further fragmentation by application of a collision energy of 35 eV (candidate 541) and a collision energy of 20 eV (candidate 557), resulting in a product ion spectrum. The QQQ settings were similar to the NLS except for the scan time which was changed to 500 ms and the MS2 scan range which was changed to m/z 15-220. Due to sensitivity issues, the candidates were enriched by collecting the fraction of 50 μg hydrolysed and chromatographically separated E. coli RNA around the elution time of the early peak five times (vide infra). After evaporation of the solvent, the dry residues of the fractions were pooled in MilliQ water.
In order to identify the exact masses of candidates 541 and 557, the Agilent MassHunter software (version 10.0) was employed in the MS mode from m/z 100 to m/z 1000 at a scan rate of 1 spectrum per s. Based on the experimentally detected mass, the sum formula calculator within the software allowed calculation of the exact mass and the corresponding sum formula.
HRMS (ESI): m/z calcd for C18H22N4O10S2: 519.0850 [M + H]+, found: 519.0852; m/z calcd for C18H22N4O10S2: 541.0670 [M + Na]+, found: 541.0674; m/z calcd for C18H22N4O10S2: 557.0409 [M + K]+, found: 557.0411.
The exact masses of the fragments (identified by pseudo-MS3 analysis) were determined by use of the Agilent MassHunter software (version 10.0) in the targeted MS2 mode. The precursor ion used in this analysis was m/z 541.0700 and the centroid mode was used for data acquisition with the following method settings: MS m/z 100–1000, MS scan rate 1 spectrum per s, MS/MS m/z 30–500, MS/MS scan rate 1 spectrum per s, max time between MS 5 s and use of fixed collision energies (35 eV).
HRMS (ESI): m/z calcd for C4H2N2OS: 126.9961 [M + H]+, found: 126.9964; m/z calcd for C4H4N2OS2: 160.9838 [M + H]+, found: 160.9839; m/z calcd for C4H4N2OS2: 182.9657 [M + Na]+, found: 182.9657; m/z calcd for C8H6N4O2S2: 255.0005 [M + H]+, found: 255.0009; m/z calcd for C8H6N4O2S2: 276.9824 [M + Na]+, found: 276.9830; m/z calcd for C13H14N4O6S2: 409.0247 [M + Na]+, found: 409.0254.
Author contributions
L. Bessler performed the majority of the experimental work except for NMR analysis which was performed by J. Groß and T. Opatz as well as HRMS measurements which were performed by C. J. Kampf. All authors discussed the results. L. Bessler and M. Helm wrote the manuscript which was reviewed and edited by all authors. M. Helm designed and supervised the work.Conflicts of interest
Mark Helm is a consultant for Moderna Inc.Acknowledgements
The authors would like to thank Dr Christina dal Magro, Dr Patrick Keller and Dr Annika Kotter for helpful discussions. This work was funded by the DFG (HE 3397/14-2 in SPP1784 and TP C03 in TRR 319; Project-ID 439669440) to M. H.References
- T. Suzuki, Nat. Rev. Mol. Cell Biol., 2021, 22, 375–392 CrossRef CAS PubMed.
- W. V. Gilbert and S. Nachtergaele, Annu. Rev. Biochem., 2023, 92, 175–198 CrossRef CAS PubMed.
- P. Boccaletto, F. Stefaniak, A. Ray, A. Cappannini, S. Mukherjee, E. Purta, M. Kurkowska, N. Shirvanizadeh, E. Destefanis, P. Groza, G. Avşar, A. Romitelli, P. Pir, E. Dassi, S. G. Conticello, F. Aguilo and J. M. Bujnicki, Nucleic Acids Res., 2022, 50, D231–D235 CrossRef CAS PubMed.
- P. Morais, H. Adachi and Y.-T. Yu, Front. Cell Dev. Biol., 2021, 9, 789427 CrossRef PubMed.
- K. D. Nance and J. L. Meier, ACS Cent. Sci., 2021, 7, 748–756 CrossRef CAS PubMed.
- I. Freund, T. Eigenbrod, M. Helm and A. H. Dalpke, Genes, 2019, 10, 92 CrossRef CAS PubMed.
- K. Karikó and D. Weissman, Curr. Opin. Drug Discovery Dev., 2007, 10, 523–532 Search PubMed.
- M. Jora, D. Corcoran, G. G. Parungao, P. A. Lobue, L. F. L. Oliveira, G. Stan, B. Addepalli and P. A. Limbach, Anal. Chem., 2022, 94, 13958–13967 CrossRef CAS PubMed.
- T. Ohira, K. Minowa, K. Sugiyama, S. Yamashita, Y. Sakaguchi, K. Miyauchi, R. Noguchi, A. Kaneko, I. Orita, T. Fukui, K. Tomita and T. Suzuki, Nature, 2022, 605, 372–379 CrossRef CAS PubMed.
- V. F. Reichle, D. P. Petrov, V. Weber, K. Jung and S. Kellner, Nat. Commun., 2019, 10, 5600 CrossRef CAS PubMed.
- C. Dal Magro, P. Keller, A. Kotter, S. Werner, V. Duarte, V. Marchand, M. Ignarski, A. Freiwald, R.-U. Müller, C. Dieterich, Y. Motorin, F. Butter, M. Atta and M. Helm, Angew. Chem., Int. Ed., 2018, 57, 7893–7897 CrossRef CAS PubMed.
- E. M. Phizicky and J. D. Alfonzo, FEBS Lett., 2010, 584, 265–271 CrossRef CAS PubMed.
- A. Nunomura, P. I. Moreira, R. J. Castellani, H.-G. Lee, X. Zhu, M. A. Smith and G. Perry, Neurotoxic. Res., 2012, 22, 231–248 CrossRef CAS PubMed.
- E. J. Wurtmann and S. L. Wolin, Crit. Rev. Biochem. Mol. Biol., 2009, 44, 34–49 CrossRef CAS PubMed.
- K. Miyauchi, S. Kimura and T. Suzuki, Nat. Chem. Biol., 2013, 9, 105–111 CrossRef CAS PubMed.
- M. Jora, K. Borland, S. Abernathy, R. Zhao, M. Kelley, S. Kellner, B. Addepalli and P. A. Limbach, Angew. Chem., Int. Ed., 2021, 60, 3961–3966 CrossRef CAS PubMed.
- N. C. de Souza-Pinto and V. A. Bohr, Int. Rev. Neurobiol., 2002, 53, 519–534 CrossRef CAS PubMed.
- M. Packer, D. Gyawali, R. Yerabolu, J. Schariter and P. White, Nat. Commun., 2021, 12, 6777 CrossRef CAS PubMed.
- J. B. Macon and R. Wolfenden, Biochemistry, 1968, 7, 3453–3458 CrossRef CAS PubMed.
- P. F. Crain, Methods Enzymol., 1990, 193, 782–790 CAS.
- S. Kaiser, S. R. Byrne, G. Ammann, P. Asadi Atoi, K. Borland, R. Brecheisen, M. S. DeMott, T. Gehrke, F. Hagelskamp, M. Heiss, Y. Yoluç, L. Liu, Q. Zhang, P. C. Dedon, B. Cao and S. Kellner, Angew. Chem., Int. Ed., 2021, 60, 23885–23893 CrossRef CAS PubMed.
- M. Tanaka, P. B. Chock and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 66–71 CrossRef CAS PubMed.
- C. M. Gedik and A. Collins, FASEB J., 2005, 19, 82–84 CrossRef CAS PubMed.
- M. Liu, X. Gong, R. K. Alluri, J. Wu, T. Sablo and Z. Li, Biol. Chem., 2012, 393, 123–132 CrossRef CAS PubMed.
- Z. Li, J. Wu and C. J. Deleo, IUBMB Life, 2006, 58, 581–588 CrossRef CAS PubMed.
- X. Shan, H. Tashiro and C. G. Lin, J. Neurosci., 2003, 23, 4913–4921 CrossRef CAS PubMed.
- X. Shan, Y. Chang and C. G. Lin, FASEB J., 2007, 21, 2753–2764 CrossRef CAS PubMed.
- L. L. Yan and H. S. Zaher, J. Biol. Chem., 2019, 294, 15158–15171 CrossRef CAS PubMed.
- J. Y. Hahm, J. Park, E.-S. Jang and S. W. Chi, Exp. Mol. Med., 2022, 54, 1626–1642 CrossRef CAS PubMed.
- T. Ishii and M. Sekiguchi, DNA Repair, 2019, 81, 102666 CrossRef CAS PubMed.
- C. L. Simms and H. S. Zaher, Cell. Mol. Life Sci., 2016, 73, 3639–3653 CrossRef CAS PubMed.
- C. L. Simms, B. H. Hudson, J. W. Mosior, A. S. Rangwala and H. S. Zaher, Cell Rep., 2014, 9, 1256–1264 CrossRef CAS PubMed.
- L. Bessler, L.-M. Vogt, M. Lander, C. Dal Magro, P. Keller, J. Kühlborn, C. J. Kampf, T. Opatz and M. Helm, Angew. Chem., Int. Ed., 2023, 62, e202217128 CrossRef CAS PubMed.
- S. Kellner, J. Neumann, D. Rosenkranz, S. Lebedeva, R. F. Ketting, H. Zischler, D. Schneider and M. Helm, Chem. Commun., 2014, 50, 3516–3518 RSC.
- B. Reichholf, V. A. Herzog, N. Fasching, R. A. Manzenreither, I. Sowemimo and S. L. Ameres, Mol. Cell, 2019, 75, 756–768.e7 CrossRef CAS PubMed.
- J. A. Schofield, E. E. Duffy, L. Kiefer, M. C. Sullivan and M. D. Simon, Nat. Methods, 2018, 15, 221–225 CrossRef CAS PubMed.
- P. Bommisetti and V. Bandarian, ACS Omega, 2022, 7, 4011–4025 CrossRef CAS PubMed.
- E. E. Duffy and M. D. Simon, Curr. Protoc. Chem. Biol., 2016, 8, 234–250 CrossRef PubMed.
- K. Schmid, M. Adobes-Vidal and M. Helm, Bioconjugate Chem., 2017, 28, 1123–1134 CrossRef CAS PubMed.
- J. Spitzer, M. Hafner, M. Landthaler, M. Ascano, T. Farazi, G. Wardle, J. Nusbaum, M. Khorshid, L. Burger, M. Zavolan and T. Tuschl, Methods Enzymol., 2014, 539, 113–161 CAS.
- R. Schaffrath and S. A. Leidel, RNA Biol., 2017, 14, 1209–1222 CrossRef PubMed.
- J. J. Fox, D. van Praag, I. Wempen, I. L. Doerr, L. Cheong, J. E. Knoll, M. L. Eidinoff, A. Bendich and G. B. Brown, J. Am. Chem. Soc., 1959, 81, 178–187 CrossRef CAS.
- S. Irie, Y. Inoue and F. Egami, J. Biochem., 1968, 63, 274–276 CrossRef CAS PubMed.
- M. N. Lipsett, J. Biol. Chem., 1967, 242, 4067–4071 CrossRef CAS PubMed.
- E. Shefter and T. I. Kalman, Biochem. Biophys. Res. Commun., 1968, 32, 878–884 CrossRef CAS PubMed.
- J. A. Carbon, L. Hung and D. S. Jones, Proc. Natl. Acad. Sci. U. S. A., 1965, 53, 979–986 CrossRef CAS PubMed.
- M. N. Lipsett and B. P. Doctor, J. Biol. Chem., 1967, 242, 4072–4077 CrossRef CAS PubMed.
- K. Karikó, M. Buckstein, H. Ni and D. Weissman, Immunity, 2005, 23, 165–175 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00221g |
| This journal is © The Royal Society of Chemistry 2024 |