
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Efficient photocatalytic carbon monoxide production from ammonia and carbon dioxide by the aid of artificial photosynthesis†
Zeai
Huang
 a,
Kentaro
Teramura
*ab,
Hiroyuki
Asakura
ab,
Saburo
Hosokawa
ab and
Tsunehiro
Tanaka
*ab
a,
Kentaro
Teramura
*ab,
Hiroyuki
Asakura
ab,
Saburo
Hosokawa
ab and
Tsunehiro
Tanaka
*ab
aDepartment of Molecular Engineering, Graduate School of Engineering, Kyoto University, Kyoto 615-8510, Japan. E-mail: teramura@moleng.kyoto-u.ac.jp; tanakat@moleng.kyoto-u.ac.jp
bElements Strategy Initiative for Catalysts and Batteries, Kyoto University, Kyoto 615-8510, Japan
First published on 19th June 2017
Abstract
Ammonium bicarbonate (NH4HCO3) was generated by the absorption of carbon dioxide (CO2) into an aqueous solution of ammonia (NH3). NH4HCO3 was successfully used to achieve highly efficient photocatalytic conversion of CO2 to carbon monoxide (CO). NH3 and/or ammonium ions (NH4+) derived from NH4HCO3 in aqueous solution were decomposed into nitrogen (N2) and hydrogen (H2). Stoichiometric amounts of the N2 oxidation product and the CO and H2 reduction products were generated when the photocatalytic reaction was carried out in aqueous NH4HCO3 solution. NH3 and/or NH4+ functioned as electron donors in the photocatalytic conversion of CO2 to CO. A CO formation rate of 0.5 mmol h−1 was obtained using 500 mg of catalyst (approximately 7500 ppm) in ambient conditions (303 K, 101.3 kPa). Our results demonstrated that NH4HCO3 is a novel inorganic sacrificial reagent, which can be used to increase the efficiency of photocatalytic CO production to achieve one step CO2 capture, storage and conversion.
Introduction
The production of chemical feedstocks and hydrocarbon fuels from CO2 is a promising approach to alleviate the global energy crisis and global warming.1 Conversion of CO2 to CO using clean and renewable solar energy is the first step to store energy in chemicals because CO can be further converted into other highly valuable chemicals using the Fischer–Tropsch process.2 A variety of heterogeneous and homogeneous photocatalysts have been reported to achieve the conversion of CO2 to CO.3–5 However, the formation rate of CO has been limited to a few tens of μmol h−1 or hundreds of μmol h−1 g−1 because of the high energy barrier to CO2 reduction and inefficient light utilization.6,7 Furthermore, CO2 is not easily adsorbed onto catalytic surfaces nor activated by photoirradiation because of its high thermodynamic stability. This further reduces the efficiency of the photocatalytic conversion of CO2.Water (H2O) is widely used as an electron donor in the photocatalytic conversion of CO2 to CO.7–12 However, the overall water splitting into H2 and O2 is more thermodynamically favorable than the reduction of CO2 in aqueous solution. Hence, CO2 reduction competes with overall water splitting. Moreover, the solubility of CO2 in pure H2O is only 0.033 mol L−1 (at 298 K and 101.3 kPa),13 which further limits the efficiency of CO2 conversion by H2O using heterogeneous photocatalysts. Therefore, it would be meaningful to find a readily available, highly efficient, and abundant in nature and industries electron donor (sacrificial reagent) other than water for the photocatalytic conversion of CO2. NH3 and NH4+ in aqueous solution can be readily oxidized to N2, NO2−, and NO3− using a photocatalyst.14–17 The decomposition of aqueous NH3 to H2 and N2 requires a standard Gibbs free energy change ΔG° of 18 kJ mol−1 (eqn (1)).18 This is significantly smaller than that required for the decomposition of H2O to H2 and O2 (237 kJ mol−1; eqn (2)).
NH3(aq) → ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) H2(g) + ½N2(g) ΔG° = 18 kJ mol−1 H2(g) + ½N2(g) ΔG° = 18 kJ mol−1 | (1) |
| H2O(l) → H2(g) + ½O2(g) ΔG° = 237 kJ mol−1 | (2) |
Because the photocatalytic oxidation of NH3 and NH4+ is significantly more favorable than the oxidation of H2O to O2,18 it is possible to use NH3 and NH4+ as electron donors in the photocatalytic conversion of CO2. Moreover, NH3 has been considered for use as an efficient post-combustion CO2 capture and storage (CCS) reagent because of its high absorption efficiency and loading capacity.19 The absorption and capture of CO2 by an aqueous solution of NH3 results in the formation of NH4HCO3.20 Other basic species, such as NaHCO3 and KHCO3, have been used to increase the solubility of CO2 in aqueous solutions.21,22 Previous reports have suggested that dissolved CO2, rather than bicarbonate (HCO3−) or carbonate (CO32−) ions, is the active species in the reduction of CO2.23,24 Correspondingly, the conversion of CO2 and/or the selectivity toward CO evolution have been significantly enhanced by the presence of bases in both photocatalytic (PC) and photoelectrochemical (PEC) cell systems.25,26 In the present study, we designed the use of NH4HCO3 for the efficient photocatalytic conversion of CO2 to CO in H2O.
Results and discussion
Flux-mediated crystal growth method shows the advantage of the synthetic control over particle sizes, morphologies, and surface features comparing with that of solid-state reaction method (SSR).27 Modification of these features as a function of flux conditions have been reported to show significant enhancements in both water splitting and CO2 photoreduction.11,27,28 Sr2KTa5O15 has been reported to show good activity and selectivity toward CO evolution when used as a photocatalyst in the conversion of CO2 by H2O in our previous work.29 In the present study, Sr2KTa5O15 was fabricated by a modified flux method, using a mixture of NaCl and KCl as the flux. The resultant catalyst was confirmed to have tetragonal tungsten bronze (TTB) structure (Fig. S1A†), and its real chemical formula was determined to be Sr1.6K0.35Na1.45Ta5O15 using ICP-OES. Its morphology was observed by SEM, and was found to consist of a mixture of nanorods and nanoparticles (Fig. S1B†).Fig. 1 shows the time courses of the photocatalytic conversion of CO2 in H2O and aqueous solutions of NaHCO3 and NH4HCO3. In pure H2O, only 16.8 μmol of CO was evolved after 5 h of photoirradiation (Fig. 1A), and the main reduction product was H2 (139.0 μmol). These results were consistent with previous reports.25,29 In this system, overall water splitting proceeded more readily than CO2 reduction, resulting in the generation of H2 as the major product, rather than CO. The amount of CO evolved in 0.1 M aqueous NaHCO3 solution after 5 h of photoirradiation (448.7 μmol) was 26.7 times higher than that evolved in pure H2O (Fig. 1B). However, the formation of H2 (94.7 μmol) was not significantly affected by NaHCO3. Thus, NaHCO3 greatly enhanced the conversion of CO2 to CO without affecting the water splitting process.25 In both pure H2O and 0.1 M aqueous NaHCO3, stoichiometric amounts of O2 were evolved continuously during the reaction, implying that H2O functioned as an electron donor in the reduction of CO2. Moreover, the evolution of CO increased dramatically in 0.1 M aqueous NH4HCO3 solution; 1600 μmol (1.6 mmol) of CO was evolved after 5 h of photoirradiation (Fig. 1C). This is 94.2 times greater than the amount evolved in pure H2O. The selectivity of the reaction toward CO evolution was calculated and the details were shown in ESI.† The selectivity toward CO evolution in 0.1 M aqueous NH4HCO3 (86.2%) was similar to that in aqueous NaHCO3 (82.5%). The production of gaseous products was negligible in blank tests conducted without either a catalyst or photoirradiation (Fig. S2A and B†). Thus, both are necessary for the photocatalytic conversion of CO2 to CO to proceed. Without Ag cocatalyst, H2 was formed as main product (Fig. S2C†), both of N2 and O2 were detected as oxidation products, however, the amount of these gases was far beyond the stoichiometric amount. Tiny amount of CO was formed after 5 hour photoirradiation (14.9 μmol). Ag cocatalysts were important in photocatalytic conversation of CO2 to CO, which is thought to be the active sites. H2, CO, and N2 were obtained without a continuous CO2 flow (Fig. S2D†). However, H2 was generated as a major product, suggesting a very low selectivity toward CO evolution (less than 30%). This suggested that CO2 presence significantly increases the selectivity of the photocatalytic conversion of CO2 toward CO evolution in NH4HCO3 solution. NH4HCO3 can be formed directly by the absorption of CO2 in an aqueous solution of NH3; wherein H2 and CO can be produced from CO2 and NH3via artificial photosynthesis. Thus, our designed system can achieve carbon capture and utilization (CCU) in a single process.
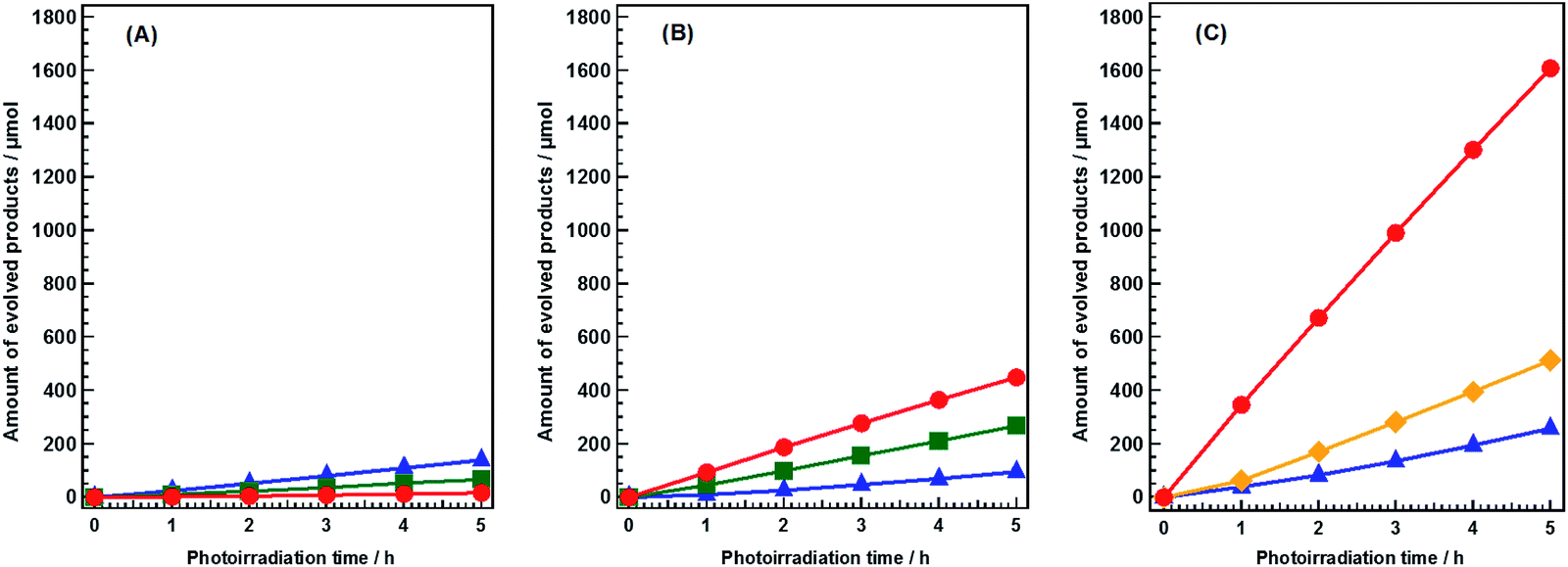 | ||
| Fig. 1 Time courses of CO (circle), O2 (square), N2 (lozenge), and H2 (triangle) evolutions during the photocatalytic conversion of CO2 over Ag-modified Sr1.6K0.35Na1.45Ta5O15. Amount of catalyst: 0.5 g; cocatalyst loading: 1.0 wt% Ag; light source: 400 W high-pressure Hg lamp; water volume: 1.0 L; CO2 flow rate: 30 mL min−1; additive: (A) none, (B) 0.1 M NaHCO3, or (C) 0.1 M NH4HCO3. | ||
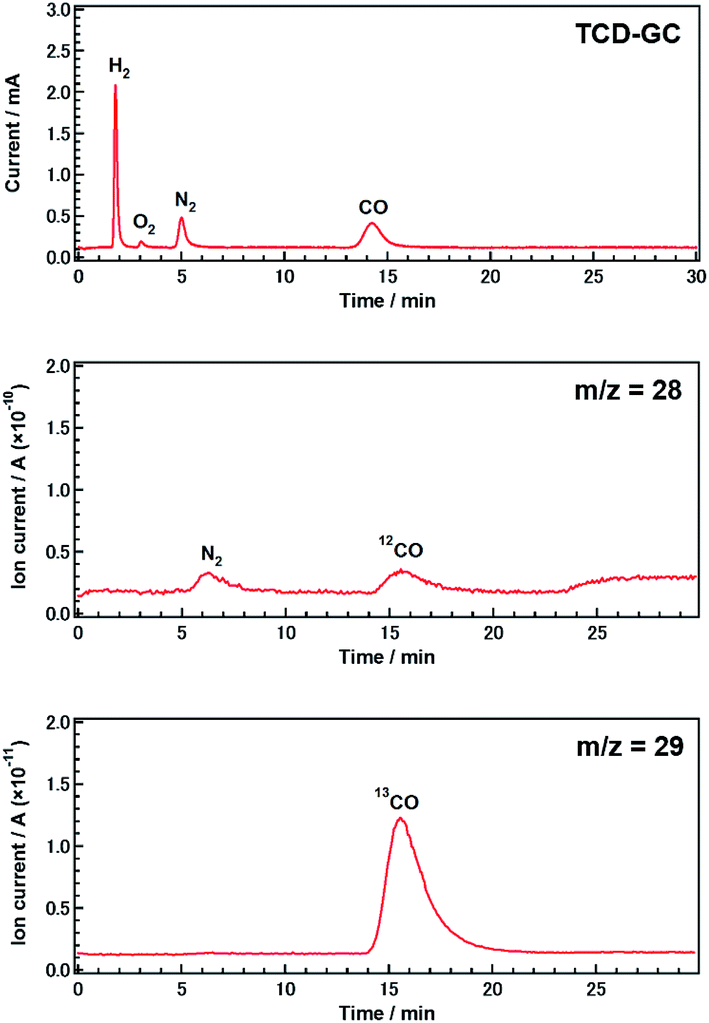 | ||
| Fig. 2 Gas chromatogram and mass spectra (m/z = 28 and 29) obtained during the photocatalytic conversion of 13CO2 using Ag-modified Sr1.6K0.35Na1.45Ta5O15. Amount of catalyst: 0.5 g; cocatalyst loading: 5.0 wt% Ag; light source: 400 W high-pressure Hg lamp; water volume: 1.0 L; CO2 flow rate: 30 mL min−1; additive: 0.5 M NH4HCO3. | ||
N2, rather than O2, was generated as the oxidation product during photoirradiation in the presence of NH4HCO3 (Fig. 1C). This demonstrated that H2O did not function as an electron donor in this system. Instead, NH3 and/or NH4+ functioned as electron donors, because the ΔG° of NH3(aq) oxidation (18 kJ mol−1) is significantly lower than that of water oxidation (237 kJ mol−1). Analysis of the liquid phase showed that neither NO2− nor NO3− were present during photoirradiation (Fig. S3†). Other gaseous NOx products, such as N2O and NO, were not detected by gas chromatography (GC). These results indicated that NH3 and/or NH4+ were oxidized only to N2 in this photocatalytic system. Hence, by using NH4HCO3, we succeeded in controlling the oxidation product, in addition to enhancing the conversion of CO2.
The ratio of electrons to holes consumed in the photocatalytic conversion of CO2 was calculated to be 2.0 after 1 h of photoirradiation (Fig. S4†). Given that the total number of electrons generated must be the same as the number of holes, this ratio indicates that significantly more electrons were consumed than holes in the initial stages of photoirradiation. We noted that the state of Ag was changed from metallic to Ag+ on the surface of catalyst measured by XPS (Fig. S5†), however, it might be not the main reason for the excess of electron consumption. We calculated that if all Ag0 was changed to Ag+ in the first hour, the consumed holes were still only 469 μmol, which was much less than the consumed electrons (770 μmol). NH4+ can be reduced to NH3 and ˙H by photogenerated electrons. Hydrazine (N2H4) has been determined to be an intermediate species in the photocatalytic decomposition of NH3 and/or NH4+ to H2 and N2 using Pt/TiO2.14 Stoichiometric amounts of products, including H2 and N2, were not obtained in the initial stages of photoirradiation due to the formation of hydrazine. In our system, it is also possible to form hydrazine at the beginning, however, hydrazine is reported to be reacted with CO2 to form zwitterionic intermediate and carbamate-type species,30 which made the detection of intermediate oxidation species much more difficult. Nevertheless, stoichiometric amounts of products were obtained after 2 h of photoirradiation (Fig. S4†), indicating that the total decomposition of NH3 and/or NH4+ occurred sooner.
The above results demonstrate that the highly efficient photocatalytic conversion of CO2 to CO was achieved in our system. The stoichiometric amounts of H2, N2 and CO generated indicated that NH3 and/or NH4+ functioned as electron donors in the photocatalytic conversion of CO2. Significantly greater photocatalytic activity was obtained using NH3 and/or NH4+, compared to reactions using H2O as an electron donor under the same conditions. NH3 and/or NH4+ are suitable for use in practical applications because NH3 is industrially produced in large quantities. Furthermore, in our photocatalytic system, NH3 and/or NH4+ can be completely decomposed to N2, which is an inert and non-toxic gas.
Table 1 shows the effects of NH4HCO3 concentration on the photocatalytic conversion of CO2. In pure H2O, overall water splitting proceeded as the dominant reaction. Hence, the evolution of CO was negligible (entry 1). When the photocatalytic reaction was carried out in 0.01 M aqueous NH4HCO3 the production of H2 resulting from water splitting was dramatically suppressed (entry 2). Because the oxidation of NH3 and/or NH4+ to N2 proceeds more readily than the oxidation of H2O to O2, the formation rate of O2 in 0.01 M aqueous NH4HCO3 was less than half that in pure H2O. Even low concentrations of NH4HCO3 (0.01 M) significantly increased the formation rate of CO, indicating that the presence of NH4HCO3 is vital to achieving high photocatalytic activity. NH4HCO3 can also be used to increase the pH of the reaction solution, to offset the decrease in pH caused by the dissolution of CO2. With CO2 flowing, the pH of the reaction solution based on pure H2O was 3.95, which increased to 5.88 with the addition of 0.01 M NH4HCO3. Increasing the pH also increases the amount of CO2 that can be dissolved in the reaction solution.31 Generally, the formation rate of CO increases with increasing pH, because the reaction rate largely depends on the concentration of substrate. Therefore, the addition of NH4HCO3 contributed to the efficient conversion of CO2 and the good selectivity toward CO evolution. Increasing the concentration of NH4HCO3 from 0.01 M to 0.05 M completely suppressed the overall water splitting reaction, since only N2 was generated as an oxidation product (entry 3). The formation rate of CO increased with the concentration of NH4HCO3. Increasing the NH4HCO3 concentration from 0.1 M to 1.0 M increased the formation rate of CO to 550.7 μmol h−1 except the selectivity toward CO evolution decreased slightly, from 86.1% to 65.5% (entry 4 to 7). As previously discussed, NH4HCO3 can be synthesized by flowing CO2 through an aqueous solution of NH3. To determine whether NH3 functions as an electron donor under a flow of CO2, we carried out the photocatalytic conversion of CO2 in an aqueous solution of NH3 (entry 8). The formation rate of CO was 547.2 μmol h−1 in ca. 0.5 M aqueous NH3, indicating that NH3 functions efficiently as an electron donor under these conditions. The ratio of photogenerated electrons to holes (e−/h+) was estimated to be around 1.0 in reactions with high concentrations of aqueous NH4HCO3 after 5 h of photoirradiation. This further supports the hypothesis that NH3 and/or NH4+ function as effective electron donors during the photocatalytic conversion of CO2.
| Entry | NH4HCO3a/M | Formation rateb/μmol h−1 | Selec.c (%) | e−/h+d | |||
|---|---|---|---|---|---|---|---|
| H2 | O2 | N2 | CO | ||||
| a Additive concentration used for CO2 conversion. b Formation rate after 5 h of irradiation. c Selectivity toward CO evolution. d Ratio of consumed electrons to holes after 5 h of irradiation. e 0.5 M aqueous NH3 solution was used as the additive, instead of NH4HCO3. | |||||||
| 1 | 0 | 35.9 | 16.3 | Trace | 3.6 | 9.2 | 1.21 |
| 2 | 0.01 | 16.9 | 7.0 | 12.3 | 54.5 | 76.3 | 1.17 |
| 3 | 0.05 | 23.8 | Trace | 42.3 | 146.7 | 86.1 | 1.34 |
| 4 | 0.1 | 48.4 | Trace | 94.3 | 270.4 | 84.8 | 1.13 |
| 5 | 0.5 | 119.8 | Trace | 193.6 | 512.9 | 81.1 | 1.09 |
| 6 | 0.8 | 175.4 | Trace | 213.3 | 520.0 | 74.8 | 1.09 |
| 7 | 1.0 | 290.1 | Trace | 258.1 | 550.7 | 65.5 | 1.09 |
| 8e | — | 235.0 | Trace | 244.9 | 547.2 | 70.0 | 1.06 |
To confirm that CO evolution originated from CO2 introduced in the gas phase, rather than from carbon contaminants, we conducted an isotopic labeling experiment. Fig. 2 shows mass spectra (m/z = 28 and 29) obtained during the photocatalytic conversion of 13CO2 in 0.5 M aqueous NH4HCO3 over Ag-modified Sr1.6K0.35Na1.45Ta5O15 after 0.5 h of photoirradiation. Gaseous samples were introduced into a mass spectrometer (MS) after separation by thermal conductivity detector-gas chromatography (TCD-GC). CO was observed in both the gas chromatogram and the mass spectra. The peak positions in the mass spectra were consistent with those in the chromatogram. The major product was 13CO, rather than 12CO. The presence of a small amount of 12CO may be due to the direct decomposition of NH4HCO3 since the decomposition of NH4HCO3 was observed in samples without a CO2 flow (Fig. S2D†). The amount of 13CO estimated by mass spectrometry was approximately equal to the amount of CO determined using a flame ionization detector (FID-GC) (Fig. S6†). These results demonstrate that CO was predominantly generated from CO2 introduced in the gas phase, rather than from other carbon resources.
The recycle test was also performed to confirm the stability and durability of our catalyst and system using the Sr1.6K0.35Na1.45Ta5O15 photocatalyst repeatedly for three times under the same conditions (Fig. S7†). In the second cycle, there is a slight loss by ca. 10% of CO evolution activity during 5 h photoirradiation as compared to the first run, however, the evolution of H2 showed no obvious changes. The slight loss of activity should be due to the change of Ag cocatalyst (Fig. S5†).29 The photocatalytic activity of CO, N2, and H2 were stabilized at ca. 0.5, 0.19 and 0.07 mmol h−1, respectively, during the second and third runs. The structure of catalyst itself was stable during the three cycles (Fig. S8†). These results suggested that the photocatalyst and the system exhibit favorable stability to form CO, N2, and H2 during the photocatalytic conversion of CO2.
To confirm the versatility of NH4HCO3 as a general electron donor in photocatalytic reactions, we carried out the photocatalytic conversion of CO2 in aqueous NH4HCO3 solution over 4 types of photocatalysts. All these photocatalysts have been already reported to show good activity and high selectivity toward CO evolution in the photocatalytic conversion of CO2 using H2O as an electron donor.25,32–34 As shown in Table 2, all the photocatalysts showed good activity for conversion of CO2 and high selectivity toward CO evolution. The activities of the photocatalysts were significantly increased in aqueous NH4HCO3 solution, compared with their reported activities in pure H2O or aqueous NaHCO3.32–34 N2 was detected as the only oxidation product and the e−/h+ ratio was approximately equal to 1.0. These results indicated that NH3 and/or NH4+ was easily decomposed to N2 gas by the photocatalysts tested.
| Entry | Catalyst | Formation ratea/μmol h−1 | Selec.b (%) | e−/h+c | ||
|---|---|---|---|---|---|---|
| H2 | N2 | CO | ||||
| a Formation rate after 5 h of irradiation. O2 was not detected in any of the samples. b Selectivity toward CO evolution. c Ratio of consumed electrons to holes after 5 h of irradiation. | ||||||
| 1 | ZnGa2O4/Ga2O3 | 125.2 | 191.4 | 532.0 | 80.9 | 1.14 |
| 2 | ZnGa2O4 | 39.4 | 94.2 | 305.4 | 88.6 | 1.22 |
| 3 | La2Ti2O7 | 5.9 | 17.4 | 41.6 | 87.6 | 0.91 |
| 4 | SrO/Ta2O5 | 2.71 | 11.5 | 42.9 | 94.1 | 1.32 |
Conclusions
We designed a highly efficient process for the photocatalytic conversion of CO2 to CO in aqueous NH4HCO3 solution. The stoichiometric formation of CO, H2, and N2 indicated that NH3 and/or NH4+ were consumed as electron donors, instead of H2O. NH4HCO3 was determined to be an effective electron donor for the photocatalytic conversion of CO2, whereby CO2 can be captured, stored, and efficiently converted into CO. This novel inorganic additive is suitable for use in carbon capture and utilization process. This new process is a promising way to control the conversion of CO2 to CO and efficiently produce H2 and CO.Acknowledgements
This study was partially supported by a Grant-in-Aid for Scientific Research on Innovative Areas “All Nippon Artificial Photosynthesis Project for Living Earth” (No. 2406) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan, the Precursory Research for Embryonic Science and Technology (PRESTO), supported by the Japan Science and Technology Agency (JST), and the Program for Elements Strategy Initiative for Catalysts & Batteries (ESICB), commissioned by the MEXT of Japan. Zeai Huang thanks the State Scholarship of China Scholarship Council, affiliated with the Ministry of Education of the P. R. China.Notes and references
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- G. Centi, E. A. Quadrelli and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 CAS.
- K. Teramura, S. Iguchi, Y. Mizuno, T. Shishido and T. Tanaka, Angew. Chem., 2012, 124, 8132–8135 CrossRef.
- R. Kuriki, K. Sekizawa, O. Ishitani and K. Maeda, Angew. Chem., Int. Ed., 2015, 54, 2406–2409 CrossRef CAS PubMed.
- R. Kuriki, H. Matsunaga, T. Nakashima, K. Wada, A. Yamakata, O. Ishitani and K. Maeda, J. Am. Chem. Soc., 2016, 138, 5159–5170 CrossRef CAS PubMed.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro and H. Garcia, ChemSusChem, 2013, 6, 562–577 CrossRef PubMed.
- X. Chang, T. Wang and J. Gong, Energy Environ. Sci., 2016, 9, 2177–2196 CAS.
- G. Liu, S. Xie, Q. Zhang, Z. Tian and Y. Wang, Chem. Commun., 2015, 51, 13654–13657 RSC.
- K. Iizuka, T. Wato, Y. Miseki, K. Saito and A. Kudo, J. Am. Chem. Soc., 2011, 133, 20863–20868 CrossRef CAS PubMed.
- M. Yamamoto, T. Yoshida, N. Yamamoto, T. Nomoto, Y. Yamamoto, S. Yagi and H. Yoshida, J. Mater. Chem. A, 2015, 3, 16810–16816 CAS.
- H. Yoshida, L. Zhang, M. Sato, T. Morikawa, T. Kajino, T. Sekito, S. Matsumoto and H. Hirata, Catal. Today, 2015, 251, 132–139 CrossRef CAS.
- H. Nakanishi, K. Iizuka, T. Takayama, A. Iwase and A. Kudo, ChemSusChem, 2017, 10, 112–118 CrossRef CAS PubMed.
- K. Hara, A. Kudo, T. Sakata and M. Watanabe, J. Electrochem. Soc., 1995, 142, L57–L59 CrossRef CAS.
- H. Yuzawa, T. Mori, H. Itoh and H. Yoshida, J. Phys. Chem. C, 2012, 116, 4126–4136 CAS.
- H. Wang, Y. Su, H. Zhao, H. Yu, S. Chen, Y. Zhang and X. Quan, Environ. Sci. Technol., 2014, 48, 11984–11990 CrossRef CAS PubMed.
- X. Zhu, S. R. Castleberry, M. A. Nanny and E. C. Butler, Environ. Sci. Technol., 2005, 39, 3784–3791 CrossRef CAS PubMed.
- S. Yamazoe, Y. Hitomi, T. Shishido and T. Tanaka, Appl. Catal., B, 2008, 82, 67–76 CrossRef CAS.
- H. Kominami, H. Nishimune, Y. Ohta, Y. Arakawa and T. Inaba, Appl. Catal., B, 2012, 111, 297–302 CrossRef.
- B. T. Zhao, Y. X. Su, W. W. Tao, L. L. Li and Y. C. Peng, Int. J. Greenhouse Gas Control, 2012, 9, 355–371 CrossRef CAS.
- X. Wang, W. Conway, D. Fernandes, G. Lawrance, R. Burns, G. Puxty and M. Maeder, J. Phys. Chem. A, 2011, 115, 6405–6412 CrossRef CAS PubMed.
- S. F. Shen, Y. N. Yang and S. F. Ren, Fluid Phase Equilib., 2014, 367, 38–44 CrossRef CAS.
- H. Zhong, K. Fujii, Y. Nakano and F. Jin, J. Phys. Chem. C, 2015, 119, 55–61 CAS.
- B. Kumar, M. Llorente, J. Froehlich, T. Dang, A. Sathrum and C. P. Kubiak, Annu. Rev. Phys. Chem., 2012, 63, 541–569 CrossRef CAS PubMed.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309–2326 RSC.
- K. Teramura, Z. Wang, S. Hosokawa, Y. Sakata and T. Tanaka, Chem.–Eur. J., 2014, 20, 9906–9909 CrossRef CAS PubMed.
- E. E. Barton, D. M. Rampulla and A. B. Bocarsly, J. Am. Chem. Soc., 2008, 130, 6342–6344 CrossRef CAS PubMed.
- J. Boltersdorf, N. King and P. A. Maggard, CrystEngComm, 2015, 17, 2225–2241 RSC.
- Y. Ham, T. Hisatomi, Y. Goto, Y. Moriya, Y. Sakata, A. Yamakata, J. Kubota and K. Domen, J. Mater. Chem. A, 2016, 4, 3027–3033 CAS.
- Z. Huang, K. Teramura, S. Hosokawa and T. Tanaka, Appl. Catal., B, 2016, 199, 272–281 CrossRef CAS.
- K. H. Lee, B. Lee, J. H. Lee, J. K. You, K. T. Park, I. H. Baek and N. H. Hur, Int. J. Greenhouse Gas Control, 2014, 29, 256–262 CrossRef CAS.
- K. Teramura, K. Hori, Y. Terao, Z. Huang, S. Iguchi, Z. Wang, H. Asakura, S. Hosokawa and T. Tanaka, J. Phys. Chem. C, 2017, 121, 8711–8721 CAS.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, J. Mater. Chem. A, 2015, 3, 11313–11319 CAS.
- Z. Wang, K. Teramura, S. Hosokawa and T. Tanaka, Appl. Catal., B, 2015, 163, 241–247 CrossRef CAS.
- K. Teramura, H. Tatsumi, Z. Wang, S. Hosokawa and T. Tanaka, Bull. Chem. Soc. Jpn., 2015, 88, 431–437 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, calculations and characterizations. See DOI: 10.1039/c7sc01851g |
| This journal is © The Royal Society of Chemistry 2017 |