 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpiers Memorial Lecture: How to do impactful research in artificial intelligence for chemistry and materials science
Austin H. Cheng abc,
Cher Tian Serabc,
Marta Skretabc,
Andrés Guzmán-Corderocd,
Luca Thiedebc,
Andreas Burgerbc,
Abdulrahman Aldossarya,
Shi Xuan Leongae,
Sergio Pablo-Garcíaf,
Felix Strieth-Kalthoffg and
Alán Aspuru-Guzik*abcfhij
abc,
Cher Tian Serabc,
Marta Skretabc,
Andrés Guzmán-Corderocd,
Luca Thiedebc,
Andreas Burgerbc,
Abdulrahman Aldossarya,
Shi Xuan Leongae,
Sergio Pablo-Garcíaf,
Felix Strieth-Kalthoffg and
Alán Aspuru-Guzik*abcfhij
aDepartment of Chemistry, University of Toronto, Toronto, Ontario M5S 3H6, Canada. E-mail: alan@aspuru.com
bDepartment of Computer Science, University of Toronto, Toronto, Ontario M5S 2E4, Canada
cVector Institute for Artificial Intelligence, Toronto, Ontario M5G 1M1, Canada
dTinbergen Institute, University of Amsterdam, Amsterdam, Netherlands
eSchool of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, Singapore 63737, Singapore
fAcceleration Consortium, Toronto, Ontario M5G 1X6, Canada
gSchool of Mathematics and Natural Sciences, University of Wuppertal, Wuppertal, Germany
hDepartment of Chemical Engineering and Applied Chemistry, University of Toronto, Canada
iDepartment of Materials Science and Engineering, University of Toronto, Canada
jLebovic Fellow, Canadian Institute for Advanced Research (CIFAR), Canada
First published on 13th September 2024
Abstract
Machine learning has been pervasively touching many fields of science. Chemistry and materials science are no exception. While machine learning has been making a great impact, it is still not reaching its full potential or maturity. In this perspective, we first outline current applications across a diversity of problems in chemistry. Then, we discuss how machine learning researchers view and approach problems in the field. Finally, we provide our considerations for maximizing impact when researching machine learning for chemistry.
1 Introduction
Machine learning (ML) has been applied in many facets of chemistry, and its use is rapidly growing. We argue in this perspective that despite this dramatic growth and impact, ML could be employed better and more extensively. Current work is still far from exhausting the potential of ML to advance theory and application in chemistry in terms of breadth, depth, and scale. In addition, the actual types of problems that ML could tackle, such as hypothesis generation or enabling internalized scientific understanding, are still areas of active research or open problems.To color a picture of the field, we begin by outlining a taxonomy of the chemical problems to which ML has been applied, ranging from prediction, generation, synthesis, force fields, spectroscopy, reaction optimization, and foundation models. Shifting gears, we then introduce types of problems in ML and show how chemical problems can be reformulated as instances of ML problems. These standard problems help organize the toolbox of algorithms and theory provided by ML. Digging further into this perspective, we examine differences in practices and values between the ML and chemistry communities and highlight where collaboration and cross-pollinating perspectives can advance both fields. Armed with the above, we can then discuss how to select impactful applications of ML in chemistry and recommend our suggested good practices for research in this area.
2 Chemistry meets data: a taxonomy of problems
Chemistry, and science in general, involves data in one form or another. Not surprisingly, then, data science is integral to chemistry. Machine learning, a subfield of data science, has become an integral tool in our domain science's arsenal. Therefore, it is crucial to begin cataloguing and organizing critical efforts to date.We suggest a taxonomy of the chemical problems to which machine learning has been applied. As shown in Fig. 1, ML has been applied to solve various chemical problems by encoding and decoding to and from chemical structure, properties, 3D structure and dynamics, and experimental data. For reasons of space, time, and focus, this is not a comprehensive review but rather an opportunity to highlight diverse applications of ML in chemistry. We will not introduce ML algorithms in detail. For exhaustive reviews, please see other works.1–6
 | ||
| Fig. 1 A taxonomy of chemical problems related to machine learning. Each arrow indicates an application of ML and signifies how all these relate to each other. Foundation models and self-driving labs touch all these areas. | ||
2.1 Structure to property: property prediction
While simple atomic and bond features required for the constructed graphs can be generated quickly,27 the properties that one wants to target for prediction are much harder to obtain – especially in higher qualities and fidelities. As learned representations typically require large amounts of data, complicated architectures do not function as well with low amounts of data gathered from typical experimental settings. To bridge this gap, molecular benchmarks were created to assess the quality of such learned representations properly. These benchmarks contain tasks gathered from literature data related to predicting biological behaviours and physicochemical or quantum chemical properties and provide a common ground on which different machine-learning architectures can harness and exploit the same data in various ways for property prediction.28
To improve the performance of such graph embeddings, they can be further tuned if there are some intuitions about how the embedding spaces should be reshaped to reflect the distances between inputs properly. These can involve strategies like making the embeddings aware of how chemical reactions should transform these embeddings29 or through strategies like contrastive learning.30 Finally, for tasks sensitive to the molecule's conformation in three dimensions, incorporating three-dimensional representations that exceed the capability of the innately deficient two-dimensional graphs has proven successful in predicting molecular properties.31
Structure-to-property models have been widely employed in screening projects, leading to experimentally verified predictions. We will discuss a few selected case studies in Section 2.2.1.
2.2 Property to structure: designing molecules in chemical space
While the rational design paradigm analyzes the relationship between structure and properties to design promising molecules, another paradigm asks: what are all the possible molecules that satisfy a given property? Solving this question is known as the inverse design problem.Chemical space is the set of all synthesizable molecules and is often cited as having an astronomical size of at least 1033 to 1060 molecules.34,35 Within this vast space are potential drugs that could cure current diseases and putative materials that could enable a sustainable future.
Similarly, VirtualFlow40 docked over 109 molecules by efficiently using thousands of CPU cores. As the size of chemical libraries grows, with the required computational resources scaling linearly, hierarchical approaches to evaluate the fitness of individual synthetic building blocks offer a way past linear scaling.41
However, another arm of ML offers a way to consider all (or a vast subset) of the chemical space. Given a dataset of molecules in a representation such as SMILES strings, generative models learn to generate strings which resemble the dataset. Because generative models can consider arbitrary strings, they could potentially generate any molecule in chemical space. They can also be conditioned to generate molecules with desired properties – essentially reversing the property prediction process.44,45 Molecular generative models have been applied with many model classes. We pioneered the use of variational autoencoders (VAEs)46 for this purpose. Other examples include autoregressive models,47 generative-adversarial networks (GANs),48 and reinforcement learning,49,50 amongst many other sampling strategies. Generative models have also been extended and shown to work well with various representations like SMILES, SELFIES,51 and Group SELFIES52 strings, as well as molecular graphs and fragments. Molecular optimization methods such as genetic algorithms53 and Bayesian optimization54 also have been sometimes called generative models despite not learning a molecular distribution per se. A recent review of different generative model classes and representations can be found in Gao et al.,55 although this is a rapidly moving field.
As more generative models were proposed, benchmarks such as GuacaMol56 and MOSES57 began evaluating and comparing different generative models based on validity, novelty, uniqueness, and goal-directed optimization. Optimization has been such a primary focus that molecular design can be regarded as a combinatorial optimization of molecular properties over the space of molecular graphs. In this way, a new benchmark emphasizes sample efficiency, which is the number of property evaluations needed to reach optimal molecules.55 In addition, more realistic benchmark tasks relying on simulation have been recently proposed by us in the Tartarus benchmark set.33 Tartarus more closely resembles real-world scenarios where computational and experimental resources are constrained.
However, by departing from chemical libraries for the entire chemical space, generative models relaxed the crucial constraint of synthesizability. Generative models can suggest molecules which are difficult to synthesize and evaluate.58 To overcome this, synthesizable generative models consider chemical synthesis pathways when generating molecules, ensuring that the generated molecules are not only theoretically valid but also practicably synthesizable.59–61 Other approaches combine virtual libraries with generative approaches to ensure that proposed molecules are always from the library.62 These methods have particular relevance for high-throughput arrays and self-driving laboratories, as predicted molecules that are not synthetically feasible with readily available platforms could slow down closed-loop approaches.
For a comprehensive overview of these advancements and the state of the art in molecule design, Du et al. provide an excellent review, summarizing the latest developments and methodologies in the field.63
Generative models have proven worthy in the recent years. Quite notably the company InSilico Medicine has employed them to generate several drugs that are undergoing clinical trials currently. In 2019, together with InSilico and Wuxi Apptec researchers, we showed the ability of generative models to develop a lead drug candidate in approximately 45 days.64 Many researchers since then have continued to show other examples of generative models in drug discovery. For example, Barzilay and co-workers have developed antibiotics using similar approaches.65
Most generative models have been developed with simple benchmarks in mind, such as predicting simple properties like log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P. However, developing using proper benchmarks (such as Tartarus) or restricting them to feasible sets of molecules, such as those synthesizable with self-driving labs (see Section 2.7), remains a challenge.
P. However, developing using proper benchmarks (such as Tartarus) or restricting them to feasible sets of molecules, such as those synthesizable with self-driving labs (see Section 2.7), remains a challenge.
2.3 Structure to structure: synthesis planning and reaction condition prediction
Synthesis planning – i.e. finding synthetic pathways that give rise to a desirable target molecule – is an open challenge that chemists have faced for over a century, particularly in the “molecular world” of drug discovery, agricultural chemistry or molecular materials chemistry. This problem is complex in two respects: first, predicting the outcome of a specific unseen reaction, given all reactants, reagents, and reaction conditions, is effectively an unsolved problem to date. Second, even with such a “reaction prediction” tool at hand, finding feasible multi-step sequences of reactions that eventually enable the synthesis of the target molecule from cheap and commercially available precursors requires searching a massive network of possible pathways. Additional challenges arise from practical demands to the synthesis planning problem: efficiency, cost, waste production, sustainability, safety, or toxicity are practical concerns, especially in an industrial setting.The past decade has seen significant progress in addressing this challenge using the toolbox of ML. In this context, the key “decision policy” has often been treated as a multi-task regression problem: given the structure of a target molecule, a ML model is trained to predict an applicable reaction out of a catalog of reactions.69–71 This symbolic approach, however, requires a pre-defined catalogue of all reaction types, often referred to as reaction “rules” or “templates”, which itself presents new obstacles. There is neither a generally accepted definition of the term “reaction rule” nor an unambiguous procedure to perform reaction rule extraction from data. Alternatively, “template-free” approaches to the one-step reaction prediction problem, predict reactions as graph edits in the starting material graph,72 or solve a sequence-to-sequence “product-to-starting-material” translation task.73,74 Notably, these models (template and template-free) can be similarly trained in the forward direction, predicting reaction products from starting materials.
These single-step prediction models have been used to build tree search models, which aim to solve the full synthesis planning problem. In this context, a Monte-Carlo tree search is usually the method of choice. Following the pioneering works from Segler et al.75 and Coley et al.,76 a number of mostly open-source systems have been released.77,78
Machine learning approaches to reaction condition optimization have thus mainly focused on regression modelling of reaction yields as a function of reaction conditions. In this context, data-driven approaches have intersected with regression techniques from physical organic chemistry, which attempt to model reaction outcomes based on mechanistic considerations. In highly constrained condition spaces, purely data-driven, supervised learning of product yields on systematically generated data from high-throughput experimentation has shown promising results.79–83 For example, our work on optimizing the E/Z ratio of a reaction relevant to pharmaceutical process chemistry showed that only with ≈100 experiments we were able to outperform what had been the state-of-the-art for this process by human-only reaction optimization.84 Meanwhile, the use of literature data for the same purpose is highly flawed,85,86 usually necessitating individual, case-by-case reaction optimization (see below for a more detailed discussion). Black-box optimization algorithms, particularly Bayesian Optimization (BO), have become increasingly prominent over the past decade.6,87 In BO, probabilistic models for predicting reaction yields are built through Bayesian inference with existing data. These models then iteratively guide decision-making throughout the optimization process. The idea of iterative, closed-loop optimization with ML-based surrogate models is discussed further in Section 2.7. For condition optimization, these iterative approaches have demonstrated remarkable success in increasingly complex synthetic reaction scenarios.87 At the same time, chemistry-specific challenges, such as the identification of conditions which are “generally applicable” to a wide variety of substrates, as opposed to just one or a few model substrates, have inspired algorithmic advances in the field.88,89 Notably, our work on the Suzuki reaction88 led to generally applicable conditions with double the yield of the previous state-of-the-art in the field.
Similar data limitations have also been discussed in the context of reaction outcome and reaction condition prediction. Patent data97 and even commercial databases are highly problematic not only because of erroneous, inconsistent or unstructured data reporting: human biases in the reported experiments, particularly the accumulation of prominent conditions and the lack of low-yielding records, have prevented predictive modelling of reaction yields from literature data.85,86 Community-driven, open source data repositories such as the Open Reaction Database98 represent an essential step towards less biased and more holistic data collection – but such initiatives require a more digitized mindset in the way data is generated, collected and reported in synthetic organic chemistry laboratories.
A further consequence of this data deficiency is the lack of representative benchmark problem sets. This applies to multi-step synthesis planning, where benchmarks are urgently needed for a more quantitative evaluation of synthesis planning performance. Similarly, optimization algorithms for chemical reactivity would benefit from representative benchmarks to evaluate how standard BO algorithms translate to the intricacies of chemical reactivity. Most importantly, such benchmarks must reflect real-life problems, as identified by expert chemists, in order to inspire and motivate algorithmic ML advances to tackle the challenges in computer-aided organic synthesis.
2.4 Structure to physics: simulation and 3D structure
Machine learning has enabled data-driven solutions to both experimental problems and computational problems. Whereas organic chemistry emphasizes molecules' 2D molecular graph structure, molecules are also grounded in 3D physical reality by the Schrödinger equation, providing a rich theory of quantum mechanics and statistical mechanics for predicting molecular properties and interactions. Simulation methods such as density functional theory (DFT) and molecular dynamics (MD) then use this theory to computationally predict molecular properties and interactions. However, despite continual increases in computing power, these simulations remain computationally costly, which has restricted simulation to small systems at short timescales. By learning from the results of many simulations, ML offers a unique opportunity to accelerate molecular simulation.A significant shift came with the introduction of neural force fields. By training neural networks on DFT data to predict energy and forces directly from 3D nuclear coordinates, molecular dynamics could now be propagated at ab initio accuracy at a much lower computational cost.103 Since forces must be equivariant to the molecule's rotation – i.e. if the molecule is rotated, the molecular forces must “rotate along with it” – this motivated the development of equivariant neural architectures to respect this symmetry.104–106 Neural force fields have been competitively benchmarked in ML, continually comparing different architectures and methods on several benchmarks. A detailed timeline of development of these equivariant architectures is given in Duval et al.107 As datasets of energy and forces have grown, such as the Open Catalyst Benchmark,108 neural force fields have started striving for universal applicability.109
Furthermore, neural networks can be used as ansätze to represent the wavefunction itself directly. In this case, the network takes as input electron coordinates, and outputs wavefunction amplitude. Using the same stochastic optimization algorithms, neural wavefunctions can be trained to minimize the variational energy and satisfy the Schrödinger equation.113–117 This approach has recently been extended to excited states.118
Alternatively, for density functional theory, neural networks can be trained to directly predict charge density given the nuclear coordinates.119–121 ML has also been applied to learn density functionals.122
The large conformational search space motivates generative models to navigate this space. Unconditional generative models such as equivariant diffusion models can generate 3D atomic positions and atom types simultaneously.123 For the problem of conformer search, which seeks stable 3D configurations for a given molecule, atom types can be held constant while generation is conditioned on the 2D molecular graph. Some approaches generate atom positions freely,124 while other approaches generate torsion angles of rotatable bonds.125,126 Recent work has shown that forgoing both torsional and rotational symmetry constraints can yield better results, but at a higher cost.127 A related task known as docking performs conformer search of a ligand inside a protein pocket, as an estimate of binding affinity. This has also been approached with diffusion models.128
In the problem of crystal structure prediction, the goal is to find the most stable periodic arrangement of atoms for a given composition. While traditional approaches search through all stable configurations of coordinates and lattice vectors to find the lowest energy structure,129 equivariant diffusion models have found a natural fit for this problem, diffusing both coordinates and lattice parameters simultaneously,130,131 while also enforcing space group constraints132 to enhance performance further. Indeed, scaling this diffusion approach to large datasets enabled inverse design to satisfy multiple desired properties simultaneously.133
In the fields related to the simulation of biomolecules, 3D structure prediction problems are abundant. The longstanding problem of predicting folded 3D protein structure from protein sequence has, to a certain extent, been solved by AlphaFold2134 and related models. Building on this approach, diffusion models have generated protein backbones represented as sequences of rigid bodies of residues.135,136 These models have been so successful that they have been used to design proteins satisfying structural constraints, which have been experimentally validated.137,138 The scope of these diffusion models has expanded to all biomolecules, with methods predicting how proteins, RNA, DNA, and ligands assemble in 3D atomistic detail,139,140 subsuming the task of docking, and hence, promising to become a de facto conditioning function for drug discovery in the future.
In coarse-graining one typically groups atoms together into so-called beads, which afford lower computational cost and the possibility to capture long timescale events. However, the forces on these coarse beads then need to be fitted to all-atom forces. To circumvent this, neural networks can be applied to learn coarse-grained force fields by predicting the gradient of the free energy, rather than the energy, and matching these predicted forces on coarse-grained beads to the all-atom forces.145–147 Flow-matching148 removes the requirement for all-atom forces, needing only equilibrium samples of coarse-grained beads. Furthermore, diffusion models can simultaneously learn a generative model and coarse-grained force field.149
While coarse-grained force fields are significantly faster to evaluate than atomistic ones, MD simulations are still limited by having to use femtosecond-level integration time steps. Alternative methods for equilibrium methods focus on accelerating molecular dynamics to reach long timescales. This can be done through “coarse-graining in time,” which trains generative models to predict the outcome of taking large timesteps.150,151
Lastly, work has been carried out towards extending models to multiple ranges of thermodynamic properties like temperature and pressure.152 This allows simulation of different environments as well as training on previously unsuitable data. Adding extra parameters like temperature to the model input, one can add the corresponding derivatives of the coarse-grained free energy function to the loss. Response properties which are higher order derivatives of the free energy can be computed via multiple backward passes. Incorporating thermodynamic parameters might be one of the key ingredients to simulate biological or industrial settings in a holistic manner.
For rare-event sampling like chemical reactions and transition state search, methods for sampling transition paths without reaction coordinates have been emerging.153,154 Alternatively, when datasets of reactants, products, and transition states are available, generative models can be directly trained to generate transition states conditioned on reactants and products.155,156
However, these issues may begin to go away as neural forces are trained on ever larger datasets in the quest for universal force fields. Though ML models are limited by the quality of their data, the fact that new data can be generated by simulation paints a promising picture for data availability and large models.
At the same time, much work remains to reach simulation at large length and time scales. The most significant challenges of proper equilibrium sampling under metastable conditions and the related problem of rare-event sampling also remain areas in need of improvement and, therefore, the focus of many recent efforts.
2.5 Structure and analysis: spectroscopy and elucidation
One natural yet underexplored area of ML application in chemistry is structure elucidation, which aims to predict 2D or 3D molecular structures from spectroscopic or other analytical data. Just as computer vision enables computers to perceive the natural world, computational spectroscopy could allow machines to perceive the molecular world through analytical instruments. The anticipated increase in the synthesis of de novo and unknown compounds through advances in experimentation automation drives the need for faster yet accurate structure elucidation to fully support these autonomous molecular and reaction discovery platforms.Combining information of many inferred functional groups has enabled structure elucidation. For NMR data, the molecular structure can be elucidated by first identifying molecular substructures and functional groups,175–177 which are then optimally assembled via beam search over possible configurations or constructed atom-by-atom,177–179 similar to the approach chemists take when interpreting NMR spectra. Similar “reconstruction-by-substructure” strategies have been employed to varying degrees of structural detail for IR180,181 and surface-enhanced Raman spectroscopy (SERS).182 However, as the number of atoms increases, this approach quickly encounters combinatorial scaling issues.
Molecular structure elucidation can also be tackled as an end-to-end problem from a deep learning perspective. In this approach, the spectra are tokenized into strings and SMILES strings are predicted; this can be viewed as a machine translation task. This approach has been applied to NMR, IR and tandem MS/MS data,183–187 showing more significant promise for scaling to larger chemical systems and de novo structure elucidation. The structure prediction problem can also be formulated as an optimization task, e.g. by formulating it as a Markov decision process.179 If we consider scenarios where we have some prior information about the chemical system at hand, such as chemical formula, known starting materials and reaction conditions, implementing this information as constraints can help the model converge on a solution more efficiently.
Moving from molecules to crystals, solving the inverse problem for X-ray spectroscopic data such as powder X-ray diffraction (PXRD) and X-ray absorption near-edge structure (XANES) spectra also poses interesting challenges for the machine learning community, where there are unique and underdeveloped opportunities for employing various deep learning models for generalizable crystal system and space group identification.188,189 Diffusion models have shown particular promise, especially given their successful application to counterpart inverse problems in text-to-image generation. In this context, we can draw parallels between text and spectra and between image generation and crystal structure prediction.190,191
In the field of rotational spectroscopy, the challenge of spectral assignment – i.e. deduce the rotational constants from a densely packed rotational spectrum – represents one of the earliest applications of ML in this domain.192 This problem is particularly well suited for deep learning techniques due to the dense yet easy-to-simulate nature of the spectra. However, the rotational constants alone do not determine the 3D structure of the molecule. The approach that we recently introduced solves this by inferring 3D structure given incomplete information as molecular formula, rotational constants, and unsigned atomic Cartesian coordinates known as substitution coordinates.193
In the realm of structural biology, advances in protein structure prediction have accompanied advances in cryo-electron microscopy. Reconstruction of protein structure from cryo-EM has been tackled using deep generative models.194,195 These methods have progressed to the point of reconstructing biomolecular dynamics from cryo-electron tomography (cryo-ET).196 Structure elucidation using CryoEM continues to show day-to-day advances. Advances in data processing have provided incredible gains in resolution197 that can only be improved by the use of ML methodologies.
For inverse spectrum-to-structure elucidation, while autonomous and de novo molecular structure determination of pure samples is indubitably essential to facilitate high-throughput reaction optimization and discovery campaigns, it is also crucial to address structure annotation of spectra from complex mixtures, which encompasses both the targeted identification of specific compounds of interest and non-targeted metabolomics. Such mixtures are standard in real-life sample matrices and are essential for various fields ranging from bio-diagnostics to forensics. Success in these tasks is highly contingent on the model's ability to disentangle and isolate individual molecular spectral signatures from the highly convoluted data. Machine learning excels in handling complex, high-dimensional data, making it well-suited for these challenging tasks.198,199 In addition, leveraging ML methods to integrate information from multiple spectral inputs could further enhance structure elucidation's accuracy and completeness.
2.6 Leveraging scale with foundational models for chemistry
With increasing computational power, machine learning models have been trained on progressively larger datasets. At scale, ML offers qualitatively different capabilities. Foundation models are large-scale models that have been trained on a broad spectrum of data and can be applied to a variety of downstream tasks. Several general-purpose foundation models – such as ChatGPT, Gemini, and Llama – are typically utilized for language and image generation; many of these are language-only models or models trained on multiple modalities. However, using these models in the chemical domain presents unique challenges, and so many have trained their models from scratch on chemical data, but this is not trivial either. In this section, we will describe the current state of foundation models in chemistry and give our perspective on remaining open questions.However, there still exists a gap in using these models out-of-the-box for discovery tasks or in domain-specific chemistry applications (at least to our knowledge),206,216 one reason being that there is not enough data to train these models in the same way that models like GPT-4 have been trained on web-scale text and images.217 One way to use these chemistry-aware language models is to finetune them on downstream tasks,218 or plug them into optimization or search frameworks as a way to provide good prior knowledge.219–222 Other works have also begun to explore scaling of both models and data.223,224
One interesting application of chemistry-aware foundation models has been the development of chemistry agents that can e.g. make use of tools225 necessary for solving chemistry problems, and/or plan chemistry experiments. Some notable examples include ChemCrow,226 Coscientist,227 our own ORGANA,228 or ChemReasoner.229 These agents have access to various chemistry-related tools, such as simulators or robots to execute chemistry experiments, and use an LLM (such as GPT-4) as a central orchestrator to decide when and how to use these tools to accomplish a user-specified goal. One longer-term goal of such agents is to develop scientific assistants that can help beyond calculating and executing to do more complex reasoning and planning by generating and refining hypotheses on their own. This has been extended to other research domains by the AI Scientist, which demonstrates autonomous machine learning research by executing experiments and writing a research paper.230
These research areas are in their infancy, so several open questions remain, including: (1) How do we effectively evaluate chemistry-aware LLMs/agents? (2) What are the use cases for these models in practice for chemists? Effective model evaluation mainly depends on developing meaningful tasks, which is currently an open problem both in terms of dataset scale and breadth. There already do exist several benchmarks in this space,28,231 which is a good start but there is room to improve them in terms of data quality and task objectives.32 More recent benchmarks have been released that are closer to real-world applications,33,232,233 and also platforms such as Polaris have made it easier for researchers to have faster access to a wide array of datasets.234 The issue with using sub-optimal benchmarks in this field has been exacerbated by the current climate in machine learning in that benchmarks are mainly used to show that a new method achieved better performance than the current state-of-the-art, without human understanding of why it improved. This is also an excellent opportunity for collaboration between chemists and the ML domain expert communities.
Language-based foundation models have also been used in other applications, including knowledge graph generation235 and knowledge extraction from chemical literature,236–239 including our own work on reaction diagram parsing,240 which is a difficult task. These efforts are essential for creating structured databases of experimental procedures, which can contribute to existing repositories such as the previously-mentioned Open Reaction Database.98
However, language models are not the only foundation models developed in the biochemical sciences. Several models have been built to universally approximate force fields and predict structures for any molecule, material, or protein.109,242–245 Perhaps the most famous example is AlphaFold2 for protein structure prediction134 and, more recently, AlphaFold3,140 which given any set of 2D biomolecules, predicts how they might assemble in 3D. To our knowledge, these models still outperform any sequence-based protein prediction models for many structural and functional tasks, especially in cases where input sequences do not have homologues in the training data.246
Another impressive example is the recent foundation model MACE-MP-0, built with the MACE equivariant architecture.109,247 MACE-MP-0 was trained on 150 thousand inorganic crystals. After a small number of task-specific examples for fine-tuning, it can be used as a force field in simulations on a wide variety of tasks, even seemingly unrelated ones such as small protein simulations. Notably, intermolecular interactions seem somewhat fuzzy in the MACE-MP-0. For example, in the aforementioned protein simulation, the model was able to capture hydrogen transfer, which is a remarkable achievement. However, the authors also opted to include D3 dispersion borrowed from classical computational chemistry, pointing to the fact that the model still needs some help to predict long-range interactions. Foundational force fields have continued to scale, with industry research labs training neural force fields on ever-larger data, such as GNoME244 and MatterSim.245
One key takeaway from these types of models is that structural information should not be ignored depending on what downstream tasks the model will be applied to, and that training models on broad, large-scale datasets (i.e., going beyond training a simple model on a single prediction task, which was the norm even a couple years ago) can help generalize better to more downstream settings. We suspect that scaling along multiple modalities concurrently is critical for building the best foundation model in chemistry – namely, training models on as many modalities as possible, such as 3D structure information, text, and spectral information.31
2.7 Closed-loop optimization and self-driving labs
Significant advancements in automated laboratory equipment have been achieved by integrating ML with computer vision,252 leading to the concept of “general chemistry robots”.253 These ML-trained robots can make decisions based on external feedback, enabling the dynamic automation of chemical operations traditionally performed by human chemists.254–256 Given the inherent challenges in training robotic equipment for active decision-making based on external feedback, a notable innovation in this area is the use of digital twins—virtual replicas of laboratory setups—that provide a robust framework for accelerating the training of robotic ML models.257 These digital twins simulate chemical scenarios with high fidelity,258 creating a realistic feedback loop that accelerates the model's learning process.
On the experimental planning side, heuristic techniques259–261 are being progressively replaced by ML optimization algorithms. When combined with chemical digitization,262 these optimization techniques can identify target chemicals and optimize reaction conditions while significantly reducing the number of experimental steps required.263 Among the various ML optimization techniques,264,265 Bayesian optimization266–268 has gained particular prominence in experimental chemistry due to its success in chemical applications.269 Machine-learning-based surrogate models, which predict the properties of chemicals and reactions,270–272 have been instrumental in this success, with documented examples in both process optimization and materials discovery.273
Moreover, the rise of LLMs has further enhanced the auxiliary components of SDLs. LLMs have been effectively used to create human–machine interfaces that bypass traditional coding,228 enabling more natural communication between chemists and laboratory systems—a significant advantage for users who may not be well versed in coding or data processing.274,275
Motor challenges. The primary hardware challenges stem from the human-centric design of chemical instruments and the lack of seamless interconnection between existing automated modules. As a result, most SDLs operate semi-automatically, requiring human intervention for tasks such as sample transfer, maintenance, and troubleshooting. Various solutions have been proposed to address these issues, including deploying mobile robots for sample transfer253 and adapting general-purpose robots to perform chemical tasks or operate instruments originally designed for human use.277–279 However, many of these methods rely on traditional algorithms that require static calibration, which is not well suited to the dynamic nature of SDLs. While computer vision coupled with AI has been proposed as a solution, laboratory equipment, particularly glassware, continues to present significant challenges that are continuously being addressed.280
Cognitive challenges. Cognitive challenges primarily arise from the difficulty in developing models that can accurately estimate the chemical output of the system. This limitation restricts the use of more general generative models, effectively reducing the amount of chemical space that experimental planners can explore. When combined with the aforementioned motor challenges, another issue becomes apparent: SDLs often operate in low-data regimes. Predictive and generative machine learning models typically require large datasets to make meaningful predictions. While generative models can be trained on existing data,219,281 deploying predictive algorithms in such low-data regimes remains a significant challenge.
Auxiliary component challenges. Regarding the auxiliary components of SDLs, the incorporation of LLMs shows promise in automating workflow creation274 and improving human–machine interfaces. However, further research is needed to ensure the safety and reliability of these processes. Additionally, while integrating bibliographic extraction into SDLs can enhance model development, its effective integration with predictive models remains an unresolved issue.
A final challenge to be addressed in the field of SDLs is the economy of scale of their development. The more SDLs the community builds, the easier it will be to build the next ones. Hence, the democratization of low-cost SDLs is crucial for the advancement of the field.282
3 Problems meet methods: a machine learning perspective on solving chemical problems
There is already a wealth of resources on how to apply the specifics of machine learning in several books, reviews, and internet resources.283–286 In this section, we provide a high-level perspective of how ML researchers and communities view and tackle problems. To start, we reclassify the diverse chemical problems introduced above as instances of well-established ML problems. To elaborate the ML perspective, we gather common themes and practices in the ML community and examine them in light of application to chemistry, highlighting points to consider related to benchmarking, the role of domain knowledge, and community values.3.1 The toolbox of machine learning
ML provides a toolbox of algorithms and theory for solving problems using data. ML has formalized a set of well-defined problems to solve diverse tasks in language, vision, audio, video, tabular data, scientific data, and other domains. Each problem establishes a set of input requirements and a desired goal, which has proved helpful for empirically benchmarking and theoretically analyzing different algorithms under a common framework. In Table 1, we lay out significant ML problems with their expected inputs and goals and reclassify different chemical problems as instances of these ML problems.| ML problem | Input | Goal | Chemical problems | Algorithms |
|---|---|---|---|---|
| Regression and classification | Paired data {(x, y)} | Predict ŷ = f(x) | • Property prediction | • Classical machine learning: linear regression, random forests, support vector machines, gradient boosting machines |
| • Neural network potentials | • Gaussian processes | |||
| • Yield prediction | • Neural networks | |||
| • Proxies for fast prediction | • Graph neural networks | |||
| • Spectra prediction | • Equivariant neural networks | |||
| • Figure segmentation | • Transformers | |||
| • (3D structure prediction) | ||||
| Generative modelling | Dataset {x}, optional conditioning {y} | Draw samples x ∼ p(x) or x ∼ p(x|y) | • Conformer search | • Variational autoencoders |
| • Docking | • Generative adversarial networks | |||
| • Crystal structure prediction | • Normalizing flows | |||
| • Transition state search | • Autoregressive models | |||
| • Structure elucidation | • Denoising diffusion and flow matching | |||
| • Forward synthesis prediction | ||||
| • (Molecular design) | ||||
| Sampling | Energy E(x) | Draw samples x ∼ p(x) ∝ e−E(x) | • Equilibrium sampling | • Markov chain Monte Carlo |
| • Transition path sampling | • Sequential Monte Carlo | |||
| • Molecular design | • GFlowNets | |||
| Gradient-based optimization | 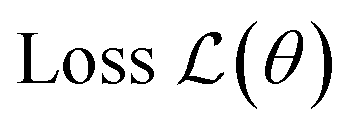 |
Optimal parameters θ* | • Neural wavefunctions | • First-order: (stochastic) gradient descent, Adam |
| • Physics-informed neural networks | • Second-order: K-FAC | |||
| • Differentiable simulation | ||||
| • (Molecular design) | ||||
| Black-box optimization | Oracle f(x) | Optimal x* | • Reaction and process optimization | • Bayesian optimization |
| • Bandit optimization | ||||
| • (Molecular design) | • Reinforcement learning | |||
| • Genetic algorithms | ||||
| Agents | Environment of states {s}, actions {a}, transitions, and reward R(s) | Draw actions from optimal policy a ∼ π*(s) | • Extracting literature data | • LLM prompting frameworks |
| • Executing simulations | • Reinforcement learning | |||
| • Question answering | ||||
| • Synthesis planning | ||||
Regression and classification aim to predict labels y from inputs x, given a dataset of paired data. Labels can be one-dimensional, such as in predicting properties, energy, or yield, but also high-dimensional, such as the ML regression problems related to force fields, spectra prediction, and segmentation. When data is small and tabular, gradient boosting machines such as XGBoost287 often perform well. Gaussian processes also work with small data and provide good uncertainties for use in Bayesian optimization.288 However, deep neural networks are the algorithm of choice for high-dimensional, complex data like images, text, and molecules. The choice of neural network architecture is informed by the problem's constraints: graph neural networks for 2D graphs and equivariant architectures for 3D data. Relatively recently, transformers289,290 have revolutionized modelling of language,289 images,291 graphs,292 and 3D molecules.134,243
Generative modelling aims to draw samples x from a distribution p(x) defined by a dataset {x}. Unconditional generative modelling tries to match the data distribution. Conditional generative modelling takes a label or prompt y and tries to learn the conditional distribution p(x|y), blurring the line between unsupervised and supervised learning. While unconditional generative modelling is rarely valuable for chemistry, conditional generative modelling is ideally suited to inverse problems or one-to-many problems. This is the case for conformer search (one 2D structure for many 3D conformers), structure elucidation (one signal could be consistent with multiple molecules), or forward synthesis prediction (given reactants, many products might be possible). Generative models are a natural fit for their ability to produce multiple quality answers to a question. On the other hand, regression will average over all the possible answers, which may not be a quality answer itself. Whereas AlphaFold2134 used regression to predict one 3D structure given one sequence, AlphaFold3140 used diffusion models to predict multiple biomolecular assemblies for the same input structures. While many generative model classes exist, such as variational autoencoders,293 generative adversarial networks,294 and normalizing flows,295 the dominant ones today are autoregressive models for language296 and diffusion/flow matching models for perceptual data like images.297 In chemistry, this translates to chemical language models of SMILES224 and diffusion models of 3D molecular structure.140 Both approaches rely on gradual generation via iterative prediction by a neural network, usually a transformer. Because an unconditional generative model learns to reproduce a data distribution, which may be a large amount of plentiful unlabeled data, training a generative model can also be thought of as compressing all this data into the network's weights, imbuing a notion of understanding. Tasks such as sampling and agent behaviour can then build on this understanding.
Sampling also aims to draw samples from a distribution but is distinguished from generative modelling because it only permits access to an energy function E(x), which defines an unnormalized probability density p(x) ∝ e−E(x). No dataset is provided, so one cannot simply train a generative model. Furthermore, generating a dataset in the first place would require drawing samples. In addition, the energy function is often computationally costly to evaluate. For these reasons, sampling problems are among the most difficult in ML and computational chemistry. Numerous sampling algorithms exist in the literature, with many originating from statistical mechanics, such as Markov chain Monte Carlo (MCMC)298 and Langevin dynamics.299 These traditional methods are beginning to incorporate ideas from modern machine learning, such as drawing inspiration from diffusion models for MCMC,300 or incorporating learnable components into sequential Monte Carlo.301 Some methods learn a bias potential to do transition path sampling,154 while other methods turn diffusion models into samplers which can solve combinatorial optimization problems.302 Sampling methods are key to solving equilibrium sampling problems, which are necessary for predicting the thermodynamics and kinetics of many chemical processes. Generative models can be used as components of sampling algorithms,303 such as in Boltzmann generators,141,144 which train both by energy and by example. Boltzmann generators have also begun to leverage generative models, transferring learning between different examples.143 Generative Flow Networks304 (GFlowNets) solve this sampling problem by learning to distribute flow in a generative graph, with a unique strength for generating diverse, discrete data. Indeed, a growing body of literature has applied GFlowNets to molecular and materials design problems.61,305–307
Gradient-based optimization seeks to optimize a smooth loss function  with respect to parameters θ, which is used to train the neural networks used to solve nearly all of the other ML problems. To do so, machine learning has developed a suite of optimization algorithms such as (stochastic) gradient descent, Adam,308 and second-order methods such as K-FAC309 which use second-derivative information. Machine learning frameworks such as PyTorch,310 JAX,311 and Tensorflow312 have implemented automatic differentiation with GPU acceleration, making it easier to optimize neural networks. The fact that neural networks can be optimized so well has motivated the use of neural networks as ansätze for finding wavefunctions to satisfy the Schrödinger equation.113 This approach, in turn, is an instance of a physics-informed neural network (PINN),313 which seeks neural network solutions to PDEs by using the PDE itself as a loss function. Automatic differentiation also enables propagating derivatives through simulation, which can learn potentials for pairwise interaction,314 bias potentials for transition path sampling,153 and perform inverse design.315
with respect to parameters θ, which is used to train the neural networks used to solve nearly all of the other ML problems. To do so, machine learning has developed a suite of optimization algorithms such as (stochastic) gradient descent, Adam,308 and second-order methods such as K-FAC309 which use second-derivative information. Machine learning frameworks such as PyTorch,310 JAX,311 and Tensorflow312 have implemented automatic differentiation with GPU acceleration, making it easier to optimize neural networks. The fact that neural networks can be optimized so well has motivated the use of neural networks as ansätze for finding wavefunctions to satisfy the Schrödinger equation.113 This approach, in turn, is an instance of a physics-informed neural network (PINN),313 which seeks neural network solutions to PDEs by using the PDE itself as a loss function. Automatic differentiation also enables propagating derivatives through simulation, which can learn potentials for pairwise interaction,314 bias potentials for transition path sampling,153 and perform inverse design.315
Black-box optimization methods try to optimize an oracle function f(x) in a derivative-free manner with as few oracle calls as possible. This is the case in many experimental problems such as optimizing reaction parameters for yield,269 device processing parameters for performance,316 or liquid handling parameters.317 To solve these problems with high sample efficiency, algorithms like Bayesian optimization and bandit optimization are applied. When sample efficiency is not a concern, families of algorithms such as reinforcement learning and metaheuristic optimization like genetic algorithms can also be applied.318 Black-box optimization can also be treated as an instance of sampling, where the target distribution is concentrated around the global optimum.
Agents solve complex multistep problems within an environment. An environment defines possible states s, actions a, transitions between states, and a reward function R(s). For example, retrosynthesis planning75 has molecules as states, chemical reactions as actions, and yield and cost as reward functions. Planning problems such as retrosynthesis planning or robotic motion planning319 are naturally solved by agent behaviour, and standard algorithms to learn optimal agent behaviour are known as reinforcement learning. Because reinforcement learning has poor sample efficiency, a common approach is to initialize agents from generative models: helpful assistants such as ChatGPT were initialized as large language models pretrained on internet-scale text, followed by finetuning to maximize a reward of satisfying human preferences.320 Prompting frameworks are a rapidly emerging set of methods for augmenting these agents' capabilities, allowing them to reason step-by-step,321 use tools,225 retrieve information,322 and execute code,323 and to continually repeat these steps.324
Each of these ML problems also has its own theoretical foundations. Mathematical theory can analyze algorithms for proofs of convergence or properties when converged, providing explanations of why certain methods work better than others. The shared problem interface also allows analysis to determine when one method is the same as another or which methods are more general than others, which helps unify a diverse literature.
The problems enumerated in Table 1 are not an exhaustive list. Other problems include uncertainty quantification, which is helpful in Bayesian optimization326 and active learning,327 federated learning for combining industrial pharmaceutical data while preserving privacy,328 representation learning for generally applicable molecular descriptors,329 causal learning, retrieval, and compression.
In chemistry, there is a tendency to treat problems as a search, like finding a needle in a haystack. Traditional docking approaches search for all feasible ligand positions, while crystal structure prediction exhaustively searches for all atom arrangements. Molecular design by virtual screening assumes there will be sufficiently good needles in a haystack of large virtual libraries. A search-based perspective is useful when available resources are sufficient to exhaustively model a space, which may be necessary to show that no good solutions exist. However, for many applications, an exhaustive search is overkill. Imagine trying to write an essay by searching over the space of all possible English texts. A helpful exercise is to ask whether a search problem has the data and algorithms available to be reframed as a generative modelling or sampling problem.
3.2 Themes and practices in the ML community
Solving chemical problems can be aided by both high-level perspectives and community practices. To contextualize ML perspectives on algorithm development, we describe common themes and practices in the ML community, such as benchmarking, extreme interdisciplinarity, and the bitter lesson of deep learning. All of these are expanded below.A prominent feature of ML research is the use of leaderboards, where proposed methods are ranked based on their performance against established benchmarks. Papers must either advance or be competitive with the state of the art to be accepted at major conferences. This process has driven notable progress in various domains, from image classification331 and machine translation332 to image generation,333 and even solving Olympiad math problems.334 Leveraging this mechanism, the Open Catalyst Project108,335,336 set a benchmark for neural network potentials to relax organic adsorbates on metal surfaces. This project provided a dataset much larger than encountered before, which motivated the continual development of more powerful equivariant architectures. From 2020 to 2023, the success rate of predicting adsorption energy grew from 1% to 14%, with current models now becoming useful in predicting adsorption.337,338 Another benchmark called Matbench Discovery339 has initiated an arms race of neural force fields on the industry level.
However, while benchmarking is a powerful tool, it is essential to be critical of its applicability to chemistry. Domain experts are uniquely positioned to define practical benchmarks that can translate to real-world outcomes in the lab.33,55 Too often, ML literature presents problem settings that, while optimized for computational performance, may be unrealistic for experimental validation. This misalignment can lead to a scenario where the focus shifts from solving the actual problem to merely advancing ML techniques. As methods mature and benchmarks become saturated, new, more relevant benchmarks must arise.
Ultimately, defining and framing problems for ML researchers is a critical task. It involves proposing important questions and calls to action in a way that is accessible to the broader ML community. By doing so, chemists can guide the development of ML tools more likely to have practical applications in experimental research. While creating datasets and benchmarks can be seen as rote work, it can spur progress on difficult problems by leveraging community efforts of the ML community. Suppose a chemical problem can be crystallized and packaged into a clearly and appropriately benchmarked ML problem. Chemists can now wonder: what new problems now become possible to solve, if these old tasks can be solved with significantly greater speed or accuracy? There are many more scientific questions in the vast set of exciting areas to work in chemistry and materials.
For chemistry, the development of graph neural networks was driven by the need to model molecular graphs.23,341 This led to practical advances in modelling other graph data like social networks, citation networks, computer programs, and databases. Graph machine learning in turn made theoretical advances, particularly in analyzing the expressivity of GNNs through the Weisfeiler–Lehman test.342,343 In addition, the need for neural networks to respect rotational symmetries of 3D space motivated the development of equivariant architectures.344 All these methodological developments in respecting symmetries have been unified with a theory of geometric deep learning,345 which shows how convolutional neural networks, graph neural networks, and transformers are actually tightly related.
Beyond theory and methods, ML researchers are also excited for the potential of ML to help tackle real-world problems like global health and climate change. This has manifested as a great eagerness to learn, as evidenced by the proliferation of blog posts,346 teaching material,286 and online reading group communities with recorded talks.347 Several workshops which focus on ML applications to chemistry are offered at main ML conferences such as NeurIPS,348–350 ICML,351,352 and ICLR.353,354 This wide availability of resources also reflects the value of openness in the ML community. Conference papers are published freely, preprints are emphasized, and sharing code is expected. Conferences even have a track for accepting blog posts.355
When speaking to ML researchers, be patient with their initial assumptions. Often, several assumptions are made in the ML literature, which ultimately pan out to lose applicability when applied to actual experiments. This occurs in molecular design neglecting the synthesizability of molecules,58 or in reaction prediction neglecting the reaction conditions.356 This reflects the different values and assumptions reviewers make in a distinct field. It is easy to view this and dismiss those approaches as naïve, and it is good to make these criticisms. But let us not throw the baby out with the bathwater: we should ask, if these additional assumptions were taken care of, could this approach help solve our problem? As ML practitioners come from different backgrounds, they will not immediately understand jargon assumptions and experimental setups in chemistry. But they are eager to learn.
In light of scaling laws, we should be careful when imposing our domain knowledge when designing algorithms. The “bitter lesson” in ML cautions against relying too heavily on domain knowledge when designing algorithms.360 While hand-crafted, domain-specific design choices can offer short-term improvements, approaches that better leverage computational scale often outperform them in the long run. Across domains like text, images, speech, chess, and Go, approaches which rely on human intuition and inductive bias have been replaced by “brute-force” approaches that can take advantage of exponential increases in computing power provided by Moore's law.
As chemists, it is joyful to develop methods that are informed by our chemical knowledge, such as by injecting quantum chemistry descriptors into regression,361 or by imposing physical constraints on the system. However, we should remind ourselves that our human understanding of a problem does not directly translate into being able to design algorithms that solve this problem. Despite extensive knowledge of linguistics in ML research, models like ChatGPT were not realized until researchers trained on massive datasets.
The power of scale can be fearful. Even beloved assumptions like enforcing equivariance in neural networks have been challenged by recent work: methods like probabilistic symmetrization362 and stochastic frame averaging363 have shown that imposing architectural constraints is not strictly necessary, while models like AlphaFold3140 and Molecular Conformer Fields127 have demonstrated that models trained with randomly rotated training examples can automatically learn rotation equivariance, but at the cost of higher computation and longer training time.
At the same time, the present-day has limited scale and data. For example, expert systems with reaction rules are still the most effective approach for synthesis planning today,90 perhaps owing to the difficulty of gathering reaction data. In addition, one can discard even more inductive bias and train language models to generate 3D molecular structure directly as .xyz files, as we did recently,364 and it can compare favourably with more hand-tailored methods for crystal structure prediction.365 Yet, as Alampara et al.241 showed, current language models cannot encode geometric information needed to represent specific material properties.
Therefore, the bitter lesson does not mean that imposing inductive bias on algorithms is never good. An optimal balance must be chosen between leveraging computational power and domain expertise. This is especially critical in chemistry: unlike domains like language and images, which are available at internet-scale, chemical data is scarce and costs real-world experiments to obtain. It is crucial to design algorithms which use this limited data most efficiently. Hand-designed algorithms can enable better predictions and faster simulations in the near-term, which can bootstrap data generation towards ultimately reaching the scale of data required for foundation models.
Another critical role of domain knowledge is determining the appropriate concept of a problem. Should we model it from first principles, like physics-based simulations, or treat it as a cheminformatics problem? How does this problem fit into the broader context of the world? For example, predicting a drug's effect on a patient could be approached by simulating the entire person, which is currently impractical, or by modelling the effects statistically or causally. At some point, these different levels of models need to align, and domain scientists are crucial in mapping out this structured hierarchy of models. They help determine when assumptions are reasonable and when they are not. While ML tools cannot solve these problems independently, they can significantly aid in integrating different model components.
4 How to tackle scientific problems?
Armed with the above toolbox and perspectives, we then make recommendations on how to choose impactful problems in ML for chemistry and introduce a high-level structure of how ML problems are tackled. We finally outline three areas for growth for research in ML for chemistry: breadth, depth, and scale.4.1 The Aspuru-Guzik/Whitesides rules for selecting important problems
When one of us (Aspuru-Guzik) started the Matter Lab then at Harvard University (2006–2018) and now at the University of Toronto (2018–), a set of rules for selecting significant problems began to emerge from intuition. In a hallway conversation with George Whitesides, who told Aspuru-Guzik he had similar guidelines, the three questions to ask before starting any research crystallized. We apply them at the Matter Lab daily to select problems. Here, we specialize in ML in chemistry, but these are widely applicable. The three questions emphasize novelty, importance, and feasibility in that order.In the context of ML, improving on benchmarks, despite providing valuable signals of progress, is not the end goal of research. This is particularly true in academic work, where research is not directly linked to profits and should be as novel as possible. Once new problems are established, the field will be opened to improve the results afterwards.
Will this work create a new connection between two areas? When a paper introduces more questions than answers, the field grows. Simply applying an ML method to a new field can be novel. But novelty can be maximized if the proposed approach offers a new perspective, such as reframing a search problem as a generative modelling problem.
For example, we introduced 3D generative modelling to the field of rotational spectroscopy,193 which has opened the question of 3D structure elucidation from rotational spectroscopy alone. This is a clear example where first beats any other research. There were no previous ML baselines to compare or benchmark our method to, because we introduced the first approach in the field!
Which audience will care? What new tasks become within reach if this task is solved with significantly greater accuracy or speed? For example, neural network potentials are significant because force fields are used in a large number of computational chemistry methods, which in turn predict properties and spectra. Solving this problem, therefore, touches a large audience.
Can the proposed method be tested experimentally if it solves a computational problem? Approaches that can be experimentally validated have a much higher impact ceiling.40,137 On the other hand, what is the worst-case scenario if the proposed approach “doesn't work”? If novelty is chosen carefully, this risk is mitigated because a method which solves an unbenchmarked problem is already state-of-the-art.
In the context of ML, it would be useful to consider the following questions: What are the available resources? Is enough data available for the desired generalization performance? Are there public code implementations? Have similar problems been solved using the same framing? For example, the success of 3D generative models in structure prediction on tasks such as conformer search and docking indicated that they can likely be successful in crystal structure prediction as well.
A crucial part of feasibility is controlling scope. What is the minimal implementation of an algorithm that can solve this problem, yet have a broad impact? How can success be evaluated within this problem scope?
4.2 The structure of data science and ML problems
Machine learning and many data science problems have a general structure, as seen in many papers. Once you begin on a chosen problem, the next considerations follow this hierarchy: (1) data, (2) problem framing, (3) method, and (4) evaluation. In our research group, we always think of these in order and in ranking. For example, without data a scientist will not be able to make progress. A publication that suggests a new method for old data will be less impactful than the publication that provided the data (and its ML application) in the first place.000 examples, generative models are more likely to generalize effectively. Problems that are repeatedly solved in the community should be considered. Can these data be routinely recorded? For instance, tasks like computing forces and conformer searches are standard in quantum chemistry, and the availability of this data has contributed to the success of neural force fields and 3D structure prediction. Additionally, data might not just be a static dataset but could include on-the-fly data acquisition, such as environments for agents or oracle functions for black-box optimization. It is because data is the ultimate resource that our group embarked on the multi-year goal of developing and employing self-driving labs. We can eat our own dog food.Another way to approach problem framing is by asking how the data will be represented. Choosing a compact, information-rich, efficient-to-compute representation is a simple way to incorporate inductive bias and accelerate learning. However, as the bitter lesson shows, it is not essential to spend too much time on designing the “perfect” representation. Deep learning can automatically find ideal representations if the input representation contains all the necessary information and is available in large enough quantities.
This is perhaps the most critical paragraph of this publication: golden advice to graduate students and postdocs, do not fall in love with the mermaids of new methodology. If older but proven methodology does the job, just use it! Focus on the scientific contributions of your work. New methods should be developed when others truly have limitations. In other words, your new fancy super-duper autoencoder will not be as impactful in the long term as if you solve an essential chemistry or materials science question with an answer that lasts for ages.
4.3 New problems: demanding impact from ML for chemistry
Applying ML to chemistry can have a greater impact in terms of breadth of application, depth of consideration, and scale of execution. In breadth, many more chemical problems can be formulated as ML problems and introduced to the ML community. In depth, proposed methods can make stronger theoretical connections between both machine learning and computational chemistry, motivating further method development in each field. Finally, at scale, ML for chemistry can aim at more significant problems requiring more data. As concerns mount about reaching the limits of internet-scale data in language and vision, chemistry stands out as a situation where more data can be “purchased” through computational simulation or high-throughput experimentation.The key is not to “force” ML into these areas but to consider whether existing or novel tasks could be framed as ML problems listed in Table 1, facilitating iterative improvements and potentially leading to new algorithms. In some situations, there is just not enough data to apply ML, but it remains that a simple way to guarantee novelty is to consider an underexplored field.
Coming back to our previous example, we are pretty happy to have applied ML to solve an essential structural determination in rotational spectroscopy: the first application of generative models to predict the 3D structure of molecules given their substitution coordinates.193 This is an example of a typical in-breadth approach seeking multidisciplinary approaches and leaving our own comfort zone.
Many ML methods such as graph neural networks and equivariant architectures were motivated or inspired by theoretical chemistry, and they are beginning to return the favor. Diffusion models were proposed in 2015, inspired by methods in statistical mechanics,380 and have since become state-of-the-art generative models enabling high-resolution text-to-image generation.381–383 Nearly a decade later, new works have connected diffusion models to traditional tools in computational chemistry. Diffusion models can simultaneously learn both coarse-grained force fields and a generative model,149 and can also be leveraged as a means for sampling and computing free energies.384 These works would not have been possible without deeper consideration of how diffusion models relate to free energy, or of the connection between diffused distributions and the ideal gas.
Furthermore, flow matching approaches derived from diffusion models relax the constraint of noising a data distribution to a pure Gaussian distribution and can instead connect two different distributions. This has enabled learning of trajectories,117,385 which is beginning to be applied for transition path sampling of reactions.386 These works create theoretical connections that may enable more techniques to transfer from computational chemistry to machine learning and vice versa.
In addition, whereas neural network potentials treat energy computation as a black-box function to be memorized, Hamiltonian prediction111 opens the box of Hartree–Fock theory, enabling access to the wavefunction, as well as a new tradeoff between accuracy and speed. Self-consistency training112 engages with this theory by removing the requirement of providing Hamiltonian matrices as labels, which has improved the speed of DFT overall.
Aiming for a concrete design goal in collaboration with experimentalists also provides much-needed depth. Real-world problems often require the integration of ML with experimental data, and such collaborations can lead to breakthroughs that would not be possible in isolation. Large-scale collaborations between experts in quantum chemistry, machine learning, and organic materials chemistry enabled the discovery of new OLEDs.43 In that work, we were among the first to demonstrate that fingerprint-based ML methods, intelligent screening methodologies, and experimental verification could lead to novel materials in a closed-loop philosophy.
Our group, more recently, spent five years in an international collaboration involving six research groups, which led to a delocalized, asynchronous closed-loop design that led to the best organic laser material to date (to our knowledge).273 In parallel, another multidisciplinary collaboration on closed-loop design387 demonstrated that ML can teach us new chemical principles from these in-depth materials science explorations.
For chemical problems which are already formalized in ML, progress can be accelerated just by increasing the scale of data and compute of these approaches. Projects like the Open Catalyst Project demonstrate the potential of ML to drive large-scale advancements in chemistry. By purchasing new data through computation and simulation and by designing better sampling algorithms, we can improve the rate of data generation, and take aim at scale. LLM agents, for example, could execute computational simulations to generate new training data, further accelerating research.
While training foundation models is often cited as a source of significant emissions, we should also be aware of the potential for compute to reduce emissions.389 Better models could reduce the number of wet-lab experiments needed, or help design greener alternatives to current and future chemical processes, observing that the chemical industry makes up a large chunk of global emissions.
Chemical space may be small. The often-cited estimated size of chemical space as 1060 fascinates us. But from a machine learning perspective, this space may be considered small. If we only consider black-and-white 28 × 28 images, the domain of the standard MNIST dataset of handwritten digits,390 this already has a size of 228×28 ≈ 10236. Of course, the space of images is far sparser, given that the number of colour images in existence is 14.3 trillion ≈ 1013 images.391 This is what makes deep learning impressive – its ability to find structure within enormously high-dimensional spaces, just from showing a bunch of examples. In the context of language, 1060 is just the number of 10-word sentences restricted to a vocabulary of 60 words, or the number of 10-sentence paragraphs restricted to 60 possible sentences. Natural language is evidently much larger.
Could these powerful capabilities be enough to turn theoretical musings into reality? Imagine being able to atomistically simulate a cell on a macroscopic timescale, or to accurately model the effectiveness and stability of soft organic devices over years of use, or to discover new reactions ab initio. These are challenges that, until recently, seemed impossibly far beyond reach. We are impressed that nanosecond simulation of an all-atom HIV capsid at DFT accuracy is possible with neural force fields.392 If modern image generative models can generate high-quality images at 1024 × 1024 resolution and higher,393 then what really stands in the way of simulating an entire cell at biological timescales? If it is data, we are fortunate to have access to more and more complex simulations and self-driving labs which can generate high-quality data independently. If the barrier is computing power, we are lucky enough to utilize the massive increases in computing power driven by mainstream AI. If it is methods or experiments, then here is the call for action to all of us, multidisciplinary theoretical chemists of the twenty-first century: let's transform our discipline together!
Data availability
No datasets are associated with this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was undertaken thanks in part to funding provided to the University of Toronto's Acceleration Consortium from the Canada First Research Excellence Fund CFREF-2022-00042. A. A.-G. thanks Anders G. Frøseth for his generous support. A. A.-G. also acknowledges the generous support of the Canada 150 Research Chairs program. A. A. gratefully acknowledges King Abdullah University of Science and Technology (KAUST) for the KAUST Ibn Rushd Postdoctoral Fellowship.References
- C. W. Coley, N. S. Eyke and K. F. Jensen, Angew. Chem., Int. Ed., 2020, 59, 22858–22893 CrossRef CAS PubMed.
- C. W. Coley, N. S. Eyke and K. F. Jensen, Angew. Chem., Int. Ed., 2020, 59, 23414–23436 CrossRef CAS PubMed.
- H. Wang, T. Fu, Y. Du, W. Gao, K. Huang, Z. Liu, P. Chandak, S. Liu, P. Van Katwyk and A. Deac, et al., Nature, 2023, 620, 47–60 CrossRef CAS PubMed.
- A. Aldossary, J. A. Campos-Gonzalez-Angulo, S. Pablo-García, S. X. Leong, E. M. Rajaonson, L. Thiede, G. Tom, A. Wang, D. Avagliano and A. Aspuru-Guzik, Adv. Mater., 2024, 36, 2402369 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, F. Sandfort, M. H. Segler and F. Glorius, Chem. Soc. Rev., 2020, 49, 6154–6168 RSC.
- G. Tom, S. P. Schmid, S. G. Baird, Y. Cao, K. Darvish, H. Hao, S. Lo, S. Pablo-García, E. M. Rajaonson and M. Skreta, et al., Chem. Rev., 2024, 124, 9633–9732 CrossRef CAS PubMed.
- L. C. Ray and R. A. Kirsch, Science, 1957, 126, 814–819 CrossRef CAS PubMed.
- H. Kubinyi, Quant. Struct.-Act. Relat., 2002, 21, 348–356 CrossRef CAS.
- C. Hansch, P. P. Maloney, T. Fujita and R. M. Muir, Nature, 1962, 194, 178–180 CrossRef CAS.
- C. Hansch and T. Fujita, J. Am. Chem. Soc., 1964, 86, 1616–1626 CrossRef CAS.
- S. M. Free and J. W. Wilson, J. Med. Chem., 1964, 7, 395–399 CrossRef CAS PubMed.
- R. D. Cramer III, G. Redl and C. E. Berkoff, J. Med. Chem., 1974, 17, 533–535 CrossRef PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- M. Glick, J. L. Jenkins, J. H. Nettles, H. Hitchings and J. W. Davies, J. Chem. Inf. Model., 2006, 46, 193–200 CrossRef CAS PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- T. C. Bruice, N. Kharasch and R. J. Winzler, Arch. Biochem. Biophys., 1956, 62, 305–317 CrossRef CAS PubMed.
- D. Ambrose, Correlation and Estimation of Vapour-Liquid Critical Properties, National Physical Library, 1978 Search PubMed.
- Y. Nannoolal, J. Rarey, D. Ramjugernath and W. Cordes, Fluid Phase Equilib., 2004, 226, 45–63 CrossRef CAS.
- T. Gensch, G. dos Passos Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D’Addario and M. S. Sigman, et al., J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef CAS PubMed.
- C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2953–2956 CrossRef CAS.
- C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2956–2965 CrossRef CAS.
- G. Monteiro-de Castro, J. C. Duarte and I. Borges Jr, J. Org. Chem., 2023, 88, 9791–9802 CrossRef CAS PubMed.
- D. K. Duvenaud, D. Maclaurin, J. Iparraguirre, R. Bombarell, T. Hirzel, A. Aspuru-Guzik and R. P. Adams, Adv. Neural Inf. Process. Syst., 2015, 28, 2224–2232 Search PubMed.
- K. Yang, K. Swanson, W. Jin, C. Coley, P. Eiden, H. Gao, A. Guzman-Perez, T. Hopper, B. Kelley and M. Mathea, et al., J. Chem. Inf. Model., 2019, 59, 3370–3388 CrossRef CAS PubMed.
- B. K. Lee, E. J. Mayhew, B. Sanchez-Lengeling, J. N. Wei, W. W. Qian, K. A. Little, M. Andres, B. B. Nguyen, T. Moloy and J. Yasonik, et al., Science, 2023, 381, 999–1006 CrossRef CAS PubMed.
- S. Pablo-García, S. Morandi, R. A. Vargas-Hernández, K. Jorner, Ž. Ivković, N. López and A. Aspuru-Guzik, Nat. Comput. Sci., 2023, 3, 433–442 CrossRef PubMed.
- E. Heid, K. P. Greenman, Y. Chung, S.-C. Li, D. E. Graff, F. H. Vermeire, H. Wu, W. H. Green and C. J. McGill, J. Chem. Inf. Model., 2024, 64, 9–17 CrossRef CAS PubMed.
- Z. Wu, B. Ramsundar, E. N. Feinberg, J. Gomes, C. Geniesse, A. S. Pappu, K. Leswing and V. Pande, Chem. Sci., 2018, 9, 513–530 RSC.
- H. Wang, W. Li, X. Jin, K. Cho, H. Ji, J. Han and M. D. Burke, Chemical-Reaction-Aware Molecule Representation Learning, International Conference on Learning Representations, 2022, https://openreview.net/forum?id=6sh3pIzKS- Search PubMed.
- Y. Wang, J. Wang, Z. Cao and A. Barati Farimani, Nat. Mach. Intell., 2022, 4, 279–287 CrossRef.
- G. Zhou, Z. Gao, Q. Ding, H. Zheng, H. Xu, Z. Wei, L. Zhang and G. Ke, Uni-Mol: A Universal 3D Molecular Representation Learning Framework, The Eleventh International Conference on Learning Representations, 2023 Search PubMed.
- P. Walters, We Need Better Benchmarks for Machine Learning in Drug Discovery — practicalcheminformatics.blogspot.com, http://practicalcheminformatics.blogspot.com/2023/08/we-need-better-benchmarks-for-machine.html, accessed 24-08- 2024.
- A. Nigam, R. Pollice, G. Tom, K. Jorner, J. Willes, L. Thiede, A. Kundaje and A. Aspuru-Guzik, Adv. Neural Inf. Process. Syst., 2023, 36, 3263–3306 Search PubMed.
- P. G. Polishchuk, T. I. Madzhidov and A. Varnek, J. Comput.-Aided Mol. Des., 2013, 27, 675–679 CrossRef CAS PubMed.
- R. S. Bohacek, C. McMartin and W. C. Guida, Med. Res. Rev., 1996, 16, 3–50 CrossRef CAS PubMed.
- W. A. Warr, J. Chem. Inf. Comput. Sci., 1997, 37, 134–140 CrossRef CAS.
- J. Carroll, Biotechnol. Healthc., 2005, 2, 26 Search PubMed.
- W. P. Walters, M. T. Stahl and M. A. Murcko, Drug Discovery Today, 1998, 3, 160–178 CrossRef CAS.
- J. Hachmann, R. Olivares-Amaya, S. Atahan-Evrenk, C. Amador-Bedolla, R. S. Sánchez-Carrera, A. Gold-Parker, L. Vogt, A. M. Brockway and A. Aspuru-Guzik, J. Phys. Chem. Lett., 2011, 2, 2241–2251 CrossRef CAS.
- C. Gorgulla, A. Boeszoermenyi, Z.-F. Wang, P. D. Fischer, P. W. Coote, K. M. Padmanabha Das, Y. S. Malets, D. S. Radchenko, Y. S. Moroz and D. A. Scott, et al., Nature, 2020, 580, 663–668 CrossRef CAS PubMed.
- A. A. Sadybekov, A. V. Sadybekov, Y. Liu, C. Iliopoulos-Tsoutsouvas, X.-P. Huang, J. Pickett, B. Houser, N. Patel, N. K. Tran and F. Tong, et al., Nature, 2022, 601, 452–459 CrossRef CAS PubMed.
- A. V. Sadybekov and V. Katritch, Nature, 2023, 616, 673–685 CrossRef CAS PubMed.
- R. Gómez-Bombarelli, J. Aguilera-Iparraguirre, T. D. Hirzel, D. Duvenaud, D. Maclaurin, M. A. Blood-Forsythe, H. S. Chae, M. Einzinger, D.-G. Ha and T. Wu, et al., Nat. Mater., 2016, 15, 1120–1127 CrossRef PubMed.
- B. Sanchez-Lengeling and A. Aspuru-Guzik, Science, 2018, 361, 360–365 CrossRef CAS PubMed.
- A. Zunger, Nat. Rev. Chem, 2018, 2, 0121 CrossRef CAS.
- R. Gómez-Bombarelli, J. N. Wei, D. Duvenaud, J. M. Hernández-Lobato, B. Sánchez-Lengeling, D. Sheberla, J. Aguilera-Iparraguirre, T. D. Hirzel, R. P. Adams and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 268–276 CrossRef PubMed.
- M. H. Segler, T. Kogej, C. Tyrchan and M. P. Waller, ACS Cent. Sci., 2018, 4, 120–131 CrossRef CAS PubMed.
- B. Sanchez-Lengeling, C. Outeiral, G. L. Guimaraes and A. Aspuru-Guzik, Optimizing distributions over molecular space. An objective-reinforced generative adversarial network for inverse-design chemistry (ORGANIC), ChemRxiv, 2017, preprint, DOI:10.26434/chemrxiv.5309668.
- M. Olivecrona, T. Blaschke, O. Engkvist and H. Chen, J. Cheminf., 2017, 9, 48 Search PubMed.
- T. Blaschke, J. Arús-Pous, H. Chen, C. Margreitter, C. Tyrchan, O. Engkvist, K. Papadopoulos and A. Patronov, J. Chem. Inf. Model., 2020, 60, 5918–5922 CrossRef CAS PubMed.
- M. Krenn, F. Häse, A. Nigam, P. Friederich and A. Aspuru-Guzik, Mach. Learn.: Sci. Technol., 2020, 1, 045024 Search PubMed.
- A. H. Cheng, A. Cai, S. Miret, G. Malkomes, M. Phielipp and A. Aspuru-Guzik, Digital Discovery, 2023, 2, 748–758 RSC.
- J. H. Jensen, Chem. Sci., 2019, 10, 3567–3572 RSC.
- K. Korovina, S. Xu, K. Kandasamy, W. Neiswanger, B. Poczos, J. Schneider and E. Xing, International Conference on Artificial Intelligence and Statistics, 2020, pp. 3393–3403 Search PubMed.
- W. Gao, T. Fu, J. Sun and C. Coley, Adv. Neural Inf. Process. Syst., 2022, 35, 21342–21357 Search PubMed.
- N. Brown, M. Fiscato, M. H. Segler and A. C. Vaucher, J. Chem. Inf. Model., 2019, 59, 1096–1108 CrossRef CAS PubMed.
- D. Polykovskiy, A. Zhebrak, B. Sanchez-Lengeling, S. Golovanov, O. Tatanov, S. Belyaev, R. Kurbanov, A. Artamonov, V. Aladinskiy and M. Veselov, et al., Front. Pharmacol., 2020, 11, 565644 CrossRef CAS PubMed.
- W. Gao and C. W. Coley, J. Chem. Inf. Model., 2020, 60, 5714–5723 CrossRef CAS PubMed.
- J. Bradshaw, B. Paige, M. J. Kusner, M. Segler and J. M. Hernández-Lobato, Adv. Neural Inf. Process. Syst., 2019, 32, 713 Search PubMed.
- W. Gao, R. Mercado and C. W. Coley, Amortized Tree Generation for Bottom-up Synthesis Planning and Synthesizable Molecular Design, International Conference on Learning Representations, 2022, https://openreview.net/forum?id=FRxhHdnxt1 Search PubMed.
- M. Koziarski, A. Rekesh, D. Shevchuk, A. van der Sloot, P. Gaiński, Y. Bengio, C.-H. Liu, M. Tyers and R. A. Batey, arXiv, 2024, preprint, arXiv:2406.08506, DOI:10.48550/arXiv.2406.08506.
- A. Pedawi, P. Gniewek, C. Chang, B. Anderson and H. van den Bedem, Adv. Neural Inf. Process. Syst., 2022, 35, 8731–8745 Search PubMed.
- Y. Du, A. R. Jamasb, J. Guo, T. Fu, C. Harris, Y. Wang, C. Duan, P. Liò, P. Schwaller and T. L. Blundell, Nat. Mach. Intell., 2024, 6, 589–604 CrossRef.
- A. Zhavoronkov, Y. A. Ivanenkov, A. Aliper, M. S. Veselov, V. A. Aladinskiy, A. V. Aladinskaya, V. A. Terentiev, D. A. Polykovskiy, M. D. Kuznetsov and A. Asadulaev, et al., Nat. Biotechnol., 2019, 37, 1038–1040 CrossRef CAS PubMed.
- J. M. Stokes, K. Yang, K. Swanson, W. Jin, A. Cubillos-Ruiz, N. M. Donghia, C. R. MacNair, S. French, L. A. Carfrae and Z. Bloom-Ackermann, et al., Cell, 2020, 180, 688–702 CrossRef CAS PubMed.
- E. J. Corey, Angew. Chem., Int. Ed. Engl., 1991, 30, 455–465 CrossRef.
- E. J. Corey and W. T. Wipke, Science, 1969, 166, 178–192 CrossRef CAS PubMed.
- M. H. Todd, Chem. Soc. Rev., 2005, 34, 247–266 RSC.
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, ACS Cent. Sci., 2016, 2, 725–732 CrossRef CAS PubMed.
- M. H. Segler and M. P. Waller, Chem. – Eur. J., 2017, 23, 5966–5971 CrossRef CAS PubMed.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, ACS Cent. Sci., 2017, 3, 434–443 CrossRef CAS PubMed.
- C. W. Coley, W. Jin, L. Rogers, T. F. Jamison, T. S. Jaakkola, W. H. Green, R. Barzilay and K. F. Jensen, Chem. Sci., 2019, 10, 370–377 RSC.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, ACS Cent. Sci., 2019, 5, 1572–1583 CrossRef CAS PubMed.
- M. H. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- C. W. Coley, D. A. Thomas, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, Science, 2019, 365, eaax1566 CrossRef CAS PubMed.
- S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist and E. Bjerrum, J. Cheminf., 2020, 12, 70 Search PubMed.
- Y. Mo, Y. Guan, P. Verma, J. Guo, M. E. Fortunato, Z. Lu, C. W. Coley and K. F. Jensen, Chem. Sci., 2021, 12, 1469–1478 RSC.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Science, 2018, 360, 186–190 CrossRef CAS PubMed.
- J. M. Granda, L. Donina, V. Dragone, D.-L. Long and L. Cronin, Nature, 2018, 559, 377–381 CrossRef CAS PubMed.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Science, 2019, 363, eaau5631 CrossRef CAS PubMed.
- F. Sandfort, F. Strieth-Kalthoff, M. Kühnemund, C. Beecks and F. Glorius, Chem, 2020, 6, 1379–1390 CAS.
- P. Schwaller, A. C. Vaucher, T. Laino and J.-L. Reymond, Mach. Learn.: Sci. Technol., 2021, 2, 015016 Search PubMed.
- M. Christensen, L. P. Yunker, F. Adedeji, F. Häse, L. M. Roch, T. Gensch, G. dos Passos Gomes, T. Zepel, M. S. Sigman and A. Aspuru-Guzik, et al., Commun. Chem., 2021, 4, 112 CrossRef PubMed.
- W. Beker, R. Roszak, A. Wołos, N. H. Angello, V. Rathore, M. D. Burke and B. A. Grzybowski, J. Am. Chem. Soc., 2022, 144, 4819–4827 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, F. Sandfort, M. Kühnemund, F. R. Schäfer, H. Kuchen and F. Glorius, Angew. Chem., Int. Ed., 2022, 61, e202204647 CrossRef CAS PubMed.
- C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson and A. A. Lapkin, Chem. Rev., 2023, 123, 3089–3126 CrossRef CAS PubMed.
- N. H. Angello, V. Rathore, W. Beker, A. Wołos, E. R. Jira, R. Roszak, T. C. Wu, C. M. Schroeder, A. Aspuru-Guzik, B. A. Grzybowski and M. D. Burke, Science, 2022, 378, 399–405 CrossRef CAS PubMed.
- J. Y. Wang, J. M. Stevens, S. K. Kariofillis, M.-J. Tom, D. L. Golden, J. Li, J. E. Tabora, M. Parasram, B. J. Shields, D. N. Primer, B. Hao, D. Del Valle, S. DiSomma, A. Furman, G. G. Zipp, S. Melnikov, J. Paulson and A. G. Doyle, Nature, 2024, 626, 1025–1033 CrossRef CAS PubMed.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. Trice and B. A. Grzybowski, Chem, 2018, 4, 522–532 CAS.
- B. Mikulak-Klucznik, P. Gołebiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuć, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska and W. Beker, et al., Nature, 2020, 588, 83–88 CrossRef CAS PubMed.
- Y. Lin, R. Zhang, D. Wang and T. Cernak, Science, 2023, 379, 453–457 CrossRef CAS PubMed.
- A. Wołos, D. Koszelewski, R. Roszak, S. Szymkuć, M. Moskal, R. Ostaszewski, B. T. Herrera, J. M. Maier, G. Brezicki and J. Samuel, et al., Nature, 2022, 604, 668–676 CrossRef PubMed.
- B. Mikulak-Klucznik, T. Klucznik, W. Beker, M. Moskal and B. A. Grzybowski, Chem, 2024, 10, 1319–1326 CAS.
- F. Strieth-Kalthoff, S. Szymkuc, K. Molga, A. Aspuru-Guzik, F. Glorius and B. A. Grzybowski, J. Am. Chem. Soc., 2024, 146, 11005–11017 CAS.
- D. M. Lowe, PhD thesis, Apollo - University of Cambridge Repository, 2012.
- S. M. Kearnes, M. R. Maser, M. Wleklinski, A. Kast, A. G. Doyle, S. D. Dreher, J. M. Hawkins, K. F. Jensen and C. W. Coley, J. Am. Chem. Soc., 2021, 143, 18820–18826 CrossRef CAS PubMed.
- J. E. Jones, Proc. R. Soc. London, Ser. A, 1924, 106, 441–462 CAS.
- A. S. Christensen, T. Kubar, Q. Cui and M. Elstner, Chem. Rev., 2016, 116, 5301–5337 CrossRef CAS PubMed.
- M. Levitt and A. Warshel, Nature, 1975, 253, 694–698 CrossRef CAS PubMed.
- R. Iftimie, P. Minary and M. E. Tuckerman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 6654–6659 CrossRef CAS PubMed.
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef PubMed.
- K. Schütt, P.-J. Kindermans, H. E. Sauceda Felix, S. Chmiela, A. Tkatchenko and K.-R. Müller, Adv. Neural Inf. Process. Syst., 2017, 30, 992–1002 Search PubMed.
- H. Wang, L. Zhang, J. Han and E. Weinan, Comput. Phys. Commun., 2018, 228, 178–184 CrossRef CAS.
- V. G. Satorras, E. Hoogeboom and M. Welling, International Conference on Machine Learning, 2021, pp. 9323–9332 Search PubMed.
- A. Duval, S. V. Mathis, C. K. Joshi, V. Schmidt, S. Miret, F. D. Malliaros, T. Cohen, P. Lio, Y. Bengio and M. Bronstein, arXiv, 2023, preprint, arXiv:2312.07511, DOI:10.48550/arXiv.2312.07511.
- C. L. Zitnick, L. Chanussot, A. Das, S. Goyal, J. Heras-Domingo, C. Ho, W. Hu, T. Lavril, A. Palizhati, M. Riviere et al., arXiv, 2020, preprint, arXiv:2010.09435, DOI:10.48550/arXiv.2010.09435.
- I. Batatia, P. Benner, Y. Chiang, A. M. Elena, D. P. Kovács, J. Riebesell, X. R. Advincula, M. Asta, W. J. Baldwin, N. Bernstein et al., arXiv, 2023, preprint, arXiv:2401.00096, DOI:10.48550/arXiv.2401.00096.
- K. T. Schütt, M. Gastegger, A. Tkatchenko, K.-R. Müller and R. J. Maurer, Nat. Commun., 2019, 10, 5024 CrossRef PubMed.
- O. Unke, M. Bogojeski, M. Gastegger, M. Geiger, T. Smidt and K.-R. Müller, Adv. Neural Inf. Process. Syst., 2021, 34, 14434–14447 Search PubMed.
- H. Zhang, C. Liu, Z. Wang, X. Wei, S. Liu, N. Zheng, B. Shao and T.-Y. Liu, Self-Consistency Training for Density-Functional-Theory Hamiltonian Prediction, Forty-first International Conference on Machine Learning, 2024, https://openreview.net/forum?id=Vw4Yar2fmW Search PubMed.
- D. Pfau, J. S. Spencer, A. G. Matthews and W. M. C. Foulkes, Phys. Rev. Res., 2020, 2, 033429 CrossRef CAS.
- J. Hermann, Z. Schätzle and F. Noé, Nat. Chem., 2020, 12, 891–897 CrossRef CAS PubMed.
- I. von Glehn, J. S. Spencer and D. Pfau, A Self-Attention Ansatz for Ab-initio Quantum Chemistry, The Eleventh International Conference on Learning Representations, 2023 Search PubMed.
- R. Li, H. Ye, D. Jiang, X. Wen, C. Wang, Z. Li, X. Li, D. He, J. Chen and W. Ren, et al., Nat. Mach. Intell., 2024, 6(2), 209–219 CrossRef.
- K. Neklyudov, J. Nys, L. Thiede, J. Carrasquilla, Q. Liu, M. Welling and A. Makhzani, Adv. Neural Inf. Process. Syst., 2024, 36 Search PubMed , https://openreview.net/forum?id=gRwSeOm6rP.
- D. Pfau, S. Axelrod, H. Sutterud, I. von Glehn and J. S. Spencer, Science, 2024, 385, eadn0137 CrossRef CAS.
- A. Fabrizio, A. Grisafi, B. Meyer, M. Ceriotti and C. Corminboeuf, Chem. Sci., 2019, 10, 9424–9432 RSC.
- S. Gong, T. Xie, T. Zhu, S. Wang, E. R. Fadel, Y. Li and J. C. Grossman, Phys. Rev. B, 2019, 100, 184103 CrossRef CAS.
- X. Fu, A. Rosen, K. Bystrom, R. Wang, A. Musaelian, B. Kozinsky, T. Smidt and T. Jaakkola, arXiv, 2024, preprint, arXiv:2405.19276, DOI:10.48550/arXiv.2405.19276.
- J. Kirkpatrick, B. McMorrow, D. H. Turban, A. L. Gaunt, J. S. Spencer, A. G. Matthews, A. Obika, L. Thiry, M. Fortunato and D. Pfau, et al., Science, 2021, 374, 1385–1389 CrossRef CAS PubMed.
- E. Hoogeboom, V. G. Satorras, C. Vignac and M. Welling, International Conference on Machine Learning, 2022, pp. 8867–8887 Search PubMed.
- M. Xu, L. Yu, Y. Song, C. Shi, S. Ermon and J. Tang, GeoDiff: A Geometric Diffusion Model for Molecular Conformation Generation, International Conference on Learning Representations, 2022, https://openreview.net/forum?id=PzcvxEMzvQC Search PubMed.
- O. Ganea, L. Pattanaik, C. Coley, R. Barzilay, K. Jensen, W. Green and T. Jaakkola, Adv. Neural Inf. Process. Syst., 2021, 34, 13757–13769 Search PubMed.
- B. Jing, G. Corso, J. Chang, R. Barzilay and T. Jaakkola, Adv. Neural Inf. Process. Syst., 2022, 35, 24240–24253 Search PubMed.
- Y. Wang, A. A. Elhag, N. Jaitly, J. M. Susskind and M. Á. Bautista, Swallowing the Bitter Pill: Simplified Scalable Conformer Generation, Forty-first International Conference on Machine Learning, 2024 Search PubMed.
- G. Corso, H. Stärk, B. Jing, R. Barzilay and T. Jaakkola, DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking, The Eleventh International Conference on Learning Representations, 2023, https://openreview.net/forum?id=kKF8_K-mBbS Search PubMed.
- C. J. Pickard and R. Needs, J. Phys.: Condens. Matter, 2011, 23, 053201 CrossRef PubMed.
- T. Xie, X. Fu, O.-E. Ganea, R. Barzilay and T. Jaakkola, Crystal Diffusion Variational Autoencoder for Periodic Material Generation, International Conference on Learning Representations, 2022, https://openreview.net/forum?id=03RLpj-tc_ Search PubMed.
- R. Jiao, W. Huang, P. Lin, J. Han, P. Chen, Y. Lu and Y. Liu, Adv. Neural Inf. Process. Syst., 2024, 36, 17464–17497 Search PubMed.
- R. Jiao, W. Huang, Y. Liu, D. Zhao and Y. Liu, Space Group Constrained Crystal Generation, The Twelfth International Conference on Learning Representations, 2024 Search PubMed.
- C. Zeni, R. Pinsler, D. Zügner, A. Fowler, M. Horton, X. Fu, S. Shysheya, J. Crabbé, L. Sun, J. Smith et al., arXiv, 2023, preprint, arXiv:2312.03687, DOI:10.48550/arXiv.2312.03687.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, et al., Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- J. Yim, B. L. Trippe, V. De Bortoli, E. Mathieu, A. Doucet, R. Barzilay and T. Jaakkola, SE(3) diffusion model with application to protein backbone generation, Proceedings of the 40th International Conference on Machine Learning, PMLR, 2023, vol. 202, pp. 40001–40039, https://proceedings.mlr.press/v202/yim23a.html Search PubMed.
- A. J. Bose, T. Akhound-Sadegh, K. Fatras, G. Huguet, J. Rector-Brooks, C.-H. Liu, A. C. Nica, M. Korablyov, M. Bronstein and A. Tong, SE(3)-Stochastic Flow Matching for Protein Backbone Generation, The Twelfth International Conference on Learning Representations, 2024, https://openreview.net/forum?id=kJFIH23hXb Search PubMed.
- J. L. Watson, D. Juergens, N. R. Bennett, B. L. Trippe, J. Yim, H. E. Eisenach, W. Ahern, A. J. Borst, R. J. Ragotte and L. F. Milles, et al., Nature, 2023, 620, 1089–1100 CrossRef CAS PubMed.
- J. B. Ingraham, M. Baranov, Z. Costello, K. W. Barber, W. Wang, A. Ismail, V. Frappier, D. M. Lord, C. Ng-Thow-Hing and E. R. Van Vlack, et al., Nature, 2023, 623, 1070–1078 CrossRef CAS PubMed.
- R. Krishna, J. Wang, W. Ahern, P. Sturmfels, P. Venkatesh, I. Kalvet, G. R. Lee, F. S. Morey-Burrows, I. Anishchenko and I. R. Humphreys, et al., Science, 2024, 384, eadl2528 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard and J. Bambrick, et al., Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- F. Noé, S. Olsson, J. Köhler and H. Wu, Science, 2019, 365, eaaw1147 CrossRef PubMed.
- L. Klein, A. Krämer and F. Noé, Adv. Neural Inf. Process. Syst., 2024, 36, 59886–59910 Search PubMed.
- L. Klein and F. Noé, arXiv, 2024, preprint, arXiv:2406.14426, DOI:10.48550/arXiv.2406.14426.
- S. Zheng, J. He, C. Liu, Y. Shi, Z. Lu, W. Feng, F. Ju, J. Wang, J. Zhu and Y. Min, et al., Nat. Mach. Intell., 2024, 6, 558–567 CrossRef.
- J. Wang, S. Olsson, C. Wehmeyer, A. Pérez, N. E. Charron, G. de Fabritiis, F. Noé and C. Clementi, ACS Cent. Sci., 2019, 5, 755–767 CrossRef CAS PubMed.
- B. E. Husic, N. E. Charron, D. Lemm, J. Wang, A. Pérez, M. Majewski, A. Krämer, Y. Chen, S. Olsson, G. de Fabritiis, F. Noé and C. Clementi, J. Chem. Phys., 2020, 153, 194101 CrossRef CAS PubMed.
- N. E. Charron, F. Musil, A. Guljas, Y. Chen, K. Bonneau, A. S. Pasos-Trejo, J. Venturin, D. Gusew, I. Zaporozhets, A. Krämer, C. Templeton, A. Kelkar, A. E. P. Durumeric, S. Olsson, A. Pérez, M. Majewski, B. E. Husic, A. Patel, G. D. Fabritiis, F. Noé and C. Clementi, Navigating protein landscapes with a machine-learned transferable coarse-grained model, arXiv, 2023, preprint, arXiv:2310.18278, DOI:10.48550/arXiv.2310.18278.
- J. Köhler, Y. Chen, A. Krämer, C. Clementi and F. Noé, J. Chem. Theory Comput., 2023, 19, 942–952 CrossRef PubMed.
- M. Arts, V. Garcia Satorras, C.-W. Huang, D. Zugner, M. Federici, C. Clementi, F. Noé, R. Pinsler and R. van den Berg, J. Chem. Theory Comput., 2023, 19, 6151–6159 CrossRef CAS PubMed.
- X. Fu, T. Xie, N. J. Rebello, B. Olsen and T. S. Jaakkola, Simulate Time-integrated Coarse-grained Molecular Dynamics with Multi-scale Graph Networks, Transactions on Machine Learning Research, 2023 Search PubMed.
- L. Klein, A. Foong, T. Fjelde, B. Mlodozeniec, M. Brockschmidt, S. Nowozin, F. Noé and R. Tomioka, Adv. Neural Inf. Process. Syst., 2024, 36, 52863–52883 Search PubMed.
- B. R. Duschatko, X. Fu, C. Owen, Y. Xie, A. Musaelian, T. Jaakkola and B. Kozinsky, Thermodynamically Informed Multimodal Learning of High-Dimensional Free Energy Models in Molecular Coarse Graining, arXiv, 2024, preprint, arXiv:2310.18278, DOI:10.48550/arXiv.2405.19386.
- M. Sipka, J. C. Dietschreit, L. Grajciar and R. Gómez-Bombarelli, International Conference on Machine Learning, 2023, pp. 31990–32007 Search PubMed.
- L. Holdijk, Y. Du, F. Hooft, P. Jaini, B. Ensing and M. Welling, Adv. Neural Inf. Process. Syst., 2024, 36, 79540–79556 Search PubMed.
- C. Duan, Y. Du, H. Jia and H. J. Kulik, Nat. Comput. Sci., 2023, 3, 1045–1055 CrossRef PubMed.
- C. Duan, G.-H. Liu, Y. Du, T. Chen, Q. Zhao, H. Jia, C. P. Gomes, E. A. Theodorou and H. J. Kulik, arXiv, 2024, preprint, arXiv:2404.13430, DOI:10.48550/arXiv.2404.13430.
- X. Fu, Z. Wu, W. Wang, T. Xie, S. Keten, R. Gomez-Bombarelli and T. Jaakkola, Forces are not Enough: Benchmark and Critical Evaluation for Machine Learning Force Fields with Molecular Simulations, Trans. Mach. Learn. Res., 2023 Search PubMed , https://openreview.net/forum?id=A8pqQipwkt.
- A. Young, H. Röst and B. Wang, Nat. Mach. Intell., 2024, 6, 404–416 CrossRef.
- A. Young, F. Wang, D. Wishart, B. Wang, H. Röst and R. Greiner, arXiv, 2024, preprint, arXiv:2404.02360, DOI:10.48550/arXiv.2404.02360.
- F. M. Paruzzo, A. Hofstetter, F. Musil, S. De, M. Ceriotti and L. Emsley, Nat. Commun., 2018, 9, 4501 CrossRef PubMed.
- M. Cordova, E. A. Engel, A. Stefaniuk, F. Paruzzo, A. Hofstetter, M. Ceriotti and L. Emsley, J. Phys. Chem. C, 2022, 126, 16710–16720 CrossRef CAS PubMed.
- M. Lupo Pasini, K. Mehta, P. Yoo and S. Irle, Sci. Data, 2023, 10, 546 CrossRef CAS PubMed.
- S. Goldman, J. Bradshaw, J. Xin and C. W. Coley, Prefix-Tree Decoding for Predicting Mass Spectra from Molecules, Advances in Neural Information Processing Systems, ed. A. Oh, T. Naumann, A. Globerson, K. Saenko, M. Hardt and S. Levine, Curran Associates, Inc., 2023, vol. 36, pp. 48548–48572 Search PubMed.
- M. Murphy, S. Jegelka, E. Fraenkel, T. Kind, D. Healey and T. Butler, Efficiently predicting high resolution mass spectra with graph neural networks, International Conference on Machine Learning, PMLR, 2023, pp. 25549–25562 Search PubMed.
- S. Goldman, J. Li and C. W. Coley, Generating Molecular Fragmentation Graphs with Autoregressive Neural Networks, Anal. Chem., 2024, 96(8), 3419–3428 CrossRef CAS PubMed.
- R. L. Zhu and E. Jonas, Anal. Chem., 2023, 95, 2653–2663 CrossRef CAS PubMed.
- Y. Hong, S. Li, C. J. Welch, S. Tichy, Y. Ye and H. Tang, Bioinformatics, 2023, 39, btad354 CrossRef CAS PubMed.
- S. A. Al and A.-R. Allouche, Neural Network Approach for Predicting Infrared Spectra from 3D Molecular Structure, arXiv, 2024, preprint, arXiv:2405.05737, 2024, DOI:10.48550/arXiv.2405.05737.
- Z. Zou, Y. Zhang, L. Liang, M. Wei, J. Leng, J. Jiang, Y. Luo and W. Hu, Nat. Comput. Sci., 2023, 3, 957–964 CrossRef CAS PubMed.
- B. Buchanan, G. Sutherland and E. A. Feigenbaum, Org. Chem., 1969, 30, 209–254 Search PubMed.
- R. K. Lindsay, B. G. Buchanan, E. A. Feigenbaum and J. Lederberg, Artif. Intell., 1993, 61, 209–261 CrossRef.
- C. Klawun and C. L. Wilkins, J. Chem. Inf. Comput. Sci., 1996, 36, 69–81 CrossRef CAS.
- B. Curry and D. E. Rumelhart, Tetrahedron Comput. Methodol., 1990, 3, 213–237 CrossRef CAS.
- C. L. Wilkins and T. L. Isenhour, Anal. Chem., 1975, 47, 1849–1851 CrossRef CAS.
- C. Li, Y. Cong and W. Deng, Magn. Reson. Chem., 2022, 60, 1061–1069 CrossRef CAS PubMed.
- T. Specht, J. Arweiler, J. Stüber, K. Münnemann, H. Hasse and F. Jirasek, Magn. Reson. Chem., 2024, 62, 286–297 CrossRef CAS PubMed.
- Z. Huang, M. S. Chen, C. P. Woroch, T. E. Markland and M. W. Kanan, Chem. Sci., 2021, 12, 15329–15338 RSC.
- B. Sridharan, S. Mehta, Y. Pathak and U. D. Priyakumar, J. Phys. Chem. Lett., 2022, 13, 4924–4933 CrossRef CAS PubMed.
- S. Devata, B. Sridharan, S. Mehta, Y. Pathak, S. Laghuvarapu, G. Varma and U. D. Priyakumar, Digital Discovery, 2024, 3, 818–829 RSC.
- A. A. Enders, N. M. North, C. M. Fensore, J. Velez-Alvarez and H. C. Allen, Anal. Chem., 2021, 93, 9711–9718 CrossRef CAS PubMed.
- G. Jung, S. G. Jung and J. M. Cole, Chem. Sci., 2023, 14, 3600–3609 RSC.
- E. X. Tan, S. X. Leong, W. A. Liew, I. Y. Phang, J. Y. Ng, N. S. Tan, Y. H. Lee and X. Y. Ling, Nat. Commun., 2024, 15, 2582 CrossRef CAS PubMed.
- M. Alberts, F. Zipoli and A. C. Vaucher, Learning the Language of NMR: Structure Elucidation from NMR spectra using Transformer Models, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-8wxcz.
- M. Alberts, T. Laino and A. C. Vaucher, Leveraging Infrared Spectroscopy for Automated Structure Elucidation, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-5v27f.
- F. Hu, M. S. Chen, G. M. Rotskoff, M. W. Kanan and T. E. Markland, arXiv, 2024, preprint, arXiv:2408.08284, DOI:10.48550/arXiv.2408.08284.
- M. A. Stravs, K. Dührkop, S. Böcker and N. Zamboni, Nat. Methods, 2022, 19, 865–870 CrossRef CAS PubMed.
- E. E. Litsa, V. Chenthamarakshan, P. Das and L. E. Kavraki, Commun. Chem., 2023, 6, 132 CrossRef PubMed.
- Q. Lai, L. Yao, Z. Gao, S. Liu, H. Wang, S. Lu, D. He, L. Wang, C. Wang and G. Ke, arXiv, 2024, preprint, arXiv:2401.03862, DOI:10.48550/arXiv.2401.03862.
- J. E. Salgado, S. Lerman, Z. Du, C. Xu and N. Abdolrahim, npj Comput. Mater., 2023, 9, 214 CrossRef.
- Y. Song, L. Shen, L. Xing and S. Ermon, Solving Inverse Problems in Medical Imaging with Score-Based Generative Models, International Conference on Learning Representations, 2022 Search PubMed.
- H. Chung, J. Kim, M. T. Mccann, M. L. Klasky and J. C. Ye, Diffusion Posterior Sampling for General Noisy Inverse Problems, The Eleventh International Conference on Learning Representations, 2023 Search PubMed.
- D. P. Zaleski and K. Prozument, J. Chem. Phys., 2018, 149, 104106 CrossRef PubMed.
- A. H. Cheng, A. Lo, S. Miret, B. H. Pate and A. Aspuru-Guzik, J. Chem. Phys., 2024, 160, 124115 CrossRef CAS PubMed.
- E. D. Zhong, T. Bepler, B. Berger and J. H. Davis, Nat. Methods, 2021, 18, 176–185 CrossRef CAS PubMed.
- A. Levy, G. Wetzstein, J. N. Martel, F. Poitevin and E. Zhong, Adv. Neural Inf. Process. Syst., 2022, 35, 13038–13049 Search PubMed.
- R. Rangan, R. Feathers, S. Khavnekar, A. Lerer, J. D. Johnston, R. Kelley, M. Obr, A. Kotecha and E. D. Zhong, Nat. Methods, 2024, 21, 1537–1545 CrossRef CAS PubMed.
- M. T. Clabbers, J. Hattne, M. W. Martynowycz and T. Gonen, Energy filtering enables macromolecular MicroED data at sub-atomic resolution, bioRxiv, 2024, preprint, DOI:10.1101/2024.08.29.610380.
- S. Goldman, J. Wohlwend, M. Stražar, G. Haroush, R. J. Xavier and C. W. Coley, Nat. Mach. Intell., 2023, 5, 965–979 CrossRef.
- S. F. Baygi and D. K. Barupal, J. Cheminf., 2024, 16, 8 Search PubMed.
- D. Weininger, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- J. Ross, B. Belgodere, V. Chenthamarakshan, I. Padhi, Y. Mroueh and P. Das, Nat. Mach. Intell., 2022, 4, 1256–1264 CrossRef.
- S. Chithrananda, G. Grand and B. Ramsundar, arXiv, 2020, preprint, arXiv:2010.09885, DOI:10.48550/arXiv.2010.09885.
- S. Liu, W. Nie, C. Wang, J. Lu, Z. Qiao, L. Liu, J. Tang, C. Xiao and A. Anandkumar, Nat. Mach. Intell., 2023, 5, 1447–1457 CrossRef.
- Q. Pei, W. Zhang, J. Zhu, K. Wu, K. Gao, L. Wu, Y. Xia and R. Yan, BioT5: Enriching Cross-modal Integration in Biology with Chemical Knowledge and Natural Language Associations, The 2023 Conference on Empirical Methods in Natural Language Processing, 2023, https://openreview.net/forum?id=uhVJ3SLq80 Search PubMed.
- D. Christofidellis, G. Giannone, J. Born, O. Winther, T. Laino and M. Manica, International Conference on Machine Learning, 2023, pp. 6140–6157 Search PubMed.
- R. Taylor, M. Kardas, G. Cucurull, T. Scialom, A. Hartshorn, E. Saravia, A. Poulton, V. Kerkez and R. Stojnic, arXiv, 2022, preprint, arXiv:2211.09085, DOI:10.48550/arXiv.2211.09085.
- C. Edwards, T. Lai, K. Ros, G. Honke, K. Cho and H. Ji, Proceedings of the 2022 Conference on Empirical Methods in Natural Language Processing, Abu Dhabi, United Arab Emirates, 2022, pp. 375–413 Search PubMed.
- M. C. Ramos, C. J. Collison and A. D. White, arXiv, 2024, preprint, arXiv:2407.01603, DOI:10.48550/arXiv.2407.01603.
- Y. Kang and J. Kim, Nat. Commun., 2024, 15, 4705 CrossRef CAS PubMed.
- N. Yoshikawa, M. Skreta, K. Darvish, S. Arellano-Rubach, Z. Ji, L. Bjørn Kristensen, A. Z. Li, Y. Zhao, H. Xu and A. Kuramshin, et al., Auton. Robots, 2023, 47, 1057–1086 CrossRef.
- J. Choi and B. Lee, Commun. Mater., 2024, 5, 13 CrossRef.
- T. Gupta, M. Zaki, N. A. Krishnan and Mausam, npj Comput. Mater., 2022, 8, 102 CrossRef.
- J. Dagdelen, A. Dunn, S. Lee, N. Walker, A. S. Rosen, G. Ceder, K. A. Persson and A. Jain, Nat. Commun., 2024, 15, 1418 CrossRef CAS PubMed.
- M. J. Buehler, ACS Eng. Au, 2024, 4, 241–277 CrossRef CAS PubMed.
- A. M. Bran, S. Cox, O. Schilter, C. Baldassari, A. D. White and P. Schwaller, Nat. Mach. Intell., 2024, 6, 525–535 CrossRef PubMed.
- Microsoft Research AI4Science and Microsoft Azure Quantum, arXiv, 2023, preprint, arXiv:2311.07361, DOI:10.48550/arXiv.2311.07361.
- J. Achiam, S. Adler, S. Agarwal, L. Ahmad, I. Akkaya, F. L. Aleman, D. Almeida, J. Altenschmidt, S. Altman, S. Anadkat et al., arXiv, 2023, preprint, arXiv:2303.08774, DOI:10.48550/arXiv.2303.08774.
- K. M. Jablonka, P. Schwaller, A. Ortega-Guerrero and B. Smit, Nat. Mach. Intell., 2024, 6, 161–169 CrossRef.
- H. Wang, M. Skreta, C.-T. Ser, W. Gao, L. Kong, F. Streith-Kalthoff, C. Duan, Y. Zhuang, Y. Yu, Y. Zhu et al., arXiv, 2024, preprint, arXiv:2406.16976, DOI:10.48550/arXiv.2406.16976.
- A. Kristiadi, F. Strieth-Kalthoff, M. Skreta, P. Poupart, A. Aspuru-Guzik and G. Pleiss, A Sober Look at LLMs for Material Discovery: Are They Actually Good for Bayesian Optimization Over Molecules?, Proceedings of the 41st International Conference on Machine Learning, ed. R. Salakhutdinov, Z. Kolter, K. Heller, A. Weller, N. Oliver, J. Scarlett and F. Berkenkamp, PMLR, 2024, vol. 235, pp. 25603–25622, https://proceedings.mlr.press/v235/kristiadi24a.html Search PubMed.
- M. C. Ramos, S. S. Michtavy, M. D. Porosoff and A. D. White, arXiv, 2023, preprint, arXiv:2304.05341, DOI:10.48550/arXiv.2304.05341.
- P. Ma, T.-H. Wang, M. Guo, Z. Sun, J. B. Tenenbaum, D. Rus, C. Gan and W. Matusik, LLM and Simulation as Bilevel Optimizers: A New Paradigm to Advance Physical Scientific Discovery, Proceedings of the 41st International Conference on Machine Learning, ed. R. Salakhutdinov, Z. Kolter, K. Heller, A. Weller, N. Oliver, J. Scarlett and F. Berkenkamp, PMLR, 2024, pp. 33940–33962, https://proceedings.mlr.press/v235/ma24m.html Search PubMed.
- N. C. Frey, R. Soklaski, S. Axelrod, S. Samsi, R. Gomez-Bombarelli, C. W. Coley and V. Gadepally, Nat. Mach. Intell., 2023, 5, 1297–1305 CrossRef.
- J. Ross, B. Belgodere, S. C. Hoffman, V. Chenthamarakshan, Y. Mroueh and P. Das, arXiv, 2024, preprint, arXiv:2405.04912, DOI:10.48550/arXiv.2405.04912.
- T. Schick, J. Dwivedi-Yu, R. Dessì, R. Raileanu, M. Lomeli, L. Zettlemoyer, N. Cancedda and T. Scialom, Advances in Neural Information Processing Systems, Toolformer: Language Models Can Teach Themselves to Use Tools, ed. A. Oh, T. Naumann, A. Globerson, K. Saenko, M. Hardt and S. Levine, Curran Associates, Inc., 2023, vol. 36, pp. 68539–68551, https://proceedings.neurips.cc/paper_files/paper/2023/file/d842425e4bf79ba039352da0f658a906-Paper-Conference.pdf Search PubMed.
- A. M. Bran, S. Cox, O. Schilter, C. Baldassari, A. D. White and P. Schwaller, Nat. Mach. Intell., 2024, 6, 525–535 CrossRef PubMed.
- D. A. Boiko, R. MacKnight, B. Kline and G. Gomes, Nature, 2023, 624, 570–578 CrossRef CAS PubMed.
- K. Darvish, M. Skreta, Y. Zhao, N. Yoshikawa, S. Som, M. Bogdanovic, Y. Cao, H. Hao, H. Xu, A. Aspuru-Guzik et al., arXiv, 2024, preprint, arXiv:2401.06949, DOI:10.48550/arXiv.2401.06949.
- H. W. Sprueill, C. Edwards, K. Agarwal, M. V. Olarte, U. Sanyal, C. Johnston and H. Liu, H. Ji and S. Choudhury, CHEMREASONER: Heuristic Search over a Large Language Model’s Knowledge Space using Quantum-Chemical Feedback, Proceedings of the 41st International Conference on Machine Learning, ed. R. Salakhutdinov, Z. Kolter, K. Heller, A. Weller, N. Oliver, J. Scarlett and F. Berkenkamp, PMLR, 2024, vol. 235, pp. 46351–46374, https://proceedings.mlr.press/v235/sprueill24a.html Search PubMed.
- C. Lu, C. Lu, R. T. Lange, J. Foerster, J. Clune and D. Ha, The AI Scientist: Towards Fully Automated Open-Ended Scientific Discovery, arXiv, 2024, preprint, arXiv:2408.06292, DOI:10.48550/arXiv.2408.06292.
- K. Huang, T. Fu, W. Gao, Y. Zhao, Y. Roohani, J. Leskovec, C. W. Coley, C. Xiao, J. Sun and M. Zitnik, Nat. Chem. Biol., 2022, 18, 1033–1036 CrossRef CAS PubMed.
- A. Mirza, N. Alampara, S. Kunchapu, B. Emoekabu, A. Krishnan, M. Wilhelmi, M. Okereke, J. Eberhardt, A. M. Elahi, M. Greiner et al., arXiv, 2024, preprint, arXiv:2404.01475, DOI:10.48550/arXiv.2404.01475.
- J. M. Laurent, J. D. Janizek, M. Ruzo, M. M. Hinks, M. J. Hammerling, S. Narayanan, M. Ponnapati, A. D. White and S. G. Rodriques, arXiv, 2024, preprint, arXiv:2407.10362, DOI:10.48550/arXiv.2407.10362.
- Polaris — polarishub.io, https://polarishub.io/, 2024, accessed 02-09-2024.
- V. Venugopal and E. Olivetti, Sci. Data, 2024, 11, 217 CrossRef PubMed.
- A. M. Bran, Z. Jončev and P. Schwaller, Proceedings of the 1st Workshop on Language+ Molecules (L+ M 2024), 2024, pp. 74–84 Search PubMed.
- Q. Ai, F. Meng, J. Shi, B. G. Pelkie and C. W. Coley, Digital Discovery, 2024, 3, 1822–1831 RSC.
- Z. Zheng, O. Zhang, C. Borgs, J. T. Chayes and O. M. Yaghi, J. Am. Chem. Soc., 2023, 145, 18048–18062 CrossRef PubMed.
- M. Schilling-Wilhelmi, M. Ríos-García, S. Shabih, M. V. Gil, S. Miret, C. T. Koch, J. A. Márquez and K. M. Jablonka, arXiv, 2024, preprint, arXiv:2407.16867, DOI:10.48550/arXiv.2407.16867.
- S. X. Leong, S. Pablo-García, Z. Zhang and A. Aspuru-Guzik, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-7fwxv.
- N. Alampara, S. Miret and K. M. Jablonka, arXiv, 2024, preprint, arXiv:2406.17295, DOI:10.48550/arXiv.2406.17295.
- T. T. Duignan, ACS Phys. Chem. Au, 2024, 4, 232–241 CrossRef CAS PubMed.
- Y.-L. Liao, B. M. Wood, A. Das and T. Smidt, EquiformerV2: Improved Equivariant Transformer for Scaling to Higher-Degree Representations, The Twelfth International Conference on Learning Representations, 2024 Search PubMed.
- A. Merchant, S. Batzner, S. S. Schoenholz, M. Aykol, G. Cheon and E. D. Cubuk, Nature, 2023, 624, 80–85 CrossRef CAS PubMed.
- H. Yang, C. Hu, Y. Zhou, X. Liu, Y. Shi, J. Li, G. Li, Z. Chen, S. Chen, C. Zeni et al., arXiv, 2024, preprint, arXiv:2405.04967, DOI:10.48550/arXiv.2405.04967.
- M. Van Kempen, S. S. Kim, C. Tumescheit, M. Mirdita, J. Lee, C. L. Gilchrist, J. Söding and M. Steinegger, Nat. Biotechnol., 2024, 42, 243–246 CrossRef CAS PubMed.
- I. Batatia, D. P. Kovacs, G. Simm, C. Ortner and G. Csányi, Adv. Neural Inf. Process. Syst., 2022, 35, 11423–11436 Search PubMed.
- E. Weingart and A. Schukar, The New York Times, https://www.nytimes.com/2023/01/06/us/widen-highways-traffic.html, 2023 Search PubMed.
- S. Raghunathan and U. D. Priyakumar, Int. J. Quantum Chem., 2022, 122, e26870 CrossRef CAS.
- D. S. Wigh, J. M. Goodman and A. A. Lapkin, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1603 CrossRef.
- M. Meuwly, Chem. Rev., 2021, 121, 10218–10239 CrossRef CAS PubMed.
- Y. R. Wang, Y. Zhao, H. Xu, S. Eppel, A. Aspuru-Guzik, F. Shkurti and A. Garg, 2023 IEEE International Conference on Robotics and Automation (ICRA), 2023, pp. 3771–3778 Search PubMed.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, Nature, 2020, 583, 237–241 CrossRef CAS PubMed.
- Y. Nakajima, M. Hamaya, Y. Suzuki, T. Hawai, F. v. Drigalski, K. Tanaka, Y. Ushiku and K. Ono, 2022 IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS), 2022, pp. 2320–2326 Search PubMed.
- M. Kennedy, K. Schmeckpeper, D. Thakur, C. Jiang, V. Kumar and K. Daniilidis, IEEE Robot. Autom. Lett., 2019, 4, 2317–2324 Search PubMed.
- Y. Huang, J. Wilches and Y. Sun, Robotics and Autonomous Systems, 2021, 136, 103692 CrossRef.
- A. Klami, T. Damoulas, O. Engkvist, P. Rinke and S. Kaski, TechRxiv, 2022, preprint, DOI:10.36227/techrxiv.20412540.v1.
- C. Beeler, S. G. Subramanian, K. Sprague, N. Chatti, C. Bellinger, M. Shahen, N. Paquin, M. Baula, A. Dawit, Z. Yang, X. Li, M. Crowley and I. Tamblyn, ChemGymRL: An Interactive Framework for Reinforcement Learning for Digital Chemistry, arXiv, 2023, preprint, arXiv:2305.14177, DOI:10.48550/arXiv.2305.14177.
- M. A. Bezerra, Q. O. dos Santos, A. G. Santos, C. G. Novaes, S. L. C. Ferreira and V. S. de Souza, Microchem. J., 2016, 124, 45–54 CrossRef CAS.
- W. Huyer and A. Neumaier, ACM Trans. Math. Softw., 2008, 35, 9 CrossRef.
- A. Lucia and J. Xu, Comput. Chem. Eng., 1990, 14, 119–138 CrossRef CAS.
- W. P. Walters and R. Barzilay, Acc. Chem. Res., 2021, 54, 263–270 CrossRef CAS PubMed.
- C. J. Taylor, A. Pomberger, K. C. Felton, R. Grainger, M. Barecka, T. W. Chamberlain, R. A. Bourne, C. N. Johnson and A. A. Lapkin, Chem. Rev., 2023, 123, 3089–3126 CrossRef CAS PubMed.
- Z. Zhou, X. Li and R. N. Zare, ACS Cent. Sci., 2017, 3, 1337–1344 CrossRef CAS PubMed.
- G. Jastrebski and D. Arnold, 2006 IEEE International Conference on Evolutionary Computation, 2006, pp. 2814–2821 Search PubMed.
- F. Häse, L. M. Roch, C. Kreisbeck and A. Aspuru-Guzik, ACS Cent. Sci., 2018, 4, 1134–1145 CrossRef PubMed.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch and A. Aspuru-Guzik, Appl. Phys. Rev., 2021, 8, 031406 Search PubMed.
- R. Hickman, M. Sim, S. Pablo-García, I. Woolhouse, H. Hao, Z. Bao, P. Bannigan, C. Allen, M. Aldeghi and A. Aspuru-Guzik, Atlas: A Brain for Self-driving Laboratories, ChemRxiv, 2023, preprint, DOI:10.26434/chemrxiv-2023-8nrxx.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Nature, 2021, 590, 89–96 CrossRef CAS PubMed.
- N. S. Eyke, B. A. Koscher and K. F. Jensen, Trends Chem., 2021, 3, 120–132 CrossRef CAS.
- J. C. Oliveira, J. Frey, S.-Q. Zhang, L.-C. Xu, X. Li, S.-W. Li, X. Hong and L. Ackermann, Trends Chem., 2022, 4, 863–885 CrossRef CAS.
- S. Dara, S. Dhamercherla, S. S. Jadav, C. M. Babu and M. J. Ahsan, Artif. Intell. Rev., 2022, 55, 1947–1999 CrossRef PubMed.
- F. Strieth-Kalthoff, H. Hao, V. Rathore, J. Derasp, T. Gaudin, N. H. Angello, M. Seifrid, E. Trushina, M. Guy and J. Liu, et al., Science, 2024, 384, eadk9227 CrossRef CAS PubMed.
- M. Skreta, N. Yoshikawa, S. Arellano-Rubach, Z. Ji, L. B. Kristensen, K. Darvish, A. Aspuru-Guzik, F. Shkurti and A. Garg, Errors are Useful Prompts: Instruction Guided Task Programming with Verifier-Assisted Iterative Prompting, arXiv, 2023, preprint, arXiv:2303.14100, DOI:10.48550/arXiv.2303.14100.
- N. Yoshikawa, M. Skreta, K. Darvish, S. Arellano-Rubach, Z. Ji, L. Bjørn Kristensen, A. Z. Li, Y. Zhao, H. Xu, A. Kuramshin, A. Aspuru-Guzik, F. Shkurti and A. Garg, Auton. Robots, 2023, 47, 1057–1086 CrossRef.
- M. Seifrid, R. Pollice, A. Aguilar-Granda, Z. Morgan Chan, K. Hotta, C. T. Ser, J. Vestfrid, T. C. Wu and A. Aspuru-Guzik, Acc. Chem. Res., 2022, 55, 2454–2466 CrossRef CAS PubMed.
- D. Knobbe, H. Zwirnmann, M. Eckhoff and S. Haddadin, 2022 IEEE/RSJ International Conference on Intelligent Robots and Systems (IROS), 2022, pp. 2335–2342 Search PubMed.
- N. Yoshikawa, G. D. Akkoc, S. Pablo-García, Y. Cao, H. Hao and A. Aspuru-Guzik, Does one need to polish electrodes in an eight pattern? Automation provides the answer, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-ttxnr.
- Y. Jiang, H. Fakhruldeen, G. Pizzuto, L. Longley, A. He, T. Dai, R. Clowes, N. Rankin and A. I. Cooper, Digital Discovery, 2023, 2, 1733–1744 RSC.
- H. Xu, Y. R. Wang, S. Eppel, A. Aspuru-Guzik, F. Shkurti and A. Garg, Seeing Glass: Joint Point-Cloud and Depth Completion for Transparent Objects, Proceedings of the 5th Conference on Robot Learning, ed. A. Faust, D. Hsu and G. Neumann, PMLR, 2022, vol. 164, pp. 827–838, https://proceedings.mlr.press/v164/xu22b.html Search PubMed.
- D. M. Anstine and O. Isayev, J. Am. Chem. Soc., 2023, 145, 8736–8750 CrossRef CAS PubMed.
- S. Lo, S. G. Baird, J. Schrier, B. Blaiszik, N. Carson, I. Foster, A. Aguilar-Granda, S. V. Kalinin, B. Maruyama and M. Politi, et al., Digital Discovery, 2024, 3, 842–868 RSC.
- J. P. Janet and H. J. Kulik, Machine Learning in Chemistry, American Chemical Society, 2020 Search PubMed.
- J. A. Keith, V. Vassilev-Galindo, B. Cheng, S. Chmiela, M. Gastegger, K.-R. Muller and A. Tkatchenko, Chem. Rev., 2021, 121, 9816–9872 CrossRef CAS PubMed.
- N. Artrith, K. T. Butler, F.-X. Coudert, S. Han, O. Isayev, A. Jain and A. Walsh, Nat. Chem., 2021, 13, 505–508 CrossRef CAS PubMed.
- A. D. White, Comp. Mol. Sci., 2022, 3, 1499 Search PubMed.
- T. Chen and C. Guestrin, Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2016, pp. 785–794 Search PubMed.
- G. Tom, R. J. Hickman, A. Zinzuwadia, A. Mohajeri, B. Sanchez-Lengeling and A. Aspuru-Guzik, Digital Discovery, 2023, 2, 759–774 RSC.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, L. Kaiser, and I. Polosukhin, Attention is all you need, Proceedings of the 31st International Conference on Neural Information Processing Systems, Curran Associates Inc., Red Hook, NY, USA, 2017, pp. 6000–6010 Search PubMed.
- T. Lin, Y. Wang, X. Liu and X. Qiu, AI Open, 2022, 3, 111–132 CrossRef.
- A. Dosovitskiy, L. Beyer, A. Kolesnikov, D. Weissenborn, X. Zhai, T. Unterthiner, M. Dehghani, M. Minderer, G. Heigold, S. Gelly, J. Uszkoreit and N. Houlsby, An Image is Worth 16x16 Words: Transformers for Image Recognition at Scale, International Conference on Learning Representations, 2021, https://openreview.net/forum?id=YicbFdNTTy Search PubMed.
- C. Ying, T. Cai, S. Luo, S. Zheng, G. Ke, D. He, Y. Shen and T.-Y. Liu, Adv. Neural Inf. Process. Syst., 2021, 34, 28877–28888 Search PubMed.
- D. P. Kingma, arXiv, 2013, preprint, arXiv:1312.6114, DOI:10.48550/arXiv.1312.6114.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, A. Courville and Y. Bengio, Adv. Neural Inf. Process. Syst., 2014, 27, 139–144 Search PubMed.
- D. Rezende and S. Mohamed, International Conference on Machine Learning, 2015, pp. 1530–1538 Search PubMed.
- T. B. Brown, arXiv, 2020, preprint, arXiv:2005.14165, DOI:10.48550/arXiv.2005.14165.
- A. Ramesh, P. Dhariwal, A. Nichol, C. Chu and M. Chen, arXiv, 2022, preprint, arXiv:2204.06125, DOI:10.48550/arXiv.2204.06125.
- N. Metropolis, A. W. Rosenbluth, M. N. Rosenbluth, A. H. Teller and E. Teller, J. Chem. Phys., 1953, 21, 1087–1092 CrossRef CAS.
- G. Parisi, Nucl. Phys. B, 1981, 180, 378–384 CrossRef.
- W. Chen, M. Zhang, B. Paige, J. M. Hernández-Lobato and D. Barber, Diffusive Gibbs Sampling, Proceedings of the 41st International Conference on Machine Learning, ed. R. Salakhutdinov, Z. Kolter, K. Heller, A. Weller, N. Oliver, J. Scarlett and F. Berkenkamp, PMLR, 2024, vol. 235, pp. 7731–7747 Search PubMed.
- S. Zhao, R. Brekelmans, A. Makhzani and R. Grosse, Probabilistic Inference in Language Models via Twisted Sequential Monte Carlo, Proceedings of the 41st International Conference on Machine Learning, ed. R. Salakhutdinov, Z. Kolter, K. Heller, A. Weller, N. Oliver, J. Scarlett and F. Berkenkamp, PMLR, 2024, vol. 235, pp. 60704–60748 Search PubMed.
- S. Sanokowski, S. Hochreiter and S. Lehner, A Diffusion Model Framework for Unsupervised Neural Combinatorial Optimization, Proceedings of the 41st International Conference on Machine Learning, ed. R. Salakhutdinov, Z. Kolter, K. Heller, A. Weller, N. Oliver, J. Scarlett and F. Berkenkamp, PMLR, 2024, vol. 235, pp. 43346–43367, https://proceedings.mlr.press/v235/sanokowski24a.html Search PubMed.
- G. M. Rotskoff, Curr. Opin. Solid State Mater. Sci., 2024, 30, 101158 CrossRef CAS.
- E. Bengio, M. Jain, M. Korablyov, D. Precup and Y. Bengio, Adv. Neural Inf. Process. Syst., 2021, 34, 27381–27394 Search PubMed.
- M. Jain, E. Bengio, A. Hernandez-Garcia, J. Rector-Brooks, B. F. Dossou, C. A. Ekbote, J. Fu, T. Zhang, M. Kilgour, D. Zhang et al., International Conference on Machine Learning, 2022, pp. 9786–9801 Search PubMed.
- A. Hernandez-Garcia, A. Duval, A. Volokhova, Y. Bengio, D. Sharma, P. L. Carrier, M. Koziarski and V. Schmidt, Crystal-GFN: sampling crystals with desirable properties and constraints, 37th Conference on Neural Information Processing Systems (NeurIPS 2023)-AI4MAt Workshop, 2023 Search PubMed.
- Y. Zhu, J. Wu, C. Hu, J. Yan, T. Hou and J. Wu, et al., Adv. Neural Inf. Process. Syst., 2024, 36, 2672–2680 Search PubMed.
- D. P. Kingma, arXiv, preprint, arXiv:1412.6980, 2014, DOI:10.48550/arXiv.1412.6980.
- J. Martens and R. Grosse, International Conference on Machine Learning, 2015, pp. 2408–2417 Search PubMed.
- A. Paszke, S. Gross, F. Massa, A. Lerer, J. Bradbury, G. Chanan, T. Killeen, Z. Lin, N. Gimelshein and L. Antiga, et al., Adv. Neural Inf. Process. Syst., 2019, 32, 8026–8037 Search PubMed.
- J. Bradbury, R. Frostig, P. Hawkins, M. J. Johnson, C. Leary, D. Maclaurin, G. Necula, A. Paszke, J. VanderPlas, S. Wanderman-Milne and Q. Zhang, JAX: composable transformations of Python+NumPy programs, 2018, https://github.com/google/jax.
- M. Abadi, A. Agarwal, P. Barham, E. Brevdo, Z. Chen, C. Citro, G. S. Corrado, A. Davis, J. Dean, M. Devin, S. Ghemawat, I. Goodfellow, A. Harp, G. Irving, M. Isard, Y. Jia, R. Jozefowicz, L. Kaiser, M. Kudlur, J. Levenberg, D. Mané, R. Monga, S. Moore, D. Murray, C. Olah, M. Schuster, J. Shlens, B. Steiner, I. Sutskever, K. Talwar, P. Tucker, V. Vanhoucke, V. Vasudevan, F. Viégas, O. Vinyals, P. Warden, M. Wattenberg, M. Wicke, Y. Yu and X. Zheng, TensorFlow: Large-Scale Machine Learning on Heterogeneous Systems, 2015, software available from https://www.tensorflow.org/.
- M. Raissi, P. Perdikaris and G. E. Karniadakis, J. Comput. Phys., 2019, 378, 686–707 CrossRef.
- W. Wang, Z. Wu, J. C. Dietschreit and R. Gómez-Bombarelli, J. Chem. Phys., 2023, 158, 044113 CrossRef CAS PubMed.
- R. A. Vargas-Hernández, K. Jorner, R. Pollice and A. Aspuru-Guzik, J. Chem. Phys., 2023, 158, 104801 CrossRef PubMed.
- T. Osterrieder, F. Schmitt, L. Lüer, J. Wagner, T. Heumüller, J. Hauch and C. J. Brabec, Energy Environ. Sci., 2023, 16, 3984–3993 RSC.
- P. Q. Velasco, K. Y. A. Low, C. J. Leong, W. T. Ng, S. Qiu, S. Jhunjhunwala, B. Li, A. Qian, K. Hippalgaonkar and J. J. W. Cheng, Digital Discovery, 2024, 3, 1011–1020 RSC.
- A. Tripp and J. M. Hernández-Lobato, arXiv, 2023, preprint, arXiv:2310.09267, DOI:10.48550/arXiv.2310.09267.
- M. Skreta, Z. Zhou, J. L. Yuan, K. Darvish, A. Aspuru-Guzik and A. Garg, arXiv, 2024, preprint, arXiv:2401.04157, DOI:10.48550/arXiv.2401.04157.
- L. Ouyang, J. Wu, X. Jiang, D. Almeida, C. Wainwright, P. Mishkin, C. Zhang, S. Agarwal, K. Slama and A. Ray, et al., Adv. Neural Inf. Process. Syst., 2022, 35, 27730–27744 Search PubMed.
- J. Wei, X. Wang, D. Schuurmans, M. Bosma, F. Xia, E. Chi, Q. V. Le and D. Zhou, et al., Adv. Neural Inf. Process. Syst., 2022, 35, 24824–24837 Search PubMed.
- Y. Gao, Y. Xiong, X. Gao, K. Jia, J. Pan, Y. Bi, Y. Dai, J. Sun and H. Wang, arXiv, 2023, preprint, arXiv:2312.10997, DOI:10.48550/arXiv.2312.10997.
- L. Gao, A. Madaan, S. Zhou, U. Alon, P. Liu, Y. Yang and G. Neubig, PAL: Program-aided Language Models, Proceedings of the 40th International Conference on Machine Learning, ed. A. Krause, E. Brunskill, K. Cho, B. Engelhardt, S. Sabato and J. Scarlett, PMLR, 2023, vol. 202, pp. 10764–10799, https://proceedings.mlr.press/v202/gao23f.html Search PubMed.
- S. Yao, J. Zhao, D. Yu, N. Du, I. Shafran, K. Narasimhan and Y. Cao, ReAct: Synergizing Reasoning and Acting in Language Models, The Eleventh International Conference on Learning Representations, 2023, https://openreview.net/forum?id=WE_vluYUL-X Search PubMed.
- Y. Song, P. Dhariwal, M. Chen and I. Sutskever, Consistency Models, Proceedings of the 40th International Conference on Machine Learning, ed. A. Krause, E. Brunskill, K. Cho, B. Engelhardt, S. Sabato and J. Scarlett, PMLR, 2023, vol. 202, pp. 32211–2252, https://proceedings.mlr.press/v202/song23a.html Search PubMed.
- R.-R. Griffiths, L. Klarner, H. Moss, A. Ravuri, S. Truong, Y. Du, S. Stanton, G. Tom, B. Rankovic and A. Jamasb, et al., Adv. Neural Inf. Process. Syst., 2024, 36, 76923–76946 Search PubMed.
- S. J. Ang, W. Wang, D. Schwalbe-Koda, S. Axelrod and R. Gómez-Bombarelli, Chem, 2021, 7, 738–751 CAS.
- W. Heyndrickx, L. Mervin, T. Morawietz, N. Sturm, L. Friedrich, A. Zalewski, A. Pentina, L. Humbeck, M. Oldenhof and R. Niwayama, et al., J. Chem. Inf. Model., 2024, 64, 2331–2344 CrossRef CAS PubMed.
- S. Zaidi, M. Schaarschmidt, J. Martens, H. Kim, Y. W. Teh, A. Sanchez-Gonzalez, P. Battaglia, R. Pascanu and J. Godwin, Pre-training via Denoising for Molecular Property Prediction, The Eleventh International Conference on Learning Representations, 2023, https://openreview.net/forum?id=tYIMtogyee Search PubMed.
- M. Jain, T. Deleu, J. Hartford, C.-H. Liu, A. Hernandez-Garcia and Y. Bengio, Digital Discovery, 2023, 2, 557–577 RSC.
- J. Deng, W. Dong, R. Socher, L.-J. Li, K. Li and L. Fei-Fei, 2009 IEEE Conference on Computer Vision and Pattern Recognition, 2009, pp. 248–255 Search PubMed.
- O. Bojar, C. Buck, C. Federmann, B. Haddow, P. Koehn, J. Leveling, C. Monz, P. Pecina, M. Post, H. Saint-Amand et al., Proceedings of the Ninth Workshop on Statistical Machine Translation, 2014, pp. 12–58 Search PubMed.
- M. Heusel, H. Ramsauer, T. Unterthiner, B. Nessler and S. Hochreiter, Adv. Neural Inf. Process. Syst., 2017, 30, 6629–6640 Search PubMed.
- D. Hendrycks, C. Burns, S. Kadavath, A. Arora, S. Basart, E. Tang, D. Song and J. Steinhardt, arXiv, 2021, preprint, arXiv:2103.03874, DOI:10.48550/arXiv.2103.03874.
- L. Chanussot, A. Das, S. Goyal, T. Lavril, M. Shuaibi, M. Riviere, K. Tran, J. Heras-Domingo, C. Ho and W. Hu, et al., ACS Catal., 2021, 11, 6059–6072 CrossRef CAS.
- R. Tran, J. Lan, M. Shuaibi, B. M. Wood, S. Goyal, A. Das, J. Heras-Domingo, A. Kolluru, A. Rizvi and N. Shoghi, et al., ACS Catal., 2023, 13, 3066–3084 CrossRef CAS.
- J. Lan, A. Palizhati, M. Shuaibi, B. M. Wood, B. Wander, A. Das, M. Uyttendaele, C. L. Zitnick and Z. W. Ulissi, npj Comput. Mater., 2023, 9, 172 CrossRef CAS.
- Open Catalyst demo — open-catalyst.metademolab.com, https://open-catalyst.metademolab.com/, accessed 24-08-2024.
- J. Riebesell, R. E. A. Goodall, P. Benner, Y. Chiang, B. Deng, A. A. Lee, A. Jain and K. A. Persson, Matbench Discovery – A framework to evaluate machine learning crystal stability predictions, arXiv, 2024, preprint, arXiv:2308.14920, DOI:10.48550/arXiv.2308.14920.
- R. David, A. Aspuru-Guzik, B. Sara, D. Bistra, D. L. Priya, G. Marzyeh, K. Hannah, M. Claire, R. Esther, T. Milind and W. Adam, Position: Application-Driven Innovation in Machine Learning, Proceedings of the 41st International Conference on Machine Learning, ed. R. Salakhutdinov, Z. Kolter, K. Heller, A. Weller, N. Oliver, J. Scarlett and F. Berkenkamp, PMLR, 2024, vol. 235, pp. 42707–42718, https://proceedings.mlr.press/v235/rolnick24a.html Search PubMed.
- J. Gilmer, S. S. Schoenholz, P. F. Riley, O. Vinyals and G. E. Dahl, International Conference on Machine Learning, 2017, pp. 1263–1272 Search PubMed.
- K. Xu, W. Hu, J. Leskovec and S. Jegelka, How Powerful are Graph Neural Networks?, International Conference on Learning Representations, 2019, https://openreview.net/forum?id=ryGs6iA5Km Search PubMed.
- V. Delle Rose, A. Kozachinskiy, C. Rojas, M. Petrache and P. Barceló, Adv. Neural Inf. Process. Syst., 2024, 36, 9556–9573 Search PubMed.
- N. Thomas, T. Smidt, S. Kearnes, L. Yang, L. Li, K. Kohlhoff and P. Riley, arXiv, 2018, preprint, arXiv:1802.08219, DOI:10.48550/arXiv.1802.08219.
- M. M. Bronstein, J. Bruna, T. Cohen and P. Veličković, arXiv, 2021, preprint, arXiv:2104.13478, DOI:10.48550/arXiv.2104.13478.
- Molecular Simulation — ai4science101.github.io, https://ai4science101.github.io/blogs/molecular_simulation/, accessed 24-08-2024.
- Portal — portal.valencelabs.com, https://portal.valencelabs.com/, accessed 24-08-2024.
- AI4Mat, AI4Mat-NeurIPS 2024 — sites.google.com, https://sites.google.com/view/ai4mat, 2024, accessed 02-09-2024.
- MLSB, Machine Learning in Structural Biology — mlsb.io, https://www.mlsb.io/, 2024, accessed 02-09-2024.
- GenBio NeurIPS Workshop 2023 — genbio-workshop.github.io, https://genbio-workshop.github.io/, 2023, accessed 02-09-2024.
- CompBio, CompBio Workshop ICML 2023 — icml-compbio.github.io, https://icml-compbio.github.io/, 2023, accessed 02-09-2024.
- AI4Science, AI for Science: Scaling in AI for Scientific Discovery — ai4sciencecommunity.github.io, https://ai4sciencecommunity.github.io/icml24.html, 2024, accessed 02-09-2024.
- MLDD, MLDD 2023 — sites.google.com, https://sites.google.com/view/mldd-2023/, 2023, accessed 02-09-2024.
- ML4Materials, ICLR 2023 Workshop, Machine Learning for Materials — ml4materials.com, https://www.ml4materials.com/, 2023, accessed 02-09-2024.
- ICLR Blog, about — ICLR Blogposts 2024 — iclr-blogposts.github.io, https://iclr-blogposts.github.io/2024/about/, accessed 24-08-2024.
- N. Schneider, D. M. Lowe, R. A. Sayle, M. A. Tarselli and G. A. Landrum, J. Med. Chem., 2016, 59, 4385–4402 CrossRef CAS PubMed.
- A. Krizhevsky, I. Sutskever and G. E. Hinton, Adv. Neural Inf. Process. Syst., 2012, 1097–1105 Search PubMed.
- J. Kaplan, S. McCandlish, T. Henighan, T. B. Brown, B. Chess, R. Child, S. Gray, A. Radford, J. Wu and D. Amodei, arXiv, 2020, preprint, arXiv:2001.08361, DOI:10.48550/arXiv.2001.08361.
- A. Radford, J. Wu, R. Child, D. Luan, D. Amodei and I. Sutskever, et al., OpenAI Blog, 2019, 1, 9 Search PubMed.
- R. Sutton, Incomplete Ideas (Blog), 2019, 13, 38 Search PubMed.
- S.-C. Li, H. Wu, A. Menon, K. A. Spiekermann, Y.-P. Li and W. H. Green, J. Am. Chem. Soc., 2024, 146, 23103–23120 CrossRef CAS PubMed.
- J. Kim, D. Nguyen, A. Suleymanzade, H. An and S. Hong, Adv. Neural Inf. Process. Syst., 2023, 36, 18582–18612 Search PubMed.
- A. A. Duval, V. Schmidt, A. Hernández-Garcıa, S. Miret, F. D. Malliaros, Y. Bengio and D. Rolnick, International Conference on Machine Learning, 2023, pp. 9013–9033 Search PubMed.
- D. Flam-Shepherd and A. Aspuru-Guzik, arXiv, 2023, preprint, arXiv:2305.05708, DOI:10.48550/arXiv.2305.05708.
- N. Gruver, A. Sriram, A. Madotto, A. G. Wilson, C. L. Zitnick and Z. W. Ulissi, Fine-Tuned Language Models Generate Stable Inorganic Materials as Text, The Twelfth International Conference on Learning Representations, 2024 Search PubMed.
- A. Aspuru-Guzik, R. Lindh and M. Reiher, ACS Cent. Sci., 2018, 4, 144–152 CrossRef CAS PubMed.
- J. Westermayr and P. Marquetand, Chem. Rev., 2021, 121, 9873–9926 CrossRef CAS PubMed.
- S. Axelrod, E. Shakhnovich and R. Gómez-Bombarelli, Nat. Commun., 2022, 13, 3440 CrossRef CAS PubMed.
- Y. Du, C. Duan, A. Bran, A. Sotnikova, Y. Qu, H. Kulik, A. Bosselut, J. Xu and P. Schwaller, Large Language Models are Catalyzing Chemistry Education, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-h722v.
- D. Morgan, G. Pilania, A. Couet, B. P. Uberuaga, C. Sun and J. Li, Curr. Opin. Solid State Mater. Sci., 2022, 26, 100975 CrossRef CAS.
- Y. Djoumbou-Feunang, J. Wilmot, J. Kinney, P. Chanda, P. Yu, A. Sader, M. Sharifi, S. Smith, J. Ou and J. Hu, et al., Front. Chem., 2023, 11, 1292027 CrossRef CAS PubMed.
- V. Barone, S. Alessandrini, M. Biczysko, J. R. Cheeseman, D. C. Clary, A. B. McCoy, R. J. DiRisio, F. Neese, M. Melosso and C. Puzzarini, Nat. Rev. Methods Primers, 2021, 1, 38 CrossRef CAS.
- S. X. Leong, S. Pablo-García, Z. Zhang and A. Aspuru-Guzik, Automated electrosynthesis reaction mining with multimodal large language models (MLLMs), ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-7fwxv.
- Z. T. Fried, S. J. El-Abd, B. M. Hays, G. Wenzel, A. N. Byrne, L. Margulès, R. A. Motiyenko, S. T. Shipman, M. P. Horne and J. K. Jørgensen, et al., Astrophys. J. Lett., 2024, 965, L23 CrossRef.
- H. Zheng, E. Sivonxay, M. Gallant, Z. Luo, M. McDermott, P. Huck and K. A. Persson, arXiv, 2024, preprint, arXiv:2402.00177, DOI:10.48550/arXiv.2402.00177.
- C.-I. Wang, J. C. Maier and N. E. Jackson, Chem. Sci., 2024, 15, 8390–8403 RSC.
- A. Ullah, Y. Huang, M. Yang and P. O. Dral, arXiv, 2024, preprint, arXiv:2404.14021, DOI:10.48550/arXiv.2404.14021.
- S. Zhu, B. H. Nguyen, Y. Xia, K. Frost, S. Xie, V. Viswanathan and J. A. Smith, Green Chem., 2023, 25, 6612–6617 RSC.
- G. Zhao, H. Kim, C. Yang and Y. G. Chung, J. Phys. Chem. A, 2024, 128, 2399–2408 CrossRef CAS PubMed.
- J. Sohl-Dickstein, E. Weiss, N. Maheswaranathan and S. Ganguli, International Conference on Machine Learning, 2015, pp. 2256–2265 Search PubMed.
- J. Ho, A. Jain and P. Abbeel, Adv. Neural Inf. Process. Syst., 2020, 33, 6840–6851 Search PubMed.
- Y. Song, J. Sohl-Dickstein, D. P. Kingma, A. Kumar, S. Ermon and B. Poole, Score-Based Generative Modeling through Stochastic Differential Equations, International Conference on Learning Representations, 2021, https://openreview.net/forum?id=PxTIG12RRHS Search PubMed.
- T. Karras, M. Aittala, T. Aila and S. Laine, Adv. Neural Inf. Process. Syst., 2022, 35, 26565–26577 Search PubMed.
- B. Máté, F. Fleuret and T. Bereau, arXiv, 2024, preprint, arXiv:2406.02313, DOI:10.48550/arXiv.2406.02313.
- K. Neklyudov, R. Brekelmans, D. Severo and A. Makhzani, International Conference on Machine Learning, 2023, pp. 25858–25889 Search PubMed.
- Y. Du, M. Plainer, R. Brekelmans, C. Duan, F. Noe, C. P. Gomes, A. Aspuru-Guzik and K. Neklyudov, Doob’s Lagrangian: A Sample-Efficient Variational Approach to Transition Path Sampling, ICML 2024 AI for Science Workshop, 2024 Search PubMed.
- N. H. Angello, V. Rathore, W. Beker, A. Wołos, E. R. Jira, R. Roszak, T. C. Wu, C. M. Schroeder, A. Aspuru-Guzik and B. A. Grzybowski, et al., Science, 2022, 378, 399–405 CrossRef CAS PubMed.
- wwPDB consortium, Nucleic Acids Res., 2019, 47, D520–D528 CrossRef PubMed.
- O. Schilter, P. Schwaller and T. Laino, Green Chem., 2024, 26, 8669–8679 RSC.
- L. Deng, IEEE Signal Process. Mag., 2012, 29, 141–142 Search PubMed.
- M. Broz, How many pictures are there (2024): Statistics, trends, and forecasts, https://photutorial.com/photos-statistics/, accessed 27-08-2024.
- B. Kozinsky, A. Musaelian, A. Johansson and S. Batzner, Scaling the leading accuracy of deep equivariant models to biomolecular simulations of realistic size, Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis, 2023, pp. 1–12 Search PubMed.
- J. Baldridge, J. Bauer, M. Bhutani, N. Brichtova, A. Bunner, K. Chan, Y. Chen, S. Dieleman, Y. Du, Z. Eaton-Rosen et al., arXiv, 2024, preprint, arXiv:2408.07009, DOI:10.48550/arXiv.2408.07009.
| This journal is © The Royal Society of Chemistry 2024 |