
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A family of cis-macrocyclic diphosphines: modular, stereoselective synthesis and application in catalytic CO2/ethylene coupling†
Ioana
Knopf
a,
Daniel
Tofan
a,
Dirk
Beetstra
b,
Abdulaziz
Al-Nezari
b,
Khalid
Al-Bahily
b and
Christopher C.
Cummins
 *a
*a
aDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139-4307, USA. E-mail: ccummins@mit.edu; Tel: +1 617 253 5332
bSABIC CRD, Fundamental Catalysis, Thuwal 23955-6900, Saudi Arabia
First published on 11th October 2016
Abstract
A family of cis-macrocyclic diphosphines was prepared in just three steps from white phosphorus and commercial materials using a modular synthetic approach. Alkylation of bicyclic diphosphane 3,4,8,9-tetramethyl-1,6-diphosphabicyclo(4.4.0)deca-3,8-diene, or P2(dmb)2, produced phosphino-phosphonium salts [R-P2(dmb)2]X, where R is methyl, benzyl and isobutyl, in yields of 90–96%. Treatment of these salts with organolithium or Grignard reagents yielded symmetric and unsymmetric macrocyclic diphosphines of the form cis-1-R-6-R′-3,4,8,9-tetramethyl-2,5,7,10-tetrahydro-1,6-DiPhospheCine, or R,R′-DPC, in which R′ is methyl, cyclohexyl, phenyl or mesityl, in yields of 46–94%. Alternatively, symmetric diphosphine Cy2-DPC was synthesized in 74% yield from the dichlorodiphosphine Cl2P2(dmb)2. As a first application, these cis-macrocyclic diphosphines were used as ligands in the nickel-catalyzed synthesis of acrylate from CO2 and ethylene, for which they showed promising catalytic activity.
While chelating diphosphines have established a wide range of uses in areas from fundamental chemistry to catalysis,1 macrocyclic diphosphines are an underrepresented class of ligands. Their scarcity is primarily due to challenges associated with their synthesis. Macrocycles containing two or more phosphorus atoms exhibit multiple stereoisomers since the phosphines, which are locked in place by the cyclic framework, have a high barrier to inversion. Typically, syntheses of macrocyclic diphosphines yield mixtures of diastereomers;2,3 stereoselective syntheses of either cis or trans macrocyclic diphosphines are rare.4,5 While synthetically challenging, embedding one or more phosphorus atoms in a cyclic framework is desirable as it leads to more rigid and robust structures compared to their acyclic phosphine counterparts.6
cis-Macrocyclic diphosphines have been postulated to be “interesting ligands for transition metal complexes, comparable to, but perhaps usefully different from, the familiar range of chelating diphosphines”,7 yet their coordination chemistry is essentially unexplored.8 In this context, it is useful to distinguish between medium-sized (7–12-membered rings) and large (>12 membered rings) macrocyclic diphosphines. Complexes of large macrocyclic diphosphines have been synthesized as mixtures of diastereomers via ring closing metathesis and hydrogenation of preassembled metal complexes of trans-spanning monophosphines with pendant olefins.3 This synthetic strategy was also extended to prepare metal complexes of large trans-spanning macrobicyclic diphosphines.9 While both cis and trans isomers of a diphosphine embedded in a very large macrocycle can bind to a metal center, only the cis isomer can bind to a single metal center in medium-sized ring systems. The only medium-sized macrocyclic diphosphine with structurally characterized metal complexes is cis-1,5-diphenyl-1,5-diphosphacyclooctane, a ligand obtained after tedious separation of the cis and trans diastereomers produced by the lithium aluminum hydride reduction of the corresponding phosphine oxides.10
The only stereoselective synthetic route to cis medium-sized macrocyclic diphosphines was reported by Alder et al.5 We were surprised to find no coordination complexes reported for these diphosphines, despite them being synthesized two decades ago. In the Alder synthesis, the desired cis-macrocyclic diphosphines are obtained by stereoselective cleavage of the P–P bond in alkylated diphosphabicyclo[k.l.0]alkanes (k = 3–5, l = 3–4) by organometallic reagents. These diphosphabicyclo[k.l.0]alkanes are prepared via a tedious procedure from diphosphinoalkanes H2P–(CH2)k–PH2, which in turn are prepared in two steps from the corresponding dibromoalkanes. While the bicyclic diphosphane framework is key to the stereoselectivity of the entire process, its assembly represents the most cumbersome part of the synthesis. Our group has recently accessed bicyclic diphosphanes in only one step from white phosphorus and commercial dienes under photochemical conditions.11,12 For example, diphosphane 3,4,8,9-tetramethyl-1,6-diphosphabicyclo(4.4.0)deca-3,8-diene or P2(dmb)2 (1) is obtained in gram quantities directly from white phosphorus and 2,3-dimethylbutadiene.11 This synthesis is part of a greater endeavor pursued by our group13 and others14 to prepare phosphorus-containing compounds in an atom economical fashion directly from P4,15 the precursor to the more widely used phosphorus source PCl3.
We envisioned that alkylating 1 would yield phosphino-phosphonium salts that would be prone to reacting with nucleophiles to break the P–P bond. This process would afford cis-macrocyclic diphosphines in a stereoselective manner in only three steps from white phosphorus! We describe herein an expedited synthetic route to a family of cis-macrocyclic diphosphines that will facilitate investigating the coordination chemistry of these molecules and enable their use as ligands in catalysis.
We began this synthetic endeavor by investigating the alkylation of diphosphane 1 with methyl iodide (MeI). Previous studies have shown excellent selectivity in functionalizing only one of the phosphorus atoms in 1.16 As expected, phosphino-phosphonium salt [Me-P2(dmb)2]I (2) was obtained in 90% yield after treatment of 1 with methyl iodide in diethyl ether at room temperature overnight. The structure of [Me-P2(dmb)2]I was confirmed by X-ray crystallography (Fig. 2), and displayed a typical P–P single bond distance of 2.1862(17) Å. Treatment of 1 with benzyl bromide (BnBr) also proceeded smoothly to give [Bn-P2(dmb)2]Br (3) in 96% yield after 2 hours at room temperature in dichloromethane (Fig. 1). The reaction of 1 with isobutyl bromide (iBuBr) was more sluggish and required heating at 100 °C for 20 h in the presence of an excess of the alkyl halide in order to achieve full conversion to [iBu-P2(dmb)2]Br (4).17 All of these phosphino-phosphonium salts have characteristic 31P{1H} NMR spectra that show strong coupling between the two phosphorus atoms. For example, the 31P{1H} NMR shifts of [Me-P2(dmb)2]I are +44.7 ppm and −69.3 ppm, with a 1JPP = 276 Hz (see Table S1 in ESI† for a summary of all the 31P{1H} NMR chemical shifts and coupling constants).
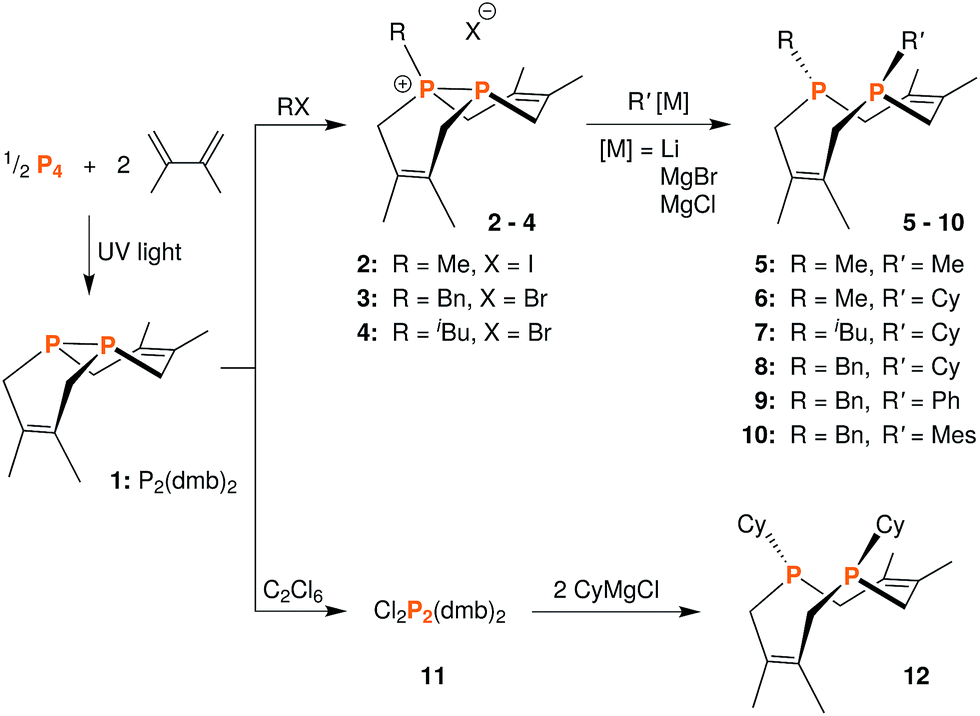 | ||
| Fig. 1 Synthetic routes to phosphino-phosphonium salts 2–4, dichloride 11, and macrocyclic diphosphines 5–10 and 12. | ||
 | ||
| Fig. 2 Solid-state structure of [Me-P2(dmb)2]I (2) with ellipsoids at the 50% probability level and disordered THF omitted for clarity. Representative interatomic distances [Å] and angles [°]: P2–C4 1.813(5), P2–C5 1.817(5), P2–C9 1.791(5), P1–P2 2.1862(17); C9–P2–C4 111.7(2), C9–P2–C5 106.9(2), C4–P2–C5 112.0(2), C9–P2–P1 114.01(19). | ||
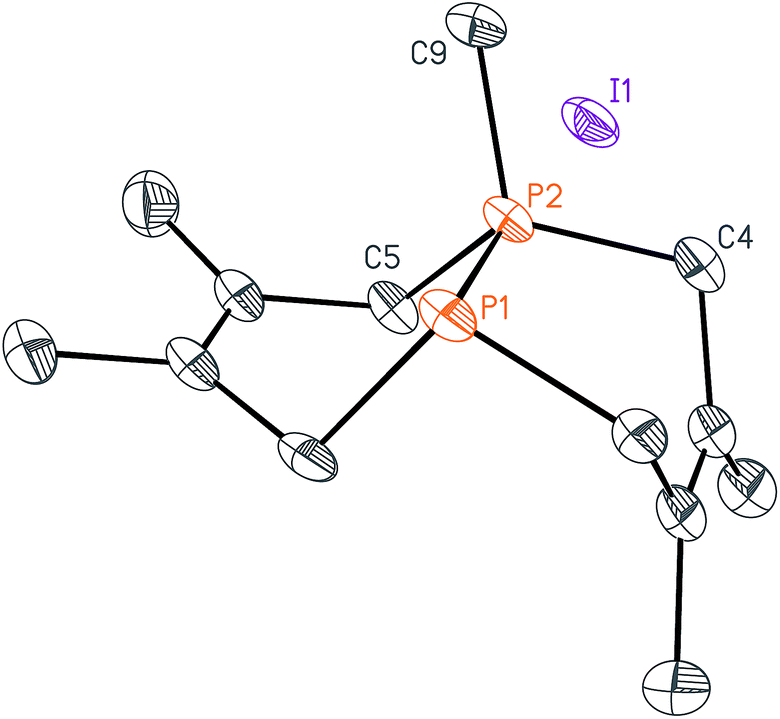
With these salts in hand, we began investigating their reactivity with organometallic reagents. Upon adding a methyllithium solution to a slurry of 2 in THF or diethyl ether at room temperature, a new signal indicative of cis-1,3,4,6,8,9-hexamethyl-2,5,7,10-tetrahydro-1,6-DiPhospheCine, or Me2-DPC (5), was observed by 31P{1H} NMR spectroscopy. A potential side reaction we had considered was deprotonation of the phosphino-phosphonium salt by methyllithium and formation of the Wittig reagent (CH2)P2(dmb)2. However, no evidence was found for the formation of such an ylide by 31P{1H} NMR spectroscopy. Diphosphine 5 was also characterized by X-ray crystallography (Fig. 3), revealing a conformation with nearly C2v symmetry in which the phosphorus lone pairs are pointed away from each other.
 | ||
| Fig. 3 Solid-state structure of Me2-DPC (5) with ellipsoids at the 50% probability level and hydrogen atoms omitted for clarity. Representative interatomic distances [Å] and angles [°]: P1–C1 1.8433(16), P1–C11 1.8583(16), P1–C18 1.8624(16), P2–C2 1.8421(17), P2–C15 1.8617(16), P2–C14 1.8630(16); C1–P1–C11 96.84(7), C1–P1–C18 98.07(7), C11–P1–C18 101.22(7), C2–P2–C15 97.03(8), C2–P2–C14 98.02(7), C15–P2–C14 101.54(7). | ||
Treatment of phosphino-phosphonium salts 2, 4, and 3 with cyclohexylmagnesium chloride led to the formation of Me,Cy-DPC (6), iBu,Cy-DPC (7), and Bn,Cy-DPC (8), respectively (Fig. 1). All of these diphosphines can be isolated in yields of 90–94% and they each show two distinct signals in their 31P{1H} NMR spectra, with small 5JPP coupling constants of ca. 5 Hz. For example, the 31P{1H} NMR spectrum of 6 shows two signals at −38.9 and −60.8 ppm. In order to demonstrate the versatility of this synthetic strategy, 3 was also treated with two aryl Grignard reagents, phenylmagnesium bromide and mesitylmagnesium bromide. The resulting diphosphines, Bn,Ph-DPC (9) and Bn,Mes-DPC (10), showed similar spectroscopic features to the dialkyl diphosphines described above.
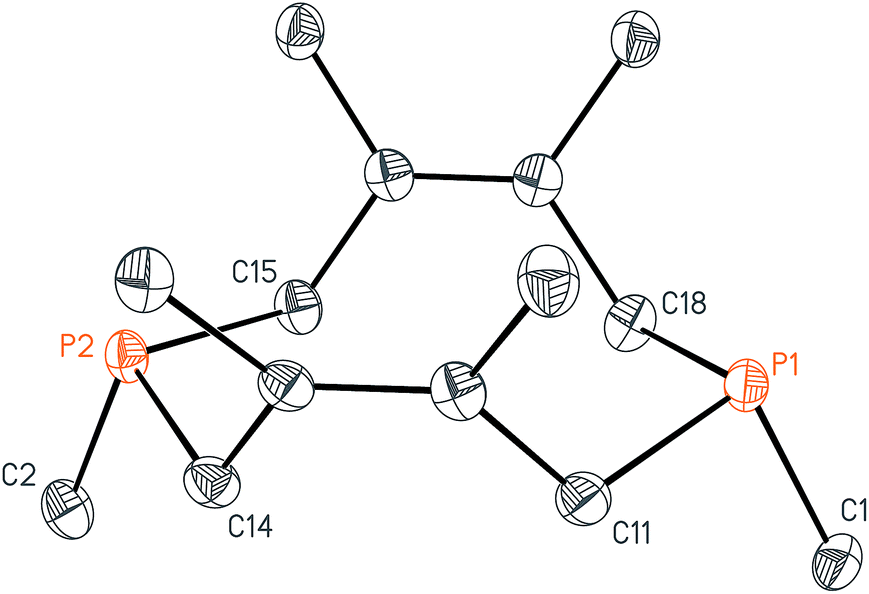
While this modular approach had already enabled the synthesis of a variety of macrocyclic diphosphines, the initial alkylation step was limited to primary halide substrates. In order to overcome this limitation, we sought to add a dihalodiphosphine to our synthetic toolbox. This intermediate would provide access to a symmetrical diphosphine upon addition of two equivalents of organometallic reagent. Treatment of 1 with iodine, a mild oxidant, did not yield a diiododiphosphine, but rather an iodine adduct, I2·1, which we were able to characterize by X-ray crystallography (Fig. 4). The elongated I–I distance of 3.4169(12) Å is similar to metrical data reported for other phosphine–iodine adducts.18 While several monophosphine–iodine adducts have been structurally characterized and described in the literature,18,19 this is an unusual example of a diphosphane–iodine adduct in which the P–P bond is still intact.
 | ||
| Fig. 4 Solid-state structure of I2·P2(dmb)2 (I2·1) with ellipsoids at the 50% probability level and disordered solvent and iodine omitted for clarity. Representative interatomic distances [Å] and angles [°]: I1–P1 2.4110(5), I1–I2 3.4169(12), P1–P2 2.1913(7); P1–I1–I2 173.21(4). | ||
The reaction of 1 with many potent halogenating agents (e.g. bromine, N-bromosuccinimide, xenon difluoride) proved to be unselective, yet oxidation of 1 with hexachloroethane produced the desired Cl2P2(dmb)2 (11) in good purity. We expected this product to have a symmetrical open structure, as the P–P bond would have been cleaved upon oxidation. However, NMR spectroscopy suggests that 11 is best described as a chloronium chloride salt in equilibrium with the open form. At room temperature, signals are broad in both the 1H NMR and 31P{1H} NMR (Δν1/2 ≈ 1700 Hz) spectra of 11; upon cooling to −40 °C, the 31P{1H} NMR spectrum shows two distinct phosphorus environments at +143.4 ppm and −48.7 ppm with a 1JPP = 291 Hz. In order to elucidate whether the broad NMR signals were due to chloride association/dissociation at the same phosphorus center versus chloride-mediated P–P bond scission/reformation, a 2D EXSY (EXchange SpectroscopY) experiment was performed.20 In the first pathway, the two phosphorus centers would remain distinct throughout the exchange process, while in the second pathway the two phosphorus centers would become chemically equivalent in the open form. The 31P{1H} 2D EXSY spectrum of 11 (Fig. 5) clearly shows the presence of exchange cross peaks, thus supporting the chemical exchange pathway depicted in the same figure. The proposed role of free chloride in the exchange was corroborated by treatment of 11 with GaCl3, which sequestered the chloride into the tetrachlorogallate [GaCl4]− anion; this inhibited the exchange process and “froze” the compound in the chloronium salt form with sharp 31P{1H} NMR resonances at room temperature.
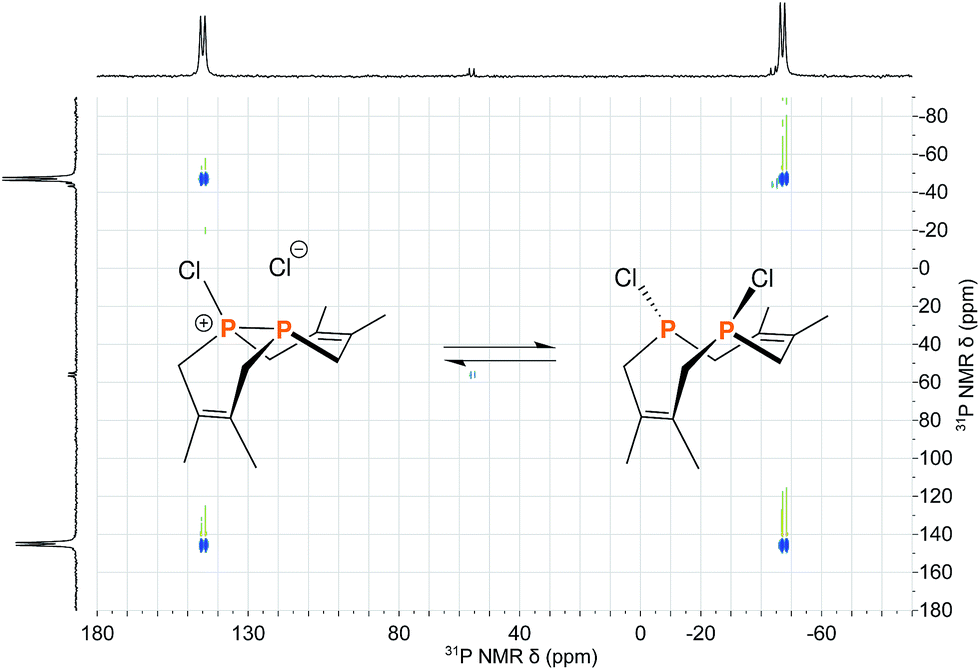 | ||
| Fig. 5 Reaction scheme depicting the equilibrium between the two structural forms of 11 overlayed on the 31P{1H} 2D EXSY spectrum of 11 acquired at 0 °C in CDCl3. | ||
Addition of two equivalents of cyclohexylmagnesium chloride to a suspension of 11 in diethyl ether or THF results in the formation of the desired Cy2-DPC (12). The 31P{1H} NMR spectrum of this product reveals a characteristic singlet resonance at −38.7 ppm for 12, but also a small amount of 1 (ca. 2%). This by-product is likely due to the reduction of 11 under the reaction conditions. While reduction was not a major reaction pathway during the formation of 12, it became problematic when attempting to prepare tBu2-DPC. Treatment of 11 with tert-butylmagnesium chloride resulted in a roughly equimolar mixture of tBu2-DPC and 1. Due to their similar solubility properties, these two products could not be separated.
While all syntheses were conducted under inert atmosphere, we wondered if these new diphosphines had any stability to air and moisture. Exposing a benzene-d6 solution of 12 to air led to no visible changes by 31P NMR spectroscopy even after 24 hours. This diphosphine proved to be remarkably robust, as less than 10% of the material had decomposed to the phosphine–phosphine oxide after 48 hours in solution.
Having accessed a family of new chelating macrocyclic diphosphines, we were interested in exploring their use as supporting ligands in a catalytic process. Given our interest in CO2 utilization,21 we turned to the coupling of CO2 and ethylene as a first application. While formation of nickelalactones from Ni(0) species, CO2 and ethylene had been known since the 1980s from the pioneering work of Hoberg and coworkers,22 the first catalytic system to produce acrylate from this coupling was only reported in 2012.23,24 The stability of nickelalactones has been one of the major challenges in assembling a catalytic process; conversion of a nickelalactone to its corresponding nickel acrylate complex requires both a base and an alkali metal Lewis acid.23,25,26 Furthermore, extensive ligand screening has shown that only a small, select group of electron-rich diphosphines gives rise to any catalytic activity whatsoever.27,28 Despite the progress made in the last few years, typical turnover numbers are in the double digits and only extensively optimized systems yield turnover numbers greater than 100.28
In order to test whether these cis-macrocyclic diphosphines could support a nickelalactone, we prepared (Fig. 6) and crystallographically characterized the nickelalactone of 12, (Cy2-DPC)Ni(CH2CH2COO) (13) (Fig. 7). The Ni1–C3 and Ni1–O1 distances of 1.982(6) and 1.899(4) Å, respectively, are similar to those reported for other nickelalactones.29 The bite angle of the diphosphine in complex 13 is 91.46(5)°, a value intermediate between that observed for the lactone of dicyclohexylphosphinoethane (dcpe), namely 88.07(5)°,26 and that observed for the lactone of diphenylphosphinobutane (dppb), namely 93.91(4)°.29 The smaller bite angle compared to dppb, a ligand that also has a four carbon atom bridge, might be due to the increased backbone rigidity of 12.
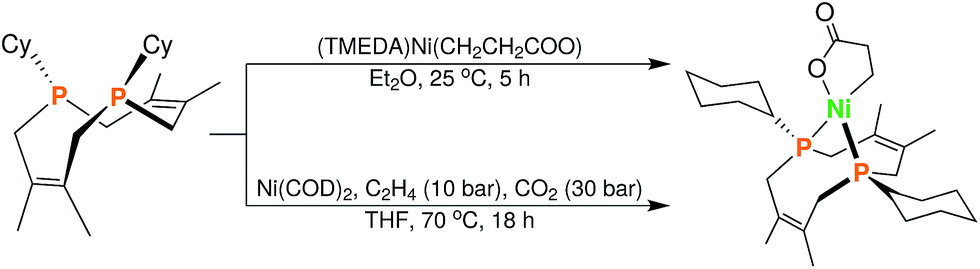 | ||
| Fig. 6 Synthesis of (Cy2-DPC)Ni(CH2CH2COO) (13) from 12via two complementary routes (see ESI† for details). | ||
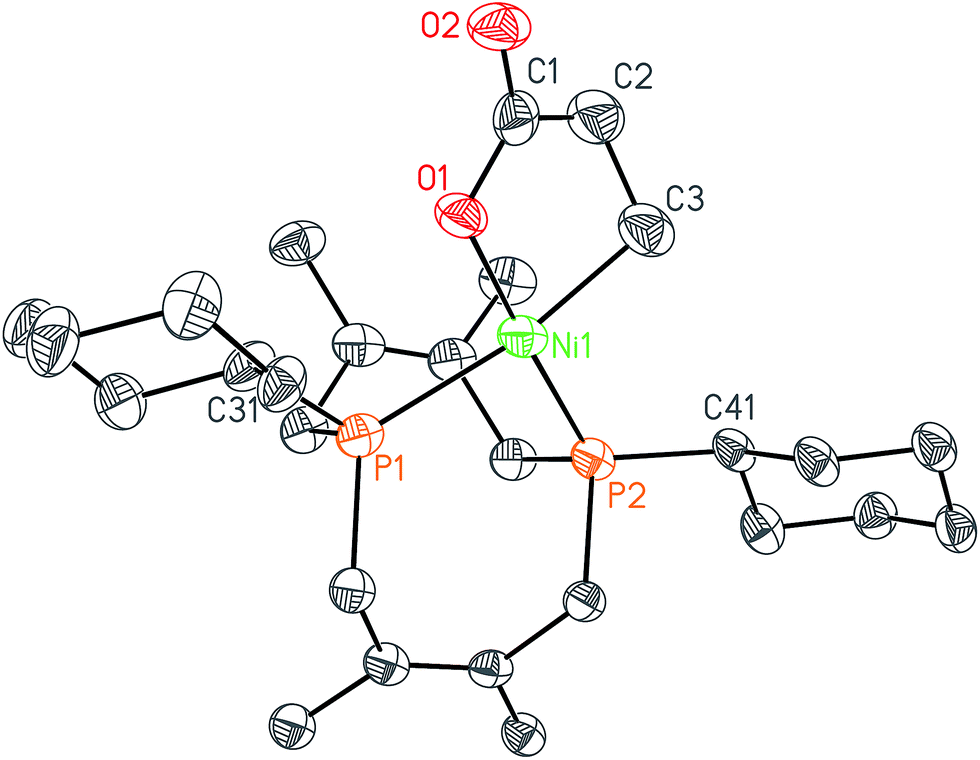 | ||
| Fig. 7 Solid-state structure of nickelalactone (Cy2-DPC)Ni(CH2CH2COO) (13) with ellipsoids at the 50% probability level and hydrogen atoms omitted for clarity. Representative interatomic distances [Å] and angles [°]: Ni1–P1 2.2454(15), Ni1–P2 2.1283(15), Ni1–C3 1.982(6), Ni1–O1 1.899(4), O1–C1 1.283(6), O2–C1 1.225(6); O1–Ni1–P1 90.86(12), O1–Ni1–C3 85.3(2), C3–Ni1–P2 92.56(19), P2–Ni1–P1 91.46(5); P1–Ni1–P2–C41–173.1(2), P2–Ni1–P1–C31 175.33(18). | ||
While helpful for comparing 12 with other ligands previously used to support nickelalactones, bite angle is an insufficient metric for describing the steric profile of 12. The cyclic backbone of this ligand renders an unusual placement of the cyclohexyl substituents in the P–Ni–P plane, a feature best described by the P1–Ni1–P2–C41 and P2–Ni1–P1–C31 torsion angles of −173.1(2)° and 175.33(18)°, respectively. In contrast, a ligand such as dcpe places its backbone linker in the P–Ni–P plane, and the cyclohexyl substituents above and below that plane (Fig. 8). The absolute values of the corresponding P–Ni–P–C torsion angles in (dcpe)Ni(CH2CH2COO) range from 96.6° to 136.7°, and average 117°.26 In effect, 12 has its cyclohexyl substituents rotated by 60° compared to a typical diphosphine such as dcpe.
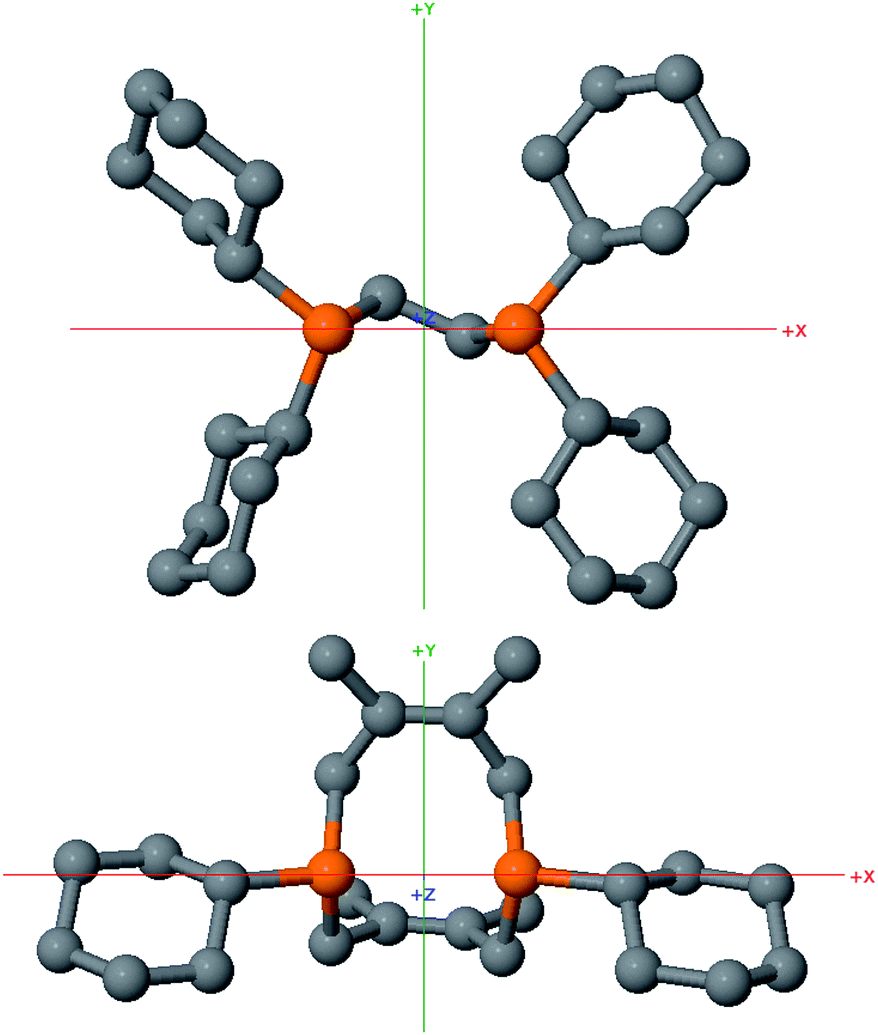 | ||
| Fig. 8 Ball and stick models of dcpe (top) and Cy2-DPC (bottom) with hydrogen atoms omitted for clarity, highlighting the differences in steric profiles between these two diphosphines. Both ligands are viewed along the Z axis, defined using SambVca 2.0 as the P–Ni–P angle bisector in their respective nickelalactone complexes. | ||
In order to further probe the comparison between dcpe and 12, their % buried volumes (%Vbur, defined as the percent volume occupied by the ligand out of the total volume of a sphere centered at the metal and typically set to have a 3.5 Å radius)30 were calculated31 using the SambVca 2.0 web application developed by Cavallo and coworkers.32 Even though dcpe has four cyclohexyl substituents and 12 has only two, these ligands have almost identical % buried volumes: 54.5% and 54.8%, respectively. This result can be in part attributed to the unusual placement of the cyclohexyl groups in 12 with respect to the metal center, but also to the significant bulk added by the protruding methyl groups of the tetramethyltetrahydroDiPhospheCine (DPC) backbone.
Having determined that nickelalactones are accessible with this new ligand platform, we proceeded in measuring the catalytic activity of diphosphines 5–10 and 12. In doing so, we used two different protocols: one adapted from the work of Vogt et al. (method A),25 and one adapted from Limbach et al. (method B).27 In a typical catalytic run, Ni(COD)2 was mixed with the ligand, along with a base, Lewis acid, and zinc as an additive. For runs using method A, the base was NEt3 and the Lewis acid was LiI, whereas for runs using method B, sodium 2-fluorophenoxide served as both the base and source of Lewis acid. As shown in Table 1, all of the new ligands but the bulky Bn,Mes-DPC (10) showed catalytic activity in this transformation. Dicyclohexylphosphinoethane (dcpe), dicyclohexylphosphinopropane (dcpp) and dicyclohexylphosphinobutane (dcpb) were used as benchmarks, as they are some of the best performing ligands reported in the literature so far.25,27 We found the turnover numbers of our most active ligands such as 12 to be comparable to or better than those of the benchmark ligands. Notably, 12 performed significantly better than dcpb, a ligand that has a similar 4-carbon atom backbone.
|
|
||
|---|---|---|
| Ligand | TONa | TONb |
| a Ni(COD)2 (0.05 mmol), ligand (0.05 mmol), Zn (2.5 mmol), LiI (1.25 mmol), NEt3 (2.5 mmol), PhCl (2 mL), pressurized with C2H4 (25 bar) and CO2 (5 bar) and heated to 50 °C for 24 h. The TONs listed are averages of two independent runs. b Ni(COD)2 (0.07 mmol), ligand (0.077 mmol), Zn (3.5 mmol), sodium 2-fluorophenoxide (3.5 mmol), THF (10 mL), pressurized with C2H4 (10 bar) and CO2 (10 bar) and heated to 100 °C for 20 h. c Complex 13 (0.07 mmol) was used as the starting nickel source in lieu of the typical mixture of Ni(COD)2 and ligand. | ||
| Me2-DPC (5) | 1 | 6 |
| Me,Cy-DPC (6) | 4 | 15 |
| i Bu,Cy-DPC (7) | 10 | 11 |
| Bn,Cy-DPC (8) | 9 | 17 |
| Bn,Ph-DPC (9) | 12 | 4 |
| Bn,Mes-DPC (10) | 0 | 0 |
| Cy2-DPC (12) | 12 | 19 (16)c |
| Cy2P–(CH2)2–PCy2 (dcpe) | 8 | 12 |
| Cy2P–(CH2)3–PCy2 (dcpp) | 18 | 7 |
| Cy2P–(CH2)4–PCy2 (dcpb) | 6 | 4 |
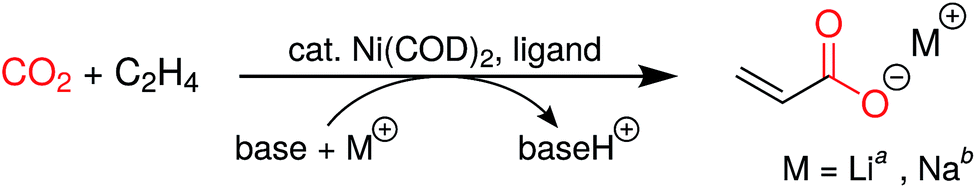
Employing this new family of cis-macrocyclic diphosphines in CO2/ethylene coupling is only the first step in exploring the catalytic relevance of these compounds. We plan to expand this ligand family using the synthetic methods disclosed herein, which have laid the groundwork for accessing phosphorus-based ligands with unique structures and steric profiles in a modular fashion. Asymmetric ligands will be easily accessible by installing chiral groups on the phosphorus centers, thus enabling numerous applications in the realm of asymmetric catalysis. Given the unusual steric profiles of the cis-macrocyclic diphosphines reported herein, our group is also actively pursuing the synthesis and characterization of a variety of metal complexes supported by these ligands in order to learn more about their coordination chemistry.
Acknowledgements
SABIC (Saudi Basic Industries Corporation) is acknowledged for funding this work. DT was funded by the National Science Foundation under CHE-1111357, renewed as CHE-1362118. Dr Bruce Adams is acknowledged for help with 2D NMR data acquisition. Prof. Dr Hansjörg Grützmacher is acknowledged for helpful discussions. X-ray diffraction data were collected on an instrument purchased with the aid of the National Science Foundation (NSF) under CHE-0946721.References
- J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science books, 1st edn, 2010 Search PubMed.
- (a) T. H. Chan and B. S. Ong, J. Org. Chem., 1974, 39, 1748–1752 CrossRef CAS; (b) S. N. Ignat'eva, A. S. Balueva, A. A. Karasik, D. V. Kulikov, A. V. Kozlov, S. K. Latypov, P. Lönnecke, E. Hey-Hawkins and O. G. Sinyashin, Russ. Chem. Bull., 2007, 56, 1828–1837 CrossRef; (c) L. Horner, P. Walach and H. Kunz, Phosphorus, Sulfur Silicon Relat. Elem., 1978, 5, 171–184 CrossRef CAS.
- (a) E. B. Bauer, J. Ruwwe, J. M. Martin-Alvarez, T. B. Peters, J. C. Bohling, F. A. Hampel, S. Szafert, T. Lis and J. A. Gladysz, Chem. Commun., 2000, 2261–2262 RSC; (b) T. Shima, E. B. Bauer, F. Hampel and J. A. Gladysz, Dalton Trans., 2004, 1012–1028 RSC.
- R. W. Alder, C. Ganter, C. J. Harris and A. G. Orpen, J. Chem. Soc., Chem. Commun., 1992, 1170–1172 RSC.
- (a) R. W. Alder, D. D. Ellis, J. K. Hogg, A. Martín, A. G. Orpen and P. N. Taylor, Chem. Commun., 1996, 537–538 RSC; (b) R. W. Alder, C. Ganter, M. Gil, R. Gleiter, C. J. Harris, S. E. Harris, H. Lange, A. G. Orpen and P. N. Taylor, J. Chem. Soc., Perkin Trans. 1, 1998, 1643–1656 RSC.
- (a) D. Gudat, Sci. Synth., 2009, 42, 155–220 Search PubMed; (b) A.-M. Caminade and J. P. Majoral, Chem. Rev., 1994, 94, 1183–1213 CrossRef CAS; (c) C. D. Swor and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 2860–2881 CrossRef CAS.
- R. W. Alder and D. Read, Coord. Chem. Rev., 1998, 176, 113–133 CrossRef CAS.
- The 1998 review article cited above mentions unpublished observations vis-a-vis the ligand behaviour of the cis-macrocyclic diphosphines synthesized by Alder et al..
- (a) T. Shima, F. Hampel and J. A. Gladysz, Angew. Chem., Int. Ed., 2004, 43, 5537–5540 CrossRef CAS PubMed; (b) A. J. Nawara, T. Shima, F. Hampel and J. A. Gladysz, J. Am. Chem. Soc., 2006, 128, 4962–4963 CrossRef CAS PubMed; (c) L. Wang, T. Shima, F. Hampel and J. A. Gladysz, Chem. Commun., 2006, 4075–4077 RSC; (d) G. D. Hess, F. Hampel and J. A. Gladysz, Organometallics, 2007, 26, 5129–5131 CrossRef CAS; (e) K. Skopek and J. A. Gladysz, J. Organomet. Chem., 2008, 693, 857–866 CrossRef CAS; (f) A. J. Nawara-Hultzsch, M. Stollenz, M. Barbasiewicz, S. Szafert, T. Lis, F. Hampel, N. Bhuvanesh and J. A. Gladysz, Chem.–Eur. J., 2014, 20, 4617–4637 CrossRef CAS PubMed.
- B. W. Arbuckle and W. Musker, Polyhedron, 1991, 10, 415–419 CrossRef CAS.
- D. Tofan and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 7516–7518 CrossRef CAS PubMed.
- M. Serrano-Ruiz, A. Romerosa and P. Lorenzo-Luis, Eur. J. Inorg. Chem., 2014, 1587–1598 CrossRef CAS.
- (a) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed; (b) B. M. Cossairt and C. C. Cummins, New J. Chem., 2010, 34, 1533–1536 RSC.
- (a) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed; (b) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed; (c) S. Heinl, S. Reisinger, C. Schwarzmaier, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 7639–7642 CrossRef CAS PubMed.
- C. C. Cummins, Daedalus, 2014, 143, 9–20 CrossRef.
- (a) D. Tofan, M. Temprado, S. Majumdar, C. D. Hoff and C. C. Cummins, Inorg. Chem., 2013, 52, 8851–8864 CrossRef CAS PubMed; (b) D. Tofan, Ph.D. thesis, Massachusetts Institute of Technology, 2013.
- Secondary halides such as cyclohexyl bromide did not react with 1 under similar conditions.
- (a) N. Bricklebank, S. M. Godfrey, H. P. Lane, C. A. McAuliffe, R. G. Pritchard and J.-M. Moreno, J. Chem. Soc., Dalton Trans., 1995, 2421–2424 RSC; (b) S. M. Godfrey, C. A. McAuliffe, R. G. Pritchard and J. M. Sheffield, J. Chem. Soc., Dalton Trans., 1998, 1919–1924 RSC.
- (a) S. M. Godfrey, D. G. Kelly, C. A. Mcauliffe, A. G. Mackie, R. G. Pritchard and S. M. Watson, J. Chem. Soc., Chem. Commun., 1991, 1163–1164 RSC; (b) F. Teixidor, R. Núñez, C. Viñas, R. Sillanpää and R. Kivekäs, Angew. Chem., Int. Ed., 2000, 39, 4290–4292 CrossRef CAS; (c) B. D. Ellis and C. L. B. Macdonald, Inorg. Chem., 2006, 45, 6864–6874 CrossRef CAS PubMed; (d) N. A. Barnes, S. M. Godfrey, R. T. A. Halton, I. Mushtaq and R. G. Pritchard, Dalton Trans., 2006, 4795–4804 RSC; (e) N. A. Barnes, K. R. Flower, S. A. Fyyaz, S. M. Godfrey, A. T. McGown, P. J. Miles, R. G. Pritchard and J. E. Warren, CrystEngComm, 2010, 12, 784–794 RSC; (f) N. A. Barnes, K. R. Flower, S. M. Godfrey, P. A. Hurst, R. Z. Khan and R. G. Pritchard, CrystEngComm, 2010, 12, 4240–4251 RSC; (g) N. A. Barnes, S. M. Godfrey, R. Z. Khan, A. Pierce and R. G. Pritchard, Polyhedron, 2012, 35, 31–46 CrossRef CAS.
- (a) C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935–967 CrossRef CAS; (b) H. Bircher, B. R. Bender and W. Vonphilipsborn, Magn. Reson. Chem., 1993, 31, 293–298 CrossRef CAS; (c) E. R. Civitello, P. S. Dragovich, T. B. Karpishin, S. G. Novick, G. Bierach, J. F. O'Connell and T. D. Westmoreland, Inorg. Chem., 1993, 32, 237–241 CrossRef CAS; (d) A. Gogoll, J. Örnebro, H. Grennberg and J.-E. Bäckvall, J. Am. Chem. Soc., 1994, 116, 3631–3632 CrossRef CAS.
- (a) J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 2169–2171 CrossRef CAS PubMed; (b) J. S. Silvia and C. C. Cummins, Chem. Sci., 2011, 2, 1474–1479 RSC; (c) I. Knopf, T. Ono, M. Temprado, D. Tofan and C. C. Cummins, Chem. Sci., 2014, 5, 1772–1776 RSC; (d) I. Knopf and C. C. Cummins, Organometallics, 2015, 34, 1601–1603 CrossRef CAS.
- (a) H. Hoberg and D. Schaefer, J. Organomet. Chem., 1983, 251, C51–C53 CrossRef CAS; (b) H. Hoberg, Y. Peres and A. Milchereit, J. Organomet. Chem., 1986, 307, C41–C43 CrossRef CAS; (c) H. Hoberg, Y. Peres, C. Kruger and Y.-H. Tsay, Angew. Chem., 1987, 99, 799–800 CrossRef CAS.
- M. L. Lejkowski, R. Lindner, T. Kageyama, G. É. Bódizs, P. N. Plessow, I. B. Müller, A. Schäfer, F. Rominger, P. Hofmann, C. Futter, S. A. Schunk and M. Limbach, Chem.–Eur. J., 2012, 18, 14017–14025 CrossRef CAS PubMed.
- S. Kraus and B. Rieger, Top. Organomet. Chem., 2016, 53, 199–224 CrossRef.
- C. Hendriksen, E. A. Pidko, G. Yang, B. Schäffner and D. Vogt, Chem.–Eur. J., 2014, 20, 12037–12040 CrossRef CAS PubMed.
- D. Jin, P. G. Williard, N. Hazari and W. H. Bernskoetter, Chem.–Eur. J., 2014, 20, 3205–3211 CrossRef CAS PubMed.
- N. Huguet, I. Jevtovikj, A. Gordillo, M. L. Lejkowski, R. Lindner, M. Bru, A. Y. Khalimon, F. Rominger, S. A. Schunk, P. Hofmann and M. Limbach, Chem.–Eur. J., 2014, 20, 16858–16862 CrossRef CAS PubMed.
- S. Manzini, N. Huguet, O. Trapp and T. Schaub, Eur. J. Org. Chem., 2015, 7122–7130 CrossRef CAS.
- R. Fischer, J. Langer, A. Malassa, D. Walther, H. Görls and G. Vaughan, Chem. Commun., 2006, 2510–2512 RSC.
- (a) A. Poater, F. Ragone, S. Giudice, C. Costabile, R. Dorta, S. P. Nolan and L. Cavallo, Organometallics, 2008, 27, 2679–2681 CrossRef CAS; (b) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC.
- The %Vbur for both dcpe and 12 was calculated using the default settings in SambVca 2.0, namely bond radii scaled by 1.17, sphere radius of 3.5 Å, Ni as the center of the sphere, mesh spacing for numerical integration of 0.1 Å, and H atoms not included.
- (a) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS; (b) https://www.molnac.unisa.it/OMtools/sambvca2.0/ .
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental, crystallographic and spectroscopic data. CCDC 1477899–1477902. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc03614g |
| This journal is © The Royal Society of Chemistry 2017 |