
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemical upcycling of poly(bisphenol A carbonate) to vinylene carbonates through organocatalysis†
Killian
Onida
 a,
Mohamad
Fayad
a,
Sébastien
Norsic
b,
Olivier
Boyron
b and
Nicolas
Duguet
*a
a,
Mohamad
Fayad
a,
Sébastien
Norsic
b,
Olivier
Boyron
b and
Nicolas
Duguet
*a
aUniv Lyon, Université Claude Bernard Lyon 1, CNRS, INSA-Lyon, CPE-Lyon, Institut de Chimie et Biochimie Moléculaires et Supramoléculaires (ICBMS), UMR CNRS 5246, Bâtiment Lederer, 1 rue Victor Grignard, F-69100 Villeurbanne, France. E-mail: nicolas.duguet@univ-lyon1.fr
bUniv. Lyon, Université Claude Bernard Lyon 1, CPE Lyon, CNRS UMR 5128, Laboratory of Catalysis Polymerization Processes and Materials (CP2M), Bat 308F, 43 Bd du 11 Novembre 1918, F-69616 Villeurbanne, France
First published on 4th May 2023
Abstract
The chemical depolymerisation of poly(bisphenol A carbonate) (BPA-PC) was studied using α-hydroxyketones as nucleophiles to give bisphenol A as the original monomer and the corresponding vinylene carbonates as co-products. The optimisation revealed that 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) is an efficient organocatalyst to perform this reaction under mild conditions (25 °C in 2-MeTHF). The success of TBD was attributed to the fact that it can both activate α-hydroxyketones and BPA-PC, through hydrogen bonding. The substrate scope was then investigated with a range of α-hydroxyketones to give vinylene carbonates (symmetrical and unsymmetrical, aromatic, heteroaromatic and alkyl) with moderate to high yields (30–97%, 13 examples). Scale-up experiments were also conducted to highlight the synthetic utility of this method. Finally, it was demonstrated that vinylene carbonates can be produced from waste polycarbonate contained in compact disks (CDs) and other BPA-PC containing materials, thus providing a safe and economical access to these species.
Introduction
Plastic materials are ubiquitous in our modern lives. Due to their unique and tunable properties, they have found widespread applications.1 However, their massive production and (mis)use have led to serious environmental pollution and represent a danger for animal and human health.2 Consequently, there is an urgent need to address these issues by developing biodegradable polymers3 as well as promoting the recycling of plastics4 in a circular economy.5 Currently, only a small proportion of plastics is recycled, most of it being done mechanically as it is usually the simplest option. However, it can only be done a limited number of times as it affects the quality of the material, meaning that even recycled plastics end up in landfills or are incinerated for energy recovery.In this context, the chemical recycling appears as the one of the most interesting options.6 Indeed, the recovery of the original monomer induces that the recycling could be virtually done indefinitely. Moreover, this also means that plastic waste can be considered as a feedstock, thus limiting the search and exploitation of new fossil resources.7
Poly(bisphenol A carbonate) (BPA-PC) is the most produced polycarbonates with an annual production almost reaching 5 million tons in 2021.8 BPA-PC is a thermoplastic material presenting excellent physical, chemical and mechanical properties such as high toughness and impact strength, heat resistance, transparency and lightness. Thanks to these outstanding properties, BPA-PC has found numerous applications in electronics, automotive (car headlamp lenses), construction (roofing and glazing), optic (compact disks and DVDs) and also in protection equipment (safety googles, bullet-proof glass). Similar to other polymers, a large portion of BPA-PC ends up in landfills, where it progressively degrades to bisphenol A, a known endocrine disruptor, that is contaminating air and soil and finally affecting both the health and the environment.9 In this context, the recycling of BPA-PC is crucial.10 The chemical recycling of BPA-PC can be performed by pyrolysis; however, it requires harsh reaction conditions and leads to the formation of complex mixtures.11 So, most other chemical recycling strategies of BPA-PC rely on the electrophilic character of the carbonyl group, thus involving the use of nucleophilic species as depolymerising agents. First of all, hydrolysis can be employed to recycle BPA but this strategy also produces carbon dioxide as a byproduct.12 Hydrogenation13 and hydrodeoxygenation14 are two complementary depolymerisation strategies employing hydrogen to form either BPA or dicycloalkanes, respectively. However, methanol and methane that are co-produced through these techniques are seldom recovered. Moreover, they require high temperature and high H2 pressure. In contrast, the depolymerisation of BPA-PC through alcoholysis15 (e.g. methanolysis,16 glycolysis17 and phenolysis),18 aminolysis19 and even thiolysis20 can occur under mild conditions, leading to the formation of co-products with higher added value such as (acyclic and cyclic) carbonates, ureas and dithiocarbonates. The use of mixed nucleophiles has also permitted to broaden the scope of applications to other heterocycles. For example, Xu and Wang have demonstrated that chiral aminoalcohols can be used for the depolymerisation of BPA-PC to form the corresponding oxazolidinones with retention of the chirality.21
Recently, Lee, Kwak and Kim have also shown that hydroxamic acid derivatives can be used to generate valuable dioxazolones.22 The use of mixed nucleophiles for the depolymerisation of BPA-PC is challenging as it inherently implies that different reactivities are involved. However, it offers the opportunity to form co-products high added value. To the best of our knowledge, the chemical depolymerisation of BPA-PC using α-hydroxyketones as nucleophiles has never been reported. This would lead to the formation of the original BPA monomer and unsaturated cyclic carbonates as higher added-value compounds, namely “vinylene carbonates”.
First described in 1953,23 vinylene carbonates are only recently receiving a lot of attention. For instance, unsubstituted vinylene carbonate (1,3-dioxol-2-one) is an essential electrolyte additive to increase the life span of lithium batteries.24 Moreover, it can be used in radical polymerisation to form poly(vinylene carbonate),25 a precursor of poly(hydroxymethylene, which has found applications in 3D printing.26 In organic synthesis, it was historically used in Diels–Alder reactions27 and it is now extensively used as a C1 or C2 synthon in annulation reactions promoted by C–H activation.28 Functional vinylene carbonate derivatives have also gained a lot of interest. For example, the dimethyl derivative (4,5-dimethyl-1,3-dioxol-2-one) is essential for the preparation of prodrugs based on the medoxomil group.29 More recently, a trifluoromethoxy derivative was developed as a trigger for vinylene carbonate polymerisation to form an excellent solid electrolyte interphase, resulting in high-energy-density lithium-ion batteries.30 Despite their great potential of applications, the development of the vinylene carbonate chemistry is hampered by their synthesis, that is far from being either economic or environmentally-friendly. Indeed, the preparation of vinylene carbonate itself requires the use of chlorine,31 while the access to other substituted derivatives requires the use of phosgene,32 triphosgene33 or 1,1′-dicarbonyldiimidazole (CDI).34 Recently, our group has demonstrated that diphenyl carbonate (DPC) can be used as a cheap and non-toxic carbonyl source for the preparation of vinylene carbonates from either benzoins/acyloins35 or directly from aldehydes.36 Based upon these previous studies, we now report here our results on the preparation of vinylene carbonates using poly(bisphenol A carbonate) as a carbonyl source, thus concomitantly leading to the chemical upcycling of this polymer (Fig. 1). Considering that about one third of the BPA-PC annual production relies on the CO2-based Asahi–Kasei process,37 this strategy affords an indirect CO2-derived access to vinylene carbonates. Moreover, it allows the preparation of the targeted products under safe and mild conditions, thus widening the scope of applications.
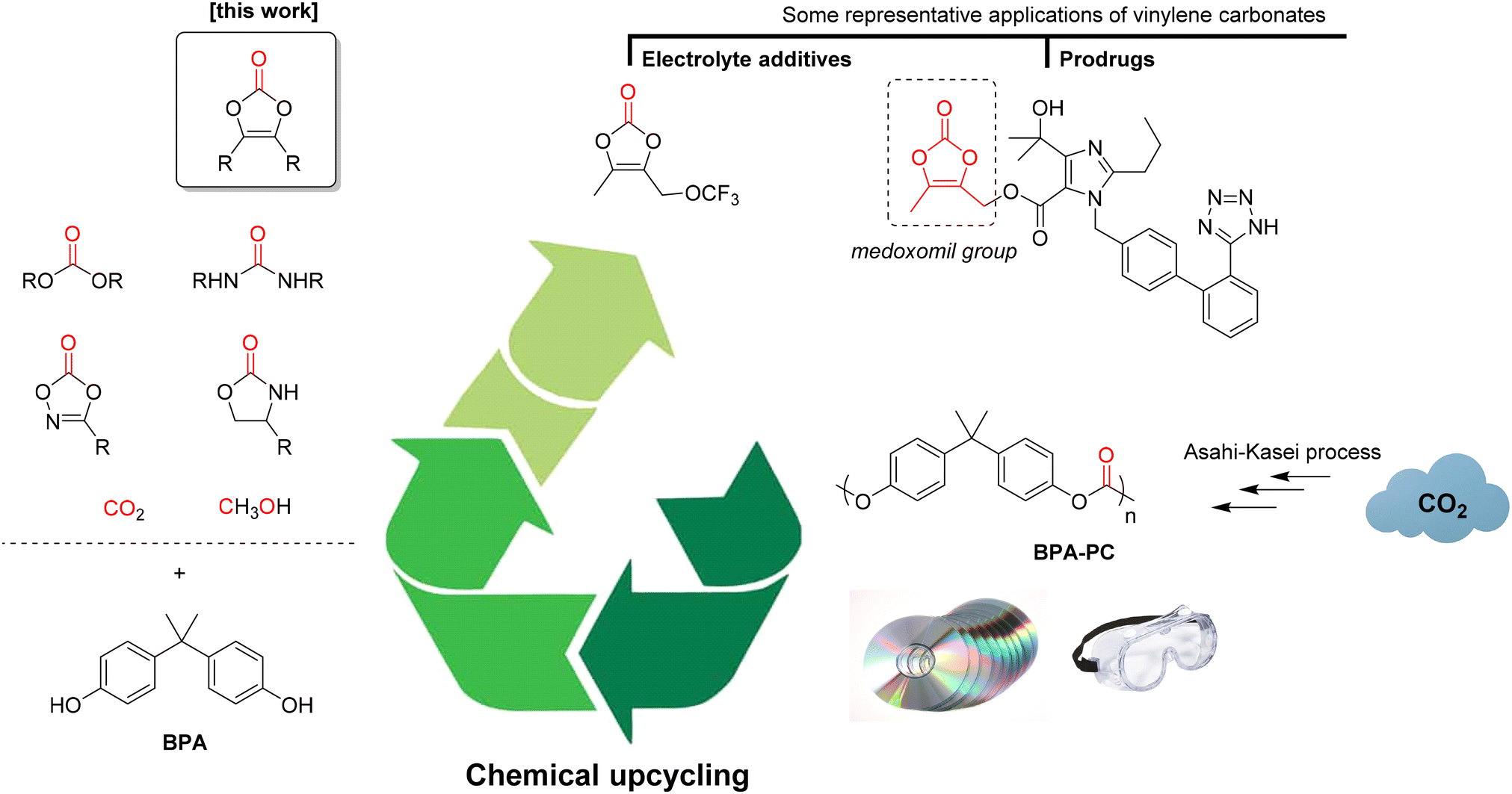 | ||
| Fig. 1 Chemical upcycling of poly(bisphenol A carbonate) to vinylene carbonates and some of their representative applications. | ||
Results and discussion
Benzoin 1 was chosen as a model substrate for the depolymerisation of a commercially available poly(bisphenol A carbonate) (Mw = 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, Đ = 1.77) to form vinylene carbonate 2. First, the catalyst screening was performed using a 10 mol% loading in THF at room temperature for 16 h (Table 1). Inorganic bases such as sodium hydroxide (NaOH) and potassium carbonate (K2CO3) were first considered and they gave moderate yields for both vinylene carbonate 2 and bisphenol A (BPA) (Table 1, entries 1 and 2). Organic base catalysts such as 4-dimethylaminopyridine (DMAP), 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were also tested but they gave lower yields of 2 (Table 1, entries 3–5). In these cases, BPA was formed in higher amount than the desired product, probably due to the competing hydrolysis of BPA-PC. With 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), the yield of 2 slightly increased to 18% but, in the same time, the yield of BPA sharply increased to 90% (Table 1, entry 6). Once again, the high yield obtained for BPA might be due to the hydrolysis of BPA-PC, promoted by the base catalyst. Encouragingly, with 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), vinylene carbonate 2 was obtained with a moderate yield of 45% whereas BPA was formed in 68% yield (Table 1, entry 7). After choosing TBD as the best catalyst, several solvents were tested under the same reaction conditions. The use of dimethylformamide (DMF) gave excellent yields for both vinylene carbonate 2 (84%) and BPA (99%), probably due to the excellent solubility of BPA-PC in DMF (Table 1, entry 8).
000 g mol−1, Đ = 1.77) to form vinylene carbonate 2. First, the catalyst screening was performed using a 10 mol% loading in THF at room temperature for 16 h (Table 1). Inorganic bases such as sodium hydroxide (NaOH) and potassium carbonate (K2CO3) were first considered and they gave moderate yields for both vinylene carbonate 2 and bisphenol A (BPA) (Table 1, entries 1 and 2). Organic base catalysts such as 4-dimethylaminopyridine (DMAP), 1,4-diazabicyclo[2.2.2]octane (DABCO), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) were also tested but they gave lower yields of 2 (Table 1, entries 3–5). In these cases, BPA was formed in higher amount than the desired product, probably due to the competing hydrolysis of BPA-PC. With 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), the yield of 2 slightly increased to 18% but, in the same time, the yield of BPA sharply increased to 90% (Table 1, entry 6). Once again, the high yield obtained for BPA might be due to the hydrolysis of BPA-PC, promoted by the base catalyst. Encouragingly, with 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), vinylene carbonate 2 was obtained with a moderate yield of 45% whereas BPA was formed in 68% yield (Table 1, entry 7). After choosing TBD as the best catalyst, several solvents were tested under the same reaction conditions. The use of dimethylformamide (DMF) gave excellent yields for both vinylene carbonate 2 (84%) and BPA (99%), probably due to the excellent solubility of BPA-PC in DMF (Table 1, entry 8).
|
|
||||
|---|---|---|---|---|
| Entry | Base catalyst (mol%) | Solvent | Yield of 2b (%) | Yield of BPAb (%) |
|
a Reaction conditions: benzoin 1 (1 mmol), BPA-PC (Mw = 45000 g mol−1, Đ = 1.77, 1 mmol based on the repeating unit), base catalyst (10 mol%), solvent (2 mL), room temperature, 16 h.
b Yields were determined by GC/FID with hexadecane as an internal standard.
c Reaction time of 32 h. DMAP: 4-dimethylaminopyridine; DABCO: 1,4-diazabicyclo[2.2.2]octane; DBU: 1,8-diazabicyclo [5.4.0]undec-7-ene; DBN: 1,5-diazabicyclo[4.3.0]non-5-ene; TBD: 1,5,7-triazabicyclo[4.4.0]dec-5-ene; THF: tetrahydrofuran; DMF: dimethylformamide; GVL: γ-butyrolactone; CPME: cyclopentyl methyl ether; 2-MeTHF: 2-methyltetrahydrofuran.
|
||||
| 1 | NaOH (10) | THF | 22 | 35 |
| 2 | K2CO3 (10) | THF | 37 | 40 |
| 3 | DMAP (10) | THF | 0 | 11 |
| 4 | DABCO (10) | THF | 3 | 27 |
| 5 | DBU (10) | THF | 6 | 28 |
| 6 | DBN (10) | THF | 18 | 90 |
| 7 | TBD (10) | THF | 45 | 68 |
| 8 | TBD (10) | DMF | 84 | 99 |
| 9 | TBD (10) | Acetone | 57 | 93 |
| 10 | TBD (10) | Ethyl acetate | 18 | 47 |
| 11 | TBD (10) | Butyl butyrate | 24 | 5 |
| 12 | TBD (10) | GVL | 47 | 43 |
| 13 | TBD (10) | CPME | 19 | 22 |
| 14 | TBD (10) | 2-MeTHF | 70 | 78 |
| 15c | TBD (5) | 2-MeTHF | 52 | 60 |
| 16c | TBD (1) | 2-MeTHF | 51 | 61 |
| 17 | — | 2-MeTHF | 3 | 4 |
|
||||
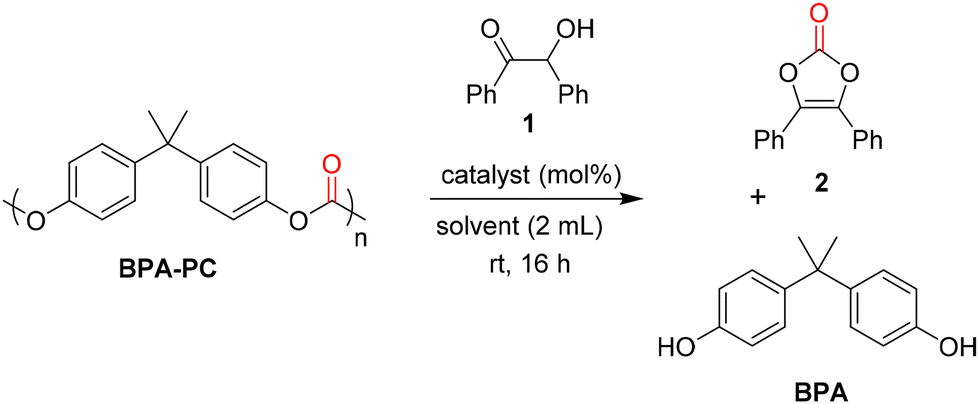
However, considering that DMF has a bad toxicity and environment profile, other solvents were sought. Acetone gave interesting results in 57% yield of 2 and 93% yield of BPA (Table 1, entry 9). Then, esters were screened such as ethyl acetate and butyl butyrate but they gave low yields (Table 1, entries 10 and 11). Interestingly, γ-valerolactone (GVL) gave 47% yield of 2 and 43% yield of BPA (Table 1, entry 12). Ethereal solvents were then considered. Cyclopentyl methyl ether (CPME) gave low yields for both products, due to the poor solubility of BPA-PC in this solvent (Table 1, entry 13). More interestingly, the use of biomass-derived 2-MeTHF led to a significant increase in yields, reaching 70% for vinylene carbonate 2 (Table 1, entry 14). Based on these results, 2-MeTHF was selected as a solvent for further optimisation. The loading of TBD was decreased to 5 and 1 mol% in order to study the limits of the method. However, under these conditions, the yield of 2 decreased to 51–52%, even after a prolonged reaction time (32 h) (Table 1, entries 15 and 16). The increase of catalyst loading was not attempted for economical and practical reasons. A control experiment was then performed without any catalyst. Under these conditions, both the yields of vinylene carbonate 2 and BPA decreased sharply to 3 and 4%, respectively, thus indicating the crucial role of TBD in the depolymerisation process and the formation of the desired product (Table 1, entry 17).
The benzoin/BPA-PC ratio was then studied in order to reach high yields of vinylene carbonates, that are considered as the products with the highest added-value in this study (Table 2). Using an equimolar ratio (1 mmol of each) of benzoin 1 and BPA-PC, vinylene carbonate 2 and BPA were obtained in 70 and 78% yields, respectively (Table 2, entry 1). Increasing the amount of BPA-PC to 1.5 equivalent led to an increase in the yield of 2 to 79% (Table 2, entry 2). Further increasing the amount of BPA-PC to 2 equivalents led to 2 in an excellent 92% yield (Table 2, entry 3).
|
|
||||
|---|---|---|---|---|
| Entry | Benzoin 1 (mmol) | BPA-PC (mmol) | Yield of 2b (%) | Yield of BPAb (%) |
| a Reaction conditions: benzoin 1, BPA-PC, TBD (10 mol%), 2-MeTHF (2 mL), rt, 16 h. b Yields were determined by GC/FID with hexadecane as an internal standard and are based on the limiting reagent. c Reaction performed in the absence of TBD. | ||||
| 1 | 1 | 1 | 70 | 78 |
| 2 | 1 | 1.5 | 79 | 85 |
| 3 | 1 | 2 | 92 | 99 |
| 4c | 1 | 2 | 9 | 10 |
| 5 | 1.5 | 1 | 62 | 89 |
| 6 | 2 | 1 | 73 | 99 |
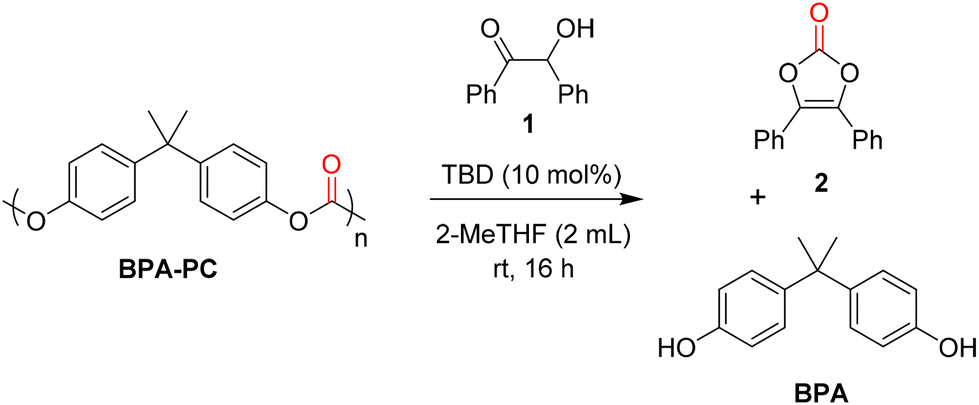
A control experiment without TBD was also repeated under these optimum conditions. As expected, a sharp decrease in yields was observed for both products, indicating the crucial role of TBD (Table 2, entry 4). Considering that the total depolymerisation of BPA-PC to BPA could also be a complementary objective, the reaction was also performed with an excess of benzoin. Using 1.5 equivalent, the yield of 2 reached 62% while the yield of BPA reached 89% (Table 2, entry 5). Finally, the full depolymerisation of BPA-PC was attained using 2 equivalents of benzoin and BPA was obtained in 99% yield (Table 2, entry 6). Under these conditions, vinylene carbonate 2 was also formed in 73% yield.
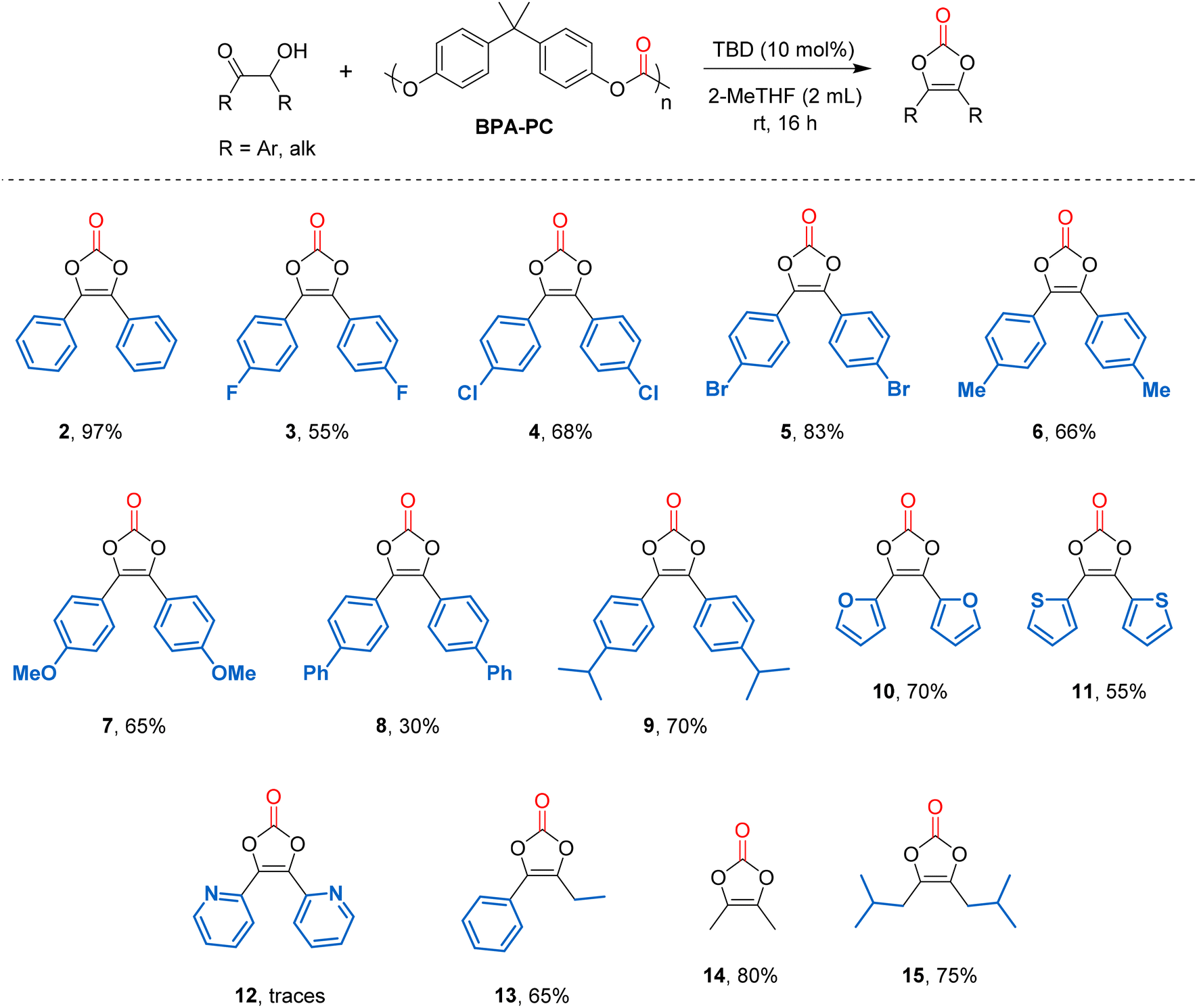
The substrate scope was then investigated using the optimized conditions (Fig. 2). Starting from symmetrical aromatic α-hydroxyketones, benzoin gave vinylene carbonate 2 in an excellent 97% yield. Benzoins bearing halogens at the para position gave vinylene carbonates 3 (X = F) in 55% yield, 4 (X = Cl) in 68% and 5 (X = Br) in 83% yields.
 | ||
| Fig. 2 Scope of vinylene carbonates (isolated yields). | ||
The progressive increase in vinylene carbonate yields could be explained by a decrease in halogen electronegativity, thus leading to an enhanced nucleophilicity of the α-hydroxyl group of benzoins. Benzoins bearing para-methyl and para-methoxy groups were then tested and gave vinylene carbonates 6 and 7 in 66 and 65% yield, respectively. A low yield (30%) was obtained for compound 8 bearing para-phenyl groups. However, it has reached 70% for compound 9 bearing para-isopropyl group. These results show that the reaction is not favoured with too sterically hindered substituents. Heterocyclic acyloins were next considered. When using furoin and 2,2′-thenoin as substrates, the corresponding vinylene carbonates 10 and 11 were isolated in 70 and 55% yield. It should be noted that the formation of these heterocyclic vinylene carbonates was not possible using diphenyl carbonate as a carbonyl source, under the previously reported conditions (neat, 90 °C).35 The fact that these compounds can be formed using BPA-polycarbonate as a carbonyl source probably lies in the use of milder conditions (2-MeTHF, 25 °C). However, when using α-pyridoin, only traces of compound 12 were detected and the corresponding diketone was obtained as the only product. An unsymmetrical aryl,alkyl-α-hydroxyketone was also considered and the corresponding vinylene carbonate 13 was isolated in 65% yield. Finally, symmetrical aliphatic α-hydroxyketones were tested and the corresponding vinylene carbonates 14 and 15 were obtained in 80 and 75% yield, respectively, showing the versatility of the method. Noteworthy, vinylene carbonate 14 is the key intermediate to prepare 4-(hydroxymethyl)-5-methyl-1,3-dioxol-2-one, known as the “medoxomil” group in prodrugs. It was previously made using phosgene as a carbonyl source.38 Thanks to the development of this methodology, it can now be produced from waste BPA-PC, which can be considered as a safer alternative.
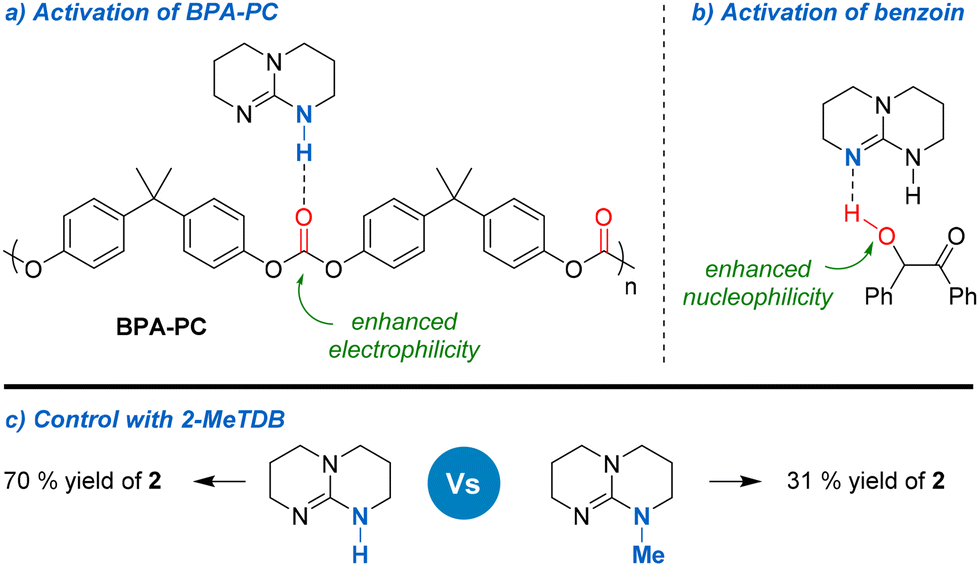
The success of TBD could be explained by the fact that, contrary to other organic bases, it could activate both the carbonyl group of BPA-PC and the hydroxyl group of benzoin 1 through hydrogen bonding, thus leading to enhanced electrophilicity and nucleophilicity, respectively (Fig. 3a and b). The important role of TBD has been well established in the synthesis of polycarbonates39 and has also been demonstrated in the context of their depolymerisation.15b,17b,40 The reaction was also carried out in the presence of 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (Me-TBD, 10 mol%). Under these conditions, vinylene carbonate 2 was only produced in 31% yield, thus confirming the important role of hydrogen bonding in the depolymerisation process (Fig. 3c).
 | ||
| Fig. 3 Role of TBD in the depolymerisation process and the formation of vinylene carbonates. | ||
The interactions between benzoin and TBD were studied by NMR by varying the ratio of each component (Fig. S4–S7 in ESI†). Progressively changing the benzoin/TBD ratio from 1:0 to 1:1 leads to a gradual shift of both the carbonyl and the CH groups of benzoin towards low field in 13C NMR. This indicates that TBD could also activate the carbonyl group of benzoin and enhance the acidic character of the CH proton. The interactions between diphenyl carbonate – used as a model for BPA-PC – and TBD were also studied by NMR (Fig. S9 and S10 in ESI†). In that case, varying the DPC/TBD ratio from 1:0 to 1:1 led to the progressive formation of a new species. This species was identified as a 1:1 DPC/TBD adduct, in which the carbonate group has been cleaved and activated by TBD. Considering that the formation of such species occurs rapidly at room temperature, this suggests that it is the predominant activation mode for the depolymerisation of BPA-PC.
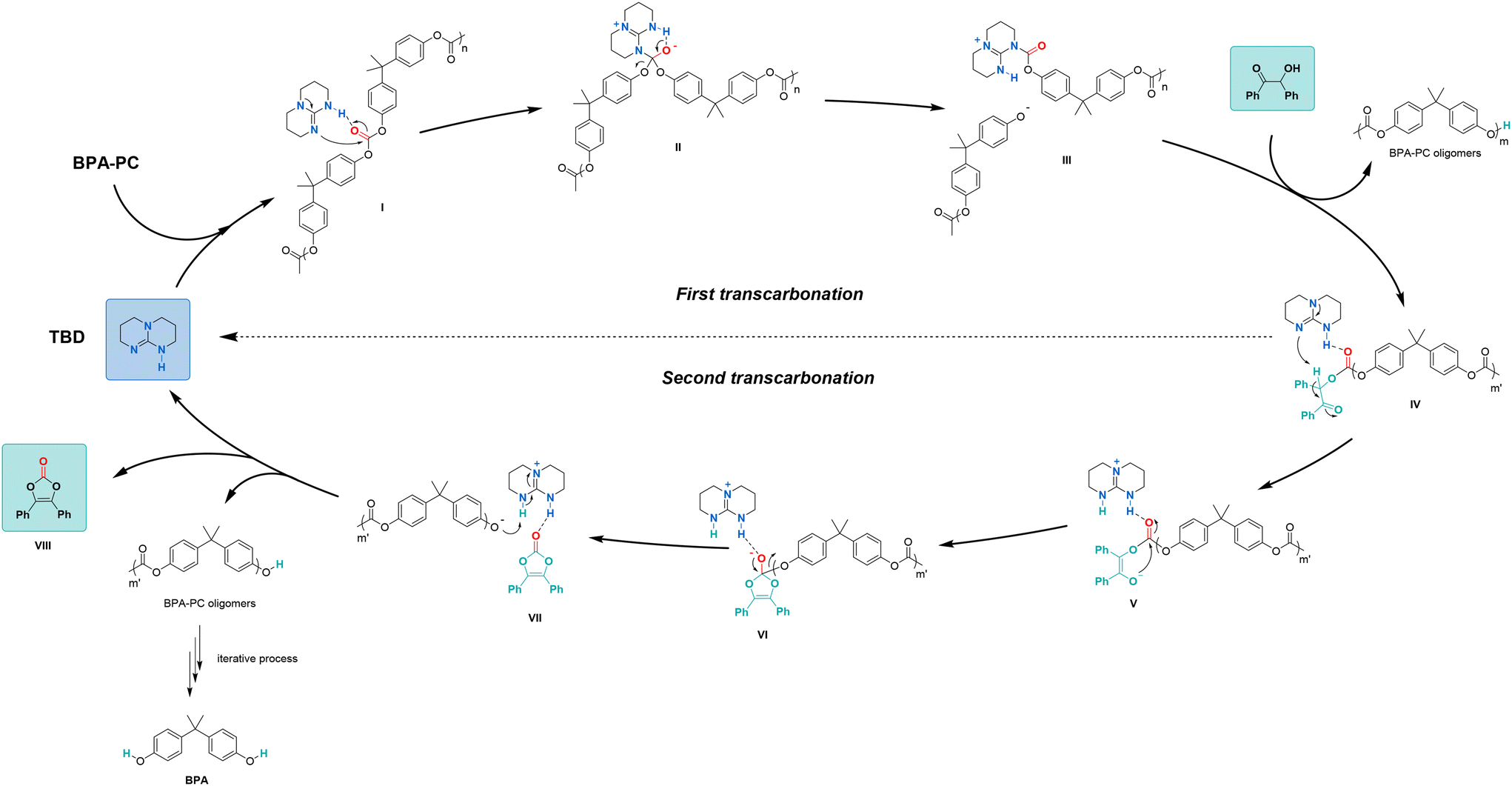
A mechanism showing all reaction intermediates leading to the formation of vinylene carbonates was then proposed based on these results (Fig. 4). First, the carbonyl group of BPA-PC could be activated by TBD through hydrogen bonding to form intermediate I. Nucleophilic addition of TBD on the activated carbonyl group of BPA-PC would lead to the formation of tetrahedral intermediate II. The release of a first phenoxy group would form intermediate III and start the depolymerisation of BPA-PC. Then, intermediate III could undergo a first trans-carbonation with benzoin to form the key intermediate IV and release oligomers.
 | ||
| Fig. 4 Mechanism proposal for TBD-catalysed depolymerisation of BPC-PC to vinylene carbonates and BPA. | ||
Noteworthy, the formation of intermediate IV could also occur through a pre-organized intermediate, in which both benzoin and BPA-PC are activated by TBD through hydrogen bonding (Fig. S12 in ESI†). In the pre-organized intermediate IV, the acidic hydrogen on the α-carbon of the keto group would be deprotonated by TBD to form the enolate V. A second nucleophilic addition on the activated carbonyl group would lead to a five-membered ring tetrahedral intermediate VI. The release of the second phenoxy group would form intermediate VII, in which the phenoxy group would be protonated by TBD(+)-H to produce the desired vinylene carbonate VIII and other oligomers. The process could be repeated until the full depolymerisation of BPA-PC to BPA.
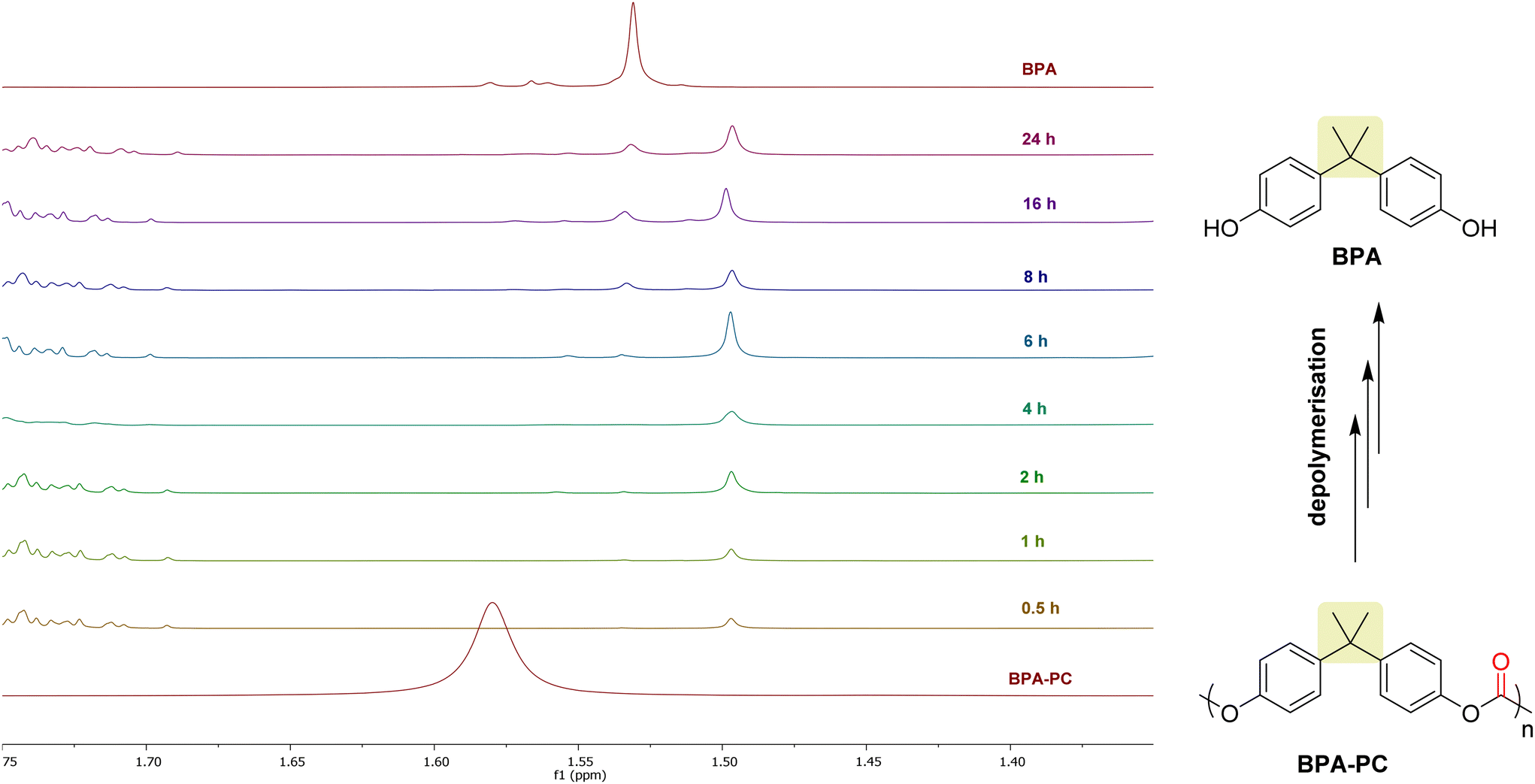
The depolymerization of BPA-PC was then studied using 1H-NMR at different intervals of time (from 30 min to 24 h) (Fig. 5). After 30 min, the BPA-PC signal around 1.58 ppm has completely disappeared showing that the depolymerization reaction is quite fast under our reaction conditions. Consequently, oligomers are rapidly formed and accumulate in the reaction mixture. However, the BPA signal is only clearly observed after 360 min and progressively increases in intensity until 1440 min (24 h). This indicates that the complete depolymerization to BPA is finally a relatively slow process under the reported conditions.
 | ||
| Fig. 5 Depolymerisation of BPA-PC followed by 1H-NMR (400 MHz, CDCl3, zoom of 1.30–1.75 ppm region corresponding to the gem-dimethyl group). Reaction conditions: benzoin 1 (1 equiv.), BPA-PC (1 equiv.), TBD (10 mol%), 2-MeTHF (2 mL), room temperature. Each aliquot is directly sampled from the reaction mixture and dissolved in CDCl3. Peaks at 1.49 ppm refers to free water found in chloroform. | ||
The depolymerisation of BPA-PC was also monitored by size-exclusion chromatography (SEC) under the optimized conditions (Fig. 6). At t = 0 min, the profile shows the presence of the polymer only (measured Mw of 42600 g mol−1, see Fig. S13† in ESI† for details). After 30 min, the high molecular weight region of BPA-PC has considerably shifted towards a small molecular weight peak identified as the BPA monomer. The presence of some small molecular weight oligomers can also be observed (shoulder on the right of the peak) but their quantity rapidly decreases, while the quantity of BPA progressively increases with reaction time. These observations indicate that the degradation mechanism of BPA-PC occurs through random chain scission rather than chain-end depolymerisation. A complementary observation was also made during these experiments.
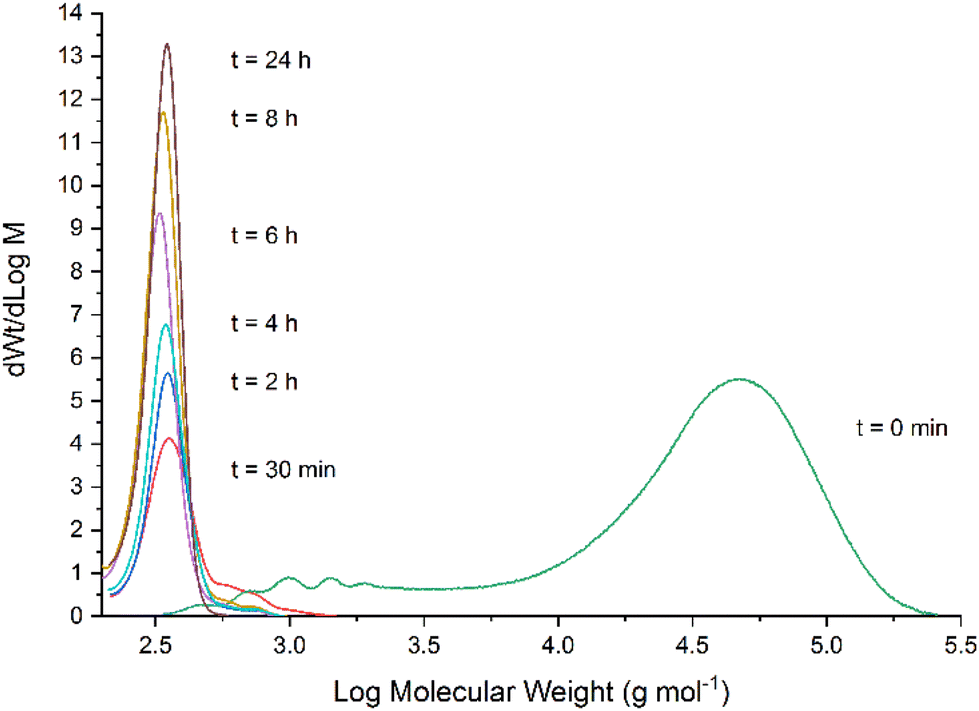 | ||
| Fig. 6 Measurement of the molecular weight distribution during the depolymerisation of BPA-PC as monitored by SEC. | ||
The original suspension of BPA-PC in 2-MeTHF turns almost instantaneously into a clear solution upon addition of TBD (benzoin was already solubilised in the reaction medium). This indicates that the soluble fraction of the polymer is rapidly degraded to oligomers and BPA, and the overall solubility of BPA-PC is driven by the depolymerisation. Therefore, this demonstrates that the organocatalyst is very effective under these conditions.
To demonstrate the synthetic utility of our protocols, two scale-up reactions (10-fold) were performed by pursuing two complementary objectives, namely, the synthesis of vinylene carbonates using BPA-PC as a carbonyl source (objective 1) and the recovery of BPA by full depolymerisation of BPA-PC using benzoin as the depolymerisation agent (objective 2). To reach the first objective, 10 mmol of benzoin were treated with 2 equivalents of BPA-PC and the desired vinylene carbonate 2 was obtained with 89% isolated yield (Fig. 7a). For the second objective, 10 mmol of BPA-PC were treated with 2 equivalents of benzoin, thus leading to the recovery of BPA in 98% yield (Fig. 7b). These results show that the ratio of reactants can be tuned to reach high yields of either vinylene carbonates or BPA, depending on which one is selected as the main target.
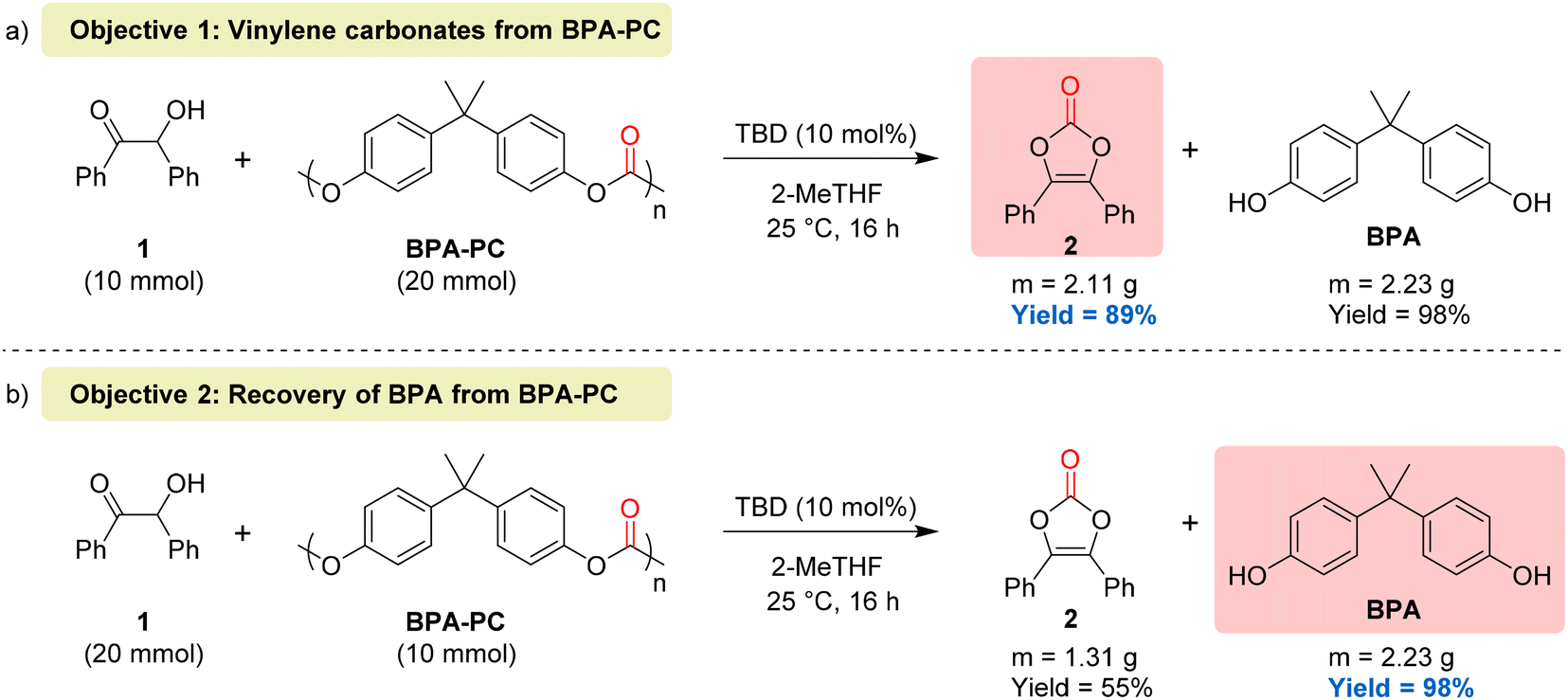 | ||
| Fig. 7 Scale-up reactions for (a) the synthesis of vinylene carbonates using BPA-PC as a carbonyl source (objective 1); (b) the recovery of BPA from BPA-PC using benzoin as the depolymerisation agent (objective 2). Yields are based on the limiting reagent. | ||
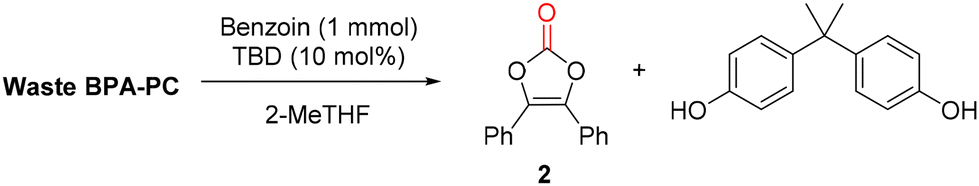
To go even further, the synthesis of vinylene carbonate 2 was attempted directly using waste poly(bisphenol A carbonate) (Mw = 32800 g mol−1, Đ = 2.03) contained in compact disks (CD) (Fig. 8). First, the CD was cut into pieces and 500 mg were dissolved in 2-MeTHF.41 The reaction was performed with benzoin 1 at 50 °C for 16 h. After reaction, the reaction mixture was treated with aqueous sodium hydroxide and extracted with EtOAc. The organic layer was separated, dried and evaporated to give vinylene carbonate 2 with 64% isolated yield, after recrystallisation. In parallel, the aqueous layer was treated with aqueous HCl, extracted with EtOAc, dried and evaporated to give BPA with 83% isolated yield, after recrystallisation. This protocol demonstrates that vinylene carbonates can be prepared from waste polycarbonate contained in CDs, in relatively mild conditions. An operationally simple separation procedure allows the recovery of both vinylene carbonate 2 and BPA without the need of column chromatography. Finally, it offers a relatively safe and economical access to vinylene carbonates, without the requirement of specialized equipment.
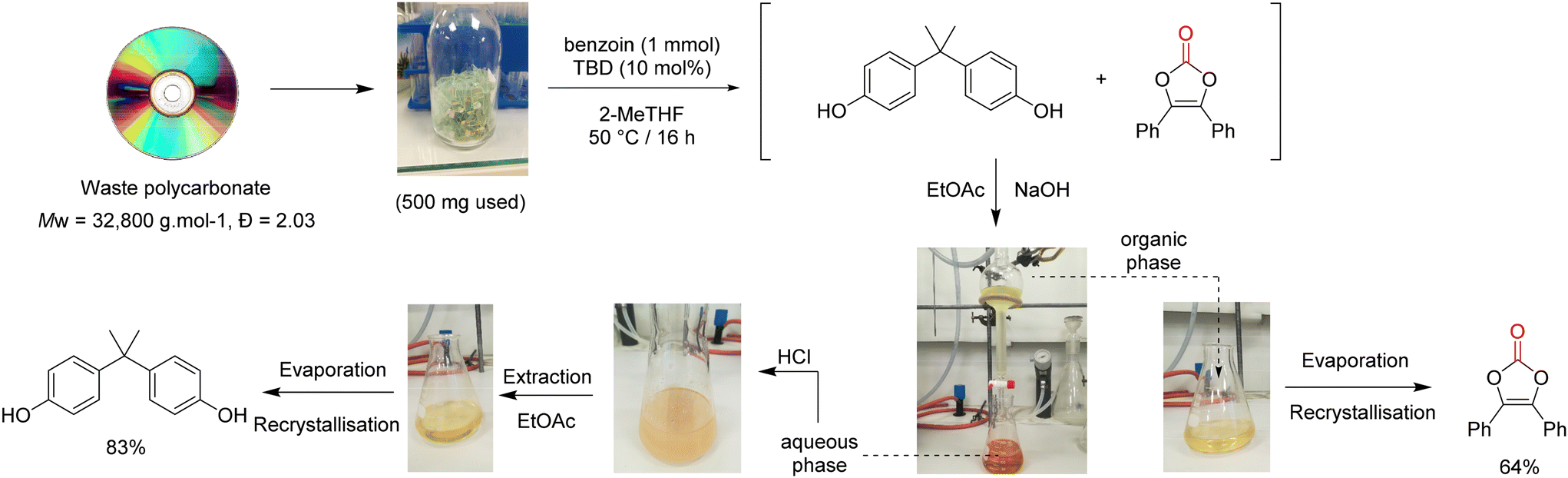 | ||
| Fig. 8 Recovery of vinylene carbonate and BPA from waste polycarbonate contained in compact disk (CD). | ||
Other waste materials containing poly(bisphenol A carbonate) were also considered (Table 3). Used safety glasses (Mw = 49200 g mol−1, Đ = 2.26) were treated at 50 °C for 16 h under the previously optimized conditions and gave vinylene carbonate 2 in 66% yield and BPA in 78% yield. These results are quite consistent with those obtained from the CD. Finally, a polycarbonate plate (Mw = 134000 g mol−1, Đ = 1.85) was depolymerised at 70 °C for 24 h. Under these conditions, vinylene carbonate 2 was only produced in 37% yield and BPA with 43% yield, thus showing that BPA-PC with higher molecular weight is much more difficult to depolymerise, notably due to solubility issues. Further optimization (e.g. harsher reaction conditions, longer reaction time) would be required to reach high yields with such materials.


Overall, the main advantage of this method is that the depolymerization of BPA-PC occurs at room temperature or at relatively mild temperature for waste materials. The recovered BPA can be recycled to the original polycarbonate or be used for preparing other polymers such as polyurethanes. The production of vinylene carbonates as co-products addresses other applications with higher added-value. For instance, vinylene carbonates can be used as electrolytes additives or as key intermediates to prepare prodrug derivatives.
Conclusions
In conclusion, we have developed an organocatalytic method for the synthesis of vinylene carbonates from benzoin and poly(bisphenol A carbonate), serving as a safe and easy-to-handle carbonyl source. The reaction is catalysed by TBD in 2-MeTHF at room temperature. The success of TBD was attributed to the fact that it can both activate the hydroxyl group of benzoin and the carbonyl group of BPA-PC, through hydrogen bonding. The reaction scope was investigated with a range of symmetrical (aryl or alkyl) and unsymmetrical α-hydroxyketones and the corresponding vinylene carbonates were obtained with 30–97% isolated yield (13 examples). It was shown that the method was amenable to scale-up (10×). Finally, it was also demonstrated that vinylene carbonates can be produced through the depolymerisation of waste BPA-polycarbonate contained in CDs and other waste materials thus offering a safer alternative to the use of phosgene.Author contributions
The project was conceptualized and supervised by N. Duguet, who also acquired the funding. K. Onida helped in the co-supervision of the project. K. Onida and M. Fayad performed the experiments and characterized the products. S. Norsic and O. Boyron performed the SEC experiments. The manuscript (original draft, review & editing) was written by N. Duguet. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the French National Agency for Research for financial support through a Ph.D. grant to K. Onida and a master grant to M. Fayad (ANR-19-CE07-0006-ThermoPESO). The authors also thank the Auvergne-Rhone-Alpes Region (SCUSI 2017 009361 01) for partial financial support.References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e170078 CrossRef PubMed
.
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed
- P. Rai, S. Mehrotra, S. Priya, E. Gnansounou and S. K. Sharma, Bioresour. Technol., 2021, 325, 124739 CrossRef CAS PubMed
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhoua and G. W. Huber, Green Chem., 2022, 24, 8899–9002 RSC
- J. Payne, P. McKeown and M. D. Jones, Polym. Degrad. Stab., 2019, 165, 170–181 CrossRef CAS
-
(a) M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS
- https://apnews.com/article/0251b75ab0de4d3080214e95a57bf0ee , data cited from “The Global Polycarbonates Market”.
- S. A. Vogel, Am. J. Public Health, 2009, 99, S559–S566 CrossRef PubMed
-
(a) R. Singh, S. Shahi and Geetanjali, ChemistrySelect, 2018, 3, 11957–11962 CrossRef CAS
-
(a) S.-J. Chiu, S.-H. Chen and C.-T. Tasi, Waste Manage., 2006, 26, 252–259 CrossRef CAS PubMed
-
(a) H. Tagaya, K. Katoh and J. Kadokawa, Polym. Degrad. Stab., 1999, 64, 289–292 CrossRef CAS
-
(a) C. Alberti, S. Eckelt and S. Enthaler, ChemistrySelect, 2019, 4, 12268–12271 CrossRef CAS
- L. Wang, G. Li, Y. Cong, A. Wang, X. Wang, T. Zhang and N. Li, Green Chem., 2021, 23, 3693–3699 RSC
-
(a) E. Quaranta, D. Sgherza and G. Tartaro, Green Chem., 2017, 19, 5422–5434 RSC
-
(a) R. Pinero, J. García and M. J. Cocero, Green Chem., 2005, 7, 380–387 RSC
-
(a) E. Quaranta, C. C. Minischetti and G. Tartaro, ACS Omega, 2018, 3, 7261–7268 CrossRef CAS PubMed
- C. Alberti, F. Scheliga and S. Enthaler, ChemistryOpen, 2019, 8, 822–827 CrossRef CAS PubMed
-
(a) E. Quaranta, A. Dibenedetto, F. Nocito and P. Fini, J. Hazard. Mater., 2021, 403, 123957 CrossRef CAS PubMed
- S. Hata, H. Goto, S. Tanaka and A. Oku, J. Appl. Polym. Sci., 2003, 90, 2959–2968 CrossRef CAS
-
(a) Z. Wang, R. Yang, G. Xu, T. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2022, 10, 4529–4537 CrossRef CAS
- H. J. Jung, S. Park, H. S. Lee, H. G. Shin, Y. Yoo, E. R. Baral, J. H. Lee, J. Kwak and J. G. Kim, ChemSusChem, 2021, 14, 4301–4306 CrossRef CAS PubMed
- M. S. Newman and R. W. Addor, J. Am. Chem. Soc., 1953, 75, 1263–1264 CrossRef CAS
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423–1439 CrossRef CAS
-
(a) L. Ding, Y. Li, Y. Li, Y. Liang and J. Huang, Eur. Polym. J., 2001, 37, 2453–2459 CrossRef CAS
-
(a) B. Stolz, F. Mönkemeyer, M. Mader, S. Schmidt, L. Volk, T. Steinberg, B. Bruchmann and R. Mülhaupt, Macromol. Chem. Phys., 2020, 221, 2000132 CrossRef
-
(a) M. E. Jung, R. B. Blum, B. J. Gnede and M. R. Gider, Heterocycles, 1989, 28, 93–97 CrossRef CAS
- For recent examples, see:
(a) J. Kitano, Y. Nishii and M. Miura, Org. Lett., 2022, 24, 5679–5683 CrossRef CAS PubMed
-
(a) K. S. Babu, M. S. Reddy, A. R. Tagore, G. S. Reddy, S. Sebastian, M. S. Varma, G. Venkateswarlu, A. Bhattacharya, P. P. Reddy and R. V. Anand, Synth. Commun., 2008, 39, 291–298 CrossRef
- S. Park, S. Y. Jeong, T. K. Lee, M. W. Park, H. Y. Lim, J. Sung, J. Cho, S. K. Kwak, S. Y. Hong and N.-S. Choi, Nat. Commun., 2021, 12, 838 CrossRef CAS PubMed
- M. S. Newman and R. W. Addor, J. Am. Chem. Soc., 1955, 77, 3789–3793 CrossRef CAS
-
(a) H.-M. Fischler, H.-G. Heine and W. Hartmann, Tetrahedron Lett., 1972, 17, 1701–1704 CrossRef
- D. P. Sahu, Indian J. Chem., 2002, 41B, 1722–1723 CAS
- M. M. Krayushkin, S. N. Ivanov, A. Yu. Martynkin, B. V. Lichitsky, A. A. Dudinov, L. G. Vorontsova, Z. A. Starikova and B. M. Uzhinov, Russ. Chem. Bull., Int. Ed., 2002, 51, 1731–1736 CrossRef CAS
- K. Onida, A. Haddleton, S. Norsic, C. Boisson, F. D'Agosto and N. Duguet, Adv. Synth. Catal., 2021, 363, 5129–5137 CrossRef CAS
- K. Onida, L. Ibrahimli and N. Duguet, Eur. J. Org. Chem., 2022, e202200153 CAS
- S. Fukuoka, I. Fukawa, T. Adachi, H. Fujita, N. Sugiyama and T. Sawa, Org. Process Res. Dev., 2019, 23, 145–169 CrossRef CAS
- C.-Q. Sun, P. T. W. Cheng, J. Stevenson, T. Dejneka, B. Brown, T. C. Wang, J. A. Robl and M. A. Poss, Tetrahedron Lett., 2002, 43, 1161–1164 CrossRef CAS
- H. Mutlu, J. Ruiz, S. C. Solleder and M. A. R. Meier, Green Chem., 2012, 14, 1728–1735 RSC
- C. Jehanno, J. Demarteau, D. Mantione, M. C. Arno, F. Ruipérez, J. L. Hedrick, A. P. Dove and H. Sardon, ACS Macro Lett., 2020, 9, 443–447 CrossRef CAS PubMed
- The calculations were done based on the hypothesis that the compact disk was composed of 100% BPA-PC, that is certainly not the case. With such hypothesis, 500 mg would correspond to approximatively 2 mmol of BPA-PC, based on the molecular weight of the repeating unit. Under these conditions, the calculated isolated yields of both vinylene carbonate and BPA are probably underestimated.
Footnote |
| † Electronic supplementary information (ESI) available: Procedures, characterisation data of products, 1H and 13C NMR spectra, mass. See DOI: https://doi.org/10.1039/d2gc04413g |
| This journal is © The Royal Society of Chemistry 2023 |