
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Durability challenges of anion exchange membrane fuel cells
William E.
Mustain
 a,
Marian
Chatenet
b,
Miles
Page
c and
Yu Seung
Kim
*d
a,
Marian
Chatenet
b,
Miles
Page
c and
Yu Seung
Kim
*d
aDepartment of Chemical Engineering, University of South Carolina, Columbia, SC, USA. E-mail: mustainw@mailbox.sc.edu
bUniv. Grenoble Alpes, Univ. Savoie Mont Blanc, CNRS, Grenoble INP (Institute of Engineering, Univ. Grenoble Alpes), LEPMI, 38000 Grenoble, France
cPO-CellTech Ltd., Caesarea, Israel
dMPA-11: Materials Synthesis & Integrated Devices, Los Alamos National Laboratory, Los Alamos, NM, USA. E-mail: yskim@lanl.gov
First published on 10th July 2020
Abstract
As substantial progress has been made in improving the performance of anion exchange membrane fuel cells (AEMFCs) over the last decade, the durability of AEMFCs has become the most critical requirement to deploy competitive energy conversion systems. Because of different operating environments from proton exchange membrane fuel cells, several AEMFC-specific component degradations have been identified as the limiting factors influencing the AEMFC durability. In this article, AEMFC durability protocol, the current status of AEMFC durability, and performance degradation mechanisms are reported based on the discussion during the US Department of Energy (DOE) Anion Exchange Membrane Workshop at Dallas, Texas, May 2019. With additional recent progress, we provide our perspectives on current technical challenges and future action to develop long-lasting AEMFCs.
 William E. Mustain | William E. Mustain is a Professor in the Department of Chemical Engineering at the University of South Carolina. He received his PhD from the Illinois Institute of Technology in 2006. He spent two years as a postdoc at Georgia Tech before beginning his academic career in 2008. He has received several prestigious awards including the DOE Early Career Award and the Fulbright Scholar Fellowship. His current research programs focus extensively on anion exchange membrane fuel cells and electrolyzers as well as high capacity materials for Li-ion batteries and catalysts for electrosynthesis. |
 Marian Chatenet | Marian Chatenet graduated as an engineer in materials-sciences and a master in electrochemistry in 1997 from Grenoble Institute of Technology (Grenoble-INP). He defended his PhD in Electrochemistry in 2000 (Grenoble-INP) and moved to the University of Minnesota as a post-doc. Appointed associate professor in electrochemistry (2002), he is professor in Grenoble-INP since 2011. He studies electrocatalysis of complex reactions and activity/durability of electrocatalysts for fuel cell/electrolyzer applications. Best young scientist in Electrochemistry of the French Chemical Society (SCF, 2009), he received the Oronzio and Niccolò De Nora Foundation Prize of the International Society of Electrochemistry on Applied Electrochemistry (ISE, 2010). |
 Miles Page | Miles Page is CTO at POCellTech, Ltd., a company commercializing AEM fuel cell technology. He holds a PhD (2003) in Physical Chemistry from The University of Sydney, Australia in surfactant self-assembly. He engaged in postdoctoral research in biomineralization, jointly with the Max-Planck-Institute for Colloids and Interfaces, Potsdam (Germany) and the Commisariat à l’Energie Atomique at Saclay (France). He undertook further postdoctoral studies at the Weizmann Institute of Science in Rehovot (Israel) in thin film solar cells before taking an R&D team leader role at pioneering AEMFC developer Cellera Technologies, on whose background IP the POCellTech fuel cell development is based. At POCellTech he has overseen development of high power density and ultra-low PGM, AEMFC membrane-electrode assembly and stack technologies. |
 Yu Seung Kim | Yu Seung Kim is a technical staff scientist at Los Alamos National Laboratory, USA. He received his BS degree from Korea University (1994) and his PhD degree from Korea Advanced Institute of Science and Technology (1999) in the field of Chemical Engineering. He spent three years as a post-doctoral fellow at the Chemistry Department of Virginia Tech before joining the fuel cell group at Los Alamos (2003). He received the Outstanding Technical Contribution and Achievements Award from US DOE Hydrogen and Fuel Cell Program (2016). His research interest is materials for fuel cells and electrolyzers. He currently leads Membrane Working Group of US DOE Hydrogen and Fuel Cell Technologies Office. |
Broader contextThe fuel cell converts the chemical energy of hydrogen to produce electricity. Cost-effective fuel cell technology has become highly desirable because hydrogen is anticipated to become an essential integrator for renewable and grid electricity. Current state-of-the-art acid-based fuel cells use expensive platinum catalysts for electrochemical reactions and therefore much of the R&D focuses on approaches that will reduce or eliminate precious metal catalysts. Anion exchange membrane fuel cells (AEMFCs) are a promising alternative since earth-abundant non-precious metal catalysts showed high activity and stability under high pH conditions. Over the past three years, the performance of AEMFCs have remarkably improved, but the durability of AEMFCs is still inferior to that of acid-based fuel cells. In this perspective article, we present the status of AEMFC durability and the degradation behaviors of AEMFCs based on both discussions at the 2019 US DOE Anion Exchange Membrane Workshop in Dallas, Texas, and additional input from other experts. We also provide comprehensive degradation mechanisms of AEMFCs and in-depth discussions on the mitigation strategies at both a single cell and system level. Lastly, we highlight current durability challenges and propose future actions to improve AEMFC durability. |
1. Introduction
This paper primarily combines contributions made in talks and discussions at the US Department of Energy (DOE) Anion Exchange Membrane Workshop (Dallas, Texas, May 2019)1 on the subject of anion-exchange membrane fuel cell (AEMFC). Furthermore, Mustain and Kim have edited the manuscript after adding more recent data, receiving additional input from other experts in this field, and highlighting the current status and challenges to provide critical insights for future actions.Over the past decade, substantial progress on AEMFC performance has been made. In fact, the performance has approached that of state-of-the-art proton exchange membrane fuel cells (PEMFCs) (≥2 W cm−2 peak power density for polyolefin-based AEMFCs at 60–80 °C2,3 and ≥1.5 W cm−2 peak power density for polyaromatic-based AEMFCs at 80–95 °C4–6). Research efforts to lowering the loading of platinum group metal (PGM) catalysts7 or implementing PGM-free catalysts8–12 have been successful as well. This research progress opens the door for the development of low-cost polymer electrolyte fuel cells.
The most significant remaining challenge of AEMFC technology is durability. The reported lifetime of the AEMFCs is significantly inferior to that of the PEMFCs.13 Most AEMFC membrane electrode assemblies (MEAs) have shown a substantial reduction in performance over the first 100–200 hours of operation.14–16 While a few reports showed a longer AEMFC lifetime (500–1000 hours) under steady-state operating conditions,17–19 the longevity of AEMFCs seemed at least one order of magnitude lower than that of the PEMFCs.20,21 In the early stages of AEMFC research before 2012, researchers had investigated the chemical stability of anion exchange membranes (AEMs), focusing on the stability of organic cation functional group because the stability of organic cations under high pH conditions is inferior to the chemical stability of organic anions under low pH conditions.22–25 However, further studies (2012–2014) revealed that the cation functionalized polymer backbone is also susceptible to degradation, particularly for aryl ether linkages (C–O–C bond), leading to the preparation of all AEMs with C–C-bond backbone.26–29 It is important to note that the development of alkaline stable aryl ether-free polymers significantly contributed to the development of cationic group stable polymers because of the difficulties in investigating the cation degradation for aryl ether containing polymers as polymer segments containing cationic groups are easily dissolved in water. As a result of studies on alkaline stable cationic groups, the most commonly used benzyl ammonium functional groups have been replaced with more stable alkyl chain tethered polymers (2013–2015)30–33 or more stable cationic functional groups such as piperidinium (2015–2017).34–40 Currently several alkaline-stable AEMs are available.41–46 However, it is important to note that the lifetime of most AEMFCs employing even alkaline-stable AEMs and stable electrocatalysts47–53 is still <1000 h. Therefore, researchers have tried to understand the degradation factors that impact the lifetime of AEMFCs. Reviewing the AEMFC degradation mechanisms at this moment is particularly desirable because not only have we accumulated substantial data regarding water management, carbonation and component stability that impact the AEMFC durability, but also the AEMFC degradation study helps to understand the longevity of other AEM-based electrochemical devices.54,55
This paper reviews the progress on AEMFC durability between 2017–2019, as earlier durability data were well documented in the previous review paper.13 In detail, we explain the AEMFC performance requirement during continuous operation at a constant condition. Then we discuss the durability test protocol of AEMFCs and the MEA components that researchers have implemented. The current status of the AEMFC durability using PGM and PGM-free catalysts is reported. Next, we discuss the AEMFC degradation behaviors that cause recoverable performance loss and MEA component degradation mechanisms that cause unrecoverable performance loss. We mostly focus our review on hydrogen-fueled AEMFCs, as liquid or other gas-fed AEMFCs have more complex operating parameters and did not have much-accumulated data to address up to date. We emphasize the transient performance change behaviors by water management and carbonation for recoverable performance loss. For the unrecoverable performance loss, we focus on the degradation of MEA components during AEMFC operations. Proposed remediation strategies are reviewed here in some details. We do not provide exhaustive discussion on alkaline stability of AEMs as excellent papers on this topic are available.46,56–59 All in all, this paper reports the progress on AEMFC durability to date, providing insight into the operation of AEMFC stacks to operate over thousands of hours, which may be an affordable option for next-generation energy conversion devices.
2. AEMFC performance requirement and test protocols
2.1 AEMFC performance requirement
In practical fuel cell applications, the fuel cell stack is designed to deliver a certain power. For example, for the automotive application, current US DOE target for the power density of AEMFCs is ≥1.0 W cm−2 at 0.76 V at 80 °C (rated power), P ≤ 250 kPa; PGM-free, under H2/air conditions.60 To assure constant power delivery, any loss in the cell performance has to be compensated with higher current density. Fig. 1a shows the performance change over time of a hypothetical single cell that illustrates the change of cell current density and voltage to generate the target power density. For the required power density of 1.0 W cm−2, the cell current density has to be increased from 1.3 to 2.1 A cm−2 for the voltage loss from 0.76 to 0.49 V. However, once the voltage reaches 0.49 V, the system fails to generate the power needed, which is indicated by the red plot on the “performance for required power density” staying above the blue polarization curve. In addition to the ultimate failure of the fuel cell system from generating required power, performance degradation leads to a decrease in the total efficiency of the fuel cell via a decrease in voltage efficiency. In the example shown in Fig. 1a, the simulated performance degradation during device life (red path) leads to a decrease in voltage efficiency at the required power density, the initial efficiency of 54% at the no-loss case to 41% in the cell operating voltage.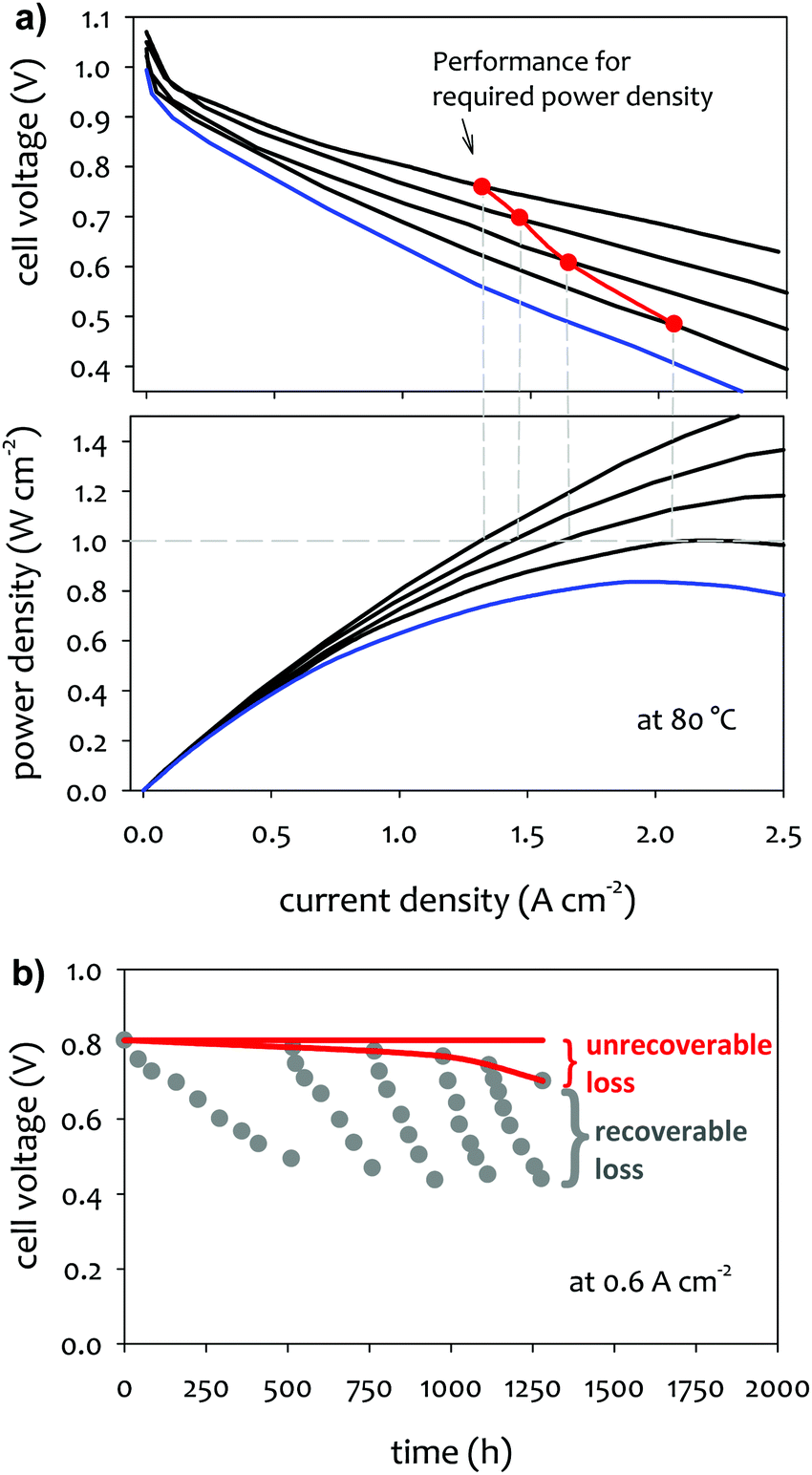 | ||
| Fig. 1 (a) Fuel cell performance loss overtime in a hypothetical fuel cell system designed to deliver the power density of 1.0 W cm−2. The initial performance starts with rated power (0.76 V at 80 °C). (b) Illustration of recoverable and unrecoverable AEMFC performance loss during constant current operation mode. | ||
2.2 Protocols for MEA durability
While the lifetime of an AEMFC system may be determined by operating the system at a required power, the constant power density mode is rarely adopted for fuel cell durability testing. Instead, the durability of an AEMFC system is usually evaluated in either constant-current or constant-voltage mode of operation. Constant current density mode is the most popular method among others, including the AEMFC durability protocol of US DOE Hydrogen and Fuel cell Technologies Office (HFTO) (Table 1).60 Constant current density mode better simulates operating conditions of a practical fuel cell system, allowing for constant consumption and generation of water in the oxygen reduction reaction (ORR) at the cathode and the hydrogen oxidation reaction (HOR) at the anode, respectively. This is suitable for studying performance degradation processes related to water management and reaction transport. Constant voltage mode, on the other hand, is more convenient to study degradation processes that depend on the electrode potentials, such as stability of electrocatalysts and electrochemical oxidation of the MEA materials.| Year | Milestone |
|---|---|
| 2022 | ≤10% voltage degradation over 1000 h at 0.6 A cm−2; T ≥ 80 °C; P ≤ 150 kPa; total PGM loading ≤0.2 mg cm−2 |
| 2023 | CO2 tolerance: ≤65 mV loss for steady state operation at 1.5 A cm−2 in H2/air scrubbed to 2 ppm CO2 |
| 2024 | Catalyst: H2/air (CO2-free) after AST ≤40% loss after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 square-wave cycles 0.6–0.95 V, PGM loading ≤0.125 mg cm−2 000 square-wave cycles 0.6–0.95 V, PGM loading ≤0.125 mg cm−2 |
| Membrane: H2 crossover ≤15 mA cm−2 (H2/N2) during 1000 h open circuit voltage (OCV) hold at 70% RH and ≥80 °C |
Under constant current density or cell voltage mode, certain performance losses incurred during the steady-state operation can be recovered by adjusting appropriate operational parameters or transient cell treatment (Fig. 1b). Such “recoverable” performance losses are associated with reversible phenomena occurring in the fuel cell, such as cell dehydration, carbonation, catalyst surface contamination, or incomplete water removal from the catalyst layer and gas diffusion layer (GDL). One common cell operational parameter change to recover AEMFC performance is cell voltage pulsing. Another common treatment is the cell replenishment by dilute alkali metal hydroxide solution, e.g., 1 M NaOH. Li et al. observed that the replenishing with 1 M NaOH made the performance recovered the cell voltage at a level of 98% after continuous run after 210 h.61 The replenishment can effectively remove carbonated species from the MEA and neutralize acidic phenol from electrochemical oxidation of phenyl groups at the fuel cell cathode. If the performance loss is related to dehydration or electrode flooding, changing the relative humidity (RH) of the supplied reactant gases can be an effective method to recover AEMFC performance.3
The AEMFC performance losses that cannot be reversed are referred to as “unrecoverable” performance losses. The magnitude of unrecoverable performance loss can be determined by subtracting the current density (or cell voltage) measured after every cell performance recovery process from the current density (or cell voltage) measured at the beginning-of-life test (Fig. 1b). More accurately, polarization curve measurements after reconditioning of the cell show the unrecoverable performance loss. They are usually caused by the degradation of MEA components, e.g., AEM degradation, catalyst nanoparticles aggregation or detachment from their support, electrochemical oxidation of ionomer, delamination of catalyst layers, or permanent impurity deposition on the catalyst surface. Several test protocols for cell components have been proposed to evaluate the component durability. Since the unrecoverable performance loss comes from permanent damage to the cell components, this is more critical to cell lifetime. However, one should also note that the operational parameter changes and transient cell treatment may lead to a shorter cell lifetime. Therefore, minimizing recoverable performance loss will be beneficial to achieve a longer life.
In some cases, AEMFC durability is carried out under the conditions that are more extreme than the expected operating conditions of a practical system to shorten the time needed for specific degradation processes to take place and manifest themselves. Two most popular accelerated stress test (AST) conditions are oxygen supply vs. CO2-free air and elevated operating temperature (>80 °C). Another AST condition that has been adopted is high voltage, voltage cycling or start/stop cycling, which rapidly degrades the electrode performance. Current US DOE HFTO component durability protocol use AST protocols for membrane and catalyst durability evaluation (Table 1).60 However, one should note that no good AEMFC lifetime prediction from ASTs yet exists and, therefore, ASTs have not been fully implemented for AEMFCs to date.
2.3 Protocols for MEA component durability
The chemical stability of AEMs is also evaluated using Fenton's reagent, ca. 4 ppm FeSO4 in 3% H2O2,64,65 which simulates a hydroxyl radical-rich environment by Fenton's reagent and provides additional information about oxidative stability of AEMs. In addition, since the formation of radicals from H2O2 decomposition is only one possible source of radicals, and not the most likely one in AEMFCs due to the very high self-dissociation of peroxide in alkaline media, other (yet to be identified) radical species resulting from the alkaline electrochemical reactions need to delineate the role and selectivity of direct and indirect potential-dependent routes.
000 cycles in dilute alkali metal hydroxide solution, ca. 0.1 M KOH.66–68 The ORR current density, onset potential or half-wave potential are measured during the potential cycling and then linked to catalyst degradation due to catalyst dissolution, agglomeration or detachment of metal particles. An alternative, and less common, stability test is to hold the RDE at a constant potential, e.g., 0.65 V vs. RHE, and to measure the current density as a function of time.51,52,68
Similar to the catalyst durability test protocols, ionomeric binder stability can be assessed by microelectrode studies.69,70 In this experimental set-up, a thin ionomer film, ca. 5 μm, is coated onto either a Pt disk or catalyst particles and then placed into contact with the reference electrode using an AEM. The ionomer stability can be measured either in dilute alkali metal hydroxide or under fixed RH conditions. Another useful durability test for the ionomeric binder is the RDE test in organic cation solutions.71–73 Organic cations such as tetramethylammonium hydroxide, tetraethylammonium hydroxide or benzyl trimethyl ammonium hydroxide can be added or replaced to the conventional alkali metal hydroxide. The advantage of this method over the microelectrode approach is simplicity. However, more complex stability behavior of ionomeric binders, such as polymer backbone degradation, cannot be properly evaluated.
3. Current status of AEMFC durability
The achievable lifetime for AEMFCs has improved significantly recently, even in just the past two years. In an earlier review, former generation AEMFCs generally suffered from very poor operational stability, even at low temperatures (40–50 °C) and current densities (ca. 0.1 A cm−2).13 Currently, AEMFC durability with >500 h tests at 65–80 °C and 0.6 A cm−2 is almost becoming routine. Fig. 2 shows the best reported durability of AEMFCs reported in the literature. Fig. 2a compares the recent AEMFC durability (2019)74 with the durability data of previously reported AEMFC (Tokuyama Corps, 2011)75 and PEMFC (2005).20 The 2019 AEMFC based on a quaternized HDPE AEM and ethylene tetrafluoroethylene (ETFE) electrode (red) achieved the lifetime (>1000 h) at 70 °C and a constant current density of 0.6 A cm−2. The cell voltage decay rate was 60 μV h−1. Though the 2011 AEMFC (blue) showed a lower voltage decay rate (34 μV h−1) at 50 °C during the first 1000 h, it should be noted that in 2019 the cell was operated at a much higher temperature and current density. Also, even at higher current density, the obtained cell voltage was higher as a result of recent efforts on AEMFC performance improvement. The substantially higher operating temperature of 2019 AEMFC may at least partially explain the much higher performance, although numerous contributing technological advancements were also incorporated including, for example, a PtRu/C anode, a very high-conductivity thin membrane, a novel electrode preparation process, and carefully optimized operational conditions. Critically, however, the 6 times higher current density at which the 2019 AEMFC result was obtained is a very significant accelerating factor (in part due to limitations of current technology) for AEMFC degradation, especially concerning chemical degradation of the membrane and ionomer, as will be discussed in Sections 5.1 and 5.2, and water management challenges, discussed in Section 4, which causes both reversible and irreversible losses. Therefore, the durability status of the 2019 MEA can be considered significantly higher than that shown in the 2011 data.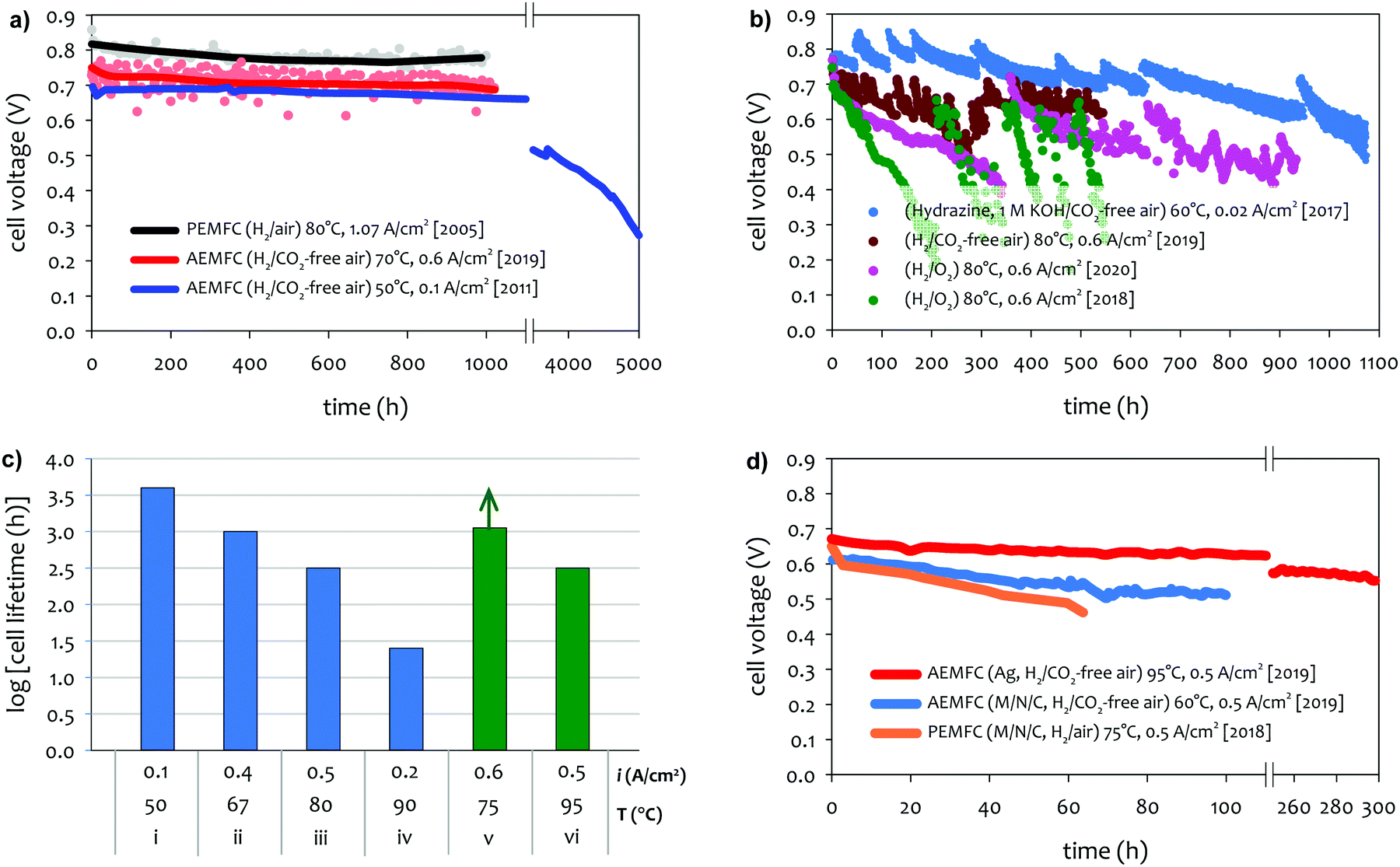 | ||
| Fig. 2 AEMFC durability reported during 2017–2020 (a) durability comparison between AEMFC (2019,74 201175) and PEMFC (200520). (b) Other significant AEMFC durability data (2017,76 2018,4 2019,3 and 202077). (c) Durability of MEA's based on materials from Tokuyama Co. (blue data set) under increasingly demanding conditions of current density and temperature, compared with current results using new ionomer chemistries (green data set). Source data: (i) Fukuta 2011;75 (ii) and (iii) PO-CellTech (unpublished data); (iv) ref. 6, ESI; (v) USC 2020 under H2/O2 conditions. This cell has been operating for 1400 h as of the submission of this article with a voltage decay rate of only 7.5 μV h−1, and the cell is still running; (vi) polyaryl-based membrane & ionomer.6 (d) Durability of PGM-free ORR catalyzed MEAs (AEMFC (Ag),6 AEMFC (M/N/C),10 and PEMFC78). | ||
Next, we compare the durability between AEMFC (2019) and PEMFC (2005). The voltage decay rate of the PEMFC (black) was slightly lower (54 μV h−1) at higher current density (1.07 A cm−2) and higher operating temperature, ca. 80 °C, indicating that the performance and durability of the PEMFC is higher than that of the AEMFC. Several other differences in catalyst loadings and operating conditions between the AEMFC and PEMFC are noted: (i) the catalyst loading for PEMFC is lower (0.43 mgPt cm−2 (PEMFC) vs. 0.6 mgPGM cm−2 (AEMFC)), (ii) AEMFC used CO2-free air vs. normal air for PEMFC, (iii) the reactant gas flowrate for AEMFC is higher (133/550 sccm for the 5 cm2 cell (PEMFC) vs. 1000/1000 sccm (AEMFC)), and (iv) some of the operating variables (reacting gas dew points and back pressurization) for the AEMFC test were dynamically changed throughout the experiment, while the PEMFC was run maintenance-free. The comparison indicated that the AEMFC durability has been improved, but is still inferior to that of the PEMFC.
Fig. 2b shows other significant AEMFC durability data reported over the last three years. Miyatake et al.76 reported >1000 h lifetime for Ni/C catalyzed hydrazine AEMFC at 60 °C and a constant current density of 0.02 A cm−2. The MEA was fabricated with quaternized perfluoroalkylene AEM and ionomer. Although the average voltage decay rate was high, ca. 300 μV h−1, the data is significant when considering they circulated 1 M KOH liquid electrolyte. An AEMFC based on a quaternized poly(phenylene) AEM30 and polyfluorene electrode4 showed a lifetime of ∼950 h at 80 °C (pink).77 The unrecoverable voltage decay rate changed throughout the durability test. For the first 350 h, the voltage decreased from 0.78 to 0.72 V (170 μV h−1). However, for the next 550 h, the voltage decreased from 0.72 to 0.56 V (290 μV h−1). Such a significantly higher voltage decay rate may be partly attributed that the test was performed using pure O2 instead of air. It was noted that the 950 h longevity was achieved from a polyfluorene ionomer with a high IEC (3.5 meq. g−1), which enabled cell operation at a reduced cathode RH. The previous MEA based on a polyfluorene electrode with a lower IEC (2.5 meq. g−1) (green) showed 550 h lifetime under 100% RH conditions.4 The effect of low cathode RH on AEMFC durability is discussed in Section 5.2.2. Another critical note is that the polyaromatics MEAs show a significant recoverable loss during the continuous operation of the cell. Kohl et al. reported the durability of an AEMFC based on a quaternized poly(norbornene) AEM and ETFE-based ionomer electrode (dark red) at 80 °C under H2/CO2-free air conditions.3 The MEA showed one of the best performances to date (peak power density of 3.4 W cm−2 under H2/O2, 80 °C conditions). The MEA showed >500 h lifetime with an overall voltage decay rate of 140 μV h−1, although it should be noted that the cell showed significant changes in voltage loss, which were recovered by changing the water content in the cell. Using a similar ETFE-based electrode, Pivovar et al. demonstrated >500 h longevity for several MEAs at temperatures between 60–70 °C.79,80
Although we reported a few selected AEMFC durability above, durability data in general is reported relatively rarely in the literature to date, and further, comparative reports employing MEA's under similar conditions with controlled variability, in high-performing MEA's, is virtually non-existent. However, surveying durability data employing commercial Tokuyama membrane and ionomer (one of the very few widely-employed and well-characterized standard membrane/ionomer materials) vs. temperature and current density, it can be surmised that the operating temperature plays a key role in AEMFC durability (Fig. 2c). Improvements over the past decade or so in other aspects of the technology have largely been realized against the backdrop of this baseline. Advances in ionomer chemistry that provide for improved chemical stability and facilitated water management, appear to be a key factor behind recent tests (Fig. 2a and b) starting to break out of that durability/current/temperature window, while further advancement is still clearly demanded.
There are a number of papers showing excellent stability of PGM-free catalysts under alkaline environments.81–85 However, there is a limited number of reports for AEMFC durability using PGM-free catalysts. In general, PGM-free catalyzed AEMFCs are known for lower durability compared to the PGM-catalyzed AEMFCs. Wang et al. compared the durability of low-density polyethylene (LDPE)-based AEMFC using three different ORR catalysts, ca. Pt/C (PGM), Ag/C (PGM-free) and FeCoPc/C (PGM-free). Although the MEAs using both PGM-free catalysts showed an excellent performance (>1 W cm−2 under H2/O2 conditions), the durability of the MEAs using the PGM-free catalysts is lower compared to that of Pt/C catalyzed MEA.8 Piana et al. prepared an MEA using in-house produced transition metal carbon-based catalyst (HYPERMEC 4020 from Acta S.p.A) on the cathode side.86 They compared the AEMFC durability between commercial Pt/C (40% Pt on Vulcan) and the carbon-based catalyst (metal loading < 10%). The performance of both MEAs decreased by more than a factor of 2 during 24 h at a constant voltage of 0.4 V, but more importantly, the decay rate of the PGM-free catalyst was higher. However, one area where best-reported AEMFCs currently outperform PEMFCs is cell stability with PGM-free cathodes, as demonstrated in Fig. 2d. Operationally, it would be expected for PGM-free cathodes to operate at higher potentials (translating to higher cell voltages) because of the intrinsically enhanced ORR activity in alkaline vs. acidic pH. Another possible advantage is that at high pH it is expected that metal dissolution, and hence electrochemical surface area (ECSA) loss, would be less. Third, hydrogen peroxide is much less chemically stable in alkaline media than acid media (in fact the decay rate is several orders of magnitude higher at alkaline vs. acidic pH). For carbon-based catalysts, this would mean much less opportunity for peroxyl attack or the formation of radicals on M–N–C catalysts (e.g., M = Fe).
For an MEA using Ag-based ORR catalyst,6 300 h of longevity was reported with an MEA based on piperidinium functionalized polyphenylene AEM and ionomer. The voltage decay rate is ∼400 μV h−1 at a constant current density of 0.5 A cm−2 and H2/CO2-free air. This result is significant mainly due to the high operating temperature of 95 °C. Indeed, it has only been in the past few years that durability tests at temperatures above 50–70 °C have been reported,14,17,87–89 and serves to showcase remarkable advances in recent years in AEM chemistry, as well as improved understanding of operating requirements especially with regard to water management in the MEA.
Several other stability studies with PGM-free MEA cathodes have been reported. Rao and Ishikawa90 reported 30 hour stability for an MEA using nitrogen-doped carbon nanotube ORR catalysts at a constant current of 20 mA cm−2. Huang et al. reported that the performance of the AEMFC using the transition metal N/S doped carbon ORR catalyst is only 16.5% of the initial value after 1 h.91 The lowest AEMFC degradation rate using PGM-free ORR catalysts was demonstrated during a 100 h test. Sanetuntikul and Shanmugam reported 8% current density loss of Fe–N–C cathode catalyzed MEA at 60 °C and a constant voltage of 0.4 V in H2/O2.92 Peng et al.10 reported 15% voltage loss of N–C–CoOx catalyzed MEA at 65 °C and a constant current density of 0.6 A cm−2. However, the power density loss of the MEA after the 100 h life test was rather significant (40%). The AEMFC durability using PGM-free HOR catalysts is even scarcer. Kabir reported that PGM-free NiMo HOR catalyzed AEMFCs showed that the current density decay from 50 to 40 mA cm−2 over ∼100 h at 60 °C and a constant voltage of 0.7 V.11 They explained that the possible performance loss may be due to the composition of the NiMo catalysts: nickel lost all metallic components and became Ni(OH)2 while molybdenum changed to a mixture of nickel molybdenum oxide and MoO3.
Based on the data shown in Fig. 2d, despite the fact that the durability of PGM-free ORR catalyzed AEMFCs presently exceeds the durability of their PEMFC counterpart, significant improvement is still needed to create a commercially viable system.
4. Degradation factors that impact AEMFC recoverable performance loss
4.1 Water management
| O2 + 2H2O + 4e− → 4OH− | (4.1) |
| 2H2 + 4OH− → 4H2O + 4e− | (4.2) |
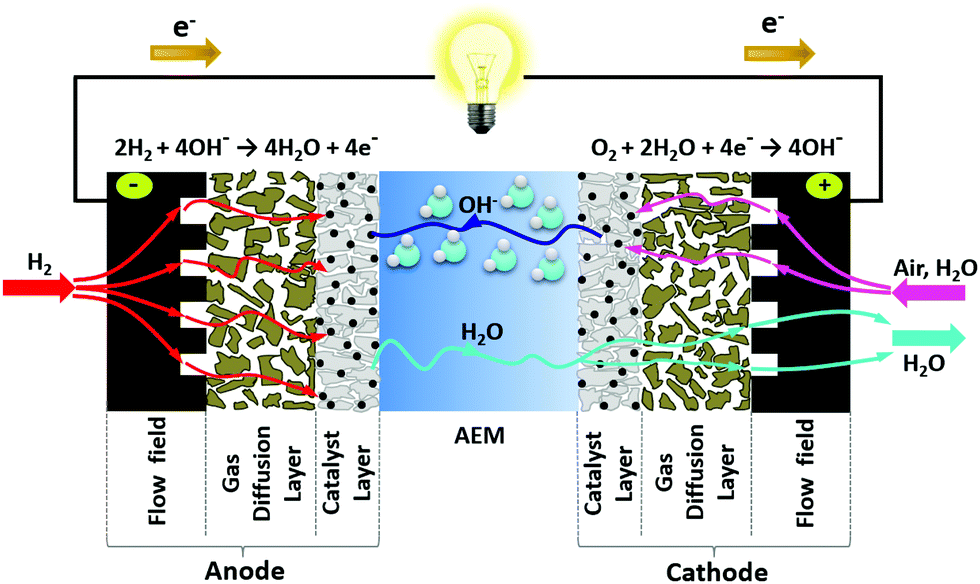 | ||
| Fig. 3 Illustration of the water dynamics in operating AEMFCs – showing the water produced and consumed by the reactions, electro-osmotic drag from the cathode to anode, and back-diffusion of water from the anode to the cathode. Reproduced with permission from ref. 93. | ||
While the HOR generates a significant amount of water, the diffusion of water through AEMFC anode is slow due to cation-hydroxide-water adsorption on HOR catalysts. Cumulative cation-hydroxide-water adsorption on the surface of HOR catalysts at the hydrogen oxidation potential, ca. 0.1 V vs. RHE was observed by several researchers.72,95–99 Surface infrared and neutron reflectometry studies indicated that the chemisorbed layer contains highly concentrated ammonium hydroxide and does not allow fast hydrogen and water transport.72,100 This means the water distribution in the AEMFC anode is non-uniform and the AEMFC anode requires high porosity for fast water transport, yet is more prone to flooding.
On the other hand, water in the cathode is consumed to generate current. What this means is that a substantial amount of the reacting water in the cathode should be supplied from the anode under high current density operation.101,102 The implications of this are twofold. First, if the water diffusivity for a given AEM is too low, the mass-transport limiting process in the cell is the diffusion of water through the AEM and the cell simply has no chance to achieve high performance. This helps to explain why the highest performing cells in the literature have also deployed very thin AEMs, 5–25 μm. That is not to say that thickness is the only important variable, as the physical chemistry of alkaline ionomers can play a significant role on their ionic conductivity, water mobility, and state of hydration.103 The second implication of low water mobility is that the cathode hydration state gets very low, and the activation energy for nucleophilic attack of the quaternary ammonium groups decreases significantly,104,105 causing irreversible physico-chemical damages to the polymer in the cathode. This also leads to irreversible performance loss, which will be discussed in detail in Section 5.2.3. Meanwhile, even subtle loss of cross-membrane water transport, that can be caused for example by partial membrane dehydration near the cathode interface, leads to a positive feedback loop causing the steadily increasing degradation rates that typically become apparent after 100's to 1000's of hours (depending on current density and temperature). This process was well captured in models by Dekel106,107 that showed quite good quantitative agreement and phenomenologically very similar voltage degradation profiles to experimental systems.
Another approach to reducing the degree of anode flooding is to redesign the electrodes themselves.2,7,115,116 In general, avoiding extremely thin electrodes is advantageous. This is because the anode ionomer provides a sink for liquid water to be absorbed. Also, increasing the hydrophobicity of the anode117 is helpful because it helps the anode layer to reject the formed liquid water. Increasing hydrophobicity also improves operational stability, and was necessary to achieve the high AEMFC operational stability for the 2019 cell in Fig. 2a.
The water conditions at the cathode are also important to determine the cell operational stability. One design criteria for the cathode that can be useful is to make the cathode hydrophobic as well. This is not intended to reject water, but to reduce the amount of liquid water that is allowed access to the cathode flowfield. Hydrophobicity can be added to the cathode catalyst layer through either the reduced RH with more hydrophilic ionomer77 or the addition of hydrophobic agents.117 Operating the cathode at a minimal degree of back-pressurization can also lead to the same effect. Another approach that has been used to overcome low cathode water by feeding the reacting gases at dew points above the cell operating temperature.110 In essence, this allows for liquid water to be fed to the cathode, which both can react and provide the necessary humidification to avoid performance loss. It is also possible to recover performance intermittently by pulsing the cell current or voltage, or making a voltage sweep; these will be further discussed later (Section 4.2).
4.2 Carbonation
| OH− + CO2 ↔ HCO3− | (4.3) |
| HCO3− + OH− ↔ CO32− + H2O | (4.4) |
The first mechanism indeed is related to the mobility of the carbonates from eqn (4.3) and (4.4). As stated above, the mobilities of (bi)carbonate are lower than OH−, which leads to an increase in the Ohmic resistance. The second mechanism is caused by the fact that the (bi)carbonate anions are not able to directly oxidize H2 in the anode at typical AEMFC anode potentials. This means that the carbonates formed at the cathode are not consumed at the anode by the reaction; hence, they are not immediately released to the anode gas flowfield as CO2 as they arrive. Instead what happens is that there is a time lag between CO2 exposure and CO2 release. During this time lag, the carbonates accumulate at the anode, causing the pH of that electrode to drop.126 As the pH drops and carbonates are accumulated, the reverse of eqn (4.3) and (4.4) occurs, resulting in the eventual release of CO2. The drop in the anode pH results in a Nerstian increase in the anode potential, reducing the overall cell voltage. The third mechanism is also related to the inability of (bi)carbonates to react directly with H2. The anode has a given IEC; therefore, the accumulation of carbonates creates a concentration gradient to manifest in the anode, and there are areas of the anode with low OH− concentrations. Combined with the fact that OH− is no longer the sole charge carrier, the anode reaction must now procure the reacting OH− anions through both migration and diffusion. This forces the anode current density to be concentrated close to the anode/AEM interface, increasing the effective local current density of the anode and forcing higher reaction overpotentials.
An AEMFC system can effectively deal with air CO2 down to a few ppm with an appropriate filtration strategy.127 But in their recent comprehensive study, Zheng and Mustain reported,74 for example, that even 5 ppm of CO2 can generate a >100 mV loss versus CO2-free air even at 1 A cm−2, with the effect significantly more pronounced still at lower current densities. Their detailed analysis confirmed that losses due to carbonation (thermodynamic and kinetic losses) are primarily the result of anode carbonation, and that performance loss due to increased Ohmic resistance across the membrane due to carbonate ion mobility are only secondary (typically <10% of the overall CO2-related voltage loss). As a CO2-loaded air stream hits the cell, near-full carbonation occurs within a few minutes to tens of minutes, depending on the air CO2 concentration, air stoichiometry, cell size and length of CO2 penetration loop.74 The process of decarbonization, meanwhile, is somewhat more complicated. Shutting off the CO2 supply results in a rapid (tens of minutes), partial recovery via “self-purging”. Cell voltage then plateaus, reaching a pseudo-steady state voltage well under the initial level. This voltage however also recovers slowly, achieving full recovery only tens of hours after removing the source of CO2 contamination.128
The remaining, long-term recovery process is a slow approach to chemical equilibrium of aqueous CO2 with the CO2-free atmosphere of hydrogen/water over the fuel cell anode.74 The question of an acceptable performance loss is outside the current scope of this discussion. However, the implication of carbonate sensitivity for cell, stack and system durability is also important to consider. We discuss here the effects of (a) a “failure event” in a system filtration unit resulting in a temporary, 400 ppm exposure leading to more or less full carbonation of the cell, and (b) a constant, few (<∼10) ppm CO2 concentration being fed to the stack.
4.2.2a Recovery from filtration failure event. It can be shown in a straightforward manner, that a filtration failure event is fully reversible,115 in contrast to CO2 buildup in alkaline liquid electrolyte fuel cells, where the process can cause irreversible damage via buildup of insoluble carbonate salts. Fig. 4a, adapted from DOE AMFC Workshop 2016,128 shows the effect of such a carbonation event occurring in a technical cell under current, and the above-described partial recovery. However, a simple perturbation of the cell operation shown in Fig. 4b, can quickly recover the remaining voltage: Following the removal of the contamination source in the air stream, a cell current above that of the normal operating window is drawn. The resulting high anode potential induces the so-called self-purge at the anode, decomposing carbonate at a greatly increased rate. The voltage recovery again stalls as described above, but at a lower carbonate concentration due to the higher anode potential at higher current density. The new steady state carbonate concentration is then lower than the steady state at the lower current density and the recovery is thus effectively complete. On return to the ‘normal’ current density, any remaining carbonate at the anode is at a far lower concentration such that the remaining cell potential loss (vs. the pre-contamination level) is negligible.
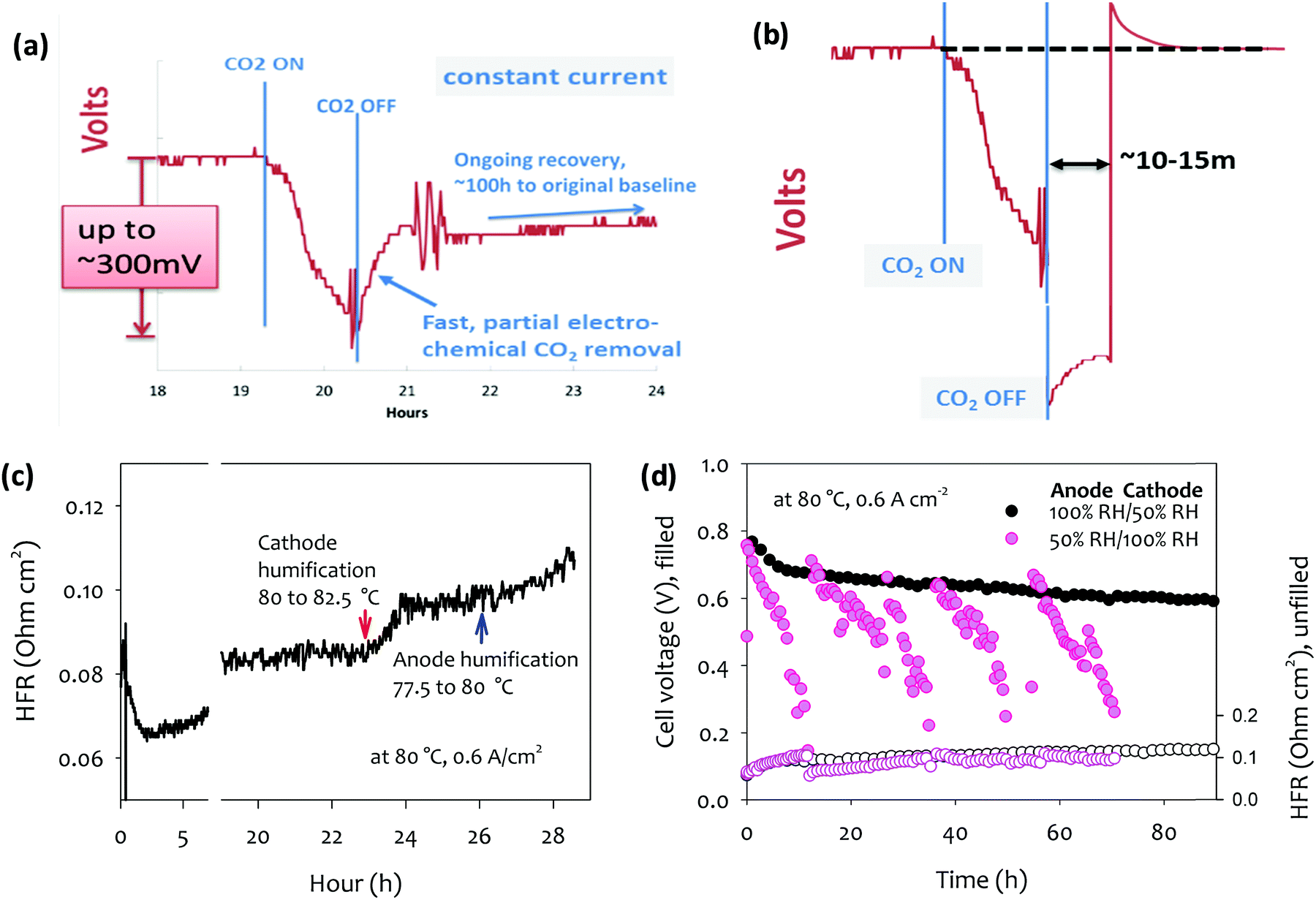 | ||
| Fig. 4 Carbonation and de-carbonation of an operating AEMFC in response to application and removal of a concentration of CO2 from the air stream (a) at constant current without perturbation to the cell load, and (b) at constant current, with a temporary applied high current for ∼15 min following the switch back to CO2-free air. Adapted from ref. 128. (c) The impact of humidification change on the cell voltage and HFR. Arrow points denote the humidification temperature change. Reproduced from ref. 61. (d) Cell voltage and HFR change of MEAs during 100 h short-term durability test.61 AEM: quaternized poly(polyphenylene) anode ionomer poly(fluorene) (IEC = 3.5 meq. g−1), cathode ionomer: poly(fluorene) (IEC = 2.5 meq. g−1), anode catalyst (Pt–Ru/C, 50 wt% Pt, 25 wt% Ru on high surface area carbon, 0.5 mgPt cm−2), cathode catalyst (Pt/C, 60 wt% Pt on high surface area carbon, 0.6 mgPt cm−2); H2/O2 AEMFC testing was performed at 80 °C and a constant current density of 0.6 A cm−2. Adapted from ref. 77. | ||
4.2.2b The effect on cell durability of continuous [CO2] in the air stream. The effects of a continuous low CO2 input are more subtle, and the effect on cell durability less clear. For the purposes of this discussion, the concentration value considered “low” is presumed to be [CO2]air < 10 ppm. It should first be recalled that the acute effect of 10 ppm CO2 on cell voltage at a given current density is far from negligible. However, the kinetics of the process is greatly dependent on [CO2] in the air feed.74 At 5 ppm, they report >60 min to full carbonation at 1 A cm−2, vs. ∼2 min at 400 ppm. A periodic, low-intensity “self-purge” process (of slightly higher current density) at sub-10 ppm concentrations is relatively straightforward in an AEMFC system (including, of course, at the startup phase of each discharge cycle). Bursts of higher currents naturally demanded by the power consumer mid-cycle also provide the same effect under normal operation. Therefore significant defense against the effects of this “background contamination” exists, and the data presented in Fig. 4a and b and in previous report74 suggest that the continuous low [CO2] feed unlikely lead to actual failure, although potential side-effects could be envisaged. For example, the less acutely important Ohmic loss component, leading to a lower conductivity/higher area-specific resistance (ASR), is a result of membrane (as opposed to anode) carbonation. In a carbonated membrane, water uptake and permeability are typically lower, which will likely affect the rate of ionomer degradation. Secondly, cationic degradation products from the ionomer in either electrode or membrane could form a carbonate solid salt and buildup in the system akin to a known liquid alkaline electrolyte degradation mode. Both these examples however, require already underlying degradation processes.
Another significant source of carbonation is electrochemical carbon oxidation reaction (COR) of cathode materials. The COR in low temperature fuel cells is a major concern as carbon in various forms is the most used electrocatalyst's support materials, ionomer, GDL and graphite plate. At a high cathode potential carbon surface is converted to CO2, hydroxyl, carbonyl and carboxyl groups according to following reactions:
| C + 2H2O → CO2 + 4H+ + 4e− | (4.5) |
| C + H2O → CO + 2H+ + 2e− | (4.6) |
| C + 2H2O → HCOOH + 2H+ + 2e− | (4.7) |
In summary, the potential contribution of background carbonation to long-term performance degradation plays a significant role in recoverable performance change, as well as being likely to amplify existing non-recoverable degradation mechanisms.
One of the most obvious variables to manipulate is the current density. In fact, even just a couple of years ago modeling work124 suggested that if AEMFCs were able to operate above 1 A cm−2, they would be able to “self-purge”; however, this has since been disproven experimentally74 and it is now known that AEMFCs remain carbonated at all operationally relevant current densities. Because the current density is related to the OH− production rate, increasing the current density can provide some relief to the CO2-related voltage loss, it only has a moderate effect. A second variable, the operating temperature, can be increased to somewhat lessen the negative effects of CO2. A third variable, the total CO2 dose to the cell can influence carbonation.135 Hence, lower cathode flowrates are preferred. Fourth, the level of cell hydration can play a role, where higher levels of water in the cell can slightly lessen the impacts of CO2. Finally, because the transport number, number of charge groups and interaction with water are all dictated by the AEM, the backbone and functional group of the AEM also can be manipulated to reduce the impacts of carbonation.
5. MEA component degradation that impacts AEMFC unrecoverable performance loss
In the early stages of the AEMFC research, it was hypothesized that durable AEMFC performance may be obtained with AEMs that show good stability under liquid alkaline conditions. However, as further research proceeded, the stability of other MEA components was shown to play a critical role in the device durability. Lu et al. observed that HFR of their MEA based on 1,4-diazabicyclo octane functionalized polybenzimidazole membrane and Pt-based electrocatalysts changed little over 100 h at a constant current density of 0.1 A cm−2; however, the charge-transfer resistance at low frequency increased from 0.7 to 1.2 Ω cm2, indicating the electrode was degraded more than the AEM during the durability test.136 This was surprising given the fact that it is well-known that Pt-based catalysts are highly stable under the test conditions. As an independent study, Liu reported that an MEA using alkaline stable long side-chain quaternized poly(phenyleneoxide) (PPO) showed poor durability compared to another MEA using less-alkaline stable benzyltrimethyl ammonium functionalized PPO, which showed an excellent fuel cell durability.16 Those two examples suggest that the alkaline stability of AEMs may be relevant to only part of the AEMFC performance degradation behavior over time. Therefore, in this section, we review the degradation of MEA components that have been shown to impact the AEMFC durability (operando stability) rather than general stability discussion of MEA components under high pH conditions.5.1 AEM degradation
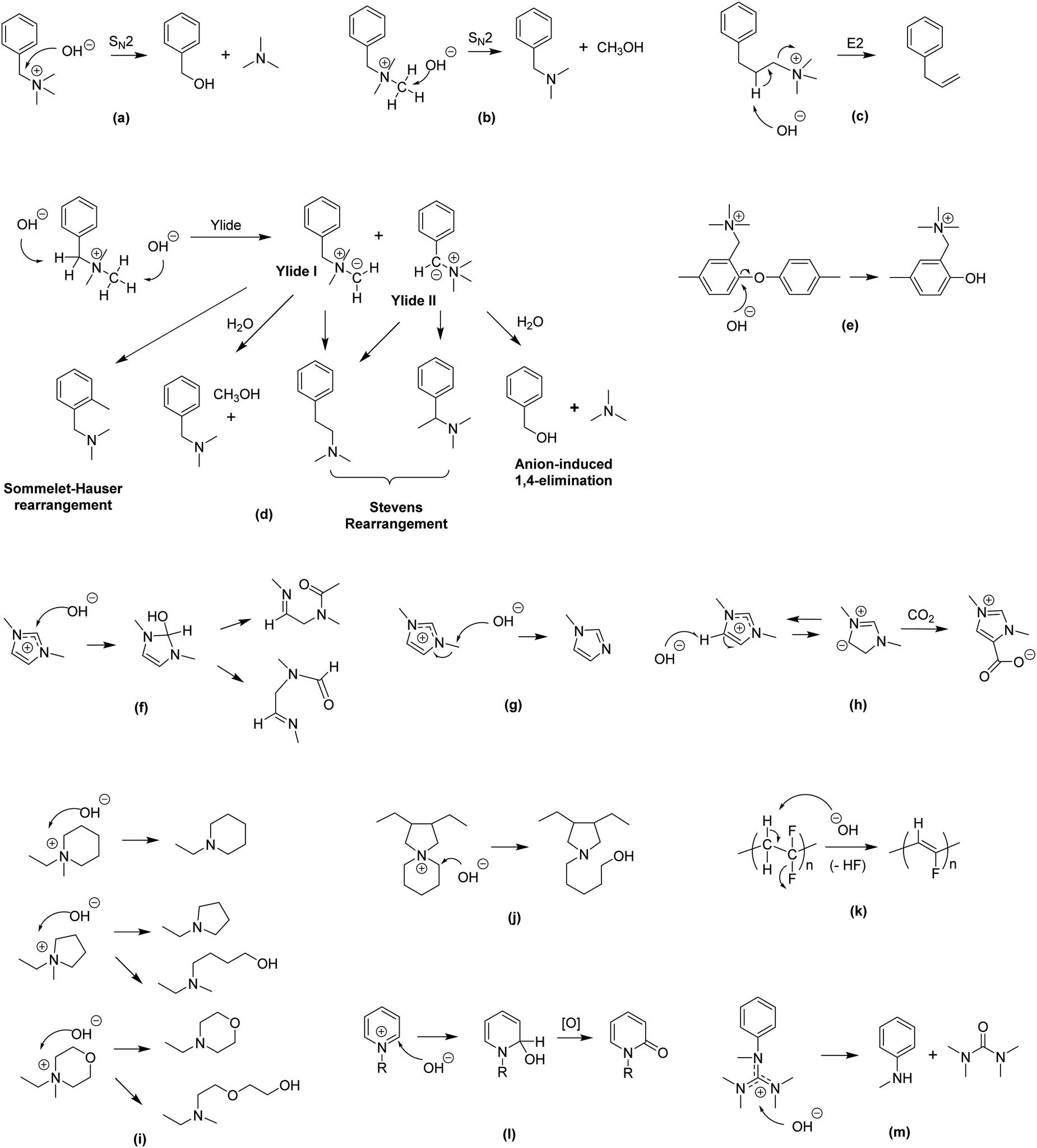 | ||
| Fig. 5 Various degradation pathways of AEMs (a) SN2 benzyl substitution,137 (b) SN2 methyl substitution,137 (c) β-elimination substitution,138 (d) ylide-intermediated rearrangements,139,140 (e) SNAr aryl ether cleavage (polymer backbone),26 (f) ring opening (imidazolium),141 (g) SN2 methyl substitution (imidazolium),141 (h) heterocycle deprotonation (imidazolium),141 (i) SN2 and ring opening (piperidinium, pyrrolidinium and morpholinium),142 (j) ring opening (N-spirocyclic ammonium),143 (k) dehydrofluorination (polymer backbone),144,145 (l) nucleophilic addition and displacement (pyridinium),146 and (m) nucleophilic degradation (guanidinium).147,148 | ||
Quaternized aryl ether-free AEMs showed good chemical stability. Mohanty et al. prepared aryl ether-free quaternized polystyrene-b-poly(ethylene-co-butylene)-b-polystyrene (SEBS) membrane.160 The AEMFC using the SEBS AEM showed voltage loss of 80 mV at 0.1 A cm−2 after running the fuel cell for 110 h at a constant voltage of 0.3 V at 60 °C. However, the chemical structure of AEM remained unchanged, as determined by elemental analysis and FTIR spectra. The cell resistance also showed stable behavior. Another stable performance of MEA using a SEBS AEM was reported by Su et al. for 100 h operation with flowing 1 M KOH solution to anode at 50 °C.161 Kuroki et al. prepared aryl ether-free spirobifluorene AEMs for direct formate alkaline fuel cells.162 The MEA stability assessed at 80 °C flowing 2 M KOH solution for 50 h indicated no AEM degradation. Kim reported the chemical stability the hexamethyl ammonium functionalized Diels Alder poly(phenylene) in an MEA at 80 °C under H2/O2 conditions.163 After 100 h test, the cationic functional group was intact with no change of cell HFR. Other aryl ether-free quaternized polyaromatics also showed stable performance for ∼100 h at 60 and 80 °C,164,165 supporting a good AEM chemical stability. Zhang et al. reported a stable fluoro-olefin-based AEM during 80 h-operation of AEMFC at 60 °C under H2/O2 conditions.166 However, it should be noted that most post mortem AEM analyses reported in the literature were performed after a relatively short time (<200 h). Therefore, it is too early to say that all current alkaline stable AEMs have enough chemical stability for long time operation of AEMFCs, and further research on this subject is necessary.
Aryl ether-containing AEMs degrade during AEMFC operation. Li's group reported several papers that analyzed the post mortem structural analysis of PPO-based AEMs after AEMFC test.16,43,167 All of the AEMs they tested did not survive >50 h during AEMFC even at relatively low temperature, ca. 60 °C. The most striking finding they made is that the degradation mechanism of the PPO-based AEMs during AEMFC operation is different from that of the AEMs in alkaline solution. They demonstrated alkaline-stable long side chain functionalized PPO degrades much faster than less-stable benzyltrimethyl ammonium functionalized PPO in fuel cell. They explained this behavior by oxidative degradation by super oxide anion radicals,168 which promotes SN2 substitution.
AEM degradation studies by radical species have been performed less extensively. While it is well known that the benzylic C–H bond of sulfonated polystyrene ionomers is susceptible to degrade by radical species,169 it seems that degradation rate of quaternized polystyrenes is much lower and at a similar level to the non-functionalized polystyrenes.64,170
Prevention of such mechanical failure of MEAs is relatively easy with several mitigating strategies. First, use an edge-protect gasket to avoid the sharp boundary between wet-dry of the AEM.173,174 Second, prepare AEMs with minimal dimensional change between wet and dry state.175–177 The minimum requirement of the dimensional change of AEMs for the durable operation of AEMFCs varies. Typically, less than 50% water uptake for rigid polyaromatics polymers and 100% water uptake for flexible polyolefinic polymers are required for stable AEMFC operation. AEMFCs employing AEMs with elongation at break of >100% show stable performance without edge-failure. Third, prepare AEMs with ductile mechanical properties.178–180 Fourth, avoid AEMs with backbone degradation. The mechanical property of quaternized poly(arylene ether) AEMs deteriorated by the aryl ether cleavage reaction.181 The degradation mechanism of aryl ether cleavage reaction is well documented in previous literature.26,158,181–183 Briefly, the electron-donating aryl ether group in the polymer backbone is destabilized by a positively charged (electron-withdrawing) ammonium cationic group close to the backbone. The energy barrier of the aryl ether cleavage in the benzyl ammonium functionalized polymer backbone is 85.8 kJ mol−1 which is lower than the energy barrier of α-carbons on benzyl trimethyl ammonium (90.8 kJ mol−1) and mechanical failure of AEMs often appears before cationic group degradation.158 The best strategy to avoid backbone degradation is to prepare aryl ether-free AEMs.164,184–188 Other mitigation strategies have also been employed including crosslinking157,189,190 incorporating cationic functional groups far from the polymer backbone aryl ether bond,79,191,192 and avoiding an electron withdrawing functional group in the polymer backbone.193,194 However, one should note that even without an electron-withdrawing functional group in the polymer backbone, e.g., quaternized PPOs, aryl ether-containing polymer backbones are not as robust as aryl ether-free polymers.184
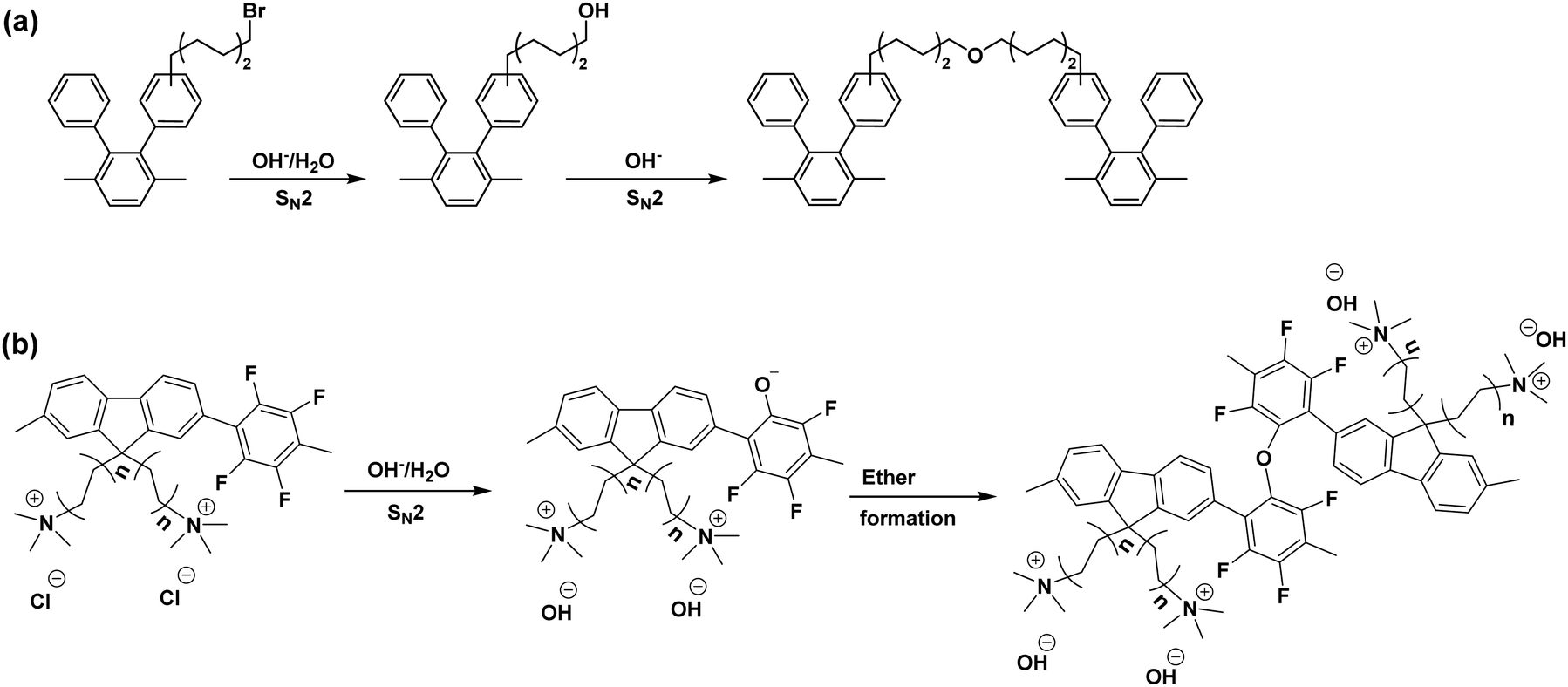 | ||
| Fig. 6 The proposed crosslinking mechanism (a) side chain crosslinking: the scheme is reproduced from ref. 195 (b) backbone crosslinking: the scheme is reproduced from ref. 197. | ||
Notable characteristics for the AEM crosslinking reaction are reported; (1) The chemical structural change by spectroscopic methods such as FTIR is insignificant, (2) the change of IEC of AEM is minimal, (3) significant decreases (30–40%) in AEM water uptake and hydroxide conductivity. The reaction rate of the crosslinking reaction depends on the concentration of the unreacted alkyl halide, which is varied from the AEM synthetic route. For example, direct polymerization of aminated monomers can minimize the amount of unreacted alkyl halide.26 For the post polymerized polymers, non-aqueous quaternization in ethyl alcohol in which the methylamine is too weak to remove a proton from ethyl alcohol; thus, the formation of alkyl hydroxyl group can be suppressed.195
Although the crosslinking reaction takes place over a few thousand hours at ca. 80 °C, the majority of the reaction may occur within the first few hundred hours of operation. Therefore, if the crosslinking reaction occurs, HFR increase accompanying fuel cell performance reduction for the early 100–200 hours is expected.
5.2 Degradation of ionomeric binder
Several approaches to mitigate the interfacial delamination including changing from a gas diffusion electrode (GDE) to catalyst-coated membrane (CCM), enhancing electrode adhesion through electrophoretic deposition, plasma treatment or wet-glue process,204–206 and reducing membrane water swelling.207
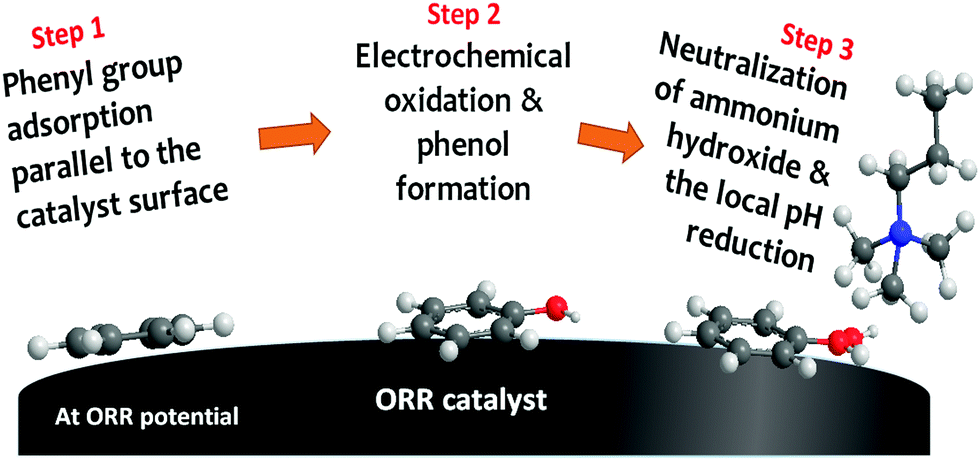 | ||
| Fig. 7 Quaternary ammonium neutralization process by electrochemical phenyl oxidation at ORR potential. The scheme is reproduced from ref. 208. | ||
There are a few critical points regarding the phenyl oxidation. First, phenyl oxidation is detrimental because the oxidation product phenol is acidic (the pKa values of 2-phenyl phenol and 2,2′-biphenol are 9.6 and 7.6, respectively), which neutralizes the basic hydroxide ion. The neutralization process negatively impacts not only hydroxide conductivity but also electrochemical activity of the ORR catalyst. Second, although the phenol concentration detected by 1H NMR is small, the local phenol concentration at the catalyst–ionomer interface is much higher and hard to remove from the interface as the phenyl group is covalently bonded to the ionomer. Third, the phenyl adsorption energy of phenyl group on the most active Pt is high. For example, the phenyl adsorption energy of the metal oxide terminated surface of PtO2 and IrO2 is −2.2 and −1.0 eV, respectively.208
Mitigating strategies for phenyl oxidation have been discussed. The first mitigating strategy is to use less-phenyl group adsorbing ionomer. Polyolefin ionomers have less phenyl group in the polymer backbone. Although some phenyl groups in the side chain can adsorb on to the ORR catalyst, the side chain phenyl group can be minimized with cationic group substitution.209 Matanovic et al. showed that some phenyl groups, e.g., polyfluorene, have relatively low phenyl adsorption energy,210 and thus, the AEMFC durability can be improved.61,211 The second mitigating strategy is to use less-phenyl group adsorbing catalysts. It has been shown that Pt-bimetallic catalysts such as PtRu, PtNi and PtMo have much less phenyl adsorption energy than pure Pt: for example, the phenyl adsorption energy in parallel to the surface of Pt(111) and PtRu(111) is −2.3 eV and −1.32 eV, respectively. Therefore, it may be considered to use a catalyst with low phenyl adsorption at the cathode, even though this could yield issues in terms of catalyst stability (as cathodes experience unavoidable potential cycling in start/stop operation – see Section 5.3). The third mitigating strategy is to operate fuel cell with low RH conditions. Since water is the reactant for phenyl oxidation, reducing cathode RH may help to reduce phenyl oxidation rate. This approach is clearly illustrated in Fig. 2b and 4d. If the MEA is operated under reduced RH at the cathode, the lifetime of MEA could increase due to the reduced rate of phenyl oxidation. However, as the ORR at the cathode requires water, operation of AEMFCs without humidification may be difficult. In this case, cell voltage should be also considered. If the cell voltage is not high enough, ca. <0.6 V, then the phenyl oxidation at the cathode may be negligible and the impact of cathode RH may be reduced. Therefore, this degradation pathway is more relevant to high cell voltage condition (high efficiency). Under high cell current conditions (high power), cathode ionomer degradation may occur with different degradation pathways, described in the next section.
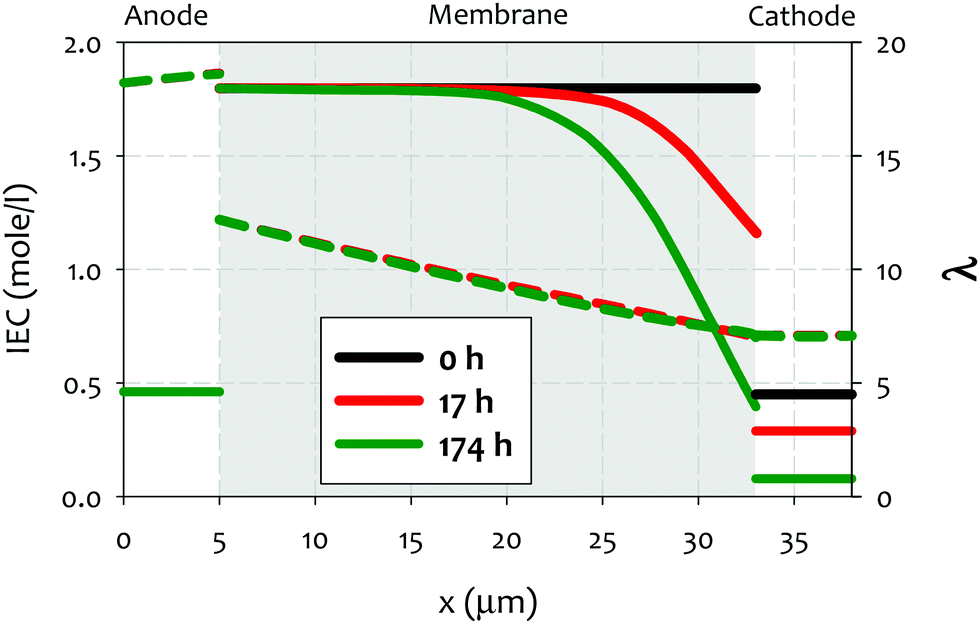 | ||
| Fig. 8 Ion exchange capacity (IEC) (full lines) and hydration number (λ) (dashed lines) across the cell, at the initial stage (t = 0 h) as well as at 17 h and 174 h after onset of AEMFC operation. The (constant) current density is 0.2 A cm−2 and the thickness of the AEM is 28 μm. Reproduced from ref. 212. | ||
A few noteworthy observations on the chemical degradation of quaternized polymers at low RH. First, all quaternized ionomers are less stable under lower RH conditions. Temperature plays a significant role in low RH stability of ionomer. Kreuer and Jannasch showed that increasing temperature from 60 to 100 °C increases the degradation at RH = 50% more than decreasing RH from 50 to 10% does.63 Second, the stability of quaternized ionomers at a given RH may be significantly influenced by the ionomer backbone structure. Quaternized ionomers with stiffer backbone may have limited swelling, thus, higher degradation rate is expected.63 Third, the extent of cation stability at lower RHs cannot be projected by alkaline stability of the cations under higher RH conditions. For example, 6-azonia-spiro undecane has a much longer half-life compared to trimethylbenzyl ammonium at λ = 9, but the much faster degradation occurs at λ = 4.107 Fourth, the degradation mechanism of cationic functional groups may be different between alkaline and low RH conditions. Park et al. showed that the degradation of alkyl ammonium under 4 M NaOH, 80 °C conditions occurs via β-elimination while the degradation under reduced humidity (10% RH, 100 °C) proceeded via SN2 methyl substitution.195 One mitigating strategy to prevent the chemical degradation of cathode ionomer is to use a thinner and highly water permeable AEM which helps the hydration of the cathode ionomer.212 Besides, operating AEMFCs under fully humidified cathode or high current generating conditions may reduce the possible cathode ionomer degradation. However, no experimental evidence that the cathode ionomer degrades during AEMFC operation has been shown in the literature, although some papers speculated such degradation might occur with their AEMs.178 This degradation pathway may become important with high power generating AEMFC system under low RH operations.212
Poor water management, considered above as a factor in recoverable losses, has a high risk in the AEMFC to lead to non-recoverable losses including chemical degradation of cathode ionomer. We propose mitigation via thinner membranes (to improve water delivery to the cathode) and improved chemical stability of the ionomer to dry conditions, while applying perturbations that could be considered “active water management”, such as periodic re-setting as described in the previous paper.102 Not considered directly by the model is possible rearrangement of polymer chains near the membrane surface leading to interfacial water transport effects. First reported in PEMFC systems,214 such effects are likely still more dominant at the cathode interface in AEMFCs, especially since it could exacerbate low-hydration induced ionomer degradation. The need for improved chemical stability in dry conditions is thus clear. A second factor to consider in a real system is that current densities and thus “cathode dryness” will vary significantly during operation. A fuel cell may spend much of its operating life, for example, at 0.2 A cm−2 while frequently being raised to much higher current densities for transient periods. The rate at which an ionomer and/or cathode electrode responds to such transients in terms of “releasing” water to reach a new steady state in response to such changes is therefore also an important consideration and can potentially be influenced by water management tools including ionomer chemistry, electrode layer morphology and additives, choice of gas diffusion media etc., as well as stack/system-level tools. A final consideration, because the proper cathode humidification is so critical to AEMFC long-term stability, is that it might be advisable to run AEMFCs at slightly higher water contents than what were previously described as “optimized” by Omasta et al.12 There, they were trying to maximize performance, which requires less water in the anode, and possibly lower water content in the cathode in order to encourage rapid back diffusion and high reaction rate. However, because of the lower water content of the cell, it is likely that the conditions that would result in the highest peak power or mass-transfer limiting current density are not the conditions that also result in the highest stability. What is needed from an experimental perspective is to develop combinations of electrodes, AEMs and operating conditions that allow for AEMFC operation near 100% RH without anode flooding or cathode dryout (or possibly flooding also). One caveat with this degradation mitigation strategy with water management is over-humidification of cathode may accelerate another degradation via electrochemical oxidation (see Section 5.2.2).
:1) at the HOR potential of ca. 0.1 V vs. RHE.100 While the concentration of the ammonium functional group of anode ionomer should be much lower due to the low mobility of the polymer tethered functional group, it is highly probable that relatively high concentration of cationic functional groups in the vicinity of the HOR catalyst surface becomes unstable at such conditions. Due to the high concentration of the ammonium hydroxide, the solubility of carbonated species significantly increases which may increase the longevity of AEMFCs. However, introducing carbonated species can further decrease the HOR activity of electrocatalysts. It was reported that introducing less cation-adsorbing ionomer such as triethyl ammonium hydroxide (TEAOH) functionalized ionomer instead of trimethylammonium hydroxide (TMAOH) functionalized ionomer may increase the stability of AEMFCs (Fig. 9).213 Less cationic group adsorbing HOR catalysts such as Pd-based catalysts may improve the durability, yet the AEMFC performance of using such catalysts is inferior to the Pt-based catalysts.18 Further research may need to identify the degradation pathway of anode ionomers.
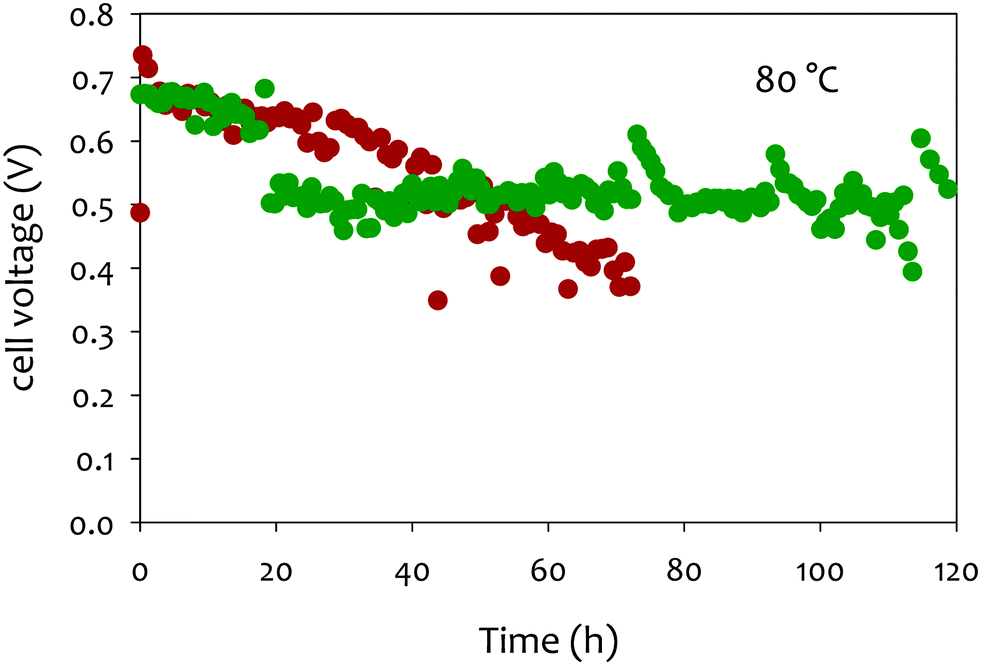 | ||
| Fig. 9 Impact of anode ionomer on AEMFC short-term stability of Pt anode catalysed MEAs. Measured the performance at 80 °C under H2/O2 (2000/300 sccm) at 147 kPa backpressure. AEM: m-TPN (∼35 μm thickness), ionomer: TMAOH functionalized poly(biphenylene) and TEAOH functionalized poly(biphenylene); anode: Pt/C (0.6 mgPt cm−2); cathode: Pt/C (0.6 mgPt cm−2), humidification: 80%. Reproduced from ref. 213. | ||
5.3 Electrocatalyst degradation
Durability is often flagged as a critical issue for AEMFC systems to compete with PEMFCs.219,220 Although the research efforts to unveil the degradation mechanisms of AEMs (and ionomers) have been intense (see the previous sections), much less effort has been devoted to the elucidation of electrocatalyst materials degradation at high pH. This stems firstly from the fact that AEMs were, until very recently, not enough durable to make electrocatalyst stability a major reason of failure for operating AEMFCs. Only very recently have long-term tests been successfully carried out with AEMFCs;221 the performances reached by several researchers were impressive, including in terms of durability in operation (>1000 h), suggesting that electrodes were stable also; however, this high stability was reached at constant polarization, i.e., not a representative test to monitor the field's durability (it will be seen below, that alternation of high/low potential can be very problematic to usual AEMFC electrocatalysts). In essence, the “common knowledge” often led (and still leads) researchers to speculate that electrocatalysts will be stable in alkaline conditions (at least more than in acidic conditions), an assumption which relies on the well-known calculated thermodynamic Pourbaix diagrams.222 In line with these predictions, “bulk structures”, like RANEY® nickel223,224 or fritted silver,225 which are used for the hydrogen evolution reaction (HER)/HOR and ORR, respectively at aerial loads of several mg cm−2, have of course shown some stability in circulating liquid electrolyte operation, because these materials are intrinsically robust (there is plenty of electrocatalyst material to oxidize/dissolve/degrade and their texture is not as “sensitive” as present AEMFC active layers); whatever these advantages, it must be pointed out that the performances reached were far inferior to the present state-of-the-art (especially when expressed per gram metal), and not stable on the long-term. Besides, materials availability and cost constraints advise the research community to look for electrode materials that consume less metals. In that context, the Holy Grail is nanostructured electrocatalysts, and the present state-of-the-art consists of nanostructured carbon-supported nanoparticles; more recently, hierarchically porous M–N–C cathode catalyst layers (M = Co or Fe)226,227 as well as active layers based on Metal Organic Frameworks (MOF)228 have also been developed, even though durability studies on these materials remain scarce.Materials stability issues are of course not less expected for supported and nanostructured electrocatalysts. Tests performed starting in the 1980s soon showed that usual carbon-supported electrocatalysts (the present standard in PEMFCs) can be unstable in alkaline conditions. The seminal work of Ross et al. for instance demonstrated that high surface area carbon materials were prone to gasification (into CO2) or dissolution (into (bi)carbonates) in strong alkaline environments, these processes being emphasized in presence of transition metal (or metal oxide) catalytic moieties at their surface.229–232 This work was essentially directed to the durability of electrode materials for circulating liquid electrolyte alkaline fuel cell (AFC), a solution that was developed with success for the space conquest but which is only marginally envisaged nowadays. Later on, Kiros and Schwarz evaluated the durability of Pt/C + Pd/C composites (C = charcoal) for the HOR in 6 M KOH at 60 °C, and discovered that the nanoparticles suffered intense coarsening upon a 3600 h-long polarization (j = 100 mA cmgeometric−2), provoking large electrochemical surface area losses and resulting in depreciated performances versus time.233 Chatenet et al. also found that a Pt/C (C = Vulcan XC72) electrocatalyst used for the ORR in 11 M NaOH at 80 °C (for brine electrolysis applications) experienced non-negligible Ostwald ripening and ECSA losses; in these conditions, the nanoparticles coarsening was inferior for AgPt/C alloyed nanoparticles.234 More recently, Olu et al. confirmed these findings, still for Vulcan XC72 carbon-supported Pd or Pt-based electrocatalysts operated in liquid alkaline electrolytes: pronounced loss of nanoparticles from the carbon substrate, agglomeration and coarsening of the remaining ones were noted after rounds of tests in direct borohydride fuel cell conditions, the degradations being more intense for Pt/C than for Pd/C.235
This short literature review shows that the benchmark electrocatalysts for AFC/AEMFC electrodes (carbon-supported Pt and Pd) suffer some degradation in operation. The group of Mayrhofer confirmed that, indeed, Pt can suffer intense dissolution in basic electrolytes when polarized below/above its surface oxidation potential,236 which suggests that classical carbon-supported Pt nanoparticles would suffer in AFC/AEMFC load cycling. On this basis, Zadick et al. initiated studies in which identical-location transmission electron microscopy (IL-TEM) was used as a tool to survey how Pt/C nanoparticles (C = Vulcan XC72) would react upon potential cycling in 0.1 M NaOH at 25 °C. Surprisingly, only 150 voltammetry cycles in the stability domain of water (0.1–1.2 V vs. RHE) is enough to dramatically alter the structure of an initially well-defined electrocatalyst; ca. 60% of its ECSA is lost, and this essentially proceeds by detachment of the Pt nanoparticles from their carbon support (neither real carbon corrosion was witnessed by XPS and Raman spectroscopy, nor Pt dissolution).237 In comparison, the same electrocatalyst is only minorly degraded in similar acidic electrolytes. More recently, Lafforgue et al. rationalized the mechanism of degradation by using in situ FTIR coupled with IL-TEM, and proved that the weak point of Pt/C in these conditions, lies in the propensity of Pt nanoparticles to assist the local corrosion of their carbon substrate into CO2 and then carbonates, thereby breaking their binding to the carbon substrate and hence provoking their detachment and agglomeration or loss.238,239 The detachment is believed to be emphasized when solid carbonates are formed at the interface between the Pt nanoparticle and the carbon substrate, as evidenced by the strong effect of the alkaline electrolyte (larger detachment in the sequence LiOH > NaOH > KOH > CsOH).238 In this process, Pt facilitates the oxidation of nearby carbon surface groups (that spontaneously form on carbon above 0.207 V vs. RHE) as soon as it nucleates water, in a process that resembles the Langmuir–Hinshelwood CO-stripping process (Fig. 10).
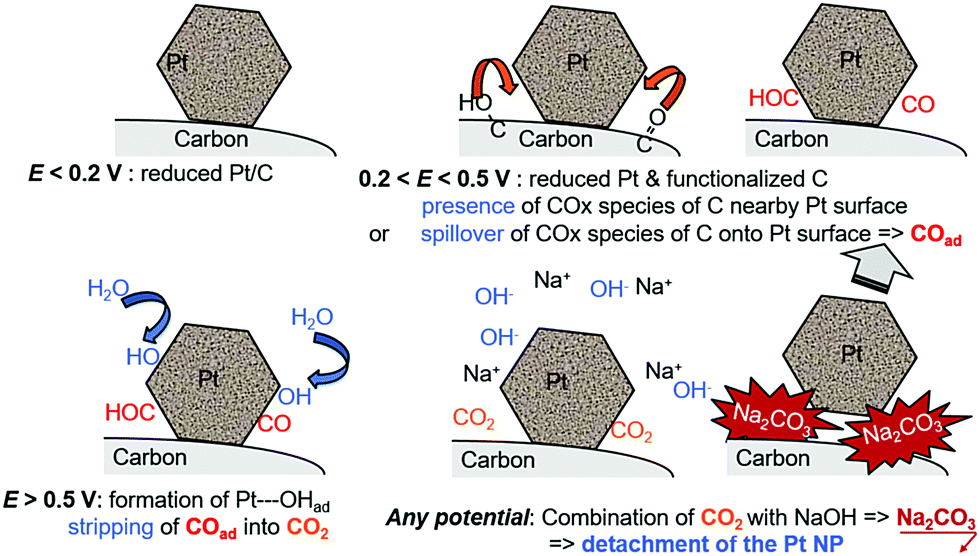 | ||
| Fig. 10 Schematic representation of the processes leading to the Pt nanoparticles’ detachment from their carbon support during repeated potential cycling from reducing (e.g., E = 0.1 V vs. RHE) to oxidizing (e.g., E = 1.23 V vs. RHE) conditions in MOH (M = Na, K, etc.) electrolytes with Pt/C electrocatalysts. The surface groups presented are indicative and do not pretend to be exhaustive. Freely adapted from ref. 238 with permission from Wiley. | ||
In that extent, it is no surprise that PtRu/C, the most-active HOR electrocatalyst in AEMFC conditions,240 but also a very active electrocatalysts to oxidize CO, shows very little durability when potential cycled in liquid alkaline environments;220 in comparison, Pd nanoparticles are subjected to smaller (but non-negligible) degradations238,239,241 (Fig. 11a).
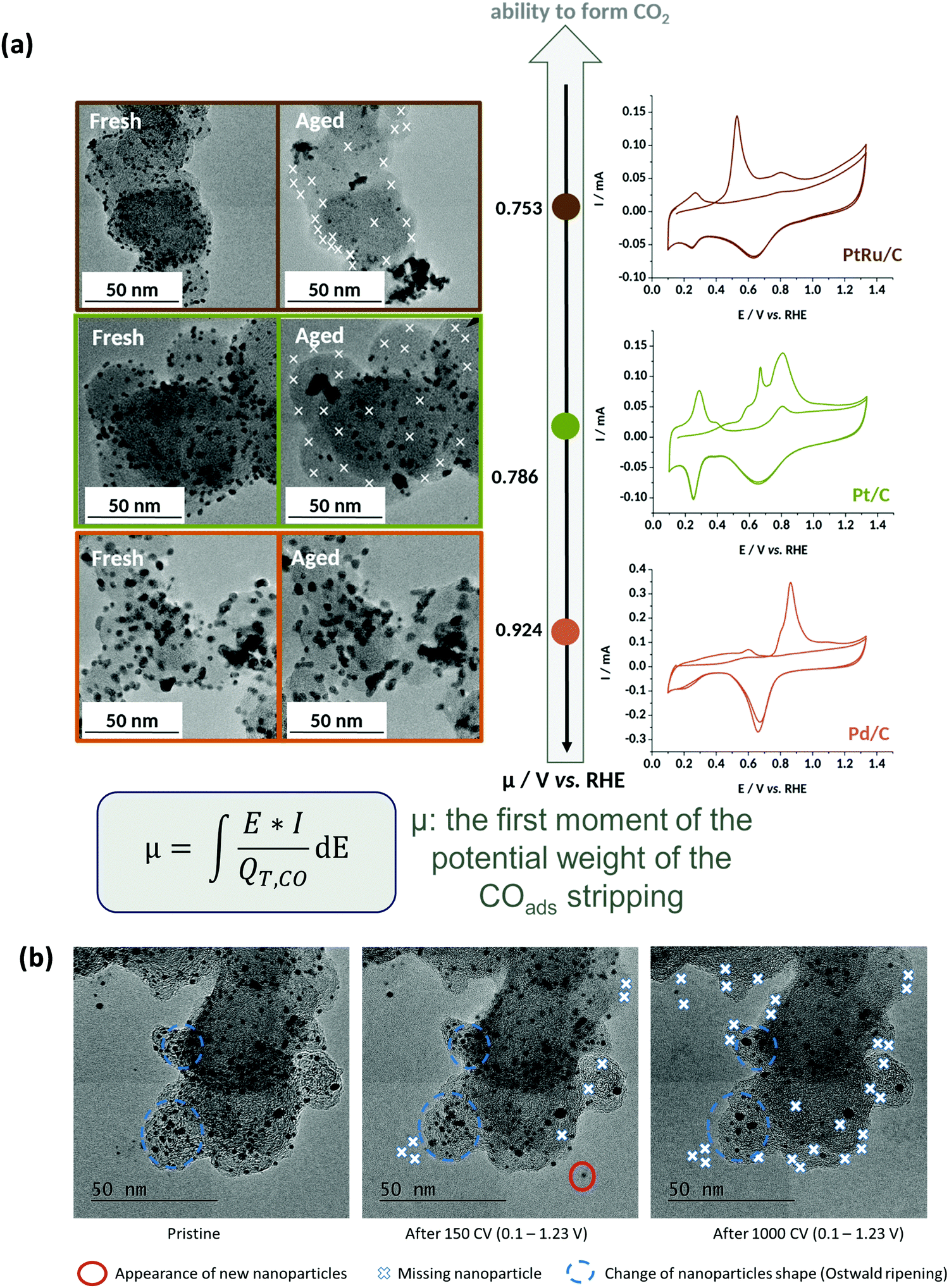 | ||
| Fig. 11 (a) IL-TEM micrographs pre-AST and post-AST and COads stripping voltammograms recorded at v = 20 mV s−1 in 0.1 M NaOH on 10 wt% PtRu/C (brown), 10 wt% Pt/C (green), and 10 wt% Pd/C (orange). The extent of nanoparticles detachment is correlated with the average COad stripping potential (first moment of the potential weight of the COads stripping). (b) Representative IL-TEM micrographs of Pt/C nanoparticles before (pristine) and after 150 or 1000 CV cycles performed at v = 100 mV s−1 between 0.1 and 1.23 V vs. RHE in interface with an anion-exchange membrane in the dry cell at T = 25 °C. The markers are not comprehensive and just illustrate the main degradation mechanisms at stake during the potential cycling procedure. Reproduced from ref. 242 with permission from the American Chemical Society. | ||
In this mechanism, it is precisely the potential cycling between a reduced and an oxidized state of surface of the electrocatalyst that is at the origin of the nanoparticles detachment. The amplitude of the potential window tested here (0.1–1.2 V vs. RHE) corresponds to that experienced by any electrocatalyst in an AFC/AEMFC operated in start/stop mode; at stop, the oxygen and the hydrogen electrodes will be at ca. 1 V vs. RHE (under air), while the anode will be close to 0 V vs. RHE in normal operation and the cathode will transiently reach close to 0 V vs. RHE in the early stages of the stop period, where the H2 present at the anode will naturally crossover to exhaust the cell. Tests performed by reducing the amplitude of the potential variations (0.1–0.6 V vs. RHE or 0.6–1.2 V vs. RHE) did not suppress these degradations, but simply lower their magnitude.242 So, even in “classical” load cycling in HOR or ORR operation, would Pt/C experience such nanoparticles detachment from the carbon support (if operated in liquid alkaline electrolytes). The size and loading of the nanoparticles, and the nature of the carbon substrate have marginal effects, both for Pt (10, 20 and 40 wt% Pt/C experience the same degradation phenomena)237–239 and Pd:241 the former is believed to influence (slightly) the propensity of the metal nanoparticles to be reduced at the lower vertex potential (the same applies to the presence of reducers in the electrolyte), while the latter mostly influences the initial definition of the nanoparticles but not so much the overall process of metal-assisted local carbon corrosion.243 In principle more robust graphitic carbon structures are not much more stable than amorphous ones, because they also get functionalized by O-containing groups above 0.207 V vs. RHE and the metal nanoparticles assist the oxidation of these groups into CO2 in an identical manner as for initially defective carbons.
Using a “buffer” layer between the metal nanoparticles and the carbon substrate (e.g., Pd/CeO2/C) can help to mitigate such degradations.244 In that case, though, the aging is not suppressed: it appears that if the nanoparticles detachment is significantly slowed down, another mechanism of degradation can proceed, like Ostwald ripening, a process that Pd (and Pt) nanostructures are prone to undergo upon potential cycling below/above the onset of their surface oxidation.245
It is also possible to minimize the effects of the metal nanoparticles detachment by using materials that cannot promote COads stripping, and Ni alloys seem a good solution in that direction.246 Mayrhofer et al. also studied the fate of nanostructured Ni-based electrocatalysts (Ni/C and bimetallic Ni3M/C, M = Co, Fe, Cu, Mo) vs. (electro)chemical oxidation/dissolution in HOR-relevant conditions.247 Whereas Mo was found to suffer intense dissolution owing to its thermodynamic instability, Cu was stable below 0.4 V vs. RHE, though it underwent non-negligible transient electrochemical dissolution above 0.4 V vs. RHE. On the contrary, Ni, Co, and Fe were found to negligibly dissolve below 0.7 V vs. RHE. The absence of dissolution must, however, not be taken as a guarantee to maintain high electrochemical activity, as all catalysts do lose their HOR properties upon incursions to potentials above 0.4 V vs. RHE, as a result of detrimental surface oxidation of the metal nanoparticles, that triggers the HOR deactivation. The authors conclude that all the electrocatalysts but Ni3Mo/C exhibit non-negligible stability in conditions relevant to an AEMFC anode in “normal operation”; however, as fuel starvation (or simply stop of the AEMFC system) would result in anodic potentials above 0.5 V vs. RHE, none of the tested materials would be robust against passivation/HOR deactivation without “system-like” strategies to control their state of surface (and these strategies are yet to be built).
Finally and importantly, the fate of Pt/C was also studied in interface with an AEM, the experiments being performed in a so-called “dry-cell”. In these conditions, the detachment of Pt nanoparticles is greatly slowed-down, but it is not suppressed248 (Fig. 11b). This shows that nucleation of solid carbonates (M2CO3, M = Li, Na, K, etc.) is not mandatory to provoke the detachment of nanoparticles; instead, the formation of soluble carbonates from the oxidation of the carbon substrate at the vicinity of the metal nanoparticles would be enough to break the binding between the Pt nanoparticles and their carbon substrate, eventually leading to their detachment. It is clear that if solid carbonates form, the detachment is emphasized, as shown in the previous sections.238–242
These selected results show that PGM/C catalysts are subjected to non-negligible degradations when operated in potential-cycling conditions, especially for large amplitudes of potential variation between reduced and oxidized states. In these conditions, the main degradation pathway consists of the detachment of the metal nanoparticles from their support following a metal-assisted local corrosion of the carbon substrate into carbonates, hence breaking the binding between the nanoparticle and the substrate. Other degradation phenomena are also possible (but slower) if this process is mitigated, and in particular Ostwald ripening; this mode of degradation will proceed in the long term if the nanoparticles detachment is “suppressed”. This highlights that reaching durable AEMFCs not only implies to work on the durability of its AEM and ionomer, but also that of its electrodes. More importantly, electrocatalyst degradation must be studied at the interface with the polymer electrolyte, if possible in AEMFC-relevant conditions. The interfaces between these two components must also be surveyed upon real operation, as it is likely that typical phases of the AEMFC usage will lead to specific (and local) degradations; start-stops are a typical example,249 in which heterogeneous operation is witnessed. Such heterogeneities of operation will likely lead to heterogeneities of aging, as was the case for PEMFCs,133,250–252 but this remains to be studied for AEMFCs.
6. Durability in AEMFC systems
Up to this point, discussion has focused mainly on component materials and fuel cell tests on small active area, so-called “differential” cells (usually ca. 5 cm2 in active area). While the fundamentals of the system do not change in “technical” scale cells, a designation typically conceded for cells of 50 cm2 or greater,253 additional variables and limitations need to be taken into account, and this is still further complicated in a multi-cell stack. A further layer of complexity is added when the stack is operated in a stand-alone system rather than a dedicated and well-controlled test bench.One of the challenges for AEMFC commercialization, is that a would-be developer of AEMFC MEAs and the component materials, cannot rely on existing PEM stack or system technology, operating procedures, control systems, etc. as a platform for testing, due to the fundamental differences especially in water management and the behavior of core material performance parameters in response to variations in, for example (but not limited to), humidity, temperature and pressure, in addition to the need for CO2 filtration in the AEM.
Therefore, we discuss some of these issues with respect to technical AEMFCs, stacks and systems. While less published data is available for these effects, we survey some of the considerations that must be applied to cell and stack operations as well as MEA design principles and fabrication requirements, and the implications of these for research requirements at the core material and differential cell levels.
Although transition to multi-cell stack and then standalone system are very significant, the switch to a large active area AEMFC is perhaps the most consequential. A feature of the impressive power densities recently achieved (up to and above 3 W cm−2 in 5 cm2 cells operating under hydrogen/oxygen3), and indeed the beginnings of successful durability tests as shown in Fig. 2, is the carefully balanced temperature, reactant flow rates and RH to tune cell operation to maintain optimum water balance in the MEA.
In the technical cell, such delicate control cannot be achieved. MEAs (and indeed flow fields) need to be designed not to one ideal condition but rather a spectrum of conditions corresponding to the intra-cell variations as well as, in an operational setting, changing external conditions, most significantly the power demand and ambient temperature.254
Fig. 12 shows a durability test of an application-sized MEA (ca. 250 cm2 active area) using the same Tokuyama materials as Fig. 2a, recorded at 0.4 A cm−2 and 67 °C. The larger active area cell results in some significant variability in output voltage, making determination of linear degradation rate somewhat difficult, but it can be seen that the cell holds to a negative slope of ∼50 μV h−1 for ∼600 hours, with a larger slope setting in over 600–1000 h. Thus, while this MEA is able to pass current for ≫1000 h at 0.4 A cm−2, performance as well as degradation rate are still inferior to the 2019 data (Fig. 2a), while the cell is operating at a lower temperature and current density. The data presented in Fig. 2 shows that, at least at moderate temperatures and current densities, an effective strategy in flow field design + operating conditions, can lead to cell durability that is comparable to that achievable in differential cells. The primary cell failure mode can be attributed with reasonable confidence to a steadily worsening water management paradigm, that can be understood when considering the AEMFC cathode as described in Section 4.1. The rate of this underlying degradation mode is likely non-linear, monotonically increasing over the whole cell lifetime, and although initially obscured by reversible voltage variability, ultimately revealing itself as it converges on catastrophic failure somewhat above 1000 h, as also seen towards 4000 h in the 2011 data of Fig. 2a.
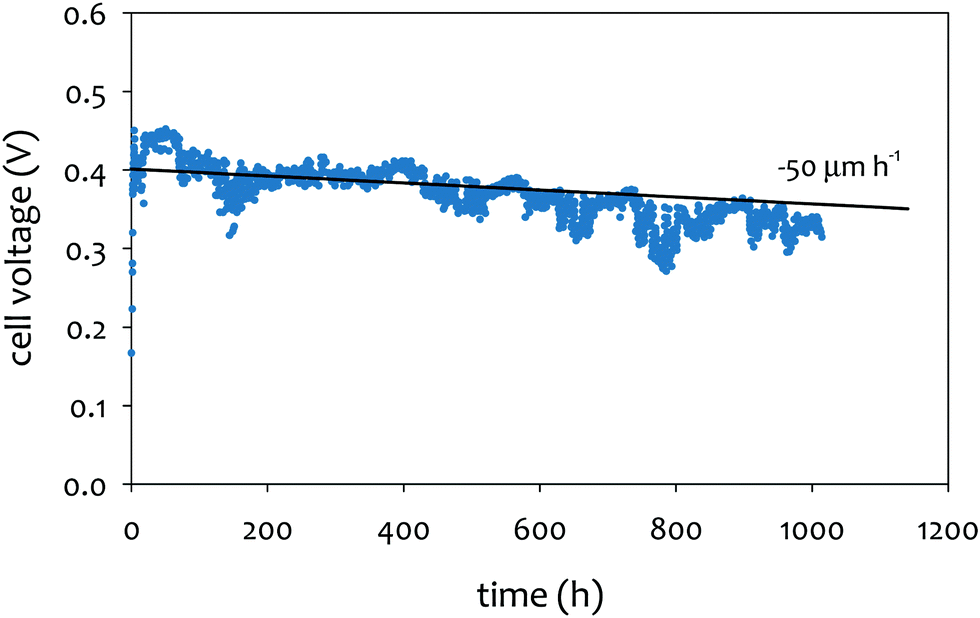 | ||
| Fig. 12 A ca. 250 cm2 MEA operated at 67 °C 0.4 A cm−2 over 1000 h. The indicated degradation line of −50 μV h−1 holds for ∼600 h. Data recorded at PO-CellTech. | ||
6.1 Water management in technical cells
Gottesfeld et al.204 described the water management challenge in a technical AEMFC cell, showing an operando neutron radiography image of a 240 cm2 device and highlighting the variations in the overall cell water content over the active area. Without covering all the details here, the important conclusion is that, in the technical cell, the delicate balance between under-hydration that leads to cathode dry-out, and over-hydration leading to anode and even cathode flooding, is recast in such a manner that both phenomena can easily be observed simultaneously and in different areas of the same cell.The question then arises as to the strategy to avoid such a situation from occurring, or at least from causing irreversible losses in cell performance. Gases entering at low humidity quickly become humidified by the cell itself. In any reasonable configuration, the outward gas streams will be fully heated and humidified. Temperature variation around the cell active area is also a feature and can be as high as 10 °C at higher current densities.
As a first order optimization, flow fields can be designed to minimize coincidence of high (or low) reactant humidity in the anode and cathode. Natural or induced temperature variations within the cell/stack can also be exploited to alleviate the issue to an extent. These technical scale water management considerations that call for hardware and system level design are of course not special to AEMFCs. However, the high sensitivity of especially the AEM – in terms of degradation195 as well as performance characteristics102 – to changes in humidity and temperature create a more significant barrier to effective operation.
Second-order strategies could include region-specific variations in gas diffusion layers or even catalyst layers – although such added complexities are undesirable (though not inconceivable) when considering device production and manufacturing costs. It is important to recognize that the very need for such approaches – the solutions to which must translate to actual costs somewhere in the value chain – stems from the ongoing limitations of this basic component of AEM technology – namely the lack of resilience of ionomers to departures from ideal hydrothermal conditions. With all the advances in the field from recent decades, this sensitivity presents itself as an emerging challenge to technology developers.
The technical water management strategy employed in Fig. 12 is to minimize the potential damage caused by drying of any part of the MEA, while accepting and addressing the resulting over-hydrated conditions in some regions of the active area.
In this approach, the humidity settings near both anode and cathode gas inlets are tailored such that the operation at these points is similar to that found in differential cell “sweet spots”, corresponding to RH values that are high but ≪100%, thus assuring that these locations which are those most susceptible to drying conditions remain well hydrated and performing optimally. This operational choice leads to a steadily increasing levels of hydration along the gas flow fields from an already well-hydrated starting point, and results in a large proportion of the active area in what would normally be called a “flooded” state.
The approach to achieving durability is then to fortify the MEA and its components against modes of degradation arising from such flooding. This can include for example, cross-linking of electrodes,255–257 and providing for effective liquid water egress via electrode formulation, selection of gas diffusion layers, and design of flow fields.
This is virtually the opposite of a common operational approach in PEMFCs, in which a portion of the MEA is allowed to remain somewhat dry, in order to avoid flooding in other parts of the active area. In the AEMFC, ionomeric degradation under dry conditions as well as the need to supply water as a reactant in the cathode, means that erring on the side of over-hydrating yields a better outcome at the present state-of-the-art of component materials. A further advantage to this direction is that the more flooded anode is normally supplied with 100% reactant, rather than the 20% found in the water-generating PEM air electrode.
There is of course a significant price to be paid in performance as a result of large portions of the MEA operating under a perpetual state of over-hydration. Choosing conditions optimized for power density, the MEA employed to generate the data in Fig. 12 is able to achieve ∼0.75 V at the same current density in a 5 cm2 differential cell at beginning of life [For example, Fig. 7 in ref. 219], and >0.7 V in the technical cell [unpublished data].
To achieve higher cell performance in such an approach, pursuing higher operating temperatures is a reasonable way to mitigate the effects of over-hydration. However, due to the sensitivity of the ionomeric material to low RH increasing, with temperature, higher RH settings are also required thus limiting the effectiveness of this approach. Higher temperature does have a positive effect on achievable power density (though presumably due more to increased catalytic activity than facilitated water management), and so the high temperature durability of component materials remains a key area for development.
6.2 Air and hydrogen stoichiometry
Assuming it should be limited to applicable operating conditions, a technical cell may not be operated at the high reactant gas stoichiometries typically employed in differential cells. Due to cost/sizing considerations of the air feed subsystem, acceptable air stoichiometry is well below 2.5, and product targets significantly lower still, especially for automotive applications where an air stoichiometry of 1.5 is targeted for PEM fuel cell systems.258The H2 feed meanwhile must approach 1.0 to avoid fuel waste, although this can be achieved by an identical fuel circulation subsystem as for a PEM device,252 albeit with modified specifications and control parameters.
In the light of the above, the high flow rates employed in the literature for differential AEMFCs may look unrealistic. However, high flows in differential cells are needed to provide a certain gas velocity to remove excess water droplets that in technical cells is achieved with much lower stoichiometries. Any given (for example) 5 cm2 portion of a technical cell sees a local reactant stoichiometry that is a function of the number of parallel gas flow channels in the given design, and the total active area of the cell, and can easily be in the range of 10–15× greater than the global stoichiometric ratio. It is therefore rather futile to try to target stoichiometry as a merit parameter in differential cells.
That said, the high stoichiometries typically used in the differential cell and the technical cell water management features described above are related, stemming from the extreme current-specific water generation rate in the anode, the fact that that water generation is on the fuel side and not the air side, and the active status of water as a reactant in the alkaline cathode. Different strategies have been reported to handle this challenging paradigm. Earlier experiments in differential cells typically employed full humidification on both anode and cathode, a reasonably effective strategy in the by then already well-studied PEM equivalents.
In attempts to improve power density by alleviating flooding, it was found perhaps counterintuitively, to be more effective to release some excess water as vapor via reduced humidity in the nominally dry cathode, rather than the nominally wet/flooded anode.259 This can be understood when considering that the most effective way to supply reactant water to the cathode is via the membrane (back diffusion), which is liable to lose a significant portion of that functionality if the anode surface is anything less than fully hydrated. Mustain et al. found that by carefully balancing humidification on both anode and cathode, and employing gas diffusion media free of microporous layer that helped prevent excessive buildup of liquid water.260 This strategy allows a strongly positive water mass balance to be maintained without strongly flooding either electrode and allows exceptional areal power density to be achieved.
In the technical cell meanwhile, a fully humidified anode at a nominal stoichiometry ≫1 can be considered a simple simulation of conditions generated in an H2 recirculation system (although a “knockout” element may be employed, with added system complexity, to operate at lower RH). Meanwhile an RH much below ∼85% (at 80 °C) at the cathode presents a very strong challenge to the durability of currently available ionomers, and can cause acute dryout near the cathode inlet of the technical cell leading to immediate (though possibly reversible) loss of performance simply due to the low concentration of reactant water. This combination leads to a strong challenge in reducing the cathode stoichiometry since either durability (due to the relatively dry operation at the cathode) or performance (due to a flooded anode from a strongly positive mass balance) is sacrificed when a “high stoichiometry/high humidity” combination is disallowed. The trade-off must be alleviated to the extent possible by electrode design and operational parameters, including the functionality of the gas diffusion media and their microporous layers, adequate hydrophobicity of catalyst layers (via the ionomer directly or via other additives),221 as well as active water management in cell operations as touched upon recently in several published durability tests.2,3
It is relevant here to survey some advanced approaches from PEM systems. Toyota Motor Company employed an innovative mesh-type air flow field (“3D fine mesh flow channels”261) that incrementally directs air flow from the flow field to the MEA, such that each portion of the cathode “feels” the same low local stoichiometry. Another such approach already employed by PEMFCs to good effect are the so-called “water transport plates” developed at United Technologies by Perry and others.262–264 Here a porous bipolar plate with hydrophilized, water-filled pores provides humidification of incoming air and H2 by allowing water exchange directly between the anode of one cell and the cathode of its adjacent cell, while simultaneously extracting excess liquid water in a combined humidification-water management-cooling system. Engineering approaches such as these, optimized for the unique requirements of the AEMFC,265 can be considered towards alleviating the simultaneous flooding/drying paradigm. Recently, an innovative GDL concept was developed at the Paul Scherrer Insitut by Forner-Cuenca and Boillat,266–268 comprising hydrophobized GDLs that were selectively hydrophilized, in patterns controlled by a masked radiation grafting process, to allow passage of liquid water through the selected regions.
Perhaps because their benefits will only be fully realized in technical cells and stacks, these above approaches have not yet been well-explored for AEMFC systems, although their beneficial effects may prove even stronger than for PEMFC because of the more stringent water management demands, and so comprise areas of significant potential for development.
6.3 AEMFC stack durability during intermittent operation
It is almost axiomatic that electrochemical device degradation will be accelerated by cycling – variations in temperature, humidity, pressure differentials etc. all provide mechano-chemical perturbations that test the hardiness of soft matter-based devices. While most of the key aspects of shutdown/restart are not fundamentally different to those of PEM systems, the peculiarities of the AEMFC system are as important to this aspect of the operational cycle as they are to long-term continuous operation. | ||
| Fig. 13 Durability of AEMFC stack during intermittent operation. Reproduced data from ref. 128. | ||
Meanwhile, as demonstrated by Kreuer,103 the cost to ionomer performance – both ion conductivity and water mobility – of low hydration levels is stronger than in PEMFC systems, and serves as positive feedback to ionomer chemical degradation in the same manner described in Section 5.2. Mitigating this process, especially at startup, shapes to be a considerable challenge for commercial operational AEMFC systems.
The choice of maximum current density accessed during a restart is an informative example: As shown above, an extended period of high current density is a strong accelerating factor for stack degradation – especially if hydration is inadequate. However, the osmotic pressure generated also helps to hydrate the cathode via delivery of water from the anode, and the high anode potential and OH− flux help to reverse any carbonation that may have occurred during the previous cycle, while the potential ‘damage’ caused by a relatively short, high-current step during startup is unclear and certainly time- and peak current dependent. Thus the design point likely affects both short-term performance and long-term durability, needing to be struck with care and tailored to the materials employed in the device in question.
Hydrogen starvation at the anode, even for a very short period of time, can cause significant and irreversible damage (Section 5.3), as can internal currents caused by coexistence of O2 and H2 in either electrode cavity. These invariably lead to catastrophic failure in just a few cycles. Extended ‘off’ periods, especially, are liable to result in such situations, either during the ‘off’ period itself or at the time of restart, and so the stack condition left at the start of the ‘off’ period is especially critical. In general, many of the degradation processes noted above for the various individual elements (e.g., catalysts, ionomer, carbon supports etc.) are more likely if “uncontrolled” potential situations are allowed to occur in the cell. However, these effects and mitigation strategies are not particular to AEMFC systems.
7. Current durability challenges and future action
7.1 Durability challenges
The history of AEMFCs development is short compared to that of PEMFCs. Over the past decade, significant progress has been made for performance and durability improvement. At the initial stage of AEMFC development (2000–2010), researchers reported hydroxide conducting materials that have the potentials to be used for AEMFCs. After this period (2011–2015), promising AEMFC performance using hydroxide-conducting materials including Tokuyama's commercialized materials (AS4 and A201) were reported. From the research point, some important findings such as polymer backbone stability,26,27 particulate ionomeric binder,260 or stable cationic functional group34,137 were reported. These results derived substantial AEMFC performance improvement during (2016–2020) particularly using Pt-Ru anode catalyst and Pt cathode catalyst. At this period, researchers realized that the AEMFC system is not just a high pH version of PEMFCs: some peculiar characteristics may impact fuel cell performance and durability. Major research topics during this period include water management, carbonation, and catalyst–ionomer interactions. Those peculiar characteristics of AEMFCs have been investigated with broader materials sections such as PGM-free catalysts, non-alkyl ammonium functionalized AEMs under extended operating conditions (higher temperature, low stoichiometries, lower RH, etc.).From the durability standpoint, researchers realized that a stable performance output for AEMFCs is considerably more challenging compared to PEMFCs. One should note that the durability challenges of AEMFCs do not only come from different HOR and ORR reaction mechanisms between AEMFCs and PEMFCs but also materials availabilities. For PEMFCs, perfluorosulfonic acids (PFSAs) are known to be chemically, and electrochemically stable and interact only minimally with electrocatalysts. However, for AEMFCs, no such materials are available for now, and thus control of available operation parameters cannot meet the targeted durability. Although recent improvement in performance and durability of AEMFCs is impressive even with hydrocarbon-based materials, the current durability of AEMFCs needs to be significantly further improved to achieve commercially viable systems.
The performance degradation mechanisms are largely related to the peculiar characteristics of AEMFCs. However, the exact degradation mechanisms of AEMFCs are still largely unknown. For the anode of AEMFCs, the degradation mechanism related to the electrode flooding should be better understood. It is still puzzling why AEMFC anode is easily flooded even with very low current generating conditions while PEMFC cathode is robust even in over-humidified conditions. This flooding issue makes it very difficult not only for development of PGM-free anode catalysts that have more hydrophilic characteristics than Pt-based catalysts but also for general water management (see Section 4.1.1). Carbonation is another important topic for anode durability (Section 4.1.2). While carbonated species of the anode can be removed by replenishing the cell with dilute caustic solutions, it is challenging to achieve a perfect carbonate-free environment. Unlike the initial assumption that carbonation would largely impact the membrane conductivity, the anode performance decrease with carbonated species may be a critical research topic. Particularly, this topic is interesting as the carbonation issue is much less significant for the hydrogen evolution electrode of alkaline water electrolyzers, that often use potassium carbonate as a liquid electrolyte.269 Possible degradation of anode ionomeric binder is unexplored and is likely related to the water management and carbonation issue (Sections 5.2.4 and 5.2.5).
7.2 Future actions
The first design strategy for advanced AEMs which has been implemented in the field is to prepare an aryl ether-free polymer backbone. Preventing aryl ether cleavage reactions for aryl ether-containing quaternized polymers is not a trivial task and has not yet been successful. Polyolefinic and aryl ether-free polyaromatics are two representative families of aryl ether-free backbone polymers. Polyolefinic AEMs such as polyethylene, polynorbornene, ethylene tetrafuoroethylene and polystyrene-block copolymers have advantages over polyaromatics-based polymers in terms of water permeability and film-forming ability. However, high temperature properties (>80 °C) of highly quaternized polyolefinic AEMs is less desirable for high temperature operations of AEMFCs. Developing dimensionally stable polyolefinic AEMs may further improve the performance and durability of AEMFCs. Possible strategies include introducing crystallinity, crosslinking, and hydrophobic cationic functional groups. Also reinforced AEMs can improve the dimensional stability. Aryl ether-free polyaromatic polymers such as polyphenylene, polyfluorene and, poly(alkyl phenylene) polymers have benefits at a higher temperature (≥80 °C). However, aryl ether-free polyaromatics are often brittle due to the absence of the flexible ether linkage in the polymer backbone and low molecular weight. Possible strategies to resolve this issue is to obtain high molecular weight, minimize chain branches and, introduce kinked structure and reinforcement.
The second design strategy is to choose suitable cationic groups. For the most popular trimethyl ammonium functional group, introducing alkyl spacers between the polymer backbone and side chain has proven to be an effective way to increase cationic group stability. Introducing more stable cationic functional groups, such as piperidinium, has been successfully implemented. Further research efforts to prepare polymers with stable cationic functional groups such as spirocyclic compounds,34 should be continued. Enhancing the stability of the known cationic groups is another area that needs continued research. Hindering the hydroxide attack center cation by Holdcroft's group270 is a good example for mitigating cationic group degradation. Even when achieving a good cationic group stability, one should note that the real benefits of adopting non-conventional ammonium group over a alkyltrimethyl ammonium group have not been clearly demonstrated yet. As both alkyl ammonium and other cationic groups showed high alkaline stability, other aspects such as conductivity at high and low RH and water transport should be examined as well.
A third design strategy is to increase water transport properties because the most high-performing AEMFCs are using high back-diffusion of water (water diffusion from anode to cathode), it is critical to develop AEMs with high water permeability and rapid water uptake. Different approaches are possible including: increasing membrane free-volume, introducing flexible polymer backbone, and increasing IEC. Particularly, preparation of mechanically stable thin-film (5–30 μm thickness) is beneficial for water transport (see Sections 5.1.2 and 5.1.3). While ex situ characterization of AEMs is simple and standardized, the relevancy between ex situ and operando AEM stability has not been established. Also, water permeability of AEMs under different hydration conditions including water concentration gradient needs to be investigated by experimental and modeling studies.
A fourth design strategy is to operate highly conducive AEMs under low RH conditions. While current low RH operation of AEMFCs is limited by electrode performance, low RH operation is desirable for automotive fuel cell applications. Similar approaches that have been implemented in PEMFCs can be used for AEMFCs. The most common approach is to enhance the phase-separated morphology of AEMs. Synthesizing multi-block copolymers or introducing hydrophobic polymer backbones may be two possibilities. It is also noted that the hydroxide conductivity of AEMs does not only depend on the concentration of cationic functional groups but also the mesoscale structure of the polymer system. In particular, order–disorder transition in the nano-phase separated domains may affect the ionic conductivity and a number of chemical events that is related to the stability of the AEMs.271–273 Further understanding on hydroxide conduction and chemical interaction at fully and partially humidified conditions may be required to develop advanced AEMs.
Lastly, AEM interaction with catalyst layers is another largely unknown field and needs to be better understood. Transporting a significant amount of water and hydroxide ions through the interface between AEM and catalyst layers requires a robust interface for long-term operation of AEMs (Section 5.2.1).
For the ionomeric binder for cathodes, the most significant concern is the electrochemical oxidation of ionomers at high electrode potentials (Section 5.2.2). The most significant element for electrochemical oxidation is the phenyl group in the ionomer because the phenyl group is converted to acidic phenol. The best solution is again to prepare phenyl-free ionomer but this approach is currently unrealistic. It is questionable how ionomers with non-adsorbing phenyl groups are effective as we observed that even ionomers with non-adsorbing phenyl groups (in parallel position) can be oxidized at high electrode potential. Possibly, phenyl group having substituent groups may help reduce the phenyl oxidation rate. Alternatively, making a stacking configuration for phenyl group can reduce the oxidation process. It has been shown that graphitic carbon (phenyl) has much less corrosion at high cell potential in PEMFCs. Also, we believe that it is urgent to have a proper method to evaluate the oxidative stability of ionomers at a high electrode potential. Another critical future action for cathode ionomer development is related to the low RH operation of AEMFCs. While low RH operation typically requires highly conductive AEMs under low RH conditions, for AEMFCs, low RH operation may be possible with proper control of water transport and an adequate cathode ionomeric binder. In this case, the two most critical issues are the chemical stability of the cathode ionomer at low RH and the proper water supply from the AEM to water deficient cathodes. A possible solution to enabling low RH AEMFC operation is ionomer with high IEC. Much efforts in developing stable cationic functional groups under low RH and high potential conditions are required for the development of these ionomers. In addition, a modeling study under high current density and low RH conditions may be desirable to correlate the ex situ degradation rate to ionomer degradation during AEMFC operations.
For PGM-free ORR catalysts, various types of catalysts, including M–N–C type,279,280 metal oxides281,282 and silver-based catalysts283 have been developed. M–N–C type of materials have shown comparable performance to Pt/C. However, the performance and durability of M–N–C catalysts should be evaluated in fuel cells, which has hardly been performed so far. The potential risks associated with poor mass-transfer in thick M–N–C catalyst layers may be mitigated by developing hybrid materials for dense oxide (or silver) catalysts. For non-carbon based metal oxide or silver-based catalysts, catalyst stability in MEAs needs evaluation in addition to the catalytic activity improvement. The ORR electrocatalyst durability studies have been performed mostly with PGM catalysts (Section 5.3), and more in-depth studies with PGM-free or low-PGM catalysts are required for durable AEMFC systems. In general, it has been shown that potential cycling of carbon-supported PGM catalyst is very detrimental to their stability, the PGM nanoparticles favoring local carbon oxidation into carbonate species, hence breaking their binding to the carbon support and provoking intense nanoparticles detachment. This process is linked to the PGM catalyst's ability to complete the oxidation of COad-like surface groups that spontaneously form over the carbon surface above 0.2 V vs. RHE. Although such degradations are very critical for PGM/C materials, non-PGM catalysts were shown to be much more resistant to this process. Unfortunately, in that case, metal dissolution and more importantly catalyst passivation (and related deactivation) are not small issues. This means that achieving durable catalysis in AEMFCs is still a very stringent challenge that requires intense research in the forthcoming years.
:1) and alcoholic dispersing agents with a high ratio of hydroxyl to methyl ratio, e.g., ethylene glycol are the preferred dispersing agents. Either CCM or GDE method can be used. The CCM method can provide better interfacial adhesion between the catalyst and the AEM; however, the CCM method requires some solubility difference between the AEM and the ionomeric binder otherwise the AEM may dissolve out during the application of the catalyst coating. Therefore, the GDE method is preferred. AEM is prepared in hydroxide form right before MEA fabrication. Typically warm 0.5 M NaOH solution is used for hydroxide form conversion. The MEA break-in process is performed at 0.6 V at 80 °C under fully-humidified conditions. The current density during the break-in process varies because catalyst activation, electrode flooding and possible CO2 formation occurs simultaneously. Break-in time also varies depending on the catalyst and the ionomer. A minimum of 2 hours of break-in time is typically required. After the break-in, replenishing cells by flowing dilute NaOH solution and a complete rinse with water is often necessary to ensure there is no carbonated species. In addition, cyclic voltammograms of anodes and cathodes are necessary to ensure that there is no contamination of electrodes by the ionomer component.
The second method is the USC/Surrey method, which was developed using quaternized ionomers with limited solubility.2,288 In this method, quaternized powders are synthesized and then ground with a mortar and pestle to limit agglomeration. Then, a small amount of water as well as the catalyst and any additives (e.g., carbon black, polytetrafluoroethylene) are introduced to the mortar and once again ground with pestle to create a slurry. Next, the slurry is transferred to a secondary vessel where additional water and 2-propanol are added, and the vessel is sonicated to create the catalyst ink dispersion. The resulting ink is then sprayed onto gas diffusion layers, creating GDEs, which are preferred to CCMs using this method. The ionomer in the GDEs is converted to the OH− form by soaking in KOH at room temperature for 60 minutes, changing the solution twice during this time. The electrodes are placed on either side of the membrane in the cell with no prior hot pressing. The cell break-in procedure begins by bringing the cell to 60 °C under H2/O2 flow at the OCV. Then, the cell voltage is held constant at 0.5 V until a stable current is observed. Next, the cell is switched to constant-current mode and the performance is improved by iteratively manipulating the anode and cathode operating dew points to balance the cell water. Finally, the cell temperature is raised to its operating value (typically 80 °C) in multiple (typically 2 or 3) steps, with the dew points being optimized at each temperature step. A typical break-in procedure takes 2–4 hours. During operation, even for 1000+ hours, cells employing the USC/Surrey method have not needed to be treated with NaOH or KOH. However, the reacting gas dew points do need to be periodically adjusted (typically ±2 °C) over long-duration experiments to ensure optimal performance. Though both of the procedures above have shown promise, it should be noted that much of the MEA fabrication work that has been done has focused mainly on MEA performance. It is therefore necessary to devote more research to understanding how MEA fabrication impacts the durability of operating AEMFCs.
When it comes to the fuel cell stack and the integrated system, several challenges with respect to durability can be identified. At the MEA level, the sensitivity of present-day MEA's to operating conditions, generally with respect to water management, is likely the most significant of these, and is amplified in commercially relevant cell active areas (Section 6.1) and with imposition of real-world limitations to reactant stoichiometry (Section 6.2). This sensitivity is partly fundamental as discussed in Section 4.1, but also antagonized by potential chemical degradation of ionomer (in catalyst layers and membranes) described in Sections 5.1 and 5.2.
Looking at the overall fuel cell system, including balance-of-plant, additional distinctions from PEM systems are somewhat less pronounced. One important subsystem does present itself with the need at least at the current state of the technology to filter CO2 from the air stream (Section 4.2). A CO2 filtration subsystem is a potential failure point, and in addition, the performance of such a filter over time would be an additional degradation trajectory, but the filter and filtration material would be replaceable. Most critical to technical viability is the understanding that an incidental carbonation event due to a system failure is fully reversible, while the effect on durability of continuous operation under a certain (presumably low) CO2 concentration is an area for further exploration, as is the sensitivity of the fuel cell anode to CO2 in the fuel stream, especially when robustness to lower purity hydrogen is required.
System operation under realistic conditions presents potential challenges again in the area of general robustness of MEAs that have not been addressed in the literature to date. Intermittent operation (Section 6.3) is one clear example where water management issues are very likely to be amplified. Behavior in response to variable power demand, system operation at various ambient temperatures, and restart from sub-freezing temperatures are other examples of commercial requirements that likely affect durability but have not yet been addressed in the literature, and these should attract further attention of researchers as the core AEMFC technology continues to mature.
8. Concluding remarks
While strong challenges clearly lie ahead in the development of AEMFC technology, especially with respect to durability as outlined in this paper, it does appear that significant advances, including an improved appreciation of the nature of these challenges, have emerged especially in the past few years. Increasing focus in the academic community on research targeting well-identified technology gaps, informed by many decades of PEMFC development, the advent of commercialized PEM systems and a growing understanding of the quirks of the AEMFC system provide significant hope that this promising technology will eventually find its place in the now rapidly emerging Hydrogen Economy.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Effort by Y. S. K. was supported by the US Department of Energy, Energy Efficiency and Renewable Energy, Hydrogen and Fuel Cell Technologies Office. Los Alamos National Laboratory is operated by Triad National Security, LLC under US Department of Energy Contract Number 89233218CNA000001. M. C. thanks the US Office of Naval Research Global (ONRG, grant number N62909-16-1-2137) as well as the French National Research Agency (ANR, grant No. ANR-16-CE05-0009-01) for funding some of this research. This work was performed within the framework of the Centre of Excellence of Multifunctional Architectured Materials “CEMAM” no. ANR-10-LABX-44-01. W. E. M. acknowledges the support from the U.S. Department of Energy for funding his effort through project DE-EE0008433.References
- B. Pivovar and Y. S. Kim, 2019 Anion Exchange Membrane Workshop Summary Report, US DOE, 2020.
- T. J. Omasta, A. M. Park, J. M. LaManna, Y. F. Zhang, X. Peng, L. Q. Wang, D. L. Jacobson, J. R. Varcoe, D. S. Hussey, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2018, 11, 551–558 RSC
.
- G. Huang, M. Mandal, X. Peng, A. C. Yang-Neyerlin, B. S. Pivovar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 166, F637–F644 CrossRef CAS
- S. Maurya, S. Noh, I. Matanovic, E. J. Park, C. N. Villarrubia, U. Martinez, J. Han, C. Bae and Y. S. Kim, Energy Environ. Sci., 2018, 11, 3283–3291 RSC
- H. G. Peng, Q. H. Li, M. X. Hu, L. Xiao, J. T. Lu and L. Zhuang, J. Power Sources, 2018, 390, 165–167 CrossRef CAS
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, B. Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS
- T. J. Omasta, Y. F. Zhang, A. M. Park, X. Peng, B. Pivovar, J. R. Varcoe and W. E. Mustain, J. Electrochem. Soc., 2018, 165, F710–F717 CrossRef CAS
- L. Q. Wang, M. Bellini, H. A. Miller and J. R. Varcoe, J. Mater. Chem. A, 2018, 6, 15404–15412 RSC
- Y. Yang, H. Q. Peng, Y. Xiong, Q. H. Li, J. T. Lu, L. Xiao, F. J. DiSalvo, L. Zhuang and H. D. Abruna, ACS Energy Lett., 2019, 4, 1251–1257 CrossRef CAS
- X. Peng, T. J. Omasta, E. Magliocca, L. Q. Wang, J. R. Varcoe and W. E. Mustain, Angew. Chem., Int. Ed., 2019, 58, 1046–1051 CrossRef CAS PubMed
- S. Kabir, K. Lemire, K. Artyushkova, A. Roy, M. Odgaard, D. Schlueter, A. Oshchepkov, A. Bonnefont, E. Savinova, D. C. Sabarirajan, P. Mandal, E. J. Crumlin, I. V. Zenyuk, P. Atanassov and A. Serov, J. Mater. Chem. A, 2017, 5, 24433–24443 RSC
- T. J. Omasta, X. Peng, H. A. Miller, F. Vizza, L. Q. Wang, J. R. Varcoe, D. R. Dekel and W. E. Mustain, J. Electrochem. Soc., 2018, 165, J3039–J3044 CrossRef CAS
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS
- N. W. Li, Y. J. Leng, M. A. Hickner and C. Y. Wang, J. Am. Chem. Soc., 2013, 135, 10124–10133 CrossRef CAS PubMed
- W. T. Lu, Z. G. Shao, G. Zhang, Y. Zhao and B. L. Yi, J. Power Sources, 2014, 248, 905–914 CrossRef CAS
- L. Liu, X. M. Chu, J. Y. Liao, Y. D. Huang, Y. Li, Z. Y. Ge, M. A. Hickner and N. W. Li, Energy Environ. Sci., 2018, 11, 435–446 RSC
- X. Q. Gao, H. M. Yu, J. Jia, J. K. Hao, F. Xie, J. Chi, B. W. Qin, L. Fu, W. Song and Z. G. Shao, RSC Adv., 2017, 7, 19153–19161 RSC
- S. Maurya, J. H. Dumont, C. N. Villarrubia, I. Matanovic, D. Li, Y. S. Kim, S. Noh, J. Y. Han, C. Bae, H. A. Miller, C. H. Fujimoto and D. R. Dekel, ACS Catal., 2018, 8, 9429–9439 CrossRef CAS
- D. R. Dekel, 2016 Alkaline Membrane Fuel Cell Workshop, 2016, https://www.energy.gov/eere/fuelcells/downloads/2016-alkaline-membrane-fuel-cell-workshop, last accessed 2020-6-19.
- J. Xie, D. L. Wood, K. L. More, P. Atanassov and R. L. Borup, J. Electrochem. Soc., 2005, 152, A1011–A1020 CrossRef
- J. R. Yu, T. Matsuura, Y. Yoshikawa, M. N. Islam and M. Hori, Electrochem. Solid-State Lett., 2005, 8, A156–A158 CrossRef CAS
- C. S. Macomber, J. M. Boncella and B. S. Pivovar, J. Therm. Anal. Calorim., 2008, 93, 225–229 CrossRef CAS
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112, 3179–3182 CrossRef CAS
- K. J. T. Noonan, K. M. Hugar, H. A. Kostalik, E. B. Lobkovsky, H. D. Abruna and G. W. Coates, J. Am. Chem. Soc., 2012, 134, 18161–18164 CrossRef CAS PubMed
- B. Qiu, B. C. Lin, Z. H. Si, L. H. Qiu, F. Q. Chu, J. Zhao and F. Yan, J. Power Sources, 2012, 217, 329–335 CrossRef CAS
- C. Fujimoto, D. S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423, 438–449 CrossRef
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS PubMed
- J. J. Han, H. Q. Peng, J. Pan, L. Wei, G. W. Li, C. Chen, L. Xiao, J. T. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2013, 5, 13405–13411 CrossRef CAS PubMed
- A. Amel, L. Zhu, M. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2014, 161, F615–F621 CrossRef CAS
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736–1742 CrossRef CAS
- Z. H. Zhang, L. Wu, J. Varcoe, C. R. Li, A. L. Ong, S. Poynton and T. W. Xu, J. Mater. Chem. A, 2013, 1, 2594–2601 Search PubMed
- H.-S. Dang, E. A. Weiber and P. Jannasch, J. Mater. Chem. A, 2015, 3, 5280–5284 RSC
- H.-S. Dang and P. Jannasch, Macromolecules, 2015, 48, 5742–5751 CrossRef CAS
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed
- K. M. Hugar, H. A. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 8730–8737 CrossRef CAS PubMed
- T. H. Pham and P. Jannasch, ACS Macro Lett., 2015, 4, 1370–1375 CrossRef CAS
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4, 11924–11938 RSC
- J. Ponce-Gonzalez, D. K. Whelligan, L. Q. Wang, R. Bnce-Soualhi, Y. Wang, Y. Q. Peng, H. Q. Peng, D. C. Apperley, H. N. Sarode, T. P. Pandey, A. G. Divekar, S. Seifert, A. M. Herring, L. Zhuang and J. R. Varcoe, Energy Environ. Sci., 2016, 9, 3724–3735 RSC
- L. Liu, S. Q. He, S. F. Zhang, M. Zhang, M. D. Guiver and N. W. Li, ACS Appl. Mater. Interfaces, 2016, 8, 4651–4660 CrossRef CAS PubMed
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS PubMed
- S. Noh, J. Y. Jeon, S. Adhikari, Y. S. Kim and C. Bae, Acc. Chem. Res., 2019, 52, 2745–2755 CrossRef CAS PubMed
- M. Mandal, G. Huang and P. A. Kohl, J. Membr. Sci., 2019, 570, 394–402 CrossRef
- X. M. Chu, L. Liu, Y. D. Huang, M. D. Guiver and N. W. Li, J. Membr. Sci., 2019, 578, 239–250 CrossRef CAS
- J. T. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306 CrossRef PubMed
- K. Yang, L. Xiaofeng, J. Guo, J. Zheng, S. Li, S. Zhang, X. Cao, T. A. Sherazi and X. Liu, J. Membr. Sci., 2020, 596, 117720 CrossRef CAS
- W. You, K. J. T. Noonan and G. W. Coates, Prog. Polym. Sci., 2020, 100, 101177 CrossRef CAS
- C. Jin, F. L. Lu, X. C. Cao, Z. R. Yang and R. Z. Yang, J. Mater. Chem. A, 2013, 1, 12170–12177 RSC
- J. Wu, Z. R. Yang, X. W. Li, Q. J. Sun, C. Jin, P. Strasser and R. Z. Yang, J. Mater. Chem. A, 2013, 1, 9889–9896 RSC
- Y. Luo, Z. J. Wang, Y. Fu, C. Jin, Q. Wei and R. Z. Yang, J. Mater. Chem. A, 2016, 4, 12583–12590 RSC
- J. Zhang, Y. M. Sun, J. W. Zhu, Z. K. Kou, P. Hu, L. Liu, S. Z. Li, S. C. Mu and Y. H. Huang, Nano Energy, 2018, 52, 307–314 CrossRef CAS
- K. Huang, J. C. Liu, L. Wang, G. Chang, R. Y. Wang, M. Lei, Y. G. Wang and Y. B. He, Appl. Surf. Sci., 2019, 487, 1145–1151 CrossRef CAS
- Y. J. Deng, B. Chi, X. L. Tian, Z. M. Cui, E. S. Liu, Q. Y. Jia, W. J. Fan, G. H. Wang, D. Dang, M. S. Li, K. T. Zang, J. Luo, Y. F. Hu, S. J. Liao, X. L. Sun and S. Mukerjee, J. Mater. Chem. A, 2019, 7, 5020–5030 RSC
- M. E. Kreider, A. Gallo, S. Back, Y. Z. Liu, S. Siahrostami, D. Nordlund, R. Sinclair, J. K. Norskov, L. A. King and T. F. Jaramillo, ACS Appl. Mater. Interfaces, 2019, 11, 26863–26871 CrossRef CAS PubMed
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378–385 CrossRef CAS
- D. A. Salvatore, D. M. Weekes, J. F. He, K. E. Dettelbach, Y. G. C. Li, T. E. Mallouk and C. P. Berlinguette, ACS Energy Lett., 2018, 3, 149–154 CrossRef CAS
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377, 1–35 CrossRef CAS
- Y. J. Wang, J. L. Qiao, R. Baker and J. J. Zhang, Chem. Soc. Rev., 2013, 42, 5768–5787 RSC
- C. G. Arges and L. Zhang, ACS Appl. Energy Mater., 2018, 1, 2991–3012 CrossRef CAS
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC
- S. T. Thompson, D. Peterson, D. Ho and D. Papageorgopoulos, J. Electrochem. Soc., 2020, 167, 084514 CrossRef
- S. Maurya, A. S. Lee, D. G. Li, E. J. Park, D. P. Leonard, S. Noh, C. Bae and Y. S. Kim, J. Power Sources, 2019, 436, 226866 CrossRef CAS
- Y. S. Kim and K. S. Lee, Polym. Rev., 2015, 55, 330–370 CrossRef CAS
- K. D. Kreuer and P. Jannasch, J. Power Sources, 2018, 375, 361–366 CrossRef CAS
- Q. H. Zeng, Q. L. Liu, I. Broadwell, A. M. Zhu, Y. Xiong and X. P. Tu, J. Membr. Sci., 2010, 349, 237–243 CrossRef CAS
- J. K. Hao, X. Q. Gao, Y. Y. Jiang, H. J. Zhang, J. S. Luo, Z. G. Shao and B. L. Yi, J. Membr. Sci., 2018, 551, 66–75 CrossRef CAS
- L. J. Zhang, Z. X. Su, F. L. Jiang, L. L. Yang, J. J. Qian, Y. F. Zhou, W. M. Li and M. C. Hong, Nanoscale, 2014, 6, 6590–6602 RSC
- V. Parthiban, B. Bhuvaneshwari, J. Karthikeyan, P. Murugan and A. K. Sahu, Nanoscale Adv., 2019, 1, 4926–4937 RSC
- J. M. Yu, Z. K. Jiang, J. G. Wang, H. Y. Fang, T. Z. Huang and S. H. Sun, Int. J. Hydrogen Energy, 2019, 44, 13345–13353 CrossRef CAS
- P. S. Khadke and U. Krewer, J. Phys. Chem. C, 2014, 118, 11215–11223 CrossRef CAS
- S. D. Yim, H. T. Chung, J. Chlistunoff, D. S. Kim, C. Fujimoto, T. H. Yang and Y. S. Kim, J. Electrochem. Soc., 2015, 162, F499–F506 CrossRef CAS
- A. L. Ong, K. K. Inglis, D. K. Whelligan, S. Murphy and J. R. Varcoe, Phys. Chem. Chem. Phys., 2015, 17, 12135–12145 RSC
- H. T. Chung, U. Martinez, J. Chlistunoff, I. Matanovic and Y. S. Kim, J. Phys. Chem. Lett., 2016, 7, 4464–4469 CrossRef CAS PubMed
- I. Matanovic, H. T. Chung and Y. S. Kim, J. Phys. Chem. Lett., 2017, 8, 4918–4924 CrossRef CAS PubMed
- Y. Zheng, T. J. Omasta, X. Peng, L. Wang, J. R. Varcoe, B. S. Pivovar and W. E. Mustain, Energy Environ. Sci., 2019, 12, 2806–2819 RSC
- K. Fukuta, 2011 Alkaline Membrane Fuel Cell Workshop, 2011.
- H. Ono, T. Kimura, A. Takano, K. Asazawa, J. Miyake, J. Inukai and K. Miyatake, J. Mater. Chem. A, 2017, 5, 24804–24812 RSC
- D. P. Leonard, S. Maurya, E. J. Park, S. Noh, C. Bae, E. D. Baca, C. Fujimoto and Y. S. Kim, J. Mater. Chem. A, 2020 10.1039/D0TA05807F
- D. Banham, T. Kishimoto, Y. J. Zhou, T. Sato, K. Bai, J. Ozaki, Y. Imashiro and S. Y. Ye, Sci. Adv., 2018, 4, 7180 CrossRef PubMed
- B. Pivovar, DOE Hydrogen and Fuel Cells Program 2019 Annual Merit Review and Peer Evaluation Meeting, https://www.hydrogen.energy.gov/pdfs/review19/fc178_pivovar_2019_o.pdf, last accessed 2020-6-9.
- B. Pivovar, DOE Hydrogen and Fuel Cells Program 2018 Annual Merit Review and Peer Evaluation Meeting, https://www.hydrogen.energy.gov/annual_review18_fuelcells.html#membranes, last accesed 2020-6-9.
- X. G. Li, G. Liu and B. N. Popov, J. Power Sources, 2010, 195, 6373–6378 CrossRef CAS
- W. Wei, Y. Tao, W. Lv, F. Y. Su, L. Ke, J. Li, D. W. Wang, B. H. Li, F. Y. Kang and Q. H. Yang, Sci. Rep., 2014, 4, 6289 CrossRef CAS PubMed
- D. S. Geng, Y. Chen, Y. G. Chen, Y. L. Li, R. Y. Li, X. L. Sun, S. Y. Ye and S. Knights, Energy Environ. Sci., 2011, 4, 760–764 RSC
- S. Ratso, I. Kruusenberg, U. Joost, R. Saar and K. Tammeveski, Int. J. Hydrogen Energy, 2016, 41, 22510–22519 CrossRef CAS
- Q. Q. Cheng, L. J. Yang, L. L. Zou, Z. Q. Zou, C. Chen, Z. Hu and H. Yang, ACS Catal., 2017, 7, 6864–6871 CrossRef CAS
- M. Piana, M. Boccia, A. Filpi, E. Flammia, H. A. Miller, M. Orsini, F. Salusti, S. Santiccioli, F. Ciardelli and A. Pucci, J. Power Sources, 2010, 195, 5875–5881 CrossRef CAS
- Y. Luo, J. Guo, C. Wang and D. Chu, Electrochem. Commun., 2012, 16, 65–68 CrossRef CAS
- G. Gupta, K. Scott and M. Mamlouk, Fuel Cells, 2018, 18, 137–147 CrossRef CAS
- X. Chu, Y. Shi, L. Liu, Y. Huang and N. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC
- C. V. Rao and Y. Ishikawa, J. Phys. Chem. C, 2012, 116, 4340–4346 CrossRef
- H. C. Huang, Y. C. Lin, S. T. Chang, C. C. Liu, K. C. Wang, H. P. Jhong, J. F. Lee and C. H. Wang, J. Mater. Chem. A, 2017, 5, 19790–19799 RSC
- J. Sanetuntikul and S. Shanmugam, Nanoscale, 2015, 7, 7644–7650 RSC
- W. E. Mustain, Curr. Opin. Electrochem., 2018, 12, 233–239 CrossRef CAS
- A. L. Roy, J. Peng and T. A. Zawodzinski, 233rd ECS Meeting, May 13,-17, 2018, Abstract No. IO4-1755, 2018.
- M. Unlu, D. Abbott, N. Ramaswamy, X. M. Ren, S. Mukerjee and P. A. Kohl, J. Electrochem. Soc., 2011, 158, B1423–B1431 CrossRef CAS
- I. T. McCrum and M. J. Janik, J. Phys. Chem. C, 2016, 120, 457–471 CrossRef CAS
- X. T. Chen, I. T. McCrum, K. A. Schwarz, M. J. Janik and M. T. M. Koper, Angew. Chem., Int. Ed., 2017, 56, 15025–15029 CrossRef CAS PubMed
- H. T. Chung, Y. K. Choe, U. Martinez, J. H. Dumont, A. Mohanty, C. Bae, I. Matanovic and Y. S. Kim, J. Electrochem. Soc., 2016, 163, F1503–F1509 CrossRef CAS
- I. T. McCrum, M. A. Hickner and M. J. Janik, J. Electrochem. Soc., 2018, 165, F114–F121 CrossRef CAS
- J. H. Dumont, A. J. Spears, R. P. Hjelm, M. Hawley, S. Maurya, D. Li, G. Yuan, E. B. Watkins and Y. S. Kim, ACS Appl. Mater. Interfaces, 2019, 12, 1825–1831 CrossRef PubMed
- M. R. Gerhardt, L. M. Pant and A. Z. Weber, J. Electrochem. Soc., 2019, 166, F3180–F3192 CrossRef CAS
- M. Mandal, G. Huang, N. Ul Hassan, X. Peng, T. Gu, A. H. Brooks-Starks, B. Bahar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2020, 167, 054501 CrossRef CAS
- M. G. Marino, J. P. Melchior, A. Wohlfarth and K. D. Kreuer, J. Membr. Sci., 2014, 2014, 61–71 CrossRef
- C. E. Diesendruck and D. R. Dekel, Curr. Opin. Electrochem., 2018, 9, 173–178 CrossRef CAS
- D. R. Dekel, M. Arnar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29, 4425–4431 CrossRef CAS
- D. R. Dekel, I. G. Rasin, M. Page and S. Brandon, J. Power Sources, 2018, 375, 191–204 CrossRef CAS
- D. R. Dekel, S. Willdorf, U. Ash, M. Amar, S. Pusara, S. Dhara, S. Srebnik and C. E. Diesendruck, J. Power Sources, 2018, 375, 351–360 CrossRef CAS
- R. K. Ahluwalia and X. H. Wang, J. Power Sources, 2008, 177, 167–176 CrossRef CAS
- T. J. Omasta, L. Wang, X. Peng, C. A. Lewis, J. R. Varcoe and W. E. Mustain, J. Power Sources, 2018, 375, 205–213 CrossRef CAS
- T. Wang, L. Shi, J. H. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell and Y. S. Yan, J. Electrochem. Soc., 2019, 166, F3305–F3310 CrossRef CAS
- Y. Oshiba, J. Hiura, Y. Suzuki and T. Yamaguchi, J. Power Sources, 2017, 345, 221–226 CrossRef CAS
- A. Carlson, P. Shapturenka, B. Eriksson, G. Lindbergh, C. Lagergren and R. W. Lindstrom, Electrochim. Acta, 2018, 277, 151–160 CrossRef CAS
- B. S. Marchado, N. Chakraborty and P. K. Das, Int. J. Hydrogen Energy, 2017, 42, 6310–6323 CrossRef
- A. Thomas, G. Maranzana, S. Didierjean, J. Dillet and O. Lottin, J. Electrochem. Soc., 2013, 160, F191–F204 CrossRef CAS
- H. Deng, D. W. Wang, X. Xie, Y. B. Zhou, Y. Yin, Q. Du and K. Jiao, Renewable Energy, 2016, 91, 166–177 CrossRef CAS
- H. Deng, D. W. Wang, R. F. Wang, X. Xie, Y. Yin, Q. Du and K. Jiao, Appl. Energy, 2016, 183, 1272–1278 CrossRef CAS
- X. Peng, D. Kulkarni, Y. Huang, T. J. Omasta, B. Ng, Y. Zheng, L. Wang, J. M. LaManna, D. S. Hussey, J. R. Varcoe, I. V. Zenyuk and W. E. Mustain, Nat. Commun. DOI:10.1038/s41467-020-17370-7
- Z. Siroma, A. Watanabe, K. Yasuda, K. Fukuta and H. Yanagi, ECS Trans., 2010, 33, 1935–1943 CrossRef CAS
- Y. Matsui, M. Saito, A. Tasaka and M. Inaba, ECS Trans., 2010, 25, 105–110 CrossRef CAS
- M. Inaba, Y. Matsui, M. Saito, A. Tasaka, K. Fukuta, S. Waanabe and H. Yanagi, Electrochemistry, 2011, 79, 322–325 CrossRef CAS
- H. Yanagi and K. Fukuta, ECS Trans., 2008, 16, 257–262 CrossRef CAS
- W. A. Rigdon, T. J. Omasta, C. A. Lewis, M. A. Hickner, J. R. Varcoe, J. N. Renner, K. E. Ayers and W. E. Mustain, J. Electrochem. Energy Convers. Storage, 2017, 14, 020701 CrossRef
- M. R. Gerhardt, L. M. Pant, H.-S. Shiau and A. Z. Weber, ECS Trans., 2018, 86, 15–24 CrossRef CAS
- U. Krewer, C. Weinzierl, N. Ziv and D. R. Dekel, Electrochim. Acta, 2018, 263, 433–446 CrossRef CAS
- J. A. Wrubel, A. A. Peracchio, B. N. Cassenti, T. D. Myles, K. N. Grew and W. K. S. Chiu, J. Electrochem. Soc., 2017, 164, F1063–F1073 CrossRef CAS
- B. P. Setzler and Y. Yan, 231st ECS Meeting, May 28-June 1, 2017, Abstract No. 1654.
-
S. Gottesfeld, US Pat., 9214691B2, 2015 Search PubMed
- M. Page and S. Gottesfeld, 2016 AMFC Workshop, https://www.energy.gov/sites/prod/files/2016/05/f32/fcto_2016_amfcw_6-page.pdf, last accessed 2020-6-9.
- K. H. Kangasniemi, D. A. Condit and T. D. Jarvi, J. Electrochem. Soc., 2004, 151, E125–E132 CrossRef CAS
- Z. Zhao, L. Dubau and F. Maillard, J. Power Sources, 2012, 217, 449–458 CrossRef CAS
- T. Asset, N. Job, Y. Busby, A. Crisci, V. Martin, V. Stergiopoulos, C. Bonnaud, A. Serov, P. Atanassov, R. Chattot, L. Dubau and F. Maillard, ACS Catal., 2018, 8, 893–903 CrossRef CAS
- N. Giordano, P. L. Antonucci, E. Passalacqua, L. Pino, A. S. Arico and K. Kinoshita, Electrochim. Acta, 1991, 36, 1931–1935 CrossRef CAS
- L. Dubau, L. Castanheira, M. Chatenet, F. Maillard, J. Dillet, G. Maranzana, S. Abbou, O. Lottin, G. De Moor, A. El Kaddouri, C. Bas, L. Flandin, E. Rossinot and N. Caqué, Int. J. Hydrogen Energy, 2014, 39, 21902–21914 CrossRef CAS
- L. Castanheira, L. Dubau, M. Mermoux, G. Berthome, N. Caque, E. Rossinot, M. Chatenet and F. Maillard, ACS Catal., 2014, 4, 2258–2267 CrossRef CAS
- Y. Zheng, G. Huang, L. Wang, J. R. Varcoe, P. A. Kohl and W. E. Mustain, J. Power Sources, 2020, 467, 228350 CrossRef CAS
- W. T. Lu, G. Zhang, J. Li, J. K. Hao, F. Wei, W. H. Li, J. Y. Zhang, Z. G. Shao and B. L. Yi, J. Power Sources, 2015, 296, 204–214 CrossRef CAS
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314–17320 RSC
- P. G. Stevens and J. H. Richmond, J. Am. Chem. Soc., 1941, 63, 3132–3136 CrossRef CAS
- G. Ghigo, S. Cagnina, A. Maranzana and G. Tonachini, J. Org. Chem., 2010, 75, 3608–3617 CrossRef CAS PubMed
- S. Chempath, J. M. Boncella, L. R. Pratt, N. Henson and B. S. Pivovar, J. Phys. Chem. C, 2010, 114, 11977–11983 CrossRef CAS
- B. Lee, D. Yun, J. S. Lee, C. H. Park and T. H. Kim, J. Phys. Chem. C, 2019, 123, 13508–13518 CrossRef CAS
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2017, 5, 21965–21978 RSC
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2020, 53(12), 4722–4732 CrossRef CAS
- T. N. Danks, R. C. T. Slade and J. R. Varcoe, J. Mater. Chem., 2002, 12, 3371–3373 RSC
- T. N. Danks, R. C. T. Slade and J. R. Varcoe, J. Mater. Chem., 2003, 13, 712–721 RSC
- A. Voge, V. Deimede and J. Kallitsis, RSC Adv., 2014, 4, 45040–45049 RSC
- D. S. Kim, A. Labouriau, M. D. Guiver and Y. S. Kim, Chem. Mater., 2011, 23, 3795–3797 CrossRef CAS
- W. W. Li, S. B. Wang, X. F. Zhang, W. P. Wang, X. F. Xie and P. C. Pei, Int. J. Hydrogen Energy, 2014, 39, 13710–13717 CrossRef CAS
- B. C. Lin, L. H. Qui, J. M. Lu and F. Yan, Chem. Mater., 2010, 22, 6718–6725 CrossRef CAS
- M. L. Guo, J. Fang, H. K. Xu, W. Li, X. H. Lu, C. H. Lan and K. Y. Li, J. Membr. Sci., 2010, 362, 97–104 CrossRef CAS
- D. Henkensmeier, H. R. Cho, H. J. Kim, C. N. Kirchner, J. Leppin, A. Dyck, J. H. Jang, E. Cho, S. W. Nam and T. H. Lim, Polym. Degrad. Stab., 2012, 97, 264–272 CrossRef CAS
- O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 10753–10756 CrossRef CAS PubMed
- F. L. Gu, H. L. Dong, Y. Y. Li, Z. H. Si and F. Yan, Macromolecules, 2014, 47, 208–216 CrossRef CAS
- S. C. Price, K. S. Williams and F. L. Beyer, ACS Macro Lett., 2014, 3, 160–165 CrossRef CAS
- A. G. Wright, J. T. Fan, B. Britton, T. Weissbach, H. F. Lee, E. A. Kitching, T. J. Peckham and S. Holdcroft, Energy Environ. Sci., 2016, 9, 2130–2142 RSC
- J. T. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6, 1089–1093 CrossRef CAS
- W. You, K. M. Hugar and G. W. Coates, Macromolecules, 2018, 51, 3212–3218 CrossRef CAS
- Y. K. Choe, C. Fujimoto, K. S. Lee, L. T. Dalton, K. Ayers, N. J. Henson and Y. S. Kim, Chem. Mater., 2014, 26, 5675–5682 CrossRef CAS
- E. J. Park, S. Maurya, M. R. Hibbs, C. H. Fujimoto, K. D. Kreuer and Y. S. Kim, Macromolecules, 2019, 52, 5419–5428 CrossRef CAS
- A. D. Mohanty, C. Y. Ryu, Y. S. Kim and C. Bae, Macromolecules, 2015, 48, 7085–7095 CrossRef CAS
- X. D. Su, L. Gao, L. Hu, N. A. Qaisrani, X. M. Yan, W. J. Zhang, X. B. Jiang, X. H. Ruan and G. H. He, J. Membr. Sci., 2019, 581, 283–292 CrossRef CAS
- H. Kuroki, S. Miyanishi, A. Sakakibara, Y. Oshiba and T. Yamaguchi, J. Power Sources, 2019, 438, 226997 CrossRef CAS
- Y. S. Kim, 2017 Annual Merit Review and Peer Evaluation Meeting, https://www.hydrogen.energy.gov/annual_review17_fuelcells.html, last accessed 2020-6-9.
- K. F. Wang, L. Gao, J. F. Liu, X. D. Su, X. M. Yan, Y. Dai, X. B. Jiang, X. M. Wu and G. H. He, J. Membr. Sci., 2019, 588, 117216 CrossRef CAS
- H. G. Peng, Q. H. Li, M. X. Hu, L. Xiao, J. T. Lu and L. Zhuang, J. Power Sources, 2018, 390, 165–167 CrossRef CAS
- X. J. Zhang, X. M. Chu, M. Zhang, M. Zhu, Y. D. Huang, Y. G. Wang, L. Liu and N. W. Li, J. Membr. Sci., 2019, 574, 212–221 CrossRef CAS
- J. D. Xue, L. Liu, J. Y. Liao, Y. H. Shen and N. W. Li, J. Mater. Chem. A, 2018, 6, 11317–11326 RSC
- Y. Z. Zhang, J. Parrondo, S. Sankarasubramanian and V. Ramani, ChemSusChem, 2017, 10, 3056–3062 CrossRef CAS PubMed
- M. A. Hickner, H. Ghassemi, Y. S. Kim, B. R. Einsla and J. E. McGrath, Chem. Rev., 2004, 104, 4587–4612 CrossRef CAS PubMed
- J. K. Hao, X. Q. Gao, Y. Y. Jiang, H. J. Zhang, J. S. Luo, Z. G. Shao and B. L. Yi, J. Membr. Sci., 2018, 551, 66–75 CrossRef CAS
- K. H. Lee, D. H. Cho, Y. M. Kim, S. J. Moon, J. G. Seong, D. W. Shin, J. Y. Sohn, J. F. Kim and Y. M. Lee, Energy Environ. Sci., 2017, 10, 275–285 RSC
- L. Q. Wang, X. Peng, W. E. Mustain and J. R. Varcoe, Energy Environ. Sci., 2019, 12, 1575–1579 RSC
- X. Y. Huang, R. Solasi, Y. Zou, M. Feshler, K. Reifsnider, D. Condit, S. Burlatsky and T. Madden, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 2346–2357 CrossRef CAS
- H. Ishikawa, T. Teramoto, Y. Ueyama, Y. Sugawara, Y. Sakiyama, M. Kusakabe, K. Miyatake and M. Uchida, J. Power Sources, 2016, 325, 35–41 CrossRef CAS
- L. Zhu, T. J. Zimudzi, N. W. Li, J. Pan, B. C. Lin and M. A. Hickner, Polym. Chem., 2016, 7, 2589 RSC
- X. Q. Wang, C. X. Lin, F. H. Liu, L. Li, Q. Yang, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Mater. Chem. A, 2018, 6, 12455–12465 RSC
- M. M. Hossain, L. Wu, X. Liang, Z. J. Yang, J. Q. Hou and T. W. Xu, J. Power Sources, 2018, 390, 234–241 CrossRef CAS
- J. Y. Jeon, S. Park, J. Han, S. Maurya, A. D. Mohanty, D. Tian, N. Saikia, M. A. Hickner, C. Y. Ryu, M. E. Tuckerman, S. J. Paddison, Y. S. Kim and C. Bae, Macromolecules, 2019, 52, 2139–2147 CrossRef CAS
- C. X. Lin, X. Q. Wang, E. N. Hu, Q. Yang, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2017, 541, 358–366 CrossRef
- P. Dai, Z. H. Mo, R. W. Xu, S. Zhang and Y. X. Wu, ACS. Appl. Mater. Interfaces, 2016, 8, 20329–20341 CrossRef CAS PubMed
- R.-A. Becerra-Arciniegas, R. Narducci, G. Ercolani, S. Antonaroli, E. Sgreccia, L. Pasquini, P. Knauth and M. L. Di Vona, Polymer, 2019, 185, 121931 CrossRef CAS
- A. Amel, L. Zhu, M. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2014, 161, F615–F621 CrossRef CAS
- S. Miyanishi and T. Yamaguchi, Phys. Chem. Chem. Phys., 2016, 18, 12009–12023 RSC
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC
- X. Wang, W. B. Sheng, Y. H. Shen, L. Liu, S. Dai and N. W. Li, J. Membr. Sci., 2019, 587, 117135 CrossRef CAS
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2019, 7, 15895–15906 RSC
- W. H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 CrossRef CAS
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS
- Q. Yang, X. L. Gao, H. Y. Wu, Y. Y. Cai, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Power Sources, 2019, 436, 226856 CrossRef CAS
- S. B. Lee, C. M. Min, J. Jang and J. S. Lee, Polymer, 2020, 122331 CrossRef
- J. J. Han, Q. Liu, X. Q. Li, J. Pan, L. Wei, Y. Wu, H. Q. Peng, Y. Wang, G. W. Li, C. Chen, L. Xiao, J. T. Lu and L. Zhuang, ACS Appl. Mater. Interfaces, 2015, 7, 2809–2816 CrossRef CAS
- J. Pan, J. J. Han, L. Zhu and M. A. Hickner, Chem. Mater., 2017, 29, 5321–5330 CrossRef CAS
- A. Amel, S. B. Smedley, D. R. Dekel, M. A. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2015, 162, F1047–F1055 CrossRef CAS
- C. G. Arges, L. H. Wang, M. S. Jung and V. Ramani, J. Electrochem. Soc., 2015, 162, F686–F693 CrossRef CAS
- E. J. Park, S. Maurya, M. R. Hibbs, C. H. Fujimoto, K. D. Kreuer and Y. S. Kim, Macromolecules, 2019, 52, 5419–5428 CrossRef CAS
- S. Miyanishi and T. Yamaguchi, New J. Chem., 2017, 41, 8036 RSC
- S. Miyanishi and T. Yamaguchi, Polym. Chem., 2020, 11, 3812–3820 RSC
- J. Prabhuram, N. N. Krishnan, B. Choi, T. H. Lim, H. Y. Ha and S. K. Kim, Int. J. Hydrogen Energy, 2010, 35, 6924–6933 CrossRef CAS
- X. G. Yang, Y. Tabuchi, F. Kagami and C. Y. Wang, J. Electrochem. Soc., 2008, 155, B752–B761 CrossRef CAS
- Y. S. Kim and B. S. Pivovar, J. Electrochem. Soc., 2010, 157, B1616–B1623 CrossRef CAS
- Y. S. Kim, M. Einsla, J. E. McGrath and B. S. Pivovar, J. Electrochem. Soc., 2010, 157, B1602–B1607 CrossRef CAS
- B. S. Pivovar and Y. S. Kim, J. Electrochem. Soc., 2007, 154, B739–B744 CrossRef CAS
- S. Kim and M. M. Mench, J. Power Sources, 2007, 174, 206–220 CrossRef CAS
- H. Morikawa, T. Mitsui, J. Hamagami and K. Kanamura, Electrochemistry, 2002, 70, 937–939 CrossRef CAS
- J. Feichtinger, J. Kerres, A. Schulz, M. Walker and U. Schumacher, J. New Mater. Electrochem. Syst., 2002, 5, 155–162 CAS
- B. Bae, D. Kim, H. J. Kim, T. H. Lim, I. H. Oh and H. Y. Ha, J. Phys. Chem. B, 2006, 110, 4240–4246 CrossRef CAS PubMed
- Y. S. Kim, M. J. Sumner, W. L. Harrison, J. S. Riffle, J. E. McGrath and B. S. Pivovar, J. Electrochem. Soc., 2004, 151, A2150–A2156 CrossRef CAS
- D. Li, I. Matanovic, A. S. Lee, E. J. Park, C. Fujimoto, H. T. Chung and Y. S. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 9696–9701 CrossRef CAS PubMed
- S. Maurya, C. H. Fujimoto, M. R. Hibbs, C. N. Villarrubia and Y. S. Kim, Chem. Mater., 2018, 30, 2188–2192 CrossRef CAS
- I. Matanovic, S. Maurya, E. J. Park, J. Y. Jeon, C. Bae and Y. S. Kim, Chem. Mater., 2019, 31, 4195–4204 CrossRef CAS
- M. R. Sturgeon, C. S. Macomber, C. Engtrakul, H. Long and B. S. Pivovar, J. Electrochem. Soc., 2015, 162, F366–F372 CrossRef CAS
- D. R. Dekel, I. G. Rasin and S. Brandon, J. Power Sources, 2019, 420, 118–123 CrossRef CAS
- E. J. Park, S. Maurya, A. S. Lee, D. P. Leonard, D. Li, J. Y. Jeon, C. Bae and Y. S. Kim, J. Mater. Chem. A, 2019, 7, 25040–25046 RSC
- N. N. Zhao, D. Edwards, Z. Q. Shi and S. Holdcroft, ECS Electrochem. Lett., 2013, 2, F22–F24 CrossRef CAS
- Y. S. Kim, C. F. Welch, N. H. Mack, R. P. Hjelm, E. B. Orler, M. E. Hawley, K. S. Lee, S.-D. Yim and C. M. Johnston, Phys. Chem. Chem. Phys., 2014, 16, 5927–5932 RSC
- T. Van Cleve, S. Khandavalli, A. Chowdhury, S. Medina, S. Pylypenko, M. Wang, K. L. More, N. Kariuki, D. J. Myers, A. Z. Weber, S. A. Mauger, M. Ulsh and K. C. Neyerlin, ACS Appl. Mater. Interfaces, 2019, 11, 46953–46964 CrossRef CAS PubMed
- Y. Yin, R. T. Li, F. Q. Bai, W. K. Zhu, Y. Z. Qin, Y. F. Chang, J. F. Zhang and M. D. Guiver, Electrochem. Commun., 2019, 109, 106590 CrossRef CAS
- Q. Li, D. Spernjak, P. Zelenay and Y. S. Kim, J. Power Sources, 2014, 271, 561–569 CrossRef CAS
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS
- A. Serov, I. V. Zenyuk, C. G. Arges and M. Chatenet, J. Power Sources, 2018, 375, 149–157 CrossRef CAS
- L. Wang, X. Peng, W. E. Mustain and J. R. Varcoe, Energy Environ. Sci., 2019, 12, 1575–1579 RSC
-
M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, National Association of Corrosion Engineers, Houston, 1979 Search PubMed
- D. Chade, L. Berlouis, D. Infield, P. T. Nielsen and T. Mathiesen, J. Electrochem. Soc., 2016, 163, F308–F317 CrossRef CAS
- Y. Kiros, M. Majari and T. A. Nissinen, J. Alloys Compd., 2003, 360, 279–285 CrossRef CAS
- I. Moussallem, S. Pinnow, N. Wagner and T. Turek, Chem. Eng. Process., 2012, 52, 125–131 CrossRef CAS
- A. Sarapuu, E. Kibena- Põldsepp, M. Borghei and K. Tammeveski, J. Mater. Chem. A, 2018, 6, 776–804 RSC
- M. M. Hossen, K. Artyushkova, P. Atanossov and A. Serov, J. Power Sources, 2018, 375, 214–221 CrossRef CAS
- J. Zhang, W. Zhu, Y. Pei, Y. Lin, Y. Qin, X. Zhang, Q. Wang, Y. Yin and M. D. Guiver, ChemSusChem, 2019, 112, 4165–4169 CrossRef
- P. N. Ross and H. Sokol, J. Electrochem. Soc., 1984, 131, 1742–1750 CrossRef CAS
- N. Staud and P. N. Ross, J. Electrochem. Soc., 1986, 133, 1079–1084 CrossRef CAS
- P. N. Ross and M. Sattler, J. Electrochem. Soc., 1988, 135, 1464–1470 CrossRef CAS
- N. Staud, H. Sokol and P. N. Ross, J. Electrochem. Soc., 1989, 136, 3570–3576 CrossRef CAS
- Y. Kiros and S. Schwartz, J. Power Sources, 2000, 87, 101–105 CrossRef CAS
- M. Chatenet, M. Aurousseau, R. Durand and F. Andolfatto, J. Electrochem. Soc., 2003, 150, D47–D55 CrossRef CAS
- P.-Y. Olu, F. Deschamps, G. Caldarella, M. Chatenet and N. Job, J. Power Sources, 2015, 297, 492–503 CrossRef CAS
- S. Cherevko, A. R. Zeradjanin, G. P. Keeley and K. J. J. Mayrhofer, J. Electrochem. Soc., 2014, 161, H822–H830 CrossRef
- A. Zadick, L. Dubau, N. Sergent, G. Berthomé and M. Chatenet, ACS Catal., 2015, 5, 4819–4824 CrossRef CAS
- C. Lafforgue, A. Zadick, L. Dubau, F. Maillard and M. Chatenet, Fuel Cells, 2018, 18, 229–238 CrossRef CAS
- C. Lafforgue, F. Maillard, V. Martin, L. Dubau and M. Chatenet, ACS Catal., 2019, 9, 5613–5622 CrossRef CAS
- Q. Li, H. Peng, Y. Wang, L. Xiao, J. Lu and L. Zhuang, Angew. Chem., Int. Ed., 2019, 58, 1442–1446 CrossRef CAS PubMed
- A. Zadick, L. Dubau, U. B. Demirci and M. Chatenet, J. Electrochem. Soc., 2016, 163, F781–F787 CrossRef CAS
- C. Lafforgue, M. Chatenet, L. Dubau and D. R. Dekel, ACS Catal., 2018, 8, 1278–1286 CrossRef CAS
- S. Kabir, A. Zadick, P. Atanassov, L. Dubau and M. Chatenet, Electrochem. Commun., 2017, 78, 33–37 CrossRef CAS
- H. A. Miller, F. Vizza, M. Marelli, A. Zadick, L. Dubau, M. Chatenet, S. Geiger, S. Cherevko, H. Doan, R. K. Pavlicek, S. Mukerjee and D. R. Dekel, Nano Energy, 2017, 33, 293–305 CrossRef CAS
- A. Zadick, L. Dubau, A. Zalineeva, C. Coutanceau and M. Chatenet, Electrochem. Commun., 2014, 48, 1–4 CrossRef CAS
- A. Zadick, L. Dubau, K. Artyushkova, A. Serov, P. Atanassov and M. Chatenet, Nano Energy, 2017, 37, 248–259 CrossRef CAS
- E. S. Davydova, F. D. Speck, M. T. Y. Paul, D. R. Dekel and S. Cherevko, ACS Catal., 2019, 9, 6837–6845 CrossRef CAS
- C. Lafforgue, M. Chatenet, L. Dubau and D. R. Dekel, ACS Catal., 2018, 8, 1278–1286 CrossRef CAS
- S. Ould-Amara, J. Dillet, S. Didierjean, M. Chatenet and G. Maranzana, J. Power Sources, 2019, 439, 227099 CrossRef CAS
- L. Dubau, J. Durst, F. Maillard, M. Chatenet, L. Guétaz, J. André and E. Rossinot, Fuel Cells, 2012, 12, 188–198 CrossRef CAS
- J. Durst, A. Lamibrac, F. Charlot, J. Dillet, L. F. Castanheira, G. Maranzana, L. Dubau, F. Maillard, M. Chatenet and O. Lottin, Appl. Catal., B, 2013, 138–139, 416–426 CrossRef CAS
- L. Dubau, L. Castanheira, F. Maillard, M. Chatenet, O. Lottin, G. Maranzana, J. Dillet, A. Lamibrac, J.-C. Perrin, E. Moukheiber, A. ElKaddouri, G. De Moor, C. Bas, L. Flandin and N. Caqué, Wiley Interdiscip. Rev.: Energy Environ., 2014, 3, 540–560 CAS
- B. Pivovar, Y. S. Kim, D. Peterson, D. Papageorgopoulos, M. Hickner, J. S. Spendelow and A. Weber, 2016 Alkaline Membrane Fuel Cell Workshop Report, 2016.
-
W. Gu, D. R. Baker, Y. Liu and H. A. Gasteiger, in Handbook of Fuel Cells, Fundamentals Technology and Applications, ed. W. Vielstich, A. Lamm, H. A. Gasteiger and H. Yokokawa, John Wiley & Sons, 2010 Search PubMed
-
D. R. Dekel, US Pat. Appl., 20110300466, 2011 Search PubMed
-
D. R. Dekel and M. Page, WIPO, WO2014195758, 2014 Search PubMed
-
D. R. Dekel and S. Gottesfeld, US Pat. Appl., 20110300466, 2014 Search PubMed
- A. Kongkanand and M. F. Mathias, J. Phys. Chem. Lett., 2016, 7, 1127–1137 CrossRef CAS PubMed
- S. Gottesfeld, M. Page and Y. Paska, 228th ECS Meeting, October, 11-15, 2015. Abstract no. 1342. Last accessed 2020-06-09.
- L. Q. Wang, E. Magliocca, E. L. Cunningham, W. E. Mustain, S. D. Poynton, R. Escudero-Cid, M. M. Nasef, J. Ponce-Gonzalez, R. Bance-Souahli, R. C. T. Slade, D. K. Whelligan and J. R. Varcoe, Green Chem., 2017, 19, 831–843 RSC
- Toyota Web Report, https://global.toyota/en/newsroom/toyota/22740159.html, Last accessed 2020-06-09.
-
B. Dufner, M. I. Perry, J. C. Trocciola, D. Yang and J. S. Yi, US Pat., 6617068B2, 2003 Search PubMed
-
J. S. Yi, D. Yang, R. D. Breault, A. P. Grasso and G. W. Scheffler, US Pat., 6794077, 2004 Search PubMed
-
S. Shimotori, Y. Ogami and M. L. Perry, US Pat., 6869709, 2005 Search PubMed
-
Y. Paska, M. Page, Y. Benjamin and S. Gottesfeld, WIPO, WO2017/163244Ai, 2016 Search PubMed
- A. Forner-Cuenca, J. Biesdorf, L. Gubler, P. M. Kristiansen, T. J. Schmidt and P. Boillat, Adv. Mater., 2015, 27, 6317 CrossRef CAS PubMed
- A. Forner-Cuenca, V. Manzi-Orezzoli, J. Biesdorf, M. El Kazzi, D. Streich, L. Gubler, T. J. Schmidt and P. Boillat, J. Electrochem. Soc., 2016, 163, F788–F801 CrossRef CAS
- A. Forner-Cuenca, J. Biesdorf, V. Manzi-Orezzoli, L. Gubler, T. J. Schmidt and P. Boillat, J. Electrochem. Soc., 2016, 163, F1389–F1398 CrossRef CAS
- H. Ito, N. Kawaguchi, S. Someya, T. Munakata, N. Miyazaki, M. Ishiba and A. Nakano, Int. J. Hydrogen Energy, 2018, 43, 17030–17039 CrossRef CAS
- A. G. Wright, T. Weissbach and S. Holdcroft, Angew. Chem., Int. Ed., 2016, 55, 4818–4821 CrossRef CAS PubMed
- T. P. Pandey, A. M. Maes, H. N. Sarode, B. D. Peters, S. Lavina, K. Vezzu, Y. Yang, S. D. Poynton, J. R. Varcoe, S. Seifert, M. W. Liberatore, V. Di Noto and A. M. Herring, Phys. Chem. Chem. Phys., 2015, 17, 4367–4378 RSC
- T. P. Pandey, H. N. Sarode, Y. T. Yang, Y. Yang, K. Vezzu, V. Di Noto, S. Seifert, D. M. Knauss, M. W. Liberatore and A. M. Herring, J. Electrochem. Soc., 2016, 163, H513–H520 CrossRef CAS
- K. Vezzu, A. M. Maes, F. Bertasi, A. R. Motz, T. H. Tsai, E. B. Coughlin, A. M. Herring and V. Di Noto, J. Am. Chem. Soc., 2018, 140, 1372–1384 CrossRef CAS PubMed
- D. Li, H. T. Chung, S. Maurya, I. Matanovic and Y. S. Kim, Curr. Opin. Electrochem., 2018, 12, 189–195 CrossRef CAS
- A. G. Oshchepkov, A. Bonnefont, S. N. Pronkin, O. V. Cherstiouk, C. Ulhaq-Bouillet, V. Papaefthimiou, V. N. Parmon and E. R. Savinova, J. Power Sources, 2018, 402, 447–452 CrossRef CAS
- A. G. Oshchepkov, A. Bonnefont, V. N. Parmon and E. R. Savinova, Electrochim. Acta, 2018, 269, 111–118 CrossRef CAS
- D. Salmazo, M. F. Juarez, A. G. Oshchepkov, O. V. Cherstiouk, A. Bonnefont, S. A. Shermukhamedov, R. R. Nazmutdinov, W. Schmickler and E. R. Savinova, Electrochim. Acta, 2019, 305, 452–458 CrossRef CAS
- R. Wang, D. Li, S. Maurya, Y. S. Kim, Y. Wu, Y. Liu, D. Strmcnik, N. M. Markovic and V. R. Stamenkovic, Nanoscale Horiz., 2020, 5, 316–324 RSC
- H. T. Chung, J. H. Won and P. Zelenay, Nat. Commun., 2013, 4, 1922 CrossRef PubMed
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2005, 109, 4707–4716 CrossRef CAS PubMed
- Y. Wang, Y. Yang, S. F. Jia, X. M. Wang, K. J. Lyu, Y. Q. Peng, H. Zheng, X. Wei, H. Ren, L. Xiao, J. B. Wang, D. A. Muller, H. D. Abruna, B. O. E. Hwang, J. T. Lu and L. Zhuang, Nat. Commun., 2019, 10, 1506 CrossRef PubMed
- M. Risch, Catalysts, 2017, 7, 154 CrossRef
- H. Erikson, A. Sarapuu and K. Tammeveski, ChemElectroChem, 2019, 6, 73–86 CrossRef CAS
- N. Job, M. Chatenet, S. Berthon-Fabry, S. Hermans and F. Maillard, J. Power Sources, 2013, 240, 294–305 CrossRef CAS
- J. Qi, Y. F. Zahai and J. St-Pierre, J. Power Sources, 2019, 413, 86–97 CrossRef CAS
- C. Welch, A. Labouriau, R. P. Hjelm, B. Orler, C. Johnston and Y. S. Kim, ACS Macro Lett., 2012, 1, 1403–1407 CrossRef CAS
- Y. S. Kim, C. F. Welch, R. P. Hjelm, N. H. Mack, A. Labouriau and E. B. Orler, Macromolecules, 2015, 48, 2161–2172 CrossRef CAS
- S. D. Poynton, R. C. T. Slade, T. Omasta, W. E. Mustain, R. E. Cid, O. Pilar and J. R. Varcoe, J. Mater. Chem. A, 2014, 2, 5124–5130 RSC
| This journal is © The Royal Society of Chemistry 2020 |