DOI:
10.1039/D2SC02573F
(Perspective)
Chem. Sci., 2022,
13, 8491-8506
Radical transformations for allene synthesis
Received
9th May 2022
, Accepted 28th June 2022
First published on 29th June 2022
Abstract
Allenes are valuable organic molecules that feature unique physical and chemical properties. They are not only often found in natural products, but also act as versatile building blocks for the access of complex molecular targets, such as natural products, pharmaceuticals, and functional materials. Therefore, many remarkable and elegant methodologies have been established for the synthesis of allenes. Recently, more and more methods for radical synthesis of allenes have been developed, clearly emphasizing the associated great synthetic values. In this perspective, we will discuss recent important advances in the synthesis of allenes via radical intermediates by categorizing them into different types of substrates as well as distinct catalytic systems. The mechanistic studies and synthetic challenges will be highlighted.
Introduction
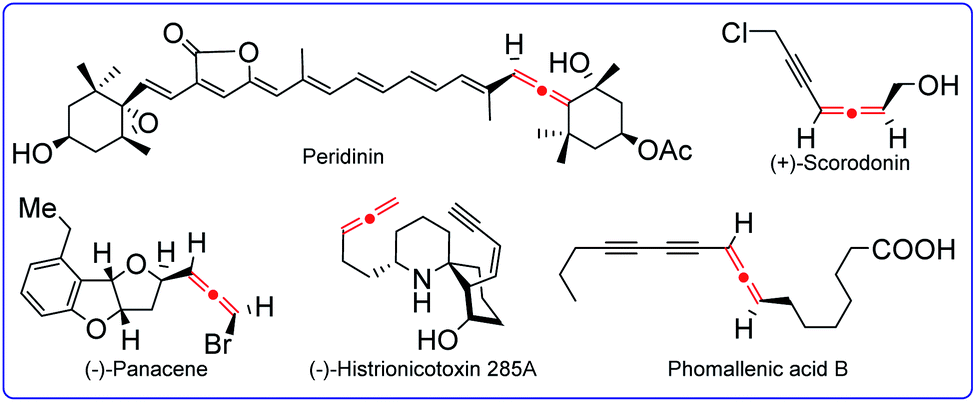
Allenes represent an important class of organic molecules that feature unique twisted orthogonal π-systems.1 They are not only key moieties found in many natural products and pharmaceuticals, but also serve as versatile synthons in the fields of modern organic synthesis and bioactive molecule design (Fig. 1).2 Since the first allene synthesis by Burton and Pechmann in 1887,3 a wide array of approaches for allene synthesis have been well documented, such as prototropic isomerization of alkynes, sigmatropic rearrangement of propargylics, addition to 1,3-enynes, 1,2-eliminations, nucleophilic substitutions, and other efficient methods. As a consequence of their rapid growth in recent decades, allene synthesis has been covered by quite some remarkable and elegant reviews.4 With the aim of developing more and more reliable methods, the applications of radical species in the synthesis of allenes have been rapidly booming, and the potential of this radical strategy has been nicely certificated recently. This perspective highlights the selected updated examples of radical synthesis of allenes to declare the current status of this area.5
 |
| | Fig. 1 Natural products containing an allenyl skeleton. | |
As an important part of the blueprint for allene synthesis, the radical strategy can be generally realized through the following approaches. (1) Radical 1,4-addition to 1,3-enynes: different from classic methods for the synthesis of allenes that usually introduce only one functional group into products, radical 1,4-difunctionalization of 1,3-enynes has the advantage to simultaneously incorporate two functional groups into allenes. (2) Radical synthesis of allenes from propargylic compounds, such as propargyl bromides, propargyl amides, propargylic carbonates, and propargylic C–H bonds. (3) Radical 1,2-eliminations: for example, radical 1,2-elimination of vinylsulfoxides by Malacria et al. about two decades ago.6 (4) Further functionalization of the allenyl C–H bond. These radical protocols have the advantages not only to expand reaction diversity by employing different type of partners and catalysts, but also to provide some remarkable virtues for the synthesis of value-added allenes that are otherwise challenging to be constructed, finally making them excellent supplements to classical non-radical strategies for the synthesis of allene.
Radical synthesis of allenes from 1,3-enynes
1,3-Enynes are common and useful building blocks in organic synthesis. For examples, with the assistance of organometallic reagents, the groups of Ma,7 Kambe,8 Yoshida,9 and Kimura10 have built elegant methods for the synthesis of allenes through nucleophilic 1,4-difunctionalization, respectively. In this part, we will disclose their applications in radical synthesis of allenes. Generally, the addition of a radical to the C–C double bond of an 1,3-enyne generates a propargylic radical species, which can resonance to an allenyl radical species, and the allene product will be sequentially produced. However, propargylic radicals were historically regarded as an intermediate with low reactivity and therefore little attention has been paid to the synthetic utility of these species.11 Till 2006, the Zard group discovered that if the propargylic radicals can be trapped intramolecularly, 1,3-enynes were excellent substrates for the synthesis of allenes.12 But, how to realize the intermolecular or multicomponent synthesis of allenes from 1,3-enynes remains a great challenge.
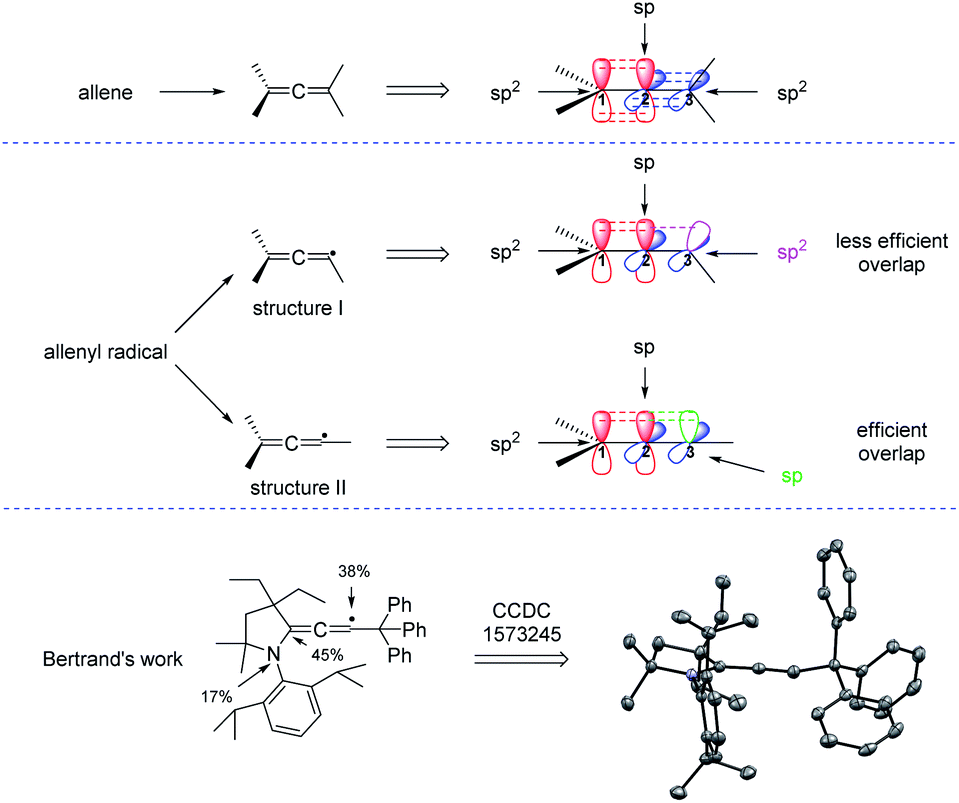
Before reviewing the recent developments on radical synthesis of allenes from 1,3-enynes, it is necessary to discuss the thermodynamically stable configuration of the allenyl radical. In the typical structure of an allene, while the two terminal carbons C1 and C3 are sp2-hybridized, the central carbon C2 adopts sp hybridization, making both the two π bonds and the terminal two carbon groups perpendicular. But, which kind of hybridization will the carbon C3 of the allenyl radical be? In literature, the allenyl radical is usually drawn in two different forms: structure I or structure II. As can be seen from Fig. 2, the carbon C3 should be sp2-hybridized in structure I and is sp-hybridized in structure II. The more efficient overlap between the π bond and the orbital of the single electron suggests that structure II is thermodynamically more stable than structure I. Actually, Bertrand et al. reported a first characterization of an allenyl radical by single crystal X-ray crystallography13 and his work supports that the allenyl radical with the configuration of structure II is more stable.
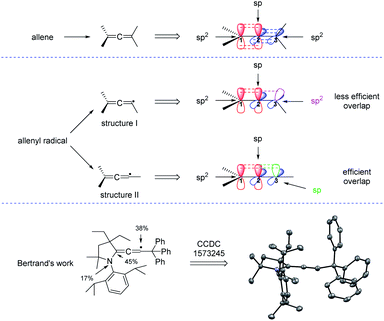 |
| | Fig. 2 Configurations and X-ray crystallography of the allenyl radical. | |
Copper catalysis
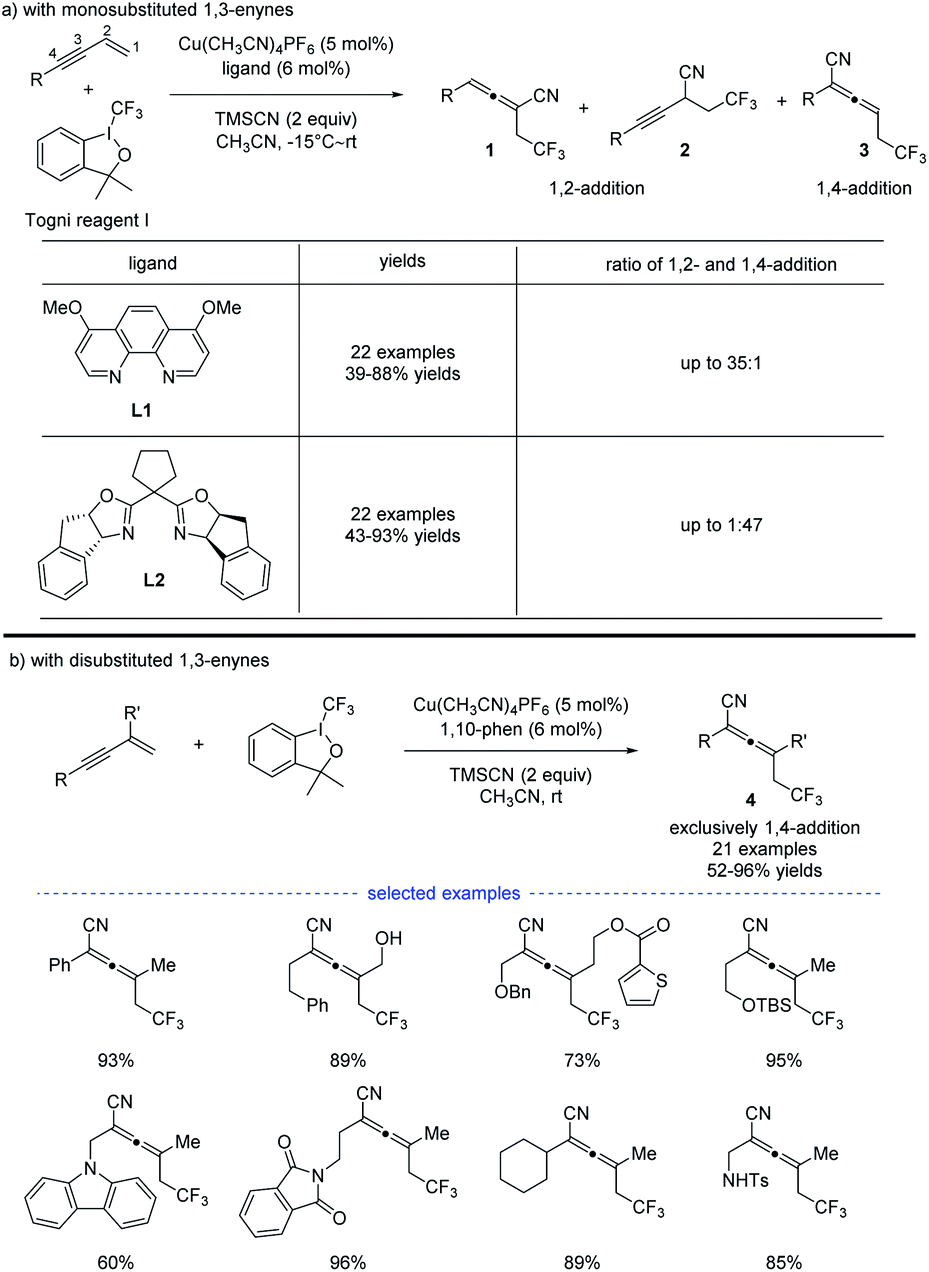
In 2018, G. Liu, Lin, and coworkers reported a copper-catalyzed divergent synthesis of allenes with an unusual ligand-controlled system (Scheme 1).14 In this three-component radical reaction, a wide range of CF3-substituted allenyl nitriles 1 and 3 can be regioselectively synthesized under mild conditions. Notably, allenyl nitriles 1 were isomerized in situ from propargyl nitriles 2.15 While a substituted 1,10-phenanthroline (1,10-phen) ligand L1 dominantly led to 1,4-adducts, the box ligand L2 was found to mainly promote the generation of 1,2-adducts. Functional groups, such as amides, esters, internal alkenyl groups, and silyl ethers, can be well tolerated to produce the corresponding products in good to excellent yields. Interestingly, when C2 position was sterically substituted, the corresponding tetrasubstituted allenes 4 were exclusively generated, and no trace of the propargyl nitriles bearing a quaternary carbon center was detected.
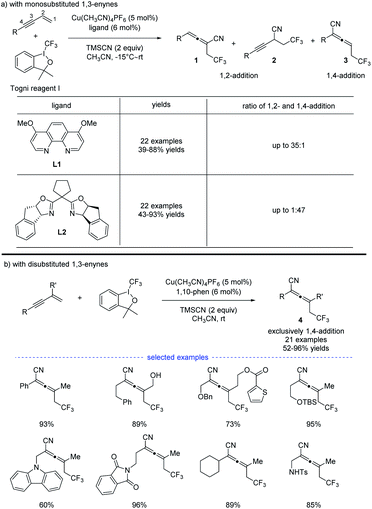 |
| | Scheme 1 Cu-catalyzed regioselective trifluoromethylcyanation of 1,3-enynes. | |
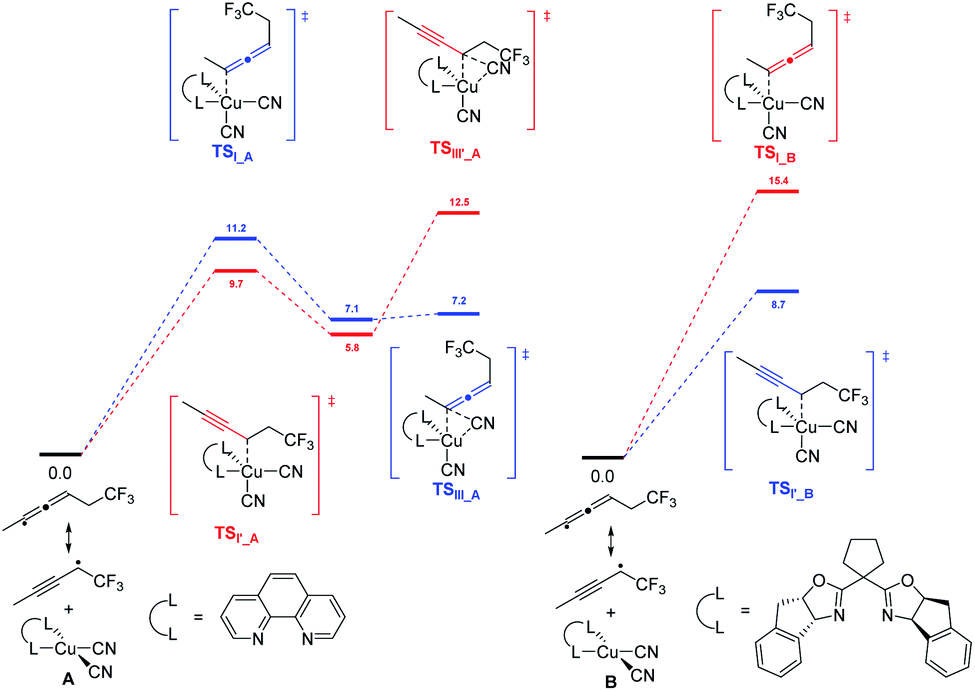
DFT calculation revealed that, in the presence of 1,10-phen as the ligand, energy barrier of the transition state for the formation of TSI–A was 1.5 kcal mol−1 higher than that of TSIʹ–A (Scheme 2). When ligand L2 was used as the ligand, energy barrier of TSI–B is 6.7 kcal mol−1 higher than that of TSIʹ–B. The result clearly indicates that the ligand 1,10-phen promotes the 1,4-adducts, while L2 favors the 1,2-adducts. Moreover, the reaction was supposed to undergo a copper(III) species and the corresponding products can be generated from sequential reductive elimination.
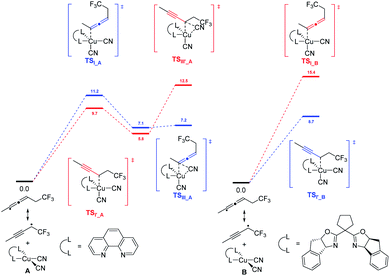 |
| | Scheme 2 Free energy profiles calculated for the reaction pathways leading to 1,4- and 1,2-additions of 1,3-enynes under different ligand conditions; (left) for 1,10-phen and (right) for L2. The favored free energy profiles are in bule. | |
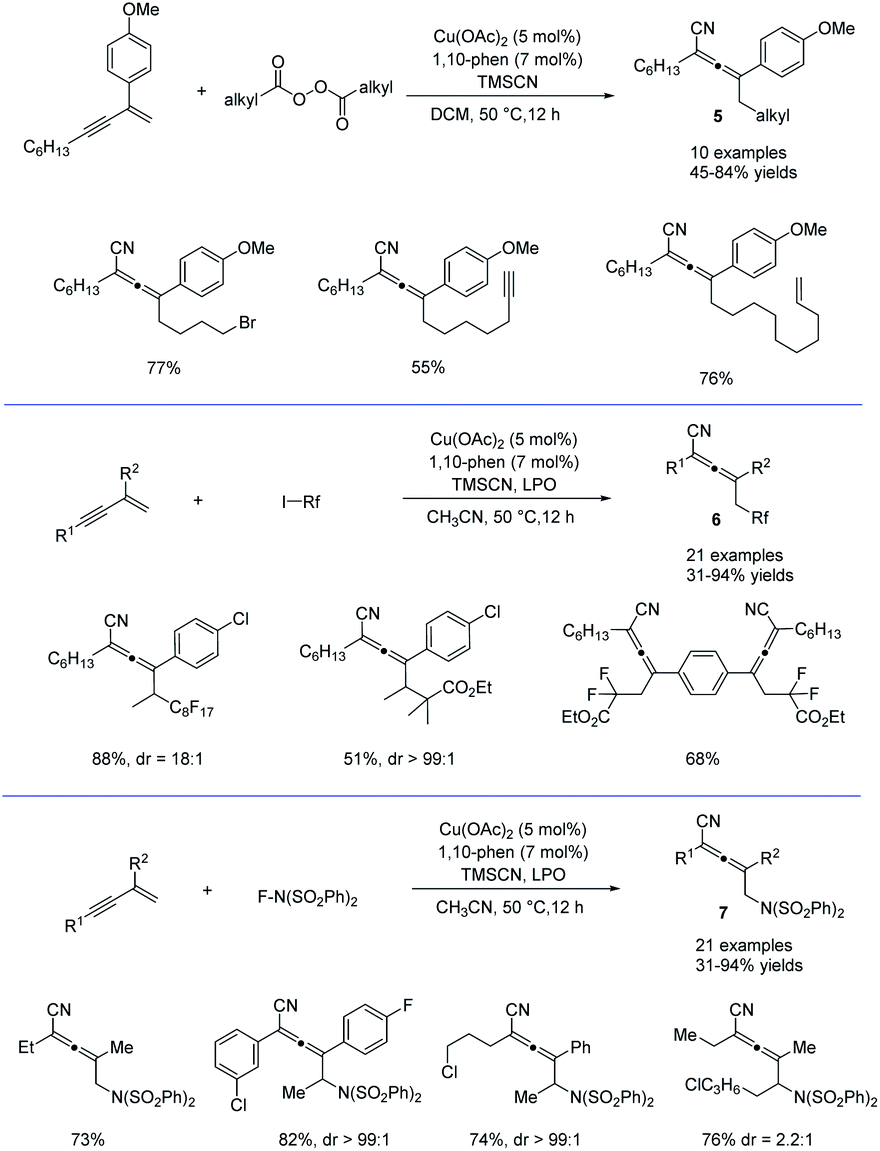
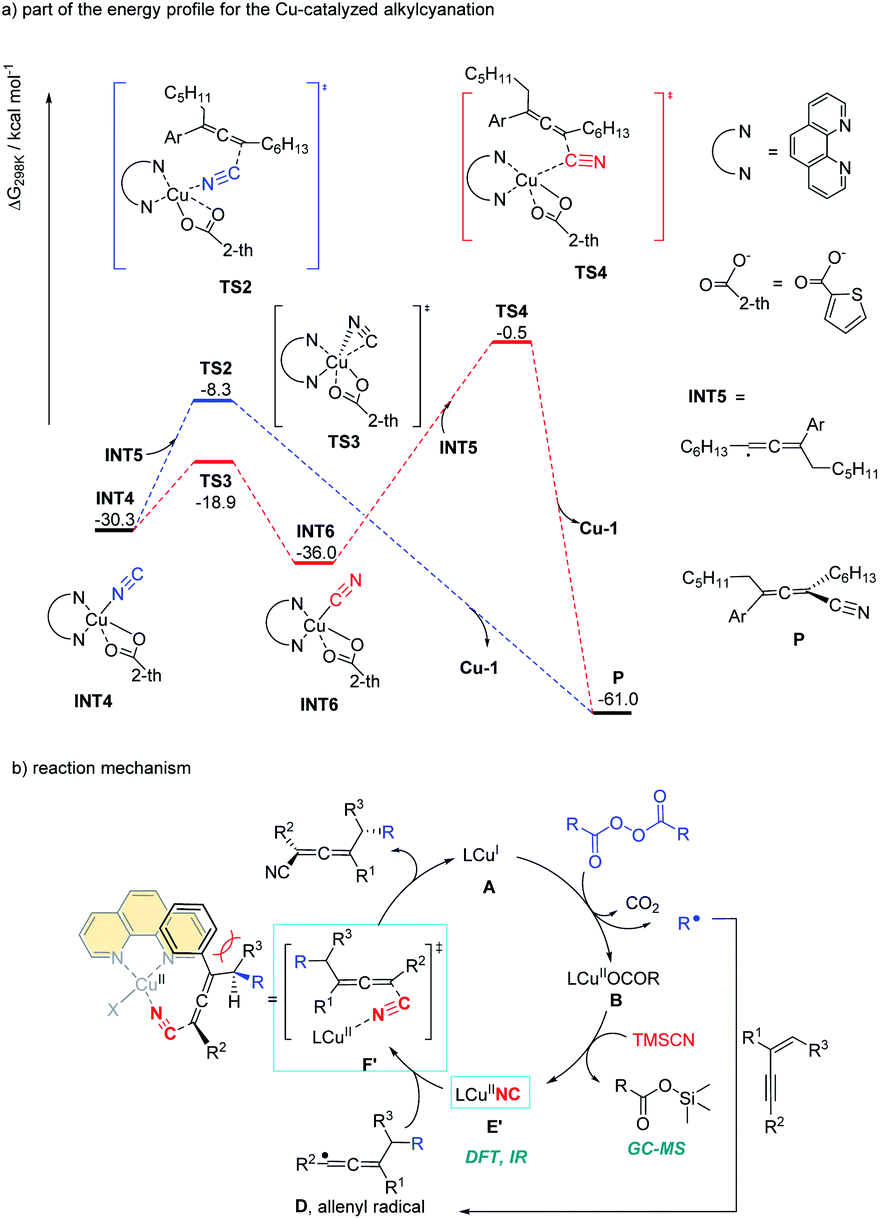
Alkyl peroxides have long-term been recognized as oxidants or radical initiators. The group of Bao discovered that alkyl peroxides actually is a type of masked trifunctional reagents: oxidants, radical initiators, and functional-group releasing agents.16 Later in 2018, Bao, Zhang, and coworkers disclosed that with alkyl diacyl peroxides as the trifunctional reagents, the copper-catalyzed intermolecular 1,4-carbocyanation of 1,3-enynes under mild conditions is a versatile method for the generation of tetrasubstituted allenes 5 and 6 (Scheme 3).17 Not only generic alkyl groups but also functionalized alkyl groups can be used as the carbon sources. Additional functionality such as bromo, carbonyl, alkenyl, alkynyl, or ester groups were well tolerated. Moreover, when activated organic iodides, such as fluorinated alkyl iodides and α-iodoacetates, were used as the substrates, the alkyl radicals generated from alkyl peroxides would abstract the iodine atom before adding to 1,3-enynes. In addition, N-fluorobenzenesulfonimide (NFSI) also performed well under mild reaction conditions and the reaction with 1,3-enynes afforded 1,4-sulfimidocyanation products 7 with good yields and excellent diastereoselectivity. The results of mechanistic studies do not support involvement of the allenyl cation and a mechanism with the formation of allenyl radicals was proposed accordingly. Interestingly, different from Liu's proposal of the CuIII(CN)2 intermediate, DTF calculation in Bao's work suggested that an isocyanocopper species CuIINC was the key intermediate and the final allene product can be produced through the abstract of the isocyano (NC) group directly from the CuIINC species by the allenyl radical without forming a high-valent copper(III) species (Scheme 4). The presence of the isocyanocopper was supported by IR spectroscopy studies. In addition, the diastereoselectivity was found to be controlled by whether π–π stacking interaction between the allenyl radical and the ligand was presented.
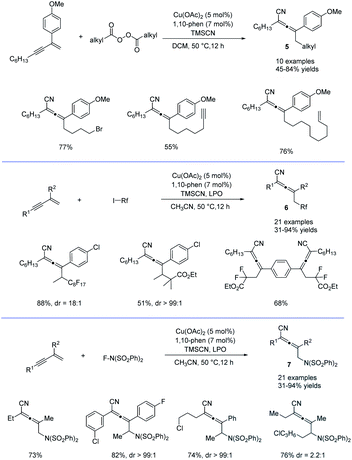 |
| | Scheme 3 Cu-catalyzed regioselective 1,4-difunctionalization of 1,3-enynes. | |
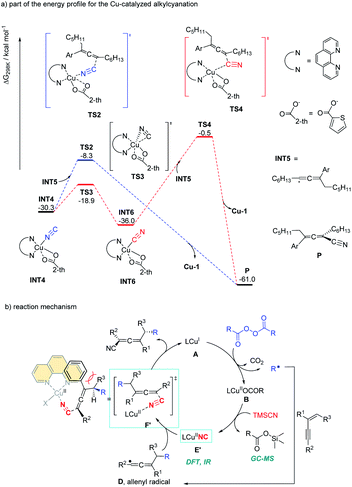 |
| | Scheme 4 Gibbs free energy profile for the Cu-catalyzed alkylcyanation and catalytic cycle for 1,4-alkylcyanation of 1,3-enynes. | |
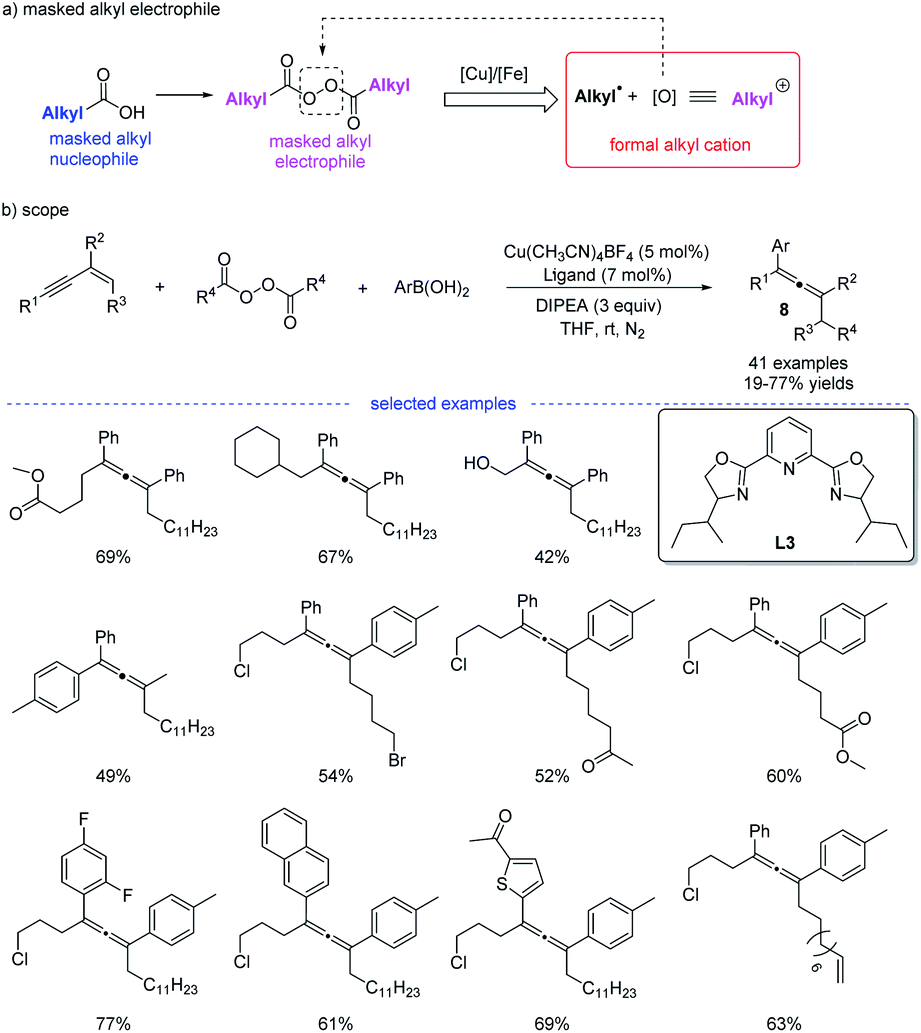
Shortly after, in continuation of their studies,18 Bao et al. developed a copper-catalyzed 1,4-alkylarylation of 1,3-enynes (Scheme 5).19 Ligand screening revealed that a tridentate Py-box ligand L3 promoted the reaction well and the reaction afforded a wide range of heavily substituted allenes 8 at room temperature. More than 40 examples have been tested in terms of alkyl diacyl peroxides, 1,3-enynes, and aryl boronic acids. The radical trapping reaction, radical clock reactions, and radical dimerization suggested that this reaction proceeded through a radical relay pathway. Moreover, in this study, it is found that alkyl diacyl peroxides acts as a kind of masked alkyl electrophiles, which are quite different from the commonly used nucleophilic organometallic reagents.
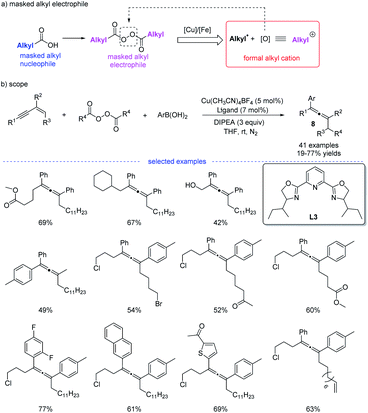 |
| | Scheme 5 Cu-catalyzed regioselective 1,4-carboarylation of 1,3-enynes. | |
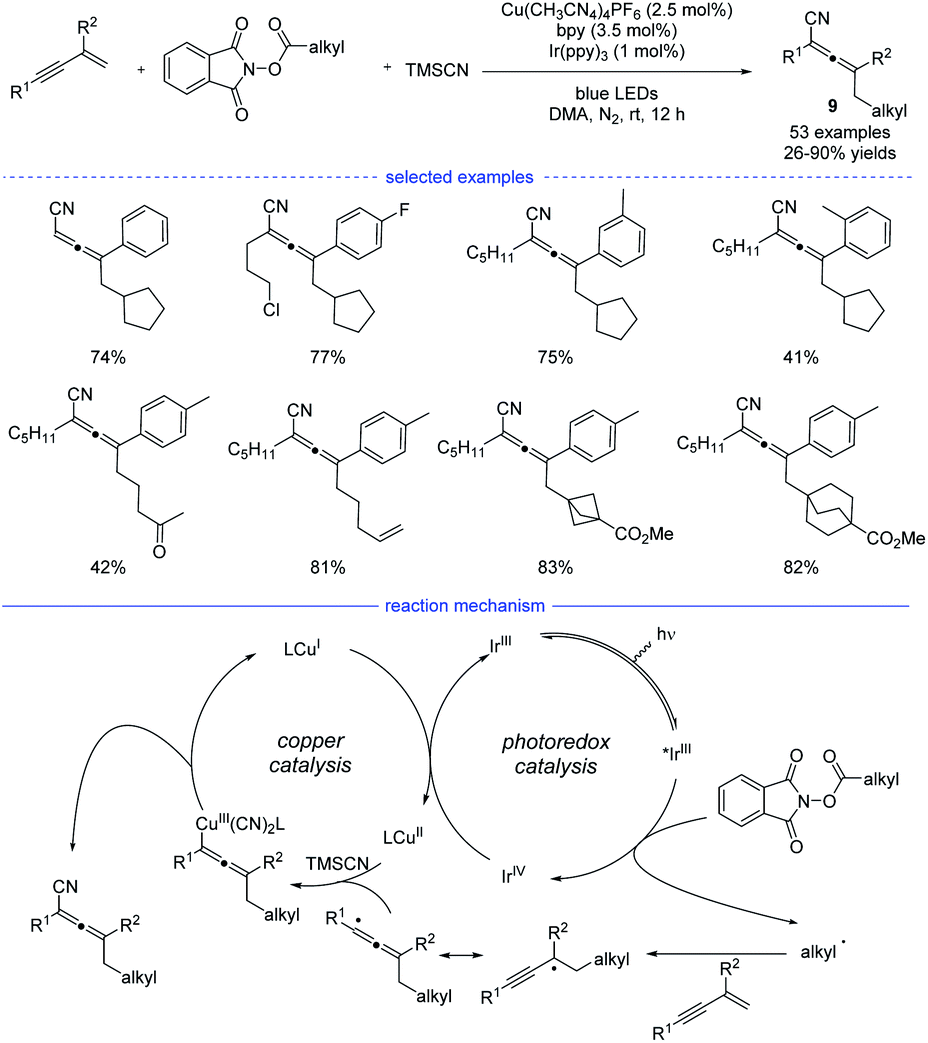
In 2021, Lu et al. reported a general strategy for 1,4-alkylcyanation of 1,3-enynes with redox-active esters by merged visible light photoredox catalysis and copper catalysis (Scheme 6).20 Alkyl N-hydroxyphthalimide esters (redox-active esters) were used as the alkyl radical precursors and a range of structurally diverse multisubstituted allenes 9 with remarkable functional group tolerance can be smoothly generated under blue light irradiation. Control experiments such as radical trapping reaction and radical ring-opening reaction suggested that the allenyl radical was involved in the catalytic cycle. It was also found that further intramolecular five-membered ring cyclization of the allenyl radical was not occurred, which presumably suggested that the cyanation step is much faster than further cyclization. Therefore, an oxidative quenching mechanism for this reaction was proposed with redox-active esters acting the fluorescent quenching reagents.
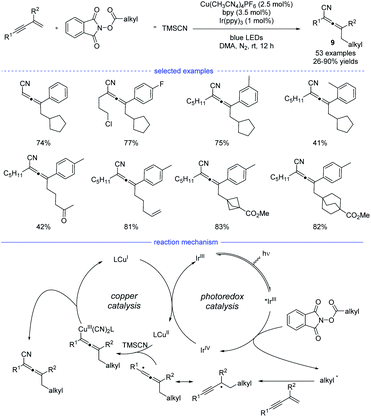 |
| | Scheme 6 Cu-catalyzed regioselective 1,4-carbocyantion of 1,3-enynes with redox-active esters. | |
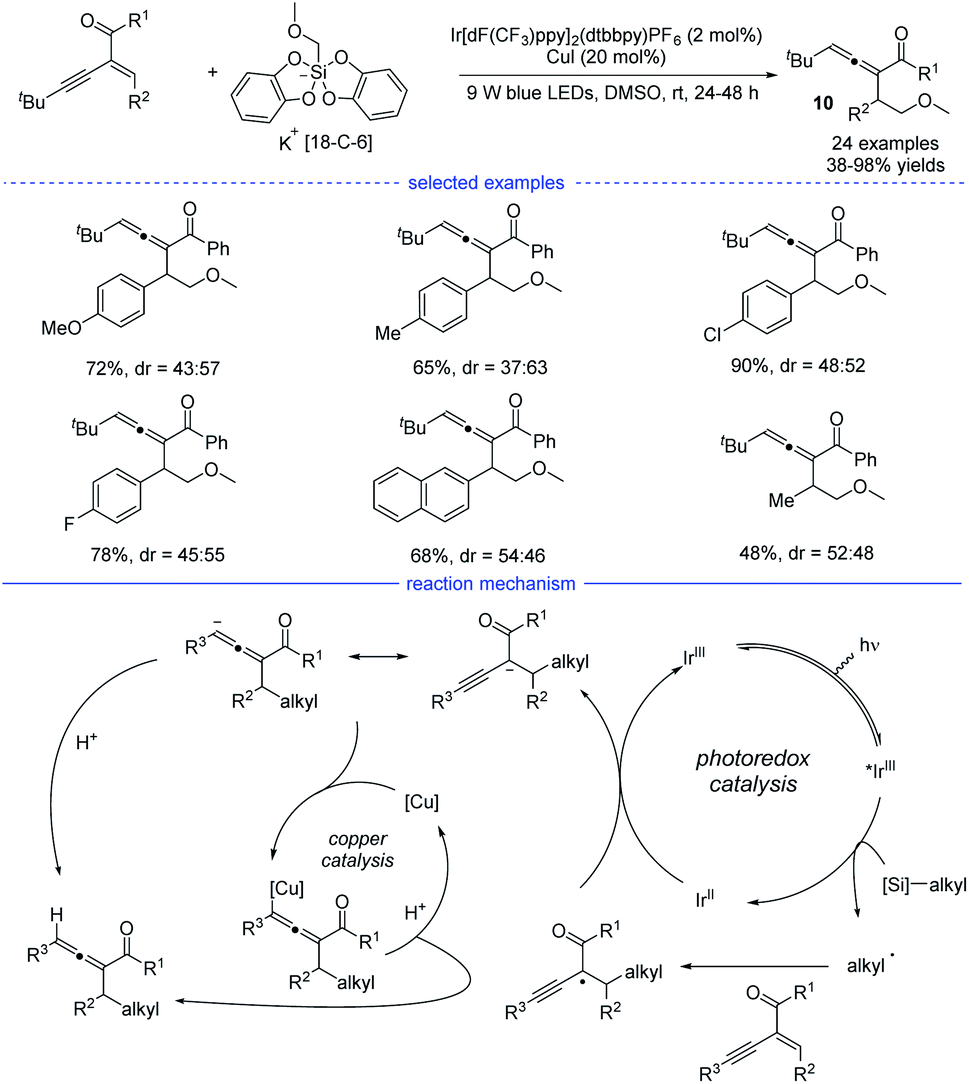
Shortly after, another examples of allene synthesis by dual photoredox/copper catalysis was developed (Scheme 7).21 A series of alkyl bis(catecholato)silicates can release the corresponding alkyl radicals under blue light irradiation and the carbonyl-activated 1,3-enynes was transformed into 1,2-allenyl ketones under mild conditions. The results of a deuteration experiment and the competition reaction between cyclopropanation and allenation suggested that a radical-polar crossover process should account for the allene generation. Therefore, a reductive quenching mechanism was proposed and the carbonyl-stabilized propargyl radical quenched the activated iridium catalyst. However, the role of the copper catalyst was not clearly illustrated.
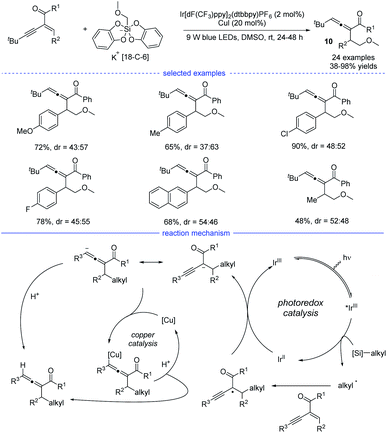 |
| | Scheme 7 Cu/Ir co-catalyzed synthesis of allenes from 1,3-enynes and alkyl bis(catecholato)silicates. | |
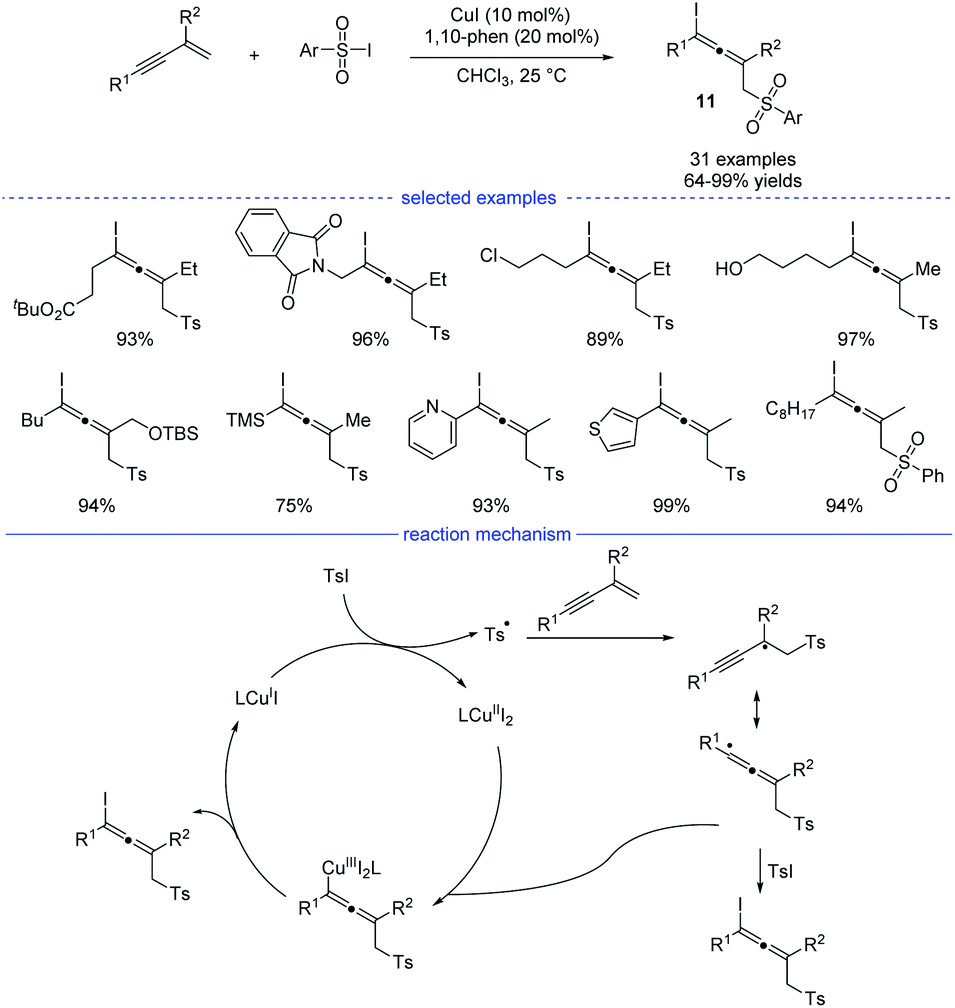
As a kind of important building blocks in organic synthesis, allenyl halides are commonly obtained from (1) SN2-type nucleophilic substitution reaction of propargyl mesylates and (2) electrophilic cyclization of 1,3-enynes bearing an intramolecular nucleophilic unit.22 In 2019, Ma and coworkers developed a copper-catalyzed efficient method for the synthesis of allenyl halides 11 under mild conditions (Scheme 8).23 Sulfonyl iodides was used as both the sulfonyl radical source and the iodine atom source. If sulfonyl bromides were used as the substrates, propargyl bromides rather than allenyl bromides would be generated, together with some amounts of the recovered sulfonyl bromides. Moreover, without the C2-substituents, the reactions with 1,3-enynes led to the formation of propargyl iodides as the major products. Under the standard conditions, many allenyl halides can be produced with the tolerance of functional groups such as ester, halide, amide, free alcohol, silyl ether, cyano, and heteroaryl. In terms of mechanism, single-electron transfer between copper catalyst LCuI and sulfonyl iodide delivered a copper(II) species and a sulfonyl radical which would lead to the formation of a propargyl radical after the addition to an 1,3-enyne. The propargyl radical would resonate to an allenyl radical and the final product formed through two possible pathways: (1) directly grab the iodine atom from the sulfonyl iodide; (2) elimination from a copper(III) species generated from the allenyl radical and LCuII.
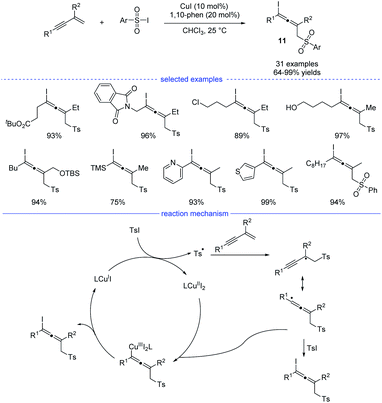 |
| | Scheme 8 Cu-catalyzed 1,4-sulfonyliodination of 1,3-enynes with sulfonyl iodides. | |
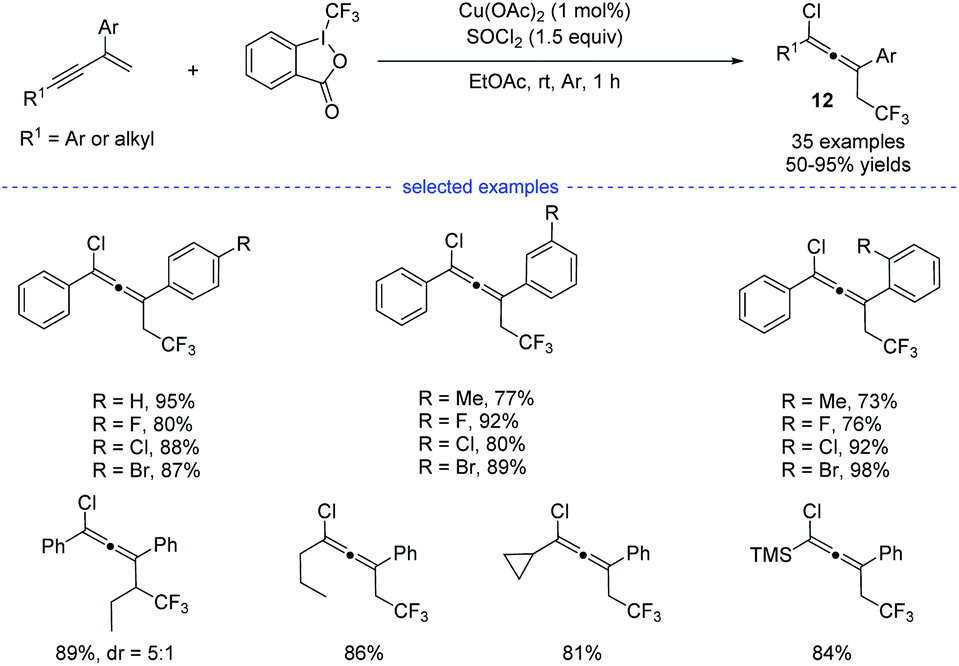
Later on, Yang et al. reported a copper-catalyzed three-component synthesis of allenyl chlorides with Togni reagent II and SOCl2 (Scheme 9).24 Solvent optimization showed that the use of ethyl acetate can maximumly prevent the formation of Heck-type side products. A wide range of 1,3-enynes were compatible with this transformation and the allenyl chlorides 12 were obtained with good to excellent yields and good functional group tolerance. Similarly, the final products were proposed to be eliminated from the high-valent copper(III) species.
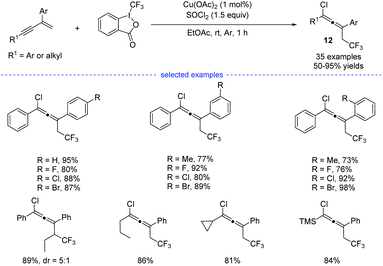 |
| | Scheme 9 Cu-catalyzed three-component 1,4-chlorotrifluoromethylation of 1,3-enynes. | |
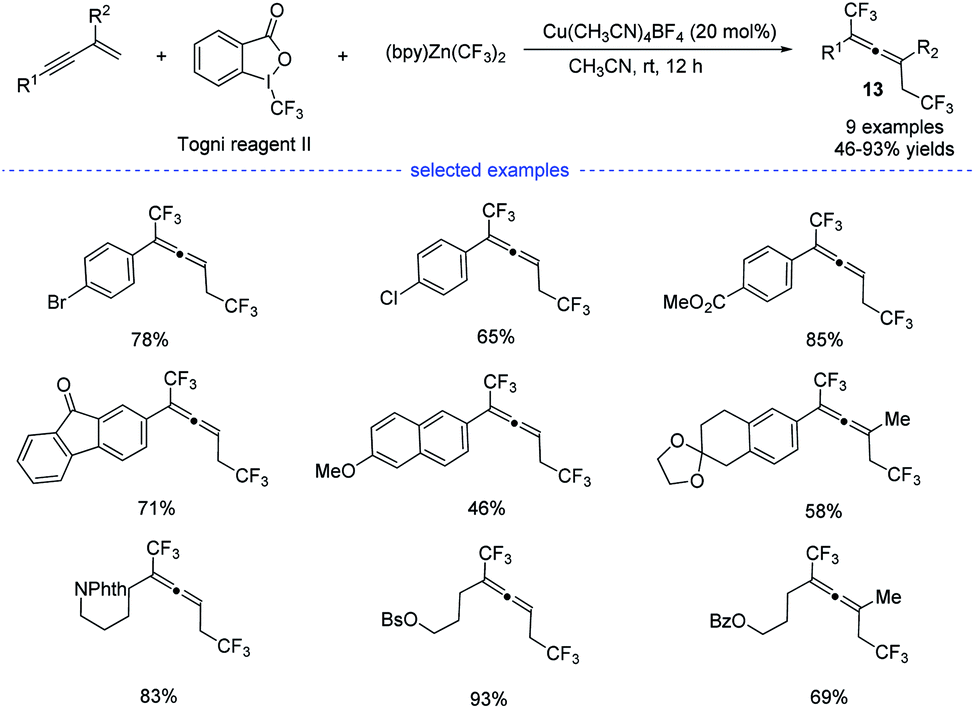
In 2019, by using Togni reagent II and (bpy)Zn(CF3)2, Li et al. disclosed a copper-catalyzed radical bis(trifluoromethylation) of 1,3-enynes at room temperature, allowing the exclusive formation of the corresponding allenes 13 (Scheme 10).25 Not only aryl-substituted 1,3-enynes but also alkyl-substituted 1,3-enynes performed well to produce trifluoromethylated allenes that typically require the use of propargyl halides, esters, or tosylates as the substrates.
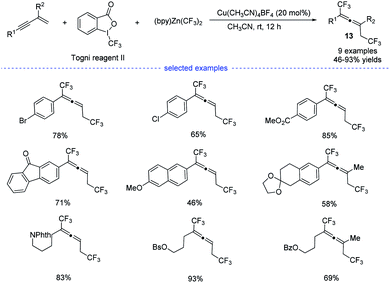 |
| | Scheme 10 Cu-catalyzed three-component 1,4-bisitrifluoromethylation of 1,3-enynes. | |
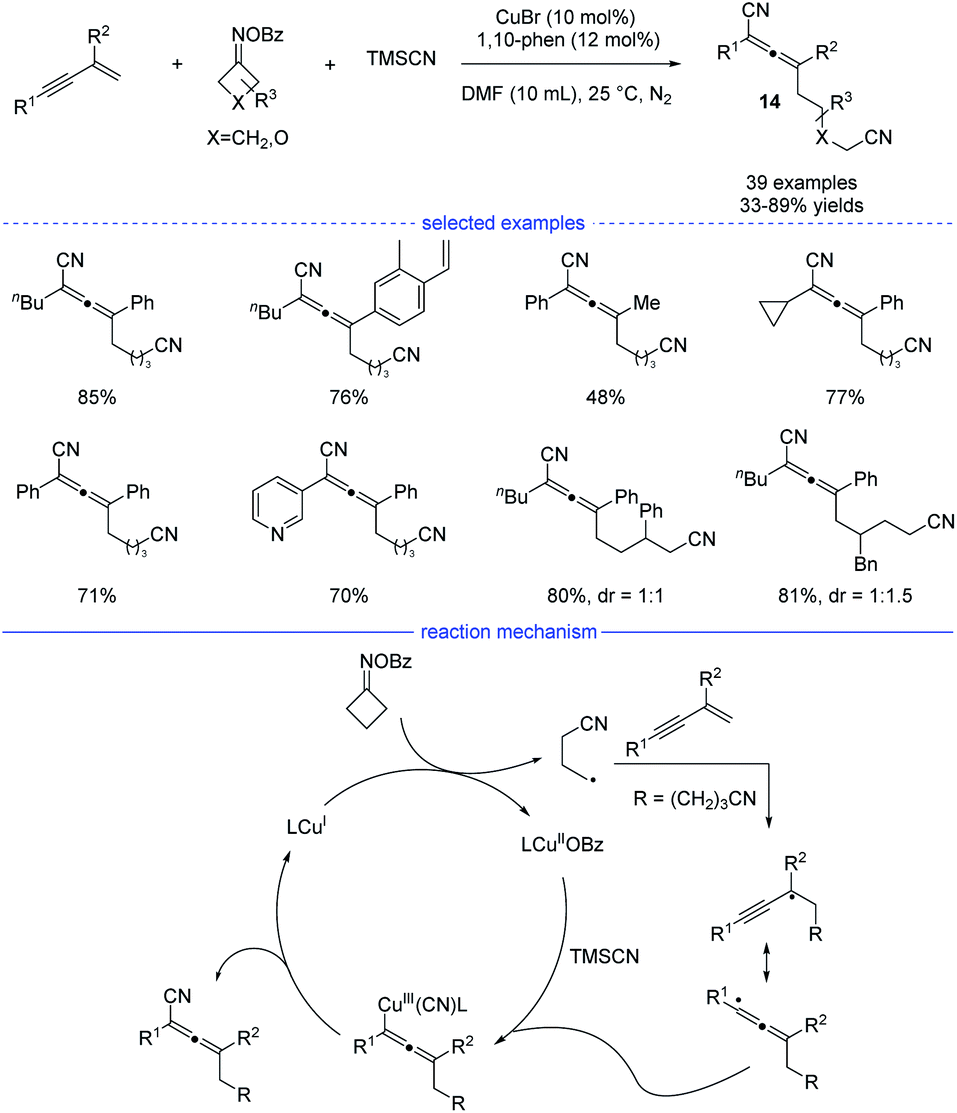
Cyclobutanone oxime esters are good precursors for the generation of cyanoalkyl radicals after single-electron transfer-induced ring-opening.26 In 2021, Ma and coworkers reported a copper-catalyzed synthesis of allenes through ring-opening of cyclobutanone oxime esters (Scheme 11).27 This mild and facile protocol features a broad substrate scope and tolerate many functional groups. Under the optimal reaction conditions, side reactions such as Heck-type alkylation, 1,2-addition, and 3,4-addition can be greatly suppressed. While a radical trapping experiment suggested the engagement of the cyanoalkyl radicals, the result of a carbanion capture experiment with d4-MeOH was negative. These results illustrate that the carbanion species was not involved in the reaction. Moreover, the optimal conditions not only work well for the generation of cyanated allenes from TMSCN, but also are suitable for the produce of trifluoromethylated allenes from TMSCF3. On the basic of the mechanistic experimental results, a possible radical mechanism was proposed. SET process between the CuI species and a cyclobutanone oxime ester afforded a CuII species and the cyanoalkyl radical. The latter one would undergo regioselective addition to form a propargyl radical that transformed into allenyl radical in a rapid manner to release the steric hinderance. The final allene product was then produced either through subsequent reductive elimination from the high-valent allenyl copper(III) species or by abstraction of the CN ligand from the CuIICN species.
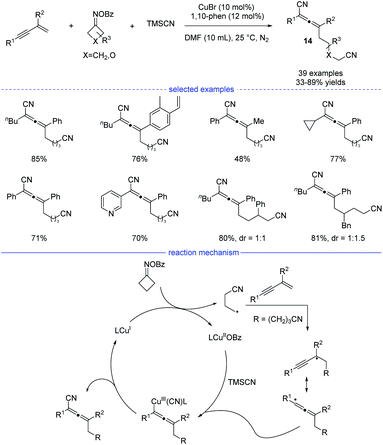 |
| | Scheme 11 Cu-catalyzed three-component synthesis of allenes from 1,3-enynes, cyclobutanone oxime esters, and TMSCN. | |
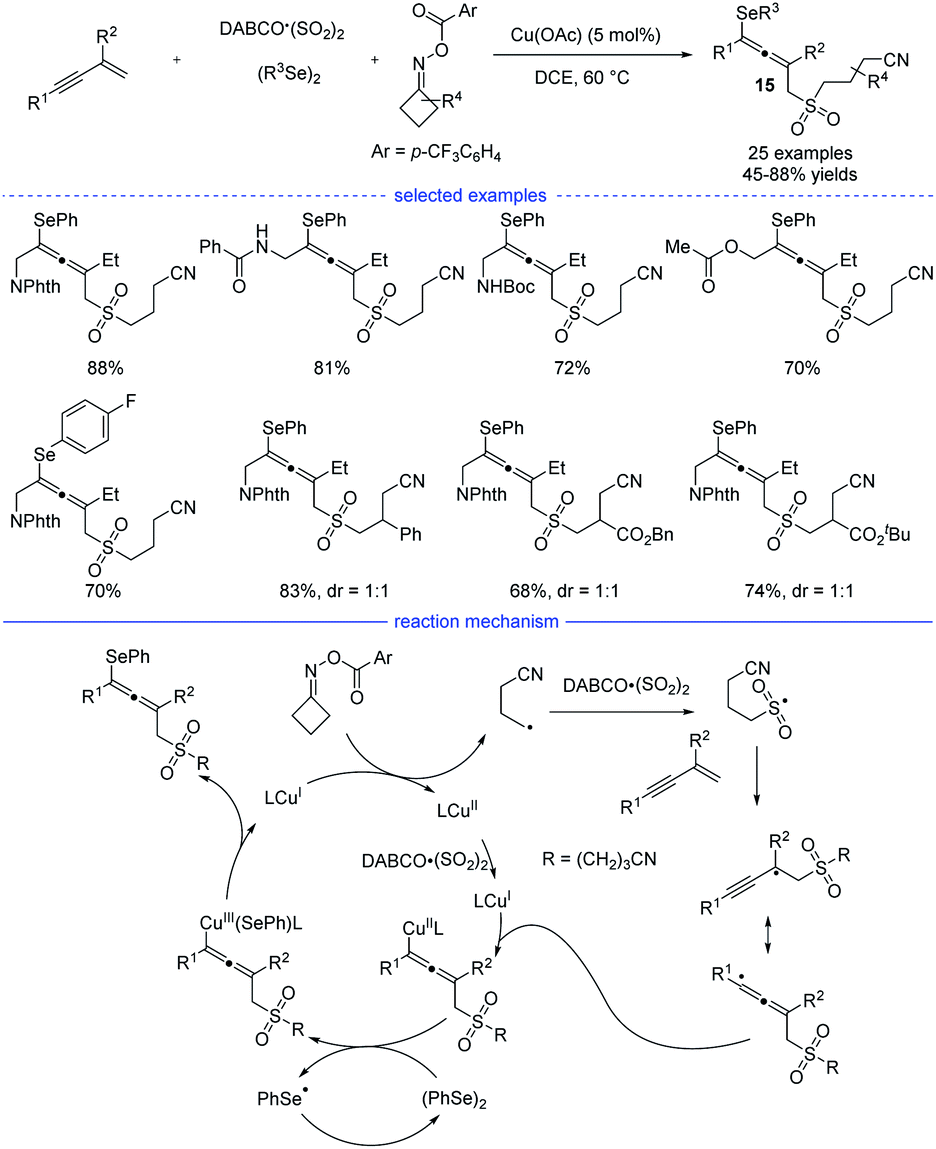
Shortly after Ma's work, Wu, He, and coworkers developed a four-component synthesis of allenes taking advantages of the sulfonyl-group insertion process (Scheme 12).28 Under the optimal copper catalysis, the reactions with 1,3-enynes, diselenides, DABCO·(SO2)2, and cyclobutanone oxime esters provided a facile access to many kind of cyanoalkylsulfonylated allenyl selenides 15 in moderate to good yields. The key step lies in sulfonyl-group insertion before the regioselective addition of the alkyl group onto the 1,3-enyne. Very recently, Qiu et al. revealed a three-component reaction of 1,3-enynes and cyclobutanone oxime esters in the presence of aryl boronic acids or organozinc reagents via the photoredox/copper or photoredox/nickel catalysis.29
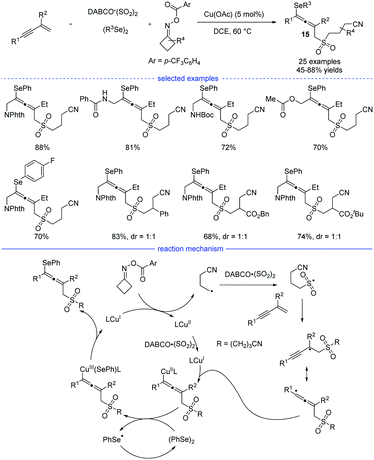 |
| | Scheme 12 Cu-catalyzed four-component synthesis of allenes from 1,3-enynes, cyclobutanone oxime esters, diselenides, and TMSCN. | |
Nickel catalysis
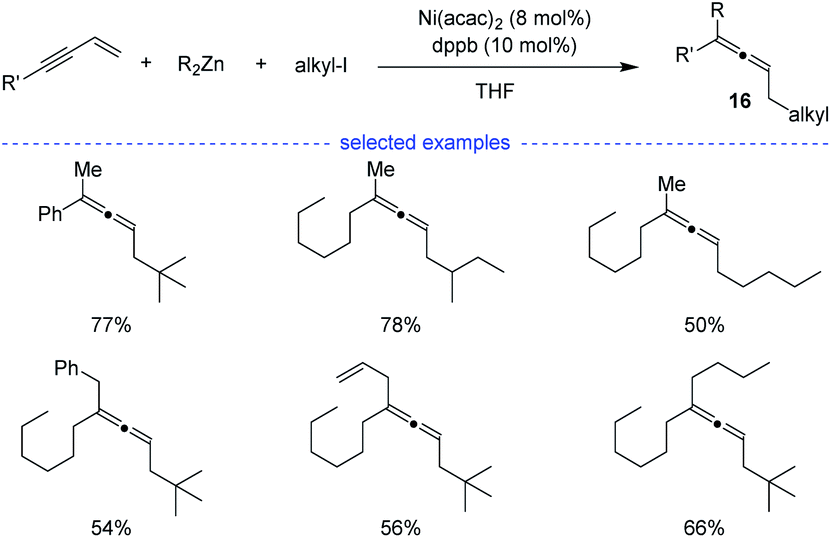
In 2009, Kambe et al. discovered that nickel salts regioselectively catalyzed three-component cross-coupling of alkyl halides, terminal alkynes with organomagnesium or organozinc reagents, affording an method for the generation of Z-selective trisubstituted allenes 16 (Scheme 13).30 In addition, this method also worked well for the cross-coupling of alkyl halides, 1,3-enynes with organozinc reagents. The alkyl radical was supposed to be generated from SET process between the nickel species and alkyl halide.
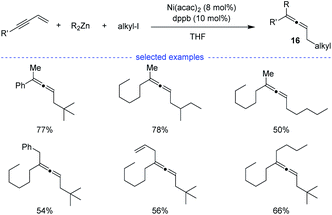 |
| | Scheme 13 Ni-catalyzed three-component synthesis of allenes from 1,3-enynes, alkyl halides, and organozinc reagents. | |
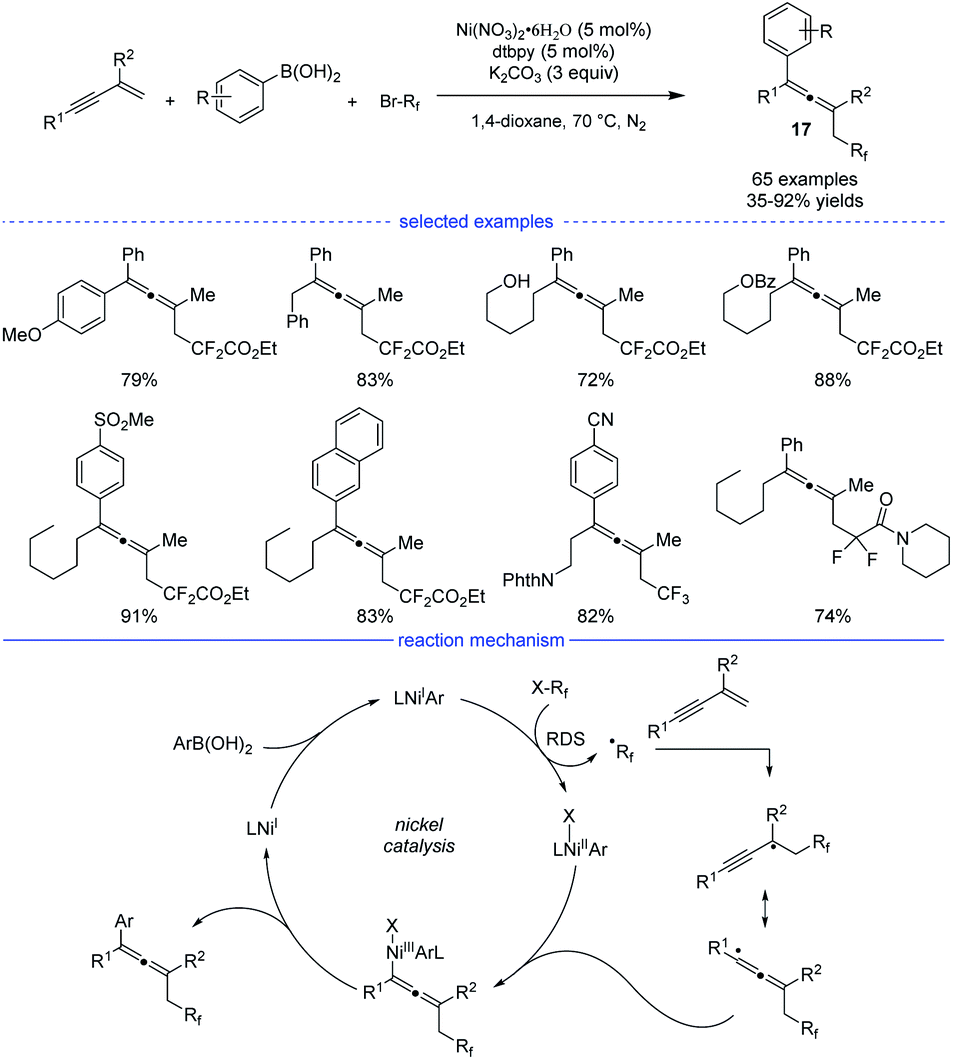
In 2019, Wang and coworkers developed an excellent nickel-catalyzed 1,4-arylfluoroalkylation of 1,3-enynes (Scheme 14).31 This three-component cross-over reaction of fluoroalkyl halides, aryl boronic acids with 1,3-enynes afforded a wide range of structurally diverse fluoroalkylated allenes 17 under mild conditions. Ligand screening revealed that an electron-rich 1,10-phen offered the best performance. Different functional groups, such as hydroxyl, chloride, ester, ether, carbonyl, cyano, Ms, and amide could be well tolerated in this catalytic system. The allene products can be transformed into various fluorinated bioactive molecules for drug discovery. Radical inhibition and radical-clock experiments suggested a radical process, while competition experiments concluded that fluoroalkyl iodide reacted fastest among the three fluoroalkyl halides (RfI > RfBr > RfCl). The reaction was supposed to start with a ligated NiIX species. Upon transmetallation of the NiIX species with an aryl boronic acid, the arylated NiIAr species was afforded, which can donate an electron to the fluoroalkyl halide to afford the NiIIAr species and the fluoroalkyl radical. The generated fluoroalkyl radical via SET process can add onto the 1,3-enyne to generate the allenyl radical that can react with NiIIAr species to afford the NiIII species. The final allene product can be produced via reductive elimination.
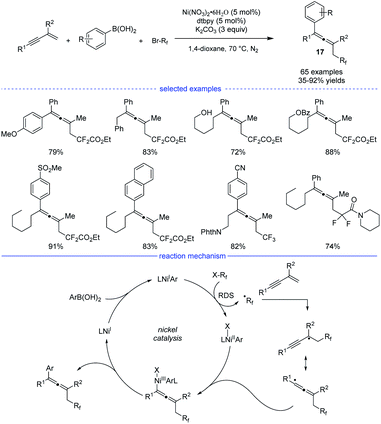 |
| | Scheme 14 Ni-catalyzed three-component synthesis of allenes from 1,3-enynes, alkyl halides, and aryl boronic acids. | |
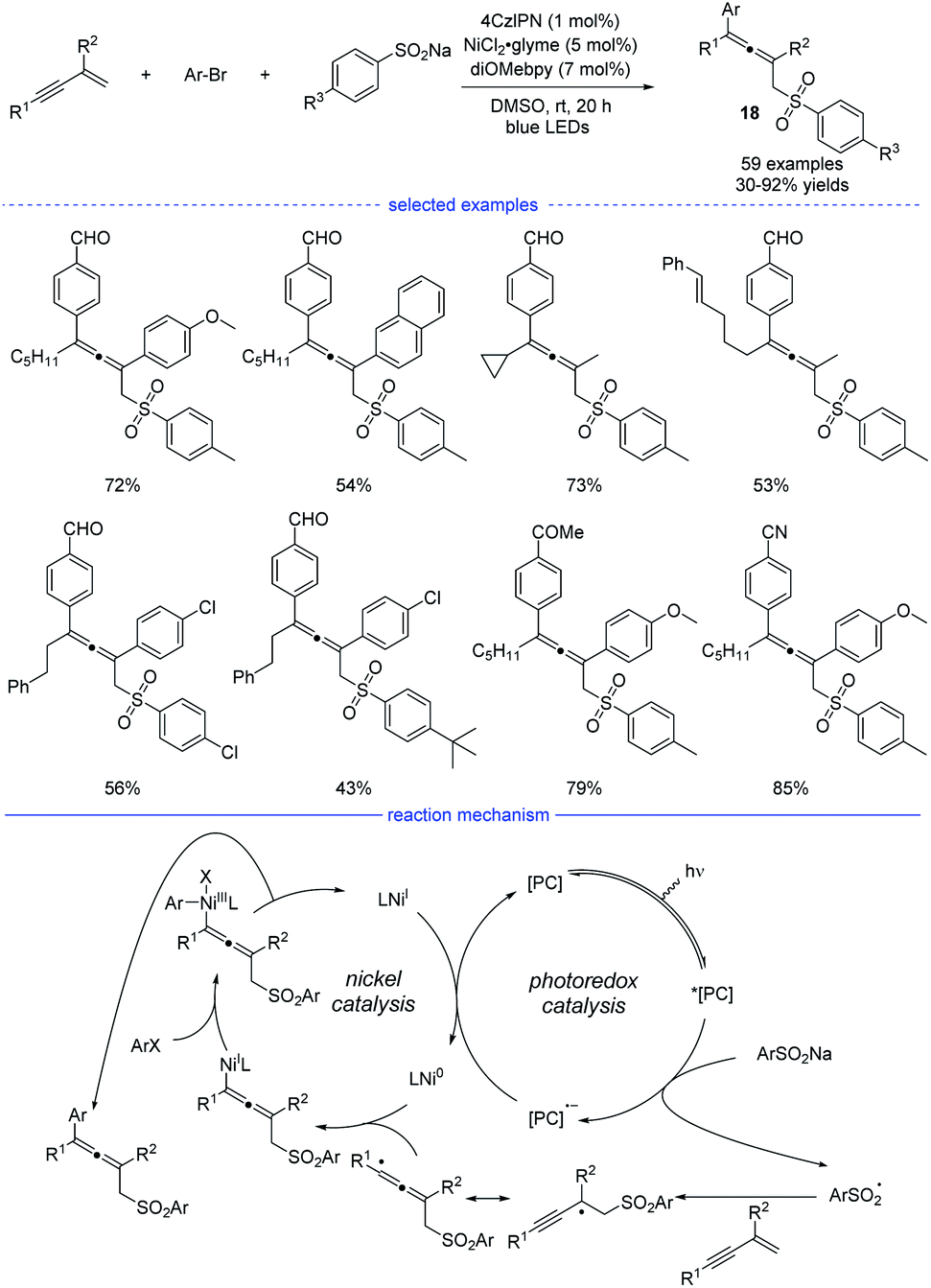
The groups of Lu,32 Li,33 and Wang34 independently disclosed parallel 1,4-sulfonylarylations of 1,3-enynes with aryl halides and sulfinate salts using cooperative photoredox/nickel catalysis. In Lu's work, 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4CzIPN) was found to be the best organic photosensitizer and bidentate diOMebpy was proved to be the best ligand for nickel catalysis (Scheme 15). This practical catalytic strategy features mild conditions, broad substrate scopes and wide functional group tolerance. Functional groups such as halide, internal C–C double bond, ketone, nitrile, trifluoromethyl, cyano, aldehyde and ester were well tolerated. Moreover, if 1,3-enynes with a terminal C–H on the alkyne side was used as the substrates, the catalytic system also worked well but afforded 3,4-adducts as the final products. In the proposed mechanism, the photocatalyst 4CzIPN (E1/2 [*PC/PC− = +1.35 V versus SCE in CH3CN]) was activated by blue light irradiation, which subsequently oxidizes sodium sulfonate (TsNa, E1/2 = +0.45 V versus SCE in CH3CN) through SET process to afford the reduced photocatalyst and the sulfonyl radical. After general steps of radical addition, resonance of the propargyl radical and the allenyl radical, and interception of the allenyl radical by Ni0 species, an allenyl nickel(I) species was generated. The 1,4-sulfonylarylated allene product would be produced through subsequent oxidative addition of aryl halide and reductive elimination. Finally, single-electron-transfer (SET) between the photocatalyst and the NiI species afforded the Ni0 catalyst and the ground-state 4CzIPN.
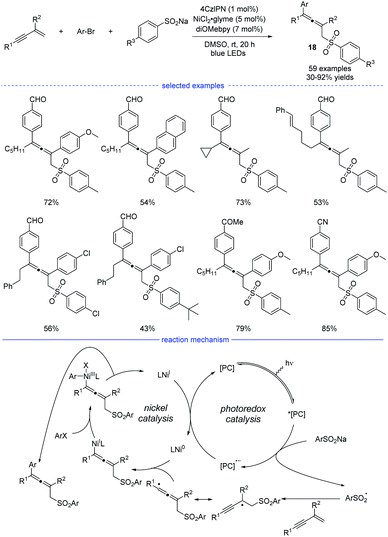 |
| | Scheme 15 Ni/4CzIPN co-catalyzed three-component synthesis of allenes from 1,3-enynes, sulfinate salts, and aryl bromides. | |
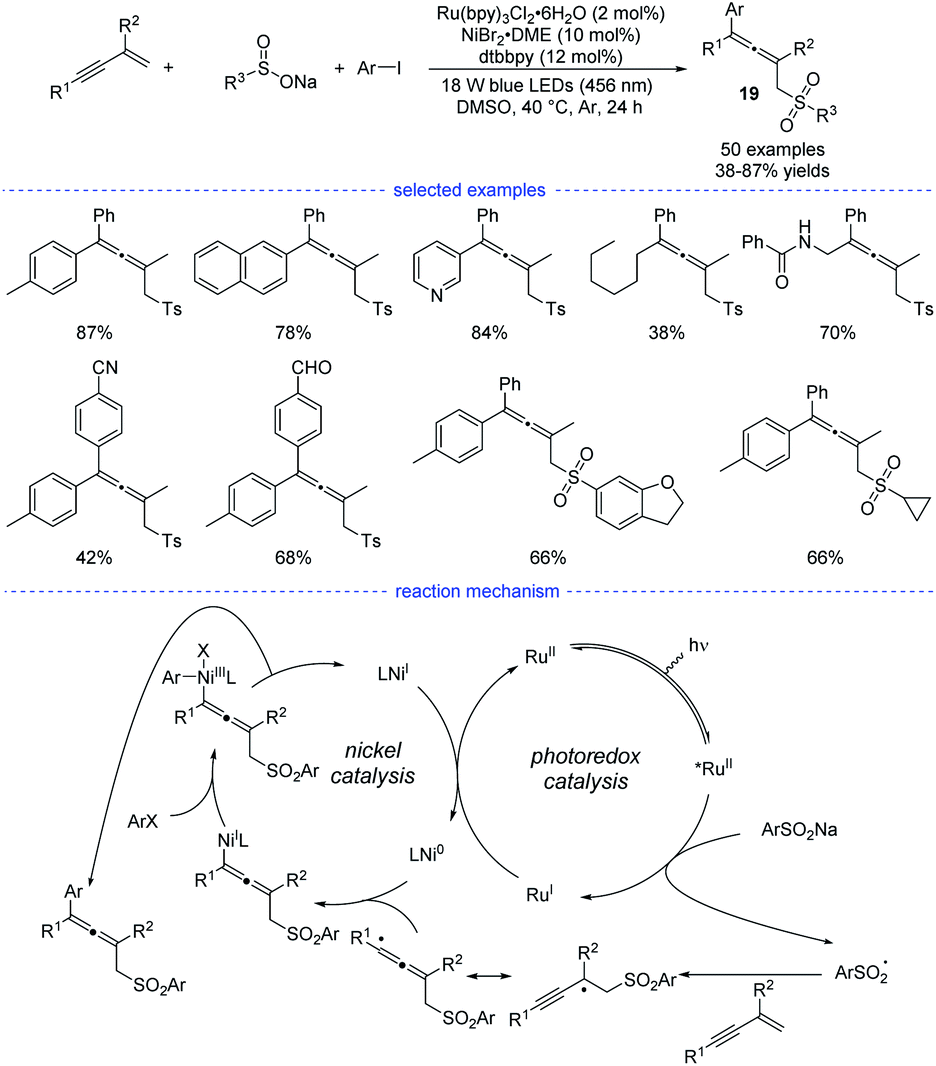
In Li's work, Ru(bpy)3Cl2·6H2O was found to be the best organic photosensitizer (Scheme 16). Bidentate dtbbpy rather than diOMebpy was found to be the best ligand for nickel catalysis. A wide range of readily accessible 1,3-enynes, sodium sulfinates, and aryl iodides were transformed into allenes 19 in moderate to good yields under mild conditions.
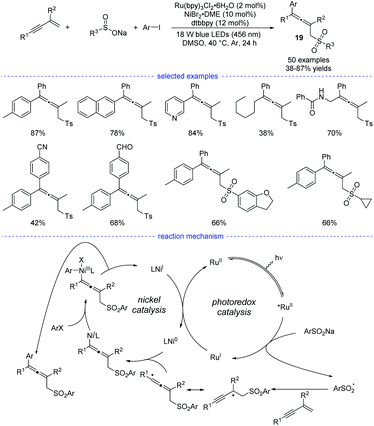 |
| | Scheme 16 Ni/Ru co-catalyzed three-component synthesis of allenes from 1,3-enynes, sulfinate salts, and aryl iodides. | |
NHC catalysis
N-heterocyclic carbenes (NHCs) have been widely applied in umpolung of aldehydes, and nucleophilic Breslow intermediates were supposed to be involved in these reactions.35 Moreover, NHC-catalyzed radical reactions received increasing attentions as well.36 Very recently, NHC catalysis has been employed for radical synthesis of allenes.
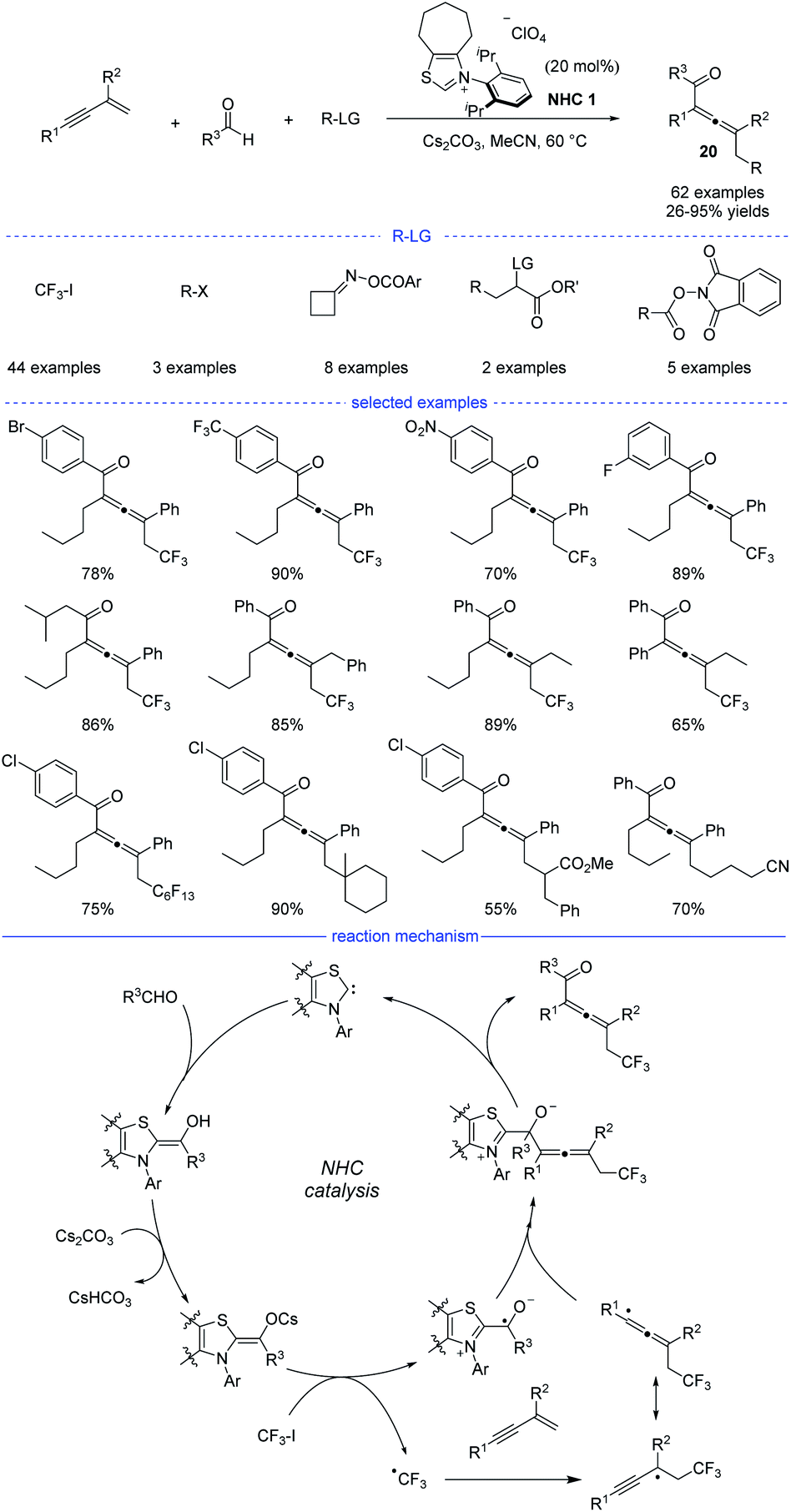
In 2021, Du et al. reported an NHC-catalyzed three-component synthesis of various tetrasubstituted allenes under mild conditions (Scheme 17).37 The structures of NHC precursors were found to be crucial to this reaction. Thiazolium precatalysts were generally much more effective than triazolium precatalysts. This strategy allows CF3I, alkyl halides, cycloketone oxime esters, and redox-active esters to be used as the different alkyl radical precursors under the standard conditions. Moreover, diverse aldehydes and various 1,3-enynes were also tested, and the tetrasubstituted allenes 20 can be obtained with high yields and excellent functional group tolerance. In addition, it should be noted that the two-component coupling reactions of the radical precursors and aldehydes did not occur in most of the cases. The results of a radical trapping reaction with TEMPO (2,2,6,6-tetramethyl piperidinyl oxy) and a radical clock experiment suggested that the reaction proceeded through a radical pathway, while the carbocation trapping experiment with the addition of three equivalents of MeOH was turned out to be negative. Therefore, a possible mechanism was proposed accordingly. First, the combination of an NHC and an aldehyde under basic conditions would form the enolate form of a Breslow intermediate. Then, a SET process between the enolate and trifluoroiodomethane provides an NHC-bound radical and a trifluoromethyl radical. Followed by radical addition on a 1,3-enyne and reversible isomerization, an allenyl radical was formed as a favorable intermediate because of the steric effect. Finally, the radical–radical coupling of the NHC-bound radical and the allenyl radical afforded the allene product and the regenerated NHC catalyst.
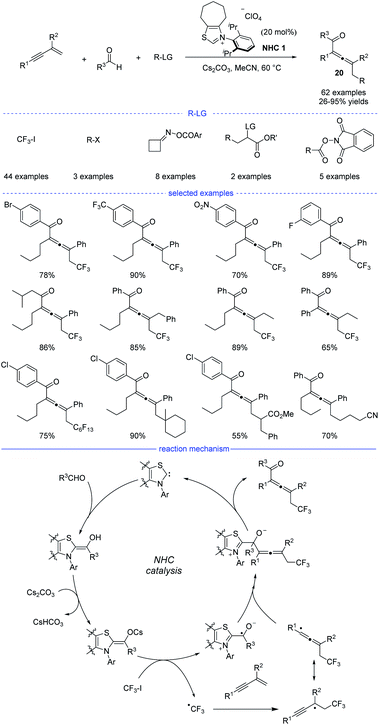 |
| | Scheme 17 NHC-catalyzed three-component 1,4-difunctionalization of 1,3-enynes. | |
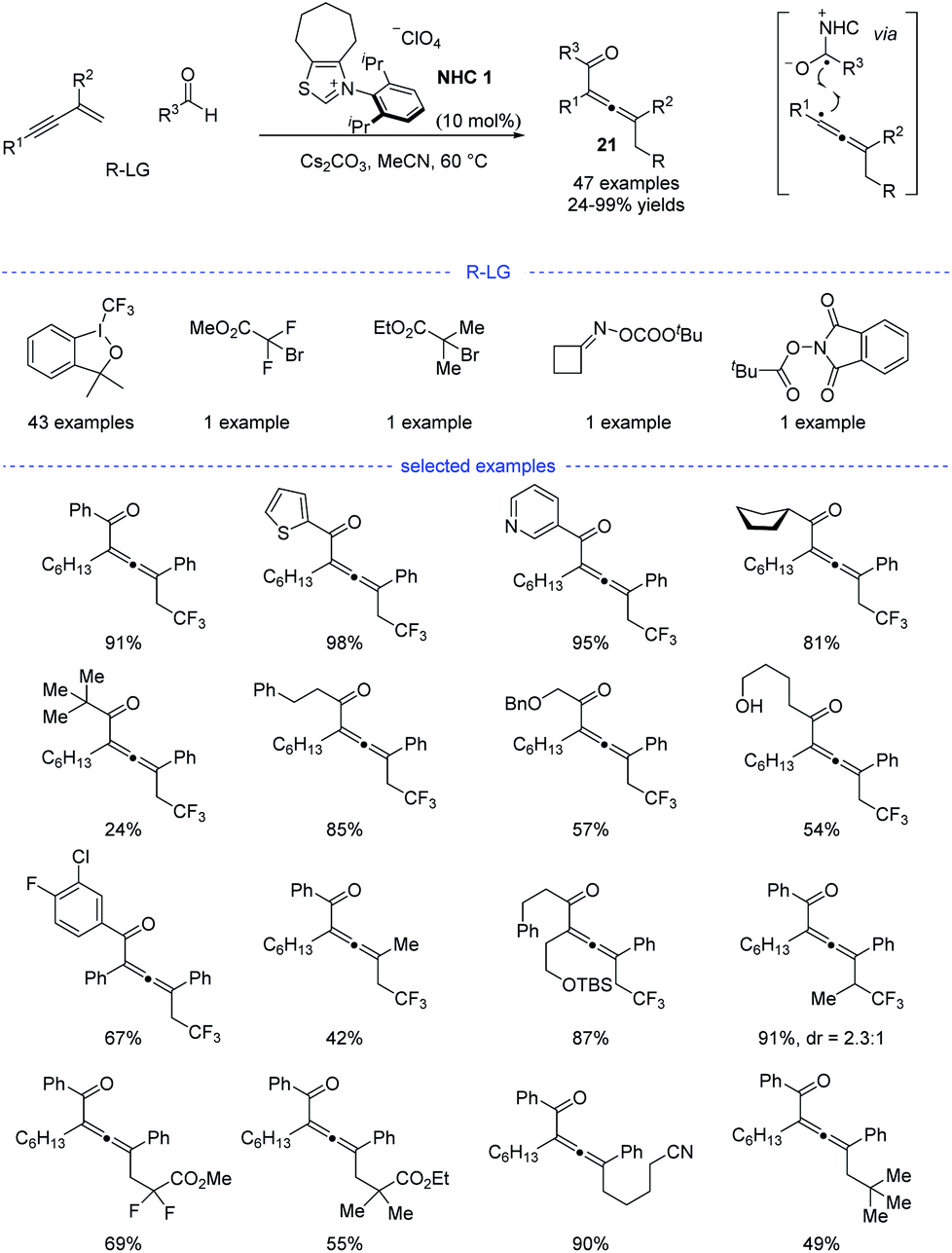
Shortly after, Huang et al. developed a parallel NHC catalysis for radical synthesis of tetrasubstituted allenes 21 (Scheme 18).38 Several kinds of radical precursors, including Togni reagent I, bromo acetates, cyclobutanone oxime esters, redox-active esters, can be used to afford the corresponding trifluoromethyl radical, stabilized alkyl radicals, and primary alkyl radicals, respectively. Moreover, both aromatic and aliphatic aldehydes performed well. Other functionalities such as halides, free alcohol, ethers, heteroaryl groups, and amides were found to be tolerated. The radical relay coupling between the allenyl radicals and the ketyl radicals generated from single-electron oxidation of the Breslow intermediates was supposed to be the key step for the success of this reaction. Very recently, Li, Han, and coworkers reported a parallel work with similar reaction partners.39
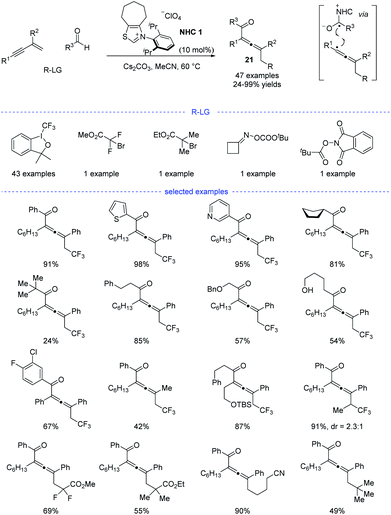 |
| | Scheme 18 NHC-catalyzed three-component 1,4-carbofunctionalization of 1,3-enynes. | |
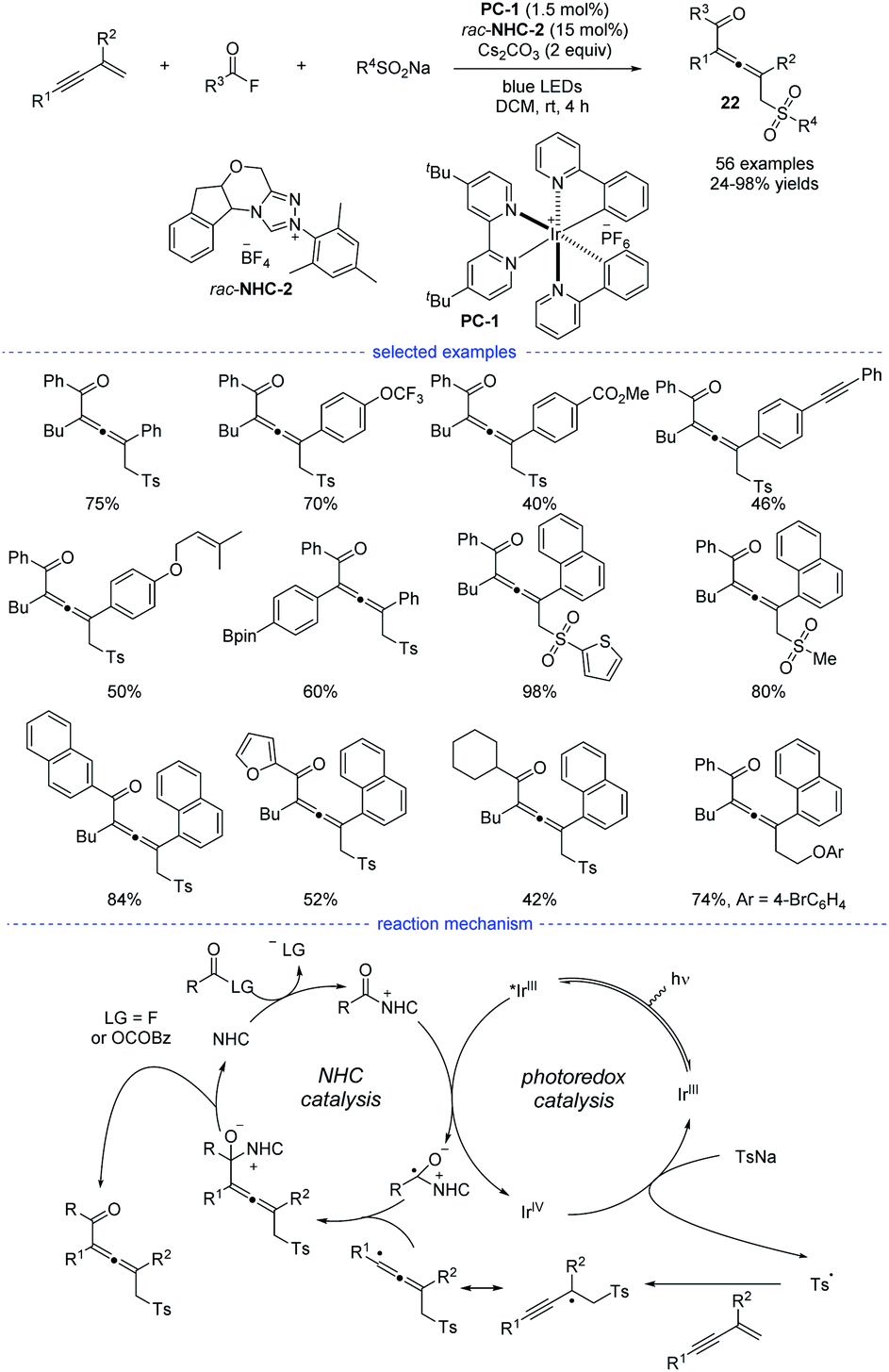
Very recently, Zhang et al. revealed a NHCs and visible light-mediated photoredox co-catalyzed radical 1,4-sulfonylacylation of 1,3-enynes (Scheme 19).40 The optimization of a range of NHC catalysts and photocatalysts found that the emerge of NHC-2 and [Ir(ppy)2(dtbbpy)]PF6 was the best choice for this transformation and the dimerized product can be greatly suppressed. Instead of using aldehydes as the carbonyl source, aroyl fluorides was turned out to be good precursors for the generation of ketyl radicals via oxidative quenching process of excited photocatalysis. On the other hand, the allenyl radicals was supposed to be stemmed from chemo-specific addition of sulfonyl radicals to 1,3-enynes. It should be noted that other than functional groups such as alkyl, methoxyl, halogen, methoxycarbonyl, trifluoromethyl, and trifluoromethoxy, the insular alkyne, vulnerable Bpin, and olefin units were all preserved after transformation. When the standard reaction was carried out in dark, or without NHC catalyst, or in the absence of photo catalyst, no corresponding product can be generated. Moreover, the acyl azolium ion was supposed to be a reaction intermediate and Stern–Volmer quenching experiments supported that acyl azolium ion quenched the excited photocatalyst. A possible mechanism for the dual NHC and photoredox catalysis was proposed accordingly. In the NHC catalysis cycle, the acyl fluoride or in situ generated bisacyl carbonate intermediate could react with NHCs to provide the acylazolium intermediate, which underwent SET with exited iridium catalyst to provide a ketyl radical. The ketyl radical then coupling with the sequential generated allenyl radical to form an NHC-bound intermediate. And the catalytic cycle finished after the disintegration of NHC-bound intermediate. In the photo catalysis cycle, an oxidative quenching process was proposed accordingly.
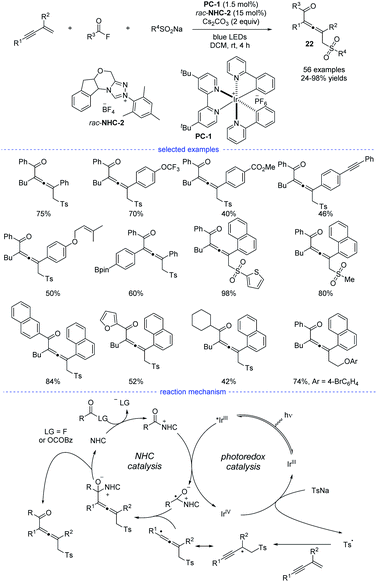 |
| | Scheme 19 NHC/Ir co-catalyzed three-component 1,4-sulfonylacylation of 1,3-enynes. | |
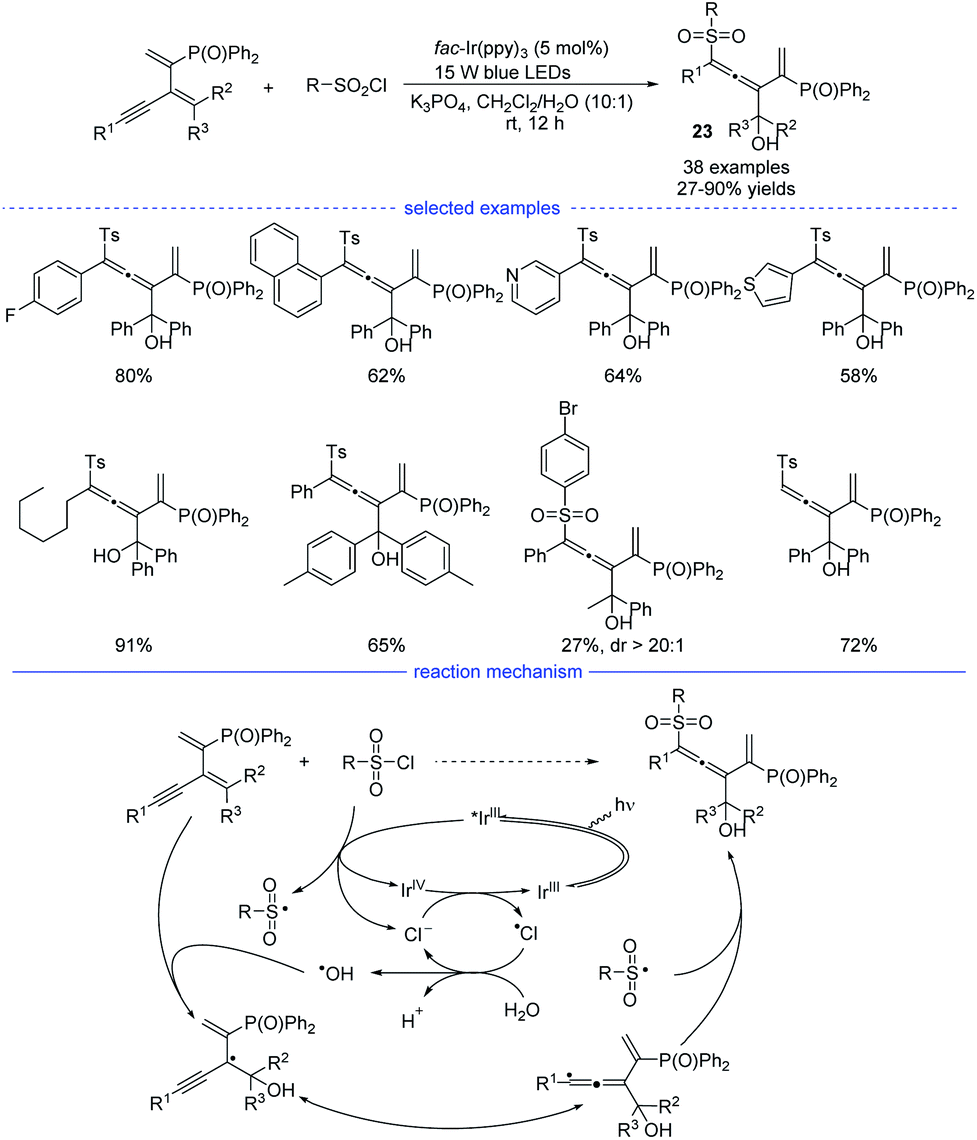
In 2021, Wu, Zhu, and coworkers developed an interesting radical synthesis of allenes. In the absence of any transition-metal catalyst, the reaction was only performed with an iridium salt as the photoredox catalyst under mild conditions (Scheme 20).41 A wide range of functionalized 1,3-enynes was found to be good substrates for the 1,4-hydroxysulfonylation system. The C–C double bond attached with the phosphine atom was crucial for the feasible allene formation. A handful of sulfonyl chlorides bearing electron-rich and electron-deficient moieties reacted well, affording the corresponding products 23 in good to excellent yields with no distinct electronic effect observed. Notably, this reaction was unexpectedly trigged by a hydroxyl radical rather than the sulfonyl radical. When N-benzoyldiallylamine was additionally added into the reaction mixture, the adducts detected by HR-MS spectrum showed the involvement of several radical species, including ˙Ts, ˙Cl, ˙OH, and the propargylic radical and/or allenic radical generated from the addition of ˙OH onto the 1,3-enyne. Further 18O-labeling experiment clearly revealed that the hydroxyl group originates from water. Accordingly, a reaction mechanism with the recycling of photocatalyst and chlorine atom was proposed.
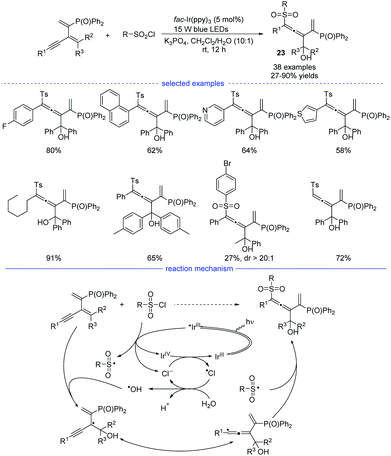 |
| | Scheme 20 Ir-catalyzed three-component 1,4-difuctionalization of activated 1,3-enynes. | |
Radical synthesis of allenes from propargylic compounds
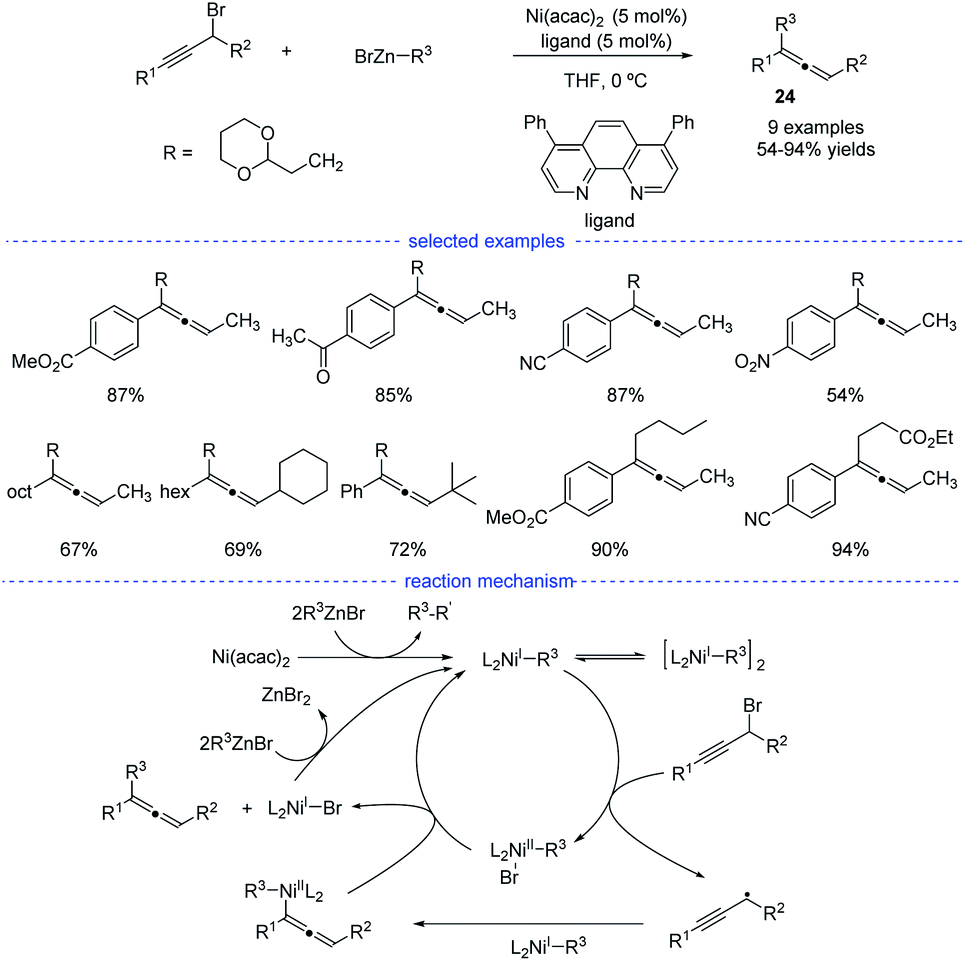
As reported by Fu et al., the Ni-catalyzed cross-coupling of propargyl halides with organozinc reagents would afford functionalized propargylic compounds.42 In 2017, Cárdenas and coworkers discovered that instead of forming propargylic compounds, the cross-coupling of alkylzinc halides and propargyl bromides catalyzed by a nickel complex offered an access to allenes 24 (Scheme 21).43 This regioselective reaction works well in the presence of a variety of substituents in moderate to excellent yields and the formation of functionalized alkynes is not observed in most of the cases. The results of mechanistic experiments showed that NiI complexes is the active species and radical intermediates were involved in the reaction. In addition, kinetic studies revealed that the reaction is first order in the electrophile, zero-order in the nucleophile, and one-half order in the metal catalyst. On the basic of the stereochemical, titration and kinetic experiments, and DFT calculations, a reaction mechanism was proposed. The extremely fast bromine abstraction by the active alkyl-NiI species led to formation of the propargyl carbon radical and allene product would be formed after reductive elimination.
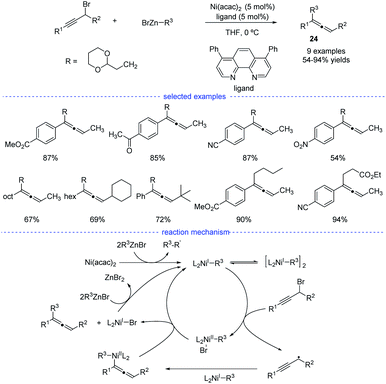 |
| | Scheme 21 Ni-catalyzed radical synthesis of allenes from propargyl bromides. | |
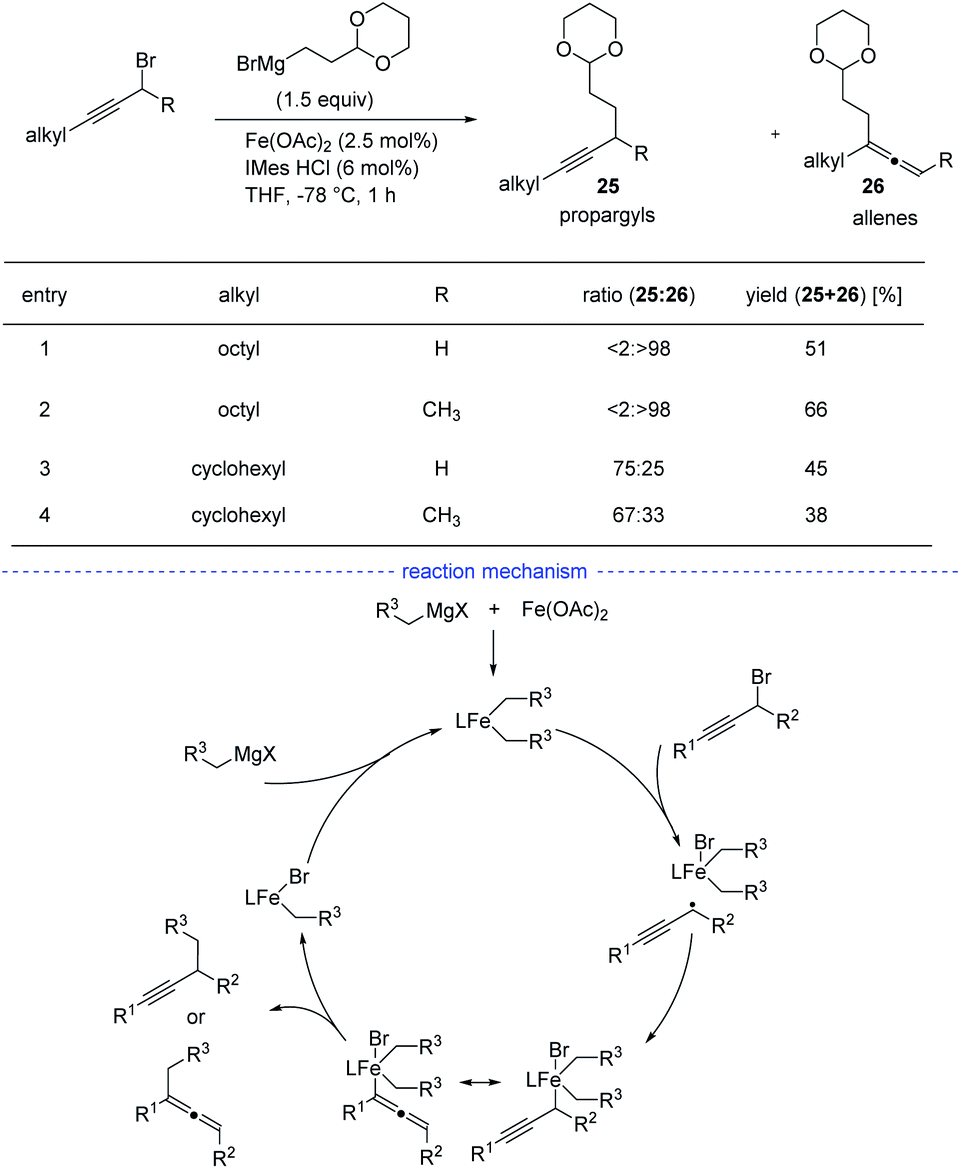
Although Fe-catalyzed cross-coupling reactions are well-established, the reaction involving propargyl electrophiles has been far less explored. In 2018, the Cárdenas group reported an iron-catalyzed Kumada-type cross-coupling reaction of propargyl halides with alkylmagnesium reagents (Scheme 22).44 However, the regioselectivity was not high enough to exclusively allow the formation allenes 26, and the propargyl coupling derivatives 25 was always produced concomitantly. Based on detection of the homocoupling compound, the formation of a propargyl radical was therefore proposed, and a possible mechanism was depicted accordingly. However, Bäckvall's studies on iron-catalyzed cross-coupling of propargyl carboxylates and Grignard reagents seem to either rule out a radical intermediate or point towards an extremely short-lived radical species.45
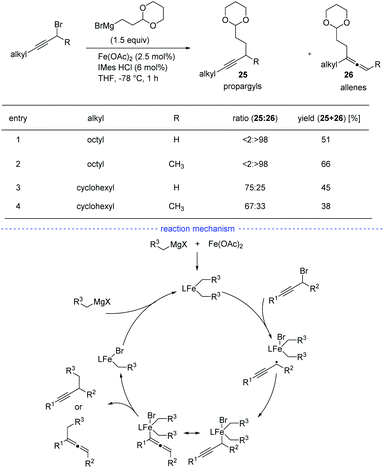 |
| | Scheme 22 Fe-catalyzed radical synthesis of allenes from propargyl bromides. | |
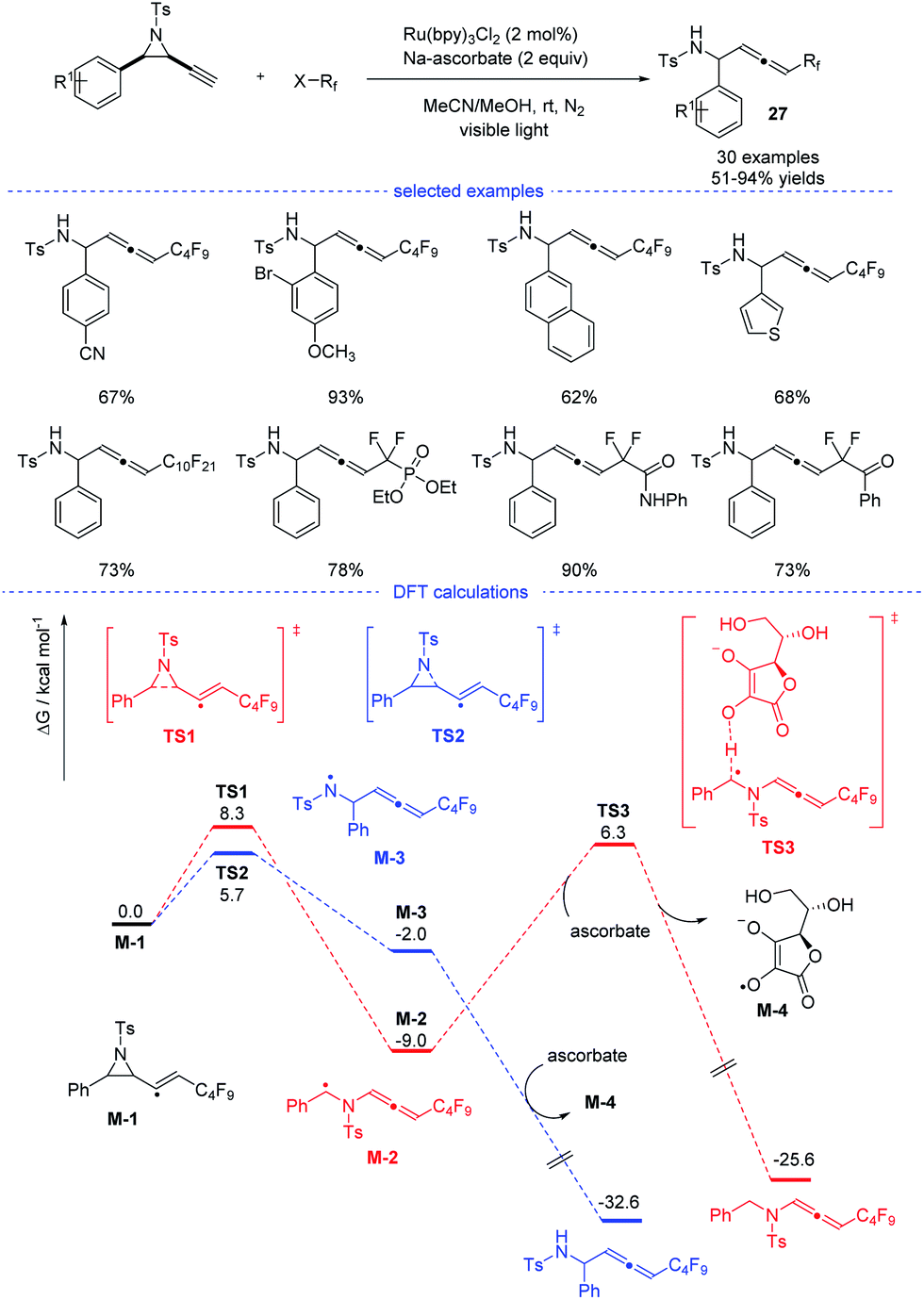
In 2020, Xu, Lan, and coworkers reported a Ru-catalyzed radical synthesis of functionalized allenes from readily available terminal alkynyl aziridines under visible-light irradiation (Scheme 23).46 A diversity of trifluoromethyl, difluoromethylene, and perfluoroalkyl halides reacted smoothly to afford the corresponding fluorinated allenes 27. Stern–Volmer fluorescence quenching experiments of photocatalyst Ru(bpy)3Cl2 reveal that the exited ruthenium catalyst was quenched by reductant sodium ascorbate. The formed stronger reductant RuI species then reacted with the perfluoroalkyl iodide to form a perfluoroalkyl radical. Further DFT calculations suggested that the formation of the observed product was kinetically favorable.
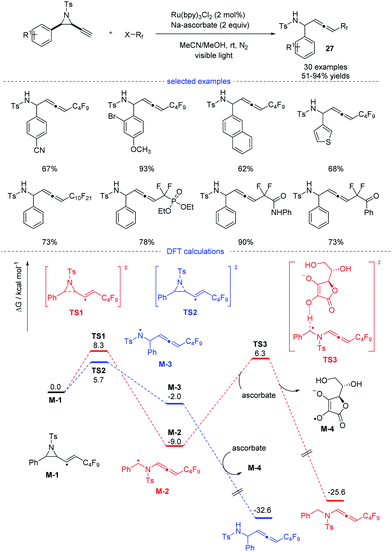 |
| | Scheme 23 Ru-catalyzed radical synthesis of allenes from propargyl amides. | |
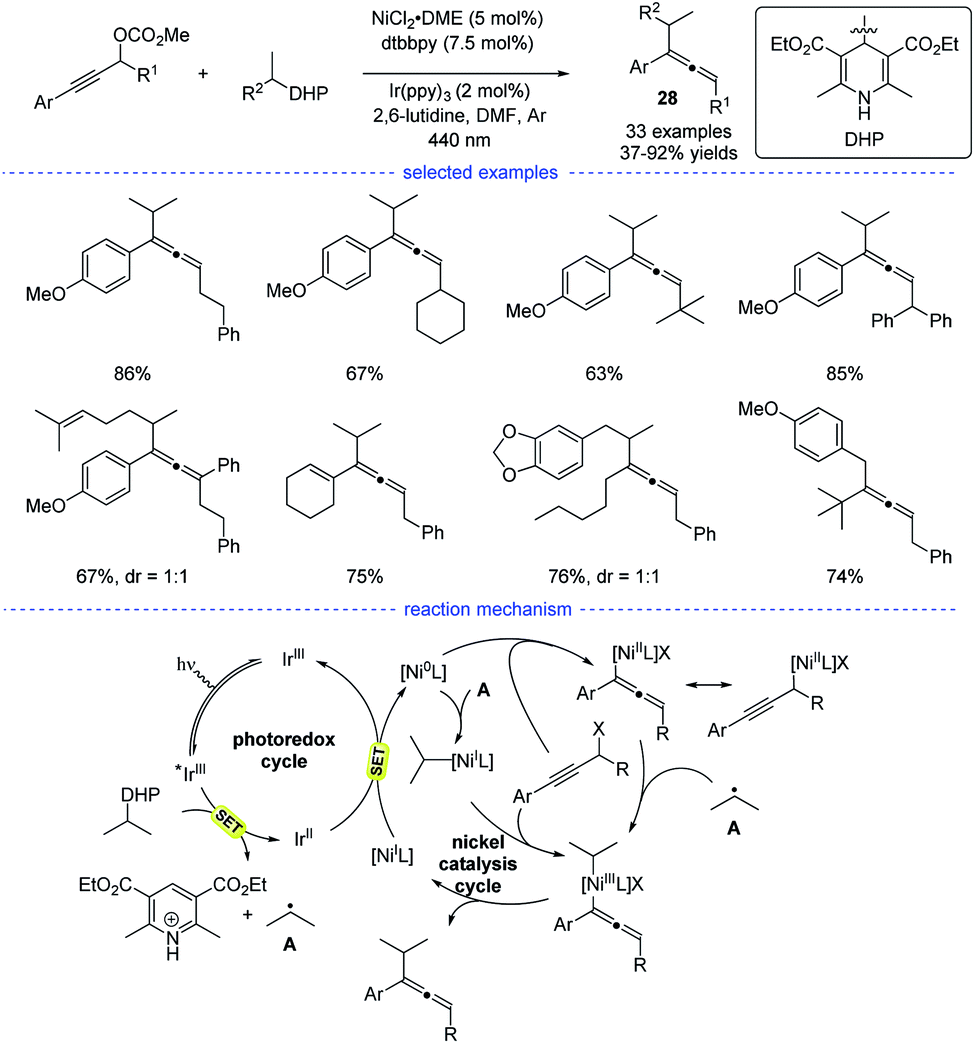
In 2021, Liang et al. disclosed a photoredox/nickel dual-catalyzed alkylation of propargylic carbonates with bench-stable alkyl 1,4-dihydropyridine derivatives (1,4-DHPs) as the alkyl radical precursors (Scheme 24).47 This highly regioselective method had good substrate scope and mild conditions. The study was initiated with the optimization of nickel catalysts, ligand, photocatalysts, and solvents. When the reaction in DMF was performed with NiCl2·DME/dtbbpy and Ir(ppy)3 as the dual catalysts, the highest yield of the desired products 28 were obtained. Based on the experimental studies and the previous work, a possible mechanism was proposed. The photoredox cycle can afford the alkyl radical and the Ni0 species through reductive quenching. An allenylic NiIII was generated from the interaction of the Ni0 species, the alkyl radical and the propargylic carbonate, either through a radical rebound/oxidative addition process or an oxidative addition/radical rebound. Finally, the allene product was afforded through reductive elimination.
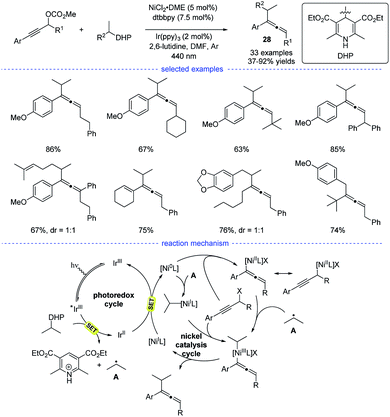 |
| | Scheme 24 Ni-catalyzed radical synthesis of allenes from propargylic carbonates. | |
Radical synthesis of allenes from allene precursors
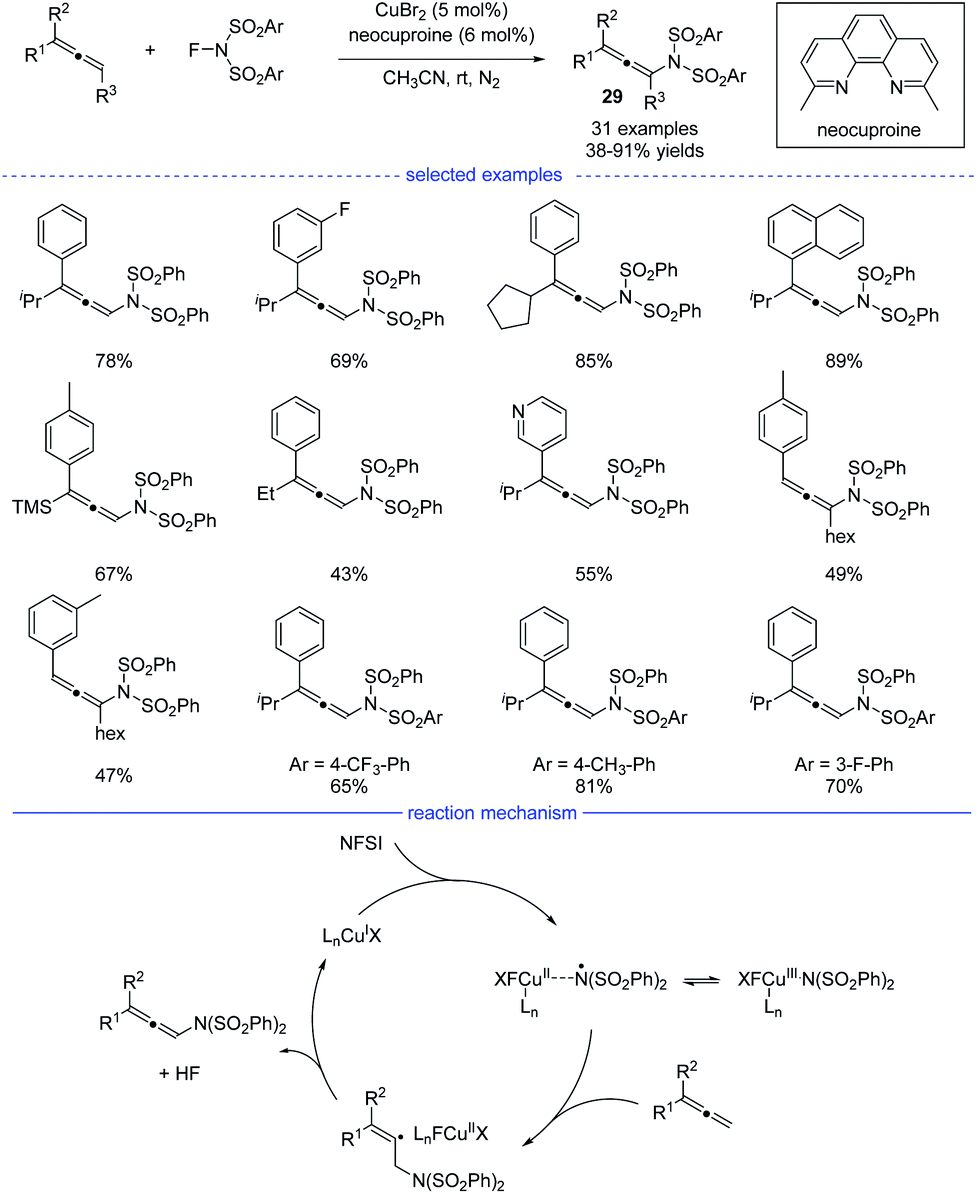
Because of the unique properties, it is no doubt that radical addition to allenes will meet the challenges of chemo-, regio-, and stereoselectivities.48 In 2015, Zhang et al. reported an excellent and interesting example on direct functionalization of the allenyl C–H bond via a radical strategy (Scheme 25).49 In the presence of a copper catalyst, highly regioselective oxidative amination of allenes with NFSI allows the formation of versatile allenamides 29 under mild reaction conditions. In addition, when AgF was used as the catalyst, fluorinated polysubstituted alkenes were found to be the final products. The reaction was initiated with the oxidation of the copper(I) species with NFSI, generating the copper(III) species that would isomerize to a Cu(II)-stabilized benzenesulfonimide radical species. The benzenesulfonimide radical then added onto the allene and a vinyl radical intermediate would be formed. Finally, the selective C(sp3)–H bond elimination process delivered the thermally disfavor allenamides and the regenerated Cu(I) species.
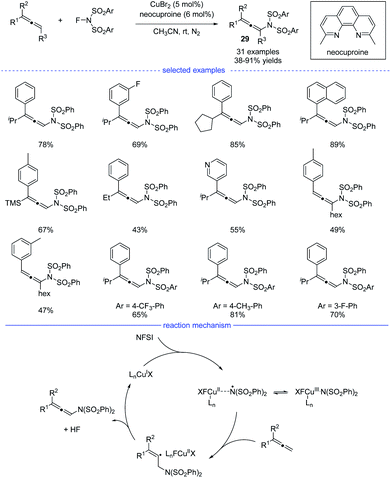 |
| | Scheme 25 Cu-catalyzed radical synthesis of allenes from allene precursors. | |
Radical asymmetric synthesis of chiral allenes
Enantioenriched allenes have an intrinsic axial chirality stemmed from the twisted orthogonal π-systems. They are important structural motifs often appeared in natural products and pharmaceuticals. In this section, recent developments on radical asymmetric synthesis of chiral allenes will be discussed. The groups of Bao, G. Liu, and X.-Y. Liu have successfully explored several radical strategies for the synthesis of enantioenriched allenes, independently.
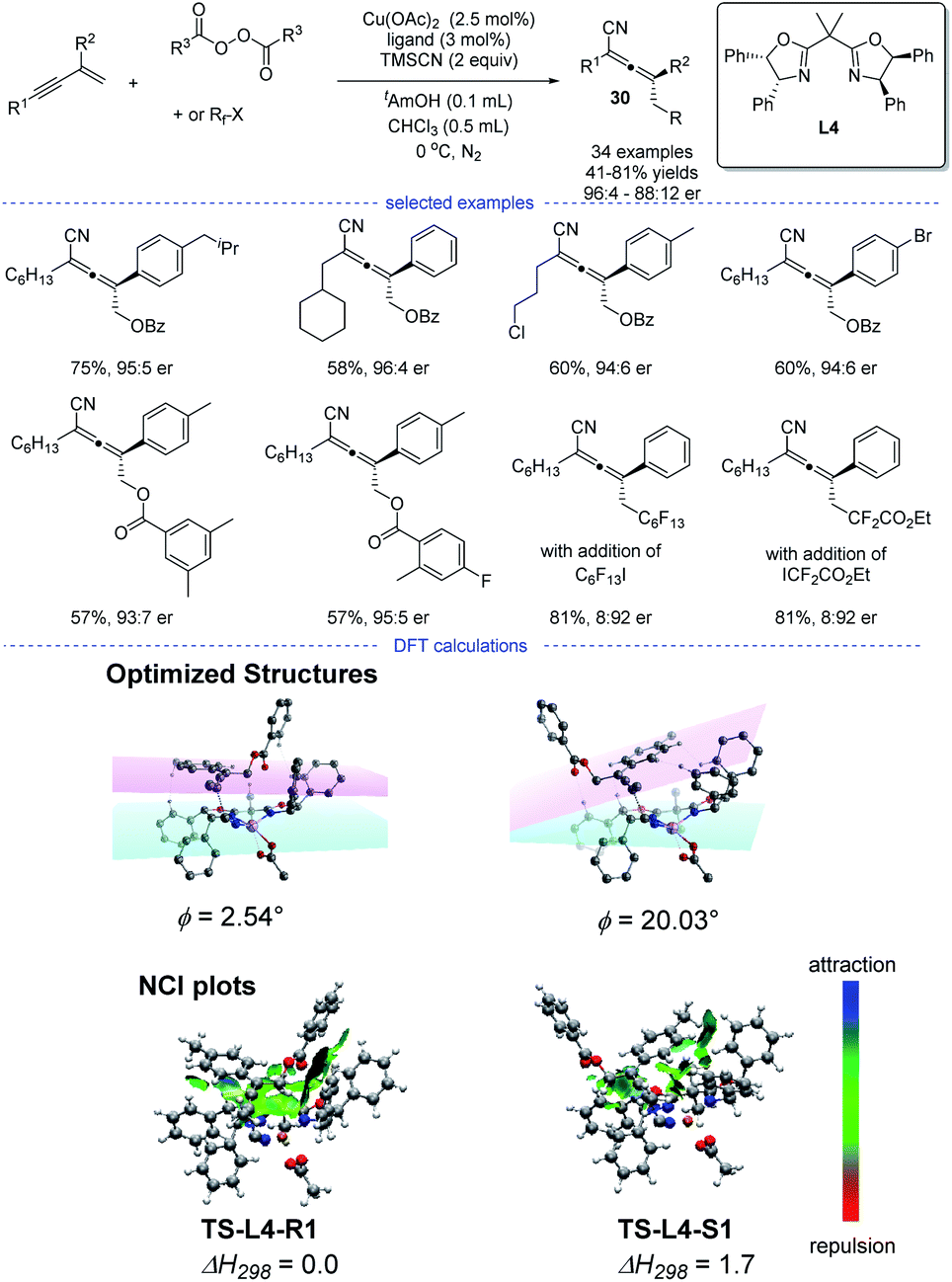
In 2020, Bao et al. reported a copper-catalyzed radical 1,4-difunctionalization of 1,3-enynes for the synthesis of chiral allenes 30 with good to excellent enantiomeric ratio (er) (Scheme 26).50 The ligand screening revealed that a bidentate box ligand with an extended planarity offered the best performance. A wide range of 1,3-enynes with different functionalities can be tolerated. Not only oxygen-centered radicals but also carbon-centered radicals reacted smoothly to afford the corresponding allenyl cyanides. The results of radical trapping experiments, carbocation trapping experiments, and radical clock experiments implied that the carbon-centered radical rather than carbocation was the reaction intermediate. A linear correlation was observed, which indicated that the active catalytic species was a monomeric copper complex bearing a single chiral ligand.
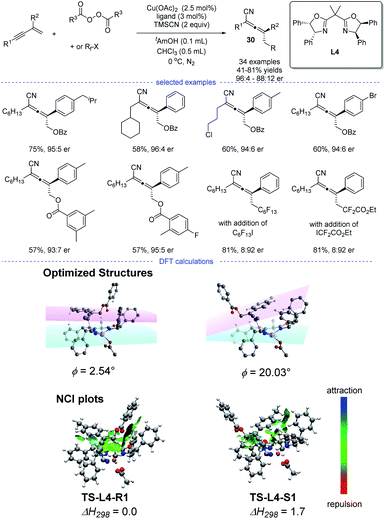 |
| | Scheme 26 Cu-catalyzed radical asymmetric synthesis of allenes from 1,3-enynes and, the optimized structures and NCI plots of TS-L4-R1 and TS-L4-S1. The dihedral angle between planes of the aromatic substituent (pink plane) and the oxazoline (cyan plane) is denoted as ϕ. | |
However, the fashion for the enantiocontrol in this case is still mysterious. Because it is not likely that the allenyl radical can get close to the chiral metal center through moderate or strong interactions such as covalent, dative, ionic and hydrogen bonds. DFT calculation suggested that weak interactions such as π–π interaction and van der Waals interaction played a vital role in the enantiocontrol and an outer-sphere group-transfer was proposed after fails in the identity of the transition state for reductive elimination from high-valent CuIII intermediates (inner sphere pathway). Specifically, transition states TS-L4-R1 and TS-L4-S1 with the optimized structures will lead to the formation of R and S enantiomers, respectively. A π−π interaction between the aromatic substituent with the plane of the oxazoline-metal complex was found in TS-L4-R1 but not in TS-L4-S1. Moreover, the analysis of noncovalent interactions (NCI) between the allenyl radical and the ligated catalyst also supported a stronger π–π interactions in TS-L4-R1.
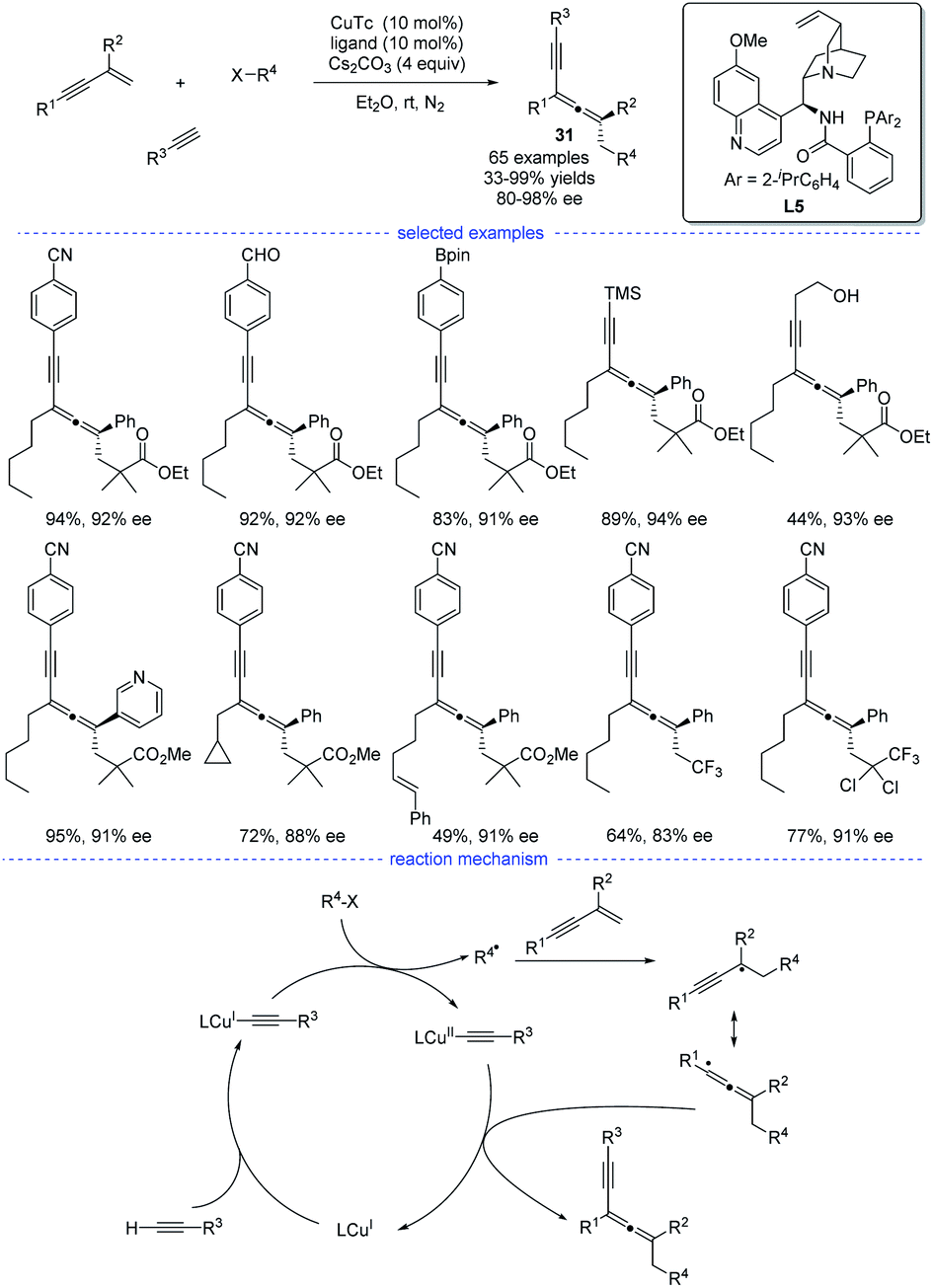
In 2021, X.-Y. Liu et al. reported a copper-catalyzed asymmetric coupling of allenyl radicals with terminal alkynes under mild conditions (Scheme 27).51 This radical 1,4-carboalkynylation of 1,3-enynes provided an feasible access to synthetically challenging chiral tetrasubstituted allenes 31 with good yields and excellent enantioselectivities. Ligand optimization found that a chiral N,N,P-ligand L5 is crucial not only for the reaction initiation but also for the enantiocontrol. The use of a variety of (hetero)aryl and alkyl alkynes, 1,3-enynes, and radical precursors with excellent functional group tolerance demonstrated the broad substrate scope of this reaction. Accordingly, a possible mechanism via either reductive elimination from CuIII species or group-transfer is proposed.
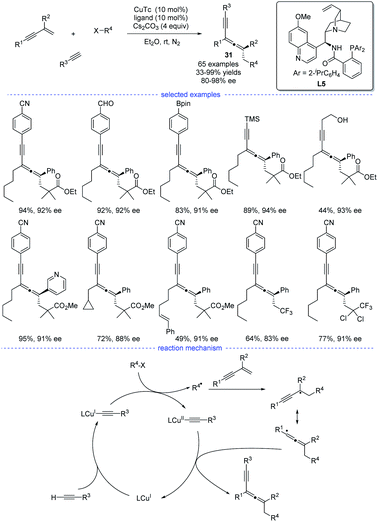 |
| | Scheme 27 Cu-catalyzed radical asymmetric synthesis of allenes from 1,3-enynes, alkyl halides, and terminal alkynes. | |
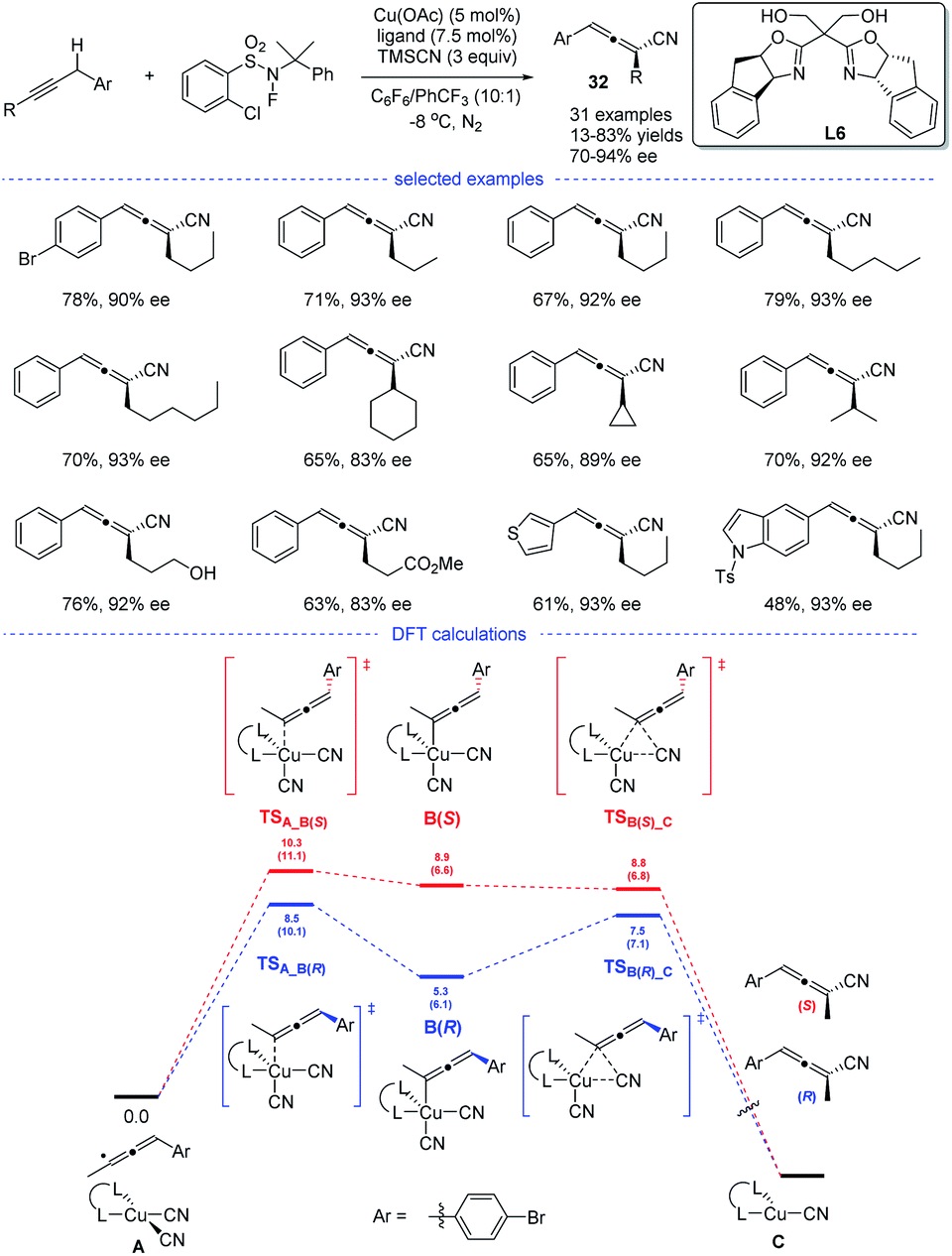
In 2018, the group of G. Liu have tentatively tried the copper-catalyzed radical 1,2- and 1,4-addition to 1,3-enynes in an enantioselective fashion (Scheme 28). However, only one example has been reported and the chiral allene can be obtained in 73% ee (enantiomeric excess).14 In 2021, G. Liu, Lin, and coworkers discovered, for the first time, an approach for the synthesis of chiral allenes 32 directly from enantioselective copper-catalyzed cyanation of propargylic C–H bonds.52 A new type of boxOTMS ligands L6 was designed and the structurally diverse chiral allenyl nitriles can be obtained in good yields and with excellent enantioselectivities. The chiral allenes can be smoothly converted into other enantioenriched organic compounds via axis-to-center chirality transfer. By considering both the Re and Si face additions, DFT calculations was carried out. It was found that the enantiodetermining step lies in the coupling of allenyl radicals with the CuII center to forming a CuIII complex, rather than previous reductive elimination of the CuIII species.53 Moreover, the energy for the enantiodetermining transition states TSA_B(R) is 1.8 kcal mol−1 lower than that of TSA_B(S).
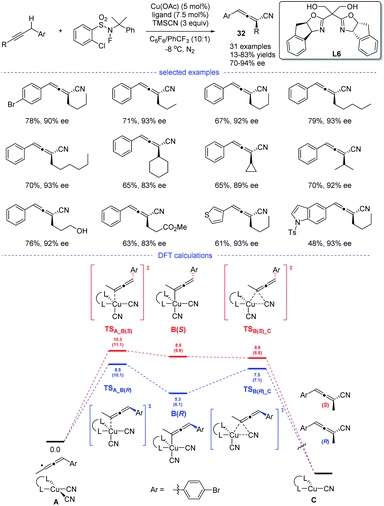 |
| | Scheme 28 Cu-catalyzed radical asymmetric cyanation of propargylic C–H bonds. | |
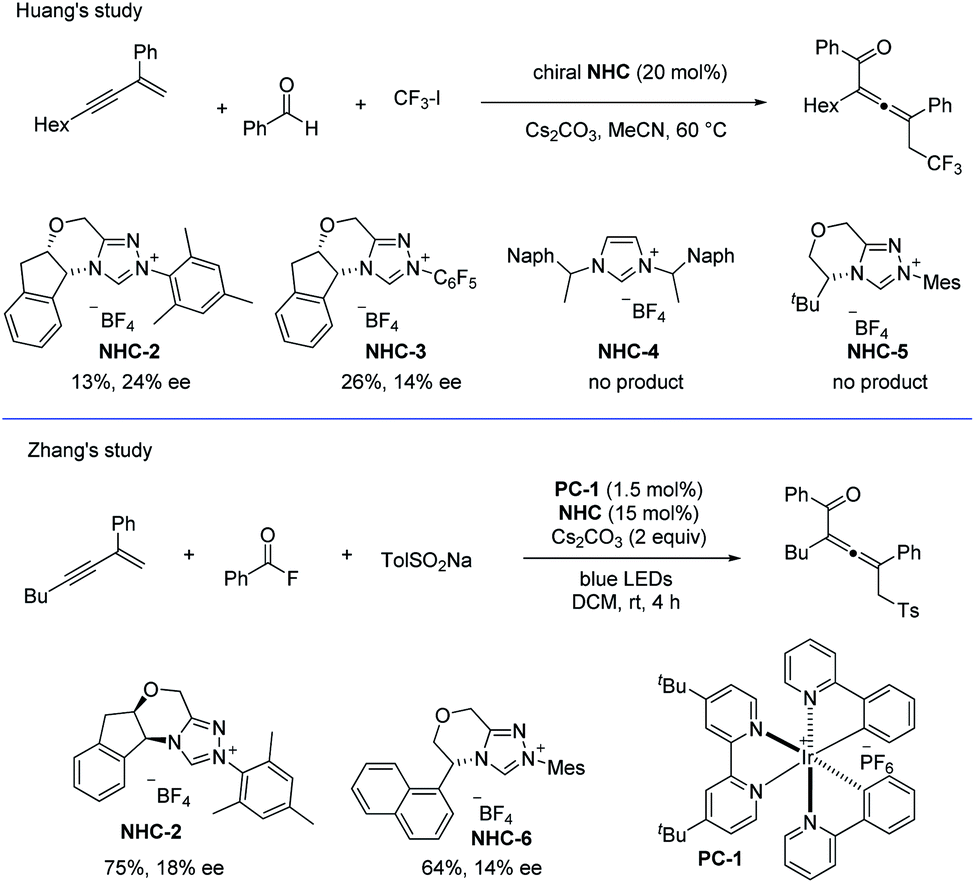
In addition, when using chiral NHCs as the catalysts, the groups of Huang38 and Zhang40 have independently studied the asymmetric coupling of allenyl radicals with the ketyl radicals (Scheme 29). In Huang's study, the chiral allene was obtained with the highest ee value of 24%, while in Zhang's work, the best ee value for the product was 18%.
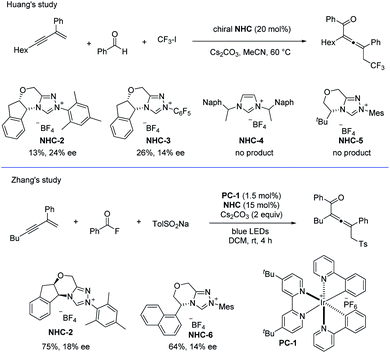 |
| | Scheme 29 NHC-catalyzed radical asymmetric synthesis of allenes by Huang and Zhang, respectively. | |
Conclusions and outlook
Allenes is a class of valuable organic molecules. Their importance in interdisciplinary research is reflected in the development of many remarkable and elegant methodologies for the synthesis of allenes that are versatile building blocks for the access of complex molecular targets, such as natural products, pharmaceuticals, and functional materials. In this perspective, we have highlighted the recent important advances in the synthesis of allenes via radical intermediates. Because rapid boom in this research area has been witnessed by the past few years and this radical strategy has greatly shown its potent ability in the synthesis of useful and otherwise difficult-to-be-synthesized allenes. Currently, they are several excellent approaches for radical synthesis of allenes, including radical 1,4-addition to 1,3-enynes, radical transformations with propargylic compounds, radical 1,2-elimination of vinylsulfoxides, and further functionalization of the allenyl C–H bond. It is worth mentioning that not only non-precious transition metals, such as copper and nickel, but organic NHCs are wonderful catalysts for these radical transformations. In addition, photoredox catalysis plays a significant role for the radical synthesis as well.
However, a great number of opportunities and challenges still exist. For example, although some progress on radical asymmetric synthesis of allenes has been achieved, there remains a significant challenge in controlling the enantioselectivity. Asymmetric reactions have the power to directly construct axial allenes that are otherwise relatively inaccessible and therefore may benefit the interdisciplinary area that requires efficient and direct strategies for chiral complex molecule assembling. In addition, the majority of examples have been obviously dominated by copper catalysis. It is necessary to develop alternatives, such as earth-abundant Fe, organocatalysts, photoredox catalysts, and others, to enrich the diversity of reactions with various mechanisms and substrates. More studies are also highly desired to explore different type of reaction substrates, which may lead to the expansion of their synthetic utilities in organic synthesis. It's expected that more and more structurally diversified allenes will spring up with the boom of new catalytic radical reactions. This will, in turn, provide an impetus for further development in this field.
Author contributions
Y. Li and H. Bao conceived the idea of the perspective and Y. Li prepared the manuscript with suggestions from H. Bao.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Supported by the NSFC (Grant Nos. 21871258,21922112, 22001251), the National Key R&D Program of China (Grant No. 2017YFA0700103), and the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDB20000000).
Notes and references
-
N. Krause and A. S. K. Hashimi, Modern Allene Chemistry, Wiley-VCH: Weinheim, 2004 Search PubMed.
-
(a) A. Hoffmann-Roder and N. Krause, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef PubMed;
(b) P. Rivera-Fuentes and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 CrossRef CAS PubMed;
(c) S. Yu and S. Ma, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS PubMed.
- B. S. Burton and H. von Pechmann, Ber. Dtsch. Chem. Ges., 1887, 20, 145–149 CrossRef.
-
(a) S. Ma, Chem. Rev., 2005, 105, 2829–2872 CrossRef PubMed;
(b) K. M. Brummond and J. E. DeForrest, Synthesis, 2007, 2007, 795–818 CrossRef;
(c) S. Ma, Acc. Chem. Res., 2009, 42, 1679–1688 CrossRef CAS PubMed;
(d) C. Aubert, L. Fensterbank, P. Garcia, M. Malacria and A. Simonneau, Chem. Rev., 2011, 111, 1954–1993 CrossRef CAS PubMed;
(e) S. Yu and S. Ma, Chem. Commun., 2011, 47, 5384–5418 RSC;
(f) D. Tejedor, G. Mendez-Abt, L. Cotos and F. Garcia-Tellado, Chem. Soc. Rev., 2013, 42, 458–471 RSC;
(g) J. Ye and S. Ma, Org. Chem. Front., 2014, 1, 1210–1224 RSC.
-
(a) F.-D. Lu, X. Jiang, L.-Q. Lu and W.-J. Xiao, Acta Chim. Sin., 2019, 77, 803–813 CrossRef CAS;
(b) Q. Dherbassy, S. Manna, F. J. T. Talbot, W. Prasitwatcharakorn, G. J. P. Perry and D. J. Procter, Chem. Sci., 2020, 11, 11380–11393 RSC;
(c) L. Fu, S. Greßies, P. Chen and G. Liu, Chin. J. Chem., 2020, 38, 91–100 CrossRef CAS;
(d) M. Muresan, H. Subramanian, M. P. Sibi and J. R. Green, Eur. J. Org. Chem., 2021, 2021, 3359–3375 CrossRef CAS.
-
(a) B. Delouvrie, E. Lacote, L. Fensterbank and M. Malacria, Tetrahedron Lett., 1999, 40, 3565–3568 CrossRef CAS;
(b) V. Mouries, B. Delouvrie, E. Lacote, L. Fensterbank and M. Malacria, Eur. J. Org. Chem., 2002, 2002, 1776–1787 CrossRef.
- S. Ma, Q. He and X. Jin, Synlett, 2005, 514–516 CrossRef CAS.
- J. Terao, F. Bando and N. Kambe, Chem. Commun., 2009, 7336–7338 RSC.
- Y. Tomida, A. Nagaki and J. Yoshida, J. Am. Chem. Soc., 2011, 133, 3744–3747 CrossRef CAS PubMed.
- Y. Mori, G. Onodera and M. Kimura, Chem. Lett., 2014, 43, 97–99 CrossRef CAS.
- B. Quiclet-Sire and S. Z. Zard, Pure Appl. Chem., 1997, 69, 645–650 CrossRef CAS.
- C. Alameda-Angulo, B. Quiclet-Sire and S. Z. Zard, Tetrahedron Lett., 2006, 47, 913–916 CrossRef CAS.
- M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed.
- F. Wang, D. Wang, Y. Zhou, L. Liang, R. Lu, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2018, 57, 7140–7145 CrossRef CAS PubMed.
- For a similar work on allene synthesis via cascade radical addition/prototropic
isomerization: M. Wang, Q. Wang, M. Ma and B. Zhao, Org. Chem. Front., 2022, 9, 1844–1849 RSC.
-
(a) L. Ge, H. Zhou, M.-F. Chiou, H. Jiang, W. Jian, C. Ye, X. Li, X. Zhu, H. Xiong, Y. Li, L. Song, X. Zhang and H. Bao, Nat. Catal., 2020, 4, 28–35 CrossRef;
(b) D. Lv, Q. Sun, H. Zhou, L. Ge, Y. Qu, T. Li, X. Ma, Y. Li and H. Bao, Angew. Chem., Int. Ed., 2021, 60, 12455–12460 CrossRef CAS PubMed;
(c) X. Ma, M.-F. Chiou, L. Ge, X. Li, Y. Li, L. Wu and H. Bao, Chin. J. Catal., 2021, 42, 1634–1640 CrossRef CAS;
(d) X. Zhu, W. Jian, M. Huang, D. Li, Y. Li, X. Zhang and H. Bao, Nat. Commun., 2021, 12, 6670 CrossRef CAS PubMed;
(e) Z. Nie, M. F. Chiou, J. Cui, Y. Qu, X. Zhu, W. Jian, H. Xiong, Y. Li and H. Bao, Angew. Chem., Int. Ed., 2022, e202202077 Search PubMed.
- X. Zhu, W. Deng, M. F. Chiou, C. Ye, W. Jian, Y. Zeng, Y. Jiao, L. Ge, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2019, 141, 548–559 CrossRef CAS PubMed.
-
(a) M. Taj Muhammad, Y. Jiao, C. Ye, M.-F. Chiou, M. Israr, X. Zhu, Y. Li, Z. Wen, A. Studer and H. Bao, Nat. Commun., 2020, 11, 416 CrossRef CAS PubMed;
(b) Q. Zhang, M.-T. Muhammad, M. F. Chiou, Y. Jiao, H. Bao and Y. Li, Org. Lett., 2020, 22, 5261–5265 CrossRef CAS PubMed.
- C. Ye, Y. Li, X. Zhu, S. Hu, D. Yuan and H. Bao, Chem. Sci., 2019, 10, 3632–3636 RSC.
- Y. Chen, J. Wang and Y. Lu, Chem. Sci., 2021, 12, 11316–11321 RSC.
- W. Lei, Y. Liu, Y. Fang, Y. Li, C. Du and J. Fang, Org. Biomol. Chem., 2021, 19, 8502–8506 RSC.
-
(a) C. J. Elsevier, J. Meijer, G. Tadema, P. M. Stehouwer, H. J. T. Bos, P. Vermeer and W. Runge, J. Org. Chem., 2002, 47, 2194–2196 CrossRef;
(b) D. Christopher Braddock, R. Bhuva, Y. Perez-Fuertes, R. Pouwer, C. A. Roberts, A. Ruggiero, E. S. Stokes and A. J. White, Chem. Commun., 2008, 1419–1421 RSC;
(c) Y. Wu and Y.-J. Jian, Synlett, 2009, 2009, 3303–3306 CrossRef;
(d) W. Zhang, H. Xu, H. Xu and W. Tang, J. Am. Chem. Soc., 2009, 131, 3832–3833 CrossRef CAS PubMed;
(e) W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, J. Am. Chem. Soc., 2010, 132, 3664–3665 CrossRef CAS PubMed.
- Y. Song, S. Song, X. Duan, X. Wu, F. Jiang, Y. Zhang, J. Fan, X. Huang, C. Fu and S. Ma, Chem. Commun., 2019, 55, 11774–11777 RSC.
- J. Huang, Y. Jia, X. Li, J. Duan, Z.-X. Jiang and Z. Yang, Org. Lett., 2021, 23, 2314–2319 CrossRef CAS PubMed.
- H. Shen, H. Xiao, L. Zhu and C. Li, Synlett, 2019, 31, 41–44 Search PubMed.
-
(a) F. Xiao, Y. Guo and Y.-F. Zeng, Adv. Synth. Catal., 2020, 363, 120–143 CrossRef;
(b) J. Chen, Y.-J. Liang, P.-Z. Wang, G.-Q. Li, B. Zhang, H. Qian, X.-D. Huan, W. Guan, W.-J. Xiao and J.-R. Chen, J. Am. Chem. Soc., 2021, 143, 13382–13392 CrossRef CAS PubMed;
(c) P.-Z. Wang, X. Wu, Y. Cheng, M. Jiang, W.-J. Xiao and J.-R. Chen, Angew. Chem., Int. Ed., 2021, 60, 22956–22962 CrossRef CAS PubMed.
- Y. Song, C. Fu and S. Ma, ACS Catal., 2021, 11, 10007–10013 CrossRef CAS.
- F.-S. He, P. Bao, F. Yu, L.-H. Zeng, W.-P. Deng and J. Wu, Org. Lett., 2021, 23, 7472–7476 CrossRef CAS PubMed.
- Q. Sun, X. P. Zhang, X. Duan, L. Z. Qin, X. Yuan, M. Y. Wu, J. Liu, S. S. Zhu, J. K. Qiu and K. Guo, Chin. J. Chem., 2022, 40, 1537–1545 CrossRef CAS.
- J. Terao, F. Bando and N. Kambe, Chem. Commun., 2009, 2009, 7336–7338 RSC.
- K.-F. Zhang, K.-J. Bian, C. Li, J. Sheng, Y. Li and X.-S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5069–5074 CrossRef CAS PubMed.
- Y. Chen, K. Zhu, Q. Huang and Y. Lu, Chem. Sci., 2021, 12, 13564–13571 RSC.
- T. Xu, S. Wu, Q.-N. Zhang, Y. Wu, M. Hu and J.-H. Li, Org. Lett., 2021, 23, 8455–8459 CrossRef CAS PubMed.
- C. Li, D.-D. Hu, R.-X. Jin, B.-B. Wu, C.-Y. Wang, Z. Ke and X.-S. Wang, Org. Chem. Front., 2022, 9, 788–794 RSC.
-
(a)
A. T. BIJU, N-Heterocyclic Carbenes in Organocatalysis, Wiley-VCH Verlag GmbH & Co. KGaA, 2018 CrossRef;
(b) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed;
(c) A. T. Biju, N. Kuhl and F. Glorius, Acc. Chem. Res., 2011, 44, 1182–1195 CrossRef CAS PubMed;
(d) S. De Sarkar, A. Biswas, R. C. Samanta and A. Studer, Chem. - Eur. J., 2013, 19, 4664–4678 CrossRef CAS PubMed;
(e) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed;
(f) H. Ohmiya, ACS Catal., 2020, 10, 6862–6869 CrossRef CAS;
(g) P. Bellotti, M. Koy, M. N. Hopkinson and F. Glorius, Nat. Rev. Chem., 2021, 5, 711–725 CrossRef CAS.
-
(a) K.-Q. Chen, H. Sheng, Q. Liu, P.-L. Shao and X.-Y. Chen, Sci. China: Chem., 2021, 64, 7–16 CrossRef CAS;
(b) T. Ishii, K. Nagao and H. Ohmiya, Chem. Sci., 2020, 11, 5630–5636 RSC;
(c) J. Liu, X.-N. Xing, J.-H. Huang, L.-Q. Lu and W.-J. Xiao, Chem. Sci., 2020, 11, 10605–10613 RSC;
(d) Q. Liu and X.-Y. Chen, Org. Chem. Front., 2020, 7, 2082–2087 RSC;
(e) L. Dai and S. Ye, Chin. Chem. Lett., 2021, 32, 660–667 CrossRef CAS;
(f) Q.-Z. Li, R. Zeng, B. Han and J.-L. Li, Chem. - Eur. J., 2021, 27, 3238–3250 CrossRef CAS PubMed.
- L. Chen, C. Lin, S. Zhang, X. Zhang, J. Zhang, L. Xing, Y. Guo, J. Feng, J. Gao and D. Du, ACS Catal., 2021, 11, 13363–13373 CrossRef CAS.
- Y. Cai, J. Chen and Y. Huang, Org. Lett., 2021, 23, 9251–9255 CrossRef CAS PubMed.
- Y. Q. Liu, Q. Z. Li, X. X. Kou, R. Zeng, T. Qi, X. Zhang, C. Peng, B. Han and J. L. Li, J. Org. Chem., 2022, 87, 5229–5241 CrossRef CAS PubMed.
- L. Wang, R. Ma, J. Sun, G. Zheng and Q. Zhang, Chem. Sci., 2022, 13, 3169–3175 RSC.
- C.-Y. Zhang, J. Zhu, S.-H. Cui, X.-Y. Xie, X.-D. Wang and L. Wu, Org. Lett., 2021, 23, 3530–3535 CrossRef CAS PubMed.
-
(a) S. W. Smith and G. C. Fu, Angew. Chem., Int. Ed., 2008, 47, 9334–9336 CrossRef CAS PubMed;
(b) S. W. Smith and G. C. Fu, J. Am. Chem. Soc., 2008, 130, 12645–12647 CrossRef CAS PubMed;
(c) A. J. Oelke, J. Sun and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 2966–2969 CrossRef CAS PubMed.
- R. Soler-Yanes, I. Arribas-Alvarez, M. Guisan-Ceinos, E. Bunuel and D. J. Cardenas, Chem.–Eur. J., 2017, 23, 1584–1590 CrossRef CAS PubMed.
- P. Domingo-Legarda, R. Soler-Yanes, M. T. Quirós-López, E. Buñuel and D. J. Cárdenas, Eur. J. Org. Chem., 2018, 2018, 4900–4904 CrossRef CAS.
- S. N. Kessler and J. E. Backvall, Angew. Chem., Int. Ed., 2016, 55, 3734–3738 CrossRef CAS PubMed.
- T. Song, L. Zhu, H. Li, C.-H. Tung, Y. Lan and Z. Xu, Org. Lett., 2020, 22, 2419–2424 CrossRef CAS PubMed.
- Z.-Z. Zhou, X.-R. Song, S. Du, K.-J. Xia, W.-F. Tian, Q. Xiao and Y.-M. Liang, Chem. Commun., 2021, 57, 9390–9393 RSC.
-
(a)
J. Hartung and T. Kopf, in Modern Allene Chemistry, ed. N. A. Krause and S. K. Hashmi, Wiley-VHC, Weinheim, Germany, 2004, pp. 701−726 Search PubMed;
(b) F. Pan, C. Fu and S. Ma, Chin. J. Org. Lett., 2004, 24, 1168–1190 CAS;
(c) G. Qiu, J. Zhang, K. Zhou and J. Wu, Tetrahedron, 2018, 74, 7290–7301 CrossRef CAS;
(d) L. Liu, R. M. Ward and J. M. Schomaker, Chem. Rev., 2019, 119, 12422–12490 CrossRef CAS PubMed.
- G. Zhang, T. Xiong, Z. Wang, G. Xu, X. Wang and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 12649–12653 CrossRef CAS PubMed.
- Y. Zeng, M. F. Chiou, X. Zhu, J. Cao, D. Lv, W. Jian, Y. Li, X. Zhang and H. Bao, J. Am. Chem. Soc., 2020, 142, 18014–18021 CrossRef CAS PubMed.
- X.-Y. Dong, T.-Y. Zhan, S.-P. Jiang, X.-D. Liu, L. Ye, Z.-L. Li, Q.-S. Gu and X.-Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 2160–2164 CrossRef CAS PubMed.
- R. Lu, T. Yang, X. Chen, W. Fan, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2021, 143, 14451–14457 CrossRef CAS PubMed.
- W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab and
Hongli
Bao
ab and
Hongli
Bao