
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral adaptive recognition with sequence specificity of aromatic dipeptides in aqueous solution by an achiral cage†
Lin
Cheng‡
,
Ping
Tian‡
,
Honghong
Duan
,
Qingfang
Li
,
Xiaowen
Song
,
Anyang
Li
 and
Liping
Cao
*
and
Liping
Cao
*
Key Laboratory of Synthetic and Natural Functional Molecule Chemistry of the Ministry of Education, Xi'an Key Laboratory of Functional Supramolecular Structure and Materials, College of Chemistry and Materials Science, Northwest University, Xi'an 710069, China. E-mail: chcaoliping@nwu.edu.cn
First published on 17th December 2022
Abstract
Sequence-specific recognition of peptides and proteins by synthetic compounds or systems remains a huge challenge in biocompatible media. Here, we report the chiral adaptive recognition (CAR) with sequence specificity of aromatic dipeptides in a purely aqueous solution using an achiral tetraphenylethene-based octacationic cage (1) as both a molecular receptor and chiroptical sensor. 1 can selectively bind and dimerize aromatic dipeptides to form 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 host–guest complexes with high binding affinity (>1010 M−2), especially up to ∼1014 M−2 for TrpTrp. Given the dynamic rotational conformation of TPE units, achiral 1 can exhibit chiral adaptive responses with mirror-symmetrical circular dichroism (CD) and circularly polarized luminescence (CPL) spectra to enantiomeric dipeptides via supramolecular chirality transfer in the host–guest complexes. Furthermore, this CAR with sequence specificity of 1 can be applied for molecular recognition of TrpTrp- or PhePhe-containing tetrapeptides, polypeptides (e.g., amyloid β-peptide1–20 and somatostatin), and proteins (e.g., human insulin) with characteristic CD responses.
:2 host–guest complexes with high binding affinity (>1010 M−2), especially up to ∼1014 M−2 for TrpTrp. Given the dynamic rotational conformation of TPE units, achiral 1 can exhibit chiral adaptive responses with mirror-symmetrical circular dichroism (CD) and circularly polarized luminescence (CPL) spectra to enantiomeric dipeptides via supramolecular chirality transfer in the host–guest complexes. Furthermore, this CAR with sequence specificity of 1 can be applied for molecular recognition of TrpTrp- or PhePhe-containing tetrapeptides, polypeptides (e.g., amyloid β-peptide1–20 and somatostatin), and proteins (e.g., human insulin) with characteristic CD responses.
Introduction
Supramolecular chemistry in water,1,2 which focuses on the synthetic mimicking of biological systems in biocompatible environments, offers a series of relatively simplified and representative biomimetic systems to achieve molecular recognition and sensing,3,4 self-assembly,5 catalysis,6 biomedical applications,7,8 and so on. Inspired by the role of hydrophobic pockets in biological systems, supramolecular chemists, over the past few decades, have designed and developed several acyclic and cyclic receptors, such as guanidinium compounds,9,10 crown ethers,11,12 cyclodextrins,13,14 cucurbiturils,15–19 coordination cages,20 and others,21,22 for the molecular recognition of peptides in aqueous solution. These host–guest recognition systems for peptides were further utilized for various applications, such as folding peptides to adjust their structural chirality,23,24 inhibiting amyloid fibrillation,25 enzyme assays and recognition.26,27 In these cases, only a few synthetic supramolecular systems could exhibit sequence-specific recognition for peptides with optical activities (e.g., UV/vis and fluorescence).12,17,22 On the other hand, most biomolecules, such as amino acids and peptides, are difficult to be analyzed due to their negligible Cotton effect on CD or CPL spectra.28–32 Despite the booming development in the molecular recognition for peptides, efficient, facile, and general methods that can offer multiple optical responses including fluorescence, CD, and CPL to recognize sequence-specific peptides in an aqueous solution are still unexplored.Tetraphenylethene (TPE) and its derivatives not only possess excellent aggregation-induced emission (AIE) properties that can show fluorescence signals, but also have chiral left-handed (M) and right-handed (P) rotational conformations that can exhibit CD and CPL signals.33–35 Based on the two direction-selective rotational conformations of the four phenyl rings in TPE, the M- and P-enantiomers of some TPE-based compounds can exhibit reliable structural chirality.36,37 Recently, our38–42 and other groups43,44 have developed some achiral TPE-based hosts including cyclophanes, molecular cages, and supramolecular organic frameworks, which can exhibit dynamic conformation chirality in host–guest recognition or self-assembly. Specifically, a water-soluble TPE-based octacationic cage (1) with two TPE faces can be an ideal candidate as both a molecular receptor and chiroptical sensor for the sequence-specific recognition of peptides in an aqueous solution. Theoretically, there are three possible conformational isomers of 1: achiral PM-1 and a pair of racemic MM-1 and PP-1, all of which have rapid and reversible conformation transformation with each other in the solution state (Fig. 1a). Among them, PM-1 is achiral and shows no CD or CPL signals. However, PP-1 exhibits negative CD and CPL signals, while MM-1 shows positive CD and CPL signals. Therefore, there are several advantages of 1 for the molecular recognition of peptides: (1) the fluorescent cage itself can exhibit optical responses without using other dyes as auxiliary chromophores. (2) The cooperation of the hydrophobic effect and electrostatic interactions arising from the large hydrophobic cavity and the eight positively charged pyridinium sites of 1 can be ideal for selectively binding to peptides with negatively charged and aromatic tryptophan (Trp), phenylalanine (Phe), or tyrosine (Tyr) residues at the C-terminal position. (3) The P- or M-rotational conformation of TPE units can offer various chiroptical responses to achieve sequence specificity based on the different binding affinities and intrinsic chiralities of various peptides via supramolecular chirality transfer in the host–guest complexes.
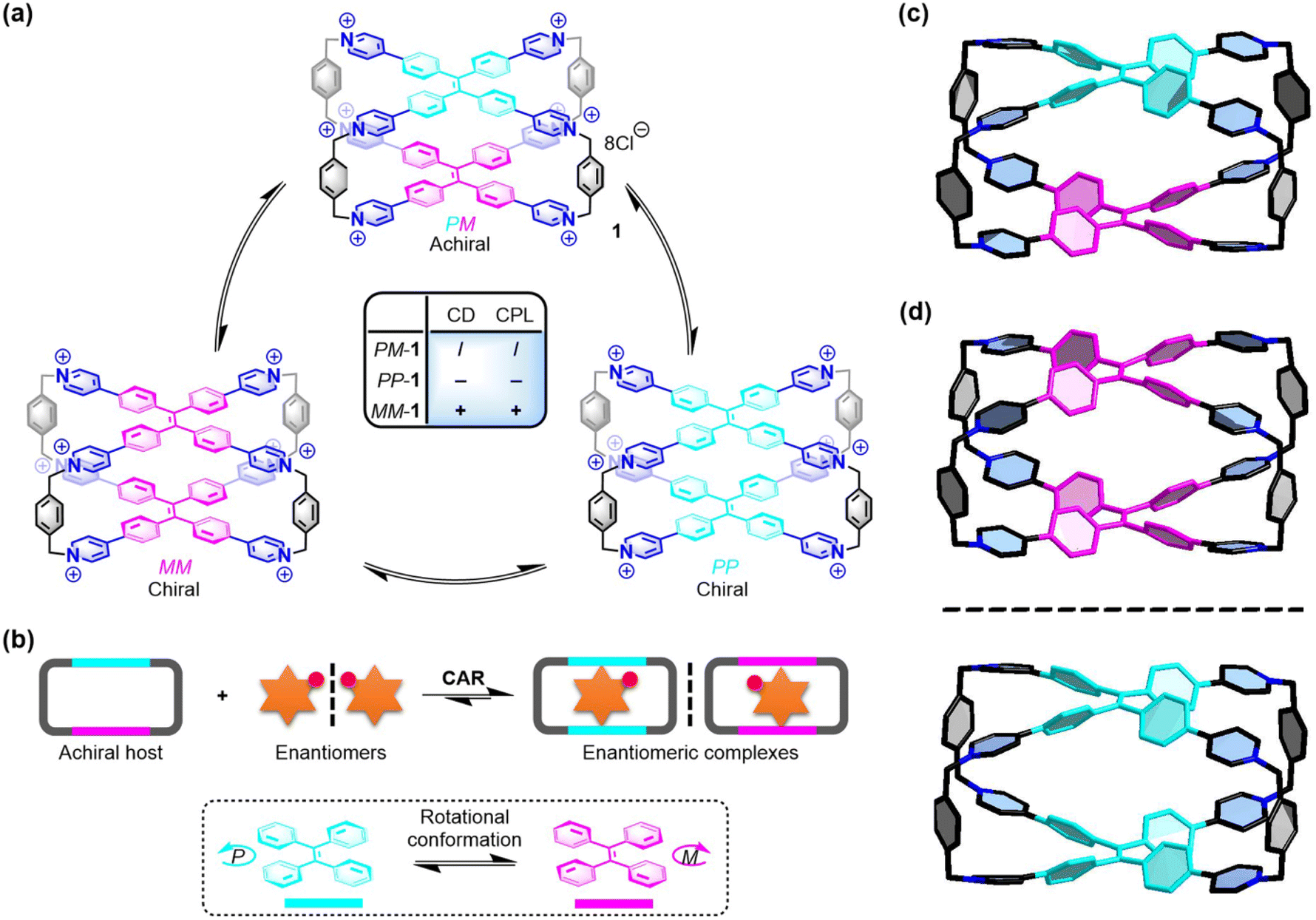 | ||
| Fig. 1 (a) Three conformational isomers of 1 and their chiroptical responses. (b) Chiral adaptive recognition of 1 for enantiomers. X-ray structures of (c) PM-1 and (d) racemic MM-1 and PP-1. Cl− counter ions and hydrogen atoms are omitted for clarity. CCDC numbers: 2100784 for PM-1; 2100668 for racemic MM-1 and PP-1.† | ||
To develop a synthetic supramolecular system with multiple chiroptical responses for the sequence-specific recognition of peptides, we propose a model of host–guest recognition for enantiomers by an achiral host (e.g., 1) with the ability of chiral adaptive responses to enantiomers, which is called chiral adaptive recognition (CAR). In traditional chiral recognition systems, chiral hosts, which have different binding affinities for enantiomers, can achieve chiral discrimination for enantiomers through non-chiral detection technologies (e.g., NMR and fluorescence).45,46 In the CAR host–guest systems, however, achiral hosts with structural flexibility (e.g., switchable P/M-rotation of TPE units in this case) can be induced by enantiomeric guests into one of their chiral conformations in the chiral host–guest complexes, respectively (Fig. 1b). As a result, these achiral hosts can gain a kind of guest-induced conformation chirality in the host–guest complexation to exhibit turn-on chiroptical signals (e.g., CD and CPL), which are distinguished, real-time, and adaptive responses to the different chiral guests and their enantiomers.47,48
Here, we develop chiral adaptive recognition (CAR) with sequence specificity of aromatic dipeptides by an achiral TPE-based cage (1) in an aqueous solution. The molecular recognition of 1 for amino acids, dipeptides, tetrapeptides, polypeptides, and even proteins is systematically proposed and investigated. Firstly, 1 exhibits selective recognition for Trp with obvious adaptive CD signals over others including glycine (Gly) and 18 pairs of enantiomeric amino acids. Secondly, 1 shows sequence-specific recognition to selectively bind and dimerize aromatic dipeptides (e.g., TrpTrp) in a 1:2 stoichiometry with high binding affinity (Ka ≈ 1010–1014 M−2). Meanwhile, dipeptide enantiomers can induce the adaptive conformation chirality of 1 in these host–guest systems: D-dipeptides induce the MM-rotational chiral conformation of 1 with positive CD and CPL signals, while L-dipeptides induce the PP-rotational chiral conformation of 1 with negative CD and CPL signals. And the CD spectra of 1 with aromatic dipeptide enantiomers are mirrored, which can be used to determine the enantiopurity of the L/D-dipeptide mixtures. Given the sequence-specific recognition for TrpTrp and PhePhe, 1 can mainly bind TrpTrp and PhePhe residues in the corresponding tetrapeptide chains regardless of these dipeptide residues at the N-terminal, middle, or C-terminal positions. Furthermore, the CAR of 1 for PhePhe-containing polypeptides and proteins, such as amyloid β-peptide1–20 (Aβ1–20), somatostatin, and human insulin, is further explored to show distinguishable CD spectral fingerprints for each valuable guest.
Results and discussion
Rotational conformation in the crystalline state
To further understand the chiral structures of the conformational isomers of 1, fortunately, we obtained two different X-ray structures of 1 by slow vapor diffusion of acetone into its aqueous solution at room temperature, which showed three conformational isomers including achiral PM-1 and a pair of racemic MM-1 and PP-1 in the crystalline state (Fig. S1 and Table S1†). In the crystal structure of achiral PM-1, the two TPE units of 1 adopt the P- and M-rotational conformation (Fig. 1c), respectively. In the crystal structure of a pair of racemic MM-1 and PP-1, however, the two TPE units of one 1 molecule simultaneously adopt the P-rotational conformation, while the two TPE units of another 1 molecule simultaneously adopt the M-rotational conformation (Fig. 1d). Although the whole structure is achiral, these two direction-selective conformational isomers of 1 including MM-1 and PP-1 in the crystalline state indicate that this achiral 1 can exhibit conformation chirality based on the rotational conformation of TPE units.Recognition of amino acids
As the basic building blocks of peptides and proteins, the selective recognition of amino acids is the key for the analysis and detection of peptides and proteins. The chiral recognition behaviors of 1 for D/L-amino acids in phosphate buffer were screened by CD experiments. Because of the coexistence of racemic MM-/PP- and achiral MP-rotational conformation of the two TPE units, this achiral 1 itself cannot show any chiral signals in the solution state. However, the CD spectra of 1 (0.10 mM) titrated with D/L-Trp showed a new absorption band in the range of 310 nm to 450 nm (Fig. S2†). More interestingly, achiral 1 exhibited chiral adaptive responses to a pair of D/L-Trp enantiomers and their mixtures, resulting in mirror-symmetrical CD spectra (Fig. S3†). These results indicated that the MM- or PP-rotational conformation of 1 could be induced by D/L-Trp, respectively. Based on our and others' previous reports, M-rotational and P-rotational TPE compounds should show positive and negative Cotton effects from long to short wavelengths, respectively.43,44 Therefore, D-Trp induces the M-rotational conformation of the two TPE units of 1 with a positive Cotton effect, while L-Trp induces the P-rotational conformation of the two TPE units of 1 with a negative Cotton effect.The CD responses of 1 to other amino acids were very weak or silent (Fig. S4–S23†), while NMR titration experiments of 1 with other amino acids showed that only some amino acids could be slightly bound inside the cavity of 1via the hydrophobic effect or interacted with the cationic surface of 1via electrostatic interactions. For aromatic amino acids including Trp, Phe and Tyr, all proton resonances of the phenyl rings showed slight upshifts owing to the shielding effect from 1, indicating the hydrophobic indolyl or phenyl rings could be inside the cavity of 1 with very weak binding affinity (Fig. S24–S26†). Besides the hydrophobic effect, the electrostatic interactions between this cationic 1 and positively or negatively charged amino acids are another non-covalent force in this aqueous recognition. Specifically, after mixing them with 1 in D2O, positively charged amino acids including histidine (His), lysine (Lys), and arginine (Arg), and negatively charged glutamic acid (Glu) showed slight downfield shifts, while negatively charged aspartic acid (Asp) and a neutral cysteine (Cys) showed slight upfield shifts, indicating that these amino acids as endo- or exo-guests could interact with the positively charged pyridinium cationic surface of 1 (Fig. S27–S32†). For neutral aliphatic amino acids including glycine (Gly), alanine (Ala), leucine (Leu), isoleucine (Ile), threonine (Thr), valine (Val), proline (Pro), serine (Ser), glutamine (Gln), methionine (Met), and asparagine (Asn), they could not exhibit any effective binding behavior with 1, owing to the lack of sufficiently strong non-covalent interactions (Fig. S33–S43†). Isothermal titration calorimetry (ITC) and UV/vis experiments proved that 1 could encapsulate two Trp molecules with weak ternary association constants of Ka = (4.97 ± 0.10) × 104 M−2 (Fig. S44†). However, the association constants between 1 and other amino acids could not be calculated by NMR or ITC experiments. Therefore, 1 exhibited a CAR with high selectivity for D/L-Trp based on their relatively strong affinity, because they have more hydrophobic indole group. This result indicates that both the strong binding affinity of the host–guest complex and the intrinsic chirality of D/L-Trp are important factors that determine the intensities and negative/positive Cotton effect of the CD spectra in the host–guest complexation.
Recognition of dipeptides
Compared with single amino acids, peptides with a specific sequence of amino acids have more important structural information to determine their function. The selective recognition of 1 for aromatic amino acids encouraged us to further investigate the sequence-specific recognition of 1 for dipeptides. 9 L-dipeptides and 3 D-dipeptides containing Trp, Phe, or Tyr residues were selected as chiral guests to investigate CAR with sequence specificity using 1 as both a molecular receptor and chiroptical sensor (Table 1). Initially, 1H NMR titration experiments were employed to investigate the host–guest chemistry between 1 and dipeptides in D2O. For example, the 1H NMR titration of 1 with L-TrpTrp showed that the proton resonances of both 1 and the guest were changed in the host–guest complexation (Fig. 2a and b and S45†). When 1.0 and 2.0 equiv. of L-TrpTrp were added to a solution of 1 in D2O, all proton resonances corresponding to L-TrpTrp showed obvious upfield shifts, while all proton resonances of 1 have slight downfield shifts, suggesting that two L-TrpTrp molecules could be encapsulated in the cavity of 1. At the same time, all proton resonances of 1 split into several peaks, resulting from the formation of an asymmetric environment upon complexation of chiral L-TrpTrp. Excess L-TrpTrp (3.0 equiv.) did not induce any obvious changes in the chemical shifts observed for 1, indicating that the host–guest stoichiometry of 1 and L-TrpTrp is 1:2. Similarly, we demonstrated that other dipeptides could also be encapsulated inside the hydrophobic cavity of 1 by 1H NMR titration experiments (Fig. S46–S53†). The ITC experiment further confirmed the 1:2 stoichiometry between 1 and L-TrpTrp with a high ternary association constant (Ka ≈ 1014 M−2) in phosphate buffer (Fig. 2c and Table 1). In the same way, 1 and other dipeptide molecules also formed 1:2 host–guest complexes (Fig. S55–S66†), whose binding model was confirmed by the X-ray structure in the next section. The association constants are summarized in Table 1. The ITC results showed that TrpTrp could have the strongest binding affinity with 1 compared to other dipeptides, which indicated that 1 has good sequence specificity for TrpTrp. In the UV/vis titration of 1 with all dipeptides, the absorbance maximum of 1 was slightly red-shifted with clean isosbestic points, resulting from the intermolecular conjugation enhancement of the TPE faces on 1 in the host–guest complexation with dipeptides (Fig. S67–S75†).
| Guests | K a (M−2) |
|---|---|
| a The data were fitted by the one sites model. In these cases, the number of binding sites was about two, indicating that the two guests bind the host with the same binding constant. b The data were fitted by the sequential binding sites model. | |
| L-TrpTrp | (1.80 ± 0.25) × 1014 |
| D-TrpTrp | (1.99 ± 0.30) × 1014 |
| L-PhePhe | (1.56 ± 0.11) × 1010 |
| D-PhePhe | (1.72 ± 0.34) × 1010 |
| L-TyrTyr | (2.36 ± 0.21) × 1011 |
| D-TyrTyr | (1.39 ± 0.11) × 1011 |
| L-PheTrpb | (4.46 ± 0.89) × 1012 |
| L-TyrTrp | (5.34 ± 1.71) × 1012 |
| L-TrpPhe | (3.55 ± 1.18) × 1013 |
| L-TyrPhe | (1.98 ± 0.31) × 1011 |
| L-TrpTyrb | (6.68 ± 0.74) × 1011 |
| L-PheTyr | (1.10 ± 0.13) × 1012 |
| D-Trp–L-Trp | (3.29 ± 0.10) × 1013 |
| L-Trp–D-Trp | (3.61 ± 0.21) × 1013 |
| TrpTrpGlyGly | (4.11 ± 0.46) × 1013 |
| GlyTrpTrpGly | (3.91 ± 0.59) × 1013 |
| GlyGlyTrpTrp | (3.36 ± 0.42) × 1013 |
| PhePheGlyGly | (9.99 ± 1.07) × 108 |
| GlyPhePheGly | (1.54 ± 0.15) × 108 |
| GlyGlyPhePhe | (4.49 ± 0.17) × 108 |
| Aβ1–20 | (2.37 ± 0.54) × 1012 |
| Somatostatin | (1.18 ± 0.06) × 109 |
| Human insulin | (1.15 ± 0.29) × 1013 |
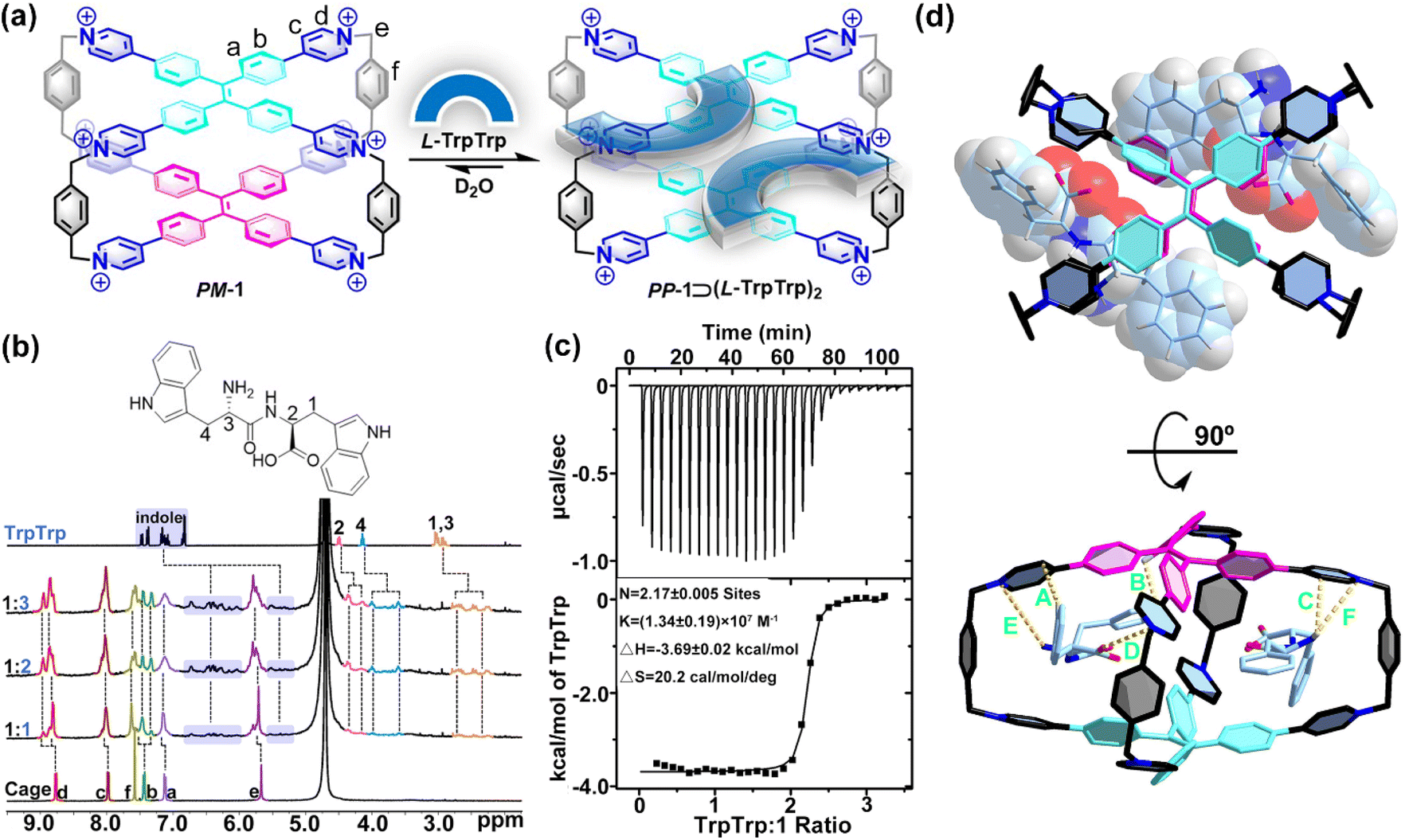 | ||
| Fig. 2 (a) Schematic representation of the host–guest complexation of PP-1⊃(L-TrpTrp)2. (b) 1H NMR spectra (400 MHz, 298 K, D2O) recorded for 1 (0.40 mM) with 1.0–3.0 equiv. of L-TrpTrp. (c) ITC of 1 with L-TrpTrp at 298 K in phosphate buffer (10 mM sodium phosphate, pH = 7.4). (d) Top and side views of the X-ray structure of PM-1⊃(L-PhePhe)2. The dashed lines are key CH⋯π interactions with distances of 2.70 Å (A), 2.35 Å (B) and 2.83 Å (C), electrostatic interactions with distances of 4.44 Å (D), and ion–dipole interactions with distances of 3.85 Å (E) and 4.97 Å (F). Br− counter ions and hydrogen atoms are omitted for clarity. CCDC number: 2155590† for PM-1⊃(L-PhePhe)2. | ||
To further understand the binding model of 1 with two dipeptide molecules, the X-ray quality crystals of 1 with L-PhePhe were obtained from their aqueous solution by slow solvent evaporation at room temperature (Fig. 2d and Table S1†). The crystallographic analysis shows that two L-PhePhe molecules are fully encapsulated inside the hydrophobic cavity of 1 through multiple noncovalent interactions along the diagonal direction of the cavity (Fig. 2d, top). And 1 can catch L-PhePhe molecules via electrostatic interaction (d(O−⋯N+) = 4.44 Å) between the carbonyl COO− and pyridinium N+ and two ion–dipole interactions (d(N⋯N+) = 3.85 and 4.97 Å) between the amido NH2 and pyridinium N+ (Fig. 2d, bottom). Therefore, the host–guest complexes of 1 and dipeptides are mainly stabilized by the cooperation of the hydrophobic effect and electrostatic/ion–dipole interactions in aqueous solution. The phenyl rings of two L-PhePhe molecules are not in the center of the cavity but at the intervals in the X-shaped molecular skeleton of 1, which indicates that the cavity of 1 is crowded for two aromatic dipeptide molecules. Therefore, the spatial proximity and steric hindrance effect between the TPE units of 1 and the phenyl rings of guests could be key factors that affect the rotational conformation of TPE units to achieve supramolecular chirality transfer in the host–guest complexes. In addition, two L-PhePhe molecules have no obvious interaction with each other. In this host–guest complex, the two TPE units of 1 adopt P- and M-rotational conformations to form PM-1⊃(L-PhePhe)2. Although the X-ray structure of PM-1⊃(L-PhePhe)2 does not show any direction selectivity on the rotational conformation of the two TPE units in the solid state, energy minimized structures of possible chiral host–guest complexes show that the selective conformation transformation from MP-1/MM-1 to PP-1 is an energy-favored process in the solution state when 1 molecule binds two L-PhePhe (Fig. S76†).
Subsequently, three pairs of aromatic dipeptide enantiomers including D/L-TrpTrp, D/L-PhePhe, and D/L-TyrTyr were selected to demonstrate the chiral adaptive responses of 1 by CD titration experiments (Fig. 3a). The CD spectra of 1 titrated with D-TrpTrp showed an obvious positive Cotton effect in the long-wavelength (310–450 nm) region, which is attributed to the M-rotational conformation of the TPE units (Fig. 3b), while the CD spectra of 1 titrated with L-TrpTrp showed negative CD signals at 310–450 nm, which corresponds to the P-rotational conformation of the TPE units (Fig. 3c). The mirror image of their CD spectra strongly confirmed that achiral 1 could exhibit CAR for D/L-TrpTrp to form a pair of enantiomeric host–guest complexes, MM-1⊃(D-TrpTrp)2 and PP-1⊃(L-TrpTrp)2, respectively. The dissymmetry factor (gabs) at 390 nm of MM-1⊃(D-TrpTrp)2 was 8.9 × 10−4, while gabs of PP-1⊃(L-TrpTrp)2 was −8.8 × 10−4. These results strongly suggest that the intrinsic chirality of D/L-TrpTrp determines the direction-selective rotational conformation transformation to generate MM-1 or PP-1, respectively. The gabs curves versus equiv. of L-TrpTrp further confirmed the 1:2 stoichiometry of PP-1⊃(L-TrpTrp)2, which is consistent with the ITC and X-ray results. Similarly, D/L-PhePhe and D/L-TyrTyr could also induce corresponding CD signals with the mirror image: one is positive for MM-1⊃(D-dipeptide)2 and the other one is negative for PP-1⊃(L-dipeptide)2, respectively (Fig. 3d and e and S77†). In addition, D/L-TrpTrp, which has a stronger binding affinity with 1, can induce a faster equilibrium based on the CD changes when compared with D/L-PhePhe and D/L-TyrTyr. This strongly suggests that the strong binding affinity of the host–guest complex is the primary factor for the chirality induction between the host and guest in the supramolecular chirality transfer process: the stronger binding between the host and guest can promote more effective chirality induction in the host–guest systems. Based on the CAR of 1 for the mixture of D/L-guests and their mirror-imaging CD spectra (Fig. 3f and S78–S80†), calibration lines with great linearity (R2 > 0.99) were obtained by plotting the CD signals of the three pairs of enantiomers at 390 nm against the enantiomeric excess (ee) values with errors of 1.22–3.68% (Fig. 3g), which can be used to determine the enantiopurity of dipeptide enantiomers. Interestingly, we also found the chiral adaptive responses of 1 are determined by the C-terminal residues of aromatic dipeptides: D-Trp–L-Trp induces the adaptive conformation chirality of PP-1, while L-Trp–D-Trp induces the adaptive conformation chirality of MM-1, which are consistent with L-TrpTrp and D-TrpTrp, respectively (Fig S81†). In these CAR host–guest systems, the association constants of achiral 1 with D/L-guests are almost same (Table 1). However, 1 can achieve chiral recognition to distinguish enantiomers and determine the enantiopurity, which mainly depend on the chiral adaptive responses with opposite CD signals to D/L-guests. The CD experiments also revealed that other L-aromatic dipeptides could also induce the conformation chirality of PP-1 to show negative CD with different intensities (Fig. S82–S87†). In addition, fluorescence titration between 1 and L-TrpTrp showed an obvious emission decrease of 1 centered at 560 nm, which could be mainly dominated by photoinduced electron transfer (PET) between electron-deficient pyridinium rings of 1 and electron-rich indole rings of L-TrpTrp (Fig. 3h).49 In contrast, the addition of L-PhePhe into the solution of 1 in phosphate buffer can induce an emission increase at around 560 nm, which could result from the restriction of intramolecular rotation (RIR) mechanism playing a dominant role when PhePhe molecules were inserted into the inner cavity of 1 (Fig. 3i).50
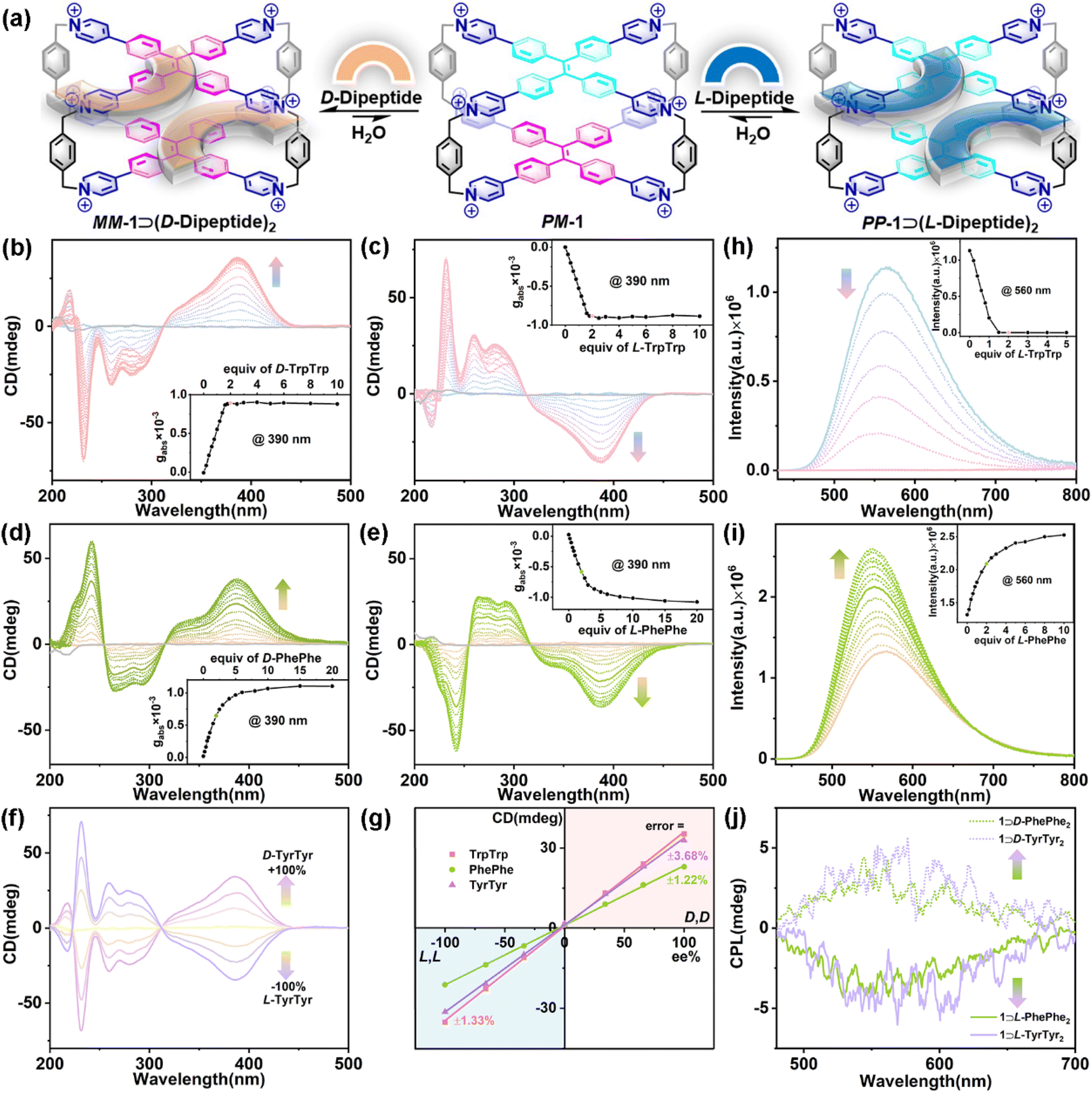 | ||
| Fig. 3 (a) Schematic representation of the CAR of 1 for D/L-dipeptides. CD spectra of 1 (20 μM) titrated with (b) D-TrpTrp, (c) L-TrpTrp, (d) D-PhePhe, and (e) L-PhePhe in phosphate buffer (10 mM sodium phosphate, pH = 7.4). (Inset) Plots of gabsversus equiv. of guests. (f) CD spectra of 1 (20 μM) in the presence of D/L-TyrTyr mixtures (40 μM) with ee ranging from −100% to +100%. (g) The calibration curve obtained for the CD signals (390 nm) upon varying ee values for D/L-dipeptides. Fluorescence spectra of 1 (10 μM) titrated with (h) L-TrpTrp and (i) L-PhePhe in phosphate buffer (10 mM sodium phosphate, pH = 7.4). (Inset) Plots of fluorescence intensity versus equiv. of guests. λex = 410 nm, ex/em slit = 1.2 nm. (j) CPL spectra of 1 (20 μM) with D/L-PhePhe (40 μM, chartreuse) and D/L-TyrTyr (40 μM, light purple) in phosphate buffer (10 mM sodium phosphate, pH = 7.4). λex = 320 nm, ex/em slit = 3000 μm. | ||
Given their adaptive conformation chirality and fluorescence, the CPL properties of the enantiomeric host–guest complexes further exhibited chiral adaptive responses to D/L-dipeptides. As shown in Fig. 3j, the CPL spectra recorded at 460–750 nm displayed positive or negative CPL induced by D-guests (e.g., D-PhePhe and D-TyrTyr) or L-guests (e.g., L-PhePhe and L-TyrTyr), respectively, which are consistent with CD experiments. Specifically, MM-1⊃(D-PhePhe)2 showed a positive CPL at around 560 nm (glum = 5.07 × 10−4), while PP-1⊃(L-PhePhe)2 displayed a negative CPL at around 560 nm (glum = −4.70 × 10−4). And the glum values of MM-1⊃(D-TyrTyr)2 and PP-1⊃(L-TyrTyr)2 were 6.40 × 10−4 and −4.10 × 10−4 at around 560 nm, respectively. The CPL needs a cooperative integration between supramolecular chirality and fluorescence in the supramolecular systems.35 Therefore, MM-1⊃(D-TrpTrp)2 and PP-1⊃(L-TrpTrp)2 hardly displayed CPL signals owing to their fluorescence quenching in the host–guest complexation (Fig. S88†).
Recognition of tetrapeptides
The above experiments demonstrate that 1 has strong binding affinity and sequence-specific recognition for aromatic dipeptides with chiroptical responses. To explore the CAR with sequence specificity of 1 for these sequence-specific dipeptides in the peptide chains, we further performed the host–guest experiments for two series of TrpTrp-containing tetrapeptides (including TrpTrpGlyGly, GlyTrpTrpGly, and GlyGlyTrpTrp) and PhePhe-containing tetrapeptides (including PhePheGlyGly, GlyPhePheGly, and GlyGlyPhePhe) with TrpTrp or PhePhe residues at the N-terminal, middle, and C-terminal positions (Fig. S89†), respectively, to study the position effect of these sequence-specific dipeptide residues on the molecular recognition and chiroptical response by 1.
1H NMR titration experiments of 1 with TrpTrp-containing tetrapeptides showed that the hydrophobic cavity of 1 preferred to encapsulate the TrpTrp residues of the peptide chains regardless of them being at the N-terminal, middle, or C-terminal positions, indicating that the hydrophobic effect is the main non-covalent force in these host–guest complexations (Fig. S90–S92†). TrpTrp-containing tetrapeptides have strong association constants (∼1013 M−2) with 1 in a 1:2 stoichiometry (Table 1). For PhePhe-containing tetrapeptides, the hydrophobic effect of PhePhe residues with 1 is much weaker than that of the TrpTrp residues. As a result, the deprotonated carboxylic groups with negative charge at the C-terminal position could offer an additional non-covalent force – electrostatic interaction – with positively charged pyridinium rings of 1, which can slightly affect the binding site between 1 and PhePhe-containing tetrapeptides. For example, the 1H NMR spectra of 1 with PhePheGlyGly showed that the proton resonances of CH2 units in the GlyGly residue and partial phenyl units in the PhePhe residue slightly shifted upfield (Δδ = −0.13 to −0.03 ppm), indicating that the PheGlyGly residue at the C-terminal position was preferentially bound inside the cavity of 1 (Fig. 4a and S93†). This is a result of the balance between the hydrophobic effect and electrostatic interaction. When the PhePhe residue is located near or at the C-terminal position in GlyPhePheGly and GlyGlyPhePhe, the positive cooperation of the hydrophobic effect and electrostatic interaction makes 1 preferentially bind with the PhePhe residue. The proton resonances of the phenyl, CH2, and CH groups of the PhePhe residue in GlyPhePheGly showed a large upfield shift (Δδ = −0.44 to −0.13 ppm) at a 1:2 ratio of host and guest, indicating that the PhePhe residue is inside the cavity of 1 (Fig. 4b and S94†). The proton resonances of phenyl, CH2, and CH groups of the PhePhe residue in GlyGlyPhePhe showed the largest upfield shifts (Δδ = −0.97 to −0.30 ppm) and the proton resonances of CH2 units in the GlyGly residue shifted downfield at a 1:2 ratio of host and guest, indicating that 1 binds the PhePhe residue (Fig. 4c and S95†). The UV/vis titration showed a slight redshift of the absorbance peak of 1 with clean isosbestic points, indicating the formation of the host–guest complexes (Fig. S96–S101†). ITC experiments further demonstrated that these tetrapeptides can be complexed with 1 in a 2:1 ratio (Fig. S102–S107†). The comparison of association constants showed that TrpTrp-containing tetrapeptides have much stronger binding affinity with 1 than PhePhe-containing tetrapeptides owing to their more hydrophobic indole groups, indicating that 1 possesses a good ability of sequence-specific recognition for TrpTrp (Table 1).
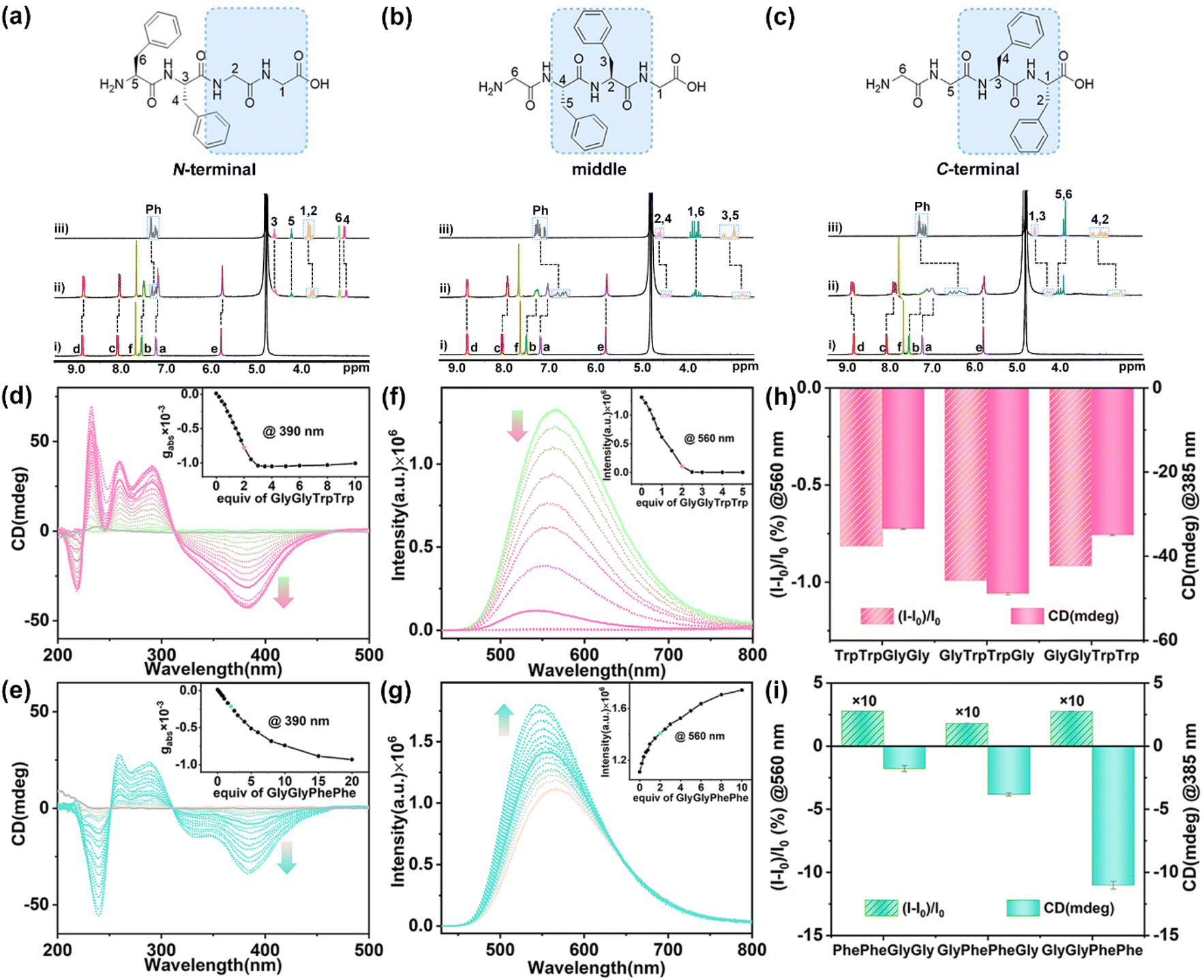 | ||
| Fig. 4 1H NMR spectra (400 MHz, 298 K, D2O) recorded for 1 (0.40 mM) with (a) PhePheGlyGly, (b) GlyPhePheGly, and (c) GlyGlyPhePhe: (i) 1, (ii) 1 and tetrapeptides (2.0 equiv.), and (iii) tetrapeptides. CD spectra of 1 (20 μM) titrated with (d) GlyGlyTrpTrp and (e) GlyGlyPhePhe in phosphate buffer (10 mM sodium phosphate, pH = 7.4). (Inset) Plots of gabsversus equiv. of guests. Fluorescence spectra of 1 (10 μM) titrated with (f) GlyGlyTrpTrp and (g) GlyGlyPhePhe (2.0 equiv.) in phosphate buffer (10 mM sodium phosphate, pH = 7.4). (Inset) Plots of fluorescence intensity versus equiv. of guests. λex = 410 nm, ex/em slit = 1.2 nm. CD and fluorescence intensities of 1 with (h) L-TrpTrp- and (i) L-PhePhe-containing tetrapeptides (2.0 equiv.). Error bars are standard deviations based on three independent measurements. | ||
When these tetrapeptides were added to the aqueous solution of 1, the characteristic CD signals of chiral PP-1 at 310–450 nm were induced. As a result, all L-type tetrapeptides induced the P-rotational conformation chirality of 1 to generate PP-1⊃(tetrapeptide)2 with negative CD signals. The TrpTrp-containing tetrapeptides containing the N-terminal, middle, and C-terminal TrpTrp residues exhibited similar shapes and intensities in the CD spectra (Fig. 4d, S108 and S109†), because 1 could bind to the same position – TrpTrp residue – in TrpTrp-containing tetrapeptides. However, when the aromatic residues are PhePhe, the chirality induction between 1 and PhePhe-containing tetrapeptides was reduced in the order of GlyGlyPhePhe to GlyPhePheGly and PhePheGlyGly, which showed a certain position selectivity (Fig. 4e, S110 and S111†). In the case of PhePhe-containing tetrapeptides, the strongest negative CD signal of 1 was induced by GlyGlyPhePhe, while the CD signals induced by GlyPhePheGly and PhePheGlyGly were much weaker. These results further confirm that the aromatic amino acid residue at the C-terminal position is mainly a structural factor for chirality induction. The bulky chiral group of the aromatic amino acid residue at the C-terminal position can effectively affect the rotational conformation of the two TPE units of 1, owing to the spatial proximity and steric hindrance effect between the C-terminal aromatic rings and the phenyl rings of TPE units in the host–guest complexes. In addition, TrpTrp-containing tetrapeptides showed fluorescence quenching (Fig. 4f, S112 and S113†), while PhePhe-containing tetrapeptides showed fluorescence enhancement, which are consistent with their dipeptides (Fig. 4g, S114 and S115†). Therefore, achiral 1 exhibited the CAR for the TrpTrp-containing tetrapeptides with strong affinity (Ka ≈ 1013 M−2) and for the PhePhe-containing tetrapeptides with distinguished CD responses, when the TrpTrp or PhePhe residues were located at different positions in their tetrapeptide chains (Fig. 4h and i).
Recognition of polypeptides and proteins
Given the chiral adaptive responses of 1 to PhePhe-containing tetrapeptides, we expected that 1 can recognize polypeptides and proteins with the PhePhe sequence to give different CD responses. Aβ1–20, somatostatin, and human insulin were selected for this purpose. Although we anticipated binding at the PhePhe position, all aromatic amino acid residues of Aβ1–20 and somatostatin could be possible binding sites based on their upfield shifts of proton resonances (Fig. S116 and S117†). Furthermore, UV/vis and fluorescence titration, and ITC experiments further gave the formation and association constants of 1⊃polypeptide/protein complexes with 1:2 stoichiometry (Fig. S118–S125† and Table 1). Due to the precipitating behavior in the host–guest complexation, the complex of 1 and human insulin could not be observed by NMR and UV/vis titration experiments (Fig. S126 and S127†). We hypothesized that 1 could simultaneously bind with two PhePhe residues of two Aβ1–20, somatostatin, or human insulin molecules in their host–guest complexes (Fig. 5a), which should be similar to the binding model of PM-1⊃(L-PhePhe)2 in the crystalline state. The CD spectra of 1 titrated with Aβ1–20, somatostatin, and human insulin showed that these polypeptides could induce a negative Cotton effect corresponding to the chiral adaptive conformation of PP-1, which is consistent with L-dipeptides (Fig. 5b–d and S128–S130†). Furthermore, these guests induced distinguishable CD spectra of 1 with different shapes and intensities. We suspect that the chirality of amino acid residues of polypeptides and proteins both inside and outside the cavity of 1 can be structural factors that cooperatively affect the different degrees of rotational conformation (e.g., rotational angles and dihedral angles) of all benzene and/or pyridinium rings on the two faces of 1, which can give distinguishable CD spectral fingerprints for different polypeptides and proteins.
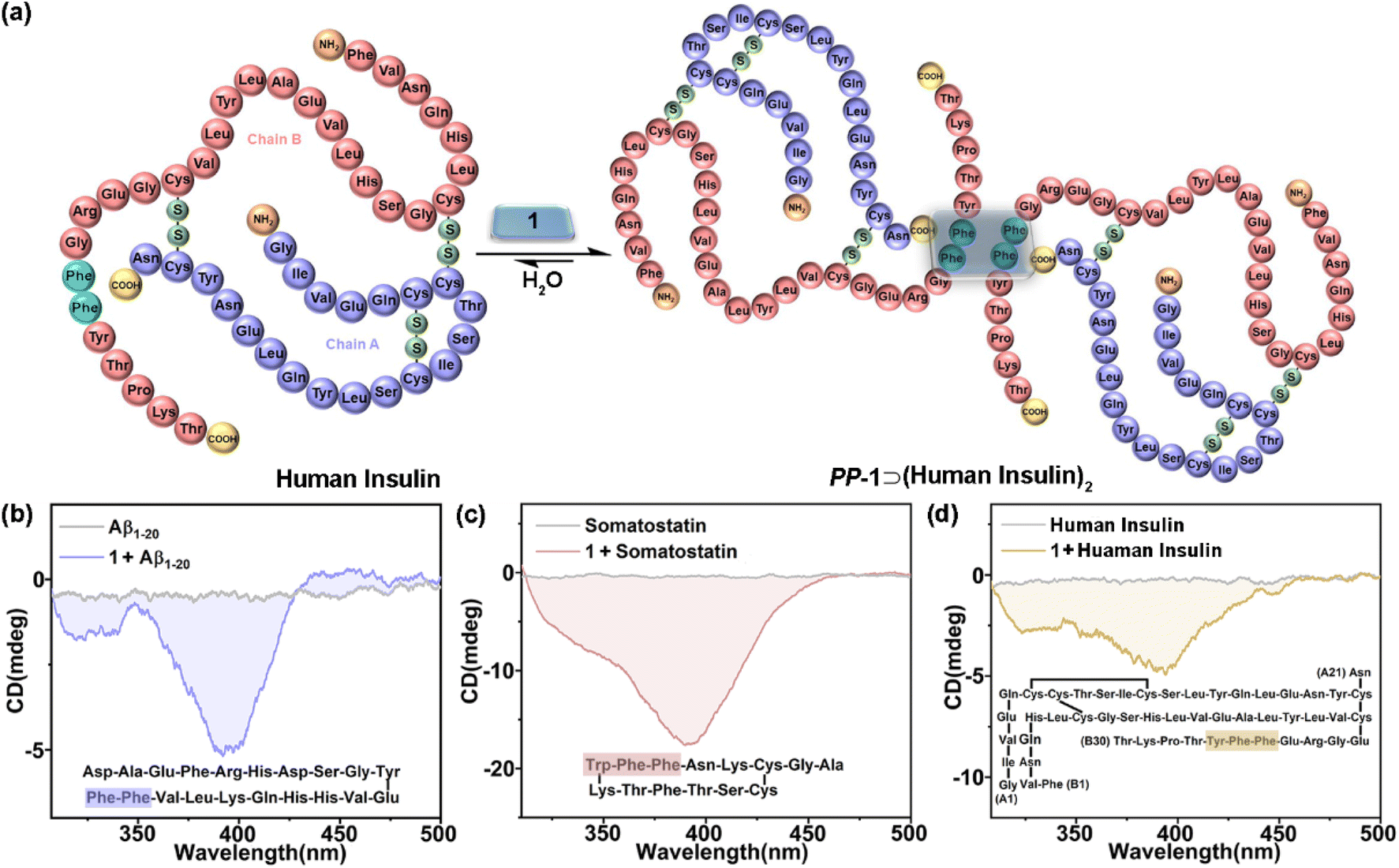 | ||
| Fig. 5 (a) Schematic representation of a possible binding model of 1⊃(human insulin)2. CD spectra of 1 (20 μM) with (b) Aβ1–20 (2.0 equiv.), (c) somatostatin (2.0 equiv.), and (d) human insulin (1.0 equiv.) in phosphate buffer (10 mM sodium phosphate, pH = 7.4). (Inset) The possible binding sites. | ||
Conclusions
In summary, we have presented the chiral adaptive recognition (CAR) with sequence selectivity of aromatic dipeptides by an achiral TPE-based octacationic cage (1) in a purely aqueous solution. 1H NMR, ITC, and X-ray analyses are used to prove that 1 and dipeptides can form 1:2 host–guest complexes. The X-ray structures confirm that the achiral 1 indeed has two chiral conformation isomers including MM-1 and PP-1, whose adaptive conformation transformation is the chiroptical-responsive mechanism in the CAR host–guest system. As a result, we systematically studied the CAR of 1 for amino acids, dipeptides, tetrapeptides, polypeptides, and proteins: (1) for amino acids, 1 shows high selectivity with adaptive CD signals for D/L-Trp. (2) 1 can selectively bind and dimerize aromatic–aromatic dipeptides with high affinity in an aqueous solution over other aliphatic–aromatic dipeptides. Meanwhile, D/L-type dipeptides induce the mirror-symmetrical CD and CPL signals of 1: D-dipeptides induce the adaptive conformation chirality of MM-1 and L-dipeptides induce the adaptive conformation chirality of PP-1, which can be applied to determine ee values and absolute configurations of the dipeptide enantiomers. (3) 1 can selectively bind the TrpTrp and PhePhe residues in tetrapeptide chains with CD responses, and further show distinguishable CD spectral fingerprints for PhePhe-containing polypeptides and proteins. Based on the CAR mechanism, therefore, this achiral TPE-based cage as a unimolecular platform can realize the molecular recognition in aqueous solution for various chiral biomolecules including amino acids, dipeptides, tetrapeptides, polypeptides, and proteins with multiple adaptive responses of fluorescence, CD, and even CPL. Our study also reveals that the key factors for supramolecular chirality transfer in the host–guest complexes mainly involve the binding affinity, spatial proximity, and steric hindrance effect between hosts with conformation-switchable units and guests with bulky chiral groups: stronger binding affinity, closer spatial position, and bigger chiral groups can improve the efficiency for chirality induction in the host–guest system. In future, this efficient, facile, and environmentally friendly CAR using an achiral host as both a molecular receptor and chiroptical sensor will provide a new approach to detect peptides and proteins, monitor their biochemical reactions, and even regulate their conformational structures and physiological functions in complex living systems.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
L. Cheng (first author): investigation, data curation, formal analysis, visualization and writing – original draft; P. Tian (co-first author): synthesis of compounds, data curation, formal analysis and writing – original draft; H. Duan: cultivation of single crystals; Q. Li: testing of single crystals; Xiaowen Song: synthesis of compounds; A. Li: DFT calculation; L. Cao (corresponding author): conceptualization, supervision, validation, writing – review & editing and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22122108 and 21971208), the Natural Science Basic Research Plan for Distinguished Young Scholars in Shaanxi Province of China (2021JC-37), and the Fok Ying Tong Education Foundation (171010).Notes and references
- J. Dong and A. P. Davis, Angew. Chem., Int. Ed., 2021, 60, 8035–8048 CrossRef CAS PubMed.
- L. Escobar and P. Ballester, Chem. Rev., 2021, 121, 2445–2514 CrossRef CAS PubMed.
- L. You, D. Zha and E. V. Anslyn, Chem. Rev., 2015, 115, 7840–7892 CrossRef CAS.
- T. J. Mooibroek, J. M. Casas-Solvas, R. L. Harniman, C. Renney, T. S. Carter, M. P. Crump and A. P. Davis, Nat. Chem., 2016, 8, 69–74 CrossRef CAS.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS.
- Y. C. Pan, X. Y. Hu and D.-S. Guo, Angew. Chem., Int. Ed., 2021, 60, 2768–2794 CrossRef CAS.
- J.-L. Lin, Z.-K. Wang, Z.-Y. Xu, L. Wei, Y.-C. Zhang, H. Wang, D.-W. Zhang, W. Zhou, Y.-B. Zhang, Y. Liu and Z.-T. Li, J. Am. Chem. Soc., 2020, 142, 3577–3582 CrossRef CAS.
- M. W. Peczuh, A. D. Hamilton, J. Sánchez-Quesada, J. de Mendoza, T. Haack and E. Giralt, J. Am. Chem. Soc., 1997, 119, 9327–9328 CrossRef CAS.
- L. G. Schmuck, J. Am. Chem. Soc., 2004, 126, 8898–8899 CrossRef.
- M. A. Hossain and H.-J. Schneider, J. Am. Chem. Soc., 1998, 120, 11208–11209 CrossRef CAS.
- K. Tsubaki, T. Kusumoto, N. Hayashi, M. Nuruzzaman and K. Fuji, Org. Lett., 2002, 4, 2313–2316 CrossRef.
- R. Ueoka, Y. Matsumoto, K. Harada, H. Akahoshi, Y. Ihara and Y. Kato, J. Am. Chem. Soc., 1992, 114, 8339–8340 CrossRef.
- M. Maletic, H. Wennemers, D. Q. McDonald, R. Breslow and W. C. Still, Angew. Chem., Int. Ed., 1996, 35, 1490–1492 CrossRef.
- M. V. Rekharsky, H. Yamamura, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, Chem. Commun., 2008, 2236–2238 RSC.
- M. V. Rekharsky, H. Yamamura, C. Inoue, M. Kawai, I. Osaka, R. Arakawa, K. Shiba, A. Sato, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2006, 128, 14871–14880 CrossRef.
- L. M. Heitmann, A. B. Taylor, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2006, 128, 12574–12581 CrossRef.
- J. M. Chinai, A. B. Taylor, L. M. Heitmann, N. D. Hargreaves, C. A. Morris, P. J. Hart and A. R. Urbach, J. Am. Chem. Soc., 2011, 133, 8810–8813 CrossRef CAS.
- Z. Hirani, H. F. Taylor, E. F. Babcock, A. T. Bockus, C. D. Varnado, C. W. Bielawski and A. R. Urbach, J. Am. Chem. Soc., 2018, 140, 12263–12269 CrossRef CAS.
- S. Tashiro, M. Tominaga, M. Kawano, B. Therrien, T. Ozeki and M. Fujita, J. Am. Chem. Soc., 2005, 127, 4546–4547 CrossRef CAS PubMed.
- S. S. Yoon and W. C. Still, J. Am. Chem. Soc., 1993, 115, 823–824 CrossRef CAS.
- C.-T. Chen, H. Wagner and W. C. Still, Science, 1998, 279, 851–853 CrossRef CAS.
- L. C. Smith, D. G. Leach, B. E. Blaylock, O. A. Ali and A. R. Urbach, J. Am. Chem. Soc., 2015, 137, 3663–3669 CrossRef CAS.
- D. E. Clarke, G. Wu, C. Wu and O. A. Scherman, J. Am. Chem. Soc., 2021, 143, 6323–6327 CrossRef CAS PubMed.
- Z. Xu, S. Jia, W. Wang, Z. Yuan, B. Jan Ravoo and D.-S. Guo, Nat. Chem., 2019, 11, 86–93 CrossRef CAS.
- G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528–7535 CrossRef CAS.
- G. Ghale, V. Ramalingam, A. R. Urbach and W. M. Nau, J. Am. Chem. Soc., 2011, 133, 7528–7535 CrossRef CAS PubMed.
- H. Zhu, Q. Li, Z. Gao, H. Wang, B. Shi, Y. Wu, L. Shangguan, X. Hong, F. Wang and F. Huang, Angew. Chem., Int. Ed., 2020, 59, 10868–10872 CrossRef CAS PubMed.
- H. Nian, L. Cheng, L. Wang, H. Zhang, P. Wang, Y. Li and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 15354–15358 CrossRef CAS PubMed.
- F. Biedermann and W. M. Nau, Angew. Chem., Int. Ed., 2014, 53, 5694–5699 CrossRef CAS PubMed.
- A. Prabodh, D. Bauer, S. Kubik, P. Rebmann, F. G. Klärner, T. Schrader, L. D. Bizzini, M. Mayor and F. Biedermann, Chem. Commun., 2020, 56, 4652–4655 RSC.
- L.-L. Wang, Z. Chen, W.-E. Liu, H. Ke, S.-H. Wang and W. Jiang, J. Am. Chem. Soc., 2017, 139, 8436–8439 CrossRef CAS PubMed.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- H.-T. Feng, Y.-X. Yuan, J.-B. Xiong, Y.-S. Zheng and B. Z. Tang, Chem. Soc. Rev., 2018, 47, 7452–7476 RSC.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef PubMed.
- L. Hu, K. Li, W. Shang, X. Zhu and M. Liu, Angew. Chem., Int. Ed., 2020, 59, 4953–4958 CrossRef PubMed.
- W. Shang, X. Zhu, T. Liang, C. Du, L. Hu, T. Li and M. Liu, Angew. Chem., Int. Ed., 2020, 59, 12811–12816 CrossRef.
- H. Duan, Y. Li, Q. Li, P. Wang, X. Liu, L. Cheng, Y. Yu and L. Cao, Angew. Chem., Int. Ed., 2020, 59, 10101–10110 CrossRef.
- L. Cheng, K. Liu, Y. Duan, H. Duan, Y. Li, M. Gao and L. Cao, CCS Chem., 2020, 2, 2749–2751 Search PubMed.
- L. Cheng, P. Tian, Q. Li, A. Li and L. Cao, CCS Chem., 2021, 3, 3608–3614 Search PubMed.
- H. Duan, F. Cao, M. Zhang, M. Gao and L. Cao, Chin. Chem. Lett., 2022, 33, 2459–2463 CrossRef CAS.
- Y. Li, Q. Li, X. Miao, C. Qin, D. Chu and L. Cao, Angew. Chem., Int. Ed., 2021, 60, 6744–6751 CrossRef CAS PubMed.
- H. Qu, Y. Wang, Z. Li, X. Wang, H. Fang, Z. Tian and X. Cao, J. Am. Chem. Soc., 2017, 139, 18142–18145 CrossRef CAS PubMed.
- J.-B. Xiong, H.-T. Feng, J.-P. Sun, W.-Z. Xie, D. Yang, M. Liu and Y.-S. Zheng, J. Am. Chem. Soc., 2016, 138, 11469–11472 CrossRef CAS PubMed.
- G. A. Hembury, V. V. Borovkov and Y. Inoue, Chem. Rev., 2008, 108, 1–73 CrossRef CAS.
- Y.-S. Zheng and C. Zhang, Org. Lett., 2004, 6, 1189–1192 CrossRef CAS.
- X. Wang, F. Jia, L. P. Yang, H. Zhou and W. Jiang, Chem. Soc. Rev., 2020, 49, 4176–4188 RSC.
- Q.-P. Hu, H. Zhou, T.-Y. Huang, Y.-F. Ao, D.-X. Wang and Q.-Q. Wang, J. Am. Chem. Soc., 2022, 144, 6180–6184 CrossRef CAS.
- J. R. Lakowicz, Mechanisms and Dynamics of Fluorescence Quenching, Principles of Fluorescence Spectroscopy, Springer, New York, 2006, ch. 9, pp. 331–348 Search PubMed.
- J. Chen, B. Xu, X. Ouyang, B. Z. Tang and Y. Cao, J. Phys. Chem. A, 2004, 108, 7522–7526 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details including NMR, ITC, UV/vis, fluorescence, CD, CPL, and X-ray data. CCDC 2100784, 2100668 and 2155590. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2sc05854e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |