
Plasma-treated gold microelectrodes for subsecond detection of Zn(II) with fast-scan cyclic voltammetry†
Anntonette N.
Perry
,
Romana
Jarosova
,
Colby E.
Witt
,
Moriah E.
Weese-Myers
 ,
Vivek
Subedi
and
Ashley E.
Ross
*
,
Vivek
Subedi
and
Ashley E.
Ross
*
University of Cincinnati, Department of Chemistry, 312 College Dr., 404 Crosley Tower, Office# 418A Rieveschl, Cincinnati, OH 45221-0172, USA. E-mail: Ashley.ross@uc.edu
First published on 7th August 2024
Abstract
The sensitivity of zinc (Zn(II)) detection using fast-scan cyclic voltammetry (FSCV) with carbon fiber microelectrodes (CFMEs) is low compared to other neurochemicals. We have shown previously that Zn(II) plates to the surface of CFME's and we speculate that it is because of the abundance of oxide functionality on the surface. Plating reduces sensitivity over time and causes significant disruption to detection stability. This limited sensitivity and stability hinders Zn(II) detection, especially in complex matrices like the brain. To address this, we developed plasma-treated gold fiber microelectrodes (AuMEs) which enable sensitive and stable Zn(II) detection with FSCV. Typically, gold fibers are treated using corrosive acids to clean the surface and this step is important for preparing the surface for electrochemistry. Likewise, because FSCV is an adsorption-based technique, it is also important for Zn(II) to adsorb and desorb to prevent irreversible plating. Because of these requirements, careful optimization of the electrode surface was necessary to render the surface for Zn(II) adsorption yet strike a balance between attraction to the surface vs. irreversible interactions. In this study, we employed oxygen plasma treatment to activate the gold fiber surface without inducing significant morphological changes. This treatment effectively removes the organic layer while functionalizing the surface with oxygen, enabling Zn(II) detection that is not possible on untreated gold surfaces. Our results demonstrate significantly improved Zn(II) detection sensitivity and stability on AuME compared to CFME's. Overall, this work provides an advance in our understanding of Zn(II) electrochemistry and a new tool for improved metallotransmitter detection in the brain.
Introduction
Fast-scan cyclic voltammetry (FSCV) is an electroanalytical technique that is used most often to detect electroactive neurotransmitters on subsecond timescales.1 Traditionally, FSCV has been used to detect neurochemicals, most notably dopamine, with carbon-fiber microelectrodes (CFMEs).2–5 Over the last several years, FSCV detection has expanded beyond traditional carbon-fiber microelectrodes6–10 and has branched out to several other classes of neurochemicals including the purines,11–15 serotonin,5,16–18 histamine,19 among many others.20–25 Previously, our group developed a novel waveform which enabled subsecond detection of Zn(II) at CFME's with FSCV.26 Despite this expansion, we noted several disadvantages to detecting Zn(II) on carbon-fiber including poor sensitivity and evidence of plating on the surface.26 Here, we provide a novel approach to detecting Zn(II) with FSCV by using oxygen (O2)-plasma treated gold-fiber microelectrodes (AuMEs). Gold is traditionally not an electrode material used with FSCV because dopamine does not adsorb as strongly to gold surfaces,7,14 however, we highlight here that careful electrode–analyte interaction studies are needed in order to optimize detection conditions for the specific analyte of interest.Chemical and electrochemical cleaning of gold is typically done to improve electrochemical detection capabilities.27 Cleaning gold electrodes is used to remove the inherent organic layer on the surface to improve charge transfer resistance and produce low peak-current potential differences.28 Strong acids and oxidizing agents are often used to clean gold; however, if mishandled, could lead to serious injury. Likewise, etching in strong acids can corrode the surface and without polishing steps, this can lead to decreased reactive surface area for electrochemistry.29 For FSCV, cylindrical electrodes are typically used due to improved sensitivity compared to disk microelectrodes for in tissue measurements;30 therefore, polishing steps aren't possible to remove the potential corrosion due to acid etching. Because of this, cleaning steps which do not corrode the surface, thus eliminating the need for polishing, were necessary for us to expand to cylinder gold microelectrodes for biological detection of Zn(II).
Alternative cleaning methods have been developed using high temperatures, and potential sweep methods in low concentrated acids.31,32 Although these methods offer excellent alternatives to cleaning the gold surface, the temperatures typically used (several hundred degrees C) can disrupt the integrity of the gold fiber leading to reduction in conductivity. Using lower concentrations of acid, for example nitric acid or hydrochloric acid mixed with hydrogen peroxide, is known to potentially contaminate the surface with carbon, chloride, and nitrogen which also can lead to loss in reactive area for electrochemistry.28 Zn(II) electrochemistry is a surface-dependent process which relies heavily on the chemistry of the electrode surface.33 Zn(II) is known to interact favorably with oxide functionality, leading to adsorption on the surface.26,34 In FSCV, the holding potential is used as a preconcentration step to promote adsorption to the surface;1 therefore, it is important to insure the surface is favorable for this interaction. Because of Zn(II)'s known interaction with oxide groups, we investigated whether functionalizing the surface with oxygen would improve electrochemical detection of Zn(II) on gold. Overall, a method to treat gold by cleaning yet maintaining some oxygen functionality with limited corrosion was necessary to insure adequate Zn(II) detection.28
Plasma etching is a well-established approach to functionalizing surfaces.11,11,35 Previously, our lab developed methods to plasma treat carbon-fiber with argon, oxygen, and nitrogen.11,36 Likewise, other materials including graphene oxide37,38 and even gold nanowires have been functionalized with various plasma gases.35,39 Oxygen (O2) plasma of gold has demonstrated removal of the organic layer from template membranes allowing for self-assembled functionalization.40 O2 plasma is highly reactive and forms volatile species on the surface which results in both cleaning and oxidation. The formation of Au2O3 on the surface results from oxygen plasma; although this can be unstable at room temperature and could rapidly degrade to other AuO constituents.35 Here, we have developed a method to O2-plasma treat gold microelectrodes to both clean and functionalize the surface for enhanced Zn(II) interaction. We show that oxide functionalities are necessary for improved Zn(II) electrochemical detection while also demonstrating that gold is an improved alternative approach (compared to the tradtional carbon-based materials) to measuring Zn(II) with FSCV.
Methods
Reagents
Chemicals were purchased from Fisher Scientific (Fair, Lawn, NJ, USA) unless noted differently. 24 mM HEPES buffer, made by dissolving 5.719 g HEPES in ultra-purified Milli-Q water (Millipore, Bilerica, MA) and adjusted to a pH of 7.4 for electrochemical characterization. Stock solutions of 10 mM Cu(NO3)2·xH2O and Zn(NO3)2 were dissolved in Milli-Q water and diluted in HEPES buffer daily for electrochemical characterization experiments. To do traditional cyclic voltammetry, solutions were made from 50 mM stock and diluted to 5 mM with 1 M KCl for electrochemical characterization. 5 mM of Ru(NH3)6Cl3 and K3[Fe(CN)6] were used in electrochemical characterization of electrode fibers. For brain tissue experiments, data was collected in artificial cerebrospinal fluid (aCSF) consisting of 2.5 mM KCl, 1.2 mM NaH2PO4, 2.4 mM CaCl2 dihydrate, 1.2 mM MgCl2 hexahydrate, 126 mM NaCl, 11 mM D-glucose, 25 mM sodium bicarbonate, and 0.4 mM ascorbic acid.Gold fiber microelectrode fabrication and plasma treatment
Cylindrical gold fiber microelectrodes (AuMEs) were made using a 25 μm in diameter gold wire purchased from Goodfellow Cambridge Limited (Huntington, England). Untreated gold fibers were pulled through a pre-pulled glass capillary tube (1.2 mm × 0.68 mm, A-M Systems, Sequim, WA) and the fiber was placed 100–150 μm in length from the end of the glass seal under an optical microscope. The glass capillaries were pulled using a vertical micropipette puller (Narishige PE-22; Tokyo, Japan). Gold fibers were treated with either O2 plasma (Nordson march, Westlake, Ohio, USA) or air plasma (SPI Plasma Prep III, SPI Supplies, West Chester, PA) and then compared. Treatment times were varied, and the experimental parameters assessed were 100 W for 200 s with 250 SCCM (O2 plasma), 100 W for 300 s with 200 SCCM (O2 plasma), and 100 W for 300 s (SCCM not controllable for air plasma instrument). To plasma treat the entire fiber surface, gold fibers were threaded through a PDMS block to expose the ends of the fiber. This is a necessity to ensure full functionalization of the fiber surface. After plasma treatment, electrodes were fabricated using the plasma-treated fibers as described above. Electrodes were then sealed with air dry epoxy (J-B Weld 50112 ClearWeld Quick-Setting Epoxy-Clear) then dipped in acetone to clean the epoxy off the fiber and cured in air overnight.Electrochemical characterization
Traditional voltammograms were collected using a CHI 620 potentiostat (CH Instruments, Bee Cave, TX, USA) with a three-electrode system. The working electrode was the cylindrical gold fiber microelectrode (AuME), the reference was a standard Ag/AgCl, and the counter electrode was a Pt wire. Square wave voltammetry (SWV) was used and included a starting potential of −1.6 V scanning to 0.4 V, with an amplitude increase of 0.025 and a frequency of 15 Hz. Traditional cyclic voltammetry (CV) was used for surface characterization. The potential waveform scanned from −0.5 V to 0.4 V at a scan rate of 0.1 mV s−1. Confirmation of a monoxide layer on the gold surface was done by scanning from −0.3 V to 1.5 V at 0.1 mV s−1 in 0.05 M H2SO4. Fast-scan cyclic voltammograms were collected using the WaveNeuro potentiostat with a 1 Ω headstage (Pine Instruments, Durham, NC). The FSCV data was collected using a National Instruments PCle-6363 interface board (Austin, TX) and HDCV software (UNC-Chapel Hill, Mark Wightman). Fast-scan cyclic voltammograms (CVs) were background subtracted to remove non-faradaic current. The waveform consisted of a holding potential at −0.1 V, a switching potential of −1.6 V, with a ramp to 0.6 V and back to the holding potential. The waveform was applied at 400 V s−1 at a frequency of 10 Hz. Electrodes were tested on a flow cell system made from a six port HPLC actuator (Valco Instruments, Houston TX), similar to prior reports.13,41 Buffer was delivered at a flow rate of 1 mL min−1 using a syringe pump (Chemyx, Stafford, TX). All experiments were conducted in a Faraday cage at room temperature.Surface characterization
Gold fibers were imaged using scanning electron microscopy (SEM) accompanied with an EDAX detector for energy dispersive spectroscopy (EDS) for elemental characterization. All images were collected on a FEI XL30 SEM with an accelerating voltage of 5.00 kV at a working distance of 12.2 mm. X-ray photoelectron spectroscopy (XPS) was utilized to determine the elemental composition of gold fiber surfaces before and after air and O2 plasma treatment using a Thermo Scientific Nexsa X-ray photoelectron spectrometer with a hemispherical analyzer and monochromatic Al Kα source (Wayne State University, Detroit, MI). Fibers were mounted using conductive Cu tape. Data was collected using a baseline pressure of 1.7 × 10−7 mbar with a flood gun utilized for surface charge neutralization.Brain slice experiments
All animal experiments were conducted in accordance with The Guide for the Care and Use of Laboratory Animals (“The Guide”) by the National Research Council and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Cincinnati. Male Sprague Dawley rats, typically weighing 170–180 g (Charles River Laboratories, Wilmington, MA, USA), were housed in a vivarium and provided food and water ad libitum. Rats were anesthetized with isoflurane (Henry Shrein, Melville, NY, USA) and euthanized via decapitation. After the brain was retrieved, it was placed in ice-cold oxygenated (95% O2 and 5% CO2) aCSF for no more than 2 min. The brain was mounted using superglue onto a vibratome stage for slicing. Sagittal slices of the hippocampus, 400 μm thick, were acquired using a VT1000S vibratome (Leica, Chicago, IL, USA) at a speed of 90 and a frequency of 3. The tissue slices were recovered in oxygenated aCSF for 1 h prior to picospritzing. For the picospritzing experiment, slices were placed in a Warner Instruments perfusion chamber set to 37 °C and perfused with oxygenated aCSF at a rate of 2 mL min−1 using a Watson-Marlow 204S peristaltic pump (Wilmington, MA, USA). The AuME was implanted into the CA1 of the hippocampus and lowered approximately 75 μm using a micromanipulator (Narishige, Tokyo, Japan). The electrode was allowed to equilibrate for 20 min prior to exogenous delivery of Zn(II). A Parker Hannifin Picospritzer III (Hollis, NH, USA) was used to deliver 200 μM Zn(II) 100 μm from the electrode by a pulled glass capillary with a 15–20 μm opening at the tip. The pressure was set to 10 psi, with 800 ms ejections. Zn(II) was delivered to a total of 3 slices.Statistics
Data was analyzed using GraphPrism V 9.0 (GraphPad Soft2ware Inc., La Jolla. CA, USA). All data were considered significant at the 95% confidence level (p < 0.05). Reported values are the mean ± the standard error of the mean (SEM), with n representing the number of electrodes or slices for each reported average.Results and discussion
Detection of Zn(II) on various plasma-treated gold surfaces
Plasma-treatment is necessary for adequate detection of Zn(II). Here, we tested the extent to which we could measure Zn(II) reduction (5 mM) on an untreated AuME (Fig. 1A, n = 6) with square wave voltammetry and observed no detectable reduction current. We speculate that this is due to the inherent organic layer that is on the surface of the gold fibers during the fabrication process, hindering available sites for Zn(II) electrochemistry.28 To improve detection, we evaluated the extent to which both air and O2 plasma influenced Zn(II) detection. Plasma was chosen as the approach for improving detection because (1) it eliminated the need to use strong, and possibly dangerous, acids, (2) avoided unnecessary corrosion on the surface which we could not be polished away (cylindrical electrode), and (3) because plasma can easily functionalize surfaces. Air and O2 plasma were compared to test whether higher oxygen content on the surface was necessary for improved detection.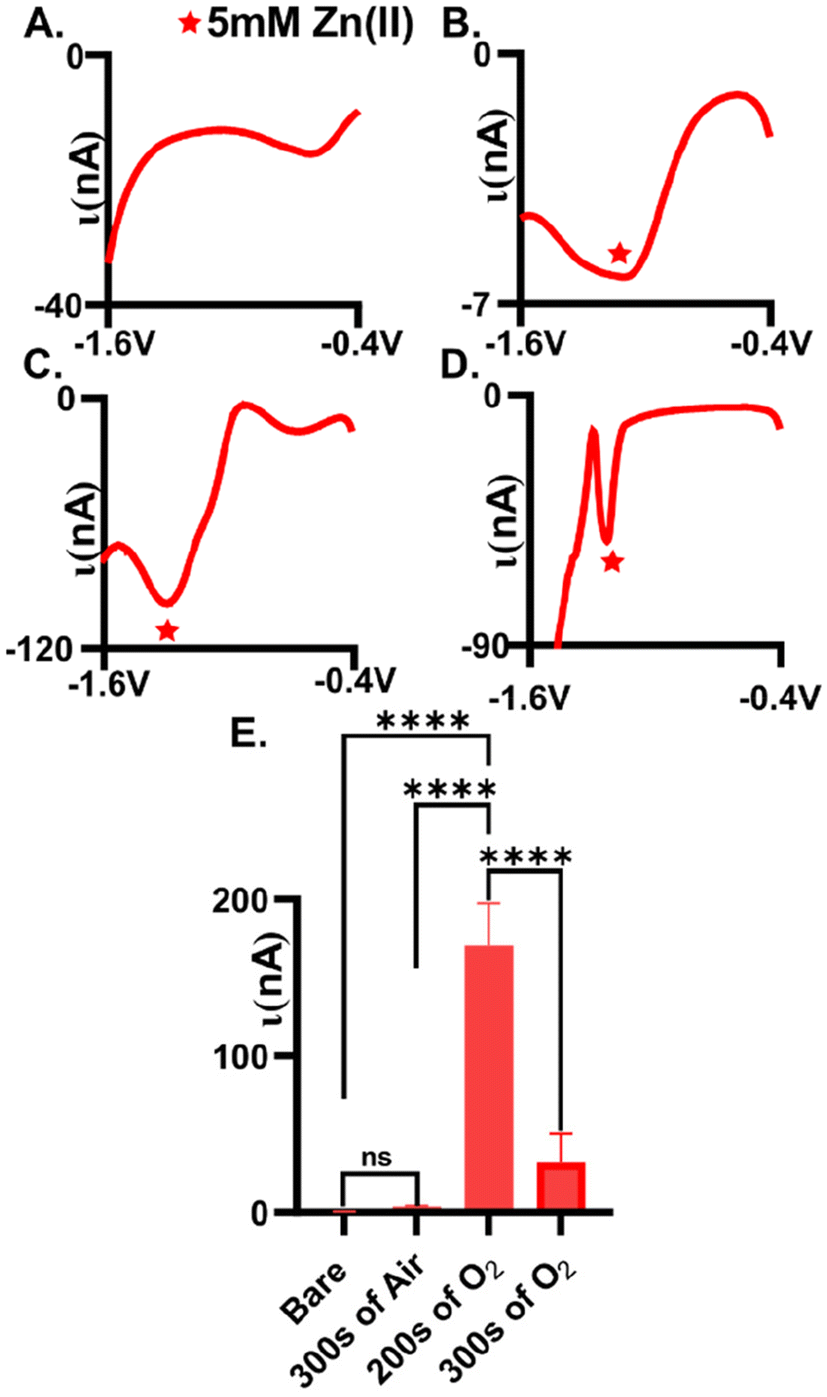 | ||
| Fig. 1 Plasma treatment with oxygen positively impacts the detection of Zn(II) at AuMEs. Measurements were made using 5 mM Zn(II) in 1 M KCl. (A) No observable reduction peak at untreated gold fibers. (B) Minimal reduction current is observed on air plasma treated fibers at −1.3 V. (C) A clear reduction peak at −1.4 V is observed for Zn(II) at oxygen plasma treated gold fibers (treatment was for 200 s). (D) Longer treatments times with oxygen plasma (300 s) resulted in less observable current for Zn(II) reduction. (E) Comparison of treatments shows an overall 442-fold (∼170.5 nA after treatment vs. 0.385 nA before treatment) increase for the detection of Zn(II) with 200 s of oxygen plasma treatment (n = 6). | ||
Oxygen plasma-treated electrodes significantly improved Zn(II) detection compared to air plasma-treated AuMEs. Square wave voltammetry, SWV, was utilized to assess peak placement and observed reduction current for plasma-treated electrodes. Gold fibers were first plasma-treated (see experimental methods for details) followed by electrode fabrication for testing. On average, we observed 3.5 ± 0.6 nA for 5 mM Zn(II) at air-plasma treated AuMEs, which is a 9.1-fold improvement compared to bare, untreated electrodes (Fig. 1B and E, n = 6). The broad peak and low current for such a large concentration indicates that low, micromolar concentrations would be infeasible for quantitation in tissue if used with FSCV. We speculated that this is because air plasma is primarily composed of nitrogen, leading to less oxygen functionality on the surface. Despite this observation, this result shows that cleaning is necessary for Zn(II) detection. Next, we compared the extent to which the O2 plasma treatment influenced Zn(II) detection. We compared a treatment consisting of a 200 s treatment with 250 SCCM and 100 W to 300 s with 200 SCCM and 100 W (Fig. 1C and D, n = 6). On average, we measured −170.5 ± 26.9 nA for 5 mM Zn(II) and this was significantly different compared to 300 s on air and oxygen plasma. The reduction peak was much more defined at these surfaces and was at approximately −1.2 V. With traditional electrochemical techniques that are used for metal detection, simultaneous detection of Cu(II) and Zn(II) has shown high sensitivity as well as simultaneous detection without interference.42–45 However, these various electrodes are organic based and cannot be compared to our novel treatment of a gold electrode. Therefore, from our observations of Cu(II) being the primary interfering analyte to Zn(II) on CFME coupled with FSCV, we tested 5 mM of Cu(II) using SWV. We discovered that Cu(II) does not need an oxide layer for detection and is more robust than 5 mM of Zn(II) when testing with traditional SWV, especially on AuMEs plasma treated for 200 s (Fig. S1,†n = 6). Previously, cleaning has shown elemental changes to the gold surface using X-ray Photoelectron Spectroscopy(XPS).28 At 200 s, we observed an approximate 442-fold increase in current compared to untreated bare gold fibers (Fig. 1E). For a 300 s treatment time, we observed an average reduction current of −31.6 ± 18.4 nA (n = 6). The peak slightly shifted in potential to approximately −1.15 V. The peak that is shown after the quantified Zn(II) peak does not grow with increasing concentration (tested 10 mM of Zn(II), data not shown), indicating that it is not inherent to Zn speciation on the surface. Longer treatment times lead to significantly less reduction current observed (one-way ANOVA, p = 0.071 Fig. 1E). We hypothesized that longer treatment times could be disrupting the integrity of the fiber, thus impacting the ability of the material to serve as an adequate electrochemical sensor. Next, fibers were surveyed using scanning electron microscopy (SEM) to visually assess the impact of plasma treatment on the morphology of the fiber.
Surface characterization
O2 plasma treatments do not significantly influence the gold fiber morphology (Fig. 2). SEM images of bare gold fibers, air, and oxygen-plasma treated fibers (both 200 and 300 s treatments) were taken and compared (Fig. 2). Bare fibers and air-treated fibers showed visible impurities on the surface (Fig. 2A and B) which were decreased or absent from O2-treated fibers (Fig. 2C and D). To validate that impurities are on the surface and that plasma treatment was functionalizing the surface with oxygen, EDAX and XPS were employed to observe elemental composition of the surface. Comparison of bare to the optimal oxygen plasma treatment demonstrates that O2 plasma treatment decreases surface carbon and increases oxygen functionalization (n = 3, Fig. S2†). XPS analysis demonstrated a decrease from 76.5% surface functionalization with adventitious carbon to 70.0%, while oxygen functionalization increased from 18.1% to 24.1% (Fig. S3†). Analysis of the oxygen peak at 530–535 eV demonstrates changes in the form of oxygen functionalization present; plasma treatment substantially increases C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O content on the fiber surface, suggesting that Zn(II) may preferentially interact at these functionalities (Fig. S4†). We show that we have a heterogeneous fiber from plasma treatment similar to previously published literature.28 Despite this observation, observed impurities do not appear to significantly impact Zn(II) detection. We additionally observe marginal impacts on surface structure from extended plasma treatment. Scattered pitting is evident on fibers treated with air plasma for 300 s (Fig. 2B). Similarly, longer exposures to O2 plasma (300 s) showed a slight increase in physical surface defects compared to the optimal 200 s treatment time (Fig. 2C and D). We speculate that extensive plasma exposure could erode the gold fiber surface, resulting in decreased current observed for Zn(II). However, it is important to note that while present, changes in surface morphology were minimal, particularly for the optimally treated fibers (Fig. S5†). Overall, plasma treatment of gold does induce noticeable changes to the fiber surface yet does not corrode it like typical acid treatments.
O content on the fiber surface, suggesting that Zn(II) may preferentially interact at these functionalities (Fig. S4†). We show that we have a heterogeneous fiber from plasma treatment similar to previously published literature.28 Despite this observation, observed impurities do not appear to significantly impact Zn(II) detection. We additionally observe marginal impacts on surface structure from extended plasma treatment. Scattered pitting is evident on fibers treated with air plasma for 300 s (Fig. 2B). Similarly, longer exposures to O2 plasma (300 s) showed a slight increase in physical surface defects compared to the optimal 200 s treatment time (Fig. 2C and D). We speculate that extensive plasma exposure could erode the gold fiber surface, resulting in decreased current observed for Zn(II). However, it is important to note that while present, changes in surface morphology were minimal, particularly for the optimally treated fibers (Fig. S5†). Overall, plasma treatment of gold does induce noticeable changes to the fiber surface yet does not corrode it like typical acid treatments.
 | ||
| Fig. 2 SEM images of gold fibers before and after treatment. (A) An example of an untreated gold fiber. (B) A representative example of a gold fiber treated with air plasma. Surface pitting is visible. (C) An example image of a fiber treated with oxygen plasma for 200 s. (D) An example image of a fiber treated with 300 s of oxygen plasma showing pitting. Scale bars are shown on each image. Red box highlights the noted pitting observed. | ||
Electrochemical characterization
Oxygen was further confirmed to be present on the fiber surface by verification using traditional cyclic voltammetry. Au oxide layers form on the surface after oxygen plasma treatment27 and here, we demonstrate that our plasma treatment generated an Au oxide layer (Fig. 3A, n = 4). Previous work showed a mixture of gold oxide monolayer species on the surface with X-ray photoelectron spectroscopy (XPS) after treatment of gold fibers in various conditions,28 which our XPS data further supports (Fig. S3 and S4†). Cyclic voltammograms (CVs) were collected in 0.05 M of H2SO4. Here, characteristic Au oxidation and reduction peaks (at 1.2 V and 1.0 V, respectively) are present on the O2-plasma treated surface indicating a surface oxide monolayer. To further explore this, we examined the extent to which two redox couples interacted with the surface: one which is surface insensitive Ru(NH3)6Cl3 (Ruhex) and one that is surface sensitive K3[Fe(CN)6] (Ferro/Ferri) (Fig. 3B and C). The ΔEp indicates the relative electron transfer and was used as a measure of electrode performance. The ΔEp for 5 mM of Ferro/Ferri was 0.33 V on a bare gold electrode compared to 0.2 V on the plasma-treated electrode. The change in ΔEp was significantly different (unpaired t-test, p = 0.012, n = 4) indicating that our plasma treatment is significantly improving Ferro/Ferri interaction at the surface, through the addition of the oxide monolayer. The shift in CV of the oxygen plasma-tread electrode could be due to the slight morphological changes of the surface that are observed with SEM after treatment (Fig. 2C). Since we have a heterogenous fiber, controlling the amount of oxygen on the surface can be difficult, this could be due to the formation of a heterogenous gold oxide layer (Fig. 3C). In contrast, 5 mM Ruhex demonstrated no change in ΔEp for the bare and plasma-treated electrode (unpaired t-test, p = 0.323, n = 3) due to its insensitivity to the chemical functionality of the electrode surface. Despite no change in electron transfer, we observe larger currents at our plasma treated surface indicating a larger surface area for electrochemistry. Comparing the untreated, or bare, to the optimal oxygen plasma treated AuMEs, we provide evidence that suggests we are functionalizing the surface with oxygen.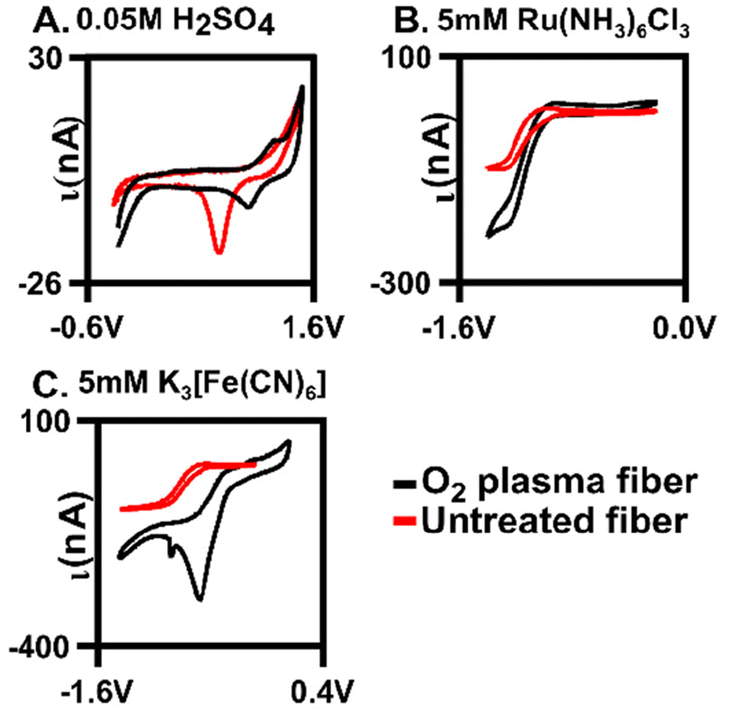 | ||
| Fig. 3 Plasma treatments influence surface-sensitive redox probes and the observed oxygen on the surface. (A) Comparison CV of bare and oxygen plasma treated AuME in 0.05 M H2SO4 showing oxide monolayer on plasma treated fibers with oxidation peak at 1.2 V and the reduction peak at 1.0 V. (B) Example CV for 5 mM Ru(NH3)6Cl3 dissolved in 1 M KCl at a scan rate of 0.1 V s−1 CV for untreated and oxygen plasma AuMEs. (C) Example CV for 5 mM K3[Fe(CN)6] aqueous solution at a scan rate of 0.1 V s−1 for untreated and oxygen plasma treated AuMEs. | ||
Fast scan cyclic voltammetry (FSCV)
Fast scan cyclic voltammetry (FSCV) is an electrochemical technique that employs higher scan rates, typically 400 V s−1, to study electroactive neurochemicals in the brain12,17,46,47 and other tissues.41,48 The typical waveform for FSCV is collected at 10 Hz, which enables 100 ms temporal resolution. When using higher scan rates, large capacitive currents are generated but are removed using background subtraction (Fig. S6†). Therefore, all CVs shown for FSCV have been background subtracted. Due to background subtraction and the use of microelectrodes with high surface area, lower limits of detection are capable compared to traditional CV. Our prior work demonstrated that Zn(II) could be detected using FSCV26 yet needs a sweep to a positive potential to aid in stripping of any plated Zn(s) on the surface. Here, we further demonstrate that Zn(II) can be measured with FSCV on a AuME. The waveform used is depicted in Fig. 4A. The observed reduction peak for Zn(II) with SWV was −1.2 V; therefore, to accommodate for the shift in potentials observed at fast scan rates, we scanned to −1.6 V. Due to the potential window of gold,7 we are limited compared to carbon-based microelectrodes that have a potential threshold of −2.0 V to 2.0 V. Despite this, gold has a substantial reduction potential range that can be utilized for the reduction of Zn(II) with a limit of −2.0 V. Because we are not scanning to high oxidizing potentials, we can eliminate the potential for water oxidation. For FSCV, we observe the reduction peak for Zn(II) at −1.2 V (Fig. 4C). An oxidation peak is observed at −0.5 V. An example false color plot is shown in Fig. 4B. Because of this, we chose to scan to 0.6 V to clearly observe both peaks for Zn(II). A negative holding potential helps to facilitate adsorption on the surface. For all characterization with FSCV, 20 μM Zn(II) was used because it is the physiological concentration of Zn(II) in the extracellular space in the brain under normal conditions.20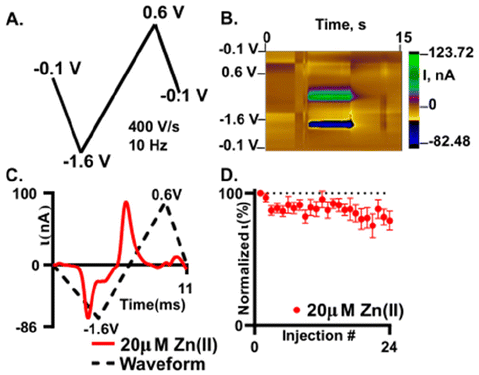 | ||
| Fig. 4 Zn(II) detection is possible and stable on an oxygen plasma treated AuME with FSCV. (A) Waveform used for the detection of Zn(II). (B) False color plot showing the redox profile of 20 μM Zn(II). Voltage is on the y-axis, time is on the x-axis, and the current is shown in the false color plot. (C) “Open” CV for 20 μM Zn(II) with a reduction peak at −1.2 V and an associated oxidation peak at −0.73 V. (D) 24 repeated injections of 20 μM Zn(II) demonstrates detection stability. Reduction current for Zn(II) was normalized to the first injection and plotted as a function of injection number (n = 6). | ||
Stability of detection is important to enable successive recording of Zn(II) release in the brain. In our stability test, we repeatedly injected 20 μM Zn(II) (Fig. 4D). The reduction current was normalized to the first injection and plotted as a function of injection number. After 24 injections of 20 μM Zn(II) we observed an overall decrease of 20 ± 1.2% with an associated RSD of 17.5% (Fig. 4D, n = 6). Here, we show that we obtain similar loss of current over time at plasma treated AuMEs to CFME's at an extended sawhorse waveform. However, the reduction current on AuMEs is higher for 20 μM Zn(II) showing improved detection ability. Our prior work demonstrated that we had to scan to high potentials (1.45 V) and hold for 3 ms to strip the plated Zn(II) off the surface.26 Here, we demonstrate that this step isn't necessary on AuMEs which shortens the waveform needed for detection.
Zn(II) interaction on gold microelectrodes
Zn(II) interacts by a combination of diffusion and adsorption at the surface of O2-plasma treated AuMEs. Analyzing the current changes as a function of scan rate can reveal the dominating interaction at the surface. The slope of the log of current vs. the log of scan rate plot reveals the dominating interaction, with slopes closer to 0.5 indicative of diffusion-limited processes and slopes closer to 1.0 indicative of adsorption-limited. The slope was 0.734 indicating that a combination of diffusion and adsorption limited processes are occurring (Fig. 5).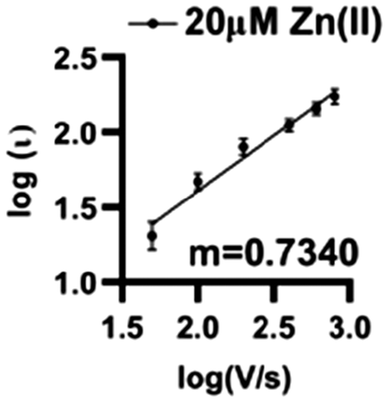 | ||
| Fig. 5 Log–log plot of current vs. scan rate demonstrates that Zn(II) is both adsorption and diffusion controlled on the surface with an associated R2 = 0.8568 and m = 0.7340 (n = 6). | ||
Sensitivity, limit of detection, and interference
A significant improvement in detection sensitivity is observed on gold compared to carbon microelectrodes for Zn(II). Previously, we reported a sensitivity of 0.203 nA μM−1 for Zn(II) with FSCV and carbon-fiber microelectrodes.26 Here, we report a sensitivity of 5.16 ± 0.66 nA μM−1 for Zn(II) on AuMEs, substantially improving sensitivity from our prior work (Fig. 6; n = 4–10). The “open” CV shows the reduction peak for 2.5 μM of Zn(II) and its reoxidation; notably, at low concentrations, large switching error is apparent at +0.6 V (Fig. 6A). Linearity is maintained from 2.5 μM to 40 μM. Current begins to taper at 80 μM but does not flatten even at 640 μM (Fig. S7†). We hypothesize this is due to the addition of a more uniform oxide layer, lending a higher amount of adsorption sites, ultimately widening where saturation at the electrode surface occurs. Alternatively, this could indicate more contribution of diffusion-limited processes at gold surfaces, leading to less saturation at the surface. This is supported by previously published work on dopamine (DA) detection using a gold electrode that showed a greater linear range compared to that of DA on CFME.49 Detection of concentrations down to 100 nM was attempted; however, at nanomolar concentrations, current was highly variable, even within a single electrode. The limit of detection (LOD) was calculated as 3σ of the noise (0.18 ± 0.67 nA) and is reported at 310 ± 29 nM. Noise was determined as the current from a set background-subtracted point at −1.2 V, 2 s prior to injection of the lowest analyte concentration. This LOD is important to determine since it has not been previously reported for Zn(II) on microelectrodes in biological buffers for FSCV. The limit of quantification (LOQ), 10σ of the noise, is 1.2 ± 0.1 μM; this is in alignment with our linear working range. This work shows that oxygen plasma treated AuMEs can increase our sensitivity for detection in tissue and hopefully move to endogenous measurements of Zn(II) within tissue on a selective electrode to compete with interfering analytes reported to be free signaling in the brain, such as. Cu(II).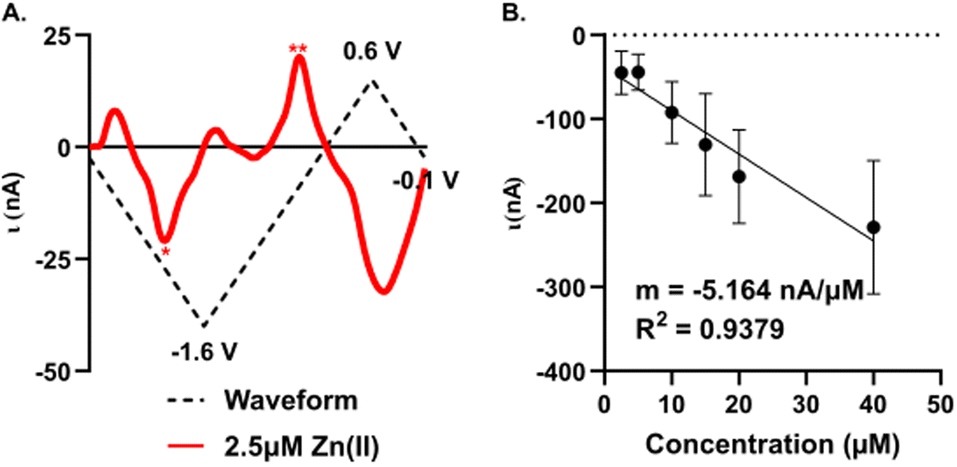 | ||
| Fig. 6 Concentration curve of 5 μM–640 μM of Zn(II) with FSCV. (A) Example “open” CV of 2.5 μM Zn(II), stars denotes reduction (*) and oxidation (*) peaks. (B) Concentration curve of Zn(II) with a slope of −5.164 nA μM−1 and an R2 value of 0.9379. | ||
As FSCV relies on direct electrochemical detection rather than a recognition element, analytes with overlapping reduction and oxidation potentials can interfere with selective detection of the target analyte. We tested four potential interferents: Cu(II), which overlaps with Zn(II) reduction; and three electroactive monoamines: dopamine (DA), norepinephrine (NE), and serotonin (5-HT), which are commonly monitored with FSCV, colocalized with Zn(II), and have oxidation peaks appearing below +0.6 V, potentially enabling codetection with Zn(II) (Fig. S8,†n = 3). A bolus of 20 μM Zn(II) and Cu(II) and 1 μM DA, NE, and 5-HT were individually applied at the Zn(II) waveform previously described. Clear reduction and oxidation peaks were observable for Zn(II) at −1.2 V and −0.5 V, respectively (Fig. S8A†). Surprisingly, Cu(II) was unstable at the modified AuMEs with FSCV and no significant reduction peaks were observed, suggesting that Cu(II) is unlikely to be a significant interferent in a tissue matrix when using fast scan rates (Fig. S8B†). DA and 5-HT showed little to no oxidation at plasma-treated AuMEs (Fig. S7C and E†); DA is known to demonstrate limited sensitivity on AuMEs with FSCV,49 while 5-HT has not been previously monitored on AuMEs. NE showed a significant oxidation at +0.55 V, which did not overlap with either zinc peak and suggests codetection is feasible (Fig. S8D†). No reduction peaks were observed for the monoamines, as they were introduced in a reduced state.
Detection of exogenous Zn(II) in the hippocampus
Detection of Zn(II) in brain tissue is possible at plasma treated AuMEs and FSCV. We pressure ejected, using a picospritzer, 200 μM of Zn(II) near our electrode to mimic transient firing in the CA1 of the hippocampus (Fig. 7, n = 3). This region was chosen because it is highly innervated with Zn(II)-containing glutamatergic cells.50Fig. 7 shows an example false color plot and associated CV of exogenous Zn(II) detection in the tissue. The false color plot shows a 30 s time file of the exogenous application of Zn(II). The Zn(II) reduction peak is circled in red, and the injection time of exogenous application is noted by the black arrow (Fig. 7A). The associated CV is an “open” CV to show the reduction peak more clearly (Fig. 7B). The reduction peak for Zn(II) is at −1.4 V with an associated current of −350 nA, this shift in potential is likely an artefact of the complex tissue matrix, similar to other analytes observed with FSCV in tissue.12–14 Additionally, the extra peak observed on the back scan is non-faradaic and due to drift from pressure ejection near the electrode. The current events last on average 10 seconds implying reuptake of the applied Zn(II). This shows feasibility for endogenous measurements which will enable pharmacological exploration and establishing a mechanism for Zn(II) release and clearance in the brain in the future, with much higher sensitivity than traditional CFMEs. In this work, we demonstrate a more robust detection of Zn(II) by utilizing oxygen plasma treated AuMEs to prove oxygen species are necessary for the detection of Zn(II) with FSCV. | ||
| Fig. 7 Exogenous detection of 200μM Zn(II). (A) False color plot showing injection of Zn(II) and duration of event. (B) “Open” CV of associated false color plot showing reduction of Zn(II) denoted by a red star. n = 3. | ||
Conclusion
In this work we have developed a new method of cleaning the organic layer from gold fibers as a safer alternative to the usual acid treatment practices and have used these plasma-treated surfaces for improved Zn(II) detection. Traditional cyclic voltammetry demonstrates fast electron transfer due to having oxygen on the surface which improves the adsorption of Zn(II). We validate that Zn(II) is a mixture of adsorption and diffusion at AuME surfaces, similar to the interactions observed on CFMEs. Utilizing these oxygen plasma treated AuMEs, we show improved sensitivity for the detection of Zn(II) which enables a lower limit of detection. Stability of Zn(II) detection on AuMEs with FSCV is comparable to carbon fiber microelectrodes coupled with the extended sawhorse waveform (ESW). The use of AuME's with FSCV eliminates the need to scan to high potentials to renew the surface thus shortening the applied waveform and ultimately helping to eliminate some biological interferences. We also demonstrate the utility of these new electrodes to measure exogenously delivered Zn(II) in the hippocampus. In conclusion, we show an improved method for the detection of Zn(II) that will hopefully lead to important investigations into the mechanism of Zn(II) signaling in selective areas of the brain.Data availability
The data supporting this article have been included within the manuscript and ESI.† Any additional raw data files will be made available upon request to the corresponding author.Conflicts of interest
No conflict of interest to declare.Acknowledgements
This work is supported by the National Science Foundation under Award 2143520, and the Alfred P. Sloan Foundation (Grant #FG-2022-18400). The is solely the responsibility of the authors and does not reflect the official views of the NSF or Alfred P. Sloan Foundation.References
- B. Jill Venton and Q. Cao, Fundamentals of Fast-Scan Cyclic Voltammetry for Dopamine Detection, Analyst, 2020, 145(4), 1158–1168, 10.1039/C9AN01586H.
- A. Hermans, R. B. Keithley, J. M. Kita, L. A. Sombers and R. M. Wightman, Dopamine Detection with Fast-Scan Cyclic Voltammetry Used with Analog Background Subtraction, Anal. Chem., 2008, 80(11), 4040–4048, DOI:10.1021/ac800108j.
- D. L. Robinson, B. J. Venton, M. L. A. V. Heien and R. M. Wightman, Detecting Subsecond Dopamine Release with Fast-Scan Cyclic Voltammetry in Vivo, Clin. Chem., 2003, 49(10), 1763–1773 CrossRef PubMed.
- M. L. A. V. Heien, P. E. M. Phillips, G. D. Stuber, A. T. Seipel and R. M. Wightman, Overoxidation of Carbon-Fiber Microelectrodes Enhances Dopamine Adsorption and Increases sensitivityElectronic Supplementary Information (ESI) Available: National Instruments Data Acquisition System, Analyst, 2003, 128(12), 1413, 10.1039/b307024g , See: https://Www.Rsc.Org/Suppdata/an/B3/B307024g/.
- E. C. Dankoski, K. L. Agster, M. E. Fox, S. S. Moy and R. M. Wightman, Facilitation of Serotonin Signaling by SSRIs Is Attenuated by Social Isolation, Neuropsychopharmacology, 2014, 39(13), 2928–2937, DOI:10.1038/npp.2014.162.
- Y. Li, R. Jarosova, M. E. Weese-Myers and A. E. Ross, Graphene-Fiber Microelectrodes for Ultrasensitive Neurochemical Detection, Anal. Chem., 2022, 94(11), 4803–4812, DOI:10.1021/acs.analchem.1c05637.
- M. K. Zachek, A. Hermans, R. M. Wightman and G. S. McCarty, Electrochemical Dopamine Detection: Comparing Gold and Carbon Fiber Microelectrodes Using Background Subtracted Fast Scan Cyclic Voltammetry, J. Electroanal. Chem., 2008, 614(1–2), 113–120, DOI:10.1016/j.jelechem.2007.11.007.
- B. J. Ostertag, M. T. Cryan, J. M. Serrano, G. Liu and A. E. Ross, Porous Carbon Nanofiber-Modified Carbon Fiber Microelectrodes for Dopamine Detection, ACS Appl. Nano Mater., 2022, 5(2), 2241–2249, DOI:10.1021/acsanm.1c03933.
- Q. Jia, B. J. Venton and K. H. DuBay, Structure and Dynamics of Adsorbed Dopamine on Solvated Carbon Nanotubes and in a CNT Groove, Molecules, 2022, 27(12), 3768, DOI:10.3390/molecules27123768.
- Y. Li, A. L. Keller, M. T. Cryan and A. E. Ross, Metal Nanoparticle Modified Carbon-Fiber Microelectrodes Enhance Adenosine Triphosphate Surface Interactions with Fast-Scan Cyclic Voltammetry, ACS Meas. Sci. Au, 2022, 2(2), 96–105, DOI:10.1021/acsmeasuresciau.1c00026.
- Y. Li and A. E. Ross, Plasma-Treated Carbon-Fiber Microelectrodes for Improved Purine Detection with Fast-Scan Cyclic Voltammetry, Analyst, 2020, 145(3), 805–815, 10.1039/C9AN01636H.
- M. D. Nguyen and B. J. Venton, Fast-Scan Cyclic Voltammetry for the Characterization of Rapid Adenosine Release, Comput. Struct. Biotechnol. J., 2015, 13, 47–54, DOI:10.1016/j.csbj.2014.12.006.
- M. T. Cryan and A. E. Ross, Subsecond Detection of Guanosine Using Fast-Scan Cyclic Voltammetry, Analyst, 2018, 144(1), 249–257, 10.1039/C8AN01547C.
- B. E. K. Swamy and B. J. Venton, Subsecond Detection of Physiological Adenosine Concentrations Using Fast-Scan Cyclic Voltammetry, Anal. Chem., 2007, 79(2), 744–750, DOI:10.1021/ac061820i.
- Nafion–CNT coated carbon-fiber microelectrodes for enhanced detection of adenosine - Analyst (RSC Publishing) 10.1039/C2AN35297D. https://pubs-rsc-org.uc.idm.oclc.org/en/content/articlehtml/2012/an/c2an35297d (accessed 2023-04-23).
- A. Abdalla, C. W. Atcherley, P. Pathirathna, S. Samaranayake, B. Qiang, E. Peña, S. L. Morgan, M. L. Heien and P. Hashemi, In Vivo Ambient Serotonin Measurements at Carbon-Fiber Microelectrodes, Anal. Chem., 2017, 89(18), 9703–9711, DOI:10.1021/acs.analchem.7b01257.
- K. M. Wood and P. Hashemi, Fast-Scan Cyclic Voltammetry Analysis of Dynamic Serotonin Reponses to Acute Escitalopram, ACS Chem. Neurosci., 2013, 4(5), 715–720, DOI:10.1021/cn4000378.
- K. E. Dunham and B. J. Venton, Improving Serotonin Fast-Scan Cyclic Voltammetry Detection: New Waveforms to Reduce Electrode Fouling, Analyst, 2020, 145(22), 7437–7446, 10.1039/D0AN01406K.
- K. Pihel, S. Hsieh, J. W. Jorgenson and R. M. Wightman, Electrochemical Detection of Histamine and 5-Hydroxytryptamine at Isolated Mast Cells, Anal. Chem., 1995, 67(24), 4514–4521, DOI:10.1021/ac00120a014.
- T. Siriwardhane, Y. Ou, P. Pathirathna and P. Hashemi, Analysis of Electrochemically Elusive Trace Metals with Carbon Fiber Microelectrodes, Anal. Chem., 2018, 90(20), 11917–11924, DOI:10.1021/acs.analchem.8b02210.
- P. Pathirathna, Y. Yang, K. Forzley, S. P. McElmurry and P. Hashemi, Fast-Scan Deposition-Stripping Voltammetry at Carbon-Fiber Microelectrodes: Real-Time, Subsecond, Mercury Free Measurements of Copper, Anal. Chem., 2012, 84(15), 6298–6302, DOI:10.1021/ac301358r.
- Y. Yang, A. A. Ibrahim, P. Hashemi and J. L. Stockdill, Real-Time, Selective Detection of Copper(II) Using Ionophore-Grafted Carbon-Fiber Microelectrodes, Anal. Chem., 2016, 88(14), 6962–6966, DOI:10.1021/acs.analchem.6b00825.
- P. Hashemi, E. C. Dankoski, J. Petrovic, R. B. Keithley and R. M. Wightman, Voltammetric Detection of 5-Hydroxytryptamine Release in the Rat Brain, Anal. Chem., 2009, 81(22), 9462–9471, DOI:10.1021/ac9018846.
- A. E. Ross and B. J. Venton, Sawhorse Waveform Voltammetry for Selective Detection of Adenosine, ATP, and Hydrogen Peroxide, Anal. Chem., 2014, 86(15), 7486–7493, DOI:10.1021/ac501229c.
- Y. Yang, P. Pathirathna, T. Siriwardhane, S. P. McElmurry and P. Hashemi, Real-Time Subsecond Voltammetric Analysis of Pb in Aqueous Environmental Samples, Anal. Chem., 2013, 85(15), 7535–7541, DOI:10.1021/ac401539f.
- A. N. Perry, M. T. Cryan and A. E. Ross, Extended Sawhorse Waveform for Stable Zinc Detection with Fast-Scan Cyclic Voltammetry, Anal. Bioanal. Chem., 2021, 413(27), 6727–6735, DOI:10.1007/s00216-021-03529-8.
- F. Widdascheck, M. Kothe, A. A. Hauke and G. Witte, The Effect of Oxygen Plasma Treatment of Gold Electrodes on the Molecular Orientation of CuPc Films, Appl. Surf. Sci., 2020, 507, 145039, DOI:10.1016/j.apsusc.2019.145039.
- L. M. Fischer, M. Tenje, A. R. Heiskanen, N. Masuda, J. Castillo, A. Bentien, J. Émneus, M. H. Jakobsen and A. Boisen, Gold Cleaning Methods for Electrochemical Detection Applications, Microelectron. Eng., 2009, 86(4–6), 1282–1285, DOI:10.1016/j.mee.2008.11.045.
- F. L. Almeida and S. G. Santos-Filho, Surface Activation of Gold Electrodes Using Electrochemical Conditioning, in 28th Symposium on Microelectronics Technology and Devices (SBMicro 2013), IEEE, Curitiba, Brazil, 2013, pp. 1–5. DOI:10.1109/SBMicro.2013.6676168.
- B. J. Venton and R. M. Wightman, Psychoanalytical Electrochemistry: Dopamine and Behavior, Anal. Chem., 2003, 75(19), 414 A–421 A, DOI:10.1021/ac031421c.
- T. Izumi, I. Watanabe and Y. Yokoyama, Activation of a Gold Electrode by Electrochemical Oxidation-Reduction Pretreatment in Hydrochloric Acid, J. Electroanal. Chem. Interfacial Electrochem., 1991, 303(1–2), 151–160, DOI:10.1016/0022-0728(91)85122-6.
- A. R. Heiskanen, C. F. Spégel, N. Kostesha, T. Ruzgas and J. Emnéus, Monitoring of Saccharomyces Cerevisiae Cell Proliferation on Thiol-Modified Planar Gold Microelectrodes Using Impedance Spectroscopy, Langmuir, 2008, 24(16), 9066–9073, DOI:10.1021/la800580f.
- M. B. Gumpu, S. Sethuraman, U. M. Krishnan and J. B. B. Rayappan, A Review on Detection of Heavy Metal Ions in Water – An Electrochemical Approach, Sens. Actuators, B, 2015, 213, 515–533, DOI:10.1016/j.snb.2015.02.122.
- T. Siriwardhane, Y. Ou, P. Pathirathna and P. Hashemi, Analysis of Electrochemically Elusive Trace Metals with Carbon Fiber Microelectrodes, Anal. Chem., 2018, 90(20), 11917–11924, DOI:10.1021/acs.analchem.8b02210.
- D. Berman and J. Krim, Impact of Oxygen and Argon Plasma Exposure on the Roughness of Gold Film Surfaces, Thin Solid Films, 2012, 520(19), 6201–6206, DOI:10.1016/j.tsf.2012.06.033.
- A. J. Syeed, Y. Li, B. J. Ostertag, J. W. Brown and A. E. Ross, Nanostructured Carbon-Fiber Surfaces for Improved Neurochemical Detection, Faraday Discuss., 2022, 233(0), 336–353, 10.1039/D1FD00049G.
- Plasma Treatment of Graphene Oxide|IntechOpen. https://www.intechopen.com/chapters/61726 (accessed 2023-05-11).
- J. Meng, W. Nie, K. Zhang, F. Xu, X. Ding, S. Wang and Y. Qiu, Enhancing Electrochemical Performance of Graphene Fiber-Based Supercapacitors by Plasma Treatment, ACS Appl. Mater. Interfaces, 2018, 10(16), 13652–13659, DOI:10.1021/acsami.8b04438.
- S. A. Sapp, D. T. Mitchell and C. R. Martin, Using Template-Synthesized Micro- and Nanowires as Building Blocks for Self-Assembly of Supramolecular Architectures, Chem. Mater., 1999, 11(5), 1183–1185, DOI:10.1021/cm990001u.
- M. Ramasamy and J. W. Ha, Influence of Oxygen Plasma Treatment on Structural and Spectral Changes in Gold Nanorods Immobilized on Indium Tin Oxide Surfaces, J. Chem. Phys., 2022, 157(1), 014702, DOI:10.1063/5.0097220.
- A. L. Hensley, A. R. Colley and A. E. Ross, Real-Time Detection of Melatonin Using Fast-Scan Cyclic Voltammetry, Anal. Chem., 2018, 90(14), 8642–8650, DOI:10.1021/acs.analchem.8b01976.
- J. Kudr, H. V. Nguyen, J. Gumulec, L. Nejdl, I. Blazkova, B. Ruttkay-Nedecky, D. Hynek, J. Kynicky, V. Adam and R. Kizek, Simultaneous Automatic Electrochemical Detection of Zinc, Cadmium, Copper and Lead Ions in Environmental Samples Using a Thin-Film Mercury Electrode and an Artificial Neural Network, Sensors, 2015, 15(1), 592–610, DOI:10.3390/s150100592.
- M. F. de Oliveira, A. A. Saczk, L. L. Okumura, A. P. Fernandes, M. de Moraes and N. R. Stradiotto, Simultaneous Determination of Zinc, Copper, Lead, and Cadmium in Fuel Ethanol by Anodic Stripping Voltammetry Using a Glassy Carbon–Mercury-Film Electrode, Anal. Bioanal. Chem., 2004, 380(1), 135–140, DOI:10.1007/s00216-004-2733-8.
- N. M. Thanh, N. Van Hop, N. D. Luyen, N. H. Phong and T. T. Tam Toan, Simultaneous Determination of Zn(II), Cd(II), Pb(II), and Cu(II) Using Differential Pulse Anodic Stripping Voltammetry at a Bismuth Film-Modified Electrode, Adv. Mater. Sci. Eng., 2019, 2019, e1826148, DOI:10.1155/2019/1826148.
- W. Wu, M. Jia, Z. Wang, W. Zhang, Q. Zhang, G. Liu, Z. Zhang and P. Li, Simultaneous Voltammetric Determination of Cadmium(II), Lead(II), Mercury(II), Zinc(II), and Copper(II) Using a Glassy Carbon Electrode Modified with Magnetite (Fe3O4) Nanoparticles and Fluorinated Multiwalled Carbon Nanotubes, Microchim. Acta, 2019, 186(2), 97, DOI:10.1007/s00604-018-3216-5.
- C. J. Meunier and L. A. Sombers, Fast-Scan Voltammetry for In Vivo Measurements of Neurochemical Dynamics, Brain Reward Syst., 2021, 93–123, DOI:10.1007/978-1-0716-1146-3_5.
- E. Castagnola, K. Woeppel, A. Golabchi, M. McGuier, N. Chodapaneedi, J. Metro, I. M. Taylor and X. T. Cui, Electrochemical Detection of Exogenously Administered Melatonin in the Brain, Analyst, 2020, 145(7), 2612–2620, 10.1039/D0AN00051E.
- Subsecond spontaneous catecholamine release in mesenteric lymph node ex vivo - Lim - 2020 - Journal of Neurochemistry - Wiley Online Library. https://onlinelibrary-wiley-com.uc.idm.oclc.org/doi/10.1111/jnc.15115 (accessed 2023-05-11).
- M. K. Zachek, A. Hermans, R. M. Wightman and G. S. McCarty, Electrochemical Dopamine Detection: Comparing Gold and Carbon Fiber Microelectrodes Using Background Subtracted Fast Scan Cyclic Voltammetry, J. Electroanal. Chem., 2008, 614(1), 113–120, DOI:10.1016/j.jelechem.2007.11.007.
- B. Jackson, S. Harper, L. Smith and J. Flinn, Elemental Mapping and Quantitative Analysis of Cu, Zn, and Fe in Rat Brain Sections by Laser Ablation ICP-MS, Anal. Bioanal. Chem., 2006, 384(4), 951–957, DOI:10.1007/s00216-005-0264-6.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4an00307a |
| This journal is © The Royal Society of Chemistry 2024 |