
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN 4-Allylcytidine: a new nucleoside analogue for RNA labelling and chemical sequencing†
Tengwei
Li
a,
Xiao
Shu
a,
Minsong
Gao
a,
Chenyang
Huang
a,
Ting
Li
a,
Jie
Cao
ab,
Xiner
Ying
a,
Donghong
Liu
a and
Jianzhao
Liu
 *ab
*ab
aMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Yuhangtang Road 866, Hangzhou 310058, Zhejiang Province, China. E-mail: liujz@zju.edu.cn
bLife Sciences Institute, Zhejiang University, Yuhangtang Road 866, Hangzhou 310058, Zhejiang Province, China
First published on 22nd November 2023
Abstract
RNA labelling has become indispensable in studying RNA biology. Nucleoside analogues with a chemical sequencing power represent desirable RNA labelling molecules because precise labelling information at base resolution can be obtained. Here, we report a new nucleoside analogue, N4-allylcytidine (a4C), which is able to tag RNA through both in vitro and in vivo pathways and further specifically reacts with iodine to form 3, N4-cyclized cytidine (cyc-C) in a catalyst-free, fast and complete manner. Full spectroscopic characterization concluded that cyc-C consisted of paired diastereoisomers with opposite chiral carbon centers in the fused 3, N4-five-membered ring. During RNA reverse transcription into complementary DNA, cyc-C induces base misincorporation due to the disruption of canonical hydrogen bonding by the cyclized structure and thus can be accurately identified by sequencing at single base resolution. With the chemical sequencing rationale of a4C, successful applications have been performed including pinpointing N4-methylcytidine methyltransferases’ substrate modification sites, metabolically labelling mammalian cellular RNAs, and mapping active cellular RNA polymerase locations with the chromatin run-on RNA sequencing technique. Collectively, our work demonstrates that a4C is a promising molecule for RNA labelling and chemical sequencing and expands the toolkit for studying sophisticated RNA biology.
Introduction
In order to reveal diverse facets of cellular RNA biology,1 RNA labelling has emerged as an indispensable tool for visualizing RNA locations, monitoring RNA dynamics and studying its interactions with other cellular components, thereby shedding light on its functional intricacies.2 The in vitro solid phase synthesis allows for efficient and site-specific attachment of functional tags to RNA oligonucleotides;3 however, some modified phosphonamidite substrates do not withstand the harsh chemical conditions and the length of RNA oligonucleotides synthesized is restricted due to a progressive decrease in the coupling efficiency with each iteration of nucleotide addition.4 Alternatively, the in vitro transcription method utilizes RNA polymerases to introduce functional modifications on RNA,5 but it does not facilitate site-specific labelling.6 Moreover, enzymes can be used to label RNA post-synthetically.7,8 For instances, N6-methyladenosine methyl transferases and demethylases, named METTL3–METTL14, METTL16 and FTO, have been utilized to label RNA by introducing functional groups to the N6-position of adenosine at a specific location of the RNA chain.9–12RNA metabolic labelling strategies have gained extensive attention in recent years.13 Nucleoside analogues are incorporated into newly transcribed RNA through the nucleotide salvage pathway. From the structural point of view, these analogues act as functional tags because they are either immunoprecipitable or capable of reacting with reporters via click reactions. For instance, 5-bromouridine (5-BrU)14 is employed as an immunoprecipitable tag, and specific antibodies facilitate the selective capture of 5-BrU-labelled messenger RNA (mRNA) from total RNA. 4-Thiouridine (4sU)-labelled RNAs can be post-modified by a 2-pyridylthio-activated disulfide of biotin15 or methylthiosulfonate-activated biotin16 for further affinity-based purification via biotin–avidin interaction. The analogues for fluorescent labelling generally contain an alkyne group, such as 5-ethynyluridine,17 2-azidocytidine18 and N6-propargyladenosine.19–21 RNAs labelled with these analogues can further be conjugated with an azide-decorated fluorophore via copper-catalyzed azide–alkyne cycloaddition and be quantified by fluorescence assays.
Among the available analogues, the ones with a chemical sequencing power offer distinct advantages as they provide base resolution labelling information from RNA sequencing in an enrichment-free manner. Typically, they undergo chemical treatments to become another form that induce base misincorporation during RNA reverse transcription (RT), enabling accurate identification of labelled transcripts in next-generation sequencing data. Chemical treatments of 4sU through thiol-linked alkylation,22 oxidative-nucleophilic-aromatic substitution,23 or thiol–ene addition24 have led to significant U-to-C mutations in RNA sequencing.25,26 In the same way, 6-thioguanosine (6sG) can be converted to 2-aminoadenosine by the oxidative-nucleophilic-aromatic substitution chemistry.27,28 In addition, N6-allyladenosine (a6A) can be transformed into 1, N6-cyclized adenosine (cyc-A) under mild iodine treatment, and the resultant cyc-A induces base mismatch during RNA RT.29,30 To date, there have been very few examples of nucleosides with chemical sequencing capability, thus it is necessary to expand the toolkit for miscellaneous applications of RNA labelling in different biological contexts.
Here we developed a cytidine analogue named N4-allylcytidine (a4C) with a chemical sequencing power and demonstrated its RNA labelling applications. Under mild iodine treatment without a catalyst, a4C was specifically, efficiently and completely transformed into 3, N4-cyclized cytidine (cyc-C), which was structurally characterized as a pair of diastereomers and could induce base mismatch during RNA RT. With the above rationale for chemical sequencing of RNA a4C label at single base resolution, successful applications were performed including pinpointing N4-methylcytodine (m4C) methyltransferases’ substrate modification sites, metabolically labelling mammalian cellular RNAs, and mapping active cellular RNA polymerase locations with the chromatin run-on RNA sequencing technique.
Results and discussion
Chemical synthesis and structural characterization of a4C and a4CTP.
The synthetic route of a4C and a4C triphosphate (a4CTP) is shown in Fig. 1A. First, 2′,3′,5′-triacetyluridine (1) was reacted with tetrazole to produce 4-(tetrazol-1-yl)-1-(2′,3′,5′-tri-O-acetyl-β-D-ribofuranosyl) pyrimidine-2-(1H)-one (2) in 80% yield. Afterwards, compound 2 was subjected to an alkaline-mediated nucleophilic substitution reaction in the presence of allylamine hydrochloride, leading to the formation of 2′,3′,5′-tri-O-acetyl-N4-allylcytidine (3) in 74% yield. Compound 3 underwent deacetylation through treatment with 2 M NH3 in methanol to generate a4C (4) in a quantitative yield. a4C was then reacted with POCl3 and tributyl ammonium pyrophosphate (TBAPP) to afford N4-allylcytidine triphosphate (a4CTP, 5) in 51% yield following the previously reported procedure. All these compounds were thoroughly characterized by standard spectroscopies (Fig. S1–S10, ESI†) including 1H nuclear magnetic resonance spectroscopy (NMR), 13C NMR and high-resolution mass spectrometry (HRMS), and proved to be structurally correct.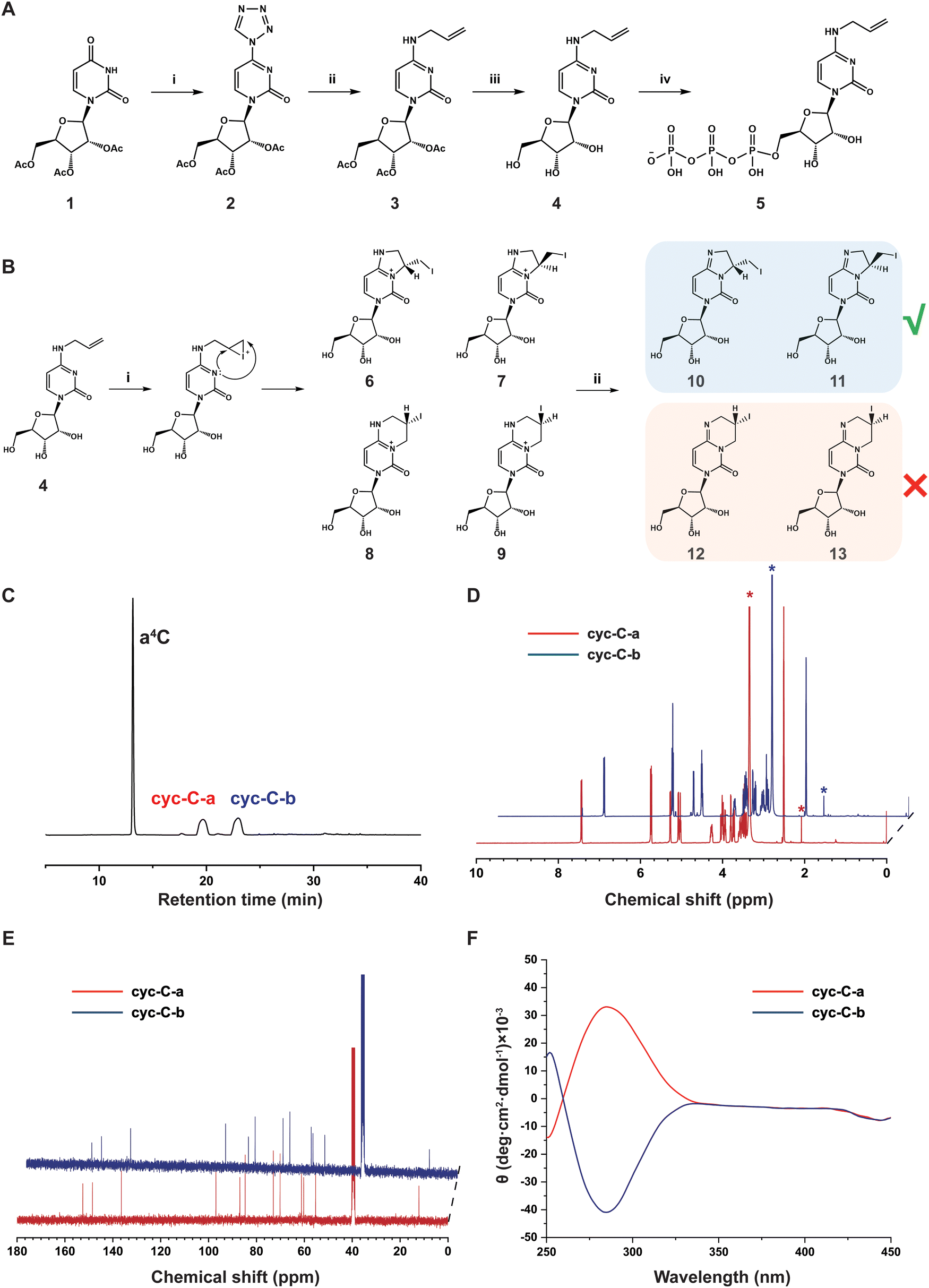 | ||
| Fig. 1 Synthesis of N4-allylcytidine (a4C) derivatives and their structural characterization. (A) Synthesis of a4C (4) and a4CTP (5). Reagents and conditions: (i) tetrazole, p-toluenesulfonyl chloride (TsCl), diphenyl phosphate, pyridine, room temperature (rt), 24 h; (ii) allylamine hydrochloride, KOH, Et3N, CH3CN, rt, 24 h; (iii) 2 M NH3 in CH3OH, rt, overnight; (iv) POCl3, (MeO)3PO, 0 °C, 3 h; TBAPP, DMF, 0 °C, 1 h; triethylammonium bicarbonate (TEAB), rt, 5 min. (B) Model reactions for conversion of a4C into 3, N4-cyclized cytidine (cyc-C). Reagents and conditions: (i) I2, KI, 40 °C, 1 h. (ii) Na2S2O3, Na2CO3, 40 °C, 30 min. (C) HPLC traces of iodination-induced cyclization products of a4C. The two peaks (cyc-C-a and cyc-C-b) correspond to two cyclized diastereomers. (D) 1H NMR spectra (400 MHz, DMSO-d6) of cyc-C-a and cyc-C-b. The asterisks (*) denote the signals from H2O and acetonitrile. (E) 13C NMR spectra (100 MHz, DMSO-d6) of cyc-C-a and cyc-C-b. (F) Circular dichroism (CD) spectra of cyc-C-a and cyc-C-b in dimethyl sulfoxide (DMSO) at a concentration of 0.5 mg mL−1. | ||
Iodination-mediated conversion of a4C into cyc-C and spectroscopic characterization of the cyc-C structure
As shown in Fig. 1B, it was anticipated that the vinyl group would be easily iodinated via the iodonium intermediate, leading to the generation of cyc-C through nucleophilic attack by the nitrogen at position 3 in the pyrimidine ring on the iodonium-connected carbons. Given that both the iodonium-connected carbons could serve as the potential reaction sites, two distinct types of precursor cyc-C might be formed with five- (6/7) and six-membered (8/9) rings, respectively. Notably, each type of precursor cyc-C was expected to exhibit a pair of diastereomers with opposite chiral carbon centres in the fused 3, N4-ring of cytidine. Under Na2CO3 treatment, deprotonation of precursor cyc-C (6/9) would lead to the stable formation of cyc-C, which corresponds to either diastereomer pair 10/11 with a fused five-membered ring or 12/13 with a fused six-membered ring. In order to determine the exact structure of cyc-C, high-performance liquid chromatography (HPLC) was first employed to analyze the composition of the reaction product. Two retention peaks labelled as cyc-C-a and cyc-C-b were observed in the chromatogram using an ultraviolet detector (Fig. 1C). Each fraction was collected and lyophilized for further NMR and HRMS characterization (Fig. S11–S18, ESI†). Based on the result of HRMS, these two compounds displayed identical molecular weight (Fig. S13 and S17, ESI†). Initially, we hypothesized that cyc-C-a and cyc-C-b corresponded to structures featuring five- and six-membered rings. However, upon examination of the standard 1H and 13C NMR spectra, it was found that cyc-C-a and cyc-C-b exhibited the same spectral characteristics (Fig. 1D and E), thereby indicating that our initial hypothesis was incorrect. The distortionless enhancement by polarization transfer (DEPT) spectra of cyc-C-a and cyc-C-b (Fig. S14 and S18, ESI†) also showed the same, and the peak at around 12 ppm in the DEPT corresponded to the secondary carbon atom bonded to the iodine atom, suggestive of the formation of a five-membered ring. Based on the above-mentioned evidence, both compounds were inferred to possess the same chemical constitution but have different configurations in the formed five-membered ring. To prove this possibility, the circular dichroism (CD) spectra were obtained. The data indeed revealed that cyc-C-a and cyc-C-b represented a diastereomer pair and preferentially absorbed left- and right-handed light, respectively (Fig. 1F). Up to now, the a4C-to-cyc-C conversion has been successfully characterized in detail by comprehensive spectroscopic techniques.Chemical sequencing assay for a4C in RNA
The chemical sequencing power of a4C label in RNA was explored. Previous studies have provided evidence that RNA polymerases could efficiently accommodate N4-acetylcytidine nucleotide derivatives as substrates.31 It is comprehensible that the single substitution at the N4-position of cytosine has little interference in the hydrogen bonding of cytosine with guanine. In contrast, from a structural point of view, as the hydrogen bonding sites of cyc-C were occupied, base misincorporation at its opposite site can be readily induced during RT and further detected in complementary DNA (cDNA) sequencing. It was also reported that the cellular RNA base lesion product 3, N4-ethenocytosine (εC) had similar five-membered ring compared with cyc-C and could induce RT mismatch.32 Based on these facts, we hypothesized that a4C and cyc-C could exhibit completely different base pairing behaviors, and the a4C-to-cyc-C axis could be utilized to identify RNA labelling sites in a reliable way.Next, we studied the base mismatch pattern of a4C and cyc-C in model RNA probes. The flowchart of a4C chemical sequencing assay is shown in Fig. 2A. Using the in vitro transcription method, an a4C-containing 327-nt RNA probe (a4C-probe) was synthesized by substituting CTP with a4CTP. The obtained a4C-probe was digested into nucleosides and analyzed by HPLC. Compared to the cytosine-containing probe (C-probe), the a4C-probe showed an obvious a4C peak, indicating the successful incorporation of the a4C probe (Fig. S19, ESI†). A portion of the obtained a4C-probe was subjected into iodination-induced cyclization to generate the cyc-C-probe. The C-probe, a4C-probe and cyc-C-probe were reversely transcribed into cDNA using commercial HIV recombinant reverse transcriptase (HIV) and its mutant HIV 733.33 The cDNAs were then amplified by polymerase chain reaction (PCR) and sent for Sanger sequencing. An approximate 30% mismatch rate was observed at the cyc-C site in the presence of commercial HIV during RT, while HIV 733 exhibited 75% mismatch rate with C-to-T mutation being predominant (Fig. 2B). The C-probe and a4C-probe showed no mutation signals. As the HIV 733 RT enzyme displayed better performance in terms of mismatch rate, it was used in the later experiments unless specified. The selected Sanger sequencing profiles using the HIV 733 RT enzyme are listed in Fig. 2C. Together, these results proved that a4C is an effective chemical sequencing tag for RNA.
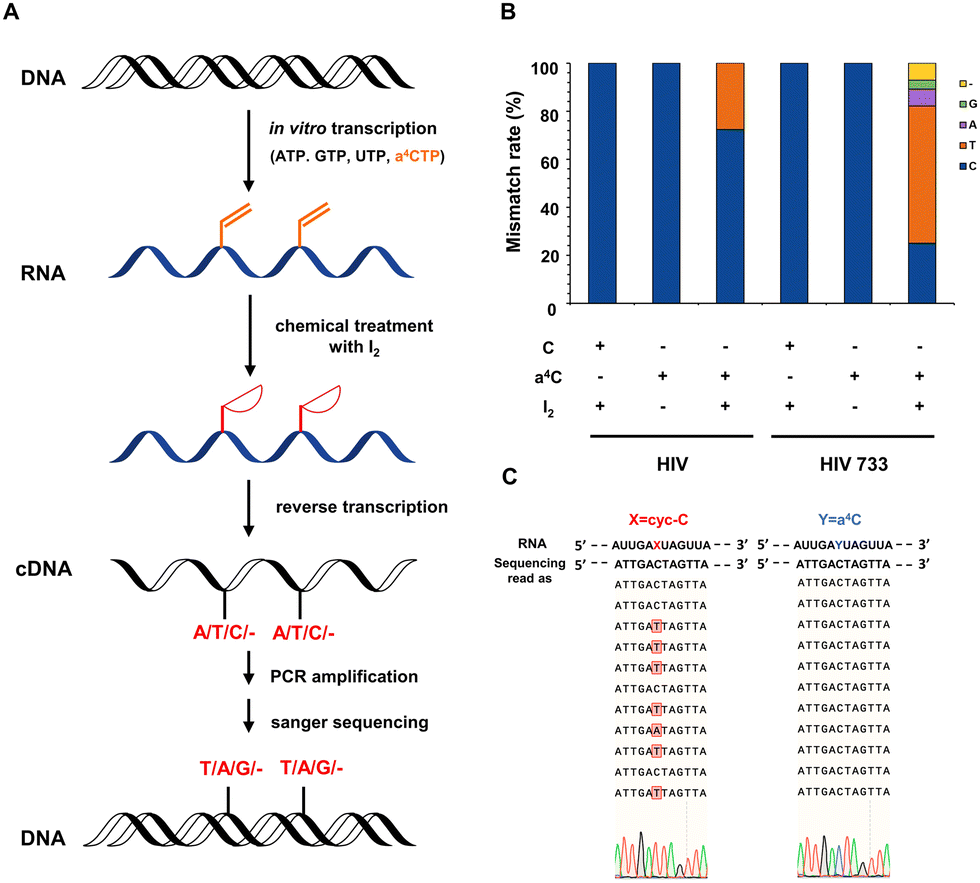 | ||
| Fig. 2 Chemical sequencing assay to pinpoint a4C labelling site on RNA. (A) Schematic illustration of identifying the a4C site on the in vitro transcribed RNA. a4C can be incorporated into the RNA probe through in vitro transcription, and iodination of a4C leads to the formation of cyc-C, which induces base mismatch in the synthesis of complementary DNA (cDNA) during RT. (B) Base mismatch rates of a4C and cyc-C in the presence of commercial HIV recombinant reverse transcriptase (HIV) or its mutant HIV 733. (C) The selected Sanger sequencing results of amplified cDNAs using HIV 733. | ||
Precise identification of modification sites on substrates of RNA N4-methyltransferase METTL15 based on a4C labelling and chemical sequencing
RNA modifications are highly dynamic and diverse, with over 170 different types identified to date.34 They occur in all major classes of RNA, including mRNA, transfer RNA (tRNA), ribosomal RNA (rRNA), and non-coding RNA (ncRNA), and are generally conserved across species.35 The presence of m4C in RNA was originally discovered in bacterial rRNA, where it was found to play a significant role in ribosome function and protein synthesis.36 Recent studies have revealed the presence of m4C modifications in eukaryotic RNA and have identified methyltransferase like 15 (METTL15) as the enzyme responsible for the m4C installation. METTL15 is responsible for 12S mitochondrial ribosomal RNA methylation at m4C839 both in vivo and in vitro, which is crucial for efficient mitochondrial protein synthesis and respiratory function.37 Although m4C839 has been identified as a modification site for METTL15, more substrate modification sites still need to be validated to enhance our understanding of METTL15's biological function as a methyltransferase. In our recent work, we have developed an enzyme-assisted chemical labelling assay capable of accurately pinpointing the substrate methylation sites of human RNA N6-methyladenosine methyltransferases METTL3, METTL14 and METTL16.38 Inspired by these results, we intended to combine enzyme-assisted chemical labelling with subsequent RNA sequencing to precisely identify m4C sites on substrates of METTL15.The anticipated process involved the transfer of the allyl group from the engineered cofactor allyl-substituted selenium (Se)-based donor analogue (allyl-SeAM)39,40 to the RNA substrate of METTL15, resulting in the formation of a4C modification (Fig. 3A). This modified base would then be subjected to base resolution sequencing using the aforementioned chemical sequencing assay. First of all, a 150-nt RNA probe was adapted from human 12S mt-rRNA and in vitro transcribed, with its predicted secondary structure shown in Fig. 3B. The target RNA probe was subjected to a series of reactions in one pot, which included: (i) methionine adenosyl transferase (MAT)-catalyzed formation of allyl-SeAM in the presence of precursor Se-allyl-L-selenohomocysteine (SeAHC); (ii) methylthioadenosine nucleosidase (MTN)-promoted degradation of Se-adenosylhomocysteine produced in the reaction (i); (iii) METTL15-catalyzed allyl transfer to potential m4C sites of the target RNA probe.
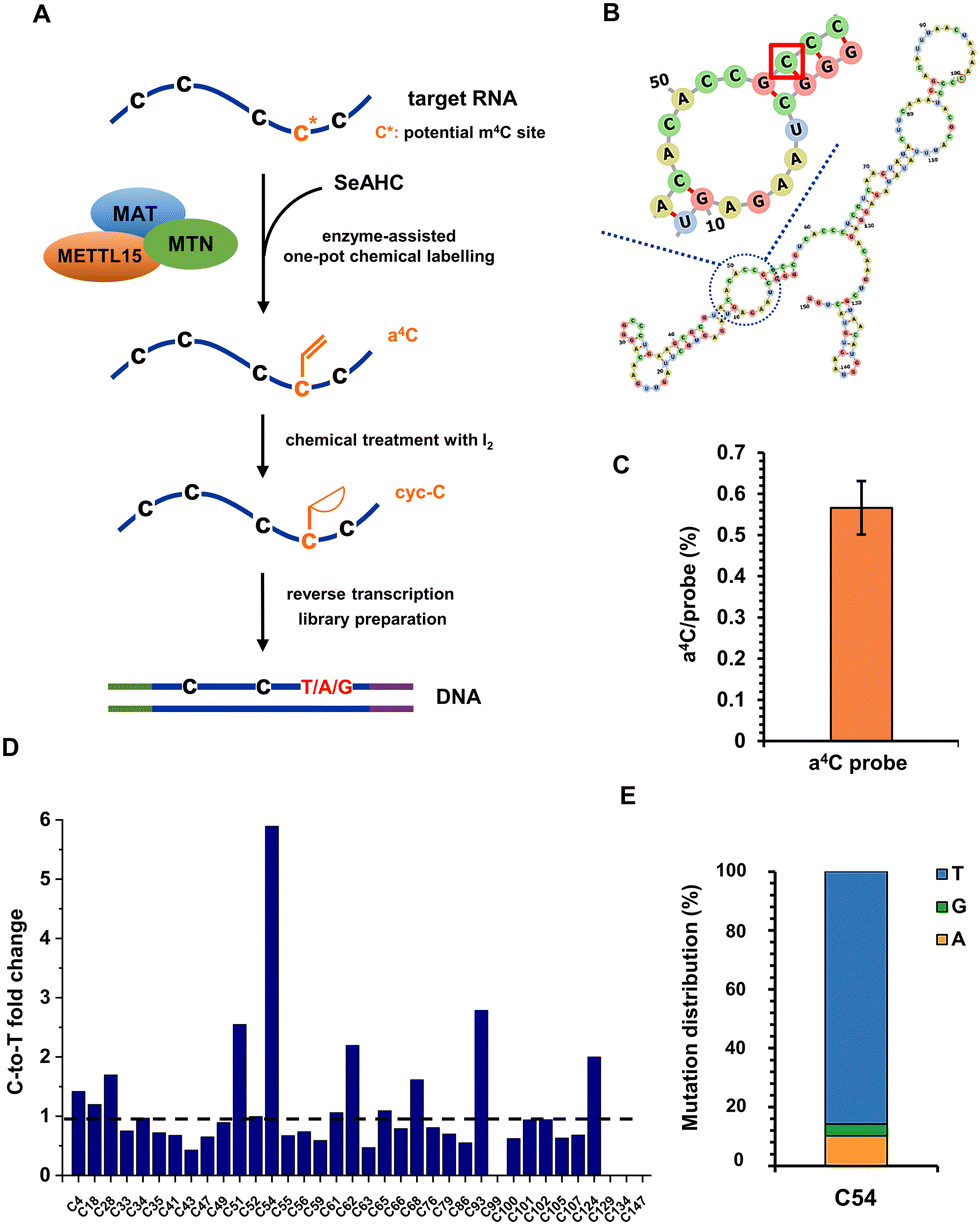 | ||
| Fig. 3 Identification of the substrate methylation sites of human mitochondrial 12S rRNA m4C methyltransferase METTL15. (A) Schematic illustration of the biochemical method to detect methyltransferase modification sites using an allyl-substituted methyltransferase cofactor and a4C chemical sequencing. MAT, methionine adenosyl transferase; MTN, methylthioadenosine nucleosidase; SeAHC, Se-allyl-L-selenohomocysteine. (B) Predicted secondary structure of the in vitro transcribed RNA probe adapted from human 12S rRNA. The potential m4C site is highlighted in rectangle. (C) The methyltransferase-assisted a4C labelling yield of the RNA probe characterized by UHPLC-QQQ-MS/MS. (D) The C-to-T mutation fold changes between iodine treated and untreated a4C-labelled RNA probes at each C site. (E) Statistics of C-to-T/G/A mutations for C54 in the RNA probe. | ||
Following the one-pot reaction, the target RNA probe was indeed modified with an allyl group, yielding approximately 0.6% a4C per probe measured by ultra-high performance liquid chromatography-triple quadrupole mass spectrometry (UHPLC-QQQ-MS/MS) (Fig. 3C). The relatively low yield indicated that the catalytic pocket of METTL15 might not efficiently accommodate the allyl-SeAM cofactor, but this did not influence the enzyme substrate specificity. In principle, the low labelling yield increases the sequencing cost as more reads are needed, however, the reads cost is fully acceptable with the current sequencing technique. With this, the recovered RNA from the above reaction was treated with iodine and reverse transcribed into cDNA, followed by PCR amplification, library preparation and next-generation high-throughput sequencing. Given the predominance of C-to-T mutation, we performed a targeted statistical analysis focusing on the occurrence of C-to-T mutation to investigate its frequency and distribution in the RNA probe. The C-to-T fold changes between iodine treated and untreated a4C-labelled RNA probes at each C site are shown in Fig. 3D. As expected, out of the investigated C sites, the m4C candidate site C54 displayed the most significant fold change, consistent with the reported result.40Fig. 3E shows detailed statistics of C-to-T/G/A mutations for C54, and around 86%, 10% and 4% of signals belonged to C-to-T, C-to-A and C-to-G, respectively. All these data provide solid evidence to prove that enzyme-assisted a4C labelling is a simple and powerful tool to accurately pinpoint m4C modification sites within the substrate RNAs of METTL15, which overcomes the disadvantages of traditional methods largely involving multistep enzyme-based digestions, radioactive labelling, thin layer or column chromatography, and mass spectrometry.
Metabolic labelling of mammalian cellular RNA using a4C
Next, we explored the utility of a4C for metabolic labelling of RNA in mammalian cells. The nucleotide salvage pathway is a critical metabolic pathway that facilitates the recycling and reutilization of nucleosides derived from the degradation of nucleic acids. In mammalian cells, uridine-cytidine kinase (UCK2) plays a crucial role in the salvage pathway for pyrimidine nucleosides. UCK2 is responsible for catalyzing the phosphorylation of cytidine and its analogues, converting them into their respective nucleotide monophosphates (Fig. 4A). Subsequently, these monophosphates can undergo further enzymatic conversions, facilitated by cytidine monophosphate kinase (CMPK) and nucleoside diphosphate kinase (NDPK), to generate nucleoside diphosphates and nucleoside triphosphates, respectively.41 The resultant triphosphates are then recognized and utilized by RNA polymerase during the process of RNA synthesis.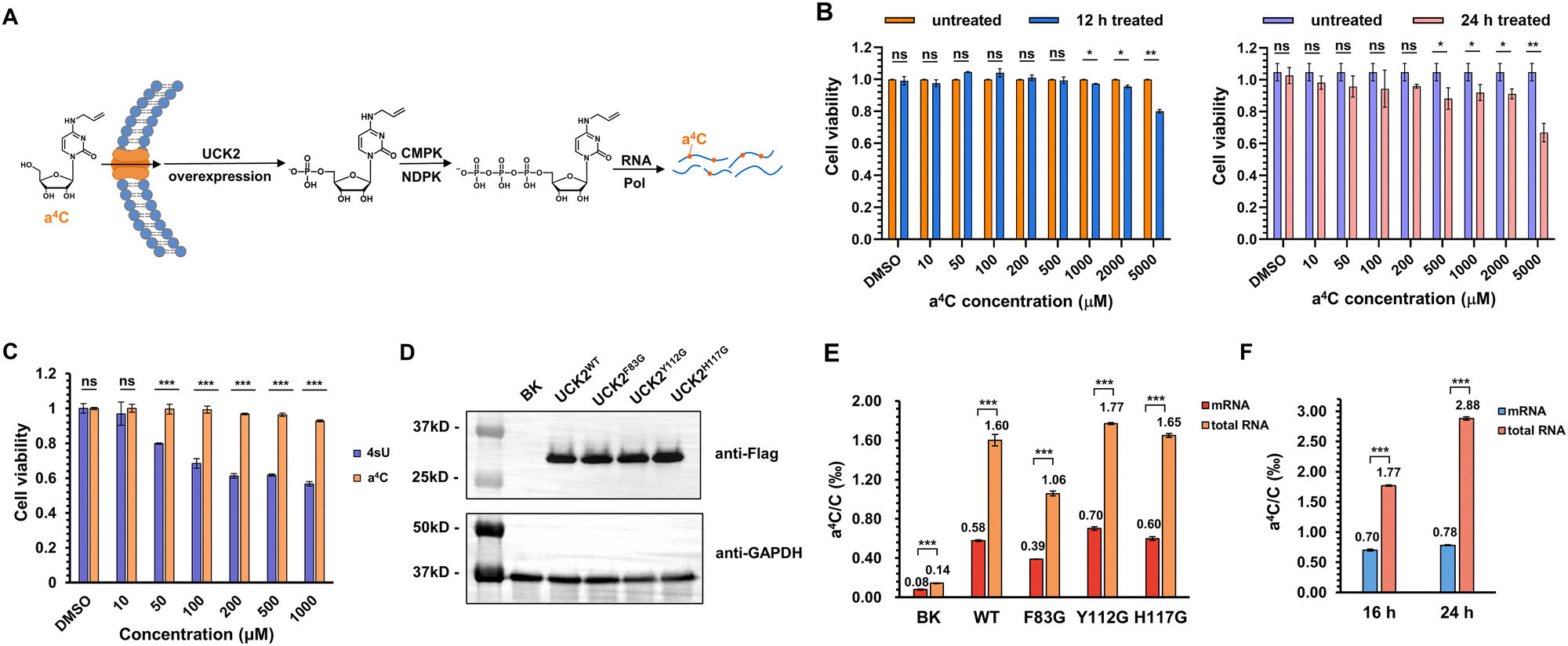 | ||
| Fig. 4 Metabolic labelling of cellular RNAs by a4C. (A) A schematic diagram for incorporation of a4C into RNA through the nucleotide salvage pathway. (B) The cell viability of HEK293T cells fed with different concentrations of a4C for 12 h (left) and 24 h (right), respectively. n = 3, two-tailed Student's test for statistical analysis. *P < 0.05, **P < 0.01, ns means not significant with P > 0.05. (C) The cell viability of HEK293T cells fed with different concentrations of 4sU and a4C, respectively, for 24 h. (D) Western blotting of expressed Flag-tagged UCK2 (WT) and its mutants. GAPDH was used as internal control. (E) The RNA a4C labelling levels in HEK293T cells expressed with UCK2 WT and different mutants. BK stands for expression of blank plasmid. (F) The effect of incubation time on RNA a4C labelling levels in UCK2Y112G-expressed HEK293T cells. n = 3, two-tailed Student's test is used for statistical analysis. ***P < 0.001. | ||
Prior to the labelling experiment, the viability of HEK293T cells was tested in the presence of different concentrations of a4C ranging from 10 μM to 5 mM under 12 h or 24 h treatment (Fig. 4B). The data revealed that the cell viability treated with a4C for 12 h did not exhibit a significant decrease until the a4C concentration reached 1000 μM. When the incubation period was extended to 24 h, the decrease in viability was observed at 500 μM. In addition, the cytotoxicity comparison of a4C with the golden standard 4sU was conducted in a parallel manner (Fig. 4C). Within the commonly used concentration range from 10 to 1000 μM,23 a4C exhibited superior performance to 4sU starting from 50 μM. All these results suggested that a4C possessed an excellent biocompatibility and a low cytotoxicity. Eventually, 500 μM concentration in between 12 and 24 h was chosen as the condition for metabolic labelling experiment.
Previous work showed that phosphorylation by UCK2 is the bottleneck for the metabolic incorporation of modified pyrimidine nucleosides.42 It was expected that the overexpression of UCK2 or its mutants in cellular systems would enhance RNA labelling efficiency with a4C. The X-ray crystal structure of UCK2 complexed with the substrate analogue cytidine monophosphate revealed that the active site exhibited limited capacity to accommodate alkyl modification at the C4 position of cytosine (Fig. S20, ESI†), which led us to speculate that the UCK2 mutant with an enlarged pocket might more efficiently accommodate a4C and enable its phosphorylation. For this purpose, Flag-tagged UCK2 (UCK2WT) and its variants containing substitution of amino acids Phe83, Tyr112 or His117 with smaller glycine (UCK2F83G, UCK2Y112G, and UCK2H117G) were constructed and expressed inside cells. After incubation with cells for 16 h, the expression levels of these variants and RNA a4C labelling levels were assessed via western blotting and UHPLC-QQQ-MS/MS, respectively. A blank plasmid (BK) was used as a control group. The western blotting results revealed that the expression levels of UCK2 mutants were comparable to that of the wild type (WT) (Fig. 4D). In UCK2 WT and mutants expressed cells, a4C levels were 5–13 fold higher than that of the BK group (Fig. 4E), revealing a positive correlation of UCK2 expression with the a4C labelling level. Among these groups, UCK2Y112G gave the highest labelling rate of 1.77‰ in total RNA and 0.7‰ in mRNA, but these mutants did not show much improved performance relative to the WT, suggesting that additional factor was involved (Fig. 4E). Interestingly, the labelling rate of a4C in total RNA is notably higher than that in mRNA, which indicated a4CTP was preferentially recognized by RNA polymerase I (RNPI) and incorporated into rRNA transcripts. In order to further explore the effect of incubation time on a4C labelling level, an additional group of UCK2Y112G-expressed cells was incubated with a4C for 24 h. As shown in Fig. 4F, the a4C labelling level in total RNA increased from 1.77‰ to 2.88‰, while mRNA labelling showed a slight increase, consistent with the above assumption that RNPI exhibited a higher tolerance towards a4CTP than RNA polymerase II (RNPII). Together, these results concluded that a4C could be metabolically incorporated into cellular RNAs and be potentially used to track RNA dynamics.
Detection of RNA polymerase transcription activity through a4C labelling on the chromatin run-on RNA transcripts and post chemical sequencing
The chromatin run-on and sequencing (ChRO-seq) technique has been widely used to map the locations of active RNA polymerases across the genome.43 The biotin-labelled nucleotide triphosphate (NTP) is commonly employed to label the run-on RNA transcripts and is further immunoprecipitated by affinitive avidin for downstream high-throughput RNA sequencing. It will be ideal to invent new NTP analogues with a chemical sequencing power for ChRO-seq application because complicated and expensive enrichment step is free and false positive signals brought by immunoprecipitation is largely avoided.44 The above results reveal that: (i) a4CTP is an NTP analogue with chemical sequencing capacity; (ii) a4CTP is able to be recognized and utilized by cellular RNA polymerase machineries. Therefore, we proposed that a4C-based ChRO-seq (a4C-ChRO-seq) would fulfil the hope of enrichment-free characterization of genome-wide RNA polymerase transcription activities.The flowchart of a4C-ChRO-seq is shown in Fig. 5A. The intact HeLa cell chromatin was isolated and the in situ polymerase run-on reactions were performed under different ratios of a4CTP/CTP and different run-on times. The chromatin run-on RNA was then extracted and its a4C level was quantified by UHPLC-QQQ-MS/MS. When the a4CTP/(a4CTP + CTP) ratio was increased from 0 to 100% with an interval of 20%, the RNA a4C level linearly increased from 0 to 3.60‰ (Fig. S21A, ESI†). With the run-on time extension from 5 min to 20 min, the a4C labelling rate only exhibited around 1.4 fold increase (Fig. S21B, ESI†). These results suggested that a4C was successfully incorporated into chromatin run-on nascent RNA transcripts within a short period of time. In the subsequent experiments, full replacement of CTP with a4CTP and 5 min of run-on time were selected as the conditions. In general, rRNA occupies around 30% of nuclear RNA.45 In order to improve library quality, rRNA was depleted from extracted run-on RNA through an RNase H-based degradation method before library construction. The rRNA level was measured by real-time quantitative polymerase chain reaction (RT-qPCR), and the result showed that the rRNA was reduced to less than 1/10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 of its original amount (Fig. S22, ESI†). The rRNA-depleted RNA was continuously subjected to iodine treatment, reverse transcription and library preparation. Finally, a4C-ChRO-seq libraries were sequenced with a paired-end 150 bp mode. Notably, both HIV and HIV 733 RT enzymes were used in parallel.
000 of its original amount (Fig. S22, ESI†). The rRNA-depleted RNA was continuously subjected to iodine treatment, reverse transcription and library preparation. Finally, a4C-ChRO-seq libraries were sequenced with a paired-end 150 bp mode. Notably, both HIV and HIV 733 RT enzymes were used in parallel.
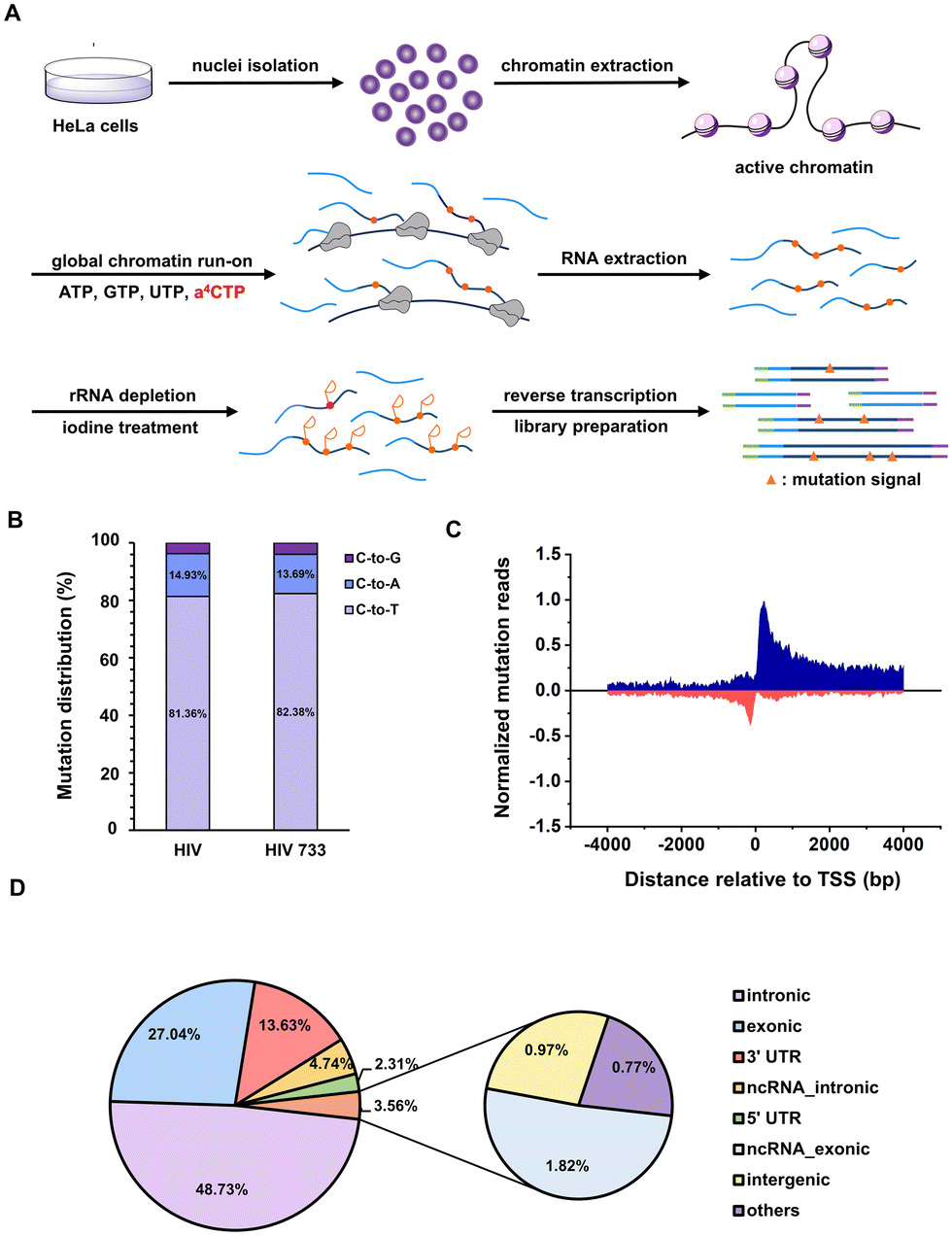 | ||
| Fig. 5 a4C-ChRO-seq detects RNA polymerase transcription activity in an enrichment-free manner. (A) Flowchart of a4C-ChRO-seq. The in situ chromatin RNA run-on reaction is performed in the presence of a4CTP, during which a4C was incorporated into newly transcribed RNA fragments. Through RNA extraction and purification, chemical transformation of a4C into cyc-C, cDNA library construction, and high-throughput sequencing, the genome-wide active RNA polymerase transcription sites are identified by counting the significant C mutation reads. (B) Statistics of C-to-T/G/A mutations using HIV (left) and HIV 733 (right) RT enzymes in a4C-ChRO-seq. (C) The normalized cumulative C-to-T mutation reads around RefSeq TSS by 50-bp windows in a4C-ChRO-seq. The signals in both sense and antisense directions relative to the direction of gene transcription were shown in blue and red, respectively. TSS, transcription start site. (D) Distribution of identified C-to-T mutation sites in different genome segments. UTR, untranslated region; ncRNA, noncoding RNA. | ||
Next, the sequencing data was analyzed in order to map the genome-wide RNA polymerase transcription activity. We extracted the aligned reads harboring C-to-T/A/G mutation sites, and analyzed the distribution of different types of mutations. The mutation patterns generated by HIV and HIV 733 were nearly identical (Fig. 5B). Given that the C-to-T mutation represented the major mutation type, further analysis was conducted on the subset of reads harboring C-to-T mutations. Through alignment of the a4C-ChRO-seq signals with respect to the transcription start site (TSS) annotated in RefSeq, a prominent and distinct peak in normalized C-to-T mutation reads was observed in both the sense and antisense directions, specifically within a range of approximately ±50 bp surrounding the TSS (Fig. 5C). These data corroborated the well-accepted principle that divergent initiation and promoter-proximal pausing occur in the cellular gene transcription process.46 The genome-wide analysis of mutation sites from a4C-ChRO-seq showed that the intronic, exonic, untranslated region (UTR), non-coding RNA (ncRNA), intergenic and other regions occupied 48.73, 27.04, 15.94, 7.56, 0.97 and 0.77%, respectively (Fig. 5D). This distribution suggested that most of the a4C-ChRO-seq signals came from nascent RNA. Additionally, we calculated the cumulative C-to-T mutation site numbers around RefSeq TSS by 50 bp-windows in a4C-ChRO-seq (Fig. S23A, ESI†). It could be found that the mutation sites were concentrated in the vicinity of the TSS and there was excellent consistency between two independent biological replicates. The cumulative mutation number of CAMK1D gene was given as an example (Fig. S23B, ESI†). These data supported that a4C was introduced into the newly synthesized RNA. Together, a4C-ChRO-seq was proved to be a new and simple tool for mapping the genome-wide RNA polymerase activities.
Conclusions
In summary, we successfully developed a4C as a new biocompatible nucleoside analogue for RNA labelling and chemical sequencing. The chemical reaction of a4C-to-cyc-C conversion is catalyst-free, specific and efficient, and is also compatible with the common RNA isolation and purification system. The structure of cyc-C was thoroughly characterized as diastereomeric 3, N4-five-memberred ring-fused cytosines, which could induce base mismatches during the RNA RT process. These advantages have contributed to the achievements of a4C RNA labelling applications including identification of m4C substrate modification site of METTL15, metabolic labelling of cellular RNA, and development of a4C-ChRO-seq for mapping cellular RNA polymerase activity across genome. To understand the complicated inter- and intra-cellular biology across temporal and spatial scales, RNA labelling/tagging with orthogonal nucleoside analogues is highly needed. For instances, different types of cells can be labelled with different chemical sequencing analogues, thus each cellular RNA dynamics can be characterized by its own sequencing mutation signal pattern. Taken together, nucleoside analogues with a chemical sequencing power offer new opportunity to elucidate the intricate mechanisms underlying RNA dynamics, modifications and functions.Materials and methods
General
All chemicals and reagents were used as purchased, without further purification. The cell lines utilized in this study (human HEK293T and HeLa cells) were obtained from the American Type Culture Collection (ATCC). 1H and 13C NMR spectra were measured on a Bruker AVANCE III 400 NMR spectrometer using DMSO-d6 as the solvent. High-resolution mass spectra (HRMS) were recorded on an AB Triple TOF 5600 plus (AB SCIEX, Framingham, USA) Mass spectrometer. HPLC profiles were acquired using a Waters e2695 module (flow rate of 3 ml min−1) with an Atlantis T3 OBD Prep Column (100 Å, 5 μm, 10 mm × 250 mm, 1 per pkg; cat. no. 186008205). Human methyltransferase like 15 (METTL15),40 human methionine adenosyltransferase (MAT2A)47 and 5′-methylthioadenosine (MTN)37 were expressed and purified using the previously published procedures. Se-allyl-L-selenohomocysteine was synthesized following the previously reported protocol.12 UHPLC-QQQ-MS/MS results were acquired using a Waters TQ MS triple-quadrupole LC spectrometer. Ultracentrifugation was carried out using an Optima L-90K ultra centrifuge supplied with an SW40Ti swing rotor.Chemical synthesis
:2) to give white powder (1.01 g, 80%). 1H NMR (400 MHz, DMSO-d6), δ (ppm): 10.30 (s, 1H), 8.60 (d, J = 7.3 Hz, 1H), 7.28 (d, J = 7.2 Hz, 1H), 6.04 (d, J = 3.4 Hz, 1H), 5.59 (dd, J = 3.3, 6.1 Hz, 1H), 5.39 (t, J = 6.5 Hz, 1H), 4.40 (d, J = 10.1 Hz, 2H), 4.27–4.35 (m, 1H), 2.11 (s, 3H), 2.08 (s, 3H), 2.07 (s, 3H). 13C NMR (100 MHz, DMSO-d6), δ (ppm): 170.06, 169.24, 157.48, 153.25, 150.32, 142.19, 95.58, 91.00, 79.54, 72.87, 69.25, 62.58, 20.56, 20.25. HRMS (ESI), m/z 457.0882 ([M + Cl]−, calcd 457.0880). See Fig. S1–S3 (ESI†) for detailed NMR and HRMS spectra.
:4) to give white powder (968.7 mg, 74%). 1H NMR (400 MHz, DMSO-d6), δ (ppm): 8.05 (t, J = 5.6 Hz, 1H), 7.61 (d, J = 7.5 Hz, 1H), 5.76–5.97 (m, 3H), 5.37–5.47 (m, 1H), 5.33 (t, J = 5.9 Hz, 1H), 5.18 (dd, J = 1.7, 17.2 Hz, 1H), 5.11 (dd, J = 1.7, 10.2 Hz, 1H), 4.31 (d, J = 8.5 Hz, 1H), 4.13–4.26 (m, 2H), 3.86–3.98 (m, 2H), 2.07 (s, 3H), 2.05 (s, 6H). 13C NMR (100 MHz, DMSO-d6), δ (ppm): 170.06, 169.41, 169.36, 163.36, 154.64, 141.49, 134.59, 115.72, 95.36, 89.23, 78.64, 72.19, 69.97, 63.21, 41.96, 20.52, 20.30, 20.27. HRMS (ESI), m/z 410.1547 ([M + H] +, calcd 410.1558). See Fig. S4–S6 (ESI†) for detailed NMR and HRMS spectra.
Model reaction of a4C nucleoside
a4C (113.2 mg, 0.4 mmol) was dissolved in 2 mL DMSO, and iodine (508 mg, 2 mmol) was added. Potassium iodide (664 mg, 4 mmol) was dissolved in 2 mL ddH2O. This mixture was vortexed to mix the two solutions thoroughly. The mixture was stirred at 40 °C for 1 h. Afterwards, saturated Na2S2O3 solution was titrated into the solution in order to remove the excess iodine, and then Na2CO3 (424 mg, 4 mmol) was added. The resultant mixture was further stirred at 40 °C for 30 min. The crude product was purified by reverse-phase HPLC using a gradient of 95% H2O and 5% acetonitrile to 100% acetonitrile. Fractions that contained the desired product were collected and lyophilized to afford 3, N4-cyclized cytidine (cyc-C-a, cyc-C-b). NMR and MS spectra for cyc-C-a and cyc-C-b are shown in Fig. S11–S18 (ESI†).Chemical sequencing assay for a4C in RNA
The a4C containing RNA probe was prepared through in vitro transcription using the HiScribe T7 Yield RNA Synthesis Kit (NEB, cat. no. E2040S). A reaction mixture (20 μL) containing 500 ng DNA template, 4 μL 5× reaction buffer, 8 μL 100 mM NTPs (100 mM for each a4CTP/UTP/ATP/GTP), 2 μL T7 RNA polymerase mix and 0.5 μL RNase inhibitor (Takara, 40 U μL−1) was incubated at 37 °C for 12 h. After incubation, to remove template DNA, 70 μL nuclease-free water, 10 μL of 10× DNase I buffer, and 2 μL of DNase I (RNase-free) were added and the resultant mixture was further incubated for 15 minutes at 37 °C. The a4C-incorporated RNA probe was purified using RNA Clean & Concentrator-25 (Zymo Research, cat. no. R1017). Afterwards, the a4C-incorporated RNA was subjected to the protocol shown in Scheme S1 (ESI†).Enzyme-assisted chemical labelling assay
The in vitro enzymatic labelling reactions were carried out in a volume of 50 μL with 2 mM SeAHC, 1 mM ATP (NEB, 10 mM), 5 μg RNA probes, 5 μM MAT2A, 10 μM MTN, 5 μM RNA methyltransferase METTL15 in reaction buffer containing 25 mM Tris buffer (pH 8.0), 5 mM MgCl2, 50 mM KCl, 0.05 mM ZnCl2 and 0.2 U μL−1 RNase inhibitor (Takara, 40 U μL−1). Prior to the reaction, the RNA probes were denatured and annealed with a program of: (i) 90 °C for 5 min, (ii) −0.1 °C s−1 down to 4 °C within 20 min and (iii) 4 °C for 5 min. After that, other components were added and the reactions were incubated at 37 °C for 4 h. Reactions were quenched by inactivating the enzyme at 70 °C for 15 min. The resultant RNA probes were recovered by acid phenol/chloroform (pH = 4.5) extraction followed by isopropanol precipitation. About 200 ng of recovered RNAs were digested into individual nucleosides by nuclease P1 and alkaline phosphatase for UHPLC-QQQ-MS/MS analysis while others were chemically treated and subjected to subsequent RNA sequencing.Quantification of a4C in RNA by UHPLC-QQQ-MS/MS
RNA probes, total RNAs, and mRNAs were digested into nucleosides and the amount of a4C was measured by reverse-phase UHPLC on a T3 column with online MS detection using a Waters TQ MS triple-quadrupole LC spectrometer in positive electrospray ionization mode and was calculated based on the standard curve generated by pure standards. For each sample, around 500 ng RNA was digested by using 1 U nuclease P1 (Wako) in 30 μL reaction mixture containing 20 mM NH4OAc at 42 °C for 2 h. Afterwards, 1 μL rSAP (NEB) and 3.5 μL Cutsmart buffer (NEB) were added and the reaction mixture was incubated at 37 °C for 2 h. Samples were filtered through a 0.22 μM filter (Millipore) and diluted to 100 μL. A 2 μL volume of the solution was injected into UHPLC-QQQ-MS/MS. The a4C nucleoside was quantified by using the nucleoside to base ion mass transition of 284 to 152.Cell culture and RNA a4C metabolic labelling
HEK293T cells were cultured in DMEM/high-glucose medium (HyClone, cat. no. SH30243.01) supplemented with 10% fetal bovine serum (Gibco, cat. no. 10270) and 1% penicillin–streptomycin (HyClone, cat. no. SV30010), and grown at 37 °C with 5% CO2. For metabolic labelling experiments in cells overexpressing nucleoside kinases, cells at ∼80% confluence were transfected with pCDNA3-Flag plasmid, pcDNA3-Flag-UCK2WT plasmid, pcDNA3-Flag-UCK2F83G plasmid, pcDNA3-Flag-UCK2Y112G plasmid and pcDNA3-Flag-UCK2H117G plasmid, respectively. 8 hours after transfection, fresh medium containing 500 μM a4C was added and incubated for 16 hours. Cellular total RNAs were isolated using the TRIzol (Invitrogen, cat. no. 10296010) reagent by following the manufacturer's protocol. mRNAs were then isolated from total RNA using the GenElute mRNA Purification Miniprep Kit (Sigma-Aldrich, cat. no. MRN10-1KT) by following manufacturer's protocol.The effect of a4C on the cell viability
HEK293T cells were seeded separately in a 96-well plate at a density of 7 × 104 cells per well in DMEM/high-glucose medium (HyClone, cat. no. SH30243.01) supplemented with 10% fetal bovine serum (Gibco, cat. no. 10270) and 1% penicillin–streptomycin (HyClone, cat. no. SV30010) at 37 °C with 5% CO2. After being incubated overnight, HEK293T cells at ∼80% confluence were treated with different concentration of a4C dissolved in DMSO. The final concentration of a4C varied from 10 μM to 50 μM, 100 μM, 200 μM, 500 μM, 1000 μM, 2000 μM and 5000 μM. For control experiments, 1% DMSO was added. After being cultured for another 12 h or 24 h, the viability of HEK293T cells was measured using the CellTiter-Glo® 2.0 Assay (Promega).Isolation of nuclei
HeLa cells were cultured in DMEM/high-glucose medium (HyClone, cat. no. SH30243.01) supplemented with 10% fetal bovine serum (Gibco, cat. no. 10270) and 1% penicillin–streptomycin (HyClone, cat. no. SV30010), and grown at 37 °C with 5% CO2. The nuclei were isolated as previously described.44Chromatin run-on reaction and RNA extraction
Briefly, 100 μL HeLa nuclei (∼5 × 106) were mixed with 10 μL 5 M NaCl by pipetting up and down until the solution became clear. Afterwards, an equal volume of nuclease-free water was added, and chromatin was pelleted by centrifugation at 12000 rpm for 30 s. The chromatin pellet was washed three times with 500 μL 50 mM Tris–HCl (pH = 7.5), followed by resuspension in 100 μL glycerol storage buffer. An equal volume of 2× chromatin run-on buffer (10 mM Tris–HCl, pH = 7.4, 5 mM MgCl2, 1 mM DTT, 300 mM KCl, 500 μM UTP, 500 μM ATP, 500 μM GTP, 500 μM a4CTP, 4 U μL−1 RRI, and 1% (w/v) Sarkosyl) was added, and the reaction mixture was incubated at 37 °C for 5 min with gentle mixing. TRIzol (Invitrogen, cat. no. 10296010) was added to terminate the reaction and extract RNA, followed by DNA depletion by DNase I digestion and isopropanol precipitation. The resultant a4C-labelled chromatin RNA was resuspended in nuclease-free water.
Library construction
rRNA was depleted using the NEBNext® rRNA Depletion Kit (NEB, E0350), and the resultant RNA was resuspended in 20 μL nuclease-free water. The efficiency of rRNA depletion was measured by RT-qPCR using PrimeScript™ 1st Strand cDNA Synthesis Kit (TOYOBO, 6110A) and iTaq™ S5 Universal SYBR® Green Supermix (Bio-Red, 1725124), with the target included GAPDH (with qPCR primers GAPDH-qF and GAPDH-qR), 5.8s rRNA (with qPCR primers rRNA5.8S-qF and rRNA5.8S-qR), 18s rRNA (with qPCR primers rRNA18S-qF and rRNA18S-qR), and 28s rRNA (with qPCR primers rRNA28S-qF and rRNA28S-qR). Then, the rRNA-depleted RNA samples were diluted to 26 μL and sequentially treated with I2, Na2S2O3 and Na2CO3 as described above. The library was constructed using the NEBNext® Ultra II Directional RNA Library Prep Kit (NEB, E7760), with several modifications. Briefly, RNA was resuspended in 5 μL nuclease-free water, and fragmented with 4 μL NEBNext First Strand Synthesis Reaction Buffer and 1 μL Random Primers for 7 min at 94 °C. Reverse transcription was performed by adding 8 μL NEBNext Strand Specificity Reagent, 5 μL 5× RT reaction buffer, and 1 μL recombinant HIV reverse transcriptase (Worthington Biochemical Corporation) or HIV 733, for 10 min at 25 °C, 1 h at 37 °C, and 15 min at 70 °C, and finally held at 4 °C. Then the following steps were performed following the manufacture's protocol. The library sizes were measured using an Agilent 2100 Bioanalyzer and sequenced using an Illumina Hiseq with paired-end 2 × 150 bp read length.Author contributions
T. L. (Tengwei Li) designed and performed most of the experiments. X. Shu provided help with data analysis. M. G. helped in nuclei isolation and chromatin extraction. C. H. worked on the synthesis of Se-allyl-L-selenohomocysteine. T. L. (Ting Li) worked on the expression and purification of HIV 733. D. Liu provided assistance with chemical synthesis. J. Cao helped in library construction. X. Ying helped with the enzyme-assisted chemical labelling reaction. T. L. (Tengwei Li) wrote the manuscript. J. L. designed and supervised the whole project, and wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from the National Key Research and Development Program of China (2022YFA1103702), the National Natural Science Foundation of China (22022702, 21977087 and 91853110), the Zhejiang Provincial Natural Science Foundation of China under Grant No. LZ23B020004, the Fundamental Research Funds for the Central Universities, and MOE Key Laboratory of Macromolecular Synthesis and Functionalization, Zhejiang University (2022MSF04). We thank B. Dickinson at The University of Chicago for kindly providing the HIV 733 plasmid.Notes and references
- C. Carrieri, L. Cimatti, M. Biagioli, A. Beugnet, S. Zucchelli, S. Fedele, E. Pesce, I. Ferrer, L. Collavin, C. Santoro, A. R. Forrest, P. Carninci, S. Biffo, E. Stupka and S. Gustincich, Nature, 2012, 491, 454–457 CrossRef CAS PubMed.
- J. Mattay, M. Dittmar and A. Rentmeister, Curr. Opin. Chem. Biol., 2021, 63, 46–56 CrossRef CAS PubMed.
- S. Verma, S. Jager, O. Thum and M. Famulok, Chem. Rec., 2003, 3, 51–60 CrossRef CAS PubMed.
- H. Rao, A. A. Sawant, A. A. Tanpure and S. G. Srivatsan, Chem. Commun., 2012, 48, 498–500 RSC.
- G. Gosselin, ChemBioChem, 2006, 7, 389 CrossRef CAS.
- I. Hirao, Curr. Opin. Chem. Biol., 2006, 10, 622–627 CrossRef CAS PubMed.
- N. Klocker, F. P. Weissenboeck and A. Rentmeister, Chem. Soc. Rev., 2020, 49, 8749–8773 RSC.
- J. M. Holstein and A. Rentmeister, Methods, 2016, 98, 18–25 CrossRef CAS PubMed.
- Y. Wang, Y. Xiao, S. Dong, Q. Yu and G. Jia, Nat. Chem. Biol., 2020, 16, 896–903 CrossRef CAS PubMed.
- A. Ovcharenko, F. P. Weissenboeck and A. Rentmeister, Angew. Chem., Int. Ed., 2021, 60, 4098–4103 CrossRef CAS PubMed.
- J. Cao, X. Shu, X.-H. Feng and J. Liu, Curr. Opin. Chem. Biol., 2021, 63, 28–37 CrossRef CAS PubMed.
- X. Shu, J. Cao, M. Cheng, S. Xiang, M. Gao, T. Li, X. Ying, F. Wang, Y. Yue, Z. Lu, Q. Dai, X. Cui, L. Ma, Y. Wang, C. He, X. Feng and J. Liu, Nat. Chem. Biol., 2020, 16, 887–895 CrossRef CAS PubMed.
- H. Tani and N. Akimitsu, RNA Biol., 2014, 9, 1233–1238 CrossRef PubMed.
- H. Tani, R. Mizutani, K. A. Salam, K. Tano, K. Ijiri, A. Wakamatsu, T. Isogai, Y. Suzuki and N. Akimitsu, Genome Res., 2012, 22, 947–956 CrossRef CAS PubMed.
- M. R. Miller, K. J. Robinson, M. D. Cleary and C. Q. Doe, Nat. Methods, 2009, 6, 439–441 CrossRef CAS PubMed.
- E. E. Duffy, M. Rutenberg-Schoenberg, C. D. Stark, R. R. Kitchen, M. B. Gerstein and M. D. Simon, Mol. Cell, 2015, 59, 858–866 CrossRef CAS PubMed.
- C. Y. Jao and A. Salic, Proc. Natl. Acad. Sci. U.S.A., 2002, 105(42), 15779–15784 Search PubMed.
- D. Wang, Y. Zhang and R. E. Kleiner, J. Am. Chem. Soc., 2020, 142, 14417–14421 CrossRef CAS PubMed.
- X. Gao, X. Shu, Y. Song, J. Cao, M. Gao, F. Wang, Y. Wang, J. Z. Sun, J. Liu and B. Z. Tang, Chem. Commun., 2019, 55, 8321–8324 RSC.
- M. Grammel, P. Luong, K. Orth and H. C. Hang, J. Am. Chem. Soc., 2011, 133, 17103–17105 CrossRef CAS PubMed.
- M. Grammel, H. Hang and N. K. Conrad, ChemBioChem, 2012, 13, 1112–1115 CrossRef CAS PubMed.
- V. A. Herzog, B. Reichholf, T. Neumann, P. Rescheneder, P. Bhat, T. R. Burkard, W. Wlotzka, A. von Haeseler, J. Zuber and S. L. Ameres, Nat. Methods, 2017, 14, 1198–1204 CrossRef CAS PubMed.
- J. A. Schofield, E. E. Duffy, L. Kiefer, M. C. Sullivan and M. D. Simon, Nat. Methods, 2018, 15, 221–225 CrossRef CAS PubMed.
- Y. Chen, F. Wu, Z. Chen, Z. He, Q. Wei, W. Zeng, K. Chen, F. Xiao, Y. Yuan, X. Weng, Y. Zhou and X. Zhou, Adv. Sci., 2020, 7, 1900997 CrossRef CAS PubMed.
- C. Riml, T. Amort, D. Rieder, C. Gasser, A. Lusser and R. Micura, Angew. Chem., Int. Ed., 2017, 56, 13479–13483 CrossRef CAS PubMed.
- F. Erhard, A.-E. Saliba, A. Lusser, C. Toussaint, T. Hennig, B. K. Prusty, D. Kirschenbaum, K. Abadie, E. A. Miska, C. C. Friedel, I. Amit, R. Micura and L. Dölken, Nat. Rev. Methods Primers, 2022, 2, 77 CrossRef CAS.
- C. Gasser, I. Delazer, E. Neuner, K. Pascher, K. Brillet, S. Klotz, L. Trixl, M. Himmelstoss, E. Ennifar, D. Rieder, A. Lusser and R. Micura, Angew. Chem., Int. Ed., 2020, 59, 6881–6886 CrossRef CAS PubMed.
- L. Kiefer, J. A. Schofield and M. D. Simon, J. Am. Chem. Soc., 2018, 140, 14567–14570 CrossRef CAS PubMed.
- X. Shu, Q. Dai, T. Wu, I. R. Bothwell, Y. Yue, Z. Zhang, J. Cao, Q. Fei, M. Luo, C. He and J. Liu, J. Am. Chem. Soc., 2017, 139, 17213–17216 CrossRef CAS PubMed.
- X. Shu, C. Huang, T. Li, J. Cao and J. Liu, Fundam. Res., 2023, 3, 657–664 CrossRef CAS.
- D. Arango, D. Sturgill and S. Oberdoerffer, Bio-Protoc., 2019, 9, e3278 CAS.
- A. Calabretta and C. J. Leumann, Biochemistry, 2013, 52, 1990–1997 CrossRef CAS PubMed.
- H. Zhou, S. Rauch, Q. Dai, X. Cui, Z. Zhang, S. Nachtergaele, C. Sepich, C. He and B. C. Dickinson, Nat. Methods, 2019, 16, 1281–1288 CrossRef CAS PubMed.
- M. Helm and Y. Motorin, Nat. Rev. Genet., 2017, 18, 275–291 CrossRef CAS PubMed.
- Z. Bao, T. Li and J. Liu, Molecules, 2023, 28, 1517 CrossRef CAS PubMed.
- S. Kimura and T. Suzuki, Nucleic Acids Res., 2010, 38, 1341–1352 CrossRef CAS PubMed.
- I. Laptev, E. Shvetsova, S. Levitskii, M. Serebryakova, M. Rubtsova, V. Zgoda, A. Bogdanov, P. Kamenski, P. Sergiev and O. Dontsova, Nucleic Acids Res., 2020, 48, 8022–8034 CrossRef CAS PubMed.
- S. Xiang, M. Gao, J. Cao, X. Shu, M. Cheng, F. Wang, T. Deng and J. Liu, Chem. Commun., 2021, 57, 2499–2502 RSC.
- J. Ren, X. Shu, Y. Wang, D. Wang, G. Wu, X. Zhang, Q. Jin, J. Liu, Z. Wu, Z. Xu, C.-Z. Li and H. Li, Chin. Chem. Lett., 2022, 33, 1650–1658 CrossRef CAS.
- H. Chen, Z. Shi, J. Guo, K. J. Chang, Q. Chen, C. H. Yao, M. C. Haigis and Y. Shi, J. Biol. Chem., 2020, 295, 8505–8513 CrossRef CAS PubMed.
- D. Wang, A. Shalamberidze, A. E. Arguello, B. W. Purse and R. E. Kleiner, J. Am. Chem. Soc., 2022, 144, 14647–14656 CrossRef CAS PubMed.
- Y. Zhang and R. E. Kleiner, J. Am. Chem. Soc., 2019, 141, 3347–3351 CrossRef CAS PubMed.
- T. Chu, E. J. Rice, G. T. Booth, H. H. Salamanca, Z. Wang, L. J. Core, S. L. Longo, R. J. Corona, L. S. Chin, J. T. Lis, H. Kwak and C. G. Danko, Nat. Genet., 2018, 50, 1553–1564 CrossRef CAS PubMed.
- M. Gao, Y. Li, X. Shu, P. Dai, J. Cao, Y. An, T. Li, Y. Huang, F. Wang, Z. Lu, F. L. Meng, X. H. Feng, L. Ma and J. Liu, ACS Chem. Biol., 2022, 17, 768–775 CrossRef CAS PubMed.
- M. Rabani, J. Z. Levin, L. Fan, X. Adiconis, R. Raychowdhury, M. Garber, A. Gnirke, C. Nusbaum, N. Hacohen, N. Friedman, I. Amit and A. Regev, Nat. Biotechnol., 2011, 29, 436–442 CrossRef CAS PubMed.
- L. J. Core, J. J. Waterfall and J. T. Lis, Science, 2008, 322, 1845–1848 CrossRef CAS PubMed.
- R. Wang, K. Islam, Y. Liu, W. Zheng, H. Tang, N. Lailler, G. Blum, H. Deng and M. Luo, J. Am. Chem. Soc., 2013, 135, 1048–1056 CrossRef CAS PubMed.
- S. K. Mahto and C. S. Chow, Bioorg. Med. Chem., 2008, 16, 8795–8800 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cb00189j |
| This journal is © The Royal Society of Chemistry 2024 |