
Highly polar stacking interactions wrap inorganics in organics: lone-pair–π-hole interactions between the PdO4 core and electron-deficient arenes†
Yury V.
Torubaev
 *a,
Ivan V.
Skabitsky
a,
Anton V.
Rozhkov
b,
Bartomeu
Galmés
c,
Antonio
Frontera
*c and
Vadim Yu.
Kukushkin
*bd
*a,
Ivan V.
Skabitsky
a,
Anton V.
Rozhkov
b,
Bartomeu
Galmés
c,
Antonio
Frontera
*c and
Vadim Yu.
Kukushkin
*bd
aN. S. Kurnakov Institute of General and Inorganic Chemistry, of Russian Academy of Sciences, Moscow, 119991 Russian Federation. E-mail: torubaev@igic.ras.ru
bInstitute of Chemistry, Saint Petersburg State University, Universitetskaya Nab. 7/9, Saint Petersburg, 199034 Russian Federation. E-mail: v.kukushkin@spbu.ru
cDepartment of Chemistry, Universitat de les IllesBalears, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
dInstitute of Chemistry and Pharmaceutical Technologies, Altai State University, 656049 Barnaul, Russian Federation
First published on 6th October 2021
Abstract
Cocrystallization of the palladium acetate cluster Pd3(OAc)6 (abbreviated as [Pd3]) with electron-deficient iodine(I)-based perfluoroarenes (ArFI: iodopentafluorobenzene, 4-iodoheptafluorotoluene, 1,4-diiodotetrafluorobenzene, 1,2-diiodotetrafluorobenzene, and octafluoro-4,4′-diiodo-1,1-biphenyl) and iodine-free octafluoronaphthalene gave a series of six cocrystals [Pd3]·(arene) studied by single-crystal X-ray diffractometry. Significant intermolecular noncovalent interactions were verified by a density functional theory study, molecular electrostatic potential and Hirshfeld surface analyses, a combined QTAIM/NCPlot approach, and NBO and energy framework calculations. In five out of six structures, the aromatic rings coupled with cluster [Pd3] via lone pair–π-(ArF)-hole interactions. The iodine-containing arenes, ArFI, were additionally involved in halogen bonding with carboxylate O centers, but inorganic–organic stacking still remained the structure-determining interaction. In the stacking, the electron-rich PdO4 plane behaved as a five-center nucleophile providing oxygen lone pairs in addition to the dz2-PdII orbital; this plane complemented the π-acidic surface of the arenes, affording highly polar circular stacking, where organics wrapped inorganics.
1. Introduction
The past few years have seen precipitous growth in the number of publications related to various aspects of weak interactions, both in chemistry and biology. The cutting-edge nature of this topic has been highlighted in a special issue of Chemical Reviews1 devoted exclusively to noncovalent interactions and in a number of subsequent topical reviews and book considerations.2–5 Despite the low energy of noncovalent interactions, in many cases, they act collectively, and the sum of their actions can play a significant role in various transformations of chemical compounds, synthesis and catalysis,4,6–11 including organic noncovalent catalysis.Stacking is a well-known subclass of intermolecular interactions that plays a significant role in the crystal packing of aromatic organics and organometallics and, in this way, affects the properties of the formed functional materials. Organic molecules with extended aromatic systems are most often applied as stacking components. In addition to the classic π-stacking of two aromatic systems with aromatic-faced, interaction planes (including heteroaromatics), numerous examples of hetero π-stacking involve various inorganic stacking partners.
Such stacking has been identified for (i) XH (X = B, C, N, O, halogen)5 centers with a lone-pair (LP) pointing to the centroid of an aromatic ring, (ii) ions,12,13 (iii) LPs of metal14,15 and nonmetal centers.16 Among these cases, π-stacking involving metal-containing inorganic planes is very rare. They include Hg3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17–22 and Hg423 cores of various anti-crowns, AuI3 clusters,24–26 MO427,28 and M(C∩N)O229 cores in the reverse sandwich systems that we reported recently, and PdCl430–33 and PtCl434 entities in mixed inorganic–organic systems. In the last two cases, a purely inorganic face consisting of Pd or Pt chlorides was cocrystallized with and wrapped by electron-deficient aromatic rings.
17–22 and Hg423 cores of various anti-crowns, AuI3 clusters,24–26 MO427,28 and M(C∩N)O229 cores in the reverse sandwich systems that we reported recently, and PdCl430–33 and PtCl434 entities in mixed inorganic–organic systems. In the last two cases, a purely inorganic face consisting of Pd or Pt chlorides was cocrystallized with and wrapped by electron-deficient aromatic rings.
Regarding π-stacking with metal-containing inorganic planes, we recently reported27 that acetylacetonate PdII complexes (Fig. 1, left panel), featuring a PtO4 core as a part of an organic system, cocrystallize with perfluorinated aromatics to give columnar stacking. As a continuation of this project, we attempted to verify whether purely inorganic planes are capable of forming stacks with pure organics. For this study, we addressed the trigonal Pd3(OAc)6 cluster (abbreviated as [Pd3]; Fig. 1, right panel) featuring three Pd atoms in the square plane of four Lewis-basic carboxyl groups. These ligands in [Pd3] are arranged in such a way that the three PdO4 planes are located on the cluster surface and they are not shielded by the sterically hindered organic fragments of the acetate ligand. In contrast to the previous work,27 it becomes possible to study the interaction of a purely inorganic group with organic electron-deficient molecules. The Pd centers, each exhibiting a dz2 orbital, and the carboxylic oxygens (having LPs) could function as acceptors toward π- and σ-holes in electron-deficient aromatics and halide-based halogen bond (HaB) donors, respectively.35 It was assumed that the availability of the three unhindered five-center PdO4 nucleophilic entities could be useful for utilization in crystal engineering and, eventually, in the design of solid materials.
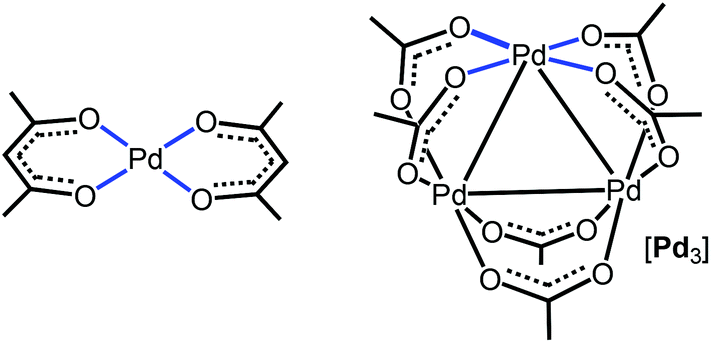 | ||
| Fig. 1 Acetylacetonate PdII complex (left panel) and the trigonal Pd3(OAc)6 cluster (right panel) exhibiting the PdO4 nucleophilic entities. | ||
We found that each of the PdO4 cores in [Pd3] behaves as an integrated 5-center nucleophile providing oxygen lone pairs in addition to the dz2-PdII orbital. This planar nucleophile couples with various electron-deficient arenes via LP–π-hole interactions to give highly polar stacking, where three inorganic PdO4 faces are wrapped by the arenes. All our results are detailed in the following sections.
Notably, hereinafter, we assign all X⋯Y noncovalent interactions, where X is an electropositive component standing before the three central dots (i.e., H for hydrogen bonding, HB; and halogen for halogen bonding, HaB), while Y is an electronegative partner standing after the dots.36 However, for LP–π-hole interactions, the electronegative/positive partners are given in the reverse order, as conventionally accepted for these types of interactions.5,37
2. Experimental section
2.1. Materials
Solvents were purified, dried, and distilled under an argon atmosphere before use. Commercially available FN, IFB, 1,4-IFB, and 1,2-IFB were used as received without additional purification; IFTol and 4,4′-IBph were prepared as previously reported.38 The cluster Pd3(OAc)3 was synthesized according to the reported procedure39 and recrystallized from glacial acetic acid before use.2.2. Cocrystal growth
All cocrystals suitable for single-crystal X-ray diffraction (SCXRD) studies were prepared by vapor diffusion of n-pentane into a DCM solution of a mixture of [Pd3] (7 mg, 0.1 mmol) and an appropriate perfluoroarene (0.5 mmol) (for a detailed scheme see the ESI†). A uniform dark-orange crystalline material was used for SCXRD without additional workup.2.3. Crystal structure determinations
A Bruker D8 Venture diffractometer equipped with graphite-monochromated Mo Kα radiation (0.71070 Å) was used for the cell determination and intensity data collection for the cocrystals. The data were collected by the standard ‘phi–omega’ scan techniques, and were reduced using SAINT v8.37A (Bruker, 2015). The SADABS (Bruker, 2016) software was used for scaling and absorption correction for [Pd3]·(FN), [Pd3]·(1,2-IFB), and [Pd3]·(1,4-IFB), and TWINABS (Bruker, 2012) for twins [Pd3]·(IFB), [Pd3]·(IFTol), and [Pd3]·(4,4′-IBph)·(CH2Cl2) respectively. The structures were solved by direct methods and refined by full-matrix least squares against F2 using Olex2 and SHELXTL software.40,41 Non-hydrogen atoms were refined with anisotropic thermal parameters. All hydrogen atoms were geometrically fixed and refined using a riding model. Atomic coordinates and other structural parameters of the reported cocrystals have been deposited at the Cambridge Crystallographic Data Center CCDC 2082585 for [Pd3]·(FN), 2082583 [Pd3]·(IFB), 2082586 [Pd3]·(IFTol), 2082588 [Pd3]·(1,4-IFB), 2082587 [Pd3]·(1,2-IFB), and 2082584† [Pd3]·(4,4′-IBph)·(CH2Cl2).2.4. Computational details
Intermolecular interaction energy calculation and subsequent energy framework generation were performed in Crystal Explorer 17.5 (TONTO, B3LYP-DGDZVP)42 for all unique molecular pairs in the first coordination sphere of a molecule (5 Å) using experimental crystal geometries.The interaction energies of the HaB and LP⋯π-hole stacking dimers of all cocrystals were calculated using the PBE0 functional49,50 together with Grimme's D3 dispersion correction51 and Becke–Johnson damping.52 They were computed as the difference between the energy of the supramolecular dimer and the sum of energies of the isolated monomers. The def2-TZVP basis set was utilized in calculations. The combined QTAIM/NCIPlot analyses of the halogen bonded and LP⋯π-hole complexes were performed using the AIMAll program;53 all expected critical points of electron density were found and the characteristic set was found to satisfy the Poincare–Hopf relation. The estimation of the individual noncovalent interaction energies was performed using the potential energy density [V(r)] values at the bond critical points, using the following equations: Eint = 0.778V(r) for halogen bonds54,55 and Eint = 0.5V(r) for hydrogen bonds.56 The MEP surfaces were computed at the PBE0-D3/def2-TZVP level of theory using the 0.001 au isosurface (Fig. 3). The energetic results were computed using the Turbomole 7.0 program. The wavefunctions were obtained using the Gaussian-16 program and utilized for the calculation of the MEP surfaces, QTAIM, and NCIPlot analyses. NBO calculations were computed at the same level of theory using the Gaussian-16 program.
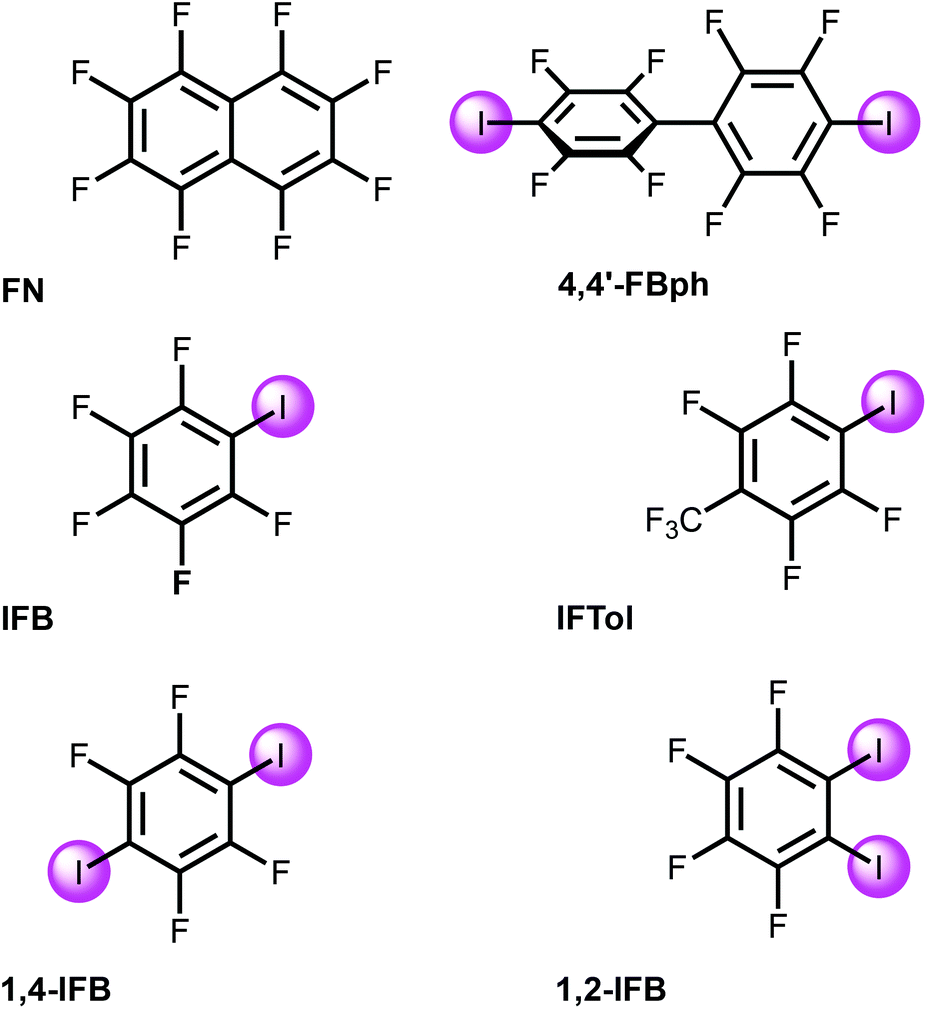 | ||
| Fig. 2 Studied electron-deficient arenes. | ||
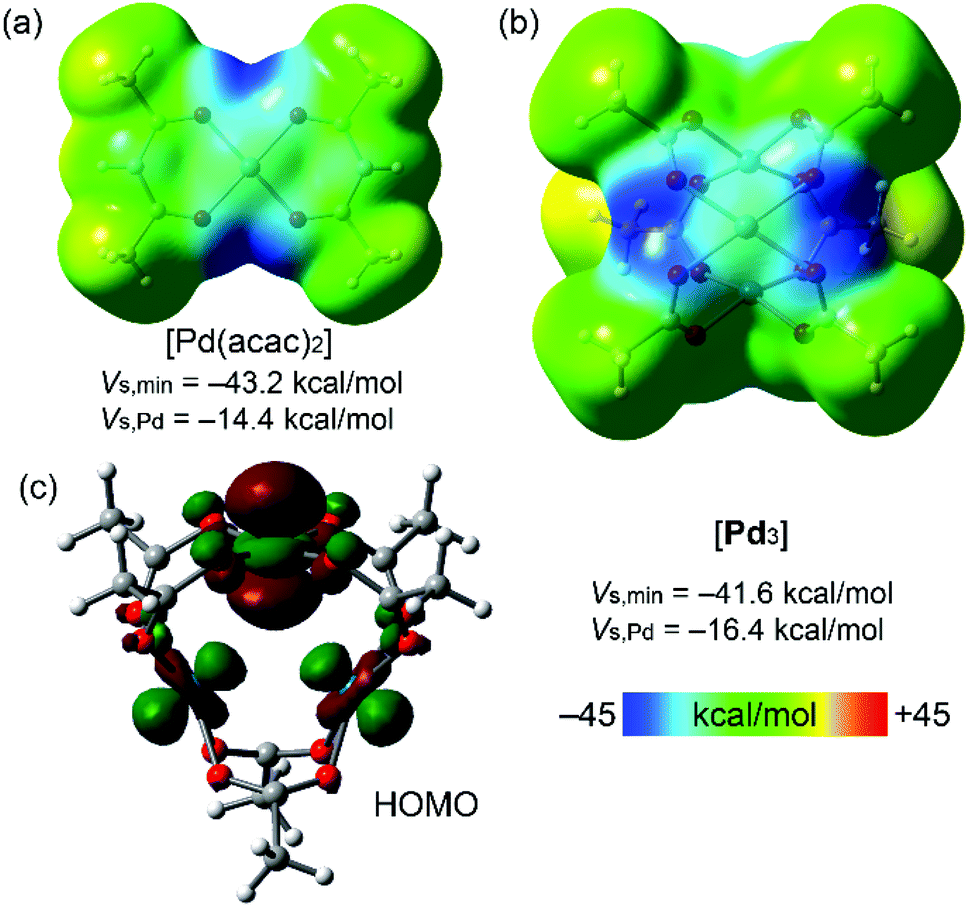 | ||
| Fig. 3 MEP plots for Pd(acac)2 (a) and cluster [Pd3] (b) (0.001 au electron density isosurface). HOMO plot of cluster [Pd3] showing the nucleophilic contribution of the dz2[Pd] atomic orbitals (c). | ||
3. Results and discussion
3.1. Noncovalent interaction partners
In [Pd3], each of the three dz2-PdII centers along with LPs on the carboxylic O centers could function as acceptors toward π- and σ-holes in electron-deficient aromatics and halide-based HaB donors, respectively.35The MEP values at the σ- and π-holes of the electron-deficient arenes represented in Fig. 2 are summarized in Table 1. The most π-acidic arene is FN (+21.6 kcal mol−1 over the centers of the rings), followed by IFB (+18.3 kcal mol−1). The rest of the arenes exhibit similar MEP values at the π-hole (+14.1 to +15.6 kcal mol−1). The deepest σ-hole corresponds to IFB (+39.6 kcal mol−1), followed by IFTol (+35.4 kcal mol−1) and 4,4′FBph (+34.1 kcal mol−1). Vs,max corresponds to the σ-hole in the iodoarenes, thus anticipating a higher ability to establish HaBs than LP–π interactions. However, the size of the σ-hole is small compared with the extended π-acidic surfaces of the arene. Therefore, depending on the electron donor, π-hole interactions can be stronger than σ-hole interactions.
| Arene | σ-Hole (iodine) | π-Hole (arene) |
|---|---|---|
| FN | — | +21.6 |
| 4,4′FBph | +34.1 | +15.6 |
| IFB | +39.6 | +18.3 |
| IFTol | +35.4 | +14.4 |
| 1,4-IFB | +31.4 | +14.1 |
| 1,2-IFB | +30.8 | +15.4 |
Fig. 3 shows the MEP surfaces of the [Pd(acac)2] complex (for comparison purposes; Fig. 1) and cluster [Pd3]. The MEP minima in both systems are in the PdO4 plane along the bisectrix of the O–Pd–O angle (dark blue regions). There is a large region over the PdO4 plane where MEP is negative, including the palladium(II) atom. Interestingly, the MEP value over the Pd center turns out to be more negative in [Pd3] than in the [Pd(acac)2] model system, thus suggesting that the intramolecular Pd⋯Pd contacts enhance the nucleophilicity of all Pd atoms. Although the Pd environment in the model system and the [Pd3] cluster is different (chelating vs. bridging ligands), the proposed enhancement of nucleophilicity in the latter is consistent with our previous data indicating the dz2-nucleophilicity enhancement of palladium(II) centers in PdII2 dimers.43 Moreover, the HOMO plot for the [Pd3] cluster (Fig. 3c) shows the dominant participation of the dz2 atomic orbitals of the Pd-atoms. This MEP surface analysis indicates that the electron-rich PdO4 plane nicely complements the π-acidic surface of the arenes and that the σ-hole of iodine complements the strong nucleophilic region between the O atoms in the PdO4 plane.
3.2. Cocrystal growth
[Pd3] was cocrystallized with iodine(I)-based arenes and one iodine-free perfluorinated arene (octafluoronaphthalene). All structures and abbreviations are shown in Fig. 2 to give six cocrystals studied by SCXRD. Thus, the cocrystallization of [Pd3] with electron-deficient iodine(I)-based perfluoroarenes (ArFI = IFB, IFTol, 1,2-IFB, 1,4-IFB, and 4,4′-IFBph) and iodine-free FN (Fig. 2) by vapor diffusion of n-pentane into a DCM solution of a mixture of [Pd3] and an appropriate arene afforded a series of [Pd3]·(arene) cocrystals (Fig. 4).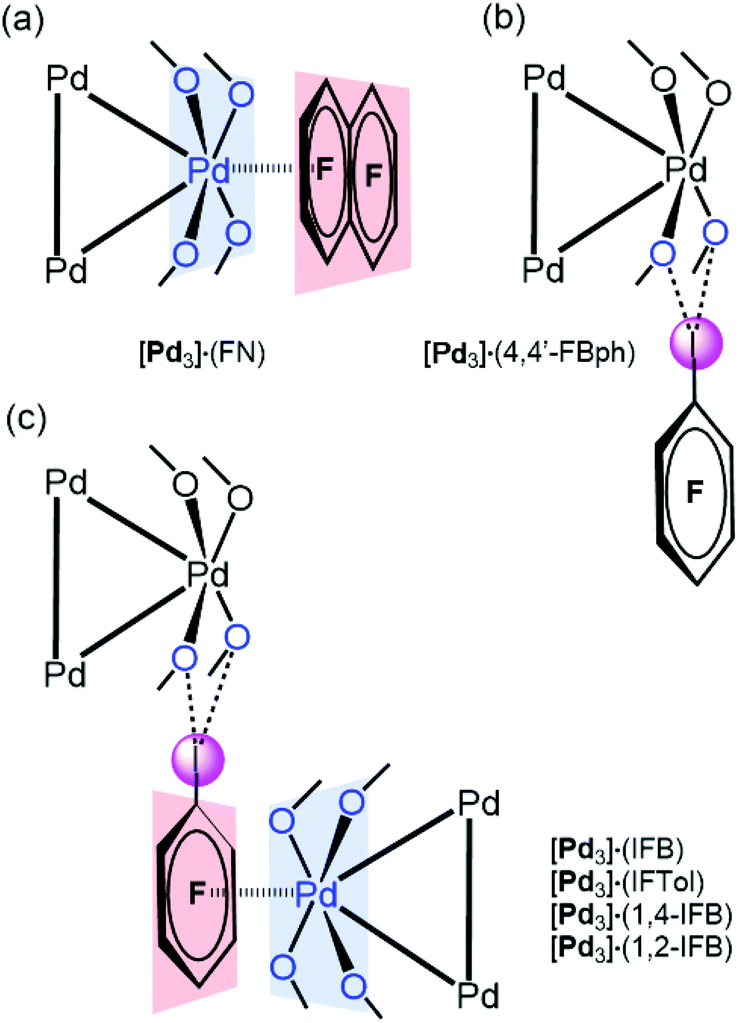 | ||
| Fig. 4 Types of the observed structure-directing noncovalent interactions: PdO4⋯π-hole (a), I⋯O HaB interactions (b), combination of both PdO4⋯π-hole and I⋯O HaB interactions (c). | ||
Some of us recently reported that robust columnar or layered supramolecular architectures may not only reoccur in the cocrystal in the same form as in the native crystal of a σ-hole donor coformer but can be split into lower dimension fragments, in agreement with their energy framework, and following a proverb, “a thread usually breaks where it is thinnest”, thus allowing the prediction of some packing pattern features.35,44,45 We indeed proved that the native packing of the structure of [Pd3]39,46 may undergo different depths of fragmentation depending on the nature of the cocrystallization partner (for crystal packings, see Fig. 5, S1 and S2†).
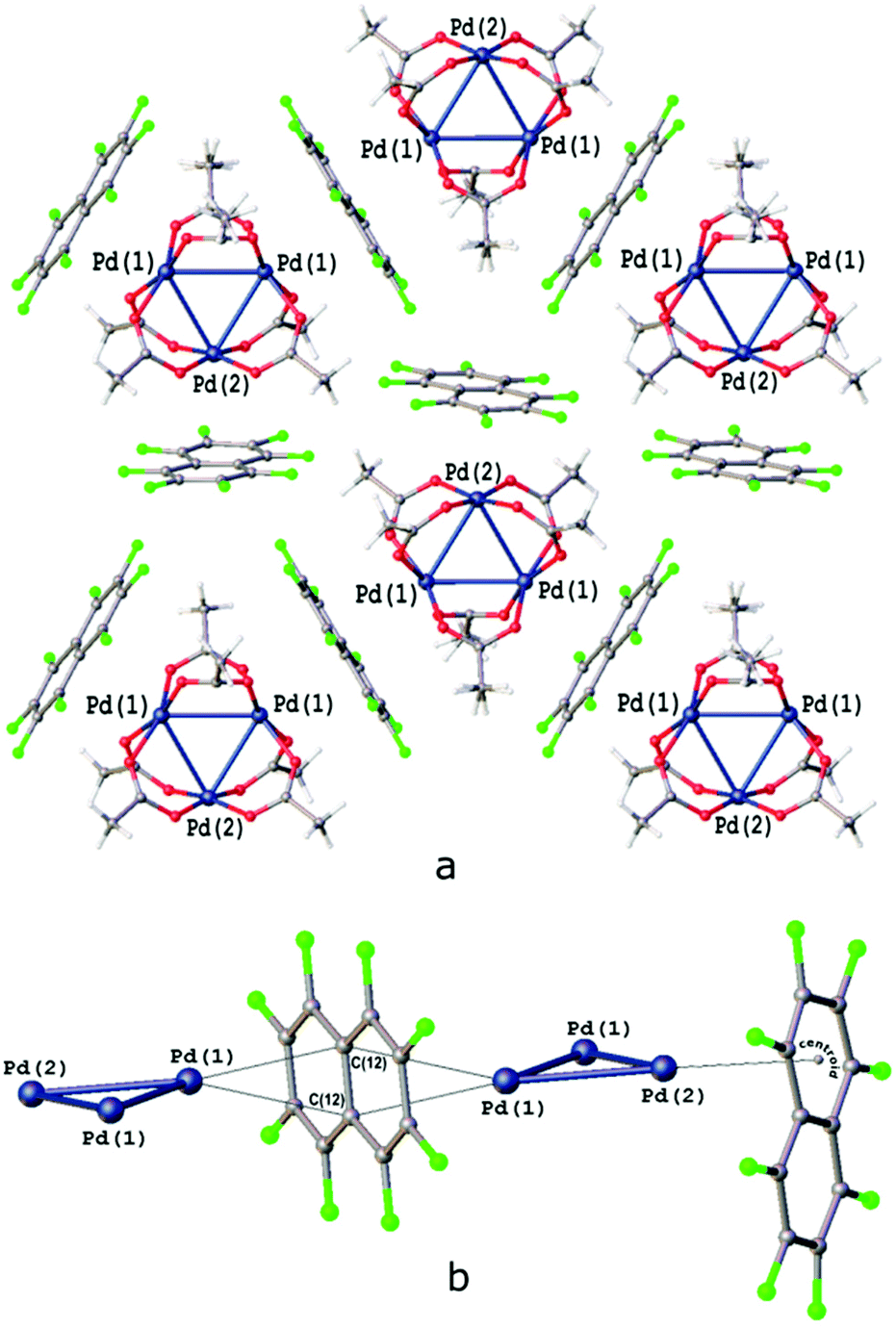 | ||
| Fig. 5 (a) Fragment of the crystal structure of [Pd3]·(FN); (b) two coordination modes of OFN. Pd1⋯C(12) 3.353(2)–3.547(2) Å, Pd⋯C6 centroid 3.328 Å; the angle between the Pd3 triangle and the FN planes in the zig-zag chain is 89.83°; the angle between the PdO4 and C10 planes is 1.65°; and the angle between the Pd3 triangle and FN plane is 86°. | ||
In five out of six structures, arenes wrap the columns of [Pd3]. According to the nature of the noncovalent interactions observed between [Pd3] and arenes, the structures of these cocrystals can be classified in increasing order of complexity (Fig. 4): (a) stabilized by the PdO4⋯π-hole donating ring system ([Pd3]·(FN); section 3.3); (b) stabilized by I⋯O HaB interactions ([Pd3]·(4,4′-IBph; section 3.4)); and (c) displaying a combination of both PdO4⋯π-hole and I⋯O HaB interactions ([Pd3]·(IFB), [Pd3]·(IFTol), [Pd3]·(1,4-IFB), and [Pd3]·(1,2-IFB); section 3.5).
3.3. Lone-pair–π-hole interactions between the PdO4 core and perfluoronaphthalene
The cocrystal [Pd3]·(FN) is a successful outcome of cocrystallizations with iodine-free perfluoroarenes; and the arenes C6F5H, C6F5CF3, and FN were applied, but only in the last case we succeeded in isolating cocrystals of SCXRD quality. The crystal packing pattern of [Pd3]·(FN) is characterized by PdO4⋯π-(FN)-hole bonding for each of the three PdO4 planes (Fig. 5a). This noncovalent interaction gives rise to planar sheets, which are parallel to the triangular Pd3 plane (Fig. 5a and c). The PdO4 planes are parallel to the FN plane, giving highly polar inorganic–organic stacking. The [Pd3] moieties are fully shielded from each other by the wrapping arenes.Three FN molecules approach [Pd3] from the three sides in two different modes: one FN appears between two apexes of the Pd3 triangles (Fig. 5); therefore, the central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond between the two rings in FN is orthogonal to the Pd3 plane (Fig. 5b), and it is the fragment of FN, which is the closest to a Pd center (Pd⋯C 3.547–3.353 Å). These bridging FNs give rise to ([Pd3]⋯FN⋯[Pd3]⋯FN)n stacking; the resulting stacks are similar to those observed in [Pd3]·(1,4-IFB), [Pd3]·(1,2-IFB), [Pd3]·(IFB), and [Pd3]·(IFTol) (section 3.5). The other FN molecule approaches the apical position of [Pd3] in such a way that the central C bond in FN is in line with a Pd3 plane and one of the FN rings is the closest to a Pd atom (Pd⋯C6-centroid of FN 3.328 Å; Fig. 5b).
C bond between the two rings in FN is orthogonal to the Pd3 plane (Fig. 5b), and it is the fragment of FN, which is the closest to a Pd center (Pd⋯C 3.547–3.353 Å). These bridging FNs give rise to ([Pd3]⋯FN⋯[Pd3]⋯FN)n stacking; the resulting stacks are similar to those observed in [Pd3]·(1,4-IFB), [Pd3]·(1,2-IFB), [Pd3]·(IFB), and [Pd3]·(IFTol) (section 3.5). The other FN molecule approaches the apical position of [Pd3] in such a way that the central C bond in FN is in line with a Pd3 plane and one of the FN rings is the closest to a Pd atom (Pd⋯C6-centroid of FN 3.328 Å; Fig. 5b).
3.4. Cocrystals predominantly stabilized by I⋯O interactions
The second group of cocrystals, featuring I⋯O HaBs as structure-directing interactions, is represented by [Pd3]·(4,4′-FBph)·(CH2Cl2). Its hybrid structure is stabilized predominantly by short I⋯O HaBs (3.119(4)–3.045(4) Å). The inability of sterically demanding rod-like 4,4′-IBph to wrap [Pd3] in the same way as the other σ/π-hole donor coformers results in the reoccurrence of the bilayered [Pd3](CH2Cl2)[Pd3] architecture, which is also observed in the Pbcm polymorph of the [Pd3]·(CH2Cl2) solvate39 (Fig. 6a and b). The packing patterns of [Pd3] and CH2Cl2 in these cocrystals are similar (Fig. 6c and d).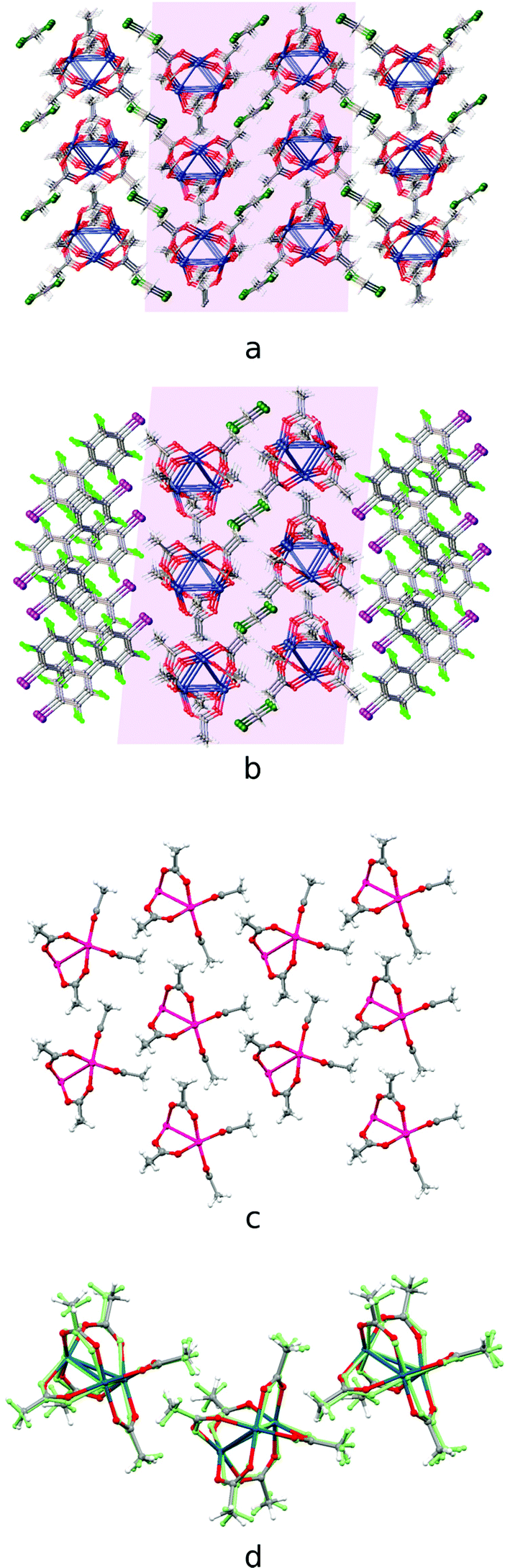 | ||
| Fig. 6 Crystal structure diagrams of (a) [Pd3] and CH2Cl2 molecules in [Pd3]·(CH2Cl2) crystal solvate [CSD refcode: SAQWUD], (b) [Pd3]·(4,4′-IBph)·(CH2Cl2) cocrystal solvate: (c) [Pd3] cluster layer in [Pd3]·(4,4′-IBph) (CH2Cl2) and (d) the overlay of the trimolecular fragments of [Pd3] layers in [Pd3]·(CH2Cl2) (fully-colored), and [Pd3]·(4,4′-IBph)·(CH2Cl2) (in green). Short I⋯O contacts are clearly seen on the Hirshfeld surface map (Fig. S4†). | ||
The highly symmetric structural pattern of [Pd3]·(FN) is broken on the formal replacement of FN by 4,4′-IBph, and substantial fragments of the [Pd3]·(CH2Cl2) solvate39 packing reappear in the [Pd3]·(4,4′-IBph)·(CH2Cl2) cocrystal.
3.5. Combined PdO4⋯π-hole and I⋯O interactions
The fluorinated iodoarenes shown in Fig. 2 are among the most common categories of synthons for HaB-involving crystal engineering. Our search and processing of the Cambridge Structural Database (CSD) for halogen bond assemblies involving various ArFI revealed that the vast majority of them function as σ-hole donating building blocks to give HaB-based systems. In these cases, application of ArFI predominantly assumes the utilization of iodine(s) σ-hole donor ability toward various nucleophiles, association with electrophilic centers via an electron belt of iodine(s), or their involvement in both types of contacts when a σ-hole is bound to a nucleophilic center, while an electron belt is associated with an electrophile.In contrast to all these observations of σ-hole donor properties of ArFI, one of us has reported47,48 that ArFI could also act as a synthon exhibiting π-hole or mixed σ-hole/π-hole donor properties and, for instance, the π-hole donor ability of PhFI (IFB) by its energetics is well comparable with the σ-hole donor properties of this arene;47 in this study we were led to the same conclusions by inspecting the MEP values given in Table 1. Hence, the π-hole donor ability of the iodoarenes (Fig. 2) is not unexpected, albeit still an unusual phenomenon that has been overlooked in the overwhelming majority of reported instances.
The application of iodoperfluorobenzenes as crystallization partners of [Pd3] leads to the most abundant subcategory of cocrystals, namely, those stabilized by both PdO4⋯π-hole and I⋯O HaBs (Fig. 4c). The packing diagram of the most representative structure of [Pd3]·(1,4-IFB) is given in Fig. 8, while the crystal packing of [Pd3]·(IFB) and [Pd3]·(IFTol) is shown in Fig. S1 and S2.†
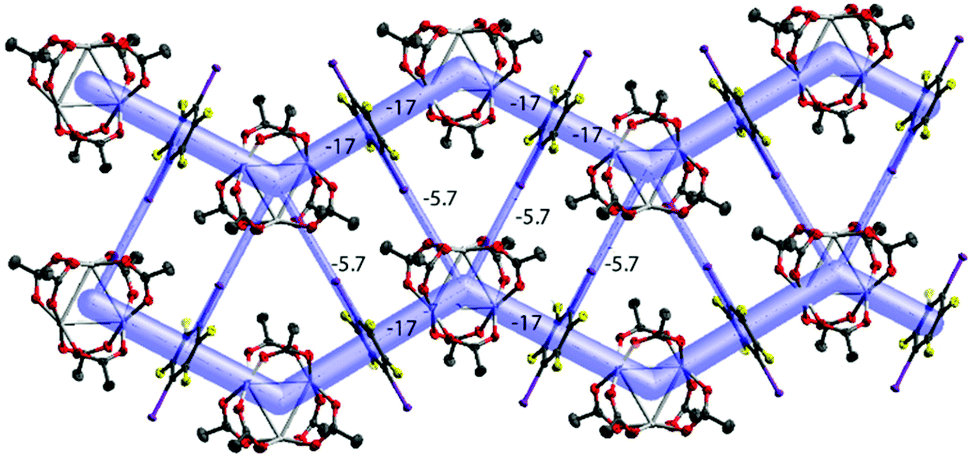 | ||
| Fig. 7 The energy framework of [Pd3]·(1,4-IFB): (100) view showing the zig-zag chains and their aggregation into the layer. Intermolecular energies are shown in kcal mol−1; cutoff 2 kcal mol−1. | ||
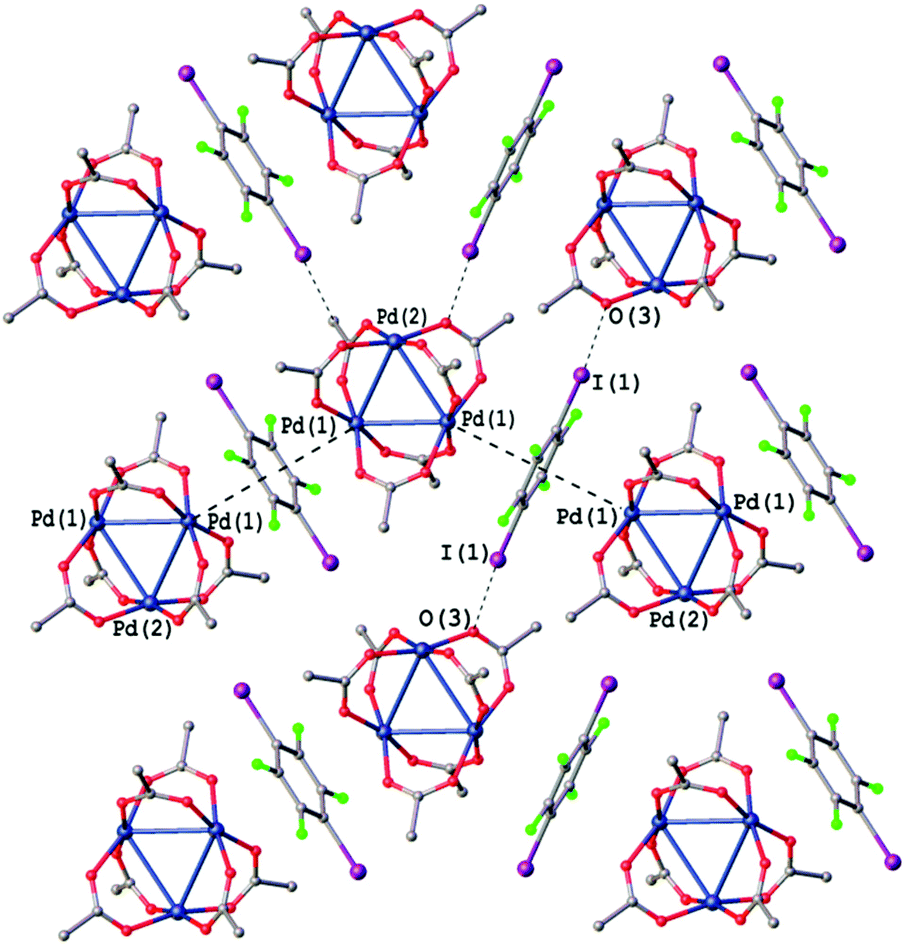 | ||
| Fig. 8 Crystal structure diagram of [Pd3]·(1,4-IFB). Selected distances (Å): I(1)⋯O(3) 3.399, Pd2⋯(1,4-IFB)centroid 3.349. Short I⋯O contacts and PdO4⋯π interactions are clearly seen in the Hirshfeld surface map (Fig. S5†). | ||
Most of the intermolecular I⋯O distances in the structures of all three [Pd3]·(ArFI) cocrystals (2.951(8)–3.469 Å, Fig. 8, S1 and S2†) are considerably shorter than the sum of Alvarez vdW49 radii (I + O = 3.54 Å). All these cocrystals exhibit the same packing pattern featuring the noncovalent π-bound arene between two [Pd3] moieties. These PdO4⋯π-hole interactions assemble [Pd3] into zig-zag chains, which are further linked into layers via I⋯O HaBs.
The packing hierarchy of [Pd3]·(1,4-IFB) (in other words, the assembly of chains into layers) is in good agreement with the energy framework of [Pd3]·(1,4-IFB), which clearly visualizes ([Pd3]⋯1,4-IFB⋯[Pd3]⋯1,4-IFB)n zig-zag chains as the strongest substructures in the cocrystal (approx. −17 kcal mol−1), associated with each other in the layers, presumably by I⋯O HaBs (approx. −5.7 kcal mol−1) (Fig. 7 and S3†). The latter interaction energy value obtained by use of the Crystal Explorer 17.5 program (CE-B3LYP DGDZVP42) is in good agreement with that calculated using the QTAIM (section 3.6), namely, −5.6 kcal mol−1 (Table 2).
| Cocrystal | CP | ρ(r) | V(r) | G(r) | E int | ∑Eint |
|---|---|---|---|---|---|---|
| [Pd3]·(IFB) | a (HaB) | 0.0166 | −0.0104 | 0.0132 | −5.0 | −7.3 |
| b (HaB) | 0.0048 | −0.0022 | 0.0032 | −1.0 | ||
| c (HB) | 0.0056 | −0.0027 | 0.0036 | −0.8 | ||
| d (HB) | 0.0036 | −0.0015 | 0.0022 | −0.5 | ||
| [Pd3]·(1,2-IFB) | a (HaB) | 0.0118 | −0.0072 | 0.0090 | −3.5 | −4.9 |
| b (HaB) | 0.0036 | −0.0015 | 0.0022 | −0.5 | ||
| c (HB) | 0.0048 | −0.0023 | 0.0031 | −0.7 | ||
| d (HB) | 0.0012 | −0.0006 | 0.0009 | −0.2 | ||
| [Pd3]·(1,4-IFB) | a (HaB) | 0.0125 | −0.0078 | 0.0096 | −3.8 | −5.6 |
| b (HaB) | 0.0038 | −0.0017 | 0.0024 | −0.8 | ||
| c (HB) | 0.0043 | −0.0021 | 0.0029 | −0.7 | ||
| d (HB) | 0.0022 | −0.0009 | 0.0014 | −0.3 | ||
| [Pd3]·(IFTol) | a (HaB) | 0.0046 | −0.0020 | 0.0030 | −1.0 | −2.8 |
| b (HaB) | 0.0046 | −0.0020 | 0.0030 | −1.0 | ||
| c (HB) | 0.0030 | −0.0013 | 0.0019 | −0.4 | ||
| d (HB) | 0.0030 | −0.0013 | 0.0019 | −0.4 | ||
| [Pd3]·(4,4′IFBph) | a (HaB) | 0.0119 | −0.0074 | 0.0092 | −3.6 | −4.2 |
| c (HB) | 0.0039 | −0.0019 | 0.0026 | −0.6 |
The I⋯O HaBs in [Pd3]·(1,2-IFB) link the chains into the layers and assemble these layers into a 3D structure (Fig. 9c). Owing to such a specific I⋯O HaB between the layers, the I⋯O HaBs in [Pd3]·(1,2-IFB) (3.06–3.23 Å; Fig. 9b) are not bifurcated.
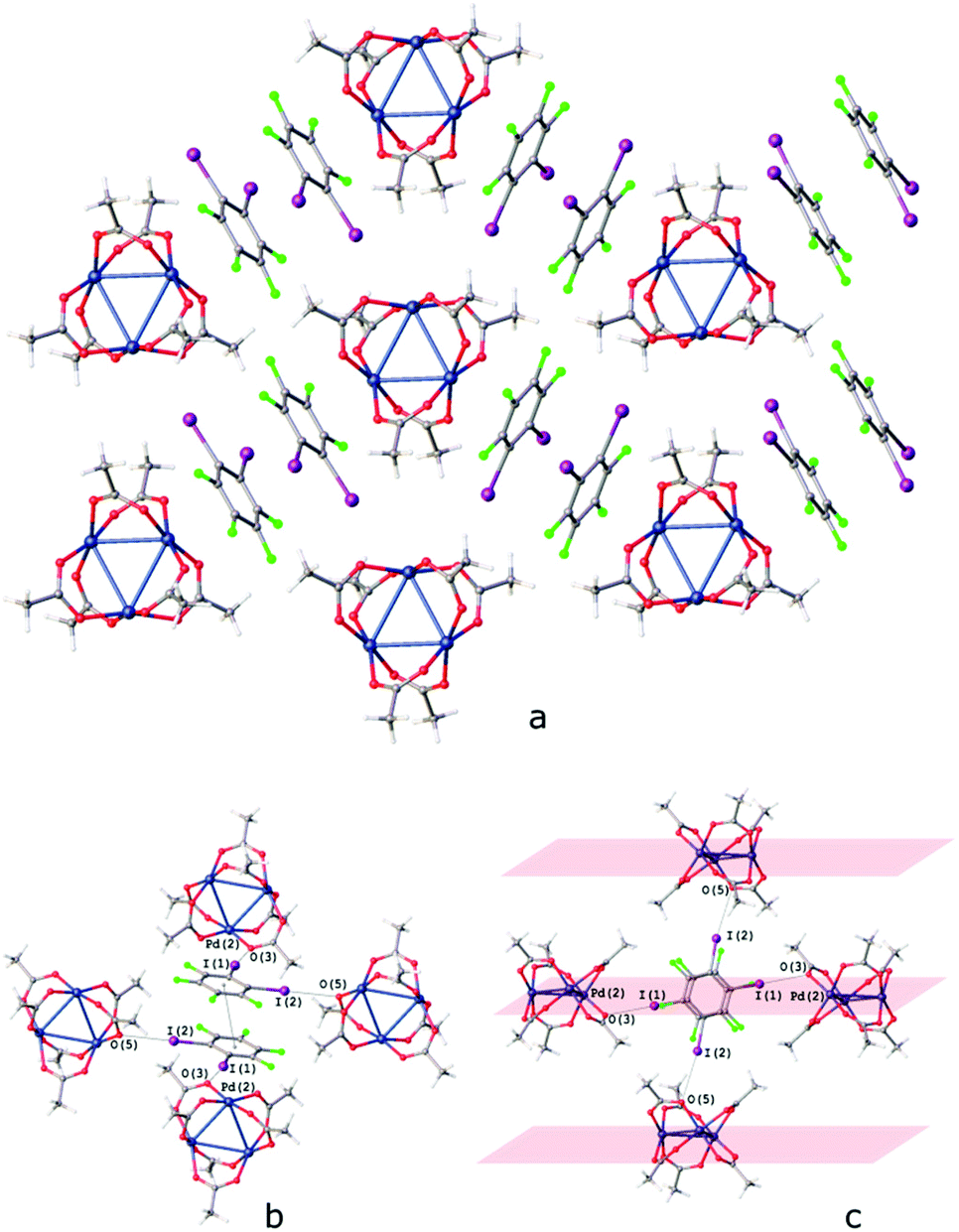 | ||
| Fig. 9 Crystal structure diagram of (a) [Pd3]·1,2-IFB; (b) π–π interaction between the 1,2-IFB molecules and (c) I⋯O interactions between the layers in [Pd3]·1,2-IFB. Selected intermolecular distances (Å): I(1)⋯O(3) – 3.116(2) Å, I(2)⋯O(5) 3.229(2) Å, Pd2⋯(1,2-IFB)centroid 3.306–3.311 Å, (1,2-IFB)centroid⋯(1,2-IFB)centroid 3.884 Å. Short I⋯O contacts and PdO4⋯π interactions are clearly seen in the Hirshfeld surface map (Fig. S6†). | ||
The I⋯OC HaBs are the most pronounced intermolecular interactions on the Hirshfeld surface map of [Pd3]·(1,4-IFB) (Fig. S5a†). The same map indicates considerable intermolecular interaction with 1,4-IFB all over the PdO4 plane (Fig. S5b†).
3.6. Theoretical study
The highly polar nature of the aforementioned PdO4⋯π-hole interactions is clearly corroborated by the MEP results shown in Fig. 3 and Table 1, which show large and positive MEP values at the π-hole or arenes and large and negative MEP values at the PdO4 face of the cluster. In addition, we studied the energetic features of the I⋯O σ-hole interactions described above and characterized them using a combination of the quantum theory of atoms in molecules and the noncovalent interaction plot index (NCIPlot). Fig. 10 shows the distribution of bond critical points (CPs) and bond paths overlapping with the NCIplot surfaces for the cocrystals [Pd3]·(IFB), [Pd3]·(1,2-IFB), [Pd3]·(1,4-IFB), [Pd3]·(IFTol), and [Pd3]·(4,4′-IFBph), along with the interaction energies.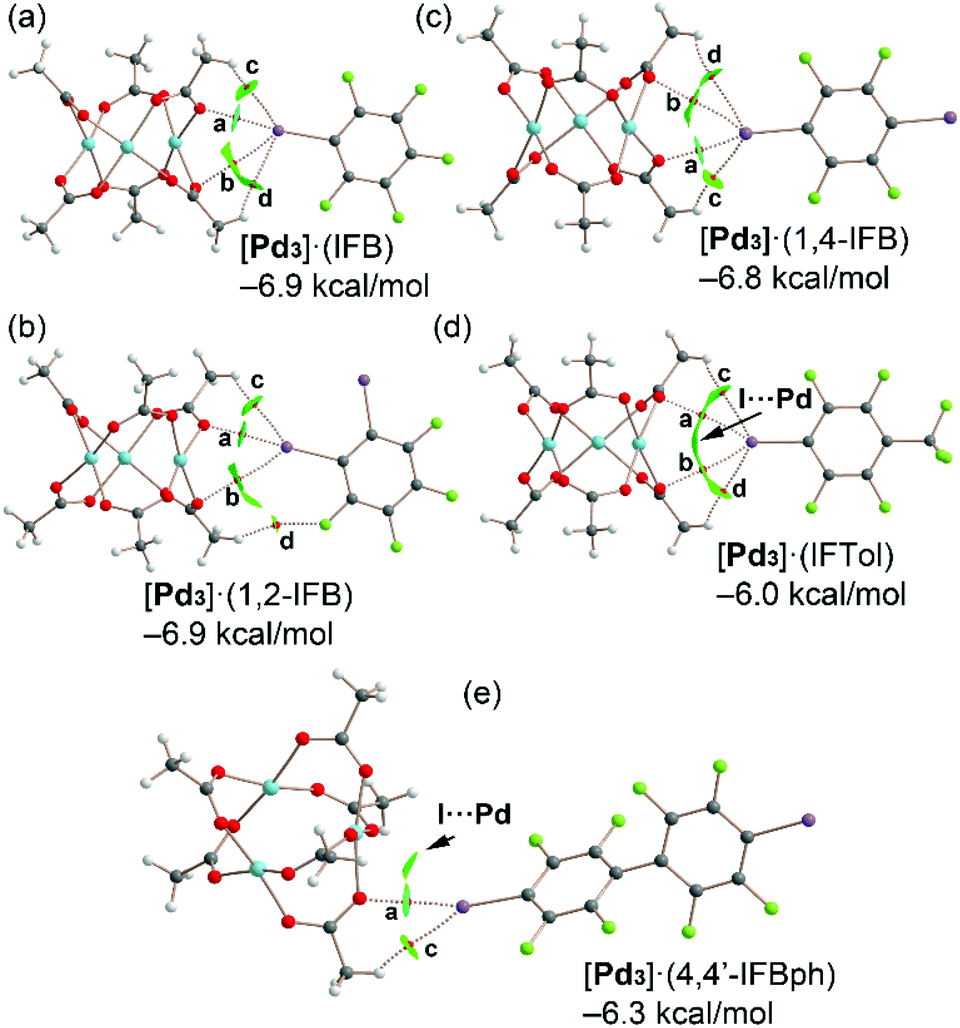 | ||
| Fig. 10 Combined QTAIM and NCIplot analysis of [Pd3]·(IFB) (a), [Pd3]·(1,2-IFB) (b), [Pd3]·(1,4-IFB) (c), [Pd3]·(IFTol) (d), and [Pd3]·(4,4′-IFBph) (e). Bond CPs are represented using red spheres. The color scale for the NCIplot is −0.035 < sign(λ2)ρ < 0.035; |RGD| isosurface 0.5; density cut-off = 0.04 au. Only intermolecular CPs and NCIplot index surfaces are given. The interaction energies of the assemblies computed using the supramolecular approach [ΔE = E(A·B) − E(A) − E(B)] are also indicated. | ||
Apart from the [Pd3]·(4,4′-IFBph) cocrystal, each dimer is characterized by four bond CPs and bond paths that interconnect both coformers. Two of them correspond to I⋯O contacts (CPs labeled as a and b), and two of them correspond to C–H⋯I(F) contacts (CPs labeled as c and d). In [Pd3]·(IFB), [Pd3]·(1,2-IFB), and [Pd3]·(1,4-IFB) dimers, one of the HaBs is directional (CP “a”) and moderately strong, while the other one presents poor directionality and is very weak (CP “b”), as further discussed below.
For cocrystal [Pd3]·(4,4′-IFBph), only two bond CPs and bond paths interconnect the (4,4′-IFBph) arene to cluster [Pd3]: one corresponds to a directional HaB (CP “a”), and the other corresponds to an ancillary HB (CP “c”). The interaction energies are similar for all dimers ranging from −6.0 kcal mol−1 in [Pd3]·(IFTol) to −6.9 kcal mol−1 in [Pd3]·(IFB) and [Pd3]·(1,2-IFB). For cocrystals [Pd3]·(IFTol) and [Pd3]·(4,4′-IFBph), an additional green NCIplot isosurface is located between the I atom and Pd atom, thus suggesting the occurrence of attractive I⋯Pd contacts.
The QTAIM parameters at the bond CPs that characterize the HaBs and HBs in cocrystals analyzed in Fig. 10 are gathered in Table 2. The Lagrangian kinetic [G(r)] energy densities are larger (in absolute value) than the potential energy densities [V(r)] at the bond CPs, thus confirming the noncovalent nature of the I⋯O and H⋯I contacts. Moreover, the Eint individual values estimated using the potential energy predictors for hydrogen bonding and HaB (for details, see section 2.4) are summarized in Table 2, and can be compared with the binding energies determined using the supramolecular approach indicated in Fig. 10. Apart from the [Pd3]·(IFB) system, ∑Eint is smaller than the binding energies, thus suggesting some contribution of long-range van der Waals interactions, as suggested by the extension of some of the green NCIplot isosurfaces, which are not characterized by bond CPs.
In fact, for the [Pd3]·(IFTol) and[Pd3]·(4,4′-IFBph) dimers, the NCIplot index analysis indicates the existence of I⋯Pd contacts, as commented above. In [Pd3]·(IFB), [Pd3]·(1,2-IFB), and [Pd3]·(1,4-IFB), the combined QTAIM/NCIplot analysis shows the existence of a moderately strong HaB (CP labeled as “a”) and a secondary and weak HaB (CP labeled as “b”), as corroborated by their Eint individual values (Table 2).
This configuration is energetically more favored than the binding mode observed in [Pd3]·(IFTol), where the HaB is perfectly bifurcated and each HaB only accounts for −1.0 kcal mol−1. The individual interaction energies of the HBs are significantly weaker than those at the HaBs, thus demonstrating that the formation of the dimers shown in Fig. 9 is clearly dominated by the HaBs. ∑Eint is generally smaller than the interaction energies given in Fig. 10, thus suggesting the existence of secondary interactions, such as I⋯Pd contacts that are not revealed by the distribution of bond CPs. In contrast, they are disclosed by the NCIplot surfaces, especially in [Pd3]·(IFTol) and[Pd3]·(4,4′-IFBph) cocrystals.
The QTAIM and NCIPlot index analyses of the LP⋯π dimers are given in Fig. 11. It can be observed that the dimerization energies are significantly larger than those of the σ-hole complexes (ranging from −12.6 to −15.6 kcal mol−1). This reveals that the combination of LP⋯π-hole interactions, CH⋯F,I contacts and dispersion forces in the assemblies of Fig. 11 is more stabilizing than the combination of HBs and HaBs in the assemblies of Fig. 10. Therefore, this intricate collection of interactions is very important in the wrapping mechanism of the inorganic cluster by the organic rings.
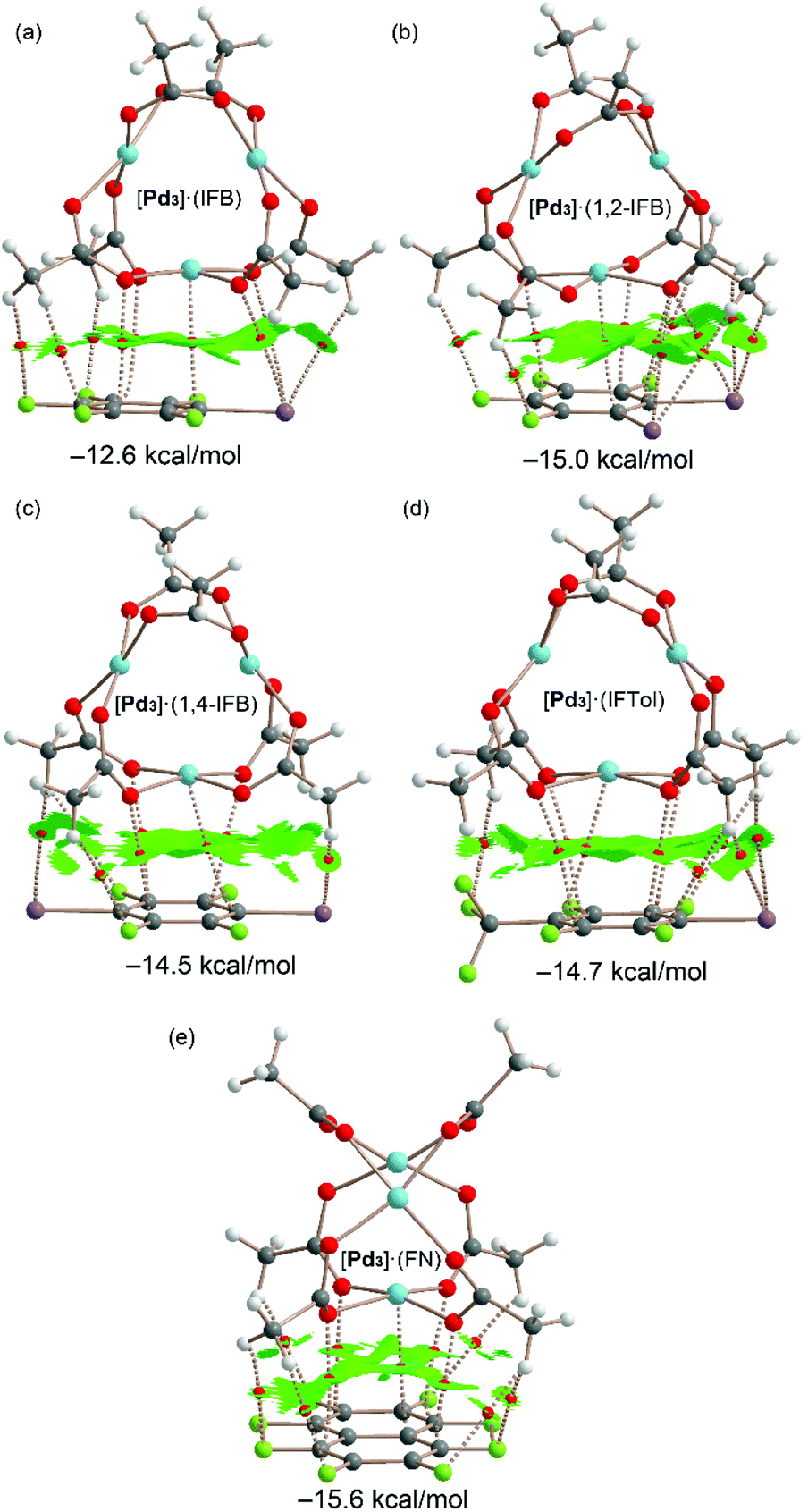 | ||
| Fig. 11 Combined QTAIM and NCIplot analysis of [Pd3]·(IFB) (a), [Pd3]·(1,2-IFB) (b), [Pd3]·(1,4-IFB) (c), [Pd3]·(IFTol) (d), and [Pd3]·(FN) (e). Bond CPs are represented using red spheres. The color scale for the NCIplot is −0.035 < sign(λ2)ρ < 0.035; |RGD| isosurface 0.5; density cut-off = 0.04 au. Only intermolecular CPs and NCIplot index surfaces are given. The interaction energies of the assemblies computed using the supramolecular approach [ΔE = E(A·B) − E(A) − E(B)] are also indicated. | ||
Possible cooperativity effects between both σ-hole and π-hole interactions have been analyzed (see ESI, section S2.1 and Table S2†), revealing that they are negligible. Moreover, the involvement of the dz2-PdII orbital in this type of complex has been further confirmed using NBO calculations, as described in the ESI (section S2.2†) evidencing a LP(Pd) → π* orbital donor–acceptor interaction that stabilizes the dimer with E(2) = 2.22 kcal mol−1 in [Pd3]·(IFB) that has been used as a representative compound.
The largest binding energy corresponds to the [Pd3]·(FN) dimer, in agreement with the more extended π-acidic aromatic surface of FN and more positive MEP value at the π-hole. These dimers are characterized by an intricate network of bond CPs and bond paths connecting both counterparts. A common feature in all dimers is the existence of a bond CP and bond path connecting one Pd atom of cluster [Pd3] to one carbon atom of the aromatic ring. This nicely confirms the participation of the Pd atom, in addition to the O atoms, in the LP⋯π-hole interaction and wrapping phenomenon.
It is noteworthy, in view of the last statement, that we previously reported that some positively charged d8-metal centers (e.g., NiII, PdII, PtII, and RhI) exhibit dz2-nucleophilicity and could function as either π-holes27,28 or HaB (see recent ref. 50 and references therein) acceptors. It is clear from this work that palladium(II) centers surrounded by other nucleophilic centers (in particular, O atoms) compose an integrated five-center PdO4 nucleophile and can potentially act as an extended square-planar inorganic nucleophile toward planar organic π-hole donors; this interaction should lead to highly polar circular inorganic–organic stacking.
All dimers present large NCIplot isosurfaces located between the π-acidic surfaces and one face of the [Pd3] cluster, in line with the large interaction energies and the ability of the organic rings used in this study to wrap the inorganic cluster. In addition to the π-surface, the NCIplot isosurfaces also embrace the substituents of the arene and CH3 groups of the cluster. In some cocrystals, the five atoms of the PdO4 core are connected via five bond CPs and bond paths to the electron-deficient arene, also supporting the nucleophilic role of the entire PdO4 and its tendency to form highly polar inorganic–organic stacking.
4. Conclusions
We observed π-stacking involving metal-containing inorganic planes with arenes to give mixed inorganic–organic systems. In five out of six studied cocrystal structures, the aromatic rings couple with cluster [Pd3] via LP–π-(ArF)-hole interactions. The iodine-containing arenes, ArFI, are additionally involved in halogen bonding with carboxylate O centers, but organic–inorganic stacking still remains the structure-determining interaction. In the stacking, the electron-rich PdO4 plane behaves as a five-center nucleophile providing oxygen LPs in addition to the dz2-PdII orbital; this plane complements the π-acidic surface of the arenes, affording highly polar circular stacking, where organics wrap inorganics.Our observations are relevant to the chemistry of Pd-containing oxometallates (also featuring the PdO4 entity). These species are of significant interest in view of their useful heterogeneous and homogeneous catalytic properties combined with solubility in various solvents, oxidative and thermal stabilities, and tunable redox activity. The square-planar PdO4 entities of oxopalladates enable their applications in catalytic CO51 and CH452,53 oxidation and in the esterification of CO to dimethyl carbonate.54
Other complexes with PdO4 planar groups (e.g., [Pd(acac)2], [Pd3(OAc)6], or [Pd(OAc)2]n) also function as ubiquitous catalysts and precatalysts for cross-coupling reactions, C–H functionalizations, carbonylation reactions, and reductive amination of aldehydes and ketones.55 It is generally believed that the first step of all these processes is substrate adsorption on Pd-spaces. In particular, for C–H activation (e.g., Fujiwara–Moritani reaction), the initial step of the catalytic cycle includes the generation of a σ/π-complex with an arene.56
In this study, we found that electron-deficient arenes can be noncovalently bound to the PdO4 inorganic space to give stable organic–inorganic associates via highly polar LP–π-hole interaction(s). We assume that the previous studies on the mechanisms of the catalytic conversion of electron-rich and electron-neutral arenes by Pd-containing oxometallates should be thoroughly revisited and that the possibility of the generation of the kinetically labile “PdO4-arene” associate should be taken into account in future studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is an integration of two diverse projects of the Russian Science Foundation grants 19-13-00338 (crystal engineering study) and 21-73-10030 (molecular design of π- and σ-hole donors). XRD experiments were performed using the equipment of the shared experimental facilities supported by N. S. Kurnakov Institute of General and Inorganic Chemistry RAS. BG and AF acknowledge the MICIU/AEI from Spain for financial support of the computational study (project numbers CTQ2017-85821-R and PID2020-115637GB-I00, FEDER funds). Prof. Dr M. N. Sokolov is thanked for valuable discussions.Notes and references
- A. R. G. Smith, P. L. Burn and B. J. Powell, Spin–Orbit Coupling in Phosphorescent Iridium(III) Complexes, ChemPhysChem, 2011, 12, 2429–2438 CrossRef CAS PubMed.
- C. B. Aakeröy and A. S. Sinha, Co-crystals, in Monographs in Supramolecular Chemistry, 2018, p. P001 Search PubMed.
- S. Scheiner, M. Michalczyk and W. Zierkiewicz, Coordination of anions by noncovalently bonded σ-hole ligands, Coord. Chem. Rev., 2020, 405, 213136 CrossRef CAS.
- K. T. Mahmudov, A. V. Gurbanov, F. I. Guseinov and M. F. C. Guedes da Silva, Noncovalent interactions in metal complex catalysis, Coord. Chem. Rev., 2019, 387, 32–46 CrossRef CAS.
- A. J. Neel, M. J. Hilton, M. S. Sigman and F. D. Toste, Exploiting non-covalent π interactions for catalyst design, Nature, 2017, 543, 637–646 CrossRef CAS PubMed.
- K. T. Mahmudov, M. N. Kopylovich, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Noncovalent Interactions in Catalysis, The Royal Society of Chemistry, 2019 Search PubMed.
- R. S. J. Proctor, A. C. Colgan and R. J. Phipps, Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates, Nat. Chem., 2020, 12, 990–1004 CrossRef PubMed.
- R. Robidas, D. L. Reinhard, C. Y. Legault and S. M. Huber, Iodine(III)-Based Halogen Bond Donors: Properties and Applications, Chem. Rec., 2021, 21, 1912–1927 CrossRef CAS PubMed.
- M. Breugst, D. von der Heiden and J. Schmauck, Novel Noncovalent Interactions in Catalysis: A Focus on Halogen, Chalcogen, and Anion-π Bonding, Synthesis, 2017, 49, 3224–3236 CrossRef CAS.
- R. L. Sutar and S. M. Huber, Catalysis of Organic Reactions through Halogen Bonding, ACS Catal., 2019, 9, 9622–9639 CrossRef CAS.
- L. Vogel, P. Wonner and S. M. Huber, Chalcogen Bonding: An Overview, Angew. Chem., Int. Ed., 2019, 58, 1880–1891 CrossRef CAS PubMed.
- I. A. Rather, S. A. Wagay and R. Ali, Emergence of anion-π interactions: The land of opportunity in supramolecular chemistry and beyond, Coord. Chem. Rev., 2020, 415, 213327 CrossRef CAS.
- A. S. Mahadevi and G. N. Sastry, Cation−π Interaction: Its Role and Relevance in Chemistry, Biology, and Material Science, Chem. Rev., 2013, 113, 2100–2138 CrossRef CAS PubMed.
- I. Caracelli, J. Zukerman-Schpector, I. Haiduc and E. R. T. Tiekink, Main group metal lone-pair⋯π(arene) interactions: a new bonding mode for supramolecular associations, CrystEngComm, 2016, 18, 6960–6978 RSC.
- E. R. T. Tiekink, Supramolecular assembly based on “emerging” intermolecular interactions of particular interest to coordination chemists, Coord. Chem. Rev., 2017, 345, 209–228 CrossRef CAS.
- T. J. Mooibroek, P. Gamez and J. Reedijk, Lone pair–π interactions: a new supramolecular bond?, CrystEngComm, 2008, 10, 1501–1515 RSC.
- K. I. Tugashov, S. M. Yunusov, E. S. Kalyuzhnaya, F. M. Dolgushin, A. S. Peregudov, Z. S. Klemenkova, M. K. Minacheva, I. A. Tikhonova and V. B. Shur, Coordination Chemistry of Anticrowns. Synthesis and Structures of Complexes of the Perfluorinated Three-Mercury Anticrown (o-C6F4Hg)3 with Monohalogenobenzenes, Eur. J. Inorg. Chem., 2019, 2019, 5018–5024 CrossRef CAS.
- K. I. Tugashov, S. M. Yunusov, E. S. Kalyuzhnaya, F. M. Dolgushin, I. A. Tikhonova and V. B. Shur, Coordination chemistry of anticrowns. An unusual complex of perfluorinated three-mercury anticrown (o-C6F4Hg)3 with p-bromotoluene and 1,2-dichloroethane, Russ. Chem. Bull., 2020, 69, 1195–1198 CrossRef CAS.
- M. Fleischmann, J. S. Jones, G. Balázs, F. P. Gabbaï and M. Scheer, Supramolecular adducts based on weak interactions between the trimeric Lewis acid complex (perfluoro-ortho-phenylene)mercury and polypnictogen complexes, Dalton Trans., 2016, 45, 13742–13749 RSC.
- M. Tsunoda, M. Fleischmann, J. S. Jones, N. Bhuvanesh, M. Scheer and F. P. Gabbaï, Supramolecular aggregation of Ni(salen) with (C6F5)2Hg and [o-C6F4Hg]3, Dalton Trans., 2016, 45, 5045–5051 RSC.
- M. Fleischmann, J. S. Jones, F. P. Gabbaï and M. Scheer, A comparative study of the coordination behavior of cyclo-P5 and cyclo-As5 ligand complexes towards the trinuclear Lewis acid complex (perfluoro-ortho-phenylene)mercury, Chem. Sci., 2015, 6, 132–139 RSC.
- M. Fleischmann, C. Heindl, M. Seidl, G. Balázs, A. V. Virovets, E. V. Peresypkina, M. Tsunoda, F. P. Gabbaï and M. Scheer, Discrete and Extended Supersandwich Structures Based on Weak Interactions between Phosphorus and Mercury, Angew. Chem., Int. Ed., 2012, 51, 9918–9921 CrossRef CAS PubMed.
- T. J. Wedge and M. F. Hawthorne, Multidentate carborane-containing Lewis acids and their chemistry: mercuracarborands, Coord. Chem. Rev., 2003, 240, 111–128 CrossRef CAS.
- A. A. Mohamed, M. A. Rawashdeh-Omary, M. A. Omary and J. J. P. Fackler, External heavy-atom effect of gold in a supramolecular acid–base π stack, Dalton Trans., 2005, 2597–2602 RSC.
- O. Elbjeirami, M. D. Rashdan, V. Nesterov and M. A. Rawashdeh-Omary, Structure and luminescence properties of a well-known macrometallocyclic trinuclear Au(i) complex and its adduct with a perfluorinated fluorophore showing cooperative anisotropic supramolecular interactions, Dalton Trans., 2010, 39, 9465–9468 RSC.
- M. A. Rawashdeh-Omary, M. A. Omary, J. P. Fackler, R. Galassi, B. R. Pietroni and A. Burini, Chemistry and Optoelectronic Properties of Stacked Supramolecular Entities of Trinuclear Gold(I) Complexes Sandwiching Small Organic Acids, J. Am. Chem. Soc., 2001, 123, 9689–9691 CrossRef CAS PubMed.
- A. V. Rozhkov, M. A. Krykova, D. M. Ivanov, A. S. Novikov, A. A. Sinelshchikova, M. V. Volostnykh, M. A. Konovalov, M. S. Grigoriev, Y. G. Gorbunova and V. Y. Kukushkin, Reverse Arene Sandwich Structures Based upon π-Hole⋯[MII] (d8M=Pt, Pd) Interactions, where Positively Charged Metal Centers Play the Role of a Nucleophile, Angew. Chem., Int. Ed., 2019, 58, 4164–4168 CrossRef CAS PubMed.
- S. V. Baykov, S. I. Filimonov, A. V. Rozhkov, A. S. Novikov, I. V. Ananyev, D. M. Ivanov and V. Y. Kukushkin, Reverse Sandwich Structures from Interplay between Lone Pair−π-Hole Atom-Directed C⋯dz2[M] and Halogen Bond Interactions, Cryst. Growth Des., 2020, 20, 995–1008 CrossRef CAS.
- A. V. Rozhkov, I. V. Ananyev, R. M. Gomila, A. Frontera and V. Y. Kukushkin, π-Hole⋯dz2[PtII] Interactions with Electron-Deficient Arenes Enhance the Phosphorescence of PtII-Based Luminophores, Inorg. Chem., 2020, 59, 9308–9314 CrossRef CAS PubMed.
- M. M. Olmstead, A. S. Ginwalla, B. C. Noll, D. S. Tinti and A. L. Balch, Supramolecular Aggregation of Pd6Cl12, a Cluster of Comparable Size to a Fullerene, with Aromatic Donors and with C60, J. Am. Chem. Soc., 1996, 118, 7737–7745 CrossRef CAS.
- S. N. Spisak, A. S. Filatov and M. A. Petrukhina, Palladium π-adduct of corannulene, J. Organomet. Chem., 2011, 696, 1228–1231 CrossRef CAS.
- M. M. Olmstead, P.-p. Wei and A. L. Balch, Solid-State Architectures of Aggregates of the Cubic Cluster [Pd6Cl12] with Polynuclear Aromatic Hydrocarbons, Chem. – Eur. J., 1999, 5, 3136–3142 CrossRef CAS.
- M. M. Olmstead, P.-p. Wei, A. S. Ginwalla and A. L. Balch, Bis(benzonitrile)palladium(II) Dihalides: Structures and Cocrystallization of the Cubic Cluster Pd6Cl12 with (E)-Stilbene and with Bis(benzonitrile)palladium(II) Dichloride, Inorg. Chem., 2000, 39, 4555–4559 CrossRef CAS.
- M. Gorlov, A. Fischer and L. Kloo, Pt6Cl12·(1,2,4-C6H3Cl3), a Structurally Characterized Cocrystallization Product of Pt6Cl12, Z. Anorg. Allg. Chem., 2005, 631, 2973–2975 CrossRef CAS.
- Y. V. Torubaev and I. V. Skabitsky, A new supramolecular heterosynthon [C–I⋯O·C(carboxylate)] at work: engineering copper acetate cocrystals, CrystEngComm, 2020, 22, 6661–6673 RSC.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Definition of the halogen bond (IUPAC Recommendations 2013), Pure Appl. Chem., 2013, 85, 1711–1713 CAS.
- M. Egli and S. Sarkhel, Lone Pair−Aromatic Interactions: To Stabilize or Not to Stabilize, Acc. Chem. Res., 2007, 40, 197–205 CrossRef CAS PubMed.
- A. V. Rozhkov, A. A. Eliseeva, S. V. Baykov, B. Galmés, A. Frontera and V. Y. Kukushkin, One-Pot Route to X-perfluoroarenes (X=Br, I) Based on FeIII-Assisted C–F Functionalization and Utilization of These Arenes as Building Blocks for Crystal Engineering Involving Halogen Bonding, Cryst. Growth Des., 2020, 20, 5908–5921 CrossRef CAS.
- V. I. Bakhmutov, J. F. Berry, F. A. Cotton, S. Ibragimov and C. A. Murillo, Non-trivial behavior of palladium(II) acetate, Dalton Trans., 2005, 1989–1992 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Energy frameworks: insights into interaction anisotropy and the mechanical properties of molecular crystals, Chem. Commun., 2015, 51, 3735–3738 RSC.
- E. A. Katlenok, M. Haukka, O. V. Levin, A. Frontera and V. Y. Kukushkin, Supramolecular Assembly of Metal Complexes by (Aryl)I⋯d [PtII] Halogen Bonds, Chem. – Eur. J., 2020, 26, 7692–7701 CrossRef CAS PubMed.
- Y. V. Torubaev and I. V. Skabitsky, The energy frameworks of aufbau synthon modules in 4-cyanopyridine co-crystals, CrystEngComm, 2019, 21, 7057–7068 RSC.
- Y. V. Torubaev, D. K. Rai, I. V. Skabitsky, S. Pakhira and A. Dmitrienko, Energy framework approach to the supramolecular reactions: interplay of the secondary bonding interaction in Ph 2 E 2 (E=Se, Te)/p -I-C 6 F 4 -I co-crystals, New J. Chem., 2019, 43, 7941–7949 RSC.
- A. C. Skapski and M. L. Smart, The crystal structure of trimeric palladium(II) acetate, J. Chem. Soc. D, 1970, 658b–6659 RSC.
- A. A. Eliseeva, D. M. Ivanov, A. S. Novikov and V. Y. Kukushkin, Recognition of the π-hole donor ability of iodopentafluorobenzene – a conventional σ-hole donor for crystal engineering involving halogen bonding, CrystEngComm, 2019, 21, 616–628 RSC.
- A. S. Novikov, D. M. Ivanov, Z. M. Bikbaeva, N. A. Bokach and V. Y. Kukushkin, Noncovalent Interactions Involving Iodofluorobenzenes: The Interplay of Halogen Bonding and Weak lp(O)⋯π-Holearene Interactions, Cryst. Growth Des., 2018, 18, 7641–7654 CrossRef CAS.
- S. Alvarez, A cartography of the van der Waals territories, Dalton Trans., 2013, 42, 8617–8636 RSC.
- A. A. Eliseeva, D. M. Ivanov, A. V. Rozhkov, I. V. Ananyev, A. Frontera and V. Y. Kukushkin, Bifurcated Halogen Bonding Involving Two Rhodium(I) Centers as an Integrated σ-Hole Acceptor, JACS Au, 2021, 1, 354–361 CrossRef CAS PubMed.
- E. J. Peterson, A. T. DeLaRiva, S. Lin, R. S. Johnson, H. Guo, J. T. Miller, J. Hun Kwak, C. H. F. Peden, B. Kiefer, L. F. Allard, F. H. Ribeiro and A. K. Datye, Low-temperature carbon monoxide oxidation catalysed by regenerable atomically dispersed palladium on alumina, Nat. Commun., 2014, 5, 4885 CrossRef CAS PubMed.
- S. Coussi, A. Gayen, M. F. Camellone, M. Boaro, J. Llorca, S. Fabris and A. Trovarelli, Nanofaceted Pd−O Sites in Pd−Ce Surface Superstructures: Enhanced Activity in Catalytic Combustion of Methane, Angew. Chem., Int. Ed., 2009, 48, 8481–8484 CrossRef PubMed.
- S. Li, Y. Zhang, J. Shi, G. Zhu, Y. Xie, Z. Li, R. Wang and H. Zhu, Catalytic Performance of Palladium Supported on Sheaf-Like Ceria in the Lean Methane Combustion, Nanomaterials, 2020, 10, 31 CrossRef CAS PubMed.
- H.-Z. Tan, Z.-N. Chen, Z.-N. Xu, J. Sun, Z.-Q. Wang, R. Si, W. Zhuang and G.-C. Guo, Synthesis of High-Performance and High-Stability Pd(II)/NaY Catalyst for CO Direct Selective Conversion to Dimethyl Carbonate by Rational Design, ACS Catal., 2019, 9, 3595–3603 CrossRef CAS.
- E. Negishi and A. de Meijere, Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley, 2003 Search PubMed.
- X. Chen, K. M. Engle, D.-H. Wang and J.-Q. Yu, Palladium(II)-Catalyzed C−H Activation/C−C Cross-Coupling Reactions: Versatility and Practicality, Angew. Chem., Int. Ed., 2009, 48, 5094–5115 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2082583–2082588. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1qi01067k |
| This journal is © the Partner Organisations 2021 |