 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting the activity of Mizoroki–Heck cross-coupling reactions with a supramolecular palladium catalyst favouring remote Zn⋯pyridine interactions†
Naba
Abuhafez
and
Rafael
Gramage-Doria
 *
*
Univ Rennes, CNRS, ISCR-UMR6226, F-35000 Rennes, France. E-mail: rafael.gramage-doria@univ-rennes1.fr
First published on 9th January 2023
Abstract
Transition metal catalysis benefitting from supramolecular interactions in the secondary coordination sphere in order to pre-organize substrates around the active site and reach a specific selectivity typically occurs under long reaction times and mild reaction temperatures with the aim to maximize such subtle effects. Herein, we demonstrate that the kinetically labile Zn⋯N interaction between a pyridine substrate and a zinc–porphyrin site serving for substrate binding is a unique type of weak interaction that enables identification of supramolecular effects in transition metal catalysis after one hour at a high reaction temperature of 130 °C. Under carefully selected reaction conditions, supramolecularly-regulated palladium-catalyzed Mizoroki–Heck reactions between 3-bromopyridine and terminal olefins (acrylates or styrenes) proceeded in a more efficient manner compared to the non-supramolecular version. The supramolecular catalysis developed here also displayed interesting substrate-selectivity patterns.
Introduction
The last decades have witnessed how merging transition metal catalysis with supramolecular chemistry has led to a remarkable number of supramolecular catalysts displaying unique reactivities.1 In fact, supramolecular catalysts aim at controlling catalyst activity and selectivity with tools that are beyond the traditional catalyst modification at the first coordination sphere (i.e. steric and electronic parameters of the ligands attached to the metal centre).2 Such approach resembles to some extent the action mode found in Nature’s catalysts, enzymes, in which dynamic binding and kinetically labile, weak interactions are at play to pre-organize substrates around an active site as well as to stabilize or destabilize the desired reaction intermediates.3 From the many weak interactions that have been employed in metal catalysts featuring substrate pre-organization, hydrogen bonding and ion-pairing have been largely explored.4 Their binding towards complementary substrates is highly predictable in view to place a specific reactive site of the substrate in close proximity to the catalytically active metal site.5 This is highly beneficial to tackle selectivity issues associated with challenging chemical reactions.6Alternatively, since many substrates are not prone to undergo hydrogen bonding or ion-pairing, it is appealing to disclose new types of weak interactions that can be explored for supramolecular substrate recognition in the context of transition metal catalysis.7 In our laboratory, we have been interested in the development of supramolecular metal catalysts comprising a zinc–porphyrin site for molecular recognition with pyridine derivatives via kinetically labile, weak Zn⋯N interactions.8 In this way, selective iridium-catalyzed C–H borylation at the meta position has been disclosed thanks to the ideal pre-organization between the active site and the substrate binding site.9 We also designed in the past a supramolecular ligand built up around a zinc–porphyrin scaffold that contains a nitrile group in the ortho position of each meso-substituted phenyl group (L, Fig. 1).10 In the presence of a palladium precursor such ligand featured interesting substrate-selectivity patterns when applied in Suzuki–Miyaura reactions between bromopyridines and phenylboronic acid at 80 °C. In this case, only the bromopyridine substrate and the ligand are soluble in toluene, an apolar and non-coordinating solvent that maximizes the binding of bromopyridine derivatives to the zinc–porphyrin molecular recognition site whereas the palladium active sites are bound to the nitrile groups in the ligand. In the present study, we decided to evaluate the compatibility of the weak Zn⋯N interactions at higher reaction temperatures and also in the presence of coupling partners that are soluble in the reaction media and can eventually compete with the bromopyridine substrates for binding the molecular recognition site. As such, we turned our attention to palladium-catalyzed Mizoroki–Heck cross-coupling reactions that typically require a significant higher reaction temperature than the Suzuki–Miyaura ones and,11 in addition, the acrylate coupling partners are usually soluble in apolar non-coordinating solvents while presenting a carbonyl ester group that is known to bind to zinc–porphyrins, at least in the solid state (Fig. 1).12 We found out that such catalytic reactions are fast (one hour time) and that acrylates did not perturb the weak Zn⋯N interaction between the substrate and the molecular recognition site even at higher reaction temperatures of 130 °C.
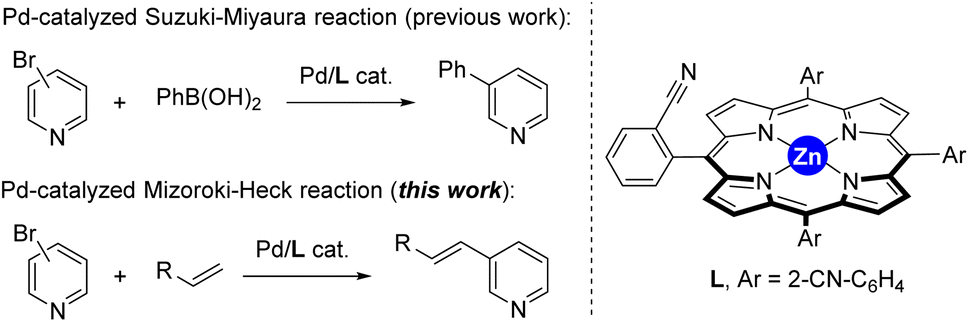 | ||
| Fig. 1 Previously developed supramolecular ligand L applied in palladium-catalyzed Suzuki–Miyaura reactions and its current application in palladium-catalyzed Mizoroki–Heck cross-coupling reactions. | ||
Results and discussion
As a model reaction we studied the reaction between 3-bromopyridine (1) with butyl acrylate (2a) in the presence of the supramolecular ligand L and catalytic amounts of Pd(OAc)2 precursor under basic conditions (Table 1). After screening some reaction conditions with different bases and catalyst loadings at different reaction temperatures (Table S1 in the ESI†), we noted that at 130 °C the expected product 3a formed in 78% yield in the presence of 10 mol% pre-catalyst precursor, 20 mol% of ligand L and 3 equivalents of both potassium carbonate and coupling partner 2a (Table 1, entry 1). In toluene solvent, the reaction was finished in one hour since longer reaction times influenced little the conversion of 1 and the formation of 3a, as was shown in a preliminary kinetic study (Fig. S12 and S13 in the ESI†). As it is well-known for Mizoroki–Heck reactions, the temperature had a dramatic effect on the activity since decreasing the temperature led to poor yields of 3a (Table S1 in the ESI†). Under identical reaction conditions, we performed a number of control experiments to assess the action mode of the supramolecular ligand L. The influence of covalently linking the nitrile groups to the zinc–porphyrin scaffold was studied by performing a catalytic reaction in the presence of unfunctionalized zinc–tetraphenylporphyrin (ZnTPP) at 20 mol% loading and benzonitrile at 80 mol% loading to provide a fair comparison with the use of 20 mol% of L in Table 1, entry 1. In this case, the product 3a formed in 28% yield (Table 1, entry 2). A similar poor reactivity was found by using only ZnTPP in the absence of benzonitrile (Table 1, entry 3) or by evaluating the zinc-free version of L, namely H2L (Table 1, entry 4). Analogously, the reaction performed in the absence of any ligand led to a poor yield of 23% under these reaction conditions (Table 1, entry 5). To further validate that the catalysis occurred with the pyridine derivative bound to the zinc–porphyrin molecular recognition site of L, we performed the standard reaction in the presence of 20 mol% of zinc–salphen (ZS), which is expected to behave as an inhibitor by coordinating with 1 since the association constant of pyridines with zinc–salphen derivatives is typically two orders of magnitude higher than with zinc–porphyrins (Table 1, entry 6).8e,13 As anticipated, the reactivity drastically dropped to 5% formation of product 3a in line with the fact that 1 is trapped by the zinc–salphen building block and cannot approach to the supramolecular palladium catalyst. Alternatively, a similar behaviour was encountered by introducing into the catalysis media a strongly coordinating 4-dimethylaminopyridine (DMAP) compound that is expected to compete with the bromopyridine 1 for binding the molecular recognition site of the supramolecular palladium catalyst (Table 1, entry 7).14a Indeed, in the presence of DMAP, the yield of product 3a also dropped to 42% confirming the inhibitory role of DMAP as well as indirectly suggesting that the catalysis likely occurs with the 3-bromopyridine 1 bound to the molecular recognition site. On the other hand, no reactivity was observed for the reaction between 3-chloropyridine and butyl acrylate (2a), whereas the 3-iodopyridine substrate behaved similarly to the 3-bromopyridine affording 60% yield of 3a using the supramolecular ligand L.|
|
||
|---|---|---|
| Entry | Deviation from above conditions | Yield of 3b (%) |
| a Reaction conditions: 1 (7.9 mg, 4.8 μL, 0.05 mmol), 2a (19.2 mg, 21.6 μL, 0.15 mmol), K2CO3 (20.7 mg, 0.15 mmol), L (7.8 mg, 0.01 mol), Pd(OAc)2 (1.12 mg, 0.005 mmol), toluene (1 mL), 130 °C, 1 h, argon. b Yields determined by GC-MS analysis. | ||
| 1 | None | 78 |
| 2 | ZnTPP (20 mol%) and benzonitrile (80 mol%) instead of L | 28 |
| 3 | ZnTPP (20 mol%) instead of L | 23 |
| 4 | H2L (20 mol%) instead of L | 24 |
| 5 | Without L | 23 |
| 6 | With addition of ZS (20 mol%) | 5 |
| 7 | With addition of DMAP (1 equiv.) | 42 |
|
||
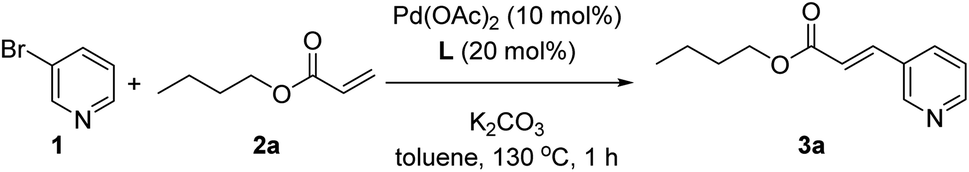
To gain insights into the binding ability of the supramolecular ligand L under conditions similar to those used in the catalysis presented above, we performed NMR experiments combining equimolar amounts of L and the starting material 3-bromopyridine (1) in the presence of excess butyl acrylate (2a) (Scheme 1). The 1H NMR spectrum of the resulting mixture showed negligible shifts in the proton signals belonging to L and the acrylate 2a (Fig. S1–S3, S6, S9, S11 in the ESI†). However, the proton signals belonging to the pyridine derivative 1 underwent an important upfield shift, which is in agreement with an interaction between 1 and Lvia Zn⋯N interaction.14 This was further evaluated by performing NMR studies at higher temperatures up to 120 °C (Fig. 2). In addition, DOSY experiments show that 3-bromopyridine (1) diffuses together with the supramolecular ligand L in the presence of acrylate 2a, which displays a different diffusion coefficient (Fig. S4–S5, S7–S8, S10 in the ESI†).15 Remarkably, the self-assembly ligand-to-substrate [L ⊂ 1] appears to be rather stable at high temperatures even in the presence of competing ester groups from 2a. It is clear that, in the present case, the ester groups are not sufficiently coordinating towards zinc to enable the displacement of the pyridine derivative 1 from the molecular recognition site of L even at high temperatures.
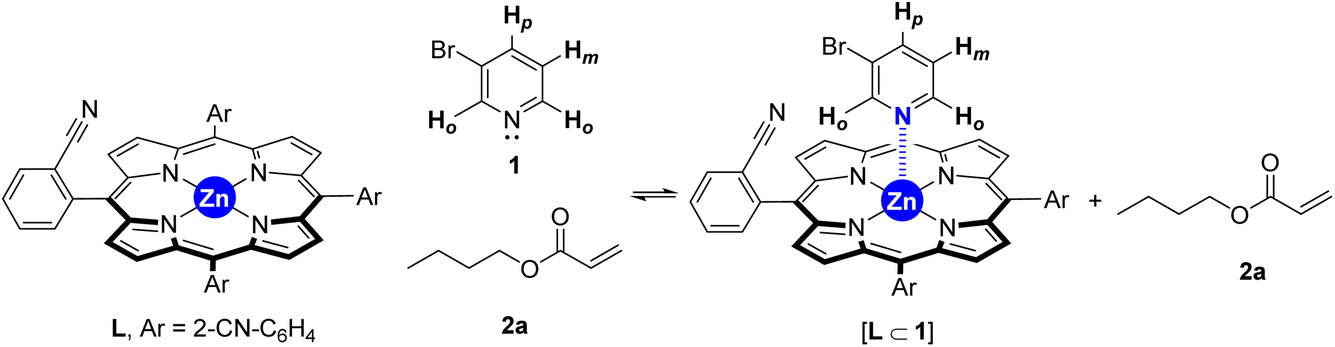 | ||
| Scheme 1 Equilibria involving the supramolecular ligand L in the presence of bromopyridine 1 and acrylate 2a. | ||
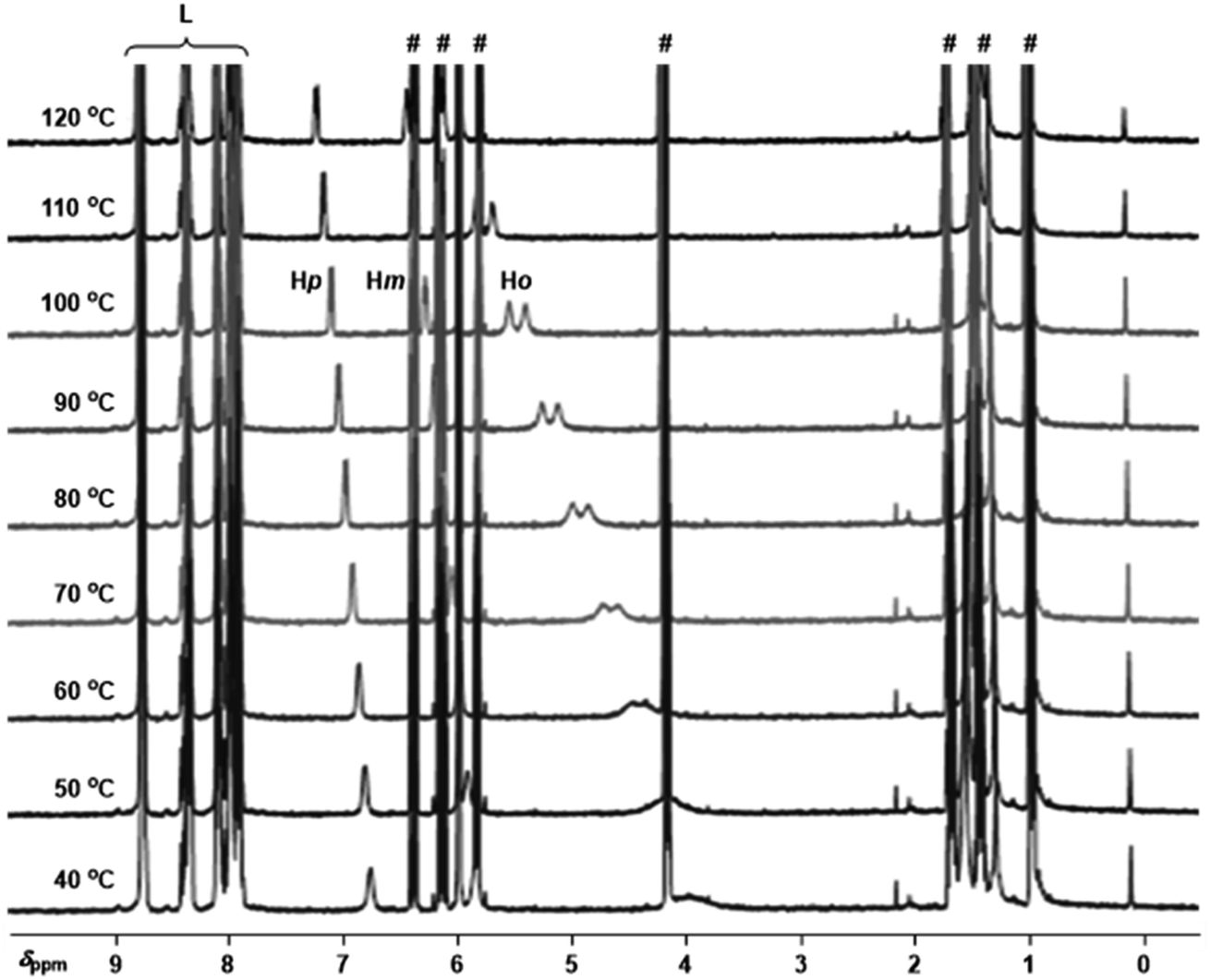 | ||
| Fig. 2 Variable temperature 1H NMR (1,1,2,2-tetrachloroethane-d2, 400 MHz) spectra for the combination of L with 1 in the presence of excess 2a (for proton numbering see Scheme 1, # marks denote the proton signals belonging to the acrylate derivative 2a and L are the proton signals belonging to the supramolecular ligand L, and for concentration details see the ESI†). | ||
In order to study the eventual possibility to cleave the Zn⋯N interaction between the pyridine substrate 1 and the supramolecular ligand L, we decided to introduce a stronger coordinating species than acrylates in the reaction medium during the palladium-catalyzed Mizoroki–Heck reaction. For that, we used N,N-dimethylformamide (DMF) as a co-solvent together with toluene with a half catalyst loading compared to that used in Table 1. As shown in Table 2, the supramolecular effect of L decreased with increasing quantities of DMF co-solvent reaching a negligible supramolecular effect when the amount of DMF was 50% in volume and beyond (Table 2, entries 4–6). With this large excess of DMF, the well-known palladium nanoparticles regime is likely operating.16 Interestingly, it is relevant to note that the supramolecular effect of L was evidenced when the amount of DMF was lower than 50% in volume (Table 2, entries 1–3). For instance, performing the catalysis with DMF at 20% volume ratio led to an almost six-fold increase in the reactivity when compared to the non-supramolecular version (Table 2, entry 1). This finding is particularly relevant since it demonstrates that it is possible to benefit from remote Zn⋯N weak interactions in metal catalysis even under such unfavourable reaction conditions regarding the nature of the solvent and the high reaction temperature, thereby enabling enlargement of the polarity window of solvents that can be used in supramolecular catalysis.
|
|
||||
|---|---|---|---|---|
| Entry |
x![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :y :y |
Yield of 3 (%) with Lb | Yield of 3 (%) without Lb | Δ yield of 3c |
| a Reaction conditions: 1 (7.9 mg, 4.8 μL, 0.05 mmol), 2a (19.2 mg, 21.6 μL, 0.15 mmol), K2CO3 (20.7 mg, 0.15 mmol), L (7.8 mg, 0.01 mol), Pd(OAc)2 (1.12 mg, 0.005 mmol), solvent (1 mL), 130 °C, 1 h, argon. b Yields determined by GC-MS analysis. c Defined as yield of 3 with L divided by yield of 3 without L and it corresponds to the manifold increase of the overall yield. | ||||
| 1 | 100:0 |
58 | 10 | 5.8 |
| 2 | 80:20 |
30 | 10 | 3 |
| 3 | 70:30 |
34 | 16 | 2.1 |
| 4 | 50:50 |
90 | 70 | 1.3 |
| 5 | 40:60 |
>99 | >99 | 1 |
| 6 | 0:100 |
>99 | >99 | 1 |
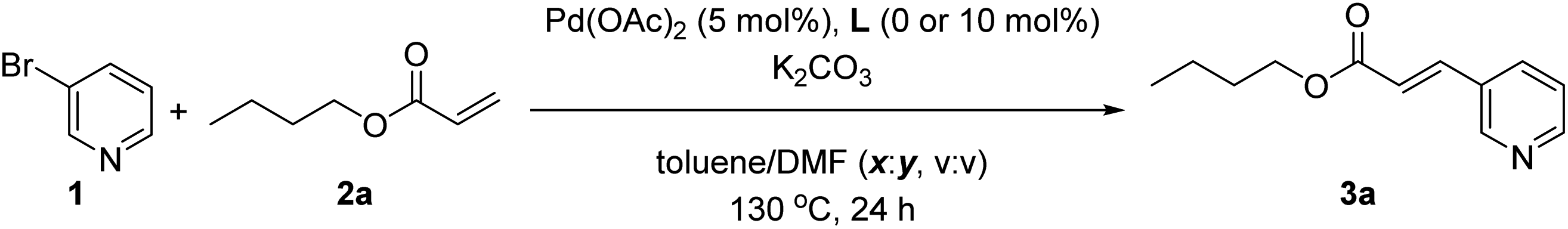
Next, the influence of the nature of the olefin derivative was studied by using different types of acrylate derivatives as well as styrene derivatives for the reaction with the 3-bromopyridine (1) under palladium catalysis in the presence or absence of the supramolecular ligand L at 130 °C for one hour in pure toluene solvent (Scheme 2). In all cases, the reactions performed in the presence of the supramolecular ligand L led to yields of olefinated products 3 that were higher than those observed for the reactions carried out in its absence. For instance, the products 3b and 3c resulting from employing methyl acrylate (2b) and tert-butylacrylate (2c), respectively, were formed in 50–59% yield in the presence of L, whereas only trace amounts (<5%) formed in the absence of L. Similarly, in the case of benzyl acrylate (2d), the supramolecular palladium catalyst afforded the corresponding product 3d in 87% yield, whilst only traces were detected in the non-supramolecular version after one hour. Olefins with a more electron donating character such as styrenes underwent a similar trend as observed for acrylates. For example, non-functionalized styrene (2e) reacted with 3-bromopyridine (1) under supramolecular palladium catalysis leading to product 3e in a virtually quantitative yield (95%). On the other hand, the catalysis performed in the absence of L led to a drop in the yield of product 3e to 15%. Styrene derivatives with electronically different substitution patterns such as methoxy and fluoride are also compatible with this supramolecular palladium catalysis affording the corresponding products 3f and 3g in 89% and 43% yields, respectively. The reactions carried out without the supramolecular ligand L led to 50% yield of 3f and 13% yield of 3g. Such supramolecular effect was also observed when employing N,N-dimethylacrylamide (2h), which afforded the olefinated product 3h in 26% yield, whereas an even lower 7% yield of 3h was obtained in the absence of L. The supramolecular effect was also observed with methyl methacrylate as the olefin partner, which gave rise to the trisubstituted olefin 3i in 50% yield whilst only 20% of 3i was formed without the supramolecular ligand L. No reactivity was observed for the challenging, unfunctionalized acrylamide.
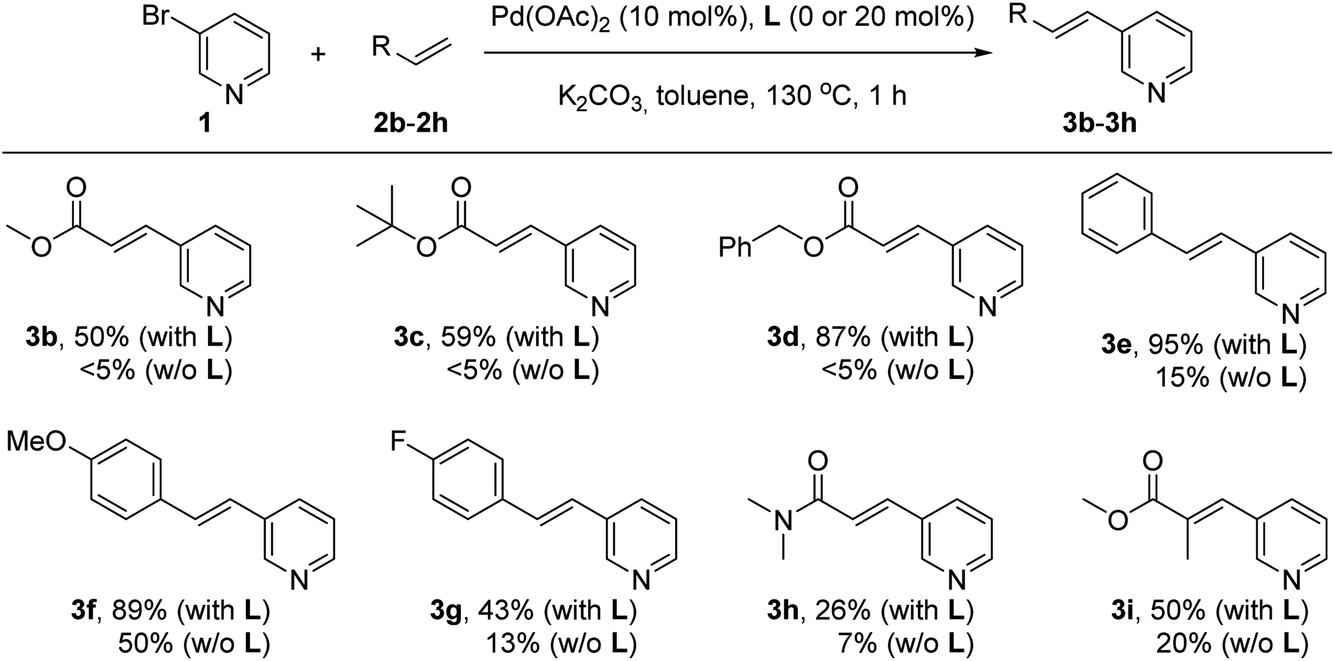 | ||
| Scheme 2 Influence of the supramolecular ligand L in the palladium-catalyzed Mizoroki–Heck cross-coupling between 3-bromopyridine and olefin derivatives. | ||
Finally we wondered whether our supramolecular palladium-catalyzed Mizoroki–Heck reactions displayed substrate selectivity to some extent. For this purpose, 2- and 4-bromopyridine were employed as substrates under the optimal reaction conditions in the presence of butyl acrylate as coupling partner (Scheme 3). Whereas the reaction with 3-bromopyridine afforded 78% yield of the corresponding product (Scheme 3, top), the yields of the olefinated products derived from the other regioisomers did not surpass 11% yield in the best case (Scheme 3, middle and bottom). As such, these observations indicate that the supramolecular palladium catalyst is more adapted for reaction with the meta isomer over the ortho or para isomers. Such observation is reminiscent of our previous findings on supramolecular palladium-catalyzed Suzuki–Miyaura reactions,10 but in the present case they are found at relatively higher reaction temperatures and short reaction times.
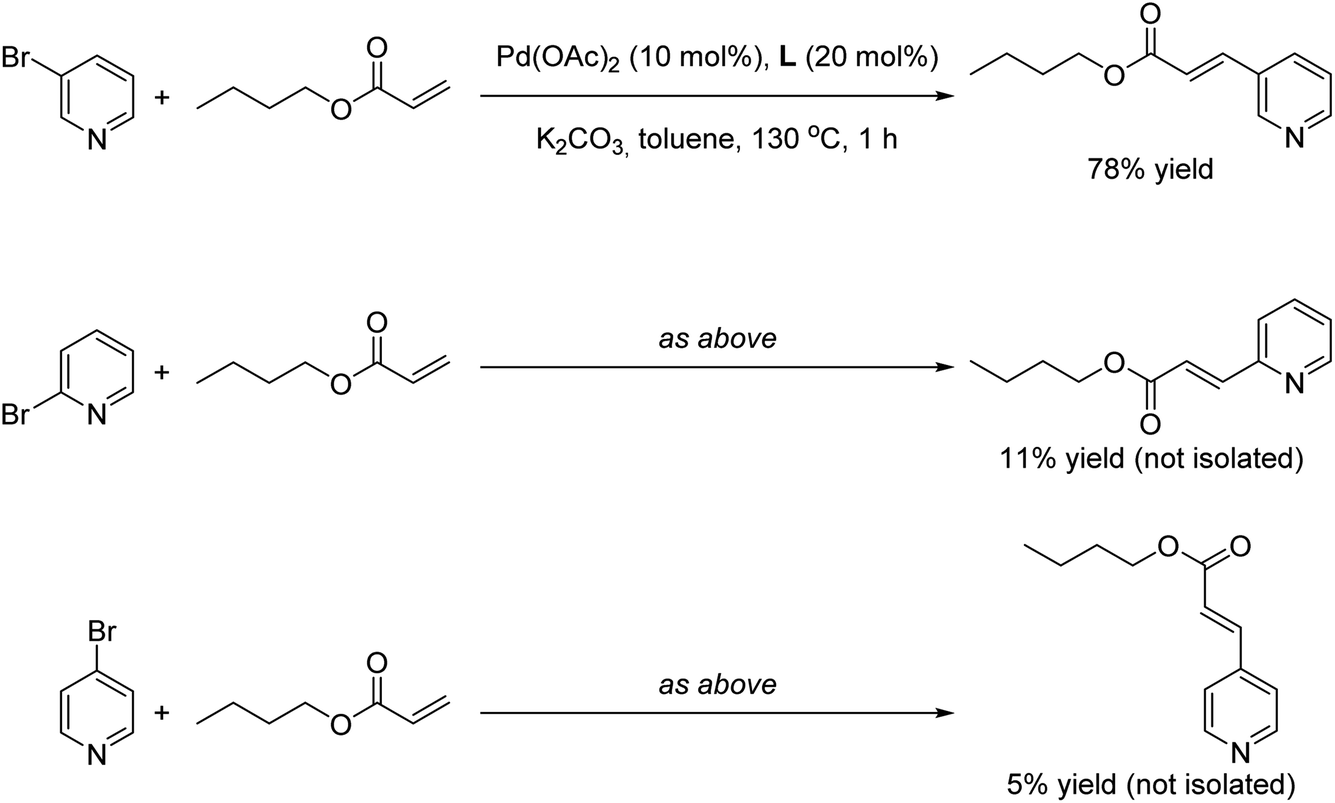 | ||
| Scheme 3 Influence of the regioisomer bromopyridine substrate in the final outcome of the supramolecular palladium-catalyzed Mizoroki–Heck cross-coupling reaction. | ||
Taking into account the well-known reaction mechanism for palladium-catalyzed Mizoroki–Heck reactions and all the above discussed observations, we propose the catalytic cycle shown in Scheme 4 for our supramolecular palladium catalyst.11,17 After initial in situ formation of palladium(0),18 the peripheral nitrile groups in the supramolecular ligand L coordinate to palladium whilst the 3-bromopyridine substrate binds to the zinc–porphyrin molecular recognition site of L (formation of species A). Oxidative addition at palladium leads to the intermediate B followed by olefin coordination and migratory insertion towards species C. A final β-hydride elimination leads to the olefinated product 3 whereas the base mediates reductive elimination at palladium, thus regenerating the active palladium(0) species. At this stage, we cannot determine whether catalyst regeneration occurs with the substrate or with the product binding to the molecular recognition site.19 This mechanistic proposal explains also the substrate selectivity encountered between the three different regioisomeric bromopyridine derivatives discussed in Scheme 3.
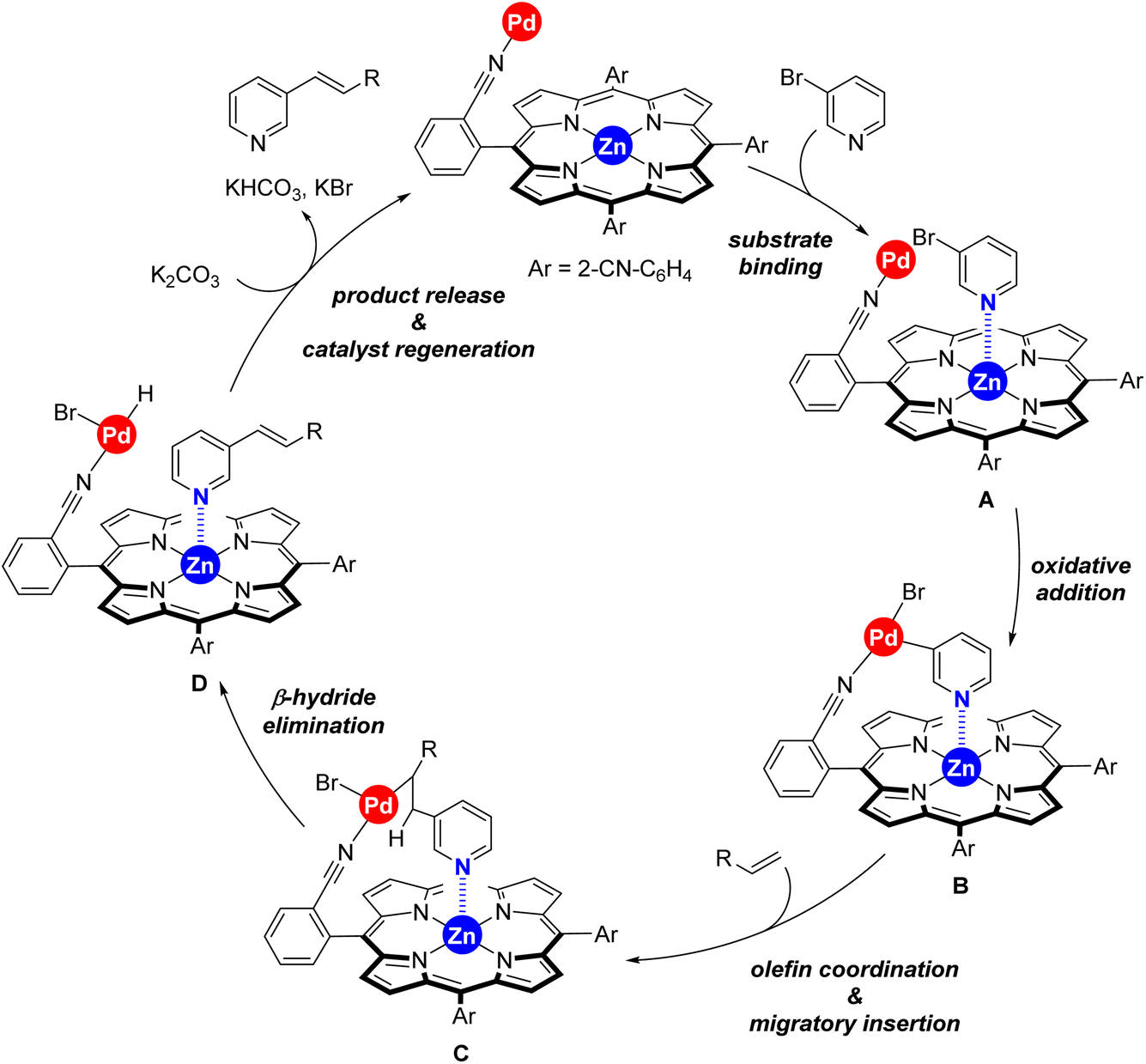 | ||
| Scheme 4 Postulated reaction mechanism for the supramolecular palladium-catalyzed Mizoroki–Heck cross-coupling reaction between 3-bromopyridine and terminal olefins exploiting remote, kinetically labile Zn⋯N weak interactions between the catalyst and the substrate. | ||
Conclusions
In summary, we have disclosed a supramolecular palladium-catalyzed Mizoroki–Heck cross-coupling reaction in which the reactivity is enhanced thanks to remote Zn⋯N interactions between the molecular recognition site in the catalyst and the 3-bromopyridine substrate. The increased reactivity of the supramolecular catalyst is evidenced in relatively short reaction times (up to one hour) and high reaction temperatures (130 °C), which is rare for supramolecular catalysts that feature substrate-recognition properties. In addition, we managed to identify that the presence of small amounts of highly polar DMF solvent are still compatible for observing such supramolecular effect, which is important for the development of future supramolecular catalysts that would require different polarity media. By means of in-depth NMR studies, this work also discloses that the kinetically labile Zn⋯N interaction between the catalyst and the substrate is compatible with potentially coordinating groups encountered in other reagents such as the ester groups from acrylate coupling partners. This supramolecular catalysis was also tolerant to typically less reactive olefins such as styrene derivatives and a certain degree of substrate selectivity was identified by evaluating all the three regioisomeric bromopyridines. We anticipate that these findings might be of interest to develop new supramolecular catalysts for challenging transformations.Experimental
The supramolecular ligand L was prepared according to a previous report.10General procedure for the supramolecular catalysis
An overnight-dried Schlenk tube was filled, under an argon atmosphere, with 3-bromopyridine 1 (7.9 mg, 4.8 μL, 0.05 mmol, 1 equiv.), the corresponding terminal olefin coupling partner (0.15 mmol, 3 equiv.), potassium carbonate (20.7 mg, 0.15 mmol, 3 equiv.), the supramolecular ligand L (7.8 mg, 0.01 mmol, 0.2 equiv.), Pd(OAc)2 (1.12 mg, 0.005 mmol, 0.1 equiv.) and toluene (1 mL). After 5 min stirring at room temperature the mixture was placed in a preheated oil bath at 130 °C and stirred for one hour. The reaction mixture was cooled down to room temperature and further analyzed by GC-MS.General procedure for the synthesis and characterization of the products resulting from the catalysis
An overnight-dried Schlenk tube was filled, under an argon atmosphere, with 3-bromopyridine 1 (7.9 mg, 4.8 μL, 0.05 mmol, 1 equiv.), the corresponding terminal olefin coupling partner (0.15 mmol, 3 equiv.), potassium carbonate (20.7 mg, 0.15 mmol, 3 equiv.), Pd(OAc)2 (1.12 mg, 0.005 mmol, 0.1 equiv.) and N,N-dimethylformamide (1 mL). After 5 min stirring at room temperature the mixture was placed in a preheated oil bath at 130 °C and stirred for 24 hours. The reaction mixture was cooled down to room temperature and the solvents evaporated under vacuum followed by purification by column chromatography (SiO2, ethyl acetate/pentane) to afford the corresponding olefinated products 3.Author contributions
N. A. designed and performed all the experiments as well as all data analysis. N. A. and R. G.-D. discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Centre National de la Recherche Scientifique (CNRS), Université de Rennes 1, the Agence Nationale de la Recherche ANR-JCJC (ANR-19-CE07-0039), Région Bretagne (ARED 2020 No. 1715) and Collège de France (PAUSE, PhD Grant to N. A.).Notes and references
- (a) Supramolecular catalysis, ed. P. W. N. M. van Leeuwen, Wiley-VCH, 2008 Search PubMed; (b) Supramolecular catalysis: New Directions and Developments, ed. P. W. N. M. van Leeuwen and M. Raynal, Wiley-VCH, 2022 Search PubMed.
- (a) T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC; (b) D. M. Vriezema, M. Comellas Aragones, J. A. A. W. Elemans, J. L. M. Cornelissen, A. E. Rowand and R. J. M. Nolte, Chem. Rev., 2005, 105, 1445–1489 CrossRef CAS PubMed; (c) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015–2041 CrossRef CAS PubMed; (d) M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed; (e) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349–358 CrossRef CAS PubMed; (f) M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed; (g) I. Nath, J. Chakraborty and F. Verpoort, Chem. Soc. Rev., 2016, 45, 4127–4170 RSC; (h) T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2013, 113, 734–777 CrossRef CAS PubMed; (i) M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969–984 CrossRef CAS.
- (a) D. Ringe and G. A. Petsko, Science, 2008, 320, 1428–1429 CrossRef CAS PubMed; (b) From enzyme models to model enzymes, ed. A. J. Kirby and F. Hollfelder, RSC, 2009 Search PubMed; (c) G. G. Hammes, S. J. Benkovic and S. Hammes-Schiffer, Biochemistry, 2011, 50, 10422–10430 CrossRef CAS PubMed; (d) X. Zhang and K. N. Houk, Acc. Chem. Res., 2005, 38, 379–385 CrossRef CAS PubMed; (e) K. N. Houk, A. G. Leach, S. P. Kim and X. Zhang, Angew. Chem., Int. Ed., 2003, 42, 4872–4897 CrossRef CAS PubMed.
- (a) B. Breit, Angew. Chem., Int. Ed., 2005, 44, 6816–6825 CrossRef CAS PubMed; (b) S. Carboni, C. Gennari, L. Pignataro and U. Piarulli, Dalton Trans., 2011, 40, 4355–4373 RSC; (c) M. R. Mote and S. H. Chikkali, Chem.–Asian J., 2018, 13, 3623–3646 CrossRef PubMed.
- (a) L. V. A. Hale and N. K. Szymczak, ACS Catal., 2018, 8, 6446–6461 CrossRef CAS; (b) J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed; (c) R. Noyori, C. A. Sandoval, K. Muniz and T. Ohkuma, Philos. Trans. R. Soc., A, 2005, 363, 901–912 CrossRef CAS PubMed; (d) A. S. Borovik, Acc. Chem. Res., 2005, 38, 54–61 CrossRef CAS PubMed.
- J. N. H. Reek, B. de Bruin, S. Pullen, T. J. Mooibroek, A. M. Kluwer and X. Caumes, Chem. Rev., 2022, 122, 12308–12369 CrossRef CAS PubMed.
- (a) P. Dydio and J. N. H. Reek, Chem. Sci., 2014, 5, 2135–2145 RSC; (b) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (c) H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC; (d) K. T. Mahmudov, A. V. Gurbanov, F. I. Guseinov and M. F. C. Guedes da Silva, Coord. Chem. Rev., 2019, 387, 32–46 CrossRef CAS; (e) G. Olivo, G. Capocasa, D. Del Giudice, O. Lanzalunga and S. Di Stefano, Chem. Soc. Rev., 2021, 50, 7681–7724 RSC; (f) J. Trouvé and R. Gramage-Doria, Chem. Soc. Rev., 2021, 50, 3565–3584 RSC; (g) Y. Jiao, X.-Y. Chen and J. F. Stoddart, Chem, 2022, 8, 414–438 CrossRef CAS; (h) B. Zhang and J. N. H. Reek, Chem.–Asian J., 2021, 16, 3851–3863 CrossRef CAS PubMed; (i) J. E. Gillespie, A. Fanourakis and R. J. Phipps, J. Am. Chem. Soc., 2022, 144, 18195–18211 CrossRef CAS PubMed; (j) R. S. J. Proctor, A. C. Colgan and R. J. Phipps, Nat. Chem., 2020, 12, 990–1004 CrossRef PubMed; (k) A. Fanourakis, P. J. Docherty, P. Chuentragool and R. J. Phipps, ACS Catal., 2020, 10, 10672–10714 CrossRef CAS PubMed; (l) A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Nature, 2017, 543, 637–646 CrossRef CAS PubMed; (m) K. Ohmatsu and T. Ooi, Top. Curr. Chem., 2019, 377, 31 CrossRef PubMed.
- (a) N. Abuhafez, A. Perennes and R. Gramage-Doria, Synthesis, 2022, 54, 3473–3481 CrossRef CAS; (b) J.-F. Longevial, S. Clément, J. A. Wytko, R. Ruppert, J. Weiss and S. Richeter, Chem.–Eur. J., 2018, 24, 15442–15460 CrossRef CAS PubMed; (c) R. P. Bonar-Law, L. G. Mackay, C. J. Walter, V. Marvaud and J. K. M. Sanders, Pure Appl. Chem., 1994, 66, 803–810 CrossRef CAS; (d) J. K. M. Sanders, Pure Appl. Chem., 2000, 72, 2265–2274 CrossRef CAS; (e) M. Kadri, J. Hou, V. Dorcet, T. Roisnel, L. Bechki, A. Miloudi, C. Bruneau and R. Gramage-Doria, Chem.–Eur. J., 2017, 23, 5033–5043 CrossRef CAS PubMed; (f) L. Poyac, S. Scoditti, X. Dumail, M. Granier, S. Clément, R. Gramage-Doria, C. H. Devillers and S. Richeter, Chem. Commun., 2022, 58, 13270–13273 RSC.
- J. Trouvé, P. Zardi, S. Al-Shehimy, T. Roisnel and R. Gramage-Doria, Angew. Chem., Int. Ed., 2021, 60, 18006–18013 CrossRef PubMed.
- P. Zardi, T. Roisnel and R. Gramage-Doria, Chem.–Eur. J., 2019, 25, 627–634 CAS.
- (a) The Mizoroki-Heck Reaction, ed. M. Oestreich, Wiley-VCH, 2009 Search PubMed; (b) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed; (c) R. F. Heck and J. P. Nolley, J. Org. Chem., 1972, 37, 2320–2322 CrossRef CAS; (d) T. Mizoroki, K. Mori and A. Ozaki, Bull. Chem. Soc. Jpn., 1971, 44, 581 CrossRef CAS; (e) W. Cabri and I. Candiani, Acc. Chem. Res., 1995, 28, 2–7 CrossRef CAS; (f) N. J. Whitcombe, K. K. Hii and S. E. Gibson, Tetrahedron, 2001, 57, 7449–7476 CrossRef CAS; (g) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2964 CrossRef CAS PubMed; (h) J. G. de Vries, Can. J. Chem., 2001, 79, 1086–1092 CrossRef CAS.
- (a) T. Hori, X. Peng, N. Aratani, A. Takagi, T. Matsumoto, T. Kawai, Z. Seok Yoon, M.-C. Yoon, J. Yang, D. Kim and A. Osuka, Chem.–Eur. J., 2008, 14, 582–595 CrossRef CAS PubMed; (b) S. Amanullah, P. Saha, R. Saha and A. Dey, Inorg. Chem., 2019, 58, 152–164 CrossRef CAS PubMed; (c) S. Gokulnath, B. S. Achary, C. K. Kumar, R. Trivedi, B. Sridhar and L. Giribabu, Photochem. Photobiol., 2015, 91, 33–41 CrossRef CAS PubMed; (d) T. Amaya, T. Ueda and T. Hirao, Tetrahedron Lett., 2010, 51, 3376–3379 CrossRef CAS; (e) M. Sankar, P. Bhyrappa, B. Varghese, K. K. Praneeth and G. Vaijayanthimala, J. Porphyrins Phthalocyanines, 2005, 9, 413–422 CrossRef CAS; (f) X. Liu, X. Ma and Y. Feng, Beilstein J. Org. Chem., 2019, 15, 1434–1440 CrossRef CAS PubMed; (g) P. Bhyrappa, G. Vaijayanthimala and B. Verghese, Tetrahedron Lett., 2002, 43, 6427–6429 CrossRef CAS; (h) X. Shi, S. R. Amin and L. S. Liebeskind, J. Org. Chem., 2000, 65, 1650–1664 CrossRef CAS PubMed; (i) M. Suzuki, S. Neya and Y. Nishigaichi, Molecules, 2016, 21, 252 CrossRef PubMed; (j) P. Bhyrappa, S. R. Wilson and K. S. Suslick, J. Am. Chem. Soc., 1997, 119, 8492–8502 CrossRef CAS; (k) R. K. Kumar, S. Balasubramanian and I. Goldberg, Inorg. Chem., 1998, 37, 541–552 CrossRef CAS PubMed; (l) M. P. Byrn, C. J. Curtis, Y. Hsiou, S. I. Khan, P. A. Sawin, S. K. Tendick, A. Terzis and C. E. Strouse, J. Am. Chem. Soc., 1993, 115, 9480–9497 CrossRef CAS; (m) I. Goldberg, H. Krupitsky, Z. Stein, Y. Hsiou and C. E. Strouse, Supramol. Chem., 1994, 4, 203–221 CrossRef CAS; (n) T. A. Dar, M. K. Chahal, R. A. Kumar and M. Sankar, J. Porphyrins Phthalocyanines, 2016, 20, 744–751 CrossRef CAS.
- (a) A. L. Singer and D. A. Atwood, Inorg. Chim. Acta, 1998, 277, 157–162 CrossRef CAS; (b) G. A. Morris, H. Zhou, C. L. Stern and S. T. Nguyen, Inorg. Chem., 2001, 40, 3222–3227 CrossRef CAS PubMed; (c) A. W. Kleij, M. Lutz, A. L. Spek, P. W. N. M. van Leeuwen and J. N. H. Reek, Chem. Commun., 2005, 3661–3663 RSC; (d) A. W. Kleij, M. Kuil, D. M. Tooke, M. Lutz, A. L. Spek and J. N. H. Reek, Chem.–Eur. J., 2005, 11, 4743–4750 CrossRef CAS PubMed.
- (a) K. M. Kadish, L. R. Shiue, R. K. Rhodes and L. A. Bottomley, Inorg. Chem., 1981, 20, 1274–1277 CrossRef CAS; (b) V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Angew. Chem., Int. Ed., 2001, 40, 4271–4274 CrossRef CAS; (c) I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed; (d) S. Belanger, M. H. Keefe, J. L. Welch and J. T. Hupp, Coord. Chem. Rev., 1999, 190–192, 29–45 CrossRef CAS; (e) H. E. Toma and K. Araki, Coord. Chem. Rev., 2000, 196, 307–329 CrossRef CAS; (f) S. Durot, J. Taesch and V. Heitz, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed; (g) J. A. A. W. Elemans, V. F. Slagt, A. E. Rowan and R. J. M. Nolte, Isr. J. Chem., 2005, 45, 271–279 CrossRef CAS; (h) A. W. Kleij and J. N. H. Reek, Chem.–Eur. J., 2006, 12, 4218–4227 CrossRef CAS PubMed; (i) K. S. Suslick, N. A. Rakow, M. E. Kosal and J.-H. Chou, J. Porphyrins Phthalocyanines, 2000, 4, 407–413 CrossRef CAS.
- (a) A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479–489 RSC; (b) L. Avrama and Y. Cohen, Chem. Soc. Rev., 2015, 44, 586–602 RSC.
- (a) T. Rosner, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 4621–4622 CrossRef CAS PubMed; (b) T. Rosner, J. Le Bars, A. Pfaltz and D. G. Blackmond, J. Am. Chem. Soc., 2001, 123, 1848–1855 CrossRef CAS PubMed; (c) A. H. M. de Vries, J. M. C. A. Mulders, J. H. M. Mommers, H. J. W. Henderickx and J. G. de Vries, Org. Lett., 2003, 5, 3285–3288 CrossRef CAS PubMed; (d) T. Nagata and Y. Obora, ACS Omega, 2020, 5, 98–103 CrossRef CAS PubMed; (e) A. M. Trzeciak and A. W. Augustyniak, Coord. Chem. Rev., 2019, 384, 1–20 CrossRef CAS; (f) A. M. Trzeciak and J. J. Ziółkowski, Coord. Chem. Rev., 2007, 251, 1281–1293 CrossRef CAS; (g) J. G. de Vries, Dalton Trans., 2006, 421–429 RSC; (h) A. S. Sigeev, A. S. Peregudov, A. V. Cheprakov and I. P. Beletskaya, Adv. Synth. Catal., 2015, 357, 417–429 CrossRef CAS; (i) M. V. Polynski and V. P. Ananikov, ACS Catal., 2019, 9, 3991–4005 CrossRef CAS.
- (a) C. S. Consorti, F. R. Flores and J. Dupont, J. Am. Chem. Soc., 2005, 127, 12054–12065 CrossRef CAS PubMed; (b) C. Amatore and A. Jutand, Acc. Chem. Res., 2000, 33, 314–321 CrossRef CAS PubMed; (c) G. T. Crisp, Chem. Soc. Rev., 1998, 27, 427–436 RSC; (d) S. Kozuch and S. Shaik, J. Am. Chem. Soc., 2006, 128, 3355–3365 CrossRef CAS PubMed; (e) A. de Meijere and F. E. Meyer, Angew. Chem., Int. Ed. Engl., 1995, 33, 2379–2411 CrossRef.
- (a) D. Ortiz, M. Blug, X.-F. Le Goff, P. Le Floch, N. Mézailles and P. Maître, Organometallics, 2012, 31, 5975–5978 CrossRef CAS; (b) A. K. de K. Lewis, S. Caddick, F. G. N. Cloke, N. C. Billingham, P. B. Hitchcock and J. Leonard, J. Am. Chem. Soc., 2003, 125, 10066–10073 CrossRef PubMed; (c) J. P. Stambuli, M. Bühl and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 9346–9347 CrossRef CAS PubMed; (d) A. H. Roy and J. Hartwig, Organometallics, 2004, 23, 194–202 CrossRef CAS; (e) C. Amatore, M. Azzabi and A. Jutand, J. Am. Chem. Soc., 1991, 113, 1670–1677 CrossRef CAS; (f) J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC; (g) K. H. Shaughnessy, Isr. J. Chem., 2020, 60, 180–194 CrossRef CAS; (h) N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- M. Tomasini, L. Caporaso, J. Trouvé, J. Poater, R. Gramage-Doria and A. Poater, Chem.–Eur. J., 2022, 28, e202201970 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2fd00165a |
| This journal is © The Royal Society of Chemistry 2023 |