
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A methodology to synthesize easily oxidized materials containing Li ions in an inert atmosphere†
Itsuki
Konuma
a,
Yosuke
Ugata
 ab and
Naoaki
Yabuuchi
*ab
ab and
Naoaki
Yabuuchi
*ab
aDepartment of Chemistry and Life Science, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan. E-mail: yabuuchi-naoaki-pw@ynu.ac.jp
bAdvanced Chemical Energy Research Center (ACERC), Institute of Advanced Sciences, Yokohama National University, 79-5 Tokiwadai, Hodogaya-ku, Yokohama 240-8501, Japan
First published on 15th April 2024
Abstract
A series of lithium-containing oxides with V3+ and Mn3+ ions are proposed as emerging high-capacity positive electrode materials for lithium-ion batteries. These oxides are typically synthesized by a calcination process in an inert atmosphere. For the successful synthesis of these materials, the strict control of oxygen partial pressure is necessary to prevent partial oxidation of materials during calcination. In this article, a simple methodology used in laboratory-scale material production, i.e., an oxygen trap by Cu foil, is described to synthesize phase pure oxides, which are easily oxidized on calcination. To demonstrate the benefit of the methodology, Li8/7Ti2/7V4/7O2 and Li8/7Ti2/7Mn4/7O2 are selected as lithium-containing oxides with V3+ and Mn3+ ions, respectively. Single phase oxides are successfully obtained from the pellets calcined with Cu foil, whereas partially oxidized phases are obtained for the pellets calcined without Cu foil. Moreover, phase-pure oxides synthesized with Cu foil show much better performance as positive electrode materials for battery applications. This methodology is also expected to be applied for material synthesis for diverse applications.
Lithium-ion batteries (LIBs), which possess the highest energy density among the commercialized rechargeable batteries, are regarded as a key technology to realize a fossil-fuel-free society and the effective use of renewable energy resources. To further improve the battery performance of LIBs, numerous efforts have been devoted to developing high performance positive electrode materials in the past three decades. Lithium cobalt oxide (LiCoO2) with a layered-type structure has been used as a positive electrode material since its use for practical applications in 1991. However, the practically available reversible capacity is limited to only 150 mA h g−1.1 In the state-of-the-art LIBs, LiNixCo1−x−yMnyO22,3 and LiNixCo1−x−yAlyO2,4 where the Co ions in LiCoO2 are partially substituted with other metal ions, are widely used owing to their large reversible capacities of 170–200 mA h g−1. Olivine-type LiFePO4 with earth abundant Fe ions is also adopted as a cost-effective positive electrode material in LIBs for electric vehicles,5,6 even though its energy density is relatively small compared with the above mentioned layered materials.7 Recently, many Li-excess metal oxides, Li1+xMe1−xO2 (Me = transition metal ions), have been developed and proposed as high-capacity positive electrode materials for LIBs. These Li-excess oxides are designed using many different transition metal ions. Among them, Li1.33Mn0.67O2 (Li2MnO3) and its derivatives have been the most intensively studied as electrode materials.8–11 Li-excess electrode materials with tetravalent Mn ions, like Li1.33Mn0.67O2, require the activation of anionic redox to obtain high-reversible capacity, but the reversibility of anionic redox is not high enough and is not suitable for practical applications. The use of anionic redox for electrode materials has also been extended to Na insertion materials.12 Another strategy for Li-excess oxides is the material design with multi-electron cationic redox, e.g., V3+/V5+, Cr3+/Cr6+, Mo3+/Mo6+ and Mn3+/Mn4+ with oxygen redox (O2−/On−).13–17 For instance, nanosized Li8/7V4/7Ti2/7O2, which is a solid solution oxide between LiVO2 and Li1.33Ti0.67O2 (Li2TiO3), has been recently proposed as a potential positive electrode material.18 Nanosized Li8/7V4/7Ti2/7O2 delivers a reversible capacity of 300 mA h g−1 based on V3+/V5+ two-electron redox with excellent reversibility and no capacity fading.
These electrode materials, oxides, phosphates, etc. are synthesized by a calcination method, which is easily scalable and thus suitable for mass production. The electrode materials with Co and Ni ions, LiCoO2, LiNixCo1−x−yMnyO2, etc., are synthesized by the calcination of precursors in air at 800–900 °C. When the Ni3+ contents are enriched in layered oxides, the oxides are calcined at slightly lower temperatures at higher oxygen partial pressures.19 The oxides containing transition metal ions, which are easily oxidized during calcination, i.e. (Fe2+, V3+, Cr3+, Mo3+, and Mn3+), can be synthesized in an inert atmosphere (N2). Ar gas is also used for the battery material synthesis in the laboratory because Ar gas is used for a glove box to avoid the reaction of metallic Li and N2 gas. However, they often suffer from the partial oxidation of materials due to a trace amount of oxygen contamination, leading to the partial oxidation of the samples and/or the formation of undesirable impurity phases. Oxygen contamination would originate from the lower purity of inert gas and/or the leakage of furnaces. LiFePO4 with Fe2+ ions is synthesized with carbon sources to increase electronic conductivity.20,21 The leakage problem is less impactful because carbons capture a trace amount of contaminated oxygen. Another methodology is the use of N2/Ar gas with a small amount of H2 gas,22,23 which serves as a reducing agent during calcination. However, these methodologies, with the addition of reducing agents, cannot be applied for the synthesis of the oxides with Mn3+ ions, e.g., Li8/7Mn4/7Ti2/7O2, which is a solid solution oxide between LiMnO2 and Li1.33Ti0.67O2,24 because Mn3+ ions are easily reduced to Mn2+ ions when carbons and/or hydrogen molecules are present. Mn3+ ions are easily oxidized to Mn4+ ions at high oxygen partial pressure and easily reduced to Mn2+ ions in the presence of reducing agents.
In this article, a simple methodology, which makes possible the strict control of oxygen partial pressure during heating, is described. This methodology, i.e., an oxygen trap by Cu foil, is a simple and suitable approach for the material synthesis in the laboratory. To prove the effectiveness of this approach, Li8/7Ti2/7V4/7O218 with V3+ ions and Li8/7Ti2/7Mn4/7O224 with Mn3+ ions have been selected, and both samples have been synthesized with or without Cu foil. Single phase Li8/7Ti2/7V4/7O2 and Li8/7Ti2/7Mn4/7O2 are successfully obtained by calcinating the precursor pellets with Cu foil because of the strict control of the oxygen partial pressure. In contrast, the samples synthesized without Cu foil show partial oxidation and/or phase segregation. Such phase purity clearly influences the electrochemical properties of these electrode materials. This approach is also beneficial for the synthesis of many different materials, which require the strict control of the oxygen partial pressure during calcination.
Li8/7Ti2/7V4/7O2 and Li8/7Ti2/7Mn4/7O2 were synthesized by calcination from mixtures of stoichiometric amounts of Li2CO3 (98.5%, Kanto Kagaku), anatase-type TiO2 (98.5%, Wako Pure Chemical Industries) and V2O3 (98%, Sigma Aldrich) or Mn2O3. Mn2O3 was prepared by heating MnCO3 (Kishida Chemical) at 850 °C for 12 h in air. These precursors were mixed by using an alumina mortar and pestle, and then ball-milled using a planetary ball mill (Pulverisette7, Fritsch) in ethanol with a zirconia pot (45 mL) and zirconia balls (15.5 g) at 300 rpm for 5 h. After drying, the mixtures of the precursors were pressed into pellets at 20 MPa. Fig. 1a summarizes a detailed protocol for the strict control of the oxygen partial pressure with Cu foil. The pellet of the precursors is placed at the center of the ceramic boat (Fig. 1a-i), and then the ceramic boat and pellet are fully covered by Cu foil, as shown in Fig. 1a-i–v. The pellet of Li8/7Ti2/7V4/7O2 covered with Cu foil was calcined at 900 °C for 12 h in Ar gas using a quartz tube furnace with Ar flow at a rate of 50 mL min−1. An additional ceramic boat is placed on the bottom of the ceramic boat covered with Cu foil to avoid the contact of the Cu foil with the quartz tube during calcination. After heating, all samples were stored in an Ar-filled glove box to prevent contact with oxygen and moisture.
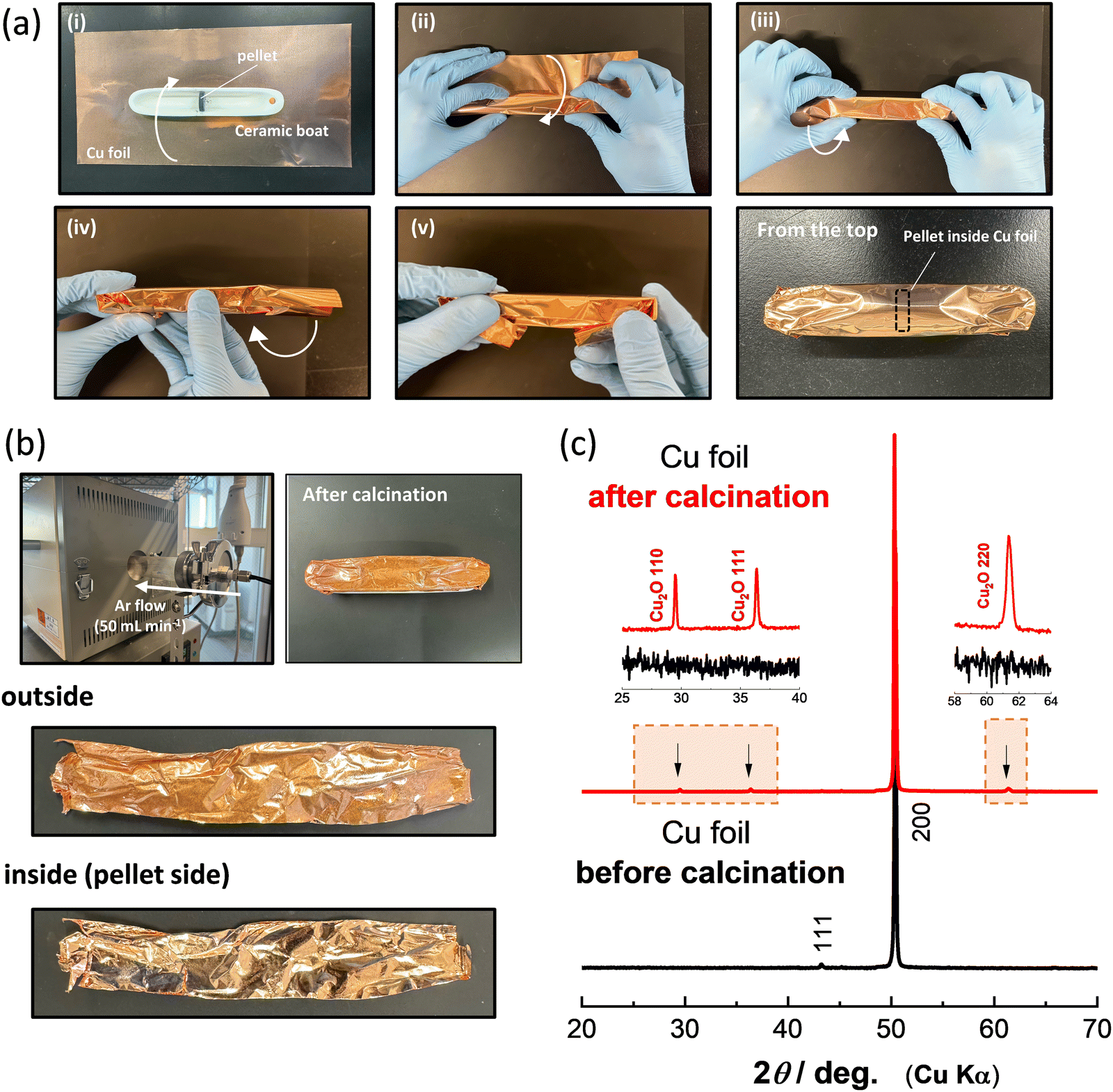 | ||
| Fig. 1 (a) A protocol of the material synthesis with the strict control of oxygen partial pressure with Cu foil. (b) Changes in the surface of Cu foil after calcination in Ar flow. The inside and outside of Cu foil after calcination are also compared. Although the metallic luster is lost for the outside of Cu foil after heating, the luster is retained for the inside of the foil. (c) XRD patterns of Cu foil before and after calcination. Cu2O was detected only for the outside of the Cu foil after calcination as expected from the loss of metallic luster and color change. | ||
The color change of the Cu foil surface after calcination is shown in Fig. 1b. The luster of the Cu metal is lost after calcination, and a color change to reddish brown is observed. Note that the color change is observed only for the outside of the Cu foil, which was exposed to Ar flow, and the luster is not lost in the inside of the Cu foil. This result demonstrates that contaminated oxygen molecules in Ar gas are effectively captured by Cu foil, and a quite low oxygen partial pressure is achieved inside the Cu foil during calcination. To further characterize the Cu foil before and after calcination, X-ray diffraction (XRD) measurement was conducted, and the XRD patterns of the Cu foil before and after calcination are shown in Fig. 1c. The XRD patterns were collected using an X-ray diffractometer (D2 PHASER; Bruker Corp., Ltd) equipped with a one-dimensional X-ray detector using Cu Kα radiation generated at 300 W (30 kV and 10 mA) with a Ni filter. The XRD data indicate that the major phase remains metallic Cu, but the presence of Cu2O25 is evidenced for the outside of the Cu foil after calcination as expected from the color change shown in Fig. 1b. During synthesis, CO2 gas is generated by the decomposition of Li2CO3 inside the Cu foil. However, the metallic luster is retained in the inside of the Cu foil, indicating that CO2 is non-reactive with Cu metal and released outside of the Cu foil. From these results, it is concluded that the use of Co foil is a beneficial approach to control oxygen partial pressure, and the oxidation of samples inside Cu foil is effectively suppressed.
To further demonstrate the effectiveness of Cu foil in material synthesis, the XRD patterns of Li8/7Ti2/7V4/7O2 and Li8/7Ti2/7Mn4/7O2 synthesized with or without Cu foil are compared in Fig. 2. Note that an airtight sample holder was used for the XRD measurement to avoid reactions with oxygen and moisture. For Li8/7Ti2/7V4/7O2 synthesized with Cu foil, the diffraction lines are indexed to an α-NaFeO2-type layered structure with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m. The peak intensity of the 003 diffraction line at 18.4 degrees is weaker than that of the 104 line at 43.9 degrees. This observation is consistent with the literature,18 and originates from approximately 10% cation mixing (anti-site defects) between lithium and transition metal ions. In contrast, for the sample synthesized without Cu foil, a clear peak shift is observed for the 003 diffraction lines. In contrast, the peak intensity of the 104 diffraction line remains unchanged. A similar trend for the diffraction profile changes is observed for the as-prepared and delithiated Li8/7−xTi2/7V4/7O2 as shown in Fig. S1 (ESI†). Moreover, no clear phase segregation is also observed in scanning electron microscopy coupled with energy dispersive X-ray spectroscopy (SEM/EDX) images (Fig. S2, ESI†). From these results, it is proposed that Li8/7Ti2/7V4/7O2 is successfully synthesized without Cu foil, but partial oxidation of the sample by contaminated oxygen is unavoidable. Indeed, the increase in the open circuit voltage associated with partial oxidation is directly evidenced as shown in the later section.
m. The peak intensity of the 003 diffraction line at 18.4 degrees is weaker than that of the 104 line at 43.9 degrees. This observation is consistent with the literature,18 and originates from approximately 10% cation mixing (anti-site defects) between lithium and transition metal ions. In contrast, for the sample synthesized without Cu foil, a clear peak shift is observed for the 003 diffraction lines. In contrast, the peak intensity of the 104 diffraction line remains unchanged. A similar trend for the diffraction profile changes is observed for the as-prepared and delithiated Li8/7−xTi2/7V4/7O2 as shown in Fig. S1 (ESI†). Moreover, no clear phase segregation is also observed in scanning electron microscopy coupled with energy dispersive X-ray spectroscopy (SEM/EDX) images (Fig. S2, ESI†). From these results, it is proposed that Li8/7Ti2/7V4/7O2 is successfully synthesized without Cu foil, but partial oxidation of the sample by contaminated oxygen is unavoidable. Indeed, the increase in the open circuit voltage associated with partial oxidation is directly evidenced as shown in the later section.
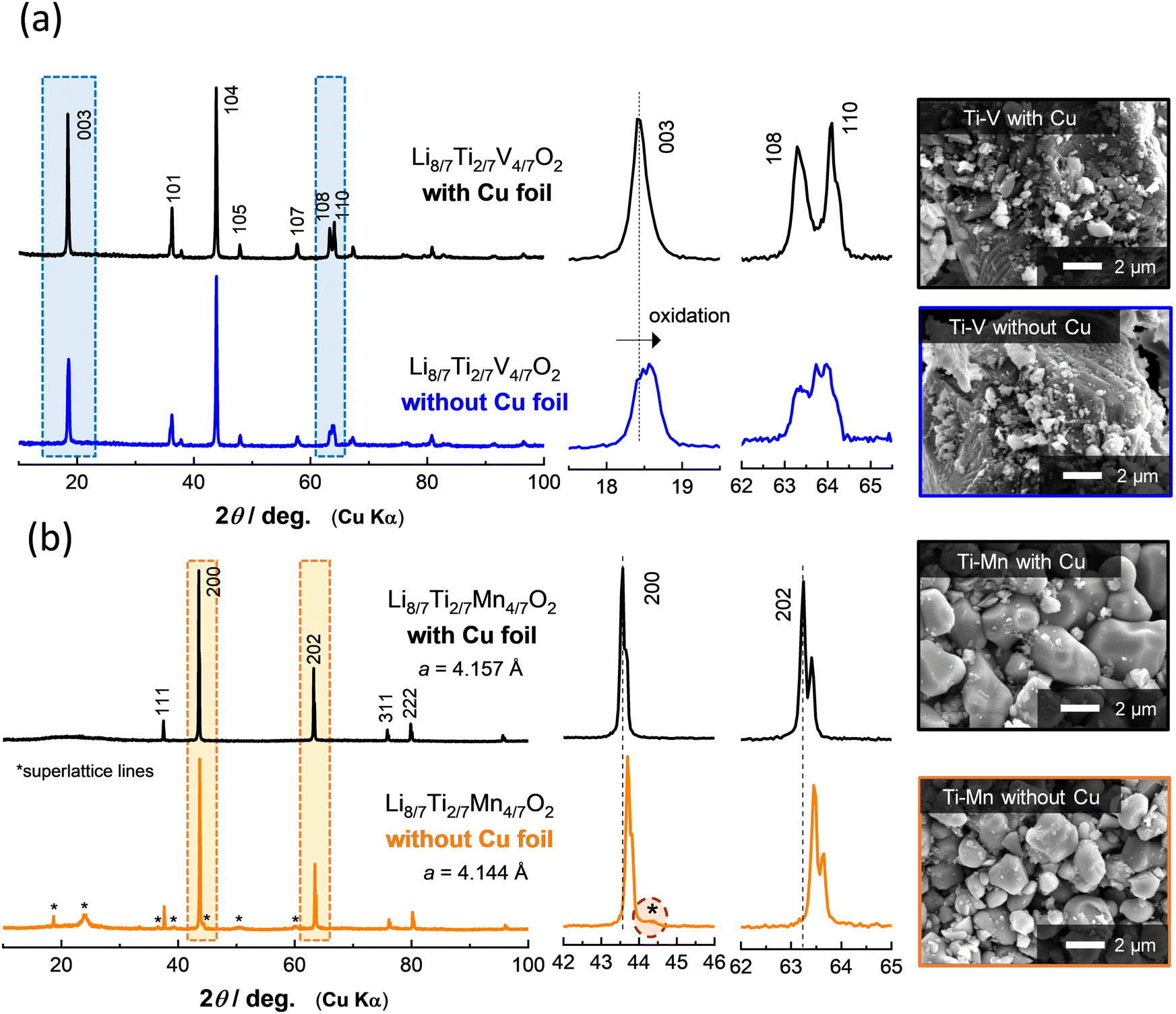 | ||
| Fig. 2 XRD patterns and SEM images of (a) Li8/7Ti2/7V4/7O2 and (b) Li8/7Ti2/7Mn4/7O2 synthesized with or without Cu foil. | ||
In the case of Li8/7Ti2/7Mn4/7O2 with Mn3+ ions, a major phase for both samples synthesized with or without Cu foil is assigned to a cation-disordered rocksalt-type structure with a space group of Fmm.24 Nevertheless, the lattice parameters of the major phase for both samples are slightly different as clearly observed by the peak shift of the 200 and 202 diffraction lines (Fig. 2b). In addition, some of the additional peaks, which cannot be assigned as the cation-disordered rocksalt phase, are observed, and these peaks can be assigned as superlattice lines based on the cubic rocksalt phase (Fig. S3–S5, ESI†), which has been derived from the ordered cation arrangement observed in LiMg0.5Mn1.5O4.26 In addition, clear evidence of phase segregation cannot be detected by SEM/EDX (Fig. S6, ESI†). These facts suggest that Mn ions are partially oxidized on heating and cation vacancies are formed, i.e., Li8/7−δ/3Ti2/7−δ/3Mn4/7−δ/3□δO2, and □ denotes vacant octahedral sites. These vacancies (and probably Li ions) have an ordered arrangement in the structure, leading to the appearance of superlattice lines.
Although no significant difference is found in the particle morphology for Li8/7Ti2/7V4/7O2 regardless of the use of Cu foil from SEM observation (Fig. 2a), the particle size growth is partly suppressed for Li8/7Ti2/7Mn4/7O2 without Cu foil compared to the sample with Cu foil (Fig. 2b). This fact suggests that oxygen contamination also influences the crystal growth process of the samples during calcination. Note that the oxygen trap by Cu foil is applicable below the melting point of Cu metal, i.e., 1085 °C. At higher temperatures, >1085 °C, high purity Fe foil would be used for the purpose of oxygen trapping.
To determine the electrochemical properties of Li8/7Ti2/7V4/7O2 and Li8/7Ti2/7Mn4/7O2 synthesized with or without Cu foil, galvanostatic charge/discharge tests were performed, and the corresponding charge/discharge curves are shown in Fig. 3a and b. The synthesized powders were mixed with acetylene black (AB, HS-100, Denka) (active material![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AB = 90:10 wt%) by using ball milling in Ar at 300 rpm for 18 h to reduce the particle size and improve the electronic conductivity. The composite electrode consisting of a ball milled sample with AB, additional AB, and poly(vinylidene fluoride) (PVdF) was pasted on an Al foil current collector. The composition for the composite electrode is active material:AB:PVdF = 76.5:13.5:10 wt%. The composite electrodes were dried at 120 °C for 12 h under vacuum. Metallic lithium (Honjo Metal) was used as a negative electrode. 1.0 M LiPF6 dissolved in ethylene carbonate and dimethyl carbonate (30:70 vol%) (battery grade, Kishida Chemical) and a polyolefin microporous membrane were used as an electrolyte solution and separator, respectively. Two-electrode cells (TJ-AC, Tomcell Japan) were assembled in an Ar-filled glovebox. The cells were cycled at a rate of 10 mA g−1 at room temperature. Galvanostatic charge/discharge tests were conducted by using a battery cycler (TOSCAT-3100, Toyo System). XRD patterns and SEM images for all samples before and after ball milling with AB are shown in Fig. S7 (ESI†). Note that the initial open circuit voltage is higher for the sample synthesized without Cu foil, suggesting the partial oxidation of V ions during calcination. Moreover, Li8/7Ti2/7V4/7O2 synthesized with Cu foil delivers a larger reversible capacity of 270 mA h g−1 and relatively higher coulombic efficiency of 96.5% at the initial cycle. In contrast, for the sample synthesized without Cu foil, the initial reversible capacity is decreased to 225 mA h g−1, and the initial Coulombic efficiency is less than 90%. In the case of Li8/7Ti2/7Mn4/7O2, a clearly reduced reversible capacity is noted for the sample synthesized without Cu foil, which probably originates from the partial oxidation of Mn ions, and the capacity based on cationic Mn3+/Mn4+ redox is decreased. Note that the reversible capacity is gradually increased for 20 continuous cycles, and this observation is expected to originate from the structural phase transition on electrochemical cycles, and a similar trend is often observed for LiMnO2 with trivalent manganese ions associated with a spinel-like phase transition.27,28
:AB = 90:10 wt%) by using ball milling in Ar at 300 rpm for 18 h to reduce the particle size and improve the electronic conductivity. The composite electrode consisting of a ball milled sample with AB, additional AB, and poly(vinylidene fluoride) (PVdF) was pasted on an Al foil current collector. The composition for the composite electrode is active material:AB:PVdF = 76.5:13.5:10 wt%. The composite electrodes were dried at 120 °C for 12 h under vacuum. Metallic lithium (Honjo Metal) was used as a negative electrode. 1.0 M LiPF6 dissolved in ethylene carbonate and dimethyl carbonate (30:70 vol%) (battery grade, Kishida Chemical) and a polyolefin microporous membrane were used as an electrolyte solution and separator, respectively. Two-electrode cells (TJ-AC, Tomcell Japan) were assembled in an Ar-filled glovebox. The cells were cycled at a rate of 10 mA g−1 at room temperature. Galvanostatic charge/discharge tests were conducted by using a battery cycler (TOSCAT-3100, Toyo System). XRD patterns and SEM images for all samples before and after ball milling with AB are shown in Fig. S7 (ESI†). Note that the initial open circuit voltage is higher for the sample synthesized without Cu foil, suggesting the partial oxidation of V ions during calcination. Moreover, Li8/7Ti2/7V4/7O2 synthesized with Cu foil delivers a larger reversible capacity of 270 mA h g−1 and relatively higher coulombic efficiency of 96.5% at the initial cycle. In contrast, for the sample synthesized without Cu foil, the initial reversible capacity is decreased to 225 mA h g−1, and the initial Coulombic efficiency is less than 90%. In the case of Li8/7Ti2/7Mn4/7O2, a clearly reduced reversible capacity is noted for the sample synthesized without Cu foil, which probably originates from the partial oxidation of Mn ions, and the capacity based on cationic Mn3+/Mn4+ redox is decreased. Note that the reversible capacity is gradually increased for 20 continuous cycles, and this observation is expected to originate from the structural phase transition on electrochemical cycles, and a similar trend is often observed for LiMnO2 with trivalent manganese ions associated with a spinel-like phase transition.27,28
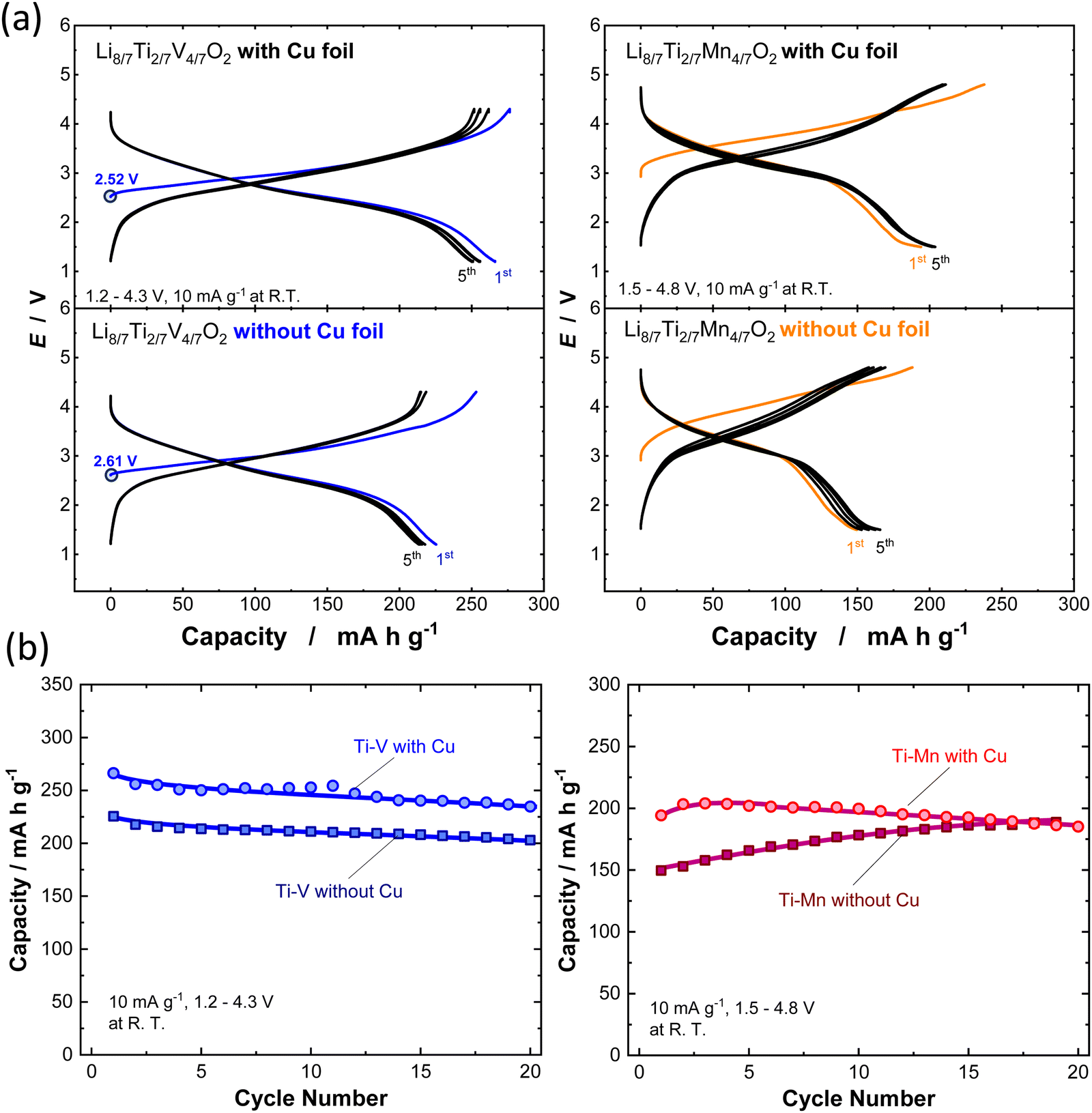 | ||
| Fig. 3 (a) Galvanostatic charge/discharge curves and (b) discharge capacity retention for 20 cycles of Li8/7−xTi2/7V4/7O2 and Li8/7−xTi2/7Mn4/7O2 synthesized with or without Cu foil at a rate of 10 mA g−1. | ||
In this study, the effectiveness of the oxygen trap by Cu foil for the synthesis of materials, which require the strict control of oxygen partial pressure, is described. Phase pure samples for Li8/7Ti2/7V4/7O2 and Li8/7Ti2/7Mn4/7O2 are successfully synthesized with high reproducibility without the addition of carbons and/or hydrogen gas. Moreover, much better electrode performance is obtained for the samples synthesized with Cu foil. This methodology is beneficial to synthesize electrode materials in an inert atmosphere in the laboratory. In addition, strict control of the oxygen partial pressure is necessary for the synthesis of some sodium29 and potassium30 insertion materials, and this process is also expected to be applied for diverse fields of inorganic material synthesis, including electronic materials, magnetic materials, catalysts, etc.
Conflicts of interest
All authors report no conflict of interest in any part of this study.Acknowledgements
NY acknowledges the partial support from JSPS, Grant-in-Aid for Scientific Research (Grant Numbers 19H05816 and 23K17954). YU also thanks the Grant-in-Aid for Scientific Research (Grant Number 23K13822) from JSPS. This work was partially supported by JST, CREST Grant Number JPMJCR21O6, Japan. This work was in part supported by MEXT Program: Data Creation and Utilization Type Materials Research and Development Project (Grant Number JPMXP1121467561). NY acknowledges the partial support by JST as part of Adopting Sustainable Partnerships for Innovative Research Ecosystem (ASPIRE), Grant Number JPMJAP2313.References
- K. Mizushima, P. C. Jones, P. J. Wiseman and J. B. Goodenogh, Mater. Res. Bull., 1980, 15, 783–789 CrossRef CAS.
- N. Yabuuchi and T. Ohzuku, J. Power Sources, 2003, 119, 171–174 CrossRef.
- H.-J. Noh, S. Youn, C. S. Yoon and Y.-K. Sun, J. Power Sources, 2013, 233, 121–130 CrossRef CAS.
- Y. Makimura, T. Sasaki, T. Nonaka, Y. F. Nishimura, T. Uyama, C. Okuda, Y. Itou and Y. Takeuchi, J. Mater. Chem. A, 2016, 4, 8350–8358 RSC.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1184–1194 Search PubMed.
- C. Delmas, M. Maccario, L. Croguennec, F. Le Cras and F. Weill, Nat. Mater., 2008, 7, 665–671 CrossRef CAS PubMed.
- B. D. L. Campéon and N. Yabuuchi, Chem. Phys. Rev., 2021, 2, 041306 CrossRef.
- A. D. Robertson and P. G. Bruce, Chem. Commun., 2002, 2790–2791, 10.1039/B207945C.
- J. R. Croy, S. H. Kang, M. Balasubramanian and M. M. Thackeray, Electrochem. Commun., 2011, 13, 1063–1066 CrossRef CAS.
- H. Koga, L. Croguennec, P. Mannessiez, M. Ménétrier, F. Weill, L. Bourgeois, M. Duttine, E. Suard and C. Delmas, J. Phys. Chem. C, 2012, 116, 13497–13506 CrossRef CAS.
- W. Wang, H. Hanzawa, K.-I. Machida, K. Miyazaki and T. Abe, Electrochemistry, 2022, 90, 017008 CrossRef CAS.
- Z.-X. Huang, X.-L. Zhang, X.-X. Zhao, H.-Y. Lü, X.-Y. Zhang, Y.-L. Heng, H. Geng and X.-L. Wu, J. Mater. Sci. Technol., 2023, 160, 9–17 CrossRef CAS.
- M. Nakajima and N. Yabuuchi, Chem. Mater., 2017, 29, 6927–6935 CrossRef CAS.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519–522 CrossRef CAS PubMed.
- H. Lee, W. Choi, W. Lee, J. H. Shim, Y. M. Kim and W. S. Yoon, Adv. Energy Mater., 2020, 11, 2002958 CrossRef.
- Y. Kobayashi, M. Sawamura, S. Kondo, M. Harada, Y. Noda, M. Nakayama, S. Kobayakawa, W. Zhao, A. Nakao, A. Yasui, H. B. Rajendra, K. Yamanaka, T. Ohta and N. Yabuuchi, Mater. Today, 2020, 37, 43–55 CrossRef CAS.
- Y. Zhang, B. D. L. Campéon and N. Yabuuchi, Electrochemistry, 2023, 91, 037004 CrossRef CAS.
- I. Konuma, D. Goonetilleke, N. Sharma, T. Miyuki, S. Hiroi, K. Ohara, Y. Yamakawa, Y. Morino, H. B. Rajendra, T. Ishigaki and N. Yabuuchi, Nat. Mater., 2023, 22, 225–234 CrossRef CAS PubMed.
- I. Konuma, N. Ikeda, B. D. L. Campeon, H. Fujimura, J. Kikkawa, H. D. Luong, Y. Tateyama, Y. Ugata, M. Yonemura, T. Ishigaki, T. Aida and N. Yabuuchi, Energy Storage Mater., 2024, 66, 103200 CrossRef.
- H. Huang, S.-C. Yin and L. F. Nazar, Electrochem. Solid-State Lett., 2001, 4, A170–A172 CrossRef CAS.
- Y. Zou, J. Cao, H. Li, W. Wu, Y. Liang and J. Zhang, Ind. Chem. Mater., 2023, 1, 254–261 RSC.
- S. Patoux, C. Wurm, M. Morcrette, G. Rousse and C. Masquelier, J. Power Sources, 2003, 119–121, 278–284 CrossRef CAS.
- P. Fu, Y. Zhao, Y. Dong, X. An and G. Shen, J. Power Sources, 2006, 162, 651–657 CrossRef CAS.
- N. Shimada, Y. Ugata, S. Nishikawa, D. Shibata, T. Ohta and N. Yabuuchi, Energy Adv., 2023, 2, 508–512 RSC.
- Z. Xu, Z. Bi and C. Shen, Cent. Eur. J. Eng., 2012, 2, 364 CAS.
- P. Strobel, A. Ibarra-Palos, M. Anne, C. Poinsignon and A. Crisci, Solid State Sci., 2003, 5, 1009–1018 CrossRef CAS.
- Y. I. Jang, B. Huang, H. Wang, D. R. Sadoway and Y. M. Chiang, J. Electrochem. Soc., 1999, 146, 3217–3223 CrossRef CAS.
- T. Sato, K. Sato, W. Zhao, Y. Kajiya and N. Yabuuchi, J. Mater. Chem. A, 2018, 6, 13943–13951 RSC.
- T. Sato, K. Yoshikawa, W. Zhao, T. Kobayashi, H. B. Rajendra, M. Yonemura and N. Yabuuchi, Energy Mater. Adv., 2021, 9857563 CAS.
- A. K. Pandey, B. D. L. Campéon, I. Konuma and N. Yabuuchi, Energy Adv., 2023, 2, 98–102 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ya00089g |
| This journal is © The Royal Society of Chemistry 2024 |