
Ultrafast photoinduced electron transfer in conjugated polyelectrolyte–acceptor ion pair complexes†
Pradeepkumar
Jagadesan
 ,
Silvano R.
Valandro
and
Kirk S.
Schanze
*
,
Silvano R.
Valandro
and
Kirk S.
Schanze
*
Department of Chemistry, University of Texas, San Antonio, One UTSA Circle, San Antonio, TX 78249, USA. E-mail: kirk.schanze@utsa.edu
First published on 18th September 2020
Abstract
Conjugated polyelectrolytes (CPEs) are a promising class of materials for photovoltaic and sensing applications due to their ability to undergo efficient photoinduced electron transfer (ET). Although water-soluble poly(p-phenylene ethynylene) (PPE) polyelectrolytes have been synthesized for various applications, no reports have clearly revealed the dynamics of the photoinduced ET process and charge transfer state of PPE-type CPE/electron acceptor systems. Herein, we investigated photoinduced ET from a water-soluble PPE based CPE functionalized with alkyl sulfonate (R–SO3−) as side groups to three cationic electron acceptors. Steady-state absorption studies showed a bathochromic shift of the absorption band for all CPE/acceptor mixtures compared to CPE as a result of the suppression of the conformational changes in the polymer caused by the ion–pair interaction. Stern–Volmer (SV) fluorescence quenching studies revealed that all of the cationic electron acceptors efficiently quenched the fluorescence of CPE with SV constants (KSV) in excess of 106 M−1. Picosecond transient absorption (TA) studies of a CPE/methyl viologen mixture revealed the absorption feature of the cation radical state (CPE˙+) of the polyelectrolyte. Kinetic measurements established that the forward electron transfer in the polyelectrolyte/acceptor mixtures occurs with rate constant kf > 1 × 1012 s−1 and the return ET occurs with kb = 3 × 1011 s−1. We also demonstrated that the electron transfer between a PPE-type polyelectrolyte and an acceptor in their assemblies could be modulated by a supramolecular approach, which revealed that the nature of the acceptor–supramolecule host–guest complexes dictates the kinetics of the electron transfer process.
Introduction
Conjugated polyelectrolytes (CPEs) are conjugated polymers featuring ionic side groups. The π-conjugated backbone in CPEs endows them with efficient light harvesting and light emitting properties, while the ionic sides chains render the polymers water-soluble.1–4 Wudl, Heeger and coworkers reported the first conjugated polyelectrolyte in 1987.5 Ever since their inception, a wide range of CPEs with diverse backbone structures and side chains have been synthesized for a spectrum of applications such as organic light emitting diodes,6–8 organic photovoltaics,9,10 drug delivery,11 antimicrobials12–15 and bioimaging.16 Moreover, the remarkable photophysical properties of CPEs such as high fluorescence quantum yields and amplified quenching make them intriguing entities for the detection and sensing of ions,17–19 molecules20–22 and biomolecules.23–25 The strikingly sensitive response in the fluorescence quenching of CPEs towards various analytes at ultralow concentrations is caused by the spatial delocalization and mobility of excitons in the polymer as a result of electron/energy transfer from the CPE to the quencher. The activity in this area has been propelled since the papers published by Swager and co-workers in 1995,26,27 in which they presented the first example of amplified quenching of a poly(p-phenylene ethynylene) (PPE) conjugated polymer by dimethyl viologen (MV2+) with a Stern–Volmer quenching constant (KSV) as high as 105 M−1. The amplified quenching effect was attributed to an effective photoinduced electron transfer (ET) process, in which a single viologen is sufficient to trap nearly the total exciton energy along one polymer chain.28Whitten, Wudl and co-workers first reported the amplified quenching effect in a water-soluble conjugated polyelectrolyte, in which they demonstrated that the fluorescence of a poly(p-phenylene vinylene) (PPV) polyelectrolyte appended with sulfonate (–SO3−) functionality was quenched by MV2+ with a KSV in excess of 107 M−1.29 Following this, Tan et al. observed that a PPE-type polyelectrolyte appended with alkyl sulfonate groups exhibited amplified quenching upon MV2+ addition in both water and methanol, with KSV values on the order of 107 M−1.30 The enhanced quenching exhibited by CPEs in comparison to Swager's neutral conjugated polymers was attributed to the ultrafast exciton migration along the polymer chain to the quencher as a result of the strong complex formation between the polyelectrolyte and quencher through ion-pairing and hydrophobic interactions.
During the past two decades, significant efforts have been devoted to the design and study of a range of CPEs for chemo- and biosensing applications that rely on amplified fluorescence quenching as the predominant signal transduction method. Among the polyelectrolytes, PPE-type CPEs have attracted considerable attention for developing fluorescence-based assays due to their rigid rod structure, high water solubility, fluorescence quantum yield and biocompatibility.31–33 The fluorescence quenching of CPEs occurs via one of three pathways, which include energy34,35 or electron transfer23,36 from the polyelectrolyte to the quencher, or through inter-chain aggregation caused quenching (ACQ).19,37 Fluorescence quenching via energy transfer can more often be easily distinguished from the others through the observation of sensitized emission from the quencher. In contrast, fluorescence quenching by an ET pathway involves the generation of the polyelectrolyte cation radical (positive polaron) and the reduced quencher from the excited state of the polymer. Subsequently, the return ET occurs non-radiatively to regenerate the ground state polyelectrolyte and acceptor. Owing to the very short lifetimes (∼ps time scale) of the polaron states, ultrafast spectroscopy is required for detection and characterization of the intermediate charge transfer (CT) state [CPE˙+, A−]. To the best of our knowledge, ultrafast transient absorption spectroscopic studies to understand the photoinduced ET process and intermediate CT state of PPE-based CPE and quencher mixtures have not been previously reported. Moreover, understanding the ET mechanism and dynamics and developing strategies to modulate the process are of paramount importance, especially for exploiting CPEs for sensing and photovoltaic applications.
The present investigation seeks to study the mechanism of quenching of a water-soluble PPE-type CPE (CPE) functionalized with alkyl sulfonate (R–SO3−) by a set of three cationic electron acceptors (MV2+, NDI2+ and NMI+, Chart 1) with the goal to observe the CT state, its spectral properties, and the dynamics for forward and return ET. For all three CPE–acceptor ion-pair complexes, the absorption bands were substantially red-shifted and their intensities are comparable to the primary absorption of CPE. Addition of NDI2+ and NMI+ to CPE resulted in amplified fluorescence quenching with KSV ∼ 107 M−1. Picosecond transient absorption (TA) spectroscopy of the CPE/MV2+ complex in water revealed the existence of the CPE cation radical (CPE˙+) state. Moreover, the observation of the absorption of reduced NDI2+ in the TA measurements of the CPE/NDI2+ complex confirms the photoinduced ET process between CPE and NDI2+. We have also shown that the ET dynamics in CPE/acceptor mixtures could be modulated using the supramolecule host cucurbit[7]uril (CB7, Chart 1).
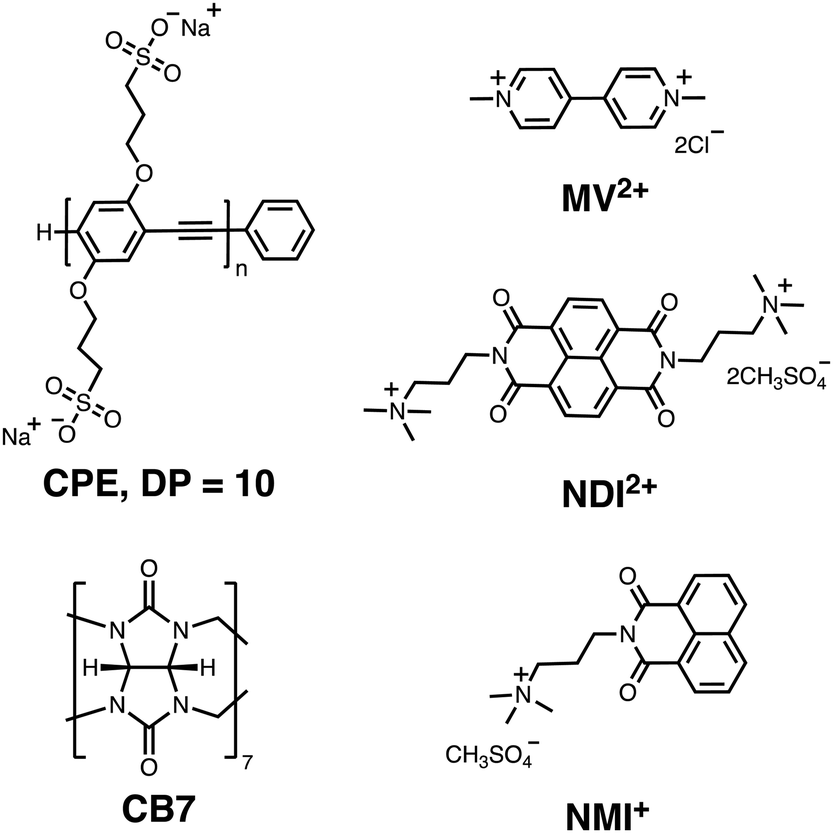 | ||
| Chart 1 Structure of CPE, MV2+, NDI2+, NMI+ and CB7. | ||
Results
Structures and thermodynamics of electron transfer
Our primary goal in this work is to elucidate the photoinduced ET mechanism between a water-soluble poly(p-phenylene ethynylene) conjugated polyelectrolyte (CPE) and cationic quenchers. Conjugated polyelectrolyte CPE featuring alkyl sulfonate (R–SO3−) was synthesized via a chain-growth polymerization method following an earlier report, and its degree of polymerization (n) was determined to be ∼10 with a polydispersity index (PDI) ∼1.2.38 Methyl viologen (MV2+) and cationic derivatives naphthalene diimide (NDI2+) and naphthalene monoimide (NMI+) were chosen as acceptors because of their capability to associate with the anionic CPE and their strong electron acceptor character. All of the quenchers are mono- or di-cations and therefore are expected to form ion-pair complexes with the anionic polyelectrolyte. Previous work has also shown that polyvalent cations associate more strongly than mono-cations, and in some cases polyvalent ions can induce the formation of aggregates.35,39,40In order to facilitate the discussion below, Fig. 1 gives a simplified excited state scheme for the ion pair complex between CPE and an acceptor, [CPE, A]. Initial excitation produces the singlet excited state, [*CPE, A], and this can be quenched by forward ET to produce a CT state, represented as [CPE+, A−]. This state then relaxes back to the ground state by charge recombination. There is also the possibility that the separation distance between the polymer and reduced acceptor could increase after forward ET occurs, giving rise to a charge-separated state, [CPE+//A−]. The thermodynamic driving force for forward ET and charge recombination (ΔGf and ΔGb, respectively) can be determined from the reduction potentials of the acceptors, and the oxidation potential and the excited state energy of CPE. The relevant potentials are listed in Table 1, along with the excited state oxidation potential of CPE, which is computed from the polymer's ground state oxidation potential, and the singlet excited state energy, E(*CPE/CPE+) = E(CPE/CPE+) − E(*CPE). From the data in Table 1, it is clear that forward ET is very exothermic for all of the acceptors, ranging from −0.5 eV for NMI+ to −1.34 eV for MV2+. Charge recombination is even more exothermic, ranging from −1.36 eV for MV2+ to −2.2 eV for NMI+.41 Given the large driving force for all of the ET processes in Fig. 1, it is predicted, assuming that the separation distance in the ion pair complexes is small (<5 Å), that both forward and return ET will occur rapidly (>109 s−1). This prediction is based on the assumption that the ET reactions will follow a Marcus dependence on the driving force and that the reorganization energy is in the range 0.5–1.0 eV.42
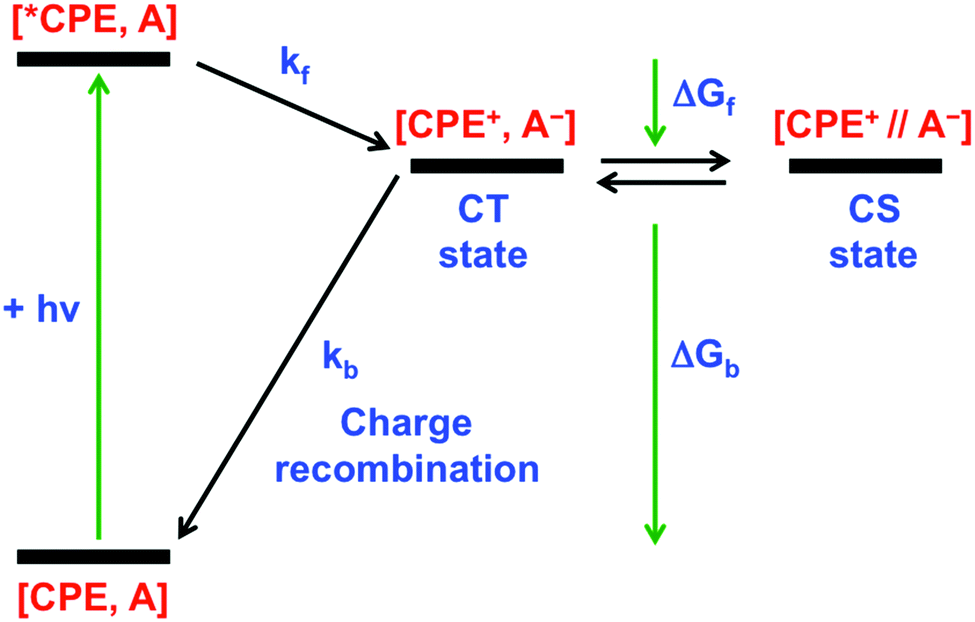 | ||
| Fig. 1 Energy diagram for photoinduced ET and charge recombination in CPE–acceptor ion-pair complexes. | ||
Steady state absorption and fluorescence spectroscopy
The steady-state absorption and fluorescence spectra of CPE and the corresponding complexes with the acceptors were studied in water as a solvent. The fluorescence spectra of the complexes were recorded by exciting at the absorption maximum (∼420 nm) of CPE, where the acceptors do not absorb (Fig. S1, ESI†).47Fig. 2 illustrates the changes in the absorption and fluorescence spectra of CPE in the presence of MV2+ in water. Addition of MV2+ (0–7.5 μM) to 5 μM (calculated based on the polymer repeat unit, PRU) CPE resulted in the significant red-shift of the absorption band, which is accompanied by a shoulder peak around 480 nm. The changes are consistent with the spectral changes observed for a CPE and 1,3-propane diammonium salt mixture.48 This indicates that the association of MV2+ with CPE induces a degree of rigidity to the polymer, restricting its ability to undergo conformational changes, which results in the extension of the conjugation length, which causes the bathochromic shift.35 Moreover, the apparent color change (yellow to brown) observed for the CPE solution upon MV2+ addition (Fig. S2, ESI†) suggests that the absorption spectrum of the CPE/MV2+ complex may have some contribution from charge transfer absorption. The weak shoulder peak that emerges around 480 nm is attributed to the absorption of CPE aggregates induced by MV2+.30,31,48 The change in CPE absorption upon MV2+ addition is accompanied by a substantial decrease in the fluorescence intensity with KSV ∼ 7.1 × 106 M−1.38 | ||
| Fig. 2 (a) Absorption and (b) fluorescence spectra (λex = 410 nm) of CPE (5 μM, PRU) in water with varying concentrations of MV2+ as indicated in the figure. | ||
The absorption and fluorescence spectra of CPE with the addition of NDI2+ and NMI+ are shown in Fig. 3. As seen in Fig. 3a and c, the absorbance bands red-shift considerably as a function of the acceptor concentration (0–10 μM). For both imide acceptors, the bathochromic shifts of the absorption peaks in the mixtures are more significant compared to CPE/MV2+, possibly due to the contribution from CT absorption (Fig. S2, ESI†). The absorption bands observed in the 330–400 nm region in Fig. 3a and c correspond to the absorption features of NDI2+ and NMI+ (Fig. S1, ESI†), respectively. Moreover, addition of NDI2+ and NMI+ to CPE also resulted in significant quenching of the CPE fluorescence (Fig. 3b and d) with KSV ∼ 7.1 × 106 M−1 and 5.5 × 106 M−1, respectively (Fig. S3, ESI†). For all of the samples, the Stern–Volmer plots are approximately linear at low concentration of the quencher but they curve upward at higher concentrations. The KSV values were estimated by extrapolating the linear region at low concentration. Note the subtle effect that the intensity ratio of the 0–0/0–1 vibronic bands changes as a function of quencher concentration, depending on whether the quencher is NDI2+ or NMI+ (Fig. 3b and d). This could be caused by the different charges on the quenchers, which leads to different ion-pair/aggregation interactions in the CPE/quencher complexes.
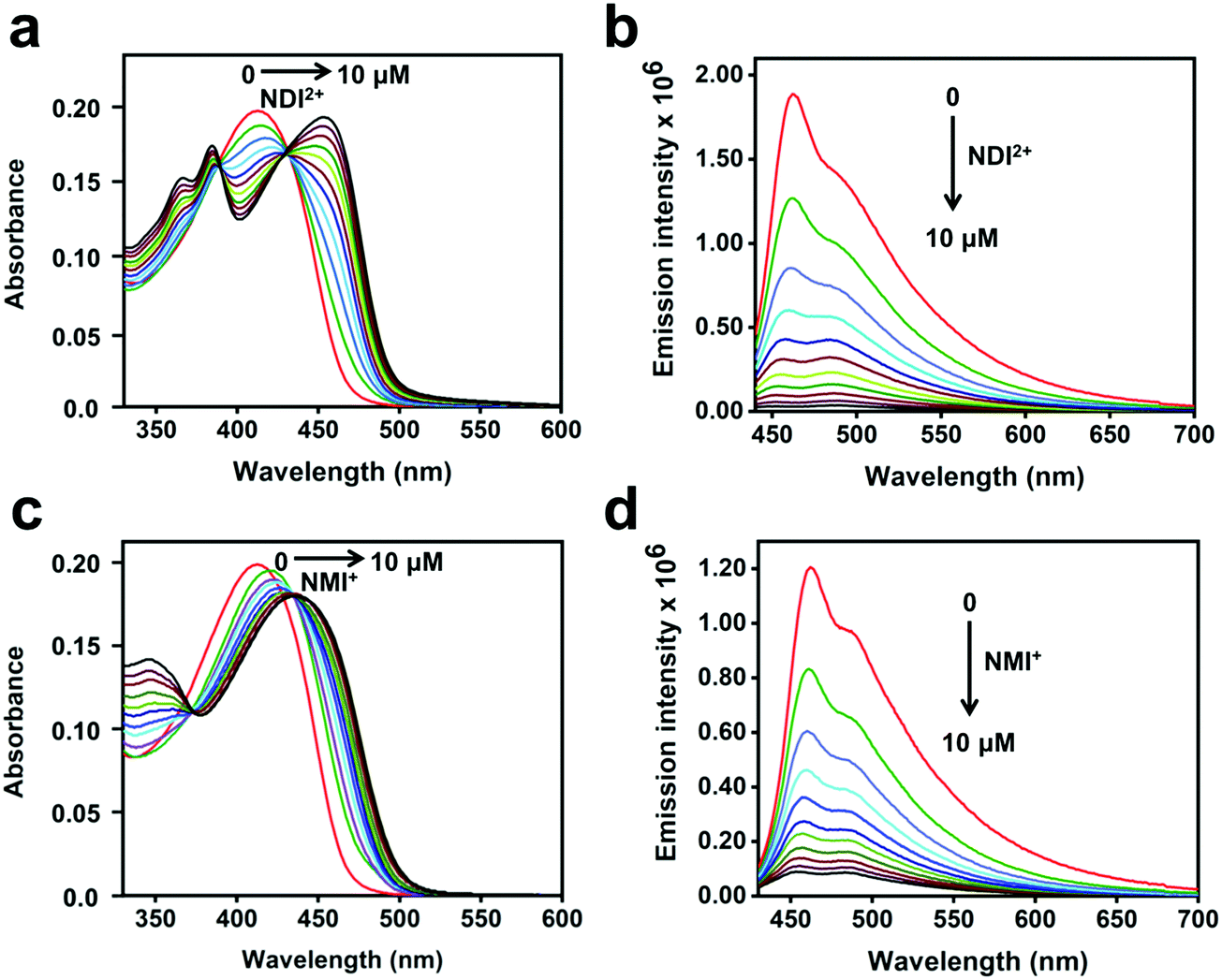 | ||
| Fig. 3 Absorption spectra of CPE (5 μM, PRU) in water with varying concentrations of (a) NDI2+ and (b) NMI+ as indicated in the figures. (b) Fluorescence spectra (λex = 410 nm) of CPE in water with varying concentrations of (a) NDI2+ and (b) NMI+. | ||
Picosecond transient absorption spectroscopy
To obtain more insight into the intermolecular ET between CPE and the cationic electron acceptors, and the absorption features of the polyelectrolyte cation radical (CPE˙+), picosecond transient absorption (TA) measurements were performed. Fig. 4a displays the TA spectra of CPE in water. The negative signal observed between 400 and 530 nm is attributed to stimulated emission (SE) from CPE, which is coupled with broad, featureless absorption in the visible region due to excited state absorption (ESA) of the CPE singlet excited-state. The TA spectrum of CPE is similar to that reported for other PPE-type conjugated polymers.49 The ESA kinetics of CPE exhibits three decay components with time constants 3, 49 and 264 ps with relative amplitudes of about 0.41, 0.30 and 0.29, respectively (Table 1). The fast decay processes (<50 ps) are likely due to initial structural relaxations followed by singlet decay via radiative and non-radiative pathways. The TA decay dynamics for CPE are in good agreement with its fluorescence decay kinetics (Fig. S4, ESI†).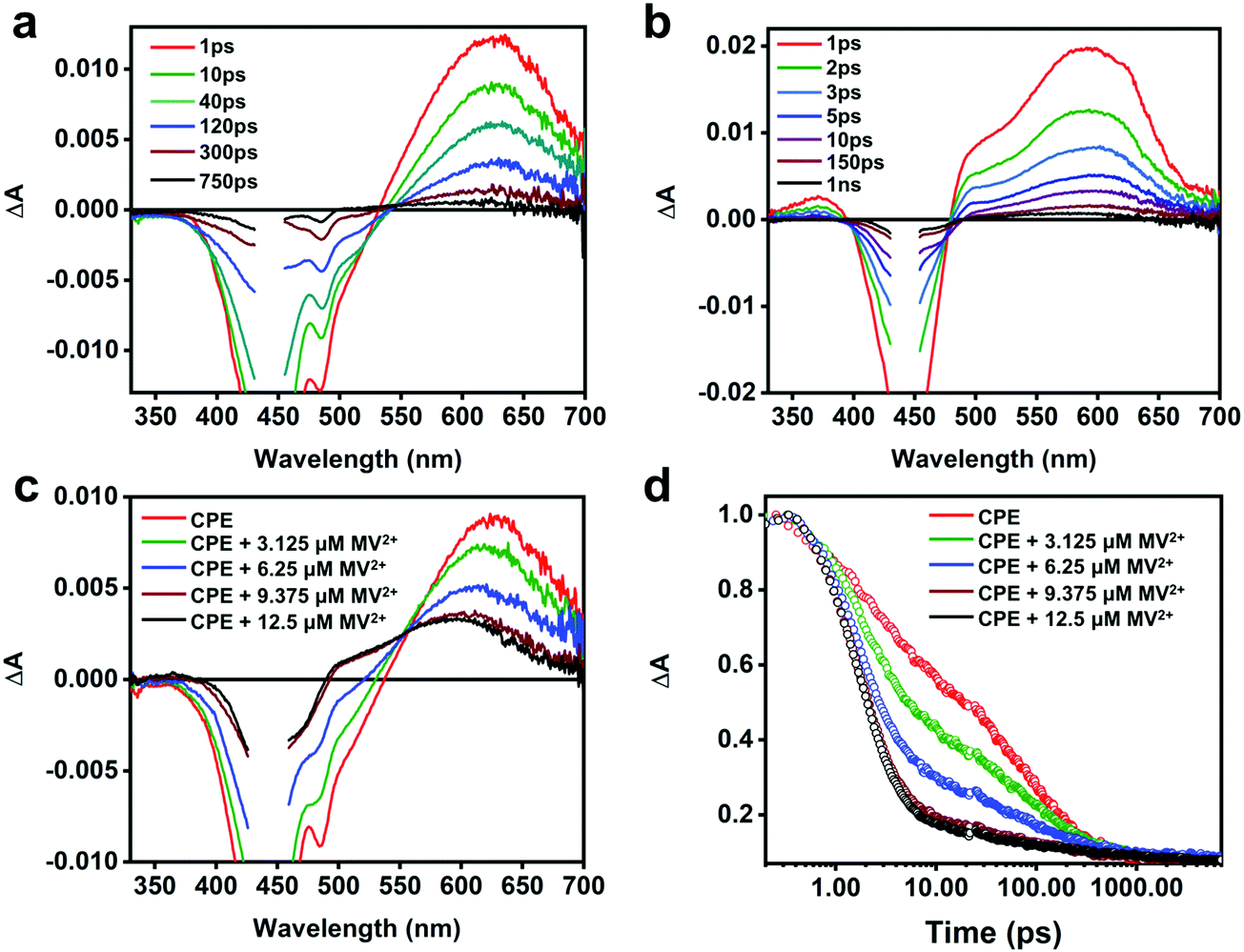 | ||
| Fig. 4 Picosecond transient absorption (psTA) spectra of (a) CPE (12.5 μM, PRU), (b) CPE + 12.5 μM MV2+ at the indicated delay times and (c) CPE in the presence of 0 μM to 12.5 μM MV2+ at 10 ps delay time in air-saturated H2O following a 440 nm laser excitation pulse (100 fs pulse width, 100 nJ per pulse). (d) Kinetic decay traces of CPE in the presence of 0 μM to 12.5 μM of MV2+ detected at 603–615 nm. | ||
The TA spectra of CPE with MV2+ (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 concentration ratio) recorded at different delay times are shown in Fig. 4b and a comparison of the spectra at 10 ps delay time for CPE with various MV2+ concentrations is shown in Fig. 4c (see Fig. S5 in the ESI† for full spectra). From these data it is clear that the TA spectra for the CPE/MV2+ complex are noticeably different from those of CPE alone. In particular, the main absorption band in the spectrum in the presence of MV2+ is blue shifted from that of CPE alone, and in addition there is substantially more absorption in the region between 330 and 500 nm (the latter is partially due to the decrease in SE due to fluorescence quenching in the CPE/MV2+ complex). The TA spectrum of the CPE/MV2+ complex is dominated by the absorption of the oxidized polymer (CPE˙+, polaron state). This assignment is supported by a previous study of a structurally-similar PPE-based polymer cation radical generated by pulse radiolysis which displayed a similar absorption-difference spectrum.50 Note that the spectrum of the oxidized polymer (CPE˙+, polaron state) is similar to that of the singlet exciton state; however, it is not unusual for exciton and polaron states of conjugated polymers to have similar spectra.51 Notable is the fact that the spectral signature of the reduced acceptor (MV˙+), which displays bands at 395 and 600 nm,47,52 is not seen in the transient absorption spectrum of the CPE/MV2+ complex. It is not clear why this is the case, but it may be due to the fact that the 395 nm band is obscured by the excitation scattering and SE, and the 600 nm band is overlapped by the significantly stronger absorption of CPE˙+.
:1 concentration ratio) recorded at different delay times are shown in Fig. 4b and a comparison of the spectra at 10 ps delay time for CPE with various MV2+ concentrations is shown in Fig. 4c (see Fig. S5 in the ESI† for full spectra). From these data it is clear that the TA spectra for the CPE/MV2+ complex are noticeably different from those of CPE alone. In particular, the main absorption band in the spectrum in the presence of MV2+ is blue shifted from that of CPE alone, and in addition there is substantially more absorption in the region between 330 and 500 nm (the latter is partially due to the decrease in SE due to fluorescence quenching in the CPE/MV2+ complex). The TA spectrum of the CPE/MV2+ complex is dominated by the absorption of the oxidized polymer (CPE˙+, polaron state). This assignment is supported by a previous study of a structurally-similar PPE-based polymer cation radical generated by pulse radiolysis which displayed a similar absorption-difference spectrum.50 Note that the spectrum of the oxidized polymer (CPE˙+, polaron state) is similar to that of the singlet exciton state; however, it is not unusual for exciton and polaron states of conjugated polymers to have similar spectra.51 Notable is the fact that the spectral signature of the reduced acceptor (MV˙+), which displays bands at 395 and 600 nm,47,52 is not seen in the transient absorption spectrum of the CPE/MV2+ complex. It is not clear why this is the case, but it may be due to the fact that the 395 nm band is obscured by the excitation scattering and SE, and the 600 nm band is overlapped by the significantly stronger absorption of CPE˙+.
As shown in Fig. 4d, addition of MV2+ to CPE leads to two notable changes in the TA decay dynamics. First, the predominant decay component is considerably more rapid in CPE/MV2+ compared to CPE. Second, less obvious is the low amplitude, long lifetime component (τ > 1000 ps) that emerges at higher MV2+ concentration (see Table 2). As discussed in more detail below, analysis of the kinetics suggests that the photoinduced ET occurs on an ultrafast timescale (kf > 1012 s−1) and the dominant decay component in the TA is due to return ET (2 ps, kb ∼ 5 × 1011 s−1), which gives rise to decay of the absorption. The long lifetime component apparent in samples with higher MV2+ concentration is due to a small component of CPE˙+ (polaron)//MV˙+ pairs that are able to undergo cage escape from the original complex geometry where CT occurs. This gives rise to a decreased rate of charge recombination (Fig. 1), which manifests as a much slower decay rate for the CPE˙+ (polaron) absorption.
| τ 1/ps | τ 2/ps | τ 3/ps | |
|---|---|---|---|
| The relative amplitudes are in parentheses. CPE = 12.5 μM, MV2+ = 12.5 μM and CB7 = 125 μM. | |||
| CPE | 3.0 ± 0.2 (0.41) | 49 ± 6 (0.30) | 264 ± 24 (0.29) |
| CPE/MV2+ | 2.0 ± 0.1 (0.89) | 115 ± 10 (0.08) | >1000 (0.03) |
| CPE/MV2+/CB7 | 4.0 ± 0.2 (0.41) | 60 ± 6 ps (0.30) | 610 ± 60 (0.29) |
In order to better under the ET process and to identify the acceptor anion radical, we utilized a naphthalene dimiide (NDI2+) electron acceptor. The spectrum of the reduced diimide (NDI+) has been well documented in previous spectroelectrochemical and transient absorption studies.53–55Fig. 5a illustrates the TA difference spectra of the [CPE, NDI2+] ion-pair complex recorded at different delay times. The spectra feature strong negative absorption features located from 330 to 470 nm that are consistent with the depopulation of the electronic ground states of CPE and NDI2+ (i.e., ground state bleaching). The positive transient absorption consists of two prominent bands. The most significant signal to emerge is the narrow absorption band at ∼490 nm, which is partially cut-off by the ground state bleach of CPE. This absorption is clearly due to the reduced diimide acceptor (e.g., NDI˙+) resulting from the photoinduced ET from CPE to NDI2+. The broad transient absorption that extends from 500–650 nm is mainly due to the CPE˙+ (polaron) state as it is similar in shape and peak wavelength compared to that observed for the polaron absorption in the CPE/MV2+ system (Fig. 4b). The dynamics for the CPE/NDI2+ system are shown in Fig. 5b; it can be seen that the charge transfer state decays very rapidly, with a dominant decay component of 3 ps (Table S2, ESI†).
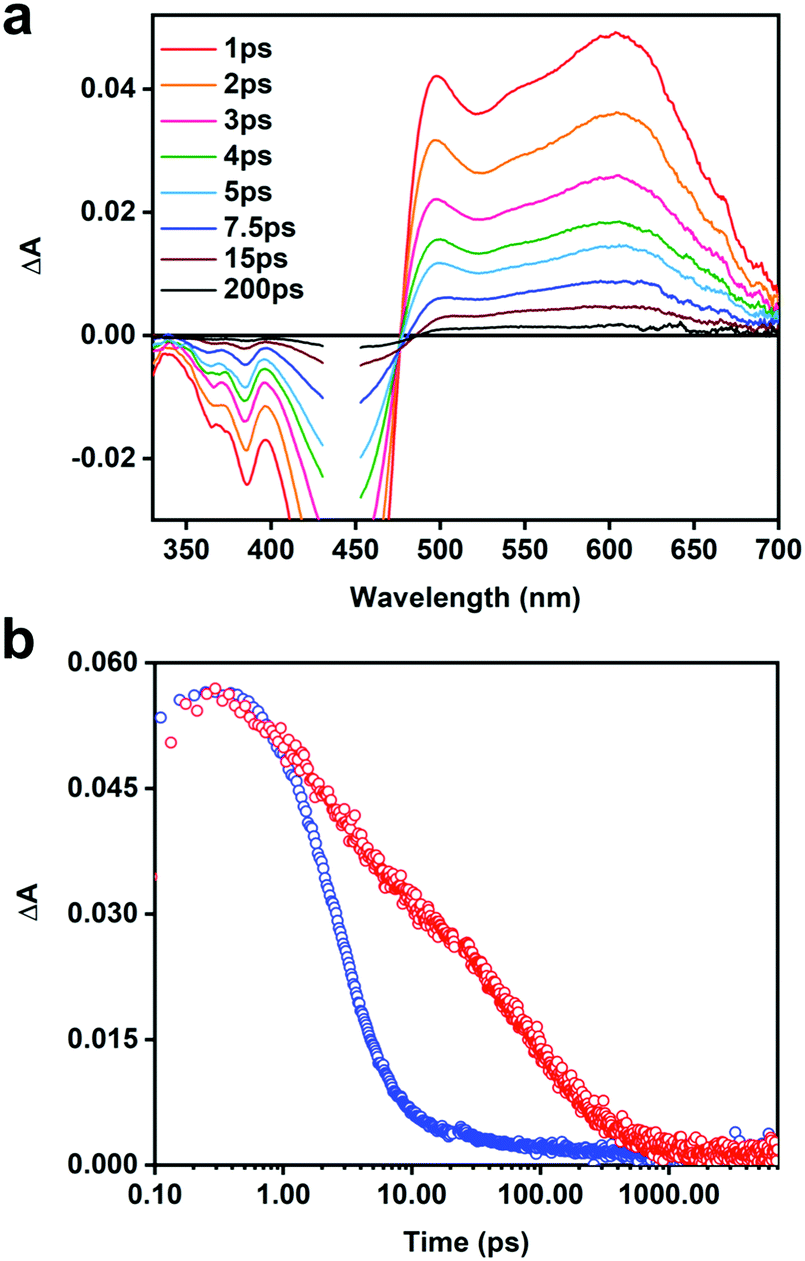 | ||
| Fig. 5 (a) Picosecond TA spectra of CPE (12.5 μM, PRU) + 12.5 μM NDI2+ in air-saturated H2O at the indicated delay times following a 440 nm laser excitation pulse (100 fs pulse width, 100 nJ per pulse). (b) Kinetic decay traces of CPE (red) and CPE + NDI2+ (blue) detected at 603 nm and 622 nm, respectively. | ||
The intermolecular ET between CPE and dicationic electron acceptors occurs very rapidly because of the strong ion-pair complex formation. To test the efficiency of the electron transfer process between CPE and a monocationic acceptor, identical experiments were carried out with a naphthalene monomiide (NMI+) derivative. The TA spectrum of CPE/NMI+ at 13 ps delay (Fig. S6a, ESI†) is qualitatively similar to the spectrum with the NDI2+ acceptor, supporting its assignment to the charge-transfer state [CPE˙+, NMI˙] produced by ultrafast photoinduced ET from CPE to the NMI+ acceptor. This species decays with single exponential kinetics with a lifetime of 35 ps (Table S3, ESI†).
Modulation of the electron transfer dynamics using cucurbit[7]uril
It is well known from previous studies that the energy and ET dynamics between donor and acceptor molecules could be modulated using supramolecules, in which one of the donor or acceptor is trapped within the cavity of a supramolecule and the other remains free in solution or electrostatically bound to the outside of its wall.56–60 Cucurbit[7]uril (CB7, Chart 1) is a water-soluble barrel-shaped organic macrocyclic host molecule composed of seven glycoluril units interlinked to each other by pairs of methylene groups.61,62 Host–guest complexation in CB7 is driven by the hydrophobic interactions of a guest molecule with its inner cavity, and electrostatic attraction of cationic groups on the guest towards C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O dipoles on CB7.63
O dipoles on CB7.63
To probe the feasibility of modulating the ET dynamics of the [CPE, MV2+] ion pair complex, CB7, which is known to strongly bind MV2+ (association constant, Ka ∼ 2 × 105 M−1)64 in aqueous medium, was used as a host. Fig. 6a and b display the changes in the absorption and fluorescence spectra of the CPE/MV2+ mixture in the presence of CB7. Upon incremental addition of CB7 (0–100 μM), the intensity of the absorption peak increased monotonically with a concomitant blue shift, which is accompanied by an increase in the fluorescence intensity. The fluorescence recovery confirms the interference of CB7 in the ET quenching process. Nevertheless, the fluorescence was not fully recovered even in the presence of 100 μM CB7 and it also resulted in a featureless fluorescence band. This suggests that CB7 may slow the rate of ET quenching (e.g., kf in Fig. 1), but not shut off ET entirely.
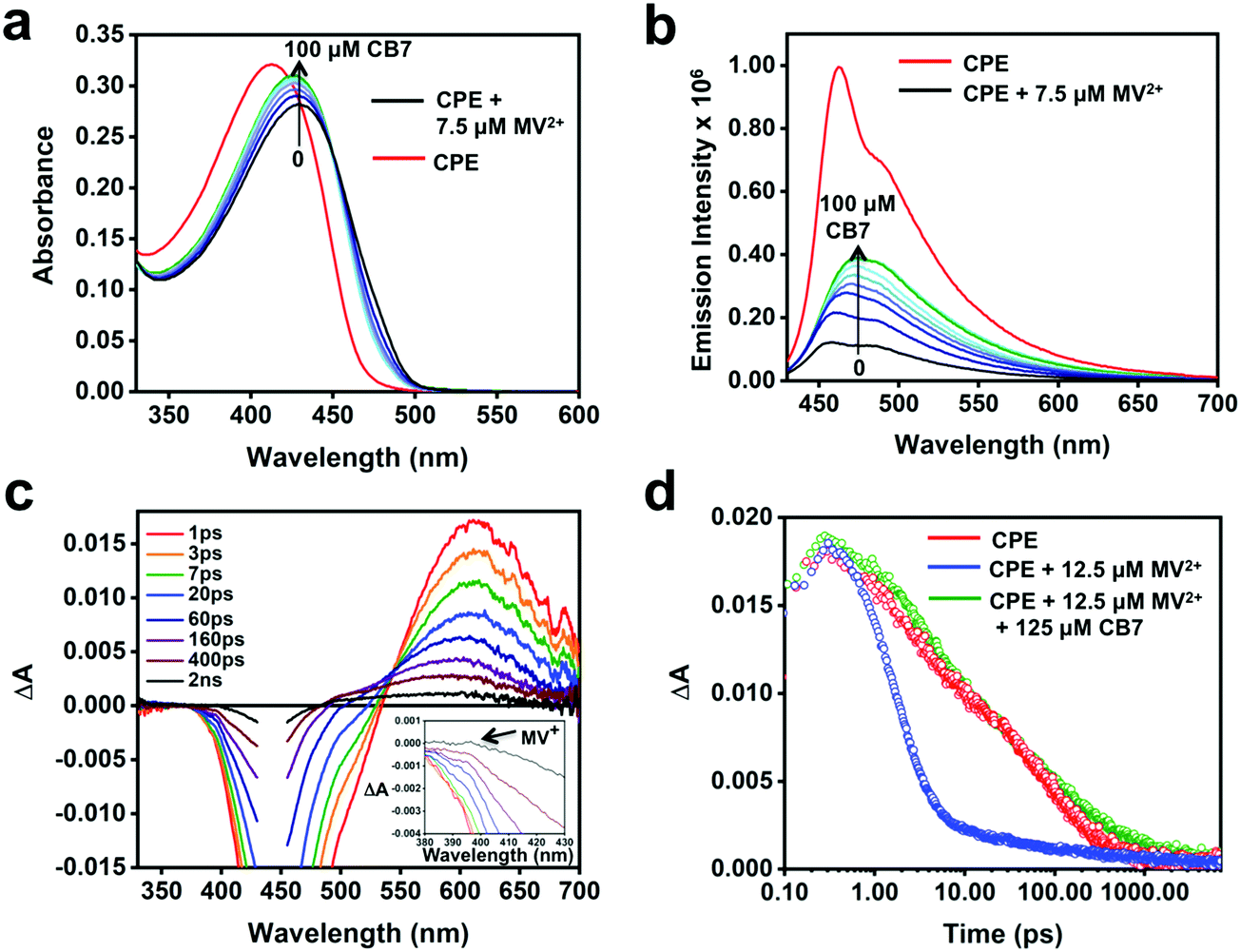 | ||
| Fig. 6 (a) Absorption and (b) fluorescence (λex = 410 nm) spectra of CPE (5 μM, PRU) + MV2+ in water with varying concentrations of CB7 as indicated in the figure. (c) Picosecond TA spectra of the CPE/MV2+/CB7 mixture (CPE = 12.5 μM, MV2+ = 12.5 μM, CB7 = 125 μM) in H2O at the indicated delay times following a 440 nm laser excitation pulse (100 fs pulse width, 100 nJ per pulse). (d) Kinetic decay traces of CPE (red), CPE/MV2+ (blue) and CPE/MV2+/CB7 (green) detected at 603 nm, 615 nm and 606 nm, respectively. | ||
Although the TA spectra (Fig. 6c) of the CPE/MV2+/CB7 mixture are complex, several important conclusions can be derived based on the spectral features and kinetics. First, the TA spectral features in the shorter time range (1 ps–60 ps) and the lifetimes (4 and 60 ps) are consistent with the spectral and decay kinetics of CPE alone (Fig. 4 and 6d, and Table 2). Secondly, the TA spectra at longer delay times blue-shift by ∼20 nm and the long lifetime component of the CPE/MV2+/CB7 decay is slower (∼610 ps) than that of CPE (∼264 ps, Table 2). Global analysis of the time-resolved spectral dynamics (Fig. S7, ESI†) clearly shows that the slowly decaying component has blue-shifted absorption that is similar to that observed for the [CPE, MV2+] complex (Fig. 4b).
Although the TA feature of CPE˙+ is not clearly visible for CPE/MV2+/CB7, careful inspection of the signals at 0.4–2 ns delay times in Fig. 6c reveals the presence of weak absorption of CPE˙+ along with very weak absorption at ∼395 nm that may be due to the reduced viologen acceptor, MV˙+ (Fig. 6c inset). Taken together, the data for the CPE/MV2+/CB7 system suggest that CB7 sequesters MV2+ from CPE, slowing the rate of forward ET due to separation of CPE from MV2+ by the CB7 wall. This gives rise to a longer lifetime for *CPE and suppressed fluorescence quenching. The rate of return ET (kb) may also be suppressed by CB7, also due to the separation imparted by the host.
The absorption and fluorescence spectra of CPE/NDI2+ in the presence of CB7 are shown in Fig. S7a and b (ESI†), respectively. Given the smaller size of the inner cavity of CB7, it is expected that NDI2+ will not form a host–guest complex with CB7. The absence of any considerable change in the absorption and fluorescence spectra of the CPE/NDI2+ mixture confirmed the inability of CB7 to alter the interaction of CPE and NDI2+.
Discussion
Following the pioneering work of Wudl and Heeger on ultrafast ET in PPV-based conjugated polymer/C60 systems and on bulk heterojunction solar cells,65,66 the rich photophysical properties of conjugated polymers/CPEs and their ability to participate in photoinduced ET processes have been exploited for their application as organic photovoltaics and sensors. Although the electron-donating feature of conjugated polyelectrolytes has been successfully utilized for a range of applications, systematic investigation of the polaron state of the polyelectrolytes resulting from photoinduced ET has not been reported previously. In this context, understanding the ET mechanism and dynamics as well as the photophysics of polaron states of conjugated systems is essential for the advancement of the field. Earlier work reported by Friend and coworkers on TA spectroscopy studies of a poly(9,9′-dioctylfluorene-alt-benzothiadiazole) based CPE revealed the generation of polaron states in the absence of electron acceptors as a result of inter-chain exciton annihilation.67 They also observed that the polarons are stabilized in the solid state by counter anions via Coulombic interaction. Clark et al. performed TA studies for a PPE-based polyelectrolyte in its aggregated state in ethanol. Their attempts to detect the polaron signals of the polyelectrolyte were unsuccessful due to the lack of effective interchain interactions.68The primary objective of this work was to study the mechanism of quenching of a water-soluble PPE-based conjugated polyelectrolyte by electron acceptors, with the aim to observe the CT state and its spectral properties and dynamics. For all three CPE/acceptor mixtures, a considerable red-shift in the absorption was readily observed in the steady-state absorption spectra and their intensities are comparable to the main band of CPE. The bathochromic shift is caused as a result of the suppression of polymer conformational changes upon association with the cationic acceptors, and also the possibility of absorption due to optical CT. Moreover, the electron acceptors quenched the fluorescence from CPE very efficiently with Stern–Volmer quenching constants (KSV) in excess of 106 M−1, consistent with the amplified quenching effect. Addition of a supporting electrolyte such as NaCl (Fig. S9, ESI†) to CPE/MV2+ resulted in the dissociation of CPE and MV2+, which is evident from the recovery of the CPE fluorescence in Fig. S9b (ESI†). Moreover, the bathochromic shift in the absorption spectra of CPE/MV2+/NaCl (Fig. S9a) (ESI†) suggests that the polymer aggregates at high ionic strength due to polyelectrolyte charge screening by NaCl.30 The distinct absorption features of the polymer aggregates and CPE/MV2+ further confirm that the CPE/MV2+ ion-pair complex mostly remains as individual chains and the binding of MV2+ to CPE suppresses the polymer conformational changes. Taken together, the pronounced changes in the absorption and fluorescence spectra of the mixtures suggest: (1) that there is tight (ion-pair) association between anionic CPE and the cationic acceptors; and (2) that photoinduced ET between the polyelectrolyte and quenchers occurs rapidly and efficiently.
The ultrafast TA studies provided some interesting details about the ET properties and the CT states of CPE and the acceptors. The TA spectra of [CPE, MV2+] (Fig. 3b) revealed the absorption feature of CPE˙+ generated as a result of the photoinduced ET to MV2+, which is unprecedented for any PPE-type polyelectrolytes. The spectrum of the polaron state of the PPE-based polymer exhibits a primary band maximum at ∼595 nm, and it is in agreement with previous studies on PPE-type oligomers and polymers.69 Moreover, the absorption feature due to the reduced acceptor, MV˙+, which is expected at ∼395 nm, was not observed for CPE/MV2+. It is unclear why, but it is possible that it is obscured by the considerably more intense bleach and absorption features due to CPE˙+. However, direct evidence for the existence of the CT state is obtained from the TA spectra of the [CPE, NDI2+] ion-pair complex (Fig. 5a). In this case, clear absorption bands due to the oxidized CPE and the reduced diimide acceptor can be seen, and these are formed essentially instantaneously following photoexcitation of CPE (<1 ps), indicating that the forward ET is ultrafast (kf > 1012 s−1).
As noted above, the forward ET in both the [CPE, MV2+] and [CPE, NDI2+] complexes is ultrafast. This is evident from the fact that the spectra of the CT states are observed at even the earliest delay times probed (1 ps, kf > 1012 s−1). In both systems, the predominant kinetic decay component for the CT state has τ ∼ 3 ps, and we attribute this process to return ET from the CT state (kb ∼ 3 × 1011 s−1). The very fast ET dynamics in these systems are readily explained by the following features. First, the distance between the polymer backbone and the acceptor is likely very short (Fig. 7a), giving rise to relatively strong electronic coupling (Vel > 50 meV).42 Second, for both the MV2+ and NDI2+ acceptors, the driving force for the forward and return ET processes is in the range of −1.3 eV, making them highly exothermic (near the activationless region of the Marcus curve). One subtle yet interesting feature is the existence of a low-amplitude, slow decay component for the CT state for the [CPE, MV2+] complex, suggesting a mechanism that gives rise to slower return ET. It is possible that the slower process is due to the formation of a “separated” CT state, where the reduced acceptor MV˙+ has diffused away from the backbone, perhaps by becoming bound to only one of the polyelectrolyte pendant anion groups (–R–SO3−), e.g., Fig. 7b.
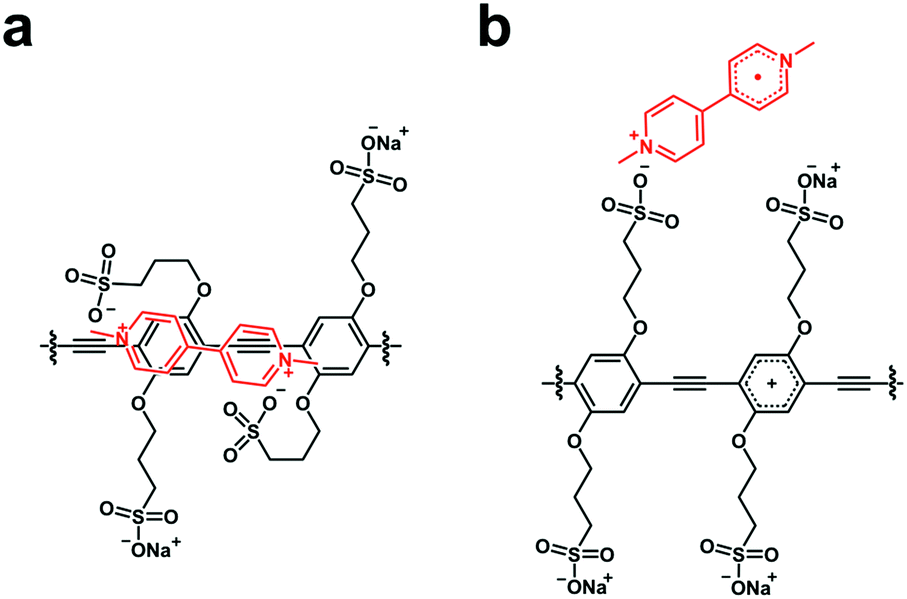 | ||
| Fig. 7 Possible structure of ion-pair complexes. (a) [CPE, MV2+] and (b) [CPE˙+, MV˙+]. | ||
Photoinduced ET between a donor and acceptor pair, especially ones that form ground-state CT complexes, is a very rapid process that occurs usually on a sub-picosecond time scale.70–72 Designing donor–acceptor systems in which the deacceleration of return ET could lead to elongation of the lifetime of the charge-separated state is essential for the efficient conversion of light to chemical energy. One of the interesting and facile approaches adopted to slow down the return ET process is through screening the donor from the acceptor or vice versa using an insulating supramolecular host.73–75 To the best of our knowledge, this strategy has not been employed for the deacceleration of the ET process in conjugated polyelectrolytes. For modulating the electron transfer process in the CPE/acceptor systems, the rigid supramolecule CB7 was employed. Our results indicate that since CB7 has the ability to fully encapsulate MV2+,64 the host–guest (MV2+/CB7) complex formed upon addition of CB7 to CPE/MV2+ remains bound to the polyelectrolyte, allowing the ET to proceed through the walls of the host. As a result, the forward and return ET processes are slowed down in the CPE/MV2+/CB7 system. By contrast, CB7 did not influence the ET process in CPE/NDI2+ due to the inability of CB7 to form a host–guest complex with NDI2+. Thus, the results provided a proof-of-concept for the modulation of the ET process in mixtures involving conjugated polyelectrolytes.
Conclusion
In this report, we investigated the photoinduced ET properties of a water-soluble poly(p-phenylene ethynylene) conjugated polyelectrolyte appended with alkyl sulfonate (R–SO3−) functionality in the presence of three strong electron acceptors. Direct observation of the red-shift in the absorption peaks revealed the ion-pair assembly formation between the polyelectrolyte and quenchers. The fluorescence of the polyelectrolyte is quenched very efficiently with Stern–Volmer constants (KSV) on the order of 106 M−1. Ultrafast transient absorption studies of the CPE/acceptor complexes revealed the unprecedented absorption feature of the cation radical species of the PPE-based polyelectrolyte. The TA spectra of the [CPE, NDI2+] complex provided further evidence for the production of anion radical species of the acceptor via photoreduction. Kinetics measurements performed on the three polyelectrolyte/acceptor mixtures indicate that the forward ET occurs with rate constant kf > 1 × 1012 s−1 and the return ET is also rapid with k ∼ 3 × 1011 s−1.Experimental section
Characterization methods
UV-visible spectra were recorded using a Shimadzu UV-2600 spectrophotometer. Corrected steady-state fluorescence spectra were obtained using an Edinburgh fluorometer. A 1 cm2 square pyrex cuvette was used for the solution spectra, and the emission was collected at 90° relative to the excitation beam. Fluorescence lifetimes were measured using a PicoQuant FluoTime 300 fluorescence lifetime spectrophotometer by time-correlated single photon counting (PicoQuant PicoHarp 300 module). A high-performance LDH-D-C-405 PicoQuant diode laser provided the excitation at 405 nm (repetition rate 40 MHz, P < 3 mW) for the sample. The instrument response function (ca. 100 ps) was measured using a Ludox scattering solution. Decays were obtained using single/biexponential fitting parameters using FluoroFit software.Femtosecond-picosecond transient absorption spectroscopy was carried out using a system consisting of a Coherent Astrella Ti:sapphire (100 fs, 1 KHz repetition) source (https://www.coherent.com/lasers/laser/astrella-ultrafast-tisapphire-amplifier) coupled with an Opera Solo OPA (https://www.coherent.com/lasers/laser/opera-solo-ultrafast-optical-parametric-amplifier) and a Ultrafast Systems Helios transient absorption spectrometer (https://ultrafastsystems.com/products/helios-fire). The first beam generated from the Coherent Astrella was directed through an optical parametric amplifier in the Opera Solo OPA, where the wavelength was tuned to 440 nm for pumping. The pump beam was then directed into a Helios fire (Ultrafast) automated femtosecond transient absorption spectrometer where the beam passed through a chopper, depolarizer and neutral density filter to tune the beam to 0.1 mW (100 nJ per pulse) of power before hitting the stirred sample (O.D. ≈ 0.2). The second beam was passed through a digitally controlled delay stage (in the Helios fire) with a maximum range of 8 ns, and afterward the beam was focused into a calcium fluoride crystal to generate a UV-visible probe ranging from 320–700 nm. The two beams overlap at the sample position with their respective electronic polarizations at the magic angle. The output signal with and without pumping (at several time delays) was detected using a fiber-coupled alignment-free spectrometer with a 1024-pixel CMOS sensor. Chirp- and time-zero corrected spectra, and multiexponential decay fits were acquired and manipulated using Surface Explorer software provided by Ultrafast Systems. OriginLab Corporation (version 9.55) data analysis software was used to plot the spectra.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Welch Foundation for support through the Welch Chair (Grant No. AX-0045-20110629).References
- H. Jiang, P. Taranekar, J. R. Reynolds and K. S. Schanze, Conjugated Polyelectrolytes: Synthesis, Photophysics, and Applications, Angew. Chem., Int. Ed., 2009, 48, 4300–4316 CrossRef CAS
.
- Y. Liu, K. Ogawa and K. S. Schanze, Conjugated Polyelectrolytes as Fluorescent Sensors, J. Photochem. Photobiol., C, 2009, 10, 173–190 CrossRef CAS
-
B. Liu and G. C. Bazan, Conjugated Polyelectrolytes Fundamentals and Applications, Wiley-VCH, Weinheim, 2013 Search PubMed
- X. Zhao, M. R. Pinto, L. M. Hardison, J. Mwaura, J. Müller, H. Jiang, D. Witker, V. D. Kleiman, J. R. Reynolds and K. S. Schanze, Variable band gap poly (arylene ethynylene) conjugated polyelectrolytes, Macromolecules, 2006, 39, 6355–6366 CrossRef CAS
- A. O. Patil, Y. Ikenoue, F. Wudl and A. J. Heeger, Water soluble conducting polymers, J. Am. Chem. Soc., 1987, 109, 1858–1859 CrossRef CAS
- J. H. Seo, E. B. Namdas, A. Gutacker, A. J. Heeger and G. C. Bazan, Conjugated Polyelectrolytes for Organic Light Emitting Transistors, Appl. Phys. Lett., 2010, 97, 043303 CrossRef
- C. Duan, L. Wang, K. Zhang, X. Guan and F. Huang, Conjugated Zwitterionic Polyelectrolytes and Their Neutral Precursor as Electron Injection Layer for High-Performance Polymer Light-Emitting Diodes, Adv. Mater., 2011, 23, 1665–1669 CrossRef CAS
- W. Lee, J. H. Seo and H. Y. Woo, Conjugated Polyelectrolytes: A New Class of Semiconducting Material for Organic Electronic Devices, Polymer, 2013, 54, 5104–5121 CrossRef CAS
- A. Duarte, K.-Y. Pu, B. Liu and G. C. Bazan, Recent Advances in Conjugated Polyelectrolytes for Emerging Optoelectronic Applications, Chem. Mater., 2011, 23, 501–515 CrossRef CAS
- J. Kesters, T. Ghoos, H. Penxten, J. Drijkoningen, T. Vangerven, D. M. Lyons, B. Verreet, T. Aernouts, L. Lutsen, D. Vanderzande, J. Manca and W. Maes, Imidazolium-Substituted Polythiophenes as Efficient Electron Transport Materials Improving Photovoltaic Performance, Adv. Energy Mater., 2013, 3, 1180–1185 CrossRef CAS
- Y. Yuan, J. Liu and B. Liu, Conjugated-Polyelectrolyte-Basedpolyprodrug:Targeted and Image-Guided Photodynamic and Chemotherapy with on-Demand Drug Release Upon Irradiation with a Single Light Source, Angew. Chem., Int. Ed., 2014, 53, 7163–7168 CrossRef CAS
-
P. Jagadesan, Y. Huang and K. S. Schanze, in Conjugated Polymers: Perspective, Theory, and New Materials, Taylor & Francis, 2019, pp. 547–587 Search PubMed
- G. Feng, C.-K. Mai, R. Zhan, G. C. Bazan and B. Liu, Narrow Band Gap Conjugated Polyelectrolytes for Photothermal Killing of Bacteria, J. Mater. Chem. B, 2015, 3, 7340–7346 RSC
- Y. Huang, H. C. Pappas, L. Zhang, S. Wang, R. Cai, W. Tan, S. Wang, D. G. Whitten and K. S. Schanze, Selective Imaging and Inactivation of Bacteria over Mammalian Cells by Imidazolium-Substituted Polythiophene, Chem. Mater., 2017, 29, 6389–6395 CrossRef CAS
- C. Zhu, Q. Yang, L. Liu, F. Lv, S. Li, G. Yang and S. Wang, Multifunctional Cationic Poly(p-phenylene vinylene) Polyelectrolytes for Selective Recognition, Imaging, and Killing of Bacteria over Mammalian Cells, Adv. Mater., 2011, 23, 4805–4810 CrossRef CAS
- K.-Y. Pu and B. Liu, Fluorescent Conjugated Polyelectrolytes for Bioimaging, Adv. Funct. Mater., 2011, 21, 3408–3423 CrossRef CAS
- B. Kim, I. H. Jung, M. Kang, H.-K. Shim and H. Y. Woo, Cationic Conjugated Polyelectrolytes-Triggered Conformational Change of Molecular Beacon Aptamer for
Highly Sensitive and Selective Potassium Ion Detection, J. Am. Chem. Soc., 2012, 134, 3133–3138 CrossRef CAS
- Y. Wu, Y. Tan, J. Wu, S. Chen, Y. Z. Chen, X. Zhou, Y. Jiang and C. Tan, Fluorescence Array-Based Sensing of Metal Ions Using Conjugated Polyelectrolytes, ACS Appl. Mater. Interfaces, 2015, 7, 6882–6888 CrossRef CAS
- Z. Li, R. Acharya, S. Wang and K. S. Schanze, Photophysics and phosphate fluorescence sensing by poly(phenylene ethynylene) conjugated polyelectrolytes with branched ammonium side groups, J. Mater. Chem. C, 2018, 6, 3722–3730 RSC
- Y. Tang, F. He, M. Yu, S. Wang, Y. Li and D. Zhu, Radical Scavenging Mediating Reversible Fluorescence Quenching of an Anionic Conjugated Polymer:Highly Sensitive Probe for Antioxidants, Chem. Mater., 2006, 18, 3605–3610 CrossRef CAS
- A. H. Malik and P. K. Iyer, Conjugated Polyelectrolyte Based Sensitive Detection and Removal of Antibiotics Tetracycline from Water, ACS Appl. Mater. Interfaces, 2017, 9, 4433–4439 CrossRef CAS
- A. S. Tanwar, S. Patidar, S. Ahirwar, S. Dehingia and P. K. Iyer, “Receptor free” inner filter effect based universal sensors for nitroexplosive picric acid using two polyfluorene derivatives in the solution and solid states, Analyst, 2019, 144, 669–676 RSC
- M. R. Pinto and K. S. Schanze, Amplified fluorescence sensing of protease activity with conjugated polyelectrolytes, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7505–7510 CrossRef CAS
- F. He, F. Feng, X. Duan, S. Wang, Y. Li and D. Zhu, Selective and Homogeneous Fluorescent DNA Detection by Target-Induced Strand Displacement Using Cationic Conjugated Polyelectrolytes, Anal. Chem., 2008, 80, 2239–2243 CrossRef CAS
- H. Li and G. C. Bazan, Conjugated Oligoelectrolyte/ssDNA Aggregates: Self-Assembled Multicomponent Chromophores for Protein Discrimination, Adv. Mater., 2009, 21, 964–967 CrossRef CAS
- Q. Zhou and T. M. Swager, Method for enhancing the sensitivity of fluorescent chemosensors: energy migration in conjugated polymers, J. Am. Chem. Soc., 1995, 117, 7017–7018 CrossRef CAS
- Q. Zhou and T. M. Swager, Fluorescent Chemosensors Based on Energy Migration in Conjugated Polymers: The Molecular Wire Approach to Increased Sensitivity, J. Am. Chem. Soc., 1995, 117, 12593–12602 CrossRef CAS
- T. M. Swager, The Molecular Wire Approach to Sensory Signal Amplification, Acc. Chem. Res., 1998, 31, 201–207 CrossRef CAS
- L. Chen, D. McBranch, H.-L. Wang, R. Helgeson, F. Wudl and D. G. Whitten, Highly sensitive biological and chemical sensors based on reversible fluorescence quenching in a conjugated polymer, Proc. Natl. Acad. Sci. U. S. A., 1999, 22, 12287–12292 CrossRef
- C. Y. Tan, M. R. Pinto and K. S. Schanze, Photophysics, aggregation and amplified quenching of a water-soluble poly(phenylene ethynylene), Chem. Commun., 2002, 446–447 RSC
- J.-M. Koenen, X. Zhu, Z. Pan, F. Feng, J. Yang and K. S. Schanze, Enhanced Fluorescence Properties of Poly(phenylene ethynylene)-Conjugated Polyelectrolytes Designed to Avoid Aggregation, ACS Macro Lett., 2014, 3, 405–409 CrossRef CAS
- H. Yue, M. Wu, C. Xue, S. Velayudham, H. Liu and D. H. Waldeck, Evolution in the Supramolecular Complexes between Poly(phenylene ethynylene)-Based Polyelectrolytes and Octadecyltrimethylammonium Bromide as Revealed by Fluorescence Correlation Spectroscopy, J. Phys. Chem. B, 2008, 112, 8218–8226 CrossRef CAS
- M. Zhang, P. Wu, W.-T. Dou, H.-H. Han, X.-P. He, C. Tan and Y. Jiang, Conjugated Polyelectrolytes with Galactose-Containing Side Chains for Targeted Hepatoma Cell Imaging, Chem. Commun., 2017, 53, 5625–5628 RSC
- K. Lee, L. K. Povlich and J. Kim, Label-Free and Self-Signal Amplifying Molecular DNA Sensors Based on Bioconjugated Polyelectrolytes, Adv. Funct. Mater., 2007, 17, 2580–2587 CrossRef CAS
- C. Tan, E. Atas, J. G. Muller, M. R. Pinto, V. D. Kleiman and K. S. Schanze, Amplified Quenching of a Conjugated Polyelectrolyte by Cyanine Dyes, J. Am. Chem. Soc., 2004, 126, 13685–13694 CrossRef CAS
- H. Huang, F. Shi, Y. Li, L. Niu, Y. Gao, S. M. Shah and X. Su, Water-soluble conjugated polymer–Cu(II) system as a turn-on fluorescence probe for label-free detection of glutathione and cysteine in biological fluids, Sens. Actuators, B, 2013, 178, 532–540 CrossRef CAS
- X. Zhao and K. S. Schanze, Fluorescent ratiometric sensing of pyrophosphate via induced aggregation of a conjugated, Chem. Commun., 2010, 46, 6075–6077 RSC
- P. Jagadesan and K. S. Schanze, Poly(phenylene ethynylene) Conjugated Polyelectrolytes Synthesized via Chain-Growth Polymerization, Macromolecules, 2019, 52, 3845–3851 CrossRef CAS
- D. Wang, J. Wang, D. Moses, G. C. Bazan and A. Heeger, Photoluminescence Quenching of Conjugated Macromolecules by Bipyridinium Derivatives in Aqueous Media: Charge Dependence, Langmuir, 2001, 17, 1262–1266 CrossRef CAS
- H. Jiang, X. Zhao and K. S. Schanze, Effects of Polymer Aggregation and Quencher Size on Amplified Fluorescence Quenching of Conjugated Polyelectrolytes, Langmuir, 2007, 23, 9481–9486 CrossRef CAS
- Calculated by the following equations, ΔGf = E(*CPE/CPE+) − E(A/A−) and ΔGb = E(A/A−) − E(CPE/CPE+).
- M. R. Wasielewski, Photoinduced electron transfer in supramolecular systems for artificial photosynthesis, Chem. Rev., 1992, 92, 435–461 CrossRef CAS
-
Y. Yang, Study on Fluorescence Quenching, Aggregation and Charge Transfer Properties of Conjugated Polymers, PhD Dissertation, University of Florida, 2019 Search PubMed
- C. R. Bock, J. A. Connor, A. R. Gutierrez, T. J. Meyer, D. G. Whitten, B. P. Sullivan and J. K. Nagle, Estimation of Excited-state Redox Potentials by Electron-transfer Quenching. Application of Electron-transfer Theory to Excited-state Redox Processes, J. Am. Chem. Soc., 1979, 101, 4815–4824 CrossRef CAS
- D. Gosztola, M. P. Niemczyk, W. Svec, A. S. Lukas and M. R. Wasielewski, Excited Doublet States of Electrochemically Generated Aromatic Imide and Diimide Radical Anions, J. Phys. Chem. A, 2000, 104, 6545–6551 CrossRef CAS
- A. Samanta and G. Saroja, Steady State and Time-resolved Studies on the Redox Behaviour of 1,8-Naphthalimide in the Excited state, J. Photochem. Photobiol., A, 1994, 84, 19–26 CrossRef CAS
- J. Peon, X. Tan, J. D. Hoerner, C. Xia, Y. F. Luk and B. Kohler, Excited State Dynamics of Methyl Viologen. Ultrafast Photoreduction in Methanol and Fluorescence in Acetonitrile, J. Phys. Chem. A, 2001, 105, 5768–5777 CrossRef CAS
- S. R. Valandro, P. Jagadesan, F. Feng and K. S. Schanze, Aggregation-Enhanced Two-Photon Absorption of Anionic Conjugated Polyelectrolytes, J. Phys. Chem. Lett., 2020, 11, 8292–8296 CrossRef CAS
- R. Dillon, Z. Pan, J. Jiang, R. Winkel, J. Papanikolas and K. S. Schanze, Ultrafast Energy Transfer in Fully Conjugated Thiophene-Benzothiadiazole Capped Poly(Phenylene Ethynylene) Molecular Wires, J. Phys. Chem. C, 2020, 124, 18920–18929 CrossRef CAS
- A. M. Funston, E. E. Silverman, J. R. Miller and K. S. Schanze, Charge Transfer through Terthiophene End-Capped Poly(arylene ethynylene)s, J. Phys. Chem. B, 2004, 108, 1544–1555 CrossRef CAS
- A. O. Patil, A. J. Heeger and F. Wudl, Optical properties of conducting polymers, Chem. Rev., 1988, 88, 183–200 CrossRef CAS
- E. M. Kosower and J. L. Cotter, Stable Free Radicals. II. The Reduction of 1-Methyl-4-cyanopyridinium Ion to Methylviologen Cation Radical, J. Am. Chem. Soc., 1964, 86, 5524–5527 CrossRef CAS
- C. Thalacker, C. Röger and F. Würthner, Synthesis and Optical and Redox Properties of Core-Substituted Naphthalene Diimide Dyes, J. Org. Chem., 2006, 71, 8098–8105 CrossRef CAS
- S. J. Langford, M. J. Latter and C. P. Woodward, Progress in Charge Transfer Systems Utilizing Porphyrin Donors and Simple Aromatic Diimide Acceptor Units, Photochem. Photobiol., 2006, 82, 1530–1540 CAS
- S. R. Greenfield, W. A. Svec, D. Gosztola and M. R. Wasielewski, Multistep Photochemical Charge Separation in Rod-like Molecules Based on Aromatic Imides and Diimides, J. Am. Chem. Soc., 1996, 118, 6767–6777 CrossRef CAS
- P. Piotrowiak, Photoinduced Electron Transfer in Molecular Systems: Recent Developments, Chem. Soc. Rev., 1999, 28, 143–150 RSC
- I. Place, A. Farran, K. Deshayes and P. Piotrowiak, Triplet Energy Transfer through the Walls of Hemicarcerands: Temperature Dependence and the Role of Internal Reorganization Energy, J. Am. Chem. Soc., 1998, 120, 12626–12633 CrossRef CAS
- Z. S. Romanova, K. Deshayes and P. Piotrowiak, Triplet Excitation Transfer through the Walls of Hemicarcerands: Depend- ence of the Electronic Coupling on the Size of the Molecular Cage, J. Am. Chem. Soc., 2001, 123, 11029–11036 CrossRef CAS
- A. J. Parola, F. Pina, E. Ferreira, M. Maestri and V. Balzani, Photoinduced Electron- and Energy-Transfer Processes of Biacetyl Imprisoned in a Hemicarcerand, J. Am. Chem. Soc., 1996, 118, 11610–11616 CrossRef CAS
- A. Das, N. Kamatham, A. M. Raj, P. Sen and V. Ramamurthy, Marcus Relationship Maintained During Ultrafast Electron Transfer Across a Supramolecular Capsular Wall, J. Phys. Chem. A, 2020, 124, 5297–5305 CrossRef CAS
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, New Cucurbituril Homologues: Syntheses, Isolation, Characterization, and X-ray Crystal Structures of Cucurbit[n]uril (n = 5, 7, and 8), J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS
- J. W. Lee, S. Samal, N. Selvapalam, H. J. Kim and K. Kim, Cucurbituril Homologues and Derivatives: New Opportunities in Supramolecular Chemistry, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS
- F. Biedermann, W. M. Nau and H.-J. Schneider, The Hydrophobic Effect Revisited-Studies With Supramolecular Complexes Imply High-Energy Water as a Noncovalent Driving Force, Angew. Chem., Int. Ed., 2014, 53, 11158 CrossRef CAS
- H.-J. Kim, W. S. Jeon, Y. H. Ko and K. Kim, Inclusion of Methylviologen in Cucurbit[7]uril, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5007–5011 CrossRef CAS
- N. S. Sariciftci, L. Smilowitz, A. J. Heeger and F. Wudl, Photoinduced Electron-Transfer from a Conducting Polymer to Buckminsterfullerene, Science, 1992, 258, 1474–1476 CrossRef CAS
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Polymer Photovoltaic Cells-Enhanced Efficiencies via a Network of Internal Donor–Acceptor Heterojunctions, Science, 1995, 270, 1789–1791 CrossRef CAS
- J. M. Hodgkiss, G. Tu, S. Albert-Seifried, W. T. S. Huck and R. H. Friend, Ion-Induced Formation of Charge-Transfer States in Conjugated Polyelectrolytes, J. Am. Chem. Soc., 2009, 131, 8913–8921 CrossRef CAS
- A. P.-Z. Clark, A. J. Cadby, C. K.-F. Shen, Y. Rubin and H. Tolbert, Synthesis and Self-Assembly of an Amphiphilic Poly(phenylene ethynylene) Ionomer, J. Phys. Chem. B, 2006, 110, 22088–22096 CrossRef CAS
- J. Jiang, A. Alsam, S. Wang, S. M. Aly, Z. Pan, O. F. Mohammed and K. S. Schanze, Effect of Conjugation Length on Photoinduced Charge Transfer in π-Conjugated Oligomer-Acceptor Dyads, J. Phys. Chem. A, 2017, 121, 4891–4901 CrossRef CAS
- J. M. Masnovi, J. K. Kochi, E. F. Hilinski and P. M. Rentzepis, Reactive ion pairs from the charge-transfer excitation of electron donor–acceptor complexes, J. Am. Chem. Soc., 1986, 108, 1126–1135 CrossRef CAS
- T. Yabe and J. K. Kochi, Contact ion pairs.Picosecond dynamics of solvent separation,
internal return, and special salt effect, J. Am. Chem. Soc., 1992, 114, 4491–4500 CrossRef CAS
- E. F. Hilinski, J. M. Masnovi, C. Amatore, J. K. Kochi and P. M. Rentzepis, Charge-transfer excitation of electron donor–acceptor complexes. Direct observation of ion pairs by time-resolved (picosecond) spectroscopy, J. Am. Chem. Soc., 1983, 105, 6167–6168 CrossRef CAS
- M. Porel, A. Klimczak, M. Freitag, E. Galoppini and V. Ramamurthy, Photoinduced Electron Transfer Across a Molecular Wall: Coumarin Dyes as Donors and Methyl viologen and TiO2 as Acceptors, Langmuir, 2012, 28, 3355–3359 CrossRef CAS
- M. Porel, C.-H. Chuang, C. Burda and V. Ramamurthy, Ultrafast Photoinduced Electron Transfer Between an Incarcerated Donor and a Free Acceptor in Aqueous Solution, J. Am. Chem. Soc., 2012, 134, 14718–14721 CrossRef CAS
- C.-H. Chuang, M. Porel, R. Choudhury, C. Burda and V. Ramamurthy, Ultrafast Electron Transfer across a Nanocapsular Wall: Coumarins as Donors, Viologen as Acceptor, and Octa Acid Capsule as the Mediator, J. Phys. Chem. B, 2018, 122, 328–337 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0qm00476f |
| This journal is © the Partner Organisations 2020 |