
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Sustainability in peptide chemistry: current synthesis and purification technologies and future challenges
Lucia
Ferrazzano
 a,
Martina
Catani
b,
Alberto
Cavazzini
b,
Giulia
Martelli
a,
Dario
Corbisiero
a,
Paolo
Cantelmi
a,
Tommaso
Fantoni
a,
Alexia
Mattellone
a,
Chiara
De Luca
b,
Simona
Felletti
b,
Walter
Cabri
*a and
Alessandra
Tolomelli
*a
a,
Martina
Catani
b,
Alberto
Cavazzini
b,
Giulia
Martelli
a,
Dario
Corbisiero
a,
Paolo
Cantelmi
a,
Tommaso
Fantoni
a,
Alexia
Mattellone
a,
Chiara
De Luca
b,
Simona
Felletti
b,
Walter
Cabri
*a and
Alessandra
Tolomelli
*a
aP4i Lab – Peptidomimetics and Peptides Targeting Protein–Protein Interactions, Department of Chemistry “Giacomo Ciamician”, Via Selmi 2, 40126, Bologna, Italy. E-mail: walter.cabri@unibo.it; alessandra.tolomelli@unibo.it
bDepartment of Chemical, Pharmaceutical and Agricultural Science, Via Luigi Borsari 46, 44121, Ferrara, Italy
First published on 7th January 2022
Abstract
Developing greener synthesis processes is an inescapable necessity to transform the industrial landscape, mainly in the pharmaceutical sector, into a long-term, sustainable reality. In this context, the renaissance of peptides as medical treatments, and the enforcement of more stringent sustainability requirements by regulatory agencies, pushed chemists toward the introduction of sustainable processes to prepare highly pure, active pharmaceutical ingredients (APIs). Innovative upstream (synthesis) and downstream (purification) methodologies have been developed during the last 5 years with the introduction and optimization of several technologies in solid-phase peptide synthesis (SPPS), liquid-phase peptide synthesis (LPPS), chemo-enzymatic peptide synthesis (CEPS), and chromatographic procedures. These innovations are also moving toward the introduction of continuous processes that represent one of the most important targets for iterative processes. This overview discusses the most recent efforts in making peptide chemistry greener. The extensive studies that were carried out on green solvents, reaction conditions, auxiliary reagents and purification technologies in the peptide segment can be useful to other fields of organic synthesis.
1. Introduction
In 1990, the scientific community began to pay attention to pollution prevention by enacting the Pollution Prevention Act.1 Through a collaboration between academia and industry, several initiatives were launched in Europe and USA to solicit the development of new technologies to industrialize environmentally benign processes.2 In this context, the paramount role played by chemistry was immediately clear, because reduction or elimination of hazardous or toxic reagents and solvents, together with specific attention to waste and its fate in the environment, represented the first challenge. Following the 12 principles of green chemistry3 and using measurable parameters (i.e., green metrics), excellent research programs have been proposed for designing green synthesis processes and virtuous approaches for scientific development.4 Presently, the urgency to protect the environment from pollution, to contain climate change, and to ensure that water and clean energy is available for future generations has transformed green chemistry from an ethical approach to an inescapable necessity. As stated during the US congressional hearing in 2019, green chemistry can be described, according to the U.S. Environmental Protection Agency's (EPA) definition, as “the design of chemical products and processes that reduce or eliminate the generation of hazardous substances”. It is quite far from the concept of sustainability, which is more connected to the “improvement of efficiency with which natural resources are used to meet human needs for chemical products and services”.5 In this context, companies manufacturing fine-chemical intermediates understood the importance of greener processes early on, both for environmental and economic reasons.6 The successful synthesis of structurally challenging molecules for pharmaceutical use has been possible thanks to reliable synthesis strategies. The need to connect the synthesis of active pharmaceutical ingredients (APIs) for clinical development with the timeline required by the market, combined with intellectual-property politics, has often justified the preference for robust methodologies rather than greener approaches. At any rate, difficulties in integrating green chemistry and engineering in the API research and development supply chain is still the challenge of this century.7To bridge the gap, in 2005, the American Chemical Society created a Green Chemistry roundtable to integrate green protocols in the practice of chemical engineering in the pharmaceutical industry (ACS GCI-PR).8 As consequence, all of the 26 member-companies, representing a relevant part of the entire industry, have been developing green-by-design processes. Through the identification of sustainable practices in all stages of the manufacturing process, the aim is to reduce their impact on the environment and meet the needs of productive plans, going from preclinical development to commercialization. The use of green metrics, like the process mass intensity (PMI), has become essential for chemists and engineers to categorize processes and guide the selection of greener alternatives, especially in the production of bioactive molecules.9 Lately, green processes must also address the issues of cost efficiency, which sometimes generates a complete supply chain revolution. In this context, new chemical entities (NCEs) should be discussed separately from generics. For NCEs, the API cost is not a major influence because the commercialization of a product under patents guarantees a large profit regardless. In contrast, when a product becomes generic, its final price goes down dramatically and API's impact on the cost is a critical consideration. For generics, the market driver is mainly the price. From the industrial point of view, the main barriers to the introduction of green processes are determined by intellectual property and cost position.
Even though the ACS GCI-PR's attention was mainly focused on small molecules initially, more recently, medium-sized molecules have emerged in importance, including peptides. In this context, great attention was paid to peptidomimetic small molecules as modulators and ligands for receptors in several biological processes. This approach was not always convenient due to the reduced potency that peptidomimetics showed when compared to the corresponding reference peptides. Examples include angiotensin II receptor blockers for hypertension, losartan and valsartan, as mimics of the peptide saralasin. In addition, although it is possible to develop small molecules to mimic short peptide sequences,10 the same approach cannot be applied to mimic longer peptides that are generally developed for protein–protein interference interactions. As recently summarized by Lau and Dunn,11 peptide therapeutics can be divided, according to their sequences, into native, analogs and heterologous forms. Native peptides have the same sequence as natural products but can be obtained through isolation, chemical synthesis, or recombinant technologies. To overcome the limitations that native sequences present in terms of short half-life, proteolytic sensitivity, or low membrane permeability, modifications to the chemical structures can be introduced, improving their pharmacological profiles, and generating a new family of peptide analogues. Finally, when an active peptide sequence is found independently from natural peptides, by library screening or the phage display method, it is classified as a heterologous peptide. Analogues and heterologous peptides generally present a better pharmaceutical profile than the native peptides in term of half-life and membrane permeability. As shown in Fig. 1, among FDA-approved therapeutic peptides,12 analogues represent the majority. Some analogues, like liraglutide and semaglutide, take advantage of the integration between recombinant technology to produce long, native sequences, and chemical modifications.13 However, for most of the other molecules, chemical synthesis to obtain them is the method of choice. The main industrial target in the last 30 years has been the development of consistent and reliable chemical technologies for peptide production. The scientific achievements in this area, mainly in solid-phase peptide synthesis (SPPS), and the lower attrition rate in peptide development, attracted several pharmaceutical companies to this modality, determining its success.11,14 There are three main areas that still need to rapidly evolve. The first is related to product quality, since several regulatory agencies are asking to lower impurity limits to avoid any risk of immunogenicity.15 In this context, increased efficiency of analytical technologies plays a key role. The second area is related to the introduction of more rigorous Environmental Health & Safety (EHS) policies. The third is due to therapeutic successes in diseases such diabetes and obesity, which require larger production volumes of drugs (>100 kg).16 During the last six years (2015–2021) several research groups from academia and industry devoted incredible efforts to improve efficacy and greenness of peptide production, as witnessed by the large number of publications and patents on the topic. The bloom of new approaches to upstream and downstream technologies is fragmented. For this reason, the target of this review is a critical evaluation of the latest publications on greening peptide synthesis and purification, to stimulate a proactive approach to the application of these techniques in peptide chemistry.
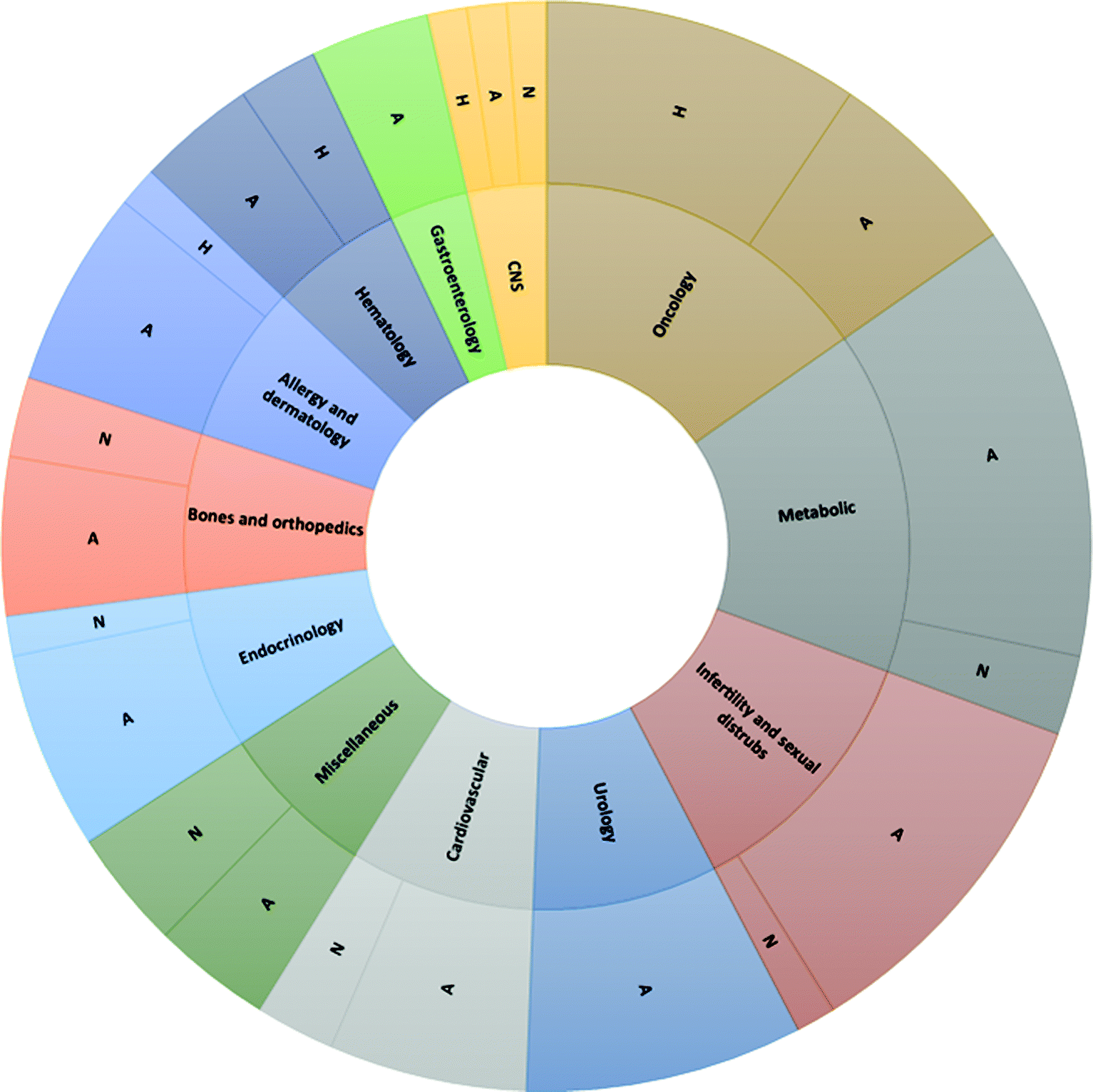 | ||
| Fig. 1 Chemical basis of peptide drugs approved by Food & Drug Administration (FDA) from 1952 to 2021. Adapted from ref. 12. Legend: A = Analogs; H = Heterologous; N = Native. | ||
2. Sustainable peptide synthesis: an inescapable necessity
2.1 Relevance of peptides in the pharmaceutical sector
Recently, the potential of peptides as therapeutics gained particular interest because of their high likelihood of approval (LoA) or probability of success (POS) in clinical trials, compared to small molecules. If the limitations of these “medium-sized” molecules, such as short half-lives and poor oral bioavailability, put a break on enthusiastic peptide drug development, novel synthesis strategies to modulate their pharmacokinetic properties and target-specificity have generated renewed appreciation for this pharmaceutical segment.11 The development of stable techniques for peptide synthesis and purification have made pharmaceutical-grade peptides more accessible, even when they have a high number of amino acids. For these reasons, peptides have received more attention in the last two decades. Today, about 80 peptide-based therapeutics (PbTs) have been approved and launched on the market, with >150 peptides in clinical development and 400–600 peptides in preclinical studies.17 From a commercial standpoint (excluding insulin), market studies forecast a possible Compound Average Growth Rate (CARG) of 10% per year in the peptide sector, from $29 billion to $51 billion between 2020 and 2026 (Fig. 2).14 Research into peptide-based drug candidates is attractive because they are smaller than proteins and can reach extracellular and intracellular targets. This contrasts with proteins, which are large molecules, generally not cell membrane-permeable, and limited to extracellular targets. Despite their success as pharmacological agents, native peptides cannot be used against many target diseases. Improvement of peptides’ pharmaceutical profiles was achieved by structural modifications, like the introduction of unnatural amino acids, cyclization, synthesis of stapled peptides and cyclotides, or conjugation with stabilizing lipids or pegylated chains.18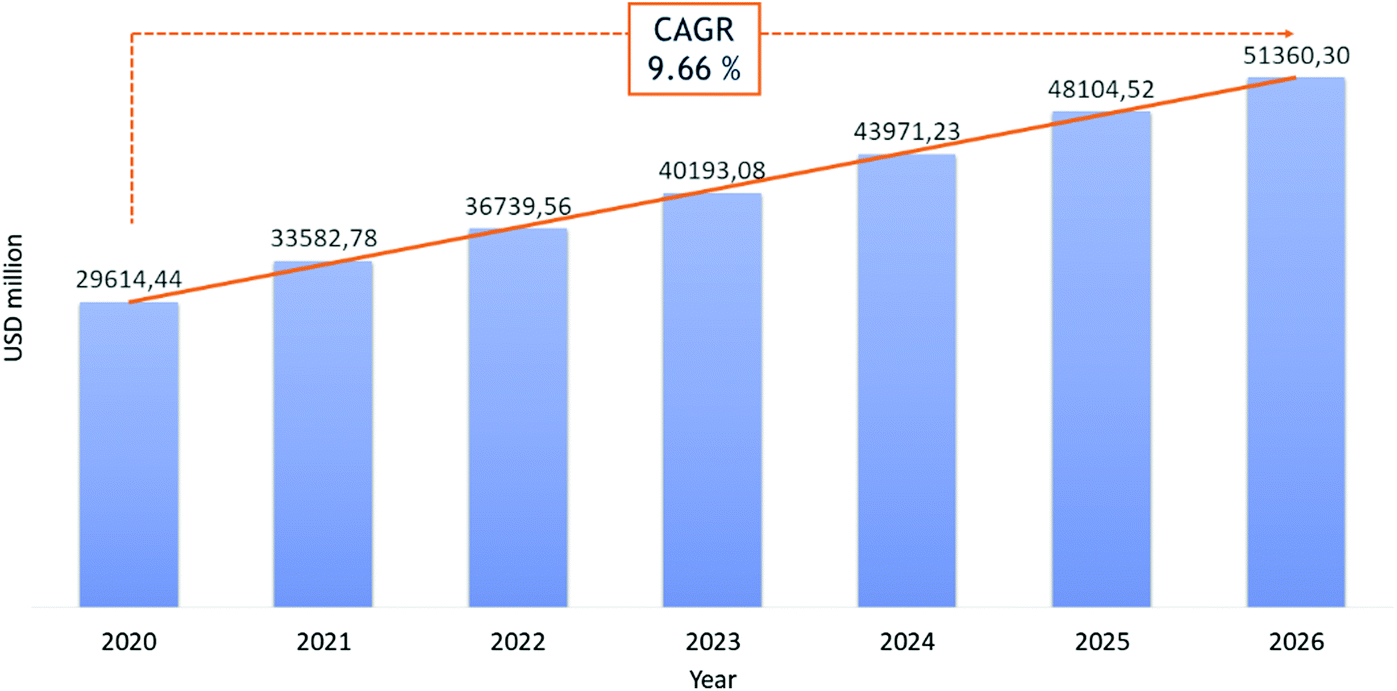 | ||
| Fig. 2 Revenue in USD million for peptide therapeutic market in the period 2020–2026. Ref. 14. | ||
Retracing the milestones in the discovery and introduction of peptide-drug therapeutics, the role of peptides as biological mediators is immediately clear, as is the impact of peptides on the pharmaceutical market (Fig. 3). Progress toward a systematic approach to peptides becoming real pharmaceutical entities required the pioneering work of du Vigneaud19 and Merrifield,20 who are responsible for the total synthesis of oxytocin and vasopressin, and the introduction of SPPS, respectively. Furthermore, the advent of recombinant technologies in the 1980s allowed an enhanced greenness score in peptide production. These important advancements enabled the manufacturing of peptides on larger scales. Examples include production of dulaglutide, liraglutide, octreotide, triptorelin, degarelix, icatibant, abaloparatide, and etelcalcetide. Looking at the frequencies of NCE approvals for peptides and the considerable innovations proposed by synthesis technologies for greener manufacturing (reported below), it is easy to predict further expansion of this pharmaceutical segment.17,21
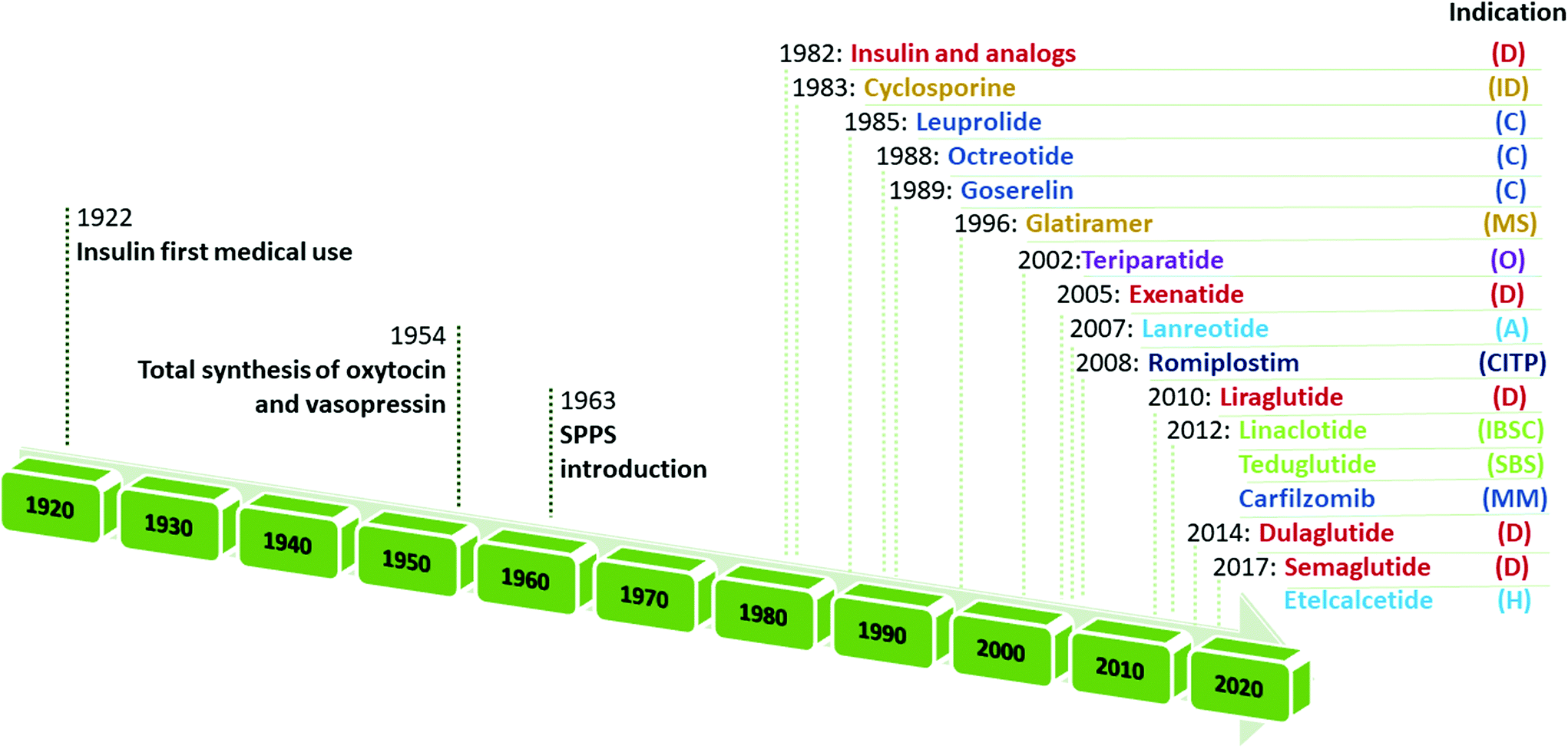 | ||
| Fig. 3 Timeline of peptide drug approvals and the top-selling peptide drugs list. Note, the year refers to the first approval. Abbreviations: D = diabetes, obesity; ID = immune diseases; C = cancer; MS = multiple sclerosis; O = osteoporosis; A = acromegaly; CITP = chronic immune thrombocytopenia; IBS = irritable bowel syndrome with constipation; SBS = short bowel syndrome; MM = multiple myeloma; and H = hyperparathyroidism. Data adapted from ref. 17. | ||
2.2 Therapeutic areas addressed by peptides
The possibility of exploiting peptide drugs for a wide range of therapeutic indications has been, in the last two decades, the main force pushing pharmaceutical companies towards investing in peptide candidates for drug discovery and development programs.11 The biggest areas of marketed therapeutic peptides are those targeting protein–protein interactions (PPIs) that regulate several diseases, called interfering peptides (IPs). In this class of drugs, the majority are applied to the treatment of cancer, diabetes and obesity, cardiovascular disease, or gastrointestinal disorders (Fig. 4).22 Recently, the necessity to replace classical antibiotics to address widespread antibiotic resistance led to rapid and continuous production of data on antimicrobial peptides (AMPs).23 Finally, the recent Covid-19 pandemic pushed forward a search for novel therapeutic peptides against SARS-CoV-2 infection and associated diseases.24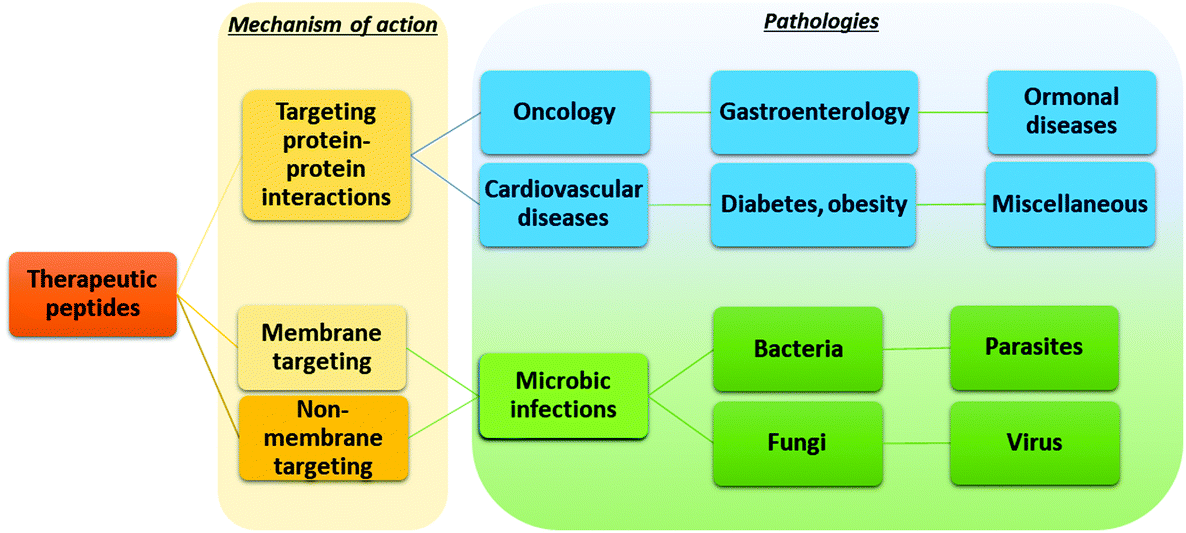 | ||
| Fig. 4 Application of peptide therapeutics according to the mechanism of action and therapeutic area. | ||
2.3 Green metrics in peptide synthesis
Considering the relevance that peptide pharmaceuticals are gaining from both economic and technological standpoints, to address the greenness of processes for peptide API manufacturing, green metrics must be introduced. Peptide chemical synthesis has some clear limitations compared to small molecules, determined by its specific, iterative nature. In fact, the formation of the peptide bond is always performed by activating the acid moiety of the protected amino acid. Therefore, although Trost's atom economy (AE),25 Wender's step count economy,26 and Baran's ideality factor (IF)27 are inefficient descriptors of peptide synthesis, there is no way to overcome this limitation. Among the different green metrics introduced in the last 25 years,28 the most useful are complete environmental factor (cEF) introduced by Sheldon, and PMI, which, considering all chemicals involved in a synthesis process, comprising water, allow a correct head-to-head comparison for peptide manufacturing technologies. Because the two metrics are easily interconnected, in this review, the PMI will be used.The techniques commonly adopted in peptide synthesis to build the sequences rely on previously optimized procedures and chemistry, making significant methodological innovations more difficult to achieve.
For example, the synthesis of a model 10-mer peptide through classical Fmoc/tBu orthogonal SPPS consists of (i) a loading step, (ii) 10 Fmoc-deprotections, (iii) 10 couplings, and (iv) final deprotection and cleavage. Of note, each step is followed by (v) several washes, with considerable consumption of solvent. Washes represent 80–90% of the total waste from the process. It has been estimated that, for a peptide with an average molecular mass between 1000 and 5000 Da, a considerable volume of waste is generated, leading to a typical PMI between 3000 and 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 kg kg−1 of API. Additionally, the use of hazardous solvents and reagents must be added to the calculation (Table 1).7
000 kg kg−1 of API. Additionally, the use of hazardous solvents and reagents must be added to the calculation (Table 1).7
| PMI impact | Main component | |
|---|---|---|
| Solvents | 80–90% | DMF, NMP, DCM |
| Base | 4–5% | Piperidine |
| Cleavage blend | 4–5% | TFA |
| Activated amino acid | 1% | Fmoc chemistry |
| Coupling blend | 1% | OximaPure/DIC |
| Resin | 0.1% | PS, PEG |
Although techniques other than SPPS can be used in peptide synthesis, the basic chemistry behind each process does not change considerably and the number of synthesis steps cannot be reduced. Moreover, the amount of water needed must be always considered for protocols that require excess removal of side products and reagents by water washes. Only by introducing green solvents and reagents, allowing for efficient and high-purity synthesis of peptides, and by using reagents for couplings and deprotections that are easily recoverable/reusable for a new process, could it be possible to achieve competitive PMI values. However, only one paper in this field reports green-metric evaluations or describes the process in detail.29
2.4 Ranking of solvent selection guides
The selection of a solvent, either as single chemical or a mixture of chemicals for a specific reaction, is strictly connected to its properties.30 In particular for synthesis purposes and the application to peptide chemistry, the solubility of all reaction components is an important parameter to consider when optimizing all steps of the process, including synthesis and purification. Furthermore, physical parameters like the maximum working range between melting and boiling points, chemical and thermal stability, viscosity, density, and heat of vaporization are important toward setting up experiments in both academic laboratories and downstream production plants.31 Focusing on the production of therapeutic peptides, target molecules can be synthesized chemically or by recombinant technology. In the first case, different approaches are possible, like solid, liquid, or hybrid-phase synthesis; microwave-assisted technology; or ligation technology. Currently, and likely to still be the case in 2030, peptides are mainly synthesized by chemical methods (as with 80% of peptide API manufacturers in 2020) due to the type of production facility adopted by pharma and related contract development and manufacturing organizations (CDMOs). Among them, liquid-phase and solid-phase synthesis are the most used (39% and 41%, respectively, Fig. 5).32 Concerning purification, the preferred approach relies on reverse-phase, high-performance liquid chromatography (RP-HPLC), which allows, with high efficiency, isolation of the target peptides from by-products (shorter sequences, isomers, and products of side-chain reactions).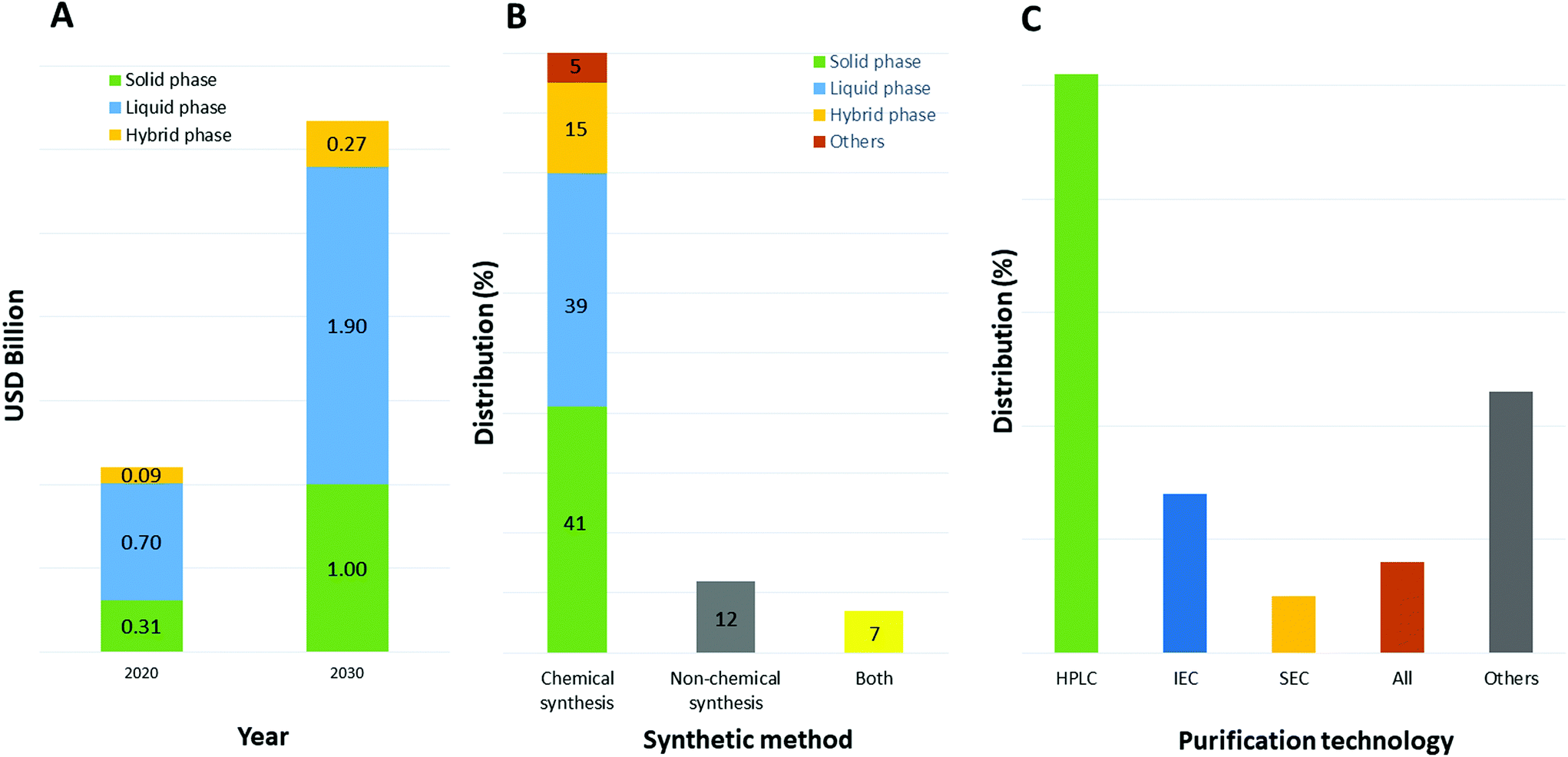 | ||
| Fig. 5 (A) Market impact of specific peptide chemical synthesis methods that are used by contract manufacturing organizations— comparison between 2020 and 2030; (B) distribution of peptide manufacturing companies by peptide synthesis method used; (C) distribution of purification technology used. Abbreviations: USD = United States Dollar; HPLC = High-Performance Liquid Chromatography; IEC = ion-exchange chromatography; SEC = size-exclusion chromatography. Ref. 32. | ||
These considerations demonstrate the important role played by solvents in making peptide production a greener reality. The solvents’ impact on the environment has additional social and economic effects that cost about $1 billion per year in technologically advanced countries, along with effects on ecosystems and health. For a solvent to be defined as “green”, it needs to conform not only to EHS assessments but also to energy demand evaluations. The latter requirements can be calculated as the net cumulative energy demand (CED) for solvent production, between the energy required to produce it and the recoverable end-of-life energy that can be achieved by incinerating it or recycling it via distillation.
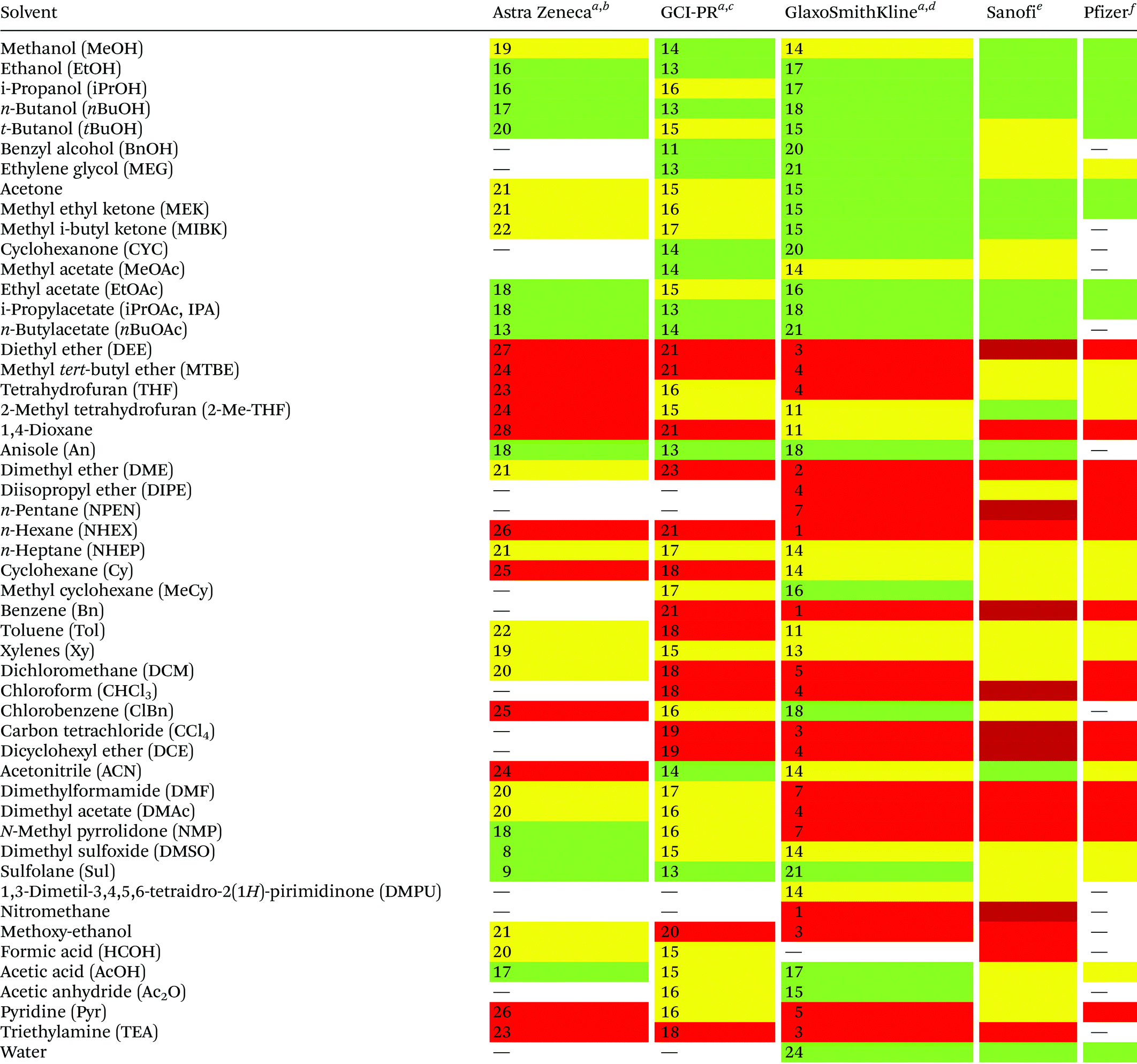
Reduction in CED values depends on the solvent and it is not defined for all of them.33 Therefore, EHS profiling of organic solvents is the main tool for the solvent greenness ranking. The ACS GCI-PR and several pharmaceutical companies (GlaxoSmithKline, Sanofi, Pfizer, AstraZeneca, etc.) have published similar solvent-selection guides to facilitate the comparison between the most-used solvents and new, potential green candidates. In particular, these selection guides cover different aspects of greenness such as (i) waste (e.g., recycling, incineration, and volatility), (ii) environmental impact, (iii) health (acute or chronic effects on humans), (iv) flammability and explosivity, (v) stability in handling and storage, and (vi) life cycle assessment (LCA).34
Pfizer was the first to develop a list of solvents classified as “preferred”, “usable”, and “undesirable”, accompanied by a list of greener alternatives for those solvents that need to be replaced.33 Together with the above reported criteria, GlaxoSmithKline's guide35 considers additional parameters related to physical properties, legislation constraints, and most hazards. The latest version of GSK's guide includes 110 solvents, and the reported information can be considered according to specific users’ needs. Each solvent is scored from 1 (red, unfavourable) to 10 (green, preferred) considering all the data obtained for each EHS parameter. Combining all the results from each category, a final EHS red flag is attributed to unfavoured solvents. Sanofi's guide offers, in addition to definitions similar to GSK's, details for solvent applications in synthesis chemistry and substitution advice (i.e., green for recommended solvents, yellow for substitution advisable, red for requested substitution, and brown for banned solvents).36 From an industrial point of view, Sanofi's guide is particularly useful because it includes legislative categories, so the advantages of the overall ranking reported by others remain unchanged. Similarly, AstraZeneca's guide has one health, two safety, and seven environmental criteria. The guide developed by ACS GCI-PR has one health, one safety, and three environmental criteria.37 For the easiest interpretation of the data, Prat and co-workers proposed a smart comparison of AstraZeneca (AZ), ACS GCI-PR, and GSK's guides for 51 solvents, limiting the analysis to three parameters: health, safety, and the environment; giving results as sum of these values, and assigning a coloured flag determined by the arithmetical mean of all EHS results (columns 2–4, Table 2).37b The data they reported are summarized in Table 2 and compared to Sanofi's and Pfizer's guide results (columns 5 and 6, Table 2).
| Sanofi and Pfizer's guides do not use numbers for solvent's greenness evaluation, but only colour codes.a Ref. 37b.b Arithmetical mean: 20.3.c Arithmetical mean: 16.3.d Arithmetical mean: 11.5.e Ref. 36.f Ref. 33; green = recommended; yellow = substitution advisable; red = unfavoured; dark red = banned. |
|---|
|
In 2016, Byrne and co-workers took the solvent guide a step further, comparing all of the reported guides with the CHEM21 report.33 The solvents were reported according to the Global Harmonized System of Classification, Labelling & Packaging (GHS-CLP) regulation. The final ranking of a solvent for each EHS parameter depended on the least green characteristic (maximum score 10). With two red scores, or a score ≥8, the solvent was classified as “hazardous”; with two yellow scores, or a score = 7, the solvent was classified as “problematic”; otherwise, it was classified as “recommended”.38
Due to difficulties in finding consensus in the categorisation of solvents, intermediate categories (“recommended or problematic” and “problematic or hazardous”) were introduced, allowing the classification of solvents with ambiguous interpretations (Fig. 6). Generally, the previously reported guides did not consider the source of the solvent as a ranking criterion. Only recently, unconventional bio-based solvents, namely glycerol and Cyrene®, were introduced in two new solvent-selection guides.39 Because both solvents have a boiling point >200 °C, they were red-flagged for experimental reasons related to solvent removal. However, this does not mean these solvents should be defined as environmentally damaging, considering their low toxicities and their sustainable production.40
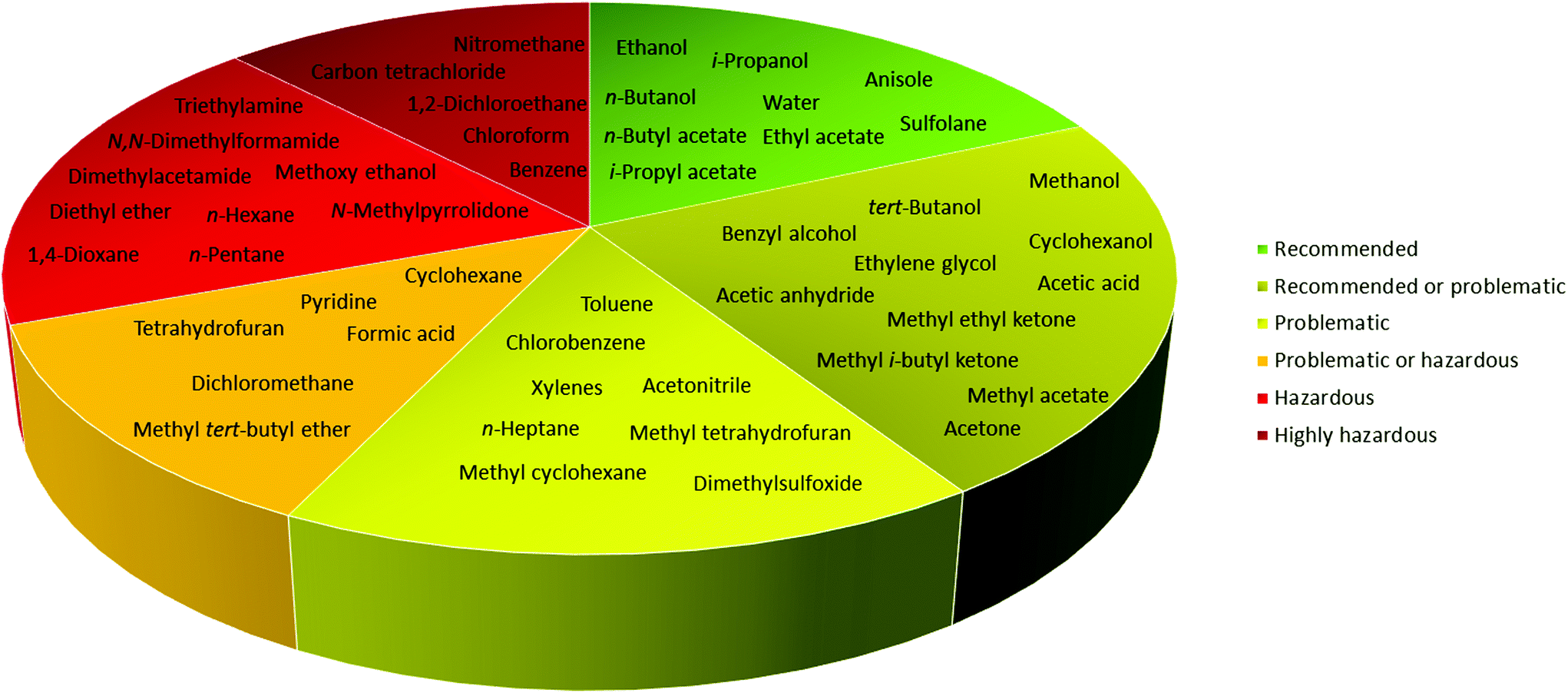 | ||
| Fig. 6 Overall ranking of solvents resulting from the comparison of most common solvent selection guides. Ref. 38. | ||
Solvent selection must always consider the specific reaction or process since it can affect reaction rates, chemical selectivity, temperature control, and efficiency in purification.41 This aspect makes it fundamental for chemists in academia and industry to use the tools reported above to make the optimization and the scale-up of pharmaceutical manufacturing greener.
3. Technologies and synthesis modifications toward “greening” peptide synthesis
The synthesis of peptides can be achieved by different methodologies, namely (i) solid-phase peptide synthesis (SPPS), (ii) liquid-phase peptide synthesis (LPPS), (iii) semi-continuous/continuous peptide synthesis, (iv) chemoenzymatic peptide synthesis (CEPS), (v) mechanochemical peptide synthesis, and (vi) recombinant production via fusion proteins. Because the last technique is used only to synthesize long peptides containing natural amino acids, it has a limited scope in modern drug discovery. It is specifically applied to the synthesis of native, pharmaceutically active peptides or to the semi-synthesis of analogues, in which the native sequence is sufficiently long (e.g., liraglutide or semaglutide). In this section, we focus on the five other synthesis technologies, reporting on the most recent improvements toward enhancing the greenness of synthesis processes for peptide production.The final goal of peptide synthesis, independent from the applied technique, is to achieve the target product with the least amount of impurities, in order to reduce downstream purification phases and impact the PMI of the whole process. For this reason, particular attention is paid to monitoring the most commonly occurring side reactions observed in peptide synthesis: racemization, diketopiperazine and aspartimide formation, arginine lactamization, etc. (see Fig. 7). Moreover, the deprotection and coupling efficiency must be always guaranteed, being critical to avoid redundant amino acid incorporation or sequence deletion.42 Accordingly, whenever a new synthesis methodology is studied, or a new solvent or reagent is introduced, the standard procedures include an evaluation of the tendency of the peptide model sequences to produce the above-mentioned impurities. Therefore, this study represents the key, common determinants in assessing the feasibility of a new protocol.
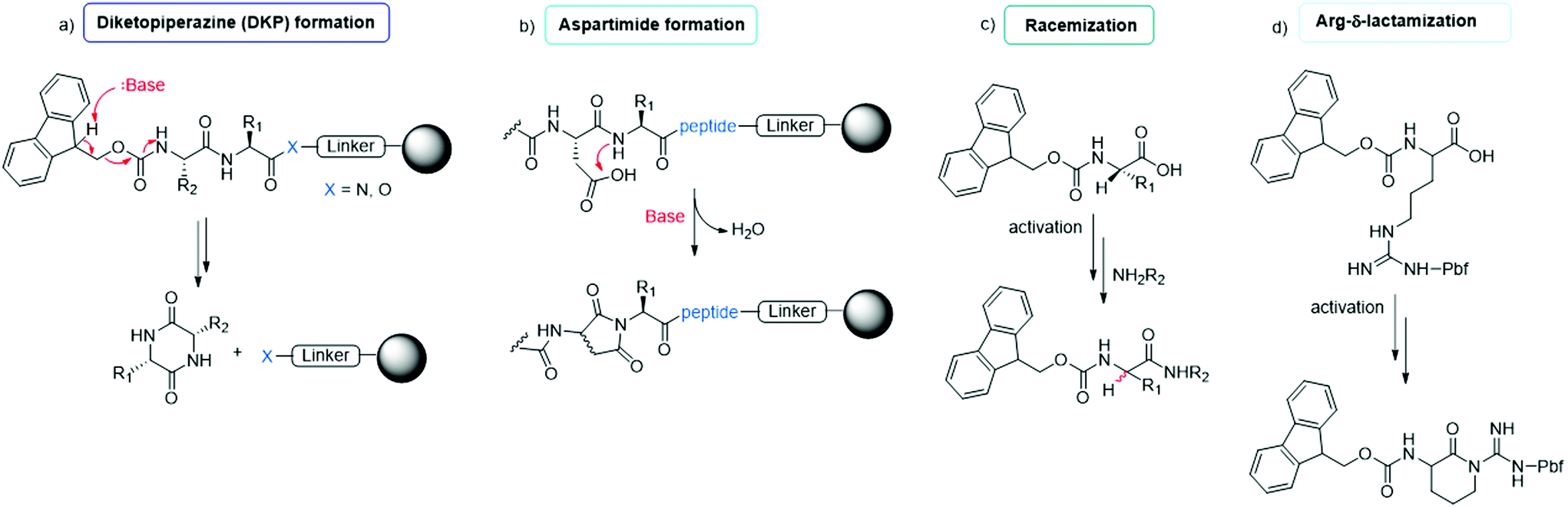 | ||
| Fig. 7 Common side reactions in peptide synthesis occurring on resin (a and b) or during Fmoc-amino acid activation (c and d). | ||
3.1. Green solid-phase peptide synthesis (GSPPS)
SPPS technology has been optimized over more than half a century around the use of N,N-dimethylformamide (DMF) as a solvent. Currently, this technology allows for the synthesis of long peptide sequences with a high throughput in purity and yield, with any kind of amino acid. SPPS is based on stepwise growth of a peptide sequence on a polymer-based, insoluble, solid support. Once the first amino acid, protected at the amino group, is anchored on the resin via the acidic terminal end, the peptide sequence is built-up by an iterative reaction sequence, consisting of amino deprotection–wash–coupling–wash steps. Then, the peptide is cleaved from the solid support and isolated by precipitation with an anti-solvent. The Fmoc/tBu orthogonal strategy42b is routinely exploited, in which the growing α-amino terminus is protected with a temporary Fmoc group that is removed at each deprotection step under basic conditions. Conversely, the amino acid side chains are protected with acid-sensitive groups, which can be removed, if required, together with the cleavage of the peptide from the resin.43 During the SPPS cycle, purification steps are not necessary because the excess of reagents or by-products are eliminated from the solid support by simple filtrations. This is not only an advantage in terms of yield, but it also saves time and introduces the possibility of automating the process. However, this approach has a high environmental impact due to the huge volume of solvents required for the synthesis protocol, and super-stoichiometric amounts of reagents introduced to push the reaction to completion and minimize the formation of impurities. In addition, when the access to the growing peptide attached to the resin is particularly hampered the procedure requires a double coupling. Peptide PMIs44 are estimated about 100 times higher than those for other synthesis procedures, justifying the great need for new, eco-friendly protocols.45Only very recently, prompted by the increasing interest in the GSPPS field by both academia and the pharmaceutical industry, the technical requirements that a solvent should fulfil in a green API manufacturing industrial setting were clearly defined as follows:6b,46
• Melting point ≤10 °C;
• Viscosity < 4 mPa s;
• Reagents’ and by-products’ solubilities ≥0.25 M;
• Starting resin's swelling: 4–7 mL g−1;
• Coupling time: 60–90 min;
• Fmoc-removal time: 30–40 min;
• Loading of the first amino acid on the resin < 120 min;
• Stability and inertia along the SPPS cycle;
• Low levels of epimerization or other side-reactions.
All of these features are required in order to obtain the best product output while reducing possible technical issues in production plants. A single candidate that can simultaneously produce optimal results in all steps may be difficult to find. For this reason, a great number of green solvents have been excluded from the reported studies over the years.
N,N-Dimethylformamide (DMF) and, to a lesser extent, N-methyl-2-pyrrolidone (NMP) and dichloromethane (DCM), are currently the solvents of choice in SPPS. These solvents match every characteristic defined above together with low cost. However, as reported in Table 2, these solvents are classified by the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) regulation as substances of very high concern (SVHC) because of their carcinogenic, mutagenic, or reproductive-toxin (CMR) properties,47 and need to be urgently replaced.48 This is emphasized when considering that, for SPPS, a solvent's impact on waste production represents >80% of the total, justifying attempts to replace the hazardous, reproductively toxic solvents, DMF and NMP, with more benign and environmentally friendly alternatives. These attempts go together with introducing methods for recycling solvents or minimizing their impact.
As several green innovations for SPPS have been introduced for single steps of the process (swelling, coupling, or washing), for this report on green solvents, only cases in which all steps of the SPPS cycle were evaluated simultaneously were considered, because a green solvent (or mixture) should be successful in the whole process to have a chance to be industrially applicable. Only a brief mention of green solvents for cleavage and precipitation steps is included, because they are outside of the repeating steps of the SPPS cycle. Finally, due to the importance of PMIs in the evaluation of synthesis and productive processes, recent methodologies on solvent reduction or recycling after SPPS are described.
Generally, SPPS protocols are performed at room temperature and the majority of green innovations introduced along the years, concerning reagents and solvents, have been applied under these conditions. Anyway, the easiest way to accelerate the synthesis process reducing its impact on the productivity programs (and on the costs associated) would be increasing the operative temperatures. In this context, the most important implementation has been the introduction of microwaves (MW) as an alternative to conventional heating.49
MW-assisted peptide synthesis has been largely used at lab scale, and only recently has the development of the technology reached a semi-industrial scale. The main technological contributions in this field came from CEM50 and CS Bio51 companies, who developed efficient MW-assisted automatic peptide synthesizers, largely employed both in small- and kilogram-scale production, with significant reduction in reaction times and costs. Papini et al. recently reported an efficient synthesis of the cyclic Eptifibatide peptide by using MW-assisted Fmoc/tBu protocol.52 Confirming the industrial relevance of this technology, in 2021 CEM Corporation and AmbioPharma Inc. signed a partnership for the manufacturing of GMP peptides up to multi-kilogram quantities.53 The MW approach requires a specific development process, and the target is always to generate high-quality crude material to simplify the real time-consuming step, that is the downstream purification. Matching the advantages of the MW technique and the use of green solvents and reagents could increase both sustainability and upstream productivity.54,55
3.1.1.1. Solvents for full GSPPS. Before 2009,56 little attention was paid to the replacement of classical, hazardous solvents in SPPS, in contrast to resins, protecting groups, and coupling reagents that were more deeply investigated. However, the real boost in the development of alternative solvents for SPPS started in 2015. Since then, it has been consistently increasing. Table 3 summarizes the last six years of evolution in this field, showing the results achieved by several research groups on the application of green solvents to a fully green SPPS. The resins used in these studies are included to demonstrate how the application of green solvents could be strongly limited by the choice of the solid support. Among the combinations of coupling reagents, N,N′-diisopropylcarbodiimide (DIC)/OxymaPure® are the most explored. Model peptides that are commonly prepared as a proof of concept generally include Leu/Aib-Enkephalin or Aib-ACP, while variegated APIs are synthesized to validate the newly developed green protocols (Table 3).
| Entry | Solventa | Full SPPS: peptide | Resin | Coupling reagent | HPLC crude purity (%) | Groupb (ref.) | Year |
|---|---|---|---|---|---|---|---|
|
a Only solvents effective for all steps of SPPS and authors’ best conditions are reported.
b Corresponding author or company is listed.
c Double loading was performed.
d Piperidine was used as deprotecting agent for the selected entries.
e 2-MeTHF/MeOH = 1:1 was used for deprotections, while 2-MeTHF was used for couplings.
f An/DMSO = 4:1 for couplings, An/EtOH = 1:1 for deprotection. RA = Rink Amide; CM = ChemMatrix; AM = amino methyl; Trt = trityl; CTC = chlorotrityl; RMG = Ramage; AMS = aminomethylstyrene; TG S = TentaGel S; AMRES = aminomethylresin; HMPB = 4-(4-hydroxymethyl-3-methoxyphenoxy)-butanoyl amide; OP = OxymaPure. |
|||||||
| 1 | THF | Aib-Enkephalin-NH2 | Fmoc-RA-AM-PEG | DIC/OP | 93.6 (DMF/DCM 53) | Albericiod (57) | 2015 |
| 2 | Aib-ACP | 69.8 (DMF/DCM 37.8) | |||||
| 3 | ACN | Aib-Enkephalin-NH2 | Fmoc-RA-AM-PEG | DIC/OP | 91.8 (DMF/DCM 53) | ||
| 4 | Aib-ACP | 49.6 (DMF/DCM 37.8) | |||||
| 5 | 2-MeTHF | Aib-Enkephalin-NH2 | CM | DIC/OP | 95 (DMF/DCM 53); 91.9 (EtOAc in deprot. steps) | Albericiod (54) | 2016 |
| 6 | Aib-ACP | 37.3 (DMF/DCM 37.8); 87.1 (with coupl/depr at 40 °C) | |||||
| 7 | Aib-Enkephalin-NH2 | Fmoc-RA-AM-PS | 41.6 (DMF/DCM 71.8); 88.6 (with coupl/depr at 40 °C) | ||||
| 8 | PC | Bradykinin | HMPB-CM | HBTU/HOBt/DIPEA | 77 (DMF 79) | Northd (67) | 2017 |
| 9 | NFM | Aib-ACP | Fmoc-RA-AM-PS | DIC/OP | 54 (DMF/DCM 93.8) | Albericiod (54) | |
| 10 | GVL | 52 (DMF/DCM 93.8) | |||||
| 11 | NBP | linear octreotide | AMRES | DIC/HOBt | 80 (DMF 86) | Novartis (68) | 2018 |
| 12 | TMU | 78 | |||||
| 13 | DMI | 78 | |||||
| 14 | DMSO | 52 | |||||
| 15 | DMPU | 51 | |||||
| 16 | GVL (Microwave) | Jung-Redemann | RA-CM | DIC/OP | 68 | Albericiod (55) | |
| 17 | ACP, ABC peptide, Thymosin | RA-CM | DIC/OP | Not reported, comparable to DMF | |||
| RA-AM-PS | |||||||
| PS Wang (ACP) | |||||||
| 18 | EtOAc/DMSO 9:1 |
Aib-ACP | Fmoc-RMG-AMS | DIC/OP | 76 | PolyPeptide (29c) | 2019 |
| 19 | NBP/EtOAc 1:1 |
Ac-Nle-DHFRWK-NH2 | Fmoc-RMG-AMS | DIC/OP | 90 | PolyPeptide (78) | |
| 20 | Anisole/DMC 7:3 |
Aib-Enkephalin | RA-CM | DIC/OP | 72.1 (DMF 53) | Tolomelli/Cabrid (80) | |
| 21 | DEC/Cyr 7:3 |
PS-Wang | 72.0c (DMF 85.8) | ||||
| 22 | DEC/Sul 7:3 |
RA-CM | 62.0 (RA-CM), 72.8 (PS-Wang) | ||||
| PS Wang | |||||||
| 23 | Anisole/DMC 7:3 |
Aib-ACP | RA-CM | 31.0 (DMF 37.8) | |||
| 24 | DEC/Sul 7:3 |
10.0 (DMF 37.8) | |||||
| 25 | Anisole/DMC 7:3 |
linear Octreotide | PS-Trt-Cl | 64.6 (DMF 88.0) | |||
| 82.3 (with Cys7 coupl at 40 °C) | |||||||
| 26e | 2-MeTHF, 2-MeTHF/MeOH 1:1 |
Leu-Enkephalin-NH2 | RA-AM-PS | DIC/HOBt | 99 (DMF 99) | Schütznerová (110a) | |
| 27e | 2-MeTHF, 2-MeTHF/MeOH 1:1 |
Triptorelin | RA-PS | DIC/HOBt | 72 (DMF 96) | Schütznerová (110b) | 2020 |
| 28 | NBP | AKDGYI-NH2 | RA-AM-PS | DIC/OP | >99 (DMF 92) at 45 °C | Albericiod (69b) | |
| 29 | DMSO/EtOAc 4:6 |
Aib-Enkephalin | RA-AM-PS | DIC/OP | 44 (DMF 69) | Bachem/NovoNordiskd (46) | 2021 |
| 30 | Jung-Redemann | 52 (DMF 57) | |||||
| 31 | Thymosin-α1 | 41 (DMF 52) | |||||
| 32 | Dasiglucagon amide | 49 (DMF 59) | |||||
| 33 | DMSO/2-MeTHF 3:7 |
Aib-Enkephalin | RA-AM-PS | 75 | |||
| 34 | Jung-Redemann | 53 | |||||
| 35 | Thymosin-α1 | 47 | |||||
| 36 | Dasiglucagon amide | 50 | |||||
| 37 | Bivalirudin | 2-CTC | 70.7 (DMF 73.8) | ||||
| 38 | DMSO/DOL 3:7 |
Aib-Enkephalin | RA-AM-PS | 59 | |||
| 39 | Jung-Redemann | 52 | |||||
| 40 | Thymosin-α1 | 42 | |||||
| 41 | Dasiglucagon amide | 53 | |||||
| 42 | Bivalirudin | 2-CTC | 72.7 | ||||
| 43 | NBP/DOL 4:6 |
Bivalirudin | 2-CTC | 62.7 | |||
| 44 | NOP/DMC 8:2 |
Aib-Enkephalin, linear Octreotide | PS Wang | DIC/OP | Aib-Enk: 97.5 (Wang); Octreotide: >99 (Trt-Cl) (DMF >99) | Tolomelli/Cabrid (29a) | |
| PS-Trt-Cl | |||||||
| 45 | NOP | Aib-Enkephalin, linear Octreotide | PS Wang | Aib-Enk: 97.7 (Wang), 97.4 (Trt-Cl); Octreotide: >99 (Trt-Cl) | |||
| PS-Trt-Cl | |||||||
| 46 | NBP | Aib-Enkephalin | PS Wang | Aib-Enk: 91.1 (Wang), 91.6 (Trt-Cl); Octreotide: >99 (Trt-Cl) | |||
| PS-Trt-Cl | |||||||
| 47 | NCP | Aib-Enkephalin | PS Wang | Aib-Enk: 88.5 (Wang, DMF 85.8); Aib-Enk: 89.9 (Trt-Cl, DMF 81.1) | |||
| PS-Trt-Cl | |||||||
| 48 | ACN | H-YIGFLYIGFL-NH2 | RA-CM | DIEA/T3P/OP | Not reported | Albericio (136) | |
| 49 | PolarClean | Leu-Enkephalin-NH2, AKDGYI-NH2, KTTKS-NH2 | RA-AM-PS | DIC/OP | Not reported, comparable to DMF | Albericiod (64) | |
| 50 | H2O/PolarClean 4:1 |
Leu-Enkephalin-NH2 | RMG-TG-S | TCFH/collidine | 86 | PolyPeptide (65) | |
| 51f | Anisole/EtOH/DMSO | Leu-Enkephalin-NH2 | RA-PS | DIC/OP | 99 (DMF 99) | Schütznerová (110c) | |
| 52f | Anisole/EtOH/DMSO | Leu-Enkephalin-OH | PS-Wang | DIC/HOBt | 93 (DMF 95) | ||
| 53f | Anisole/EtOH/DMSO | Aib-Enkephalin-NH2 | RA-PS | DIC/HOBt | 28 (DMF 26) | ||
As pioneers in this field, the Albericio group first proposed the use of acetonitrile (ACN) and tetrahydrofuran (THF) for SPPS,57 both as promising alternatives to DMF/NMP in terms of minimizing racemization and increasing the coupling yield in solution and solid-phases, with THF giving the best results (Table 3, entries 1–4). Besides their compatibility with fully PEG-based resins, ACN and THF were listed as solvents with moderate to high concerns in the GSK Solvent Sustainability guide.35b Other alternatives considered for the coupling steps were 2-methyl tetrahydrofuran (2-MeTHF) (solvent derived from biomasses)58 and cyclopentyl methyl ether (CPME), after having investigated their solubility and swelling capacities.59 From this study, 2-MeTHF was the most successful, thus its application was extended to every SPPS step and full synthesis. This was performed by matching coupling steps in 2-MeTHF with deprotections and/or wash steps in isopropanol (IPA) or ethyl acetate (EtOAc), or by using 2-MeTHF for all steps (Table 3, entries 5 and 6, respectively).60 Additionally, ChemMatrix resins gave better results than PS-based supports (Table 3, entry 5 vs. entry 7). However, 2-MeTHF is considered problematic in the green score guides.37b In addition to 2-MeTHF, EtOAc, CPME and IPA, in 2017 Albericio and co-workers also explored the use of N-formylmorpholine (NFM), isosorbide dimethyl ether, dimethyl carbonate (DMC), and γ-valerolactone (GVL) in an extensive study on swelling and piperidine-mediated Fmoc-removal.61 From the screening, GVL and NFM emerged as the best-performing green alternatives on CM/PS-resins and on CM-resin, respectively. Therefore, they were investigated in the first report of full GSPPS on PS resins (Table 3, entries 9 and 10), despite giving modest results. However, whilst NFM might have some cost and stability issues, GVL is a green, aprotic solvent derived from carbohydrate-based biomasses, it is renewable, and has low toxicity and good biodegradability. GVL was investigated by optimizing a full MW-assisted green protocol, expanding the study to the SPPS of four demanding peptides (Table 3, entries 16 and 17).54 GVL demonstrated compatibility with the automation process. Hence, it expanded the greenness of the total protocol considering its low energy demand and the reduced amount of solvent waste required by an MW technique. In subsequent published studies, Albericio explored the use of this solvent to anchor the first amino acid on a Wang resin, with satisfactory loading values,55 and in the Fmoc removal step. Unfortunately, GVL revealed a poor stability in the presence of amines.62
Very recently aiming to find other suitable green solvents to apply to SPPS, the Albericio and Rasmussen research groups simultaneously proposed the use of ethyl-5-(dimethylamino)-2-methyl-5-oxopentanoate (Rhodiasolv® PolarClean, commercialized by Solvay)63 alone64 or in a mixture with water, respectively.65 Being water-miscible, biodegradable, non-toxic, non-flammable, and non-volatile, PolarClean could satisfy the needs of GSPPS. When used as such, it demonstrated a good capacity to solubilize amino acids and coupling reagents, and to swell both PS and ChemMatrix resins, despite its high viscosity. On these bases, it was then employed in the full SPPS of model short peptides, with performances that were comparable to that of DMF (Table 3, entry 49).64
After these seminal studies, many other research groups entered into this field of research. An important contribution in the search for a unique, good, alternative solvent came from North and co-workers in 2017, who reported a systematic study on the swelling ability of 25 green solvents combined with 9 commercial resins. The experiments were followed by a modelling study to predict which green candidate would swell a particular support.66 Among the others, propylene carbonate (PC), which is cheaper than DMF, non-toxic, and used in cosmetic and topically applied formulations, was tested in a full SPPS process. The final crude purity of the peptide obtained with PC was comparable to that of DMF (Table 3, entry 8).67
Another systematic study was conducted by Lopez and co-workers from Novartis, in which 37 non-hazardous solvents were evaluated step-by-step for their resin-swelling capacities, SPPS reagent solubility, coupling efficiency, and Fmoc removal ability.68 At the end of the selection process, only a few solvents were compatible with all of the steps and were further investigated using full SPPS protocols, namely dimethyl sulfoxide (DMSO), 1,3-dimethyl-3,4,5,6-tetrahydro-2-pyrimidinone (DMPU), 1,3-dimethyl-2-imidazolidinone (DMI), N-ethylpyrrolidone (NEP) and N-butylpyrrolidone (NBP) (Table 3, entries 11–15). Among them, NBP had the best results in terms of peptide purity, proving to be a strong candidate to replace DMF.68 NBP is a high-boiling, polar, aprotic solvent. More importantly, it is non-mutagenic, non-reprotoxic, and biodegradable, in contrast to its structurally related analogue, NMP. Accordingly, it was further exploited because it limited many common SPPS side reactions, such as Arg intramolecular δ-lactamization, racemization, and aspartimide formation (see Fig. 7).69 In these cases, the lower polarity of NBP strongly outperformed DMF in racemization-prone Cys-, His-, and Ser-containing sequences, and in suppressing aspartimide formation (Table 3, entry 28).69b After NBP was validated in full SPPS processes, the class of N-alkyl pyrrolidones was further explored by Tolomelli and Cabri29a,b after having tested their promising results in green cross-coupling reactions.70 While retaining the main characteristics of the parent compound, NMP, they displayed a completely different metabolic profile in an in vitro metabolic investigation, being nontoxic and not reprotoxic. The high boiling and flash points of the above solvents represent industrially relevant features for recovery by distillation.
After first screening for swelling, solubility tests, Fmoc deprotection, and coupling efficiency, N-octylpyrrolidone (NOP) emerged as the best candidate that efficiently accomplished all of these steps. In addition, it is commonly employed as a surfactant and in cosmetic formulations, guaranteeing a safety profile and a competitive cost (approximately $1–2 per kg). A full SPPS using NOP, NBP, and N-cyclohexylpyrrolidone (NCP) showed that they outperform DMF (Table 3, entries 45 and 46), with NOP displaying the best performance (Table 3, entry 45).29a
Although these issues are far from being explored versus other key steps of SPPS, there has also been little work in recent years investigating the greenness potential of cleavage and precipitation steps. However, it is important to highlight that the attempts to upgrade the environmental profile of the whole SPPS process and to obtain greener downstream purification processes are required to guarantee prioritizing the quality and the purity of the crude peptides. Concerning cleavage steps, there is still no effective way to replace trifluoroacetic acid (TFA) in the SPPS Fmoc/tBu strategy.71 However, cleavage of side chain-protected peptides from 2-chlorotrityl chloride (CTC) resins requires a low amount of TFA (1–3%) mixed with an organic solvent. Despite DCM being routinely used to do this, recent studies reported the possibility of replacing it with green solvents. Anisole and 1,3-dimethoxybenzene emerged as excellent candidates, also allowing for better manipulation due to their higher boiling points.72 Concerning using scavengers in peptide cleavage, only a single report thus far has described the use of greener options in a cleavage cocktail, obtained by the introduction of 1,4-benzenedimethanethiol (1,4-BDMT) as a scavenger for global deprotection of exenatide from the resin. This novel thiol, besides being non-malodorous and UV detectable (contrary to standard aliphatic thiol reagents), was effective in reducing the content of critical cleavage-induced peptide impurities.73
Other studies have focused on solvents that could replace standard ethers for post-cleavage peptide precipitation. The commonly employed diethyl, diisopropyl, and tert-butyl methyl ethers (DEE, DIE, TBME, respectively) are indeed considered hazardous due to their low flash and boiling points and the tendency to form peroxides. In addition, TBME was found to induce undesired tert-butylation of peptides during precipitation74 and is considered carcinogenic for its toxic metabolites.35b,37b The replacement of these ethers is therefore recommended, and, consequently, other anti-solvents were proposed in the last few years. In this context, CPME75 and 2-MeTHF76 may be alternatives. CPME has favourable EHS features, specifically a high boiling point and higher flash point, together with a low propensity to form peroxides. Both solvents naturally display a good stability under acidic conditions77 and were tested in the precipitation of 5mer to 28mer peptides. Recovery and purity of the resulting crude products were in line with those obtained with standard DEE and methyl tertbutyl ether (MTBE). Interestingly, after these and previously described results, 2-MeTHF was shown to be suitable as a single solvent in all the steps of SPPS, with the advantage of minimizing morphological damage to the resin.59 Lastly, the Polypeptide group proposed 4-methyltetrahydropyran (MTHP) as anti-solvent for crude peptide precipitation, yielding good performance if used in a 1:4 mixture with n-heptane.78
In 2019, the Rasmussen,29c,78 North,79 and Tolomelli and Cabri80 groups simultaneously reported the use of green binary mixtures for SPPS, opening a revolutionary possibility for new solvents to successfully accomplish all steps of SPPS. Fine-tuning the chemical/physical features of solvents by using a binary mixture is a valuable instrument for obtaining the appropriate polarity and/or viscosity for successful resin swelling and reagent solubilization,81 as highlighted in the guidelines for a perfect SPPS solvent.46 The influence of different solvent mixtures on swelling was first described by the North group, who developed a computational model outlining, for the first time, that resin swelling did not correlate linearly with solvent composition. More importantly, they showed that a binary mixture could be better at achieving swelling than neat solvents; mimicking the properties of traditional, polar, aprotic solvents; and replacing them. In particular, the authors identified that mixtures of EtOAc/PC and EtOAc/2,2,5,5-tetramethyloxolane (TMO) could swell PS resins more efficiently than each solvent separately.79 The paramount importance of resin swelling is highlighted by the number of publications focused on investigating this step in green solvents,82 despite traditional solvents (DMF, NMP and DCM) maintaining the highest swelling values overall.83
The concept of binary mixtures was then expanded to other SPPS steps. The Pawlas and Rasmussen group proposed a two-dimensional, green approach, where the first dimension involves a full SPPS and the second describes examples of on-resin derivatization in neat, green solvents or in a 1:1 EtOAc/MeCN mixture (i.e., selective removal of acid-labile protecting groups and on-resin cyclization).78 Specifically, the full synthesis of model APIs was performed in 1:1 EtOAc/NBP (Table 3, entry 19)78 or 9:1 EtOAc/DMSO (but minimizing the amount of DMSO in the wash steps to 49:1, Table 3, entry 18).29c Of note, the 1:1 EtOAc/MeCN mixture was also successfully tested as a DCM-replacing solvent for cleavage from CTC resins.78
As mentioned previously, Rasmussen's group also reported the use of PolarClean as an efficient water co-solvent for the first reported Fmoc/tBu SPPS in aqueous conditions.65 The solubility of standard Fmoc-AAs was achieved by the addition of 20% PolarClean.84 In this way the full GSPPS of a model peptide was obtained with the same purity as with DMF (Table 3, entry 50), achieving a new aqueous Fmoc/tBu peptide synthesis method with minimized amounts of inexpensive and non-hazardous solvents.
The only recent innovation in this field was published in 2020. It described a so-called aqueous solid phase peptide synthesis (ASPPS), made possible by the introduction of the water-compatible 2,7-disulfo-9-fluorenylmethoxycarbonyl (Smoc) protecting group that combines a fluorenyl moiety with negatively charged sulfonic substituents. The Nα-Smoc-protected amino acids are water soluble and compatible with the lack of side chain protecting groups. Moreover, they are fluorescent, which enables real-time detection of both coupling and deprotection steps. The applicability of ASPPS was demonstrated through the synthesis of 22 biologically active peptides using water-compatible activating additives for the coupling steps.85
In 2019, Tolomelli and Cabri's group also paved the way for green solvent mixtures, when they analysed new alternatives by mixing Cyrene™ (Cyr), sulfolane (Sul), or anisole (An) with DMC or diethyl carbonate (DEC) in different ratios. After an extensive study of the selected mixtures on the ability to swell different kinds of resins, three binary mixtures (7:3 DEC/Cyr, 7:3 DEC/Sul, and 3:7 DMC/An) were further explored in a full SPPS protocol with promising results (Table 3, entries 20–25).80 Moreover, with their newly proposed N-octylpyrrolidone, they enlarged the available pattern of green solvent candidates. To overcome the issue of its viscosity (6.6 mPa s at 25 °C) to an acceptable value (<4 mPa s) for automated processes, low-viscosity DMC was added to obtain a green mixture of 8:2 NOP/DMC, which was successfully tested both in manual and automatic SPPSs of linear octreotide, obtaining excellent crude purities (Table 3, entry 44).29a
Recently, alternative solvent mixtures with a low viscosity for automated synthesis have been also explored by NovoNordisk and Bachem.46,86 Identification of the solvents’ key physical parameters allowed for the prediction of a set of potential green binary solvent mixtures that could surpass DMF's performance in the large-scale SPPS of industrially relevant peptides. The authors focused on the composition of the binary mixtures, demonstrating how single steps of SPPS could be easily customized by varying the ratio of the selected solvents. This hypothesis was first validated by assessing reagent solubility, resin swelling, and by controlling coupling and deprotection reactions in a series of sustainable neat solvents or binary combinations.46 Moreover, mixtures were investigated for minimization of frequently occurring SPPS side reactions.86 Accordingly, they confirmed how solvent viscosity and polarity are the physiochemical features that mainly affect SPPS steps and therefore need to be optimized when planning a GSPPS. Mixtures of DMSO with dioxolane (DOL), 2-MeTHF, or EtOAc resulted in efficient Fmoc-removal steps (promoted by polar solvents). Couplings could instead be addressed using NFM/DOL or NBP/DOL (promoted by non-polar solvents). After these preliminary results, the authors moved onto a full GSPPS with binary mixtures (Table 3, entries 29–43). Considering all aspects, 3:7 DMSO/DOL (Table 3, entries 38–42) and 3:7 DMSO/2-MeTHF (Table 3, entries 33–37) provided the best results when compared to DMF, while 4:6 NBP/DOL gave slightly worse results (Table 3, entry 43).46
3.1.1.2. Solvent recycling and reduction. The benchmark concept of solvent recycling in SPPS was only introduced in 2019.29c It is still not as well-considered as other aspects of SPSS, mentioned only as an encouraging possibility in most papers on green alternatives. Given the urgent demand for peptides in the pharmaceutical sector and the extensive amount of solvents being discarded as waste, applying the circular economy (CE) concept to SPPS is dutiful.87 CE sets as its priorities eliminating waste, recycling products, and saving resources and the environment.88
A short-term objective is lessening the amount of solvent, namely DMF, in SPPS processes. Minimization of solvent consumption could be achieved by introducing real-time monitoring techniques. Among the developed process analytical tools (PATs), continuous refractive index (RI) measurement of coupling, deprotection and wash solutions was recently validated as a successful technique for online monitoring of all SPPS steps. Besides providing information on the kinetics of the process, RI monitoring avoids sampling, interruption of the process, and extra-analytical procedures. In this way, the consumption of reagents and solvents, together with time and costs, could be strongly minimized, favouring automation of the process, and decreasing the waste impact (Fig. 8).89
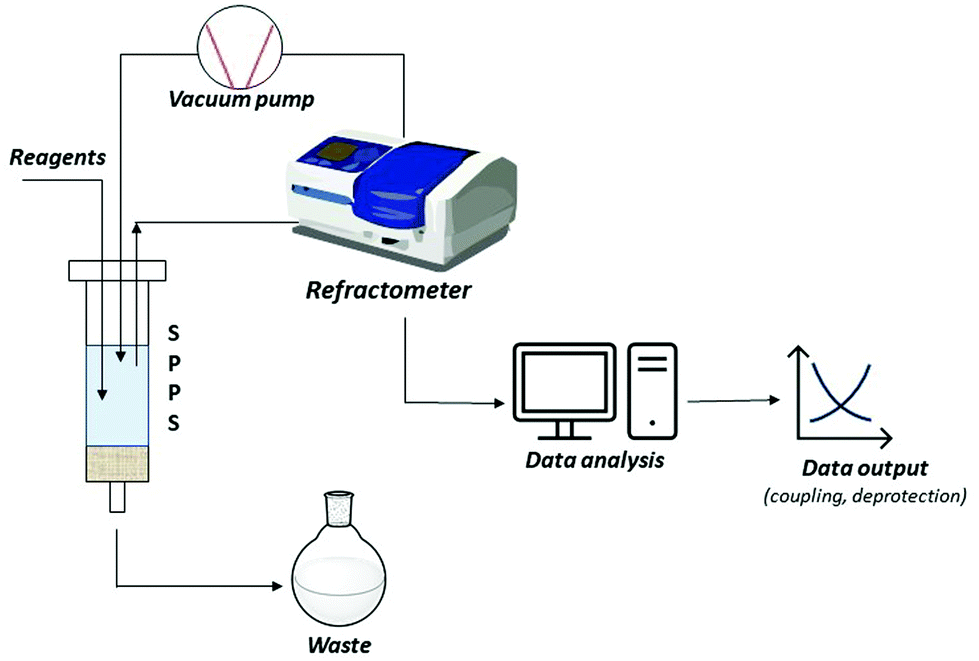 | ||
| Fig. 8 Schematic representation of SPPS real-time monitoring by refractive index. | ||
Similarly, in 2019 Polypeptide developed an automated synthesis reactor that allows real-time monitoring of the SPPS process by means of solvent percolation.90 In this context, the “tea bag” approach could also respond to CE features by lessening the amount of solvent required for SPPS. In fact, the main idea of this strategy is to perform parallel multi-syntheses of long peptides, each separately in a tea-bag (propylene bag microreactors containing the growing peptide–resin), exploiting the same solution bottles for Fmoc removal and wash steps where the bags are simultaneously immersed. In this way, washes and base solutions are re-utilized in further steps along the SPPS, allowing 25–30% less DMF volume to be used without affecting the quality of the target peptides.91 A more ambitious objective is to establish green solvents or mixtures in full SPPS protocols, which must be accompanied by a related recycling strategy in order to reduce their costs. For this long-term objective, physical features of all reagents and solvents involved in the process need to be carefully evaluated.
The ReGreen protocol proposed by Pawlas and Rasmussen29c aimed to recover reagents and solvents from the waste stream to be re-used in subsequent syntheses (Fig. 9). In particular, they focused on solvent recycling (9:1 EtOAc/DMSO) and the coupling additive (OxymaPure®), and evaluating DIC and base for Fmoc-removal (4-methylpiperidine; 4-MP), the last too reactive to be recovered as such. Then, an easy protocol was developed to distill-off the most volatile component from the waste stream, playing on the different boiling points of the various species and, hence, different temperatures/pressures of the distillation process. The recycled EtOAc, DMSO and OxymaPure® were then re-employed in a full GSPPS without affecting the results when compared to virgin materials. Importantly, the authors also evaluated the solvents from the life cycle standpoint, considering the impact of waste disposal.
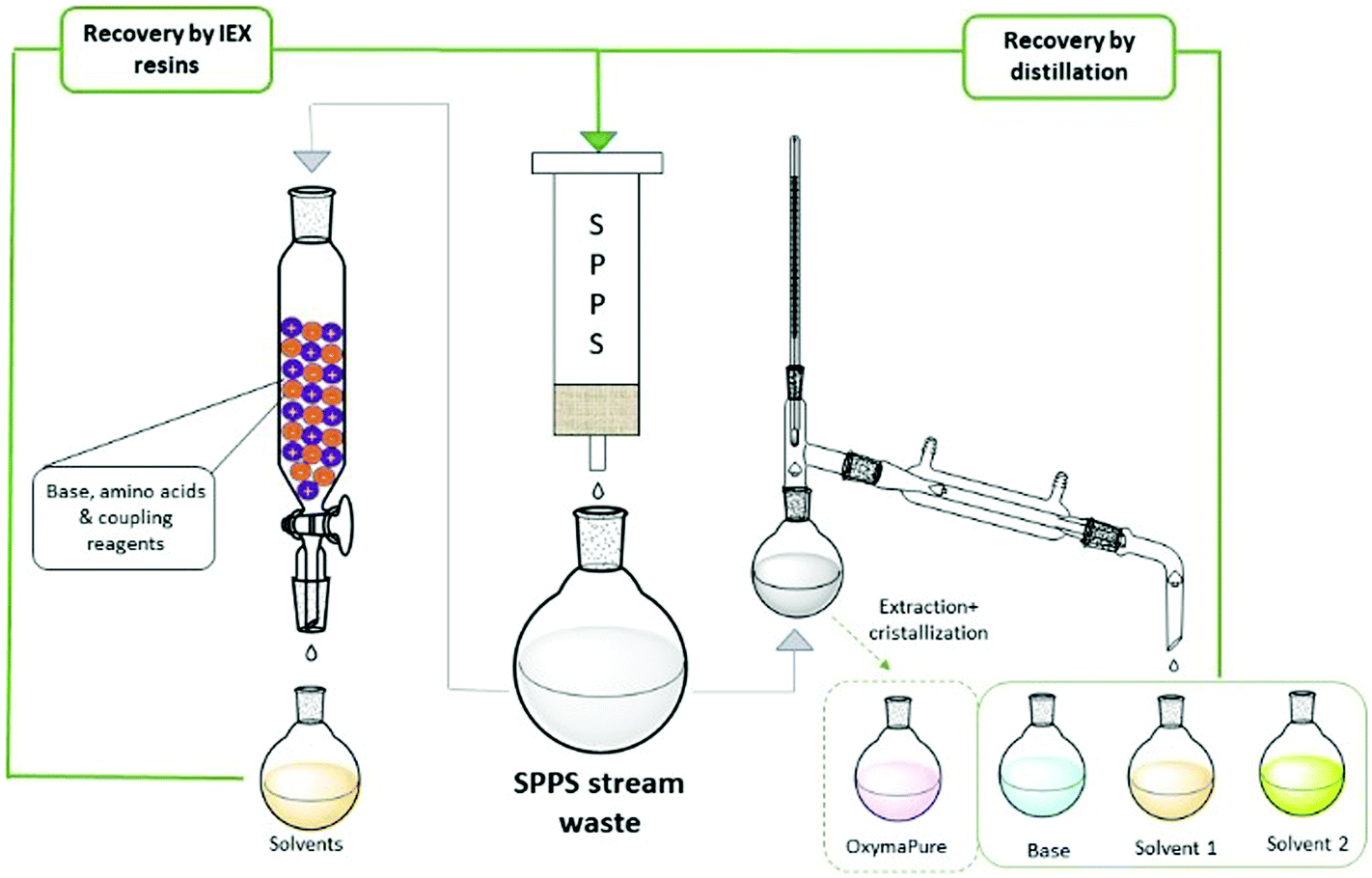 | ||
| Fig. 9 Schematic representation of solvent and reagent recovery from SPPS waste streams. Specifically, solvents coming from SPPS waste (grey arrows) can be recovered by filtration through ion exchange resin (left side), or can be distilled to separate OxymaPure from liquid components (base/solvents; right side). In both cases, recycled solvents can be reused in a new SPPS process (green arrows). | ||
From a deep analysis, it emerged, unsurprisingly, how much cEF and process costs could be greatly decreased after recovery. A different recycling method was recently disclosed by the same authors, who combined the concepts of CE and water-based SPPS. After the synthesis of Leu-Enkephalin amide in 4:1 H2O/PolarClean,65 (Table 3, entry 50), they developed a protocol for recycling the waste by filtration through a mixed ion-exchange resin (cation exchange plus anion exchange resins). They were able to capture both acid and basic species present in the waste streams (Fig. 9). The H2O/PolarClean mixture obtained was re-utilized in a full Leu-Enkephalin amide SPPS, without affecting the quality of the crude peptide by the recovered solvents.
Inspired by these concepts, Tolomelli and Cabri's group also investigated a distillation protocol for recovering NOP or a NOP/DMC mixture after the full SPPS of linear octreotide (Table 3, entries 44 and 45). For the first time, a high recovery of all liquid components (85–95% solvent recovery yields) was also considered and successfully obtained, including bases for Fmoc removal (piperidine29a or DEAPA29b with 92–95% recovery yields) (Fig. 9). Of note, coupling and deprotection waste streams were distilled separately in order to prevent base consumption by Fmoc-amino acids, which were used in excess in the coupling steps. In all cases the PMI of the process after recycling was decreased by more than 60%.
3.1.1.3. Solvents: general remarks. Several alternative solvents for SPPS have been proposed in recent years, and others are expected to be reported soon. Considering all aspects of SPPS, a single solvent is unlikely to replace the touchstone DMF. For comparison, the most important characteristics to consider in comparing the most successful green solvents for SPPS are summarized in Table 4. The use of mixtures will drive GSPPS, matching of all the properties of DMF that are otherwise impossible to achieve with neat solvents. As recently highlighted by Albericio,46 there is probably not one single mixture that will work for every peptide's synthesis.
| “–” stands for data not reported. |
|---|
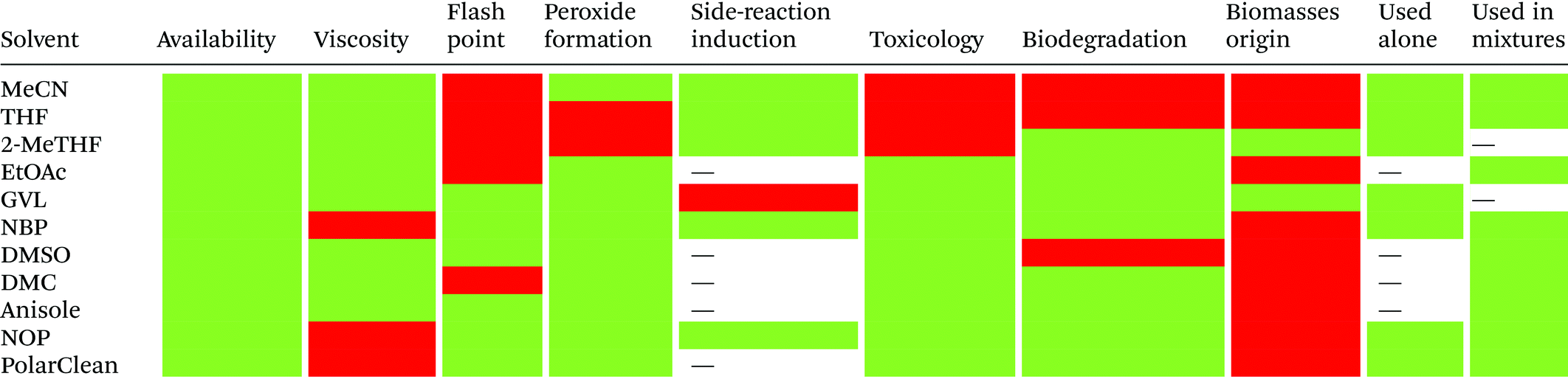
|
However, there are several hurdles to overcome to use green solvent mixtures for industrial production. These hurdles are mainly related to practical and economic considerations. In fact, by using solvent mixtures, plant supply chains will be more complex, as will the management of waste disposal. Furthermore, DMF, used in a volume exceeding 400000 tons yearly, is a very cheap solvent, but using alternative mixtures will increase production costs. In fact, the price of most of the proposed green alternatives (mixtures or not) is higher than DMF. Only if the green alternatives are universally accepted and applied industrially will the increased market demand boost volume and decrease market prices. Furthermore, regulatory authorities’ reluctance in changing synthesis protocols for already-established processes needs to be addressed.
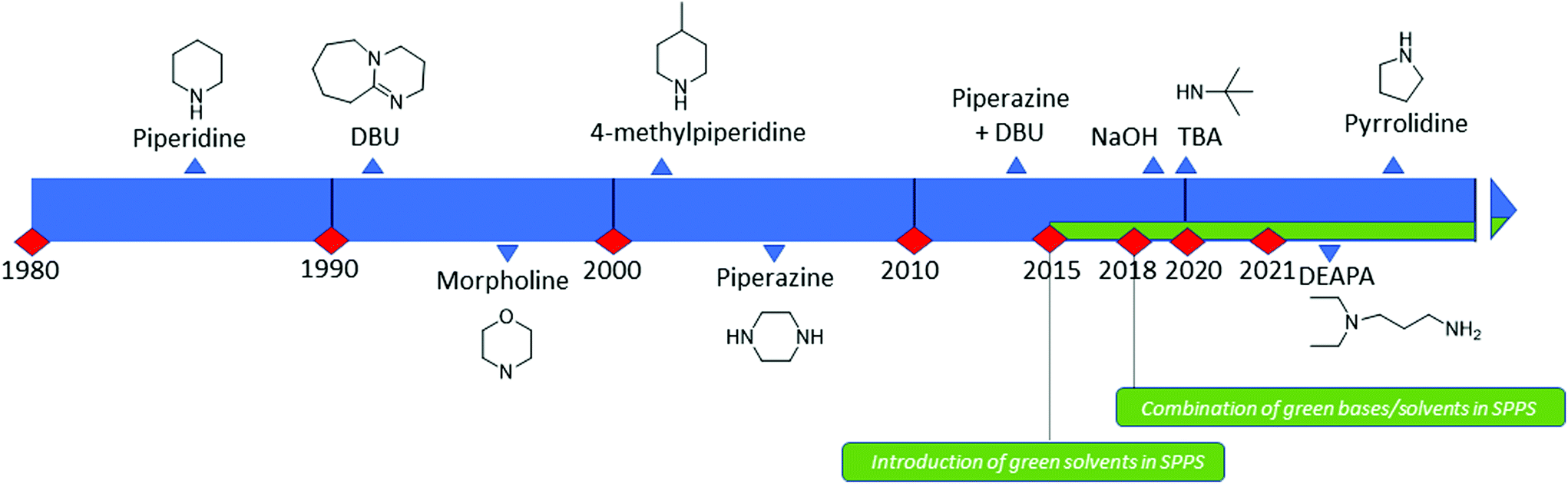 | ||
| Fig. 10 Timeline of bases introduced for Fmoc removal in SPPS. | ||
Fmoc groups can be cleaved under mild basic conditions via an E1cB β-elimination mechanism. The reaction is more efficient with bases with a high pKa and low steric hindrance in polar solvents like DMF. Being that piperidine is very efficient in both steps, the standard procedure for Fmoc removal is based on exposure of the protected peptide during SPPS or LPPS to a 20% piperidine solution in DMF, at room temperature for a few minutes.95 According to the mechanism, dibenzofulvene (DBF) is formed in the first step (Fig. 11). It is a highly reactive electrophile that could be attacked by the newly formed free alpha-amino group on the growing, anchored peptide or undergo polymerization.96
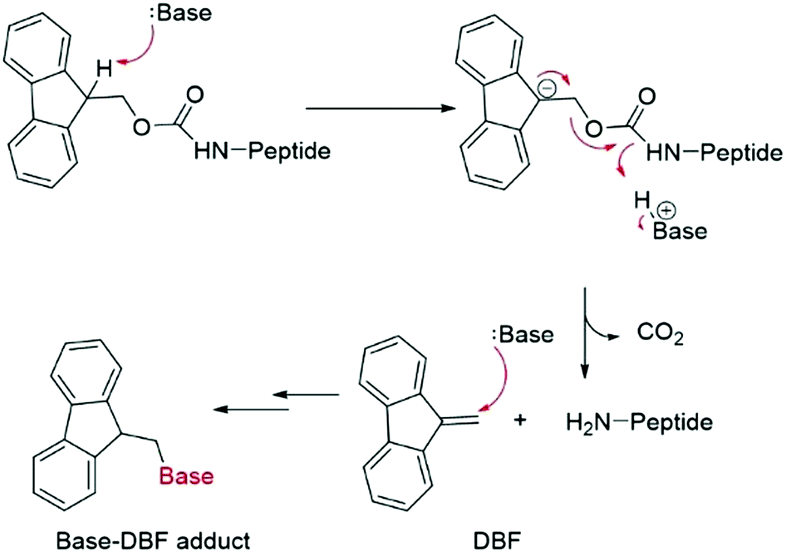 | ||
| Fig. 11 Mechanism of base-DBF adduct formation. | ||
Unfortunately, the use of piperidine is highly regulated, being a precursor in the illicit preparation of phencyclidine (PCP, also referred to as “angel dust”).97 This issue underlines the need for new bases amenable for use in the manufacturing of peptides without any usage restrictions,98 considering that the base impacts the PMI of the entire process. Besides Fmoc removal efficiency, other criteria should be used for base selection, namely EHS parameters, solubility, boiling point, recovery, and handling. This was clearly highlighted in GSK's acid and base selection guide.99
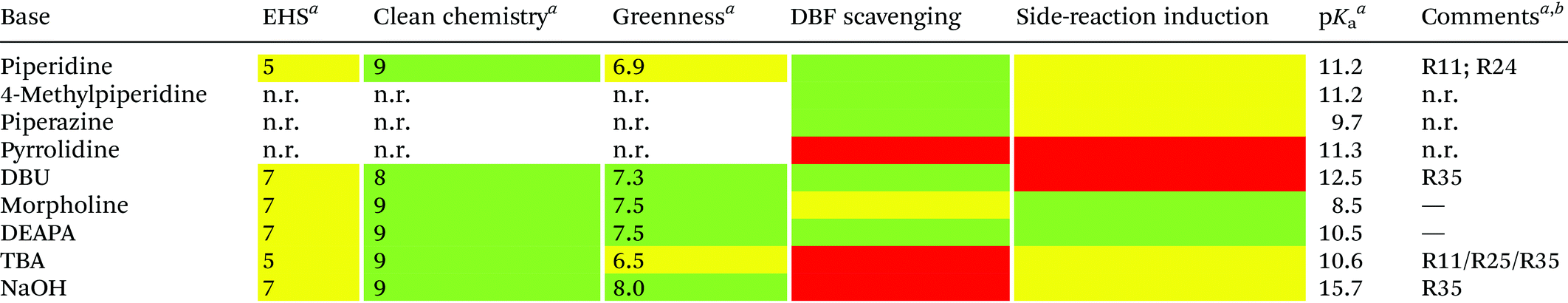
Analogous to piperidine, several secondary amines, namely piperazine,100 morpholine,95a,101 methyl-piperidines, 4-methylpiperazine,102 and pyrrolidine103 have been described to efficiently remove the Fmoc moiety, taking advantage of both basicity for the first step and nucleophilicity for DBF trapping (see Fig. 11). However, efficient deprotection has also been described using 1,8-diazabicyclo[5,4,0]undec-7-ene (DBU),104tert-butyl amine (TBA),105 and 3-diethylaminopropylamine (DEAPA).29b These bases do not efficiently react with DBF, therefore the addition of good nucleophiles like piperidine,106 piperazine,107 or thiols108 is generally required to scavenge the reactive DBF.
Piperidine was largely used in green solvent protocols for SPPS (see footnote d in Table 3) and peptide-anchored LPPS (see the corresponding paragraph in 3.2). Fine-tuning of alternative bases to piperidine has been carried out over the years, taking into consideration the potential impact on formation of impurities as described in Fig. 7. In Table 5, the introduction of bases is reported according to publication sequence, reaction solvents, the length of the target peptide, and its purity.
| Entry | Base (pKa) | Amount | Solvent | Full SPPS peptide | HPLC crude purity (%) | Groupa (ref.) | Year |
|---|---|---|---|---|---|---|---|
|
a Corresponding author or company is listed.
b The mixture was replaced by 2-MeTHF only for coupling steps.
c The mixture was replaced by An/DMSO = 4:1 for coupling steps.
|
|||||||
| 1 | Piperazine (9.8) | 10% w/v | DMF/EtOH 9:1 (microwave) |
H-FISEAIIHVLHSR-NH2 | 57.7 (Pip 43.6) | Albericio (109b) | 2016 |
| 2 | H-TLEEFSAKL-NH2 | 74.8 (Pip 83.0) | |||||
| 3 | H-KKWRWWLKALAKK-NH2 | 55.6 (Pip 59.1) | |||||
| 4 | H-VAPIAKYLATALAKWALKQGFAKLKS-NH2 | 21.4 (Pip 29.0) | |||||
| 5 | 4-(Me)piperidine (10.8) | 20% v/v | DMF | H-FISEAIIHVLHSR-NH2 | 47.6 (Pip 43.6) | ||
| 6 | H-TLEEFSAKL-NH2 | 65.1 (Pip 83.0) | |||||
| 7 | H-KKWRWWLKALAKK-NH2 | 50.4 (Pip 59.1) | |||||
| 8 | H-VAPIAKYLATALAKWALKQGFAKLKS-NH2 | 20.6 (Pip 29.0) | |||||
| 9 | 4-(Me)piperidine (10.8) | 20% v/v | Polar aprotic solvents | linear Octreotide | 86 (DMF) | Novartis (68) | 2018 |
| 80 (NBP) | |||||||
| 78 (TMU) | |||||||
| 78 (DMI) | |||||||
| 52 (DMSO) | |||||||
| 51 (DMPI) | |||||||
| 10 | Morpholine (8.4) | 50% v/v | 2-MeTHF | — | — | Schütznerová (110a) | 2019 |
| 11 | DBU (13.5) | 0.5% v/v | 2-MeTHF | — | — | ||
| 12 | NaOH (15.74) | 0.2 M | 2-MeTHF/MeOH 1:1 or 3:1b |
Leu-Enkephaline-NH2 | 99 (Pip 99) | ||
| 13 | Triptorelin | 57–72 (Pip/DMF 96) | Schütznerová (110b) | ||||
| 14 | 4-(Me)piperidine (10.8) | 5% v/v | NBP/EtOAc 1:1 |
Ac-Nle-DHFRWK-NH2 | 90 | Polypeptide (78) | |
| 15 | 4-(Me)piperidine (10.8) | 1–5% v/v | EtOAc/DMSO 9:1 |
Aib-ACP | 76 | Polypeptide (29c) | |
| 16 | TBA (10.7) | 30% v/v | DMF | Degarelix | 87.5 (Pip 83.99) | Fresenius Kabi (105b) | 2020 |
| 17 | 4-(Me)piperidine (10.8) | 2.5% v/v | DMF | KKWQWK-Ahx-RLLRRLLR | 99 | Garcia (109c) | 2020 |
| 18 | Pyrrolidine (11.3) | 20% v/v | EtOAc/DMSO 9:1 |
Aib-Enkephalin | 96 (Pip/DMF 69) | Bachem/NovoNordisk (103) | 2021 |
| 19 | Jung-Redemann | 59 (Pip/DMF 57) | |||||
| 20 | Thymosin-α1 | 56 (Pip/DMF 52) | |||||
| 21 | Dasiglucagon amide | 58 (Pip/DMF 59) | |||||
| 22 | Bivalirudin | 67 (Pip/DMF 78) | |||||
| 23 | NBP/DOL 4:6 |
Aib-Enkephalin | 97 | ||||
| 24 | Jung-Redemann | 67 | |||||
| 25 | Thymosin-α1 | 54 | |||||
| 26 | Dasiglucagon amide | 55 | |||||
| 27 | Bivalirudin | 69 | |||||
| 28 | 4-(Me)piperidine (10.8) | 5%v/v | H2O/PolarClean 4:1 |
Leu-Enkephalin amide | 86 | Polypeptide (65) | |
| 29 | TBA (10.7) | 20% v/v | NOP | Aib-Enkephalin | 97.9 (Pip 97.8) | Tolomelli/Cabri (29b) | |
| 30 | DEAPA (10.5) | 5% v/v | NOP NOP/DMC 8:2 |
Aib-Enkephalin, linear Octreotide | Aib-Enk: 97.1–97.5 (Pip 97.8) linear Octreotide: >99 (Pip >99) | ||
| 31 | Morpholine (8.4) | 50% v/v | Anisolec (microwave) | Leu-Enkephalin (OH or NH2) Aib-enkephalin-NH2 | Leu-Enk-NH2: 94 (Pip /DMF 99) | Schütznerová (110c) | |
| 32 | DBU (13.5) | 5% v/v | Anisolec | Leu-Enk-NH2: 98–99 (Pip/DMF 99) | |||
| Leu-Enk-OH: 93 (Pip/DMF 95) | |||||||
| Aib-Enk-NH2: 28 (Pip/DMF 26) | |||||||
| 33 | NaOH (15.7) | 0.2 M | An/EtOH 1:1c |
Leu-Enk-NH2: 92 (Pip/DMF 99) | |||
| 34 | 4-(Me)piperidine (10.8) | 20% v/v | ACN | H-YIGFLYIGFL-NH2 | Not reported | Albericio (136) | |
4-Methylpiperidine (4-MP) emerged as one of the most popular alternative bases to DMF (Table 5, entries 5–8 and 17)109 and it was the first to be used in a green solvent setup.68 In fact, in 2018, Lopez and coworkers combined the use of 4-MP with several green solvents (Table 5, entry 9), with NBP giving the best results.68 Rasmussen et al. successfully extended the use of 4-MP to green solvents mixtures (Table 5, entries 14, 15, 28 and 34).29c,65,78 The Tolomelli and Cabri group described the use of TBA and DEAPA in green solvents like NOP and NOP/DMC (Table 5, entries 29 and 30).29b TBA was also able to avoid the formation of the hydantoin (Hyd) impurity as rearrangement of the dihydroorotic (Hor) moiety during degarelix synthesis (Table 5, entry 16), but it was less efficient as a general base for Fmoc removal.105 Later, Novo Nordisk–Bachem reported the use of pyrrolidine in green solvent mixtures with low viscosity (Table 5, entries 18–27).103
Considering only the organic bases, the protocols generally developed in DMF can be easily transferred to green solvents. However, it is important to consider that side-products, like isomerization, aspartimide and diketopiperazine (Fig. 7), are generally determined by the pKa and the nucleophilicity of the considered base. As an example, pyrrolidine, which is less sterically demanding than piperidine, generates a higher amount of aspartimide and diketopiperazine side-products.103 In addition, less-polar solvents or solvent mixtures were efficient in reducing side-reactions. For example, the aspartimide by-product generated by piperidine or DEAPA is consistently lower in NBP and NOP than in DMF, with DEAPA giving the best results.29b
For the development of a potential industrial process, the same solvent or solvent combinations should be used for the entire synthesis to simplify their recovery. In this context, the only exception reported in Table 5 is the use of NaOH (entries 12, 13 and 33), which was introduced in protocols requiring different co-solvents according to the synthesis step (coupling or deprotection). Furthermore, NaOH is not compatible with SPPS, in which the peptide is linked with an ester. The use of alcohols to favour NaOH solubility affects resin swelling and performance.110
3.1.2.1. Bases: general remarks. To be a credible alternative to piperidine for Fmoc removal, a base should fulfil several features. In addition to possessing similar physicochemical properties (i.e., pKa and nucleophilicity), it should not be a controlled substance, it should have a robust supply chain, it should be low cost,103 and it should have a good green score.99 However, since the cost is strictly related to production volumes, the final considerations are limited to its green score and chemical performance. In fact, as summarized in Table 6, piperidine is by far the base with the highest production volume and, therefore, the cheapest. Among the potential alternatives, 4-MP has been the most investigated. Unfortunately, there are no toxicological data available for it.111 However, one can assume that piperidine and 4-MP are comparable in terms of toxicity and greenness score with their basicity and nucleophilicity almost identical. Among the other bases, DBU and DEAPA perform well and display a better greenness score than piperidine. However, with these bases, the use of a DBF-trapping reagent is advisable. Finally, concerning NaOH, it is not compatible with several resins and substrates. In addition, NaOH requires the presence of an alcohol to be sufficiently soluble in organic media. These mixtures cannot be applied to the coupling step, resulting in increased complexity of the logistic and waste management.
To date, extensive reviews have summarized the interchanging series of additives/coupling reagents actually present on the market (Fig. 12),113,114 but very few examples can be described as green reagents. The current state of the art for peptide bond formation in SPPS almost exclusively uses the carbodiimide/additive methodology,114c mainly based on DIC. Concerning additives for carbodiimides in couplings, the main options are benzotriazoles or oximes. Despite the family of benzotriazoles being the first coupling reagents introduced for SPPS (HOBt, HOAt, HBTU are prime examples, see Fig. 12), these additives have a risk of explosion and are accordingly regulated under the “Class 1 explosive category”.115 Moreover, they could induce skin sensitization after long-term exposure.116 This classification also includes all derivatives introduced as stand-alone coupling reagents, despite showing enhanced stability, a higher coupling efficiency, and a lower racemization tendency (see Fig. 12 for examples).117
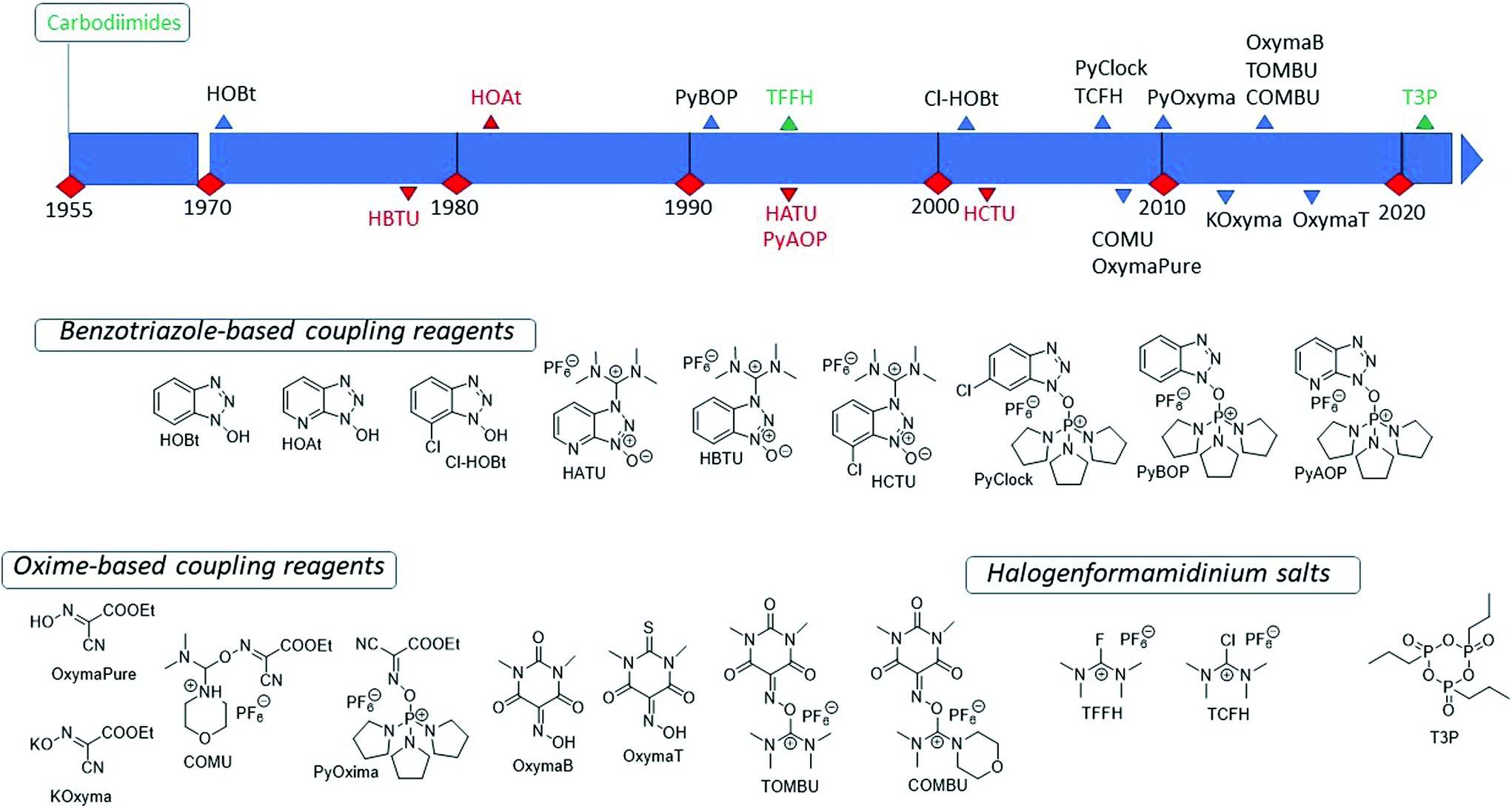 | ||
| Fig. 12 Timeline of introducing coupling reagents and additives in SPPS and relative structures. Reagents written in green or red are classified according to their thermal stability as reported in the coupling reagents selection guide by Sperry et al. (ref. 137). | ||
Concerning oximes, in 2009, Albericio and coworkers were the pioneers in introducing this class of additives,118 focusing on OxymaPure® (ethyl 2-cyano-2-(hydroxyimino)acetate)119 for its excellent properties regarding yields, low racemization levels, and increased safety compared to hydroxybenzotriazole-based coupling additives.114b From then onwards, OxymaPure® became the “first-in-class” of the category, gaining constant traction in manual, automated, and MW-assisted SPPS protocols.120 Many other analogue derivatives have flourished in recent years, with several modifications along the skeleton, namely K-Oxyma,121 COMU,122 PyOxyma,123 Oxyma B,124 TOMBU,125 COMBU,125 and Oxyma T,126 to mention some examples (see Fig. 12). Although these compounds did not possess industrial potency or applicability to green chemistry, COMU is an exception and shows great coupling efficiency accompanied by less epimerization and broad solubility in several solvents. It was recently used in liquid-phase amidations in EtOAc, 2-MeTHF, and DMC,127 and remarkably has applications in water-based SPPS. On the other hand, COMU exhibits a very low stability in DMF,128 and this, of course, limits its application in SPPS. However, in recent studies, COMU showed stability in GVL and ACN, therefore becoming an attractive coupling reagent for GSPPS.129
Notably, among the wide series of coupling reagents available for amide bond formation, DIC/OxymaPure® has undoubtedly become a benchmark in peptide synthesis due to its excellent efficacy, safety, and thermal stability,114c together with low cost and good solubility of the diisopropylurea (DIU) by-product in organic solvents. Accordingly, this reagent combination is now the coupling system of choice, and only a few protocols still involve the use of benzotriazole derivatives (see Table 3 for a comparison of coupling reagents used in GSPPS in recent years). Compatibility of DIC/OxymaPure® in the studied green solvents for SPPS is notably an established property, as demonstrated for the first time by Albericio's group in 2015 in a coupling reaction in 2-MeTHF and ACN.57
Recently, scientists from Eli Lilly pointed out a major issue with using this protocol for couplings, detecting the formation of HCN during amino acid activation.130 After deeper investigations, it was proposed that HCN was coming from the adduct formed between DIC and OxymaPure® that can hydrolyse, leading back to OxymaPure®, or undergo a nucleophilic, N-driven intramolecular cyclization on the activated C–N bond to form the intermediate oxadiazole with consequent release of HCN (Fig. 13). This report raised awareness on the need to warrant a careful evaluation of possible EHS consequences when using DIC/OxymaPure® in SPPS and in amide bond formation in general. Even if the generation of HCN was not reported in the presence of the amino counterpart, which is a reality in peptide synthesis,131 other research groups have focused their efforts on deeper investigation of this side reaction. In 2020, the Pawlas and Rasmussen group reported that HCN formation increases over time and with higher amounts of DIC.132 Interestingly, this side reaction was found to be solvent-dependent, and among several tested green alternatives, the 4:1 EtOAc/NBP mixture performed the best in less HCN formation. Also, the addition of dimethyltrisulfide (DMTS) as an HCN scavenger133 proved feasible for minimizing HCN development without affecting coupling performance.
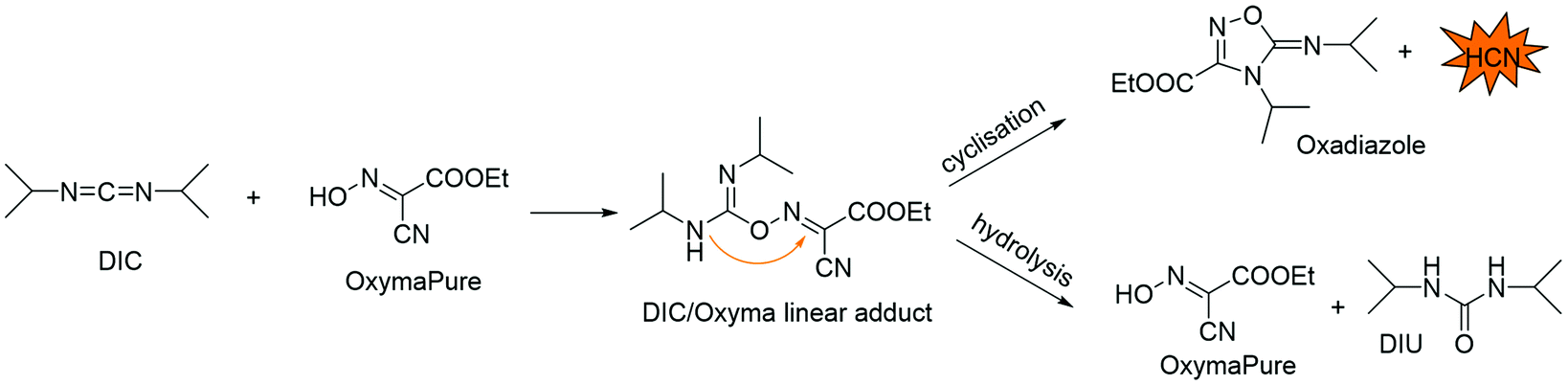 | ||
| Fig. 13 Schematic representation of reactions involving DIC and OxymaPure® to form a linear adduct and consequent possible formation of HCN. Note: DIU = diisopropyl urea. | ||
Albericio's group evaluated the effect of carbodiimides on HCN generation when coupled with OxymaPure® in SPPS protocols. Together with the most commonly used reagents, DCC, DIC, and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC), N,N′-di-sec-butylcarbodiimide (DSBC), N,N′-di-tert-butylcarbodiimide (DTBC) and N-tert-butyl-N′-ethylcarbodiimide (TBEC) were tested in solution phase in the presence of OxymaPure®. The steric hindrance of their N-substituents affected the formation of the linear carbodiimide/OxymaPure® adduct and consequently HCN release. The primary carbodiimide, EDC, and hybrid, TBEC, outperformed the other reagents with less adduct formation.134 In addition, it was discovered very recently by the same research group how the generation of HCN could be reduced or suppressed according to the sequence of adding reagents. The optimal conditions call for pre-activation of the Fmoc-amino acid with DIC, subsequent addition of the mixture onto the resin, followed by insertion of OxymaPure® directly on the resin.131
In 2019 Isidro-Llobet and coworkers introduced tetramethylfluoroformamidinium hexafluorophosphate (TFFH) and propylphosphonic anhydride (T3P®) as the most sustainable coupling reagents (Fig. 12),112 despite the fact that they are very rarely used in SPPS.
However, TFFH and tetramethylchloroformamidinium hexafluorophosphate (TCFH) have been recently tested in the water/PolarClean-based SPPS proposed by Rasmussen, with TCFH performing the best.65 However, T3P® is a cyclic reagent that maintains a discrete stability in organic solvents. It is well known for promoting amidations in liquid phase with high efficiency and no epimerization.135 Moreover, its related by-product after activation is water-soluble. Albericio's group136 investigated the use of T3P® in SPPS (Table 3, entry 48). The results did not look promising because the reaction required large excesses of OxymaPure® and diisopropyl ethyl amine (DIPEA), and the most efficient reactions were performed using T3P® as an alternative to carbodiimides.
3.1.3.1. Coupling reagents: general remarks. In summary, the concept of inherently safer process design should induce selection of reagents less prone to cause safety concerns before the development of large-scale chemical processes. In this context, Pfizer has recently ceased to develop processes containing HOBt coupling agents and derivatives, moving to safer alternatives. Scientists have compiled a peptide coupling reagents selection guide among those commonly employed in pharmaceutical manufacturing.137 It is, however, important to highlight that their classification criteria were exclusively based on thermal stability of the reagents, assessed after extensive differential-scanning-calorimetric (DSC) studies, and hence, the process-safety point of view. Of note, OxymaPure® was not included, while COMU is indicated as “least preferable”. In our opinion, other parameters should be taken into account to provide a more general classification of the “green peptide coupling reagents”, which has never been reported.
Other new strategies for peptide bond formation in short sequences have been recently reported. Organocatalysis138 and photocatalysis139 have been described with a target to overcome the issues of SPPS. However, the related chemistry will be not discussed herein since it is not suitable for industrial applications.
All of the coupling reagents reported so far are applied in the usual C-to-N building of peptide sequences. Recently, Li and coworkers shed light on the possibility of applying an inverse solid-phase peptide synthesis (ISPPS) protocol, in which N-to-C direct SPPS is performed.140 According to the authors, by using this approach, it is possible to replace the conventional Fmoc/tBu strategy with an atom-economic method free from coupling reagents and side-chain protections. However, the process is based on the use of highly toxic chemicals and the formation of potentially explosive intermediates.
3.2. Peptide-anchored, liquid-phase peptide synthesis (PA-LPPS)
Classical LPPS141 remains attractive to produce short peptides or for hybrid synthesis in combination with SPPS. LPPS, even if requiring frequent isolation steps, can be performed using standard, multi-purpose industrial plants avoiding any substantial investments in specific equipment. In addition, short peptide drugs are treated from a regulatory standpoint as small molecules. A recent example is the synthesis of peptides (up to 10 amino acids) in water or water/THF 9/1, under micellar catalysis, that was developed by Lipshutz and coworkers. The approach is a typical LPPS synthesis that required palladium-catalysed hydrogenation for the nitrogen CBz protective group removal and high dilution for solubility issues. The reported cE-factors are promising.142SPPS, conversely, does not require intermittent isolations, allowing the straightforward synthesis of long peptide sequences, and can be easily automated.143 Moreover, solvent-resin swelling and reagents’ diffusion properties are the key parameters to be considered in an SPPS's solid/liquid biphasic system. The direct consequence of this issue is the use of a large molar excess of reagents to guarantee a high conversion rate, and the use of large solvent volumes for the extensive resin washes. Instead, LPPS is a monophasic system, in which substrate and reagents solubilities are the main drivers for the solvent choice. Almost stoichiometric amounts of reagents are necessary to achieve high conversions in the coupling step.
Based on some seminal studies carried out a few decades ago,144 several research teams have focused on the development of solution-phase peptide synthesis, anchoring the peptide to an organic molecule to increase the solubility in the organic solvent. Theoretically, these new technologies lead to decreases in the PMI and excess reagents, cutting production costs (Table 7). Peptide-anchored liquid-phase peptide synthesis (PA-LPPS) tries to combine the advantages of both SPPS and LPPS. Interestingly, all the PA-LPPS technologies have been protected by patents and trademarks: molecular Hiving™ by Jitsubo,145 Ajiphase® by Ajinomoto Bio Pharma Services,146 PEPSTAR® by a team of researchers that comprise Eli Lilly and Exactmer,147 and GAP by GAPPeptides.148 In Fig. 14, the most important anchors are shown. Several papers have been published by these companies; however, it appears that the most recent and advanced procedures have been hidden to protect the inventions. Several protocol descriptions are incomplete or not suitable for industrial applications.
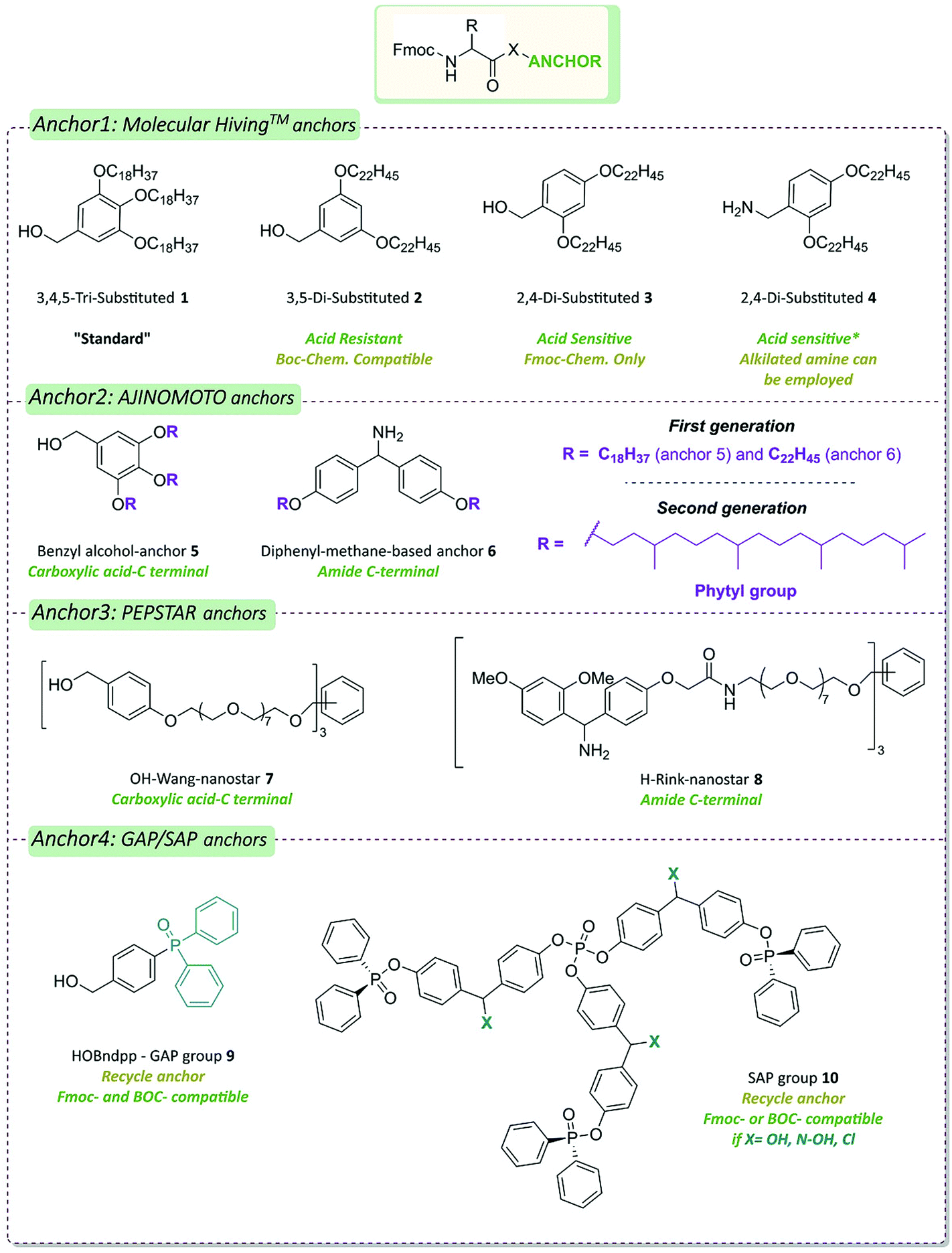 | ||
| Fig. 14 Examples of anchors developed by Jitsubo (Anchor 1: Molecular Hiving™ anchors), Ajinomoto Bio Pharma Services (Anchor 2: AJINOMOTO anchors), researchers from Eli Lilly and Exactmer (Anchor 3: PEPSTAR anchors), GAPPeptides and Northwestern Polytechnical University, China (Anchor 4: GAP anchors and SAP anchors, respectively); * Anchor 4 (MHT) is a general representation of amine anchors and some of these are more stable than the corresponding HBA under acidic conditions (TFA). | ||
| Parameters | LPPS | SPPS |
|---|---|---|
| Bold was used for the positive aspects of the technique from a greenness stand point. | ||
| Reaction medium | Solution | Gel (swollen insoluble polymer) |
| Synthesis strategy | Convergent or stepwise | Stepwise |
| Temporary protecting group | Typically Boc or Z | Typically Fmoc |
| Rapidity of synthesis | Slow | Fast |
| Consumption of amino acid derivatives | Moderate | Medium to high |
| Consumption of organic solvents | Moderate to high | High |
| In-process control | Direct monitoring applicable (e.g. HPLC) | Typically indirect monitoring applied (e.g. resin colour tests) |
| Isolation of intermediates | Typically isolated by precipitation | No isolation of intermediates |
The technology was later optimized to also produce terminal amide peptides150 (Table 8) and to improve the efficiency of the reaction sequence of the Fmoc technology, avoiding the precipitation at each reaction step. After the coupling step, the excess activated amino acid was quenched by adding a nucleophilic amine, such as propyl amine. Only after Fmoc removal with DBU/piperidine as bases was the anchored peptide precipitated as shown in Fig. 15.151 Propyl amine selectively reacts with the activated amino acid avoiding unwanted reactivity of the deprotected amine moiety. Of note, the reaction was performed in 9:1 THF/DMF. Most recently, through a collaboration with Jitsubo (as of 2008),152 Bachem claimed the industrialization of technology that avoids intermediate precipitation. The semicontinuous process was performed using aqueous washes for reagent and side product removal. In several webinars, Bachem researchers claimed a ∼60% reduced use of solvents with respect to the corresponding SPPS, without disclosing the target peptide length, the quality achieved or the final procedure.
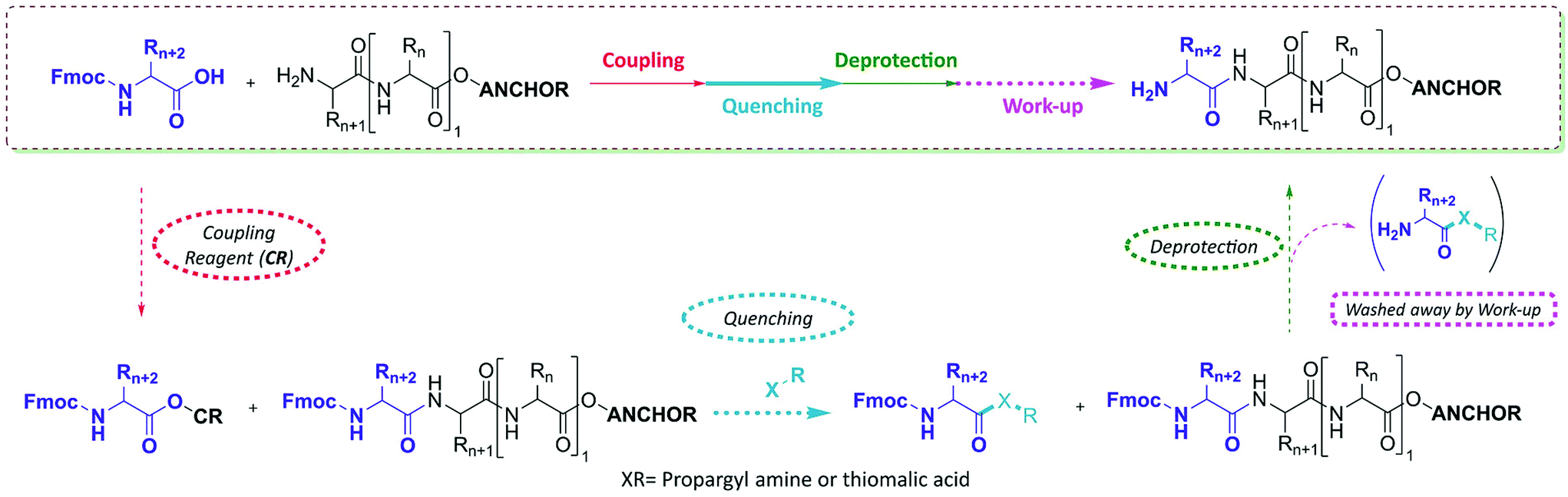 | ||
| Fig. 15 General pathway for peptide synthesis by PA-LPPS. | ||
| Entry | Peptide or PNA | No of aa/bases | Yielda (%) | Ref. | Year |
|---|---|---|---|---|---|
| a The yield does not include the reaction step for the synthesis of TAG. b Disulfide bond obtained by electrochemical reaction. c Convergent synthesis. d (H-RVYIHPI-OH). e H-AGTCAGTC-Lys-OH. f Including the purification process, the yield is 37% with purity >99%. | |||||
| 1 | Antagonist of TNF-α | 15 | 70 | 149a | 2010 |
| 2 | Somatostatin | 14 | 50 | 150b | 2011 |
| 3 | Somatostatinb | 14 | 57 | 150c | 2012 |
| 4 | Leuprolide | 9 | 40 | 149b | 2013 |
| 5 | Bivalirudinc | 20 | 44 | ||
| 6 | h-Ghrelinc | 28 | 8 | ||
| 7 | Mahafacyclin B | 7 | 39 | 150d | 2013 |
| 8 | a-Conotoxin MII | 16 | 43 | 150e | 2013 |
| 9 | Elastin | 10 | 41 | 150f | 2014 |
| 10 | ABT-510 | 9 | 66 | 150g | 2015 |
| 11 | iAβ5 | 5 | 63 | 150h | 2015 |
| 12 | Angiotensin III selective antagonistd | 7 | 73 | 149c | 2017 |
| 13 | Oligomere | 9 | 56 | 150j | 2018 |
| 14 | Icatibant acetatef | 10 | 57 | 151 | 2019 |
| 15 | Stellarin E | 7 | 29 | 150k | 2021 |
These second-generation anchors were tested in different solvents such as CHCl3 and toluene, as well in the greener solvents, EtOAc and CPME. Interestingly, the solubilities of these new branched-chain anchors in various organic solvents consistently increases. As an example, the solubility of the linear anchor in ethyl acetate increases 100-fold, from 0.2 to >25 weight%. The protocol used Fmoc-protected natural and unnatural amino acids in slight excess (1.1–1.3 eq.), and EDC·HCl/HOBt as a coupling mixture. After brine washing, Fmoc removal was performed in the presence of thiomalic acid and DBU (Fig. 15). The addition of thiomalic acid as scavenger was necessary to convert the dibenzofulvene, coming from Fmoc removal, into a water-soluble species, allowing the elimination of reagents and side products of the deprotection step by a simple wash with a mixture of aqueous sodium carbonate and DMF. The organic layer was subjected to the next coupling reaction without concentration or drying. No impurities derived from thiomalic acid or fulvene were observed in the subsequent coupling reaction. The fully protected, full-length peptides demonstrated satisfactory solubility in the organic layer, and phase separation was easily accomplished. The final anchored peptide was then precipitated by ACN addition, and the cleavage was carried out using standard methods. It is worth noting that the syntheses of two generic peptides (20-mer, Bivalirudin, anchor 5; and 10-mer, Degarelix, anchor 6), using CHCl3 as solvent, have been achieved with an HPLC purity of 84% and 89%, respectively.
The real value and greenness of AJIPHASE® technology cannot be judged using the available publications. The possibility to use alternative greener solvents, like esters, was highlighted by the solubility of the anchor with branched chains, and the large body of literature that describes ethyl acetate as an efficient solvent for peptide synthesis.
:65 THF/NMP mixture, working in the 30–35 °C temperature range with an automatic synthesizer, and employing the H-Rink-nanostar anchor. In Fig. 16, a schematic representation of the synthesizer is shown.
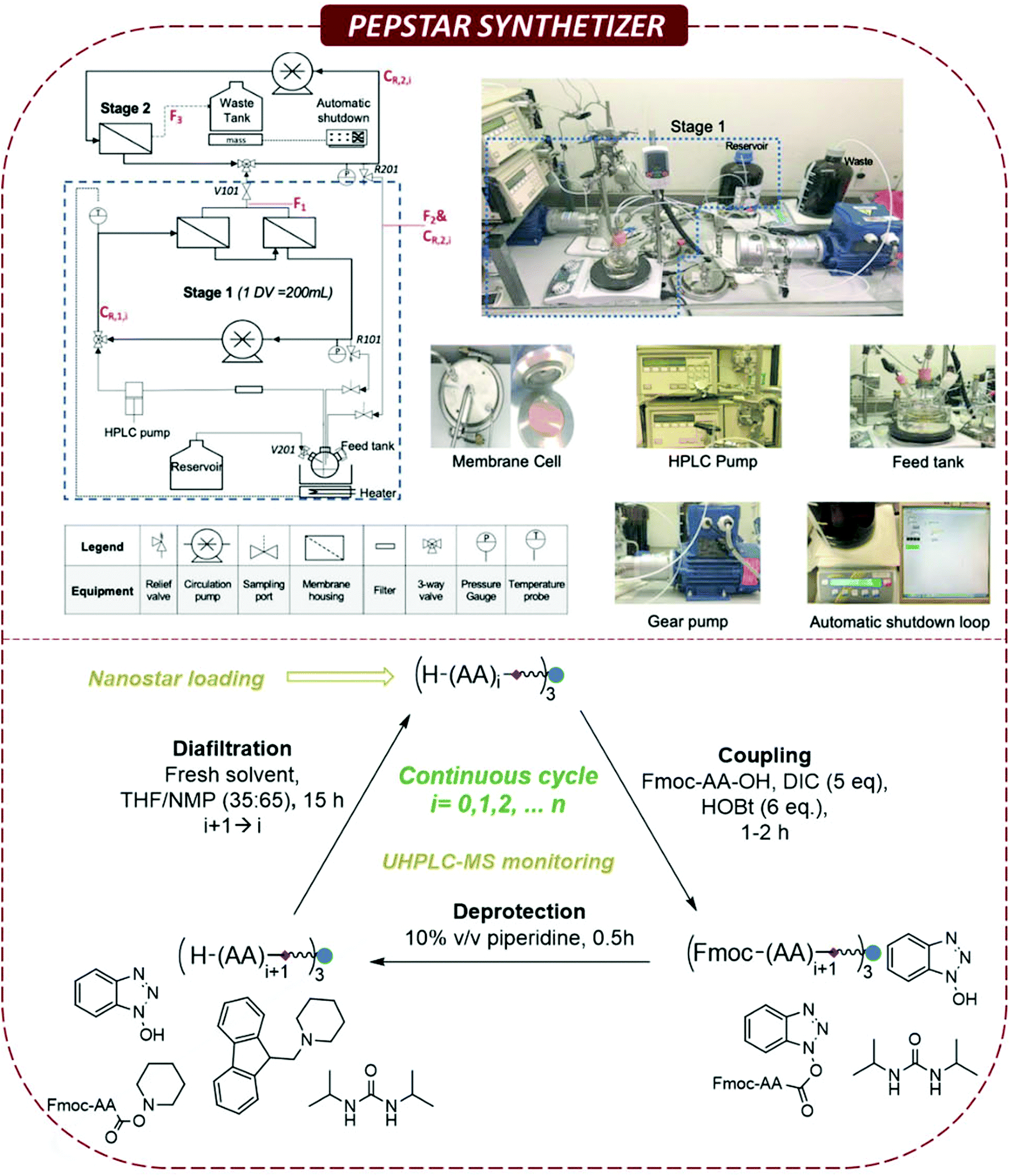 | ||
| Fig. 16 Representation of the PEPSTAR synthesizer. At the top, the scheme and photos of the instrument are shown (open access from ref. 29d); at the bottom, a general reaction scheme of the process is described (adapted from ref. 29d). | ||
Cleavage was performed using a TFA/TIS/H2O mixture, affording the crude pentapeptide H-Tyr-Ser-Ser-Phe-Leu-NH2 and the linear octreotate amide with 94% and 90% purity, respectively. PEPSTAR was a consistent improvement of the MEPS technology. However, there are still several drawbacks to be addressed. For example, the OH-Wang-nanostar, that opens the way to the synthesis of peptides with acid termini, did not perform well. In particular, the protocol requires the use of toxic NMP in mixture with THF, as the organic solvent nanofiltration (OSN) was time-demanding, and consistently decreased the productivity and increased the PMI. In fact, the impact of the OSN on the PMI was surprising. In Table 9, a comparison of the different technologies highlights the better performance of SPPS. Based on the authors’ data, SPPS outperformed MEPS and PEPSTAR, with the PMI being 5.6- and 1.7-times lower, respectively.
| g mol−1 | MEPS | PEPSTAR | SPPS |
|---|---|---|---|
| Adapted from ref. 29d. | |||
| Anchor | 5000 | 707 | 2000 |
| Amino acid | 6788 | 5770 | 10182 |
| Solvent | 9190000 |
2800000 |
1510000 |
| Yield % | 80 | 80 | 75 |
| Target peptide | 940.8 | 940.8 | 882 |
| PMI | 9783 | 2983 | 1726 |
The anchor is a very simple organic compound ((4-hydoxymethyl)phenyl)diphenylphosphine oxide, HOBndpp (Fig. 14, anchor 4). After the first amino acid is attached, the reaction sequence is performed in DCM and, after Fmoc deprotection with 30% piperidine and washes with a NH4Cl solution, the subsequent amino acid coupling is promoted by TBTU/DIPEA. After several washes, the DCM solution is evaporated, and the residue dissolved in ethyl acetate and precipitated by adding petroleum ether. The process is reiterated, and the crude peptide is isolated via a two-step process: TFA/DCM/water followed by palladium-catalysed hydrogenation. Despite this being the technology described in peer reviewed publications, several issues are associated with the use of DCM as solvent, explosive TBTU as coupling reagent, and the final cleavage that is performed in 2 steps comprising hydrogenation. Still, the technology efficiency was verified by synthesizing the pentapeptide thymopentin.
On the company's website, several documents are reported, claiming the use of sustainable alternative solvents, like 2-MeTHF as a reaction solvent and cyclopentyl methyl ether for the intermediate's precipitation. However, these two solvents are classified as problematic in several guides. The synthesis of the 20mer, Bivaluridin, obtained with an 88% purity, was also claimed on the website to be competitive in terms of PMI with respect to the corresponding SPPS process.160 However, it is not possible to make an evaluation of the technology since no raw data are available.
In 2020, Qin and coworkers focused their attention on the PA-LPPS, developing a series of novel anchors based on phosphorous derivatives: tri(4-benzoylphenyl) phosphate (TBP), tri(4-formylphenyl)phosphonate (TFP), diphenylphos-phonyloxyl diphenyl ketone (DDK), and tri(4′-diphenylphosphonyloxyl benzoyl-phenyl)phosphate (TDPBP) (in Fig. 14 only TDPBP was described).161 The supported-assisted-precipitation technology (SAP) used DCM as a solvent. The precipitation was performed at any step by adding EtOAc in a mixture with hexane, petroleum ether, or acetonitrile, depending on the anchor used. The technology can be performed using Fmoc or Boc amino acids. In this case, there is also a major issue related to the use of DCM and of the counter solvent mixtures reported above.
| Technique | Pros | Cons |
|---|---|---|
| Molecular Hiving™ Technology | - High yield and purity of products | - Use of DCM and DMF for synthesis of both anchor and peptide sequence |
| - Speed | ||
| - Intellectual property | - Coupling reagents and advisable of replacement (DMM-TM and COMU) | |
| - Synthesis up to 10mer peptide | ||
| AjJIPHASE® | - High yield and purity of products | - Use of CHCl3 for peptide synthesis and DMG in some water washes. The use of more green solvents has been claimed but not disclosed |
| - Intellectual property | ||
| - Synthesis up to 20mer peptide | - Coupling reagents are advisable of replacement (EDC/HOBt), | |
| PEPSTAR | - High yield and purity of products | - Use of NMP in peptide synthesis |
| - Synthesis up to 8mer peptide | - Coupling reagents advisable of replacement (HBTU) | |
| - PMI higher than SPPS | ||
| GAP | - High yield and purity of products | - Use of DCM for peptide synthesis – use of coupling reagents advisable of replacement (TBTU, EDC/HOBt, EDC/DMAP, DEA) |
| - Synthesis up to 5mer peptide | ||
| - The anchor can be recovered. |
From a scientific perspective this area is absolutely interesting; however, additional efforts are necessary to prove that the technology can be sustainable and suitable to deliver the same complex peptide currently produced using SPPS (Table 10).
The greatest advantage that all these techniques have in common is, undoubtedly, the possibility of considerably reducing the amount of solvent and coupling reagents needed to carry out the reactions.
However, few examples have been reported on synthesis processes involving the use of green solvents or reagents with a reduced impact on operator health, plant safety, or effect on the environment. Furthermore, evaluating the use of anchors for peptide synthesis makes it immediately clear that the possibility of obtaining good processes in terms of atom economy is already a priori excluded. However, through a relative evaluation of the process, by comparing it with SPPS, it appears that the impact of the technique is extremely limited. It is also clear that PA-SPPS techniques are poised for implementing the basic processes needed for greener solutions. Proven and successful application toward the synthesis of API peptides, therefore, creates optimal starting conditions to proceed towards a large-scale transfer of these synthesis procedures to obtain a more sustainable production chain.
3.3. Semi-continuous/continuous peptide synthesis
This section is dedicated to technologies that are based on semi-continuous/continuous processes, avoiding the use of anchors for the acid terminus, or being an evolution of the classical SPPS. The main targets of these protocols are to decrease solvent consumption and increase process efficiency and speed. Diosynth Rapid Solution Synthesis of Peptides (DioRaSSP®) by Aspen and μLOT® by SB3000 are examined, highlighting the pros and cons based on the available information, and painting a precise picture of the possible direction that continuous techniques can take for industrial synthesis of API peptides.162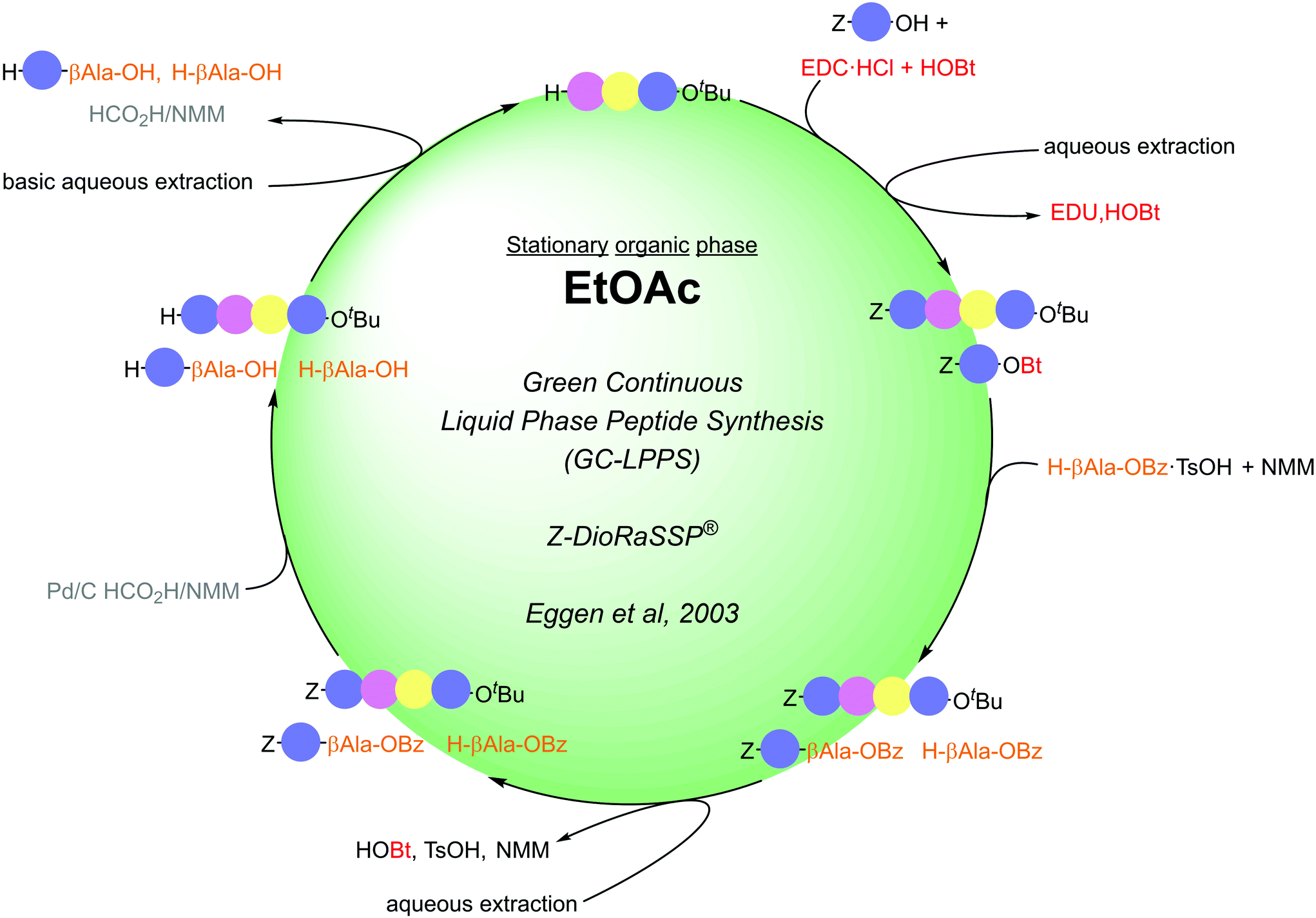 | ||
| Fig. 17 Example of a Z-DioRaSSP cycle (ref. 163). | ||
Finally, it is reasonable to assume that Aspen Oss improved the technology during the last 20 years, even though it seems to be industrially limited to the production of short peptides (<10mer).
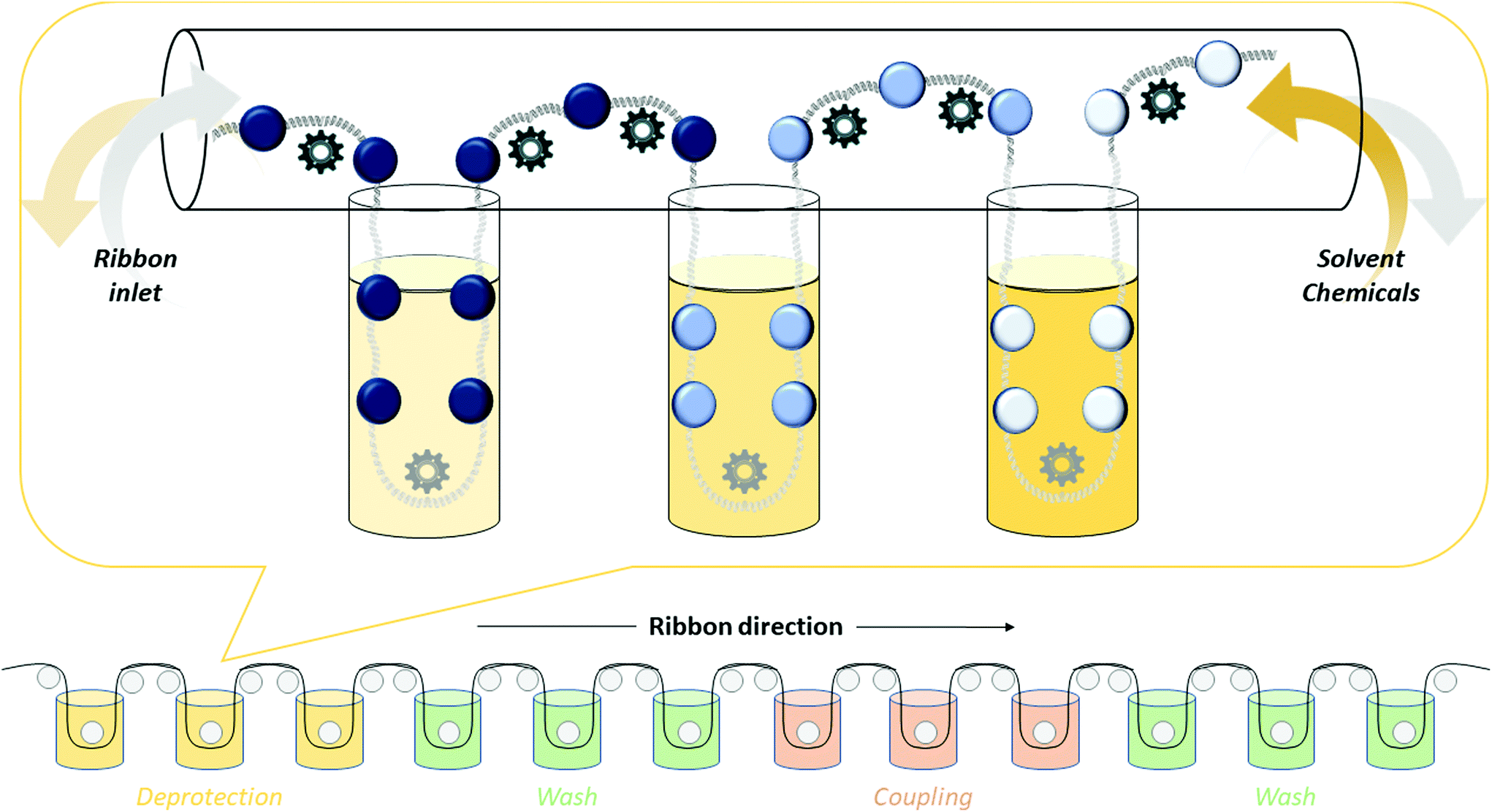 | ||
| Fig. 18 Schematic representation of μLOT® technology (ref. 162b). | ||
Unfortunately, the efficiency of μLOT® claimed by SB3000® concerning production quality, reliability, and PMI reductions is not supported by data. In fact, there are no patents or publications that allow a real evaluation of the technology. All available information has been collected from the company's website and conference presentations.169 At the TeknoScience online event on peptides held in March 2021, 90% reduced use of DMF using μLOT® compared to SPPS was claimed. Considering the technology potential, monitoring the evolution of μLOT® will certainly offer interesting information.
| Technique | Pros | Cons |
|---|---|---|
| DioRaSSP (Aspen) | - Applicability to industrial scale | - Absence of recent undisclosed information |
| - Automated process | - Suitable for short peptide sequence. | |
| - Use of green solvent without organic waste | - Hydrogenolysis deprotection (Pd metal and hydrogen formation) | |
| - Absence of intermediate isolation | - Unclear necessity of specific apparatus for hydrogen evolution | |
| - Use of aqueous work-up to remove side products | ||
| - Detection of low amount of epimers, insertion or truncated sequences | ||
| - Real time monitoring | ||
| μLOT® (SB3000®) | The lack of data does not allow a critical evaluation of the technology | |
3.4. Chemoenzymatic peptide synthesis (CEPS)
Alongside synthesis methods, biocatalysis can be considered one of the pillars of sustainable chemistry, with wide application in different industrial segments, including drug manufacturing. In chemo-enzymatic peptide synthesis (CEPS), several classes of enzymes are used in the synthesis of pharmaceutical intermediates.170 Thanks to the evolution of sophisticated technologies to adapt the enzyme for a specific use, enzymatic catalysis for peptide and protein ligations has recently emerged as a potentially sustainable alternative to chemical ligation.171 In particular, four classes of enzymes have been widely investigated: butelase, sortase, trypsiligase, and subtiligase variants, owing to their capability to generate a specific peptide bond in water.171 However, the enzymatic steps need to be fully integrated with other technologies such as GSPPS and PA-LPPS. As described in the following paragraphs, CEPS can be useful in synthesizing a wide range of linear and cyclic peptides and protein conjugates, including highly complex substrates.In the last few years, extensive research has been performed by several teams worldwide that led to the creation of engineered enzyme platforms with broad substrate scope, improved activity, and stability.
3.4.1.1. Sortases. A well-established enzyme for peptide ligation is Sortase A (SrtA), a natural enzyme that catalyses the adhesion of surface proteins to the cell wall of prokaryotes, whose use is hampered by a low catalytic efficiency. Sortase A recognises the LPXTG (where X is any amino acid) sequence on the C-terminal peptide fragment. It cleaves the glycine and generates a thioacyl-enzyme intermediate with threonine, susceptible to nucleophilic attack by the N-terminal fragment to create the peptide bond (Fig. 19).172
 | ||
| Fig. 19 Schematic representation of catalytic mechanisms of Cys-dependent enzymes, like sortases and asparaginyl endopeptidases (butilase-1), peptiligases, and recognition pockets of omniligase-1 (adapted from ref. 193b). | ||
The use of sortase for peptide ligation, defined as sortagging, is possible when the identification motif LPXT is included in the acyl-donor fragment, while a Gly is required at the N-terminus of the acyl acceptor.173 In other words, the LPXTG recognition sequence is fixed and must be part of the target peptide. In addition the reversibility of the reaction leads to low yields.
Some engineering variants have been developed to broaden the substrate scope of the reaction, but only in a few cases has the activity been improved.174,175 Interestingly, Zou et al. were able to increase the enzyme stability by the formation of a cyclized mutant of Sortase A, namely CyM6, with improved thermal stability and a good resistance against denaturating agents, such as urea.176
In summary, sortagging has a limited scope in CEPS because of the LPTXG motif sequence that must be present in the target peptide (there are no commercial peptides with that sequence) and the large amount of enzyme necessary to get good conversions.
3.4.1.2. Asparaginyl endoproteases (AEPs). Another suitable solution for enzymatic peptide ligation is offered by asparaginyl endopeptidases (AEPs). Butelase 1, one of the most important enzymes of this class, was isolated from Clitoria ternatea,177 a tropical plant in which this enzyme naturally catalyzes a head-to-tail cyclization in the biosynthesis of cyclotides.178 For this reason it was classified as a ligase rather than a protease. Butelase 1 catalyses the peptide bond via the same thioester acyl-enzyme intermediate of Sortase A (disrtupted by an amine on the N-terminal fragment, affording the ligation product). It recognises a shorter motif on acyl-donor fragment, namely NHV (Fig. 19).177 In this case, only an Asp residue is retained in the ligated sequences, differently from the longer motif of Sortase A-catalyzed ligations. Concerning the N terminal, the reaction takes place with any amino acid in the first position except Pro, Asp and Glu, and there is less tolerance for the second position, where only Ile, Leu, Val or Cys are compatible. From a green perspective, the major advantages of this enzyme are its high catalytic efficiency (0.005 molar eq. of enzyme are required) and the high yields, with cyclization preferred on intermolecular ligation. Moreover, the competitive reaction between the cleaved HV-dipeptide and the thioacyl enzyme complex is a critical aspect. Several solutions to this problem have been proposed. In linear ligation, the use of thiodepsipeptide as acyl-donor was tested successfully by Tam's group, which significantly contributed to the development and improvement of sortase applications; the thioester leaving group was not recognized by the Butelase-1, allowing high yields for the coupling steps.179 After several unsuccessful attempts, Tam and coworkers were able to successfully produce the recombinant enzyme, thus opening the possibility to produce a butelase-1 with an activity identical to the natural enzyme.180 The potential of this enzyme attracted the attention of several research groups that investigated, in parallel, the active site and the possibility of producing additional variants via recombinant technologies.181 Recently, Hemu and coworkers reported an engineered variant of butelase-2 able to work as peptide ligase.182
Other AEPs have also been identified, namely OaAEP1b/3/4,183 HeAEP3184 and VyPAL2185 with similar attributes for cyclization reactions. In particular, OaAEP1 was crystallized and several variants inspired by the natural enzyme were produced by recombinant methodologies.186 From a technological perspective, the immobilization of butelase-1 and VyPAL2 was recently explored, with satisfactory outcomes for non-covalent immobilization of butelase-1 (39% of immobilization yield, 50% of recovered activity). Interestingly, the operational stability was substantially retained over 100 runs (>90%).187 The loss of enzymatic activity after immobilization is expected, but an improvement in immobilization yield could make this technique more convenient. In summary, AEPs are interesting because of their short recognition motif and the efficiency in macrocyclization reactions. In particular, butelase-1 variants appear to be the most efficient class of AEPs enzymes thus far. The recent breakthroughs in the availability of the active site's crystal structure and recombinant production of several nature-inspired AEP enzymes open up the path to rapid optimization and evolution towards more efficient macrocyclization/ligation reactions and industrial applications.188
3.4.1.3. Subtilisin variants. Subtilisin protease was subjected to extensive modification by genetic engineering during the past few decades, generating an incredible amount of variants. This journey started with the chemical modification of the catalytic site in subtilisin protease. The Ser221 in the catalytic triad Asp-His-Ser was converted into a cysteine, increasing the ligation activity over hydrolysis (Fig. 19).189 Unfortunately, the introduction of a thiol group generated steric crowding of the catalytic site, thus a further mutation was introduced in the 1990s leading to the double mutant S221C/P225A subtiligase.171a,190 This catalyst still had some limitations, such as the necessity of high nucleophile excess (10 eq.) and a poor stability. Then peptiligase, a new variant of the Ca2+-independent subtilisin BPN’, including the previously mentioned mutations, was introduced by Toplak and coworkers.191 The impact of reagents was significantly lowered due to the theoretical 100% conversion rate with almost equimolar amount of substrates, and the side-products were minimized thanks to the high efficiency of the ligation process over hydrolysis. Despite the efforts made to reach an effective chemoenzymatic ligation, peptiligase had a narrow substrate scope, limiting its application as general peptide bond-forming catalyst. Subsequent work reported further mutations, which led to a very efficient enzyme, called omniligase-1. Thanks to engineering, the substrate scope was broadened, with two major limitations at the P4 position, where only hydrophobic or slightly polar amino acids are tolerated, and on the P1 position, where proline is not accepted. On the acyl acceptor fragment at the P1′ position, almost all amino acids except proline are tolerated, while proline and charged residues are not recommended at the P2′ position (Fig. 19).192 Furthermore, the subtiligase variants require the use of activated esters on the C-terminal fragment, usually carboxyamidomethyl (OCam) ester, improving kinetic control of the reaction.
Omniligase-1 has been applied in the synthesis of the pharmaceutical peptide exenatide. In this approach, the acyl-donor (H-1–21-O-Cam-L-NH2) and the acyl acceptor (H-22–39-NH2) fragments, produced by standard SPPS, were ligated under different conditions, in both crude and purified form, with very efficient catalytic activity. In particular, comparing the well established SPPS method with the CEPS approach, the latter proved to have a good impact on the PMI (more than halved), avoiding difficult SPPS steps, and reducing byproduct formation, thus simplifying the purification process of the target peptide. Interestingly, the ligation of purified fragments produced exenatide using only 1.1 eq. of the acyl acceptor with a very low amount of biocatalyst (0.0008 mol eq.).193 A further study on subtiligase variants demonstrated that it is possible to tailor the enzyme to a specific substrate, improving the reaction conditions and process yield compared to standard synthesis. This was the case of thymoligase, successfully applied in the synthesis of the API thymosin-α1, doubling the yields with respect to a full SPPS approach.194 Peptiligase variants are also used in the cyclization of peptides due to their high ligation efficiency (conversion >90%) in reactions involving peptides with more than 12 residues. Furthermore, they are based on a very interesting methodology since the insertion of recognition motifs in the substrates is not necessary, enabling a traceless ligation.195 More recently, the determination of the X-ray crystal structures of omniligase mutants, and the creation of computational models of the peptide binding mode, allowed a broadened substrate scope, making this class of enzymes more attractive for CEPS.192 In this context, the subtilisin variants developed by Enzypep have been used for the synthesis of at least two therapeutic peptides, namely exenatide and thymosin-α.193,194
3.4.1.4. Trypsin variants. Native trypsin and its variants have been known for decades. Bordusa and coworkers described a variant of trypsin, termed trypsiligase, for the site-specific labeling of both N- and C-termini of target peptides or proteins. Thanks to four mutations, K60E, N143H, E151H and D189 K, which conserved serine in the catalytic site, these variants can mediate ligation reactions after the activation of the enzyme by Y-RH-containing substrates.196 Trypsiligase is in equilibrium between an inactive zymogen-like conformation and an active conformation, which occurs in the presence of the YRH recognition motif and Zn2+. When the ligation occurs between a peptide or protein carrying the Y-RH motif at the C-terminus and a nucleophilic RH-peptide or protein, the competition between the RH leaving group and the RH-peptide or protein reduces the ligation efficiency and, following the Le Chatelier principle, an excess of nucleophile substrate is required.196 Trypsiligase has a good catalytic activity when only 0.1 eq. of enzyme is used, and the reaction is usually complete in a few minutes.197 The main use of tryptiligase is protein labelling even if the Y-RH recognition motif is not very common in natural proteins.197 Notably, these enzymes are not used for the CEPS approach to polypeptides (Table 12).
| Pros | Cons |
|---|---|
| - Reduced excess of reagent | - Necessity to introduce specific recognition motifs (short substrate scope) |
| - Complete conversions | - Modification of C-terminal motif to generate activated ester |
| - Absence of epimerization | - Necessity to work in combination with sustainable technologies for the chemical synthesis of fragments |
| - Use of water as reaction medium | |
| - Effective under mild conditions | |
| - Effective for peptide cyclization | |
| - Possibility to reuse the enzyme after recovery (immobilization) | |
| - Absence of protecting groups |
3.5. Mechanochemistry applied to peptide synthesis
Mechanochemical reactions, defined as chemical transformations which involve the use of mechanochemical energy to facilitate and induce reactivity, have attracted the attention of both the academic and the industrial communities.198,199 The increasing popularity and success of ball-milling techniques is due to the fact that the protocols can be carried out under solvent-free conditions or only in the presence of minimum volumes of organic solvents,200 thereby drastically reducing waste.201 Moreover, other features have led mechanochemistry to be considered as a state of art technique by the scientific community, such as fast reaction time, high efficiency protocols, stoichiometry control and unique reactivity, opening the way to the synthesis of several compounds not attainable by other synthesis techniques.202,203 Regarding peptide synthesis, the mechanochemical approach is normally limited to the use of Boc protected amino acids and only to the coupling process. In addition, there are only a few papers describing the synthesis of amino acid sequences that goes beyond dipeptides.The pioneering work in the area was reported by Lamaty and coworkers204 in 2009, who demonstrated the successful use of mechanochemical solvent-free strategies in peptide synthesis using activated urethane-protected α-amino acid N-carboxyanhydride (UNCA) derivatives. This work set the basis for the development of a mechanochemical protocol for the synthesis of Leu-Enkephalin, using a liquid-assisted-grinding (LAG) technique. The addition of EtOAc as liquid additive was critical in order to enhance the yield and the reactivity of the coupling.205
However, early developments of ball-milling peptide synthesis were based on the utilization of activated N-protected α-amino esters, but the low commercial availability of these chemicals limited the scope of the approach.204–206
For this reason, the further development of mechanochemistry has been focused on the use of commercially available amino acids and more classical coupling agents such as carbodiimides, benzotriazoles and oximes, mainly for the synthesis of dipeptides with low level of epimerization, <1%.207 Unfortunately, some of these protocols still suffer from some drawbacks, such as the use of harmful additives like DMAP, DCM, MeNO2, HOBt, cyanuric chloride and PPh3. In addition, most of the time the workup comprised the use of organic solvents, and the deprotection of the amino group, necessary for the potential elongation, was performed using gaseous HCl or was not described.
More recently, Lamaty and coworkers developed a more environmentally friendly mechanosynthesis protocol of a wide range of dipeptides, tripeptides and tetrapeptides.208–210 The optimized strategy was then utilized in the synthesis of the tetrapeptide VVIA. The coupling steps were performed by ball-milling amino ester salts (p-toluenesulfonate or hydrochloride) with Boc-AA-OH in the presence of EDC, Oxyme, NaH2PO4 as base and small amounts of EtOAc as the liquid grinding assistant. Conventional workup based on acid/basic extractions and washings was performed to achieve the desired coupling products in yields ranging from 78 to 89% (Fig. 20).
 | ||
| Fig. 20 Approaches for mechanochemical synthesis of peptides. | ||
In this case the removal of the Boc group under mechanochemical conditions was attempted, obtaining the Boc-VVIA-OBn peptide with an overall yield of 59%. In 2020, Anselmi et al.211 utilized nanocrystalline hydroxyapatite as a bio-compatible, reusable inorganic base to promote the mechanochemical solvent-free coupling synthesis of the tetrapeptide YPWF, using conventional coupling reagents and the Boc strategy already described. Furthermore, the base could be reused for several times, after a simple regeneration, with only a partial loss in activity.
As reported above, chemoenzymatic catalysis has been widely used in solution and solid-phase peptide chemistry due to the mild reaction conditions required, lower use of toxic chemicals, higher yields, minimal side-chain protections, and to the possibility to strictly control the stereoselectivity of the peptide.212
These observations quickly led to the development of mechanochemical enzymatic reactions and recently the compatibility between enzymes and mechanochemical ball-milling has been reported. In 2017 Hernández et al. developed a mechanochemical chemoenzymatic peptide and amide bond formation catalysed by papain, a cysteine protease found in papaya latex (Carica papaya).213 Despite the high energy inside the ball mill, the biocatalyst proved to be stable and highly efficient to catalyze the formation of α,α and α,β-dipeptides in good-to-high yields using Na2CO3·10H2O as base.
4. Peptide purification improvements
The interconnection between synthesis and purification is a normal practice in organic chemistry. Moving to peptide manufacturing, the complexity of the mixtures and the presence of impurities, structurally related to the main drug, determines that the upstream and downstream co-development, guided by a reliable analytical method, is the critical factor to achieve the target quality attributes. In the synthesis of any API peptide, regardless of the technique chosen, it is inevitable that a reaction crude will be characterized by the presence of various impurities, which would become increasingly significant in number as the length of the peptide increases. Regulatory agencies increased consistently their requirements by lowering the level of impurities and requesting a very sophisticated characterization of their immunogenicity and influence on product aggregation.12A preliminary evaluation on the purification method must establish the process-related impurities that cannot be eliminated by chromatography and that must be tackled in the upstream process by a fine tuning of the stoichiometry, the reaction conditions or by a change of synthesis strategy.
A possible change in strategy is the use of a hybrid SPPS/LPPS approach for the synthesis of particularly long peptides, which consists of assembling shorter sequences, optionally purified, instead of linear peptide sequences. An explicative example has been recently reported by researchers from Eli Lilly for the synthesis of the 39mer Tirzepatide. Shorter peptide fragments obtained in high purity (>97%) via SPPS and completely characterized were kept protected and then coupled via LPPS to afford the target peptide with good yield and purity (>55% and >80%, respectively) using a semi continuous approach in green solvents.214
The hybrid approach, in addition to a simplification of the chromatographic purification of the final peptide, can decrease the industrial risk and potentially generate intellectual property.215,216 Indeed, experimental conditions, such as the eluent flow rate, the temperature and, especially, the combination of stationary and mobile phase, must be adjusted case by case. In addition, it is possible to choose whether to maintain the eluent composition constant along time (isocratic mode) or to operate in gradient conditions, which most of the time is necessary when dealing with complex peptide mixtures.
Despite several benefits, including high productivity, chromatography also has some drawbacks, such as high operating costs and large solvent volumes.217 A single chromatographic step is not usually enough to achieve the desired product purity, and therefore the downstream processing usually represents the bottleneck of the entire production workflow in terms of time. This issue is the most concerning aspect related to the greenness of chromatography, since large volumes of non eco-friendly solvents, with potential risks towards environment, human health, and safety, are required.
Besides the 12 principles of green chemistry, green analytical chemistry (GAC) promotes also three Rs, that are Reduce, Replace and Recycle.3 In the past, it was believed that the only way to increase sustainability of analytical processes was through the replacement of not eco-friendly materials (such as solvents or modifiers) with greener ones, but in the last few years reduction of solvent demand has also become a viable choice thanks to the establishment of innovative continuous chromatographic techniques. These approaches, indeed, allow for internal recycling of impure fractions in the system, permitting not only to reduce wastes but also to decrease the solvent consumption.
In the following sections, current trends to increase sustainability of downstream processing of peptides are thoroughly discussed. Perspectives and pitfalls in the search for greener materials and techniques are also presented.
4.1 Greener materials
The first approach that can be pursued to increase the sustainability of purification processes is the replacement of toxic materials with more eco-friendly ones. Polar biomolecules, such as some peptides, are usually purified under ion-pair reversed-phase liquid chromatographic (RPLC) conditions by using acetonitrile/water mixtures as mobile phases, to which an ion pairing reagent is added. Its role is to mask the charge of polar biomolecules, allowing for their retention on hydrophobic adsorbents.218A greener option is ethanol (EtOH), which has several advantages over MeOH including lower toxicity, generation from biomasses and fast biodegradation besides slightly lower costs. EtOH has also higher elution strength with respect to MeOH, meaning that less EtOH is required for the elution of the target at comparable retention times. Disadvantages of the use of EtOH are related to its higher viscosity and, most importantly, to its lower vapor pressure with respect to MeOH (and ACN).220 This last condition is particularly disadvantageous in preparative chromatography because the solvent contained in the collected samples needs to be evaporated, but EtOH is particularly difficult to remove.
The employment of other alcohols, such as 2-propanol or n-butanol, is limited by their higher viscosities and boiling points in comparison with MeOH. Another trend, promoted by modern GAC and in general by green chemistry, is to shift from the use of sources coming from fossil fuels to bio-based ones, obtainable from renewable origins including agricultural wastes, forestry and wood processing, and marine biomasses.221 Except ethanol, the other bio-solvents have never, or barely ever, been applied as solvents for liquid chromatography so far. Their use is limited by the fact that a deep physico-chemical characterization of the bio-solvent/water mixture (in terms of solubility, viscosity, UV transparency, inertness, etc.) is needed before considering them suitable alternatives. Unfortunately, there are no data available on efficient purification of peptides carried out using alternative solvents, and ACN it still the most efficient one.
The most accepted alternatives are formic and acetic acids, which are ranked in a relatively high position in terms of sustainability.99 The use of these chemicals is considered to be acceptable from the environmental point of view, and acetic acid is particularly advantageous since most drugs and peptide-based therapeutics are provided as acetate salts, therefore no additional steps of counterion exchange would be needed.225
Other alternatives that are increasingly used as additive in mobile phase are quaternary ammonium salts, usually with phosphate counterion. These ion pairing reagents are characterized by UV transparency, allowing to achieve high resolution, loading and recovery yields. In addition, they are compatible with further in vitro and in vivo analysis once organic solvent has been evaporated.226
More recently the University of Berlin in collaboration with Belyntic described a chemical approach useful to pre-purify crude peptides and simplify the RP-HPLC process, identified as the “catch&release” (c&r) method.227 Other methods have been reported in the literature,228 but this appears to be the most efficient one. This approach was typically connected with the introduction at each step of the SPPS process of a capping step. The capped fragments did not react at the N terminal with the base-labile cleavable linkers, introduced in the last coupling as a catch tag to be recognized by the oxime-based and hydrazone-based ligation chemistry (Fig. 21).
 | ||
| Fig. 21 Scheme of catch-and-release method. | ||
The TFA cleavage released the peptide from the resin, removing at the same time the BOC protecting group on the oxime. The peptide was then treated with aldehyde-modified agarose beads at pH 4.5, to generate the oxime, that was extensively washed to eliminate the capped peptide fragments. The final peptide was then typically released by treatment with ethanolamine. This pre-purification process was applied to several peptides, comprising liraglutide. The elimination of some impurities should, in principle, facilitate the final purification. However, it is difficult to understand the impact on the PMI. In fact, the critical impurities, that affect the loading or the number of sequential column purification required to get the correct quality, are mainly diastereoisomers close to the main peak.
4.2 Greener technologies
Besides the replacement of non eco-friendly chemicals with greener ones, the other possible choice to improve the sustainability of the downstream processing is to move towards greener purification techniques.In batch liquid chromatography, the sample injected is completely eluted before the next injection is performed, and between two consecutive injections the column is cleaned and regenerated. The amount of sample injected is chosen to overload the column until a compromise between purity, yield and productivity is achieved. An excessive overloading of the column leads to a worsening from the point of view of the resolution between the target peak and the close-eluting species, and therefore this behavior has a direct repercussion on the product's purity. Actually, in preparative single-column chromatography, it is not uncommon to run into a purity–yield trade-off: some impurities are chemically very similar to the target product and, in consequence, their chromatographic behavior is also analogous.229 This causes their resolution to be insufficient, their peaks’ baseline not being separated, and this effect worsens at increasing loadings. The front and/or the tail of the main peak, therefore, overlap with close-eluting impurities’ peaks, and for this reason they are usually not collected in the collecting pool, which must fulfill very strict purity requirements. Excluding the overlapping windows causes the recovery to decrease unavoidably. On the other side, widening the collecting window would be beneficial for the recovery, to the detriment of the purity. In this kind of situation, purity and yield cannot be both high at the same time, and this represents a limit intrinsic to single-column chromatography often referred to as “purity–yield trade-off” (see Fig. 22).215,229 To alleviate this trade-off, it is possible to decrease the sample volume loaded or to use less steep gradients, but this would come at the cost of lower productivity and higher solvent consumption.230 This is why the downstream processing represents the manufacturing bottleneck for many biopharmaceuticals.231 Continuous chromatographic techniques can help to overcome this issue thanks to the countercurrent movement of the stationary phase with respect to the mobile phase, as will be explained in the next section. These techniques lead to a reduction in the solvent usage. Moreover, the perspectives related to the use of Supercritical Fluid Chromatography (SFC), a technique barely explored for preparative purposes but with great potential, will be illustrated. In this case, the organic solvent is replaced with an eco-friendlier one, namely carbon dioxide.
 | ||
| Fig. 22 Purity-yield trade-off scheme. (A) Front and/or tail of the main peak are discarded, affecting the yield. (B) Front and/or tail of the main peak are included in the collected fractions, affecting the overall purity. Reproduced with permission from ref. 215. | ||
In the first case, the fractions containing impure product, overlapped with close-eluting impurities, are reinjected by the operator into the same column, either with or without addition of some fresh feed. If no fresh feed is added, this two-step batch process leads to very low productivity, since the same portion of feed is reprocessed twice. On the other hand, in closed-loop the SSR fresh sample is injected into the interior of the circulating chromatographic profile, at a specific moment. Since a single column is employed (see Fig. 23),232a the injection of the feed into the instrument is not performed continuously and, therefore, SSR cannot be considered a continuous process.
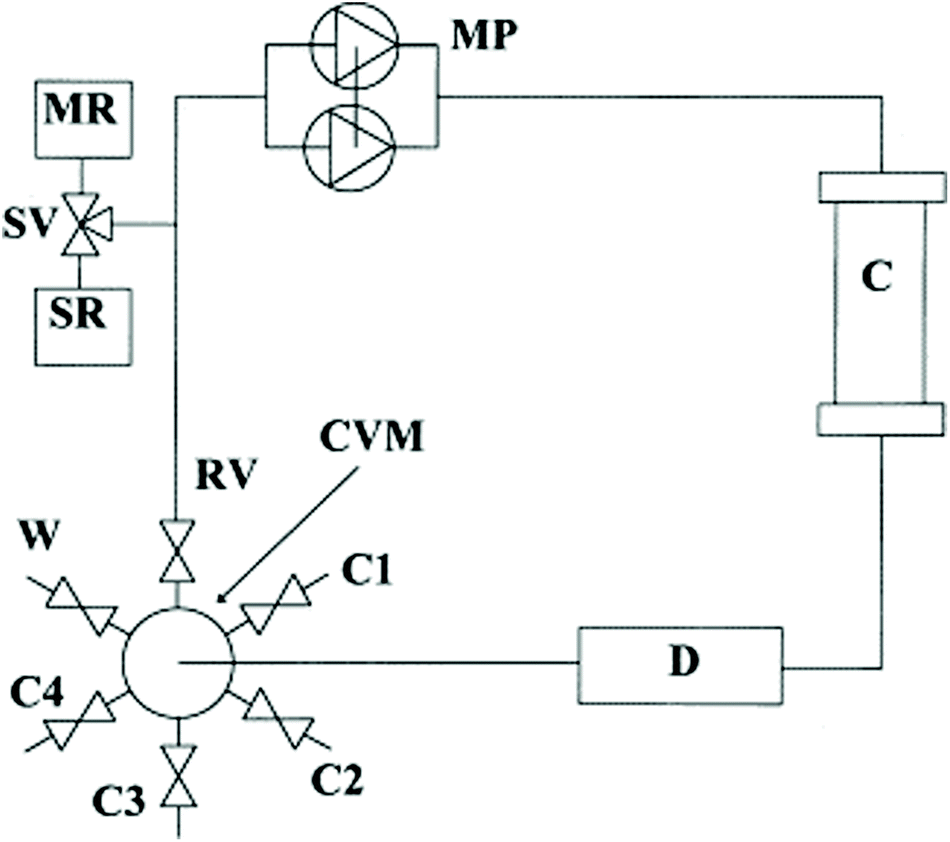 | ||
| Fig. 23 Schematic diagram of a closed-loop recycling system. Abbreviations: MP, mobile phase pump; C, column; D, detector; CVM, collection valve manifold; C1, C2, C3, C4, fraction collection valves; W, waste valve; RV, recycle valve; SR, sample reservoir; MR, mobile phase reservoir; SV, three-way selection valve. Reproduced with permission from ref. 232a. | ||
4-Zone Simulated Moving Bed (SMB), patented in early 1960s,233 follows the same concepts as SSR but works continuously, since four or more columns are used to form a circuit with two inlets and two outlets.217e,234 SMB is based on the principle of countercurrent chromatography, according to which the stationary phase moves virtually in the opposite direction with respect to the mobile phase.235 For technical reasons, the packed bed cannot be truly moved into the system or into the chromatographic column; in fact, a system of columns and valves simulates this movement. As a consequence, the faster eluting compound (more similar to the mobile phase) and the slower eluting compound (more similar to the stationary phase) can be separated in two streams, the raffinate and the extract, respectively (see Fig. 24).236 Clearly, this feature makes this technique particularly appealing for the purification of binary mixtures such as racemates. This chromatographic mode allows one to increase the stationary phase utilization and therefore achieves a much higher throughput than the batch and, furthermore, can save up to 90% of the mobile phase, which is a great result from the point of view of green chemistry, especially at commercial scale.237
 | ||
| Fig. 24 Schematic diagram of binary SMB system. Modified with permission from ref. 236b. | ||
The main disadvantage of SMB is the initial investment required, since the equipment is much more expensive than batch LC systems with similar production capacity.
At large commercial scale, the saving in solvent consumption and the increase in productivity obtained with SMB justify the significant initial cost, whereas at lab scale other solutions are often preferred. On the other hand, closed-loop SSR equipment is a preparative chromatograph for liquid chromatography, connected to a recycling valve (needed to recycle the unresolved portion) and an injection valve (to inject fresh feed at the suitable point in the profile).232a Therefore, the capital cost that must be faced for a SSR unit is not much higher than that for a traditional preparative system and is considerably lower with respect to SMB.
During the purification of industrially obtained complex peptide mixtures, the most typical situation is the so-called “center-cut (or ternary) separation”, where the target elutes as intermediate between two other groups of impurities.238 Also, SSR and SMB can only perform isocratic or step gradient elution, whereas to purify complex peptide mixtures generally a linear solvent gradient is required in order to modulate the separation, since the retention of biomolecules is greatly affected by the mobile phase composition.239
Continuous chromatography has been implemented to manage ternary separations, allowing remarkable reductions in solvent consumption during peptide purification.240 As for other chromatographic techniques based on the automatic recycling of impure fractions, and for the case of continuous ternary separations, the special feature is not the continuous nature but the countercurrent movement of the stationary phase with respect to the mobile phase.235 Two or more columns are connected in series and the positions of inlet and outlet streams are moved between the columns at precise time intervals. The switching of the valves allows one to collect suitably pure product windows, to recycle impure fractions and to discard impurities, as will be explained later.
In fact, over the years the 4-zone SMB process has been extended from binary to ternary separations, for example connecting two SMB units in series. This set-up is called “tandem SMB”. In this case, one of the streams eluting from the first SMB contains a single solute, while the other outlet stream contains two different species, that are separated into the second SMB unit. For example, this technology has been employed to separate insulin from protein aggregates and zinc chloride salt. Aggregates were removed into the first SMB unit, while insulin and salt were sent to the second SMB unit to be further separated.234,241
Using tandem SMB allowed an increase in productivity of more than 5 times and a reduction of solvent consumption of more than 3 times, while achieving high yield (99%) at steady-state. However, the compound of interest is just one, whereas the impurities to be removed are dozens, depending on the peptide size and on the number of steps performed in the upstream processing. For example, if the target peptide is produced through SPPS, the complexity of the synthesis increases with peptide length and the purity of the crude mixture decreases proportionally. Therefore, the techniques just described are not suitable for these purifications, where the use of a linear solvent gradient is of utmost necessity to increase the resolution.
To fill the gap between batch separations under solvent gradient conditions and continuous chromatography in the field of ternary mixtures, a process was developed in around 2006, called Multicolumn Countercurrent Solvent Gradient Purification (MCSGP).230,242 The working principle of MCSGP has been extensively described elsewhere.218,229,231a,243
During the purification in batch conditions, the overlap between the peptide of interest and other groups of impurities worsens, due to large injected volumes, slow mass transfer and low selectivity, from which the already mentioned yield–purity trade-off derives. The obtained batch chromatogram resembles the scheme depicted in Fig. 25: in zone 1 the feed is loaded, then the gradient starts and 4 elution zones can be identified (from 2 to 5) and, last, the column is stripped and equilibrated in zone 6. Zones 3 and 5 represent the overlapping regions that need to be recycled within the MCSGP process, whereas zone 4 is the collecting window, and in zones 2 and 6 the eluting impurities go to waste.243d The idea is to use a system of columns working either interconnected or disconnected, to recycle the impure product, collect the pure product and elute the product-free waste streams.230
 | ||
| Fig. 25 Schematic representation of a MCSGP in batch. Abbreviations: W = weak impurities, P = product, S = strong impurities. Reproduced with permission from ref. 243c. | ||
In MCSGP, from two up to six columns can be used. The version of MCSGP using six columns is the oldest and the most complex one from the point of view of the fluidics; in this case, the chromatographic process can be said to be continuous since the feed is continuously injected into the system, either in one column or in another.239c,244 In the case where the columns employed are only two, as in the most recent version of MCSGP, the system is much simpler and works cyclically and continuously.229,231b,245 The sample cannot be injected continuously into the unit since technical times are required in order to recover the purified product and discard the impurities window. Therefore, twin-column MCSGP is a semicontinuous process.
The possibility to internally recycle the impure sections of the chromatogram into the unit allows for three remarkable advantages. The first one is the automation of the whole process, which permits one to decrease the time necessary for the purification and to avoid potential errors caused by the operators.246 The second one is to overcome the yield–purity trade-off typical of the batch process, thanks to the fact that the mass of target peptide only leaves the unit when it has been purified. Finally, a net reduction in solvent consumption is achieved since the solvent eluting in the overlapping regions (and containing impure product) remains in circulation through the system.
MCSGP is a process particularly suitable for challenging purifications of several classes of biomolecule: peptides, proteins, monoclonal antibodies and, lately, also oligonucleotides and cannabinoids. In many cases, it gave successful results from the point of view of yield–purity trade-off, productivity and solvent consumption.
In the literature, some examples of MCSGP employed for peptide mixtures can be found. For instance, a comparison between batch and MCSGP processes for a peptide purification has been described.229 At a final purity of almost 99%, yield was improved by 4 times, a tenfold increase in productivity was achieved and solvent consumption decreased by 70%, from 3.5 to 1 L of solvent per gram of purified product using 3-column MCSGP. Another polypeptide, purified by MCSGP by means of reversed-phase chromatography, had an improvement of 25 times in productivity and a 60% reduction in solvent consumption.243b Recently, very good results were obtained in the purification of icatibant with MCSGP: at a purity greater than 99%, recovery and productivity increased by almost 7- and almost 6-fold, respectively, whereas the buffer consumption was reduced by more than 80% with respect to the corresponding batch set-up.231a
Ströhlein et al.247 demonstrated, through a process modeling formerly developed242c for a particularly challenging polypeptide purification conducted in reversed-phase conditions, that the batch process with no recycling led to a solvent consumption equal to 3000 L g−1 (because of the scarce yield), the batch process with ideal recycling247 to 80 L g−1 and the MCSGP to only 8 L g−1, which corresponds to an improvement of 10 times with respect to the typical process employed, corresponding to batch with recycling protocol.
The remarkable improvements obtained in productivity and solvent consumption lead undoubtedly to benefits both in the economics and in the greenness of the process. Of course, MCSGP processes are more challenging to design and operate, but Müller-Späth and Bavand248 evaluated that MCSGP would allow around 40% cost savings in the downstream processing compared to the batch chromatography scenarios, for a peptide production plant generating around 10 kg of product per year. Most of the costs in downstream processing performed in batch are indeed attributable to solvents and plant operating costs, followed by stationary phase costs and quality assurance/quality control costs. Equipment costs give the smallest contribution. This means that investing money once to implement purification systems for continuous chromatography can lead to great savings and to a decrease in the payback period, which is estimated to be around 6–18 months. Also, for the MCSGP case, stationary phase costs are negligible, since significantly smaller columns are used. Recently, the first industrial purification system based on MCSPG was installed by one of the leading companies in the peptide business, namely Bachem.249
Another advantage of preparative SFC over preparative LC is an improvement in productivity, thanks to higher used flow rates.253 In fact, supercritical fluids exhibit higher diffusivities and lower viscosities with respect to liquids, with very similar properties to gases. This translates in 3- to 4-fold higher velocity for maximum efficiency with respect to HPLC, with clear advantages in terms of time and productivity.250a
SFC is a separation technique orthogonal to RPLC, since the supercritical CO2 is rather apolar. Typical stationary phases applied in reverse and normal phase chromatography can be easily used under SFC conditions, making it a very flexible technique. The separation of a wide range of samples with different solubilities is then possible, from non-polar to polar ones. The separation and purification of highly polar compounds is also possible with the addition of water as an additive in the polar co-solvent of the mobile phase (usually 1 to 5%).254
SFC has been extensively employed in the past for the purification of chiral compounds, including chiral APIs in the pharmaceutical industry,253,255 but lately it has been used also in the field of peptides’ separations for analytical purposes, as proved by a rich literature.256 Stationary phases commonly employed are amine or cyanopropyl-bonded silica or copolymer of styrene and divinylbenzene.250a
On the other hand, very few studies report the feasibility of SFC large-scale purification of biomolecules like peptides and proteins, because this technique has started being studied only recently for this kind of preparative application.254 Schiavone et al.,257 for instance, managed to purify some peptides and proteins, among which were bradykinin, insulin, ubiquitin, cytochrome C and myoglobin, through preparative SFC using a mixture of methanol, ACN, water, TFA and CO2 as mobile phase. Recently, Ventura proposed a mobile phase made of methanol and two additives (TFA and ammonia) to separate crude synthetic therapeutic peptides using a crossed-link diol Luna® HILIC column, a stationary phase with unique selectivity properties. This separation was performed in analytical conditions but represents a good starting point for the scale-up to preparative conditions.258 Lately, Govender and coworkers demonstrated, with a proof-of-concept, that it is possible to purify, through semi-preparative SFC, samples of human insulin analogues, biosynthesized via recombinant DNA technology. Also, in that case, the organic modifier contained methanol with 5% water (added to improve peak shape) and 0.2% TFA. The biological activity of insulin was studied in vitro after the SFC purification, and it was assessed that it was not affected by the purification process.251a
On the other hand, the conformations of peptides and proteins purified and tested by Schiavone et al., except for insulin, were not preserved after the purification through SFC.257 According to Kasche et al.259 and Zagrobelny et al.,260 denaturation of some proteins (such as trypsin and α-chymotrypsin) occurs both during compression and depressurization steps. This could be a serious issue for proteins and long peptides employed as APIs, highlighting the need for a method to verify if the tridimensional structure of the compound has been irreversibly affected, and therefore if SFC is suitable for the purification under study.257
In addition, SFC also carries other drawbacks, such as the difficulty in the process scale-up from analytical to preparative conditions because of the compressibility of the mobile phase.253
In conclusion, even though the potential of SFC in the preparative purification of peptides and proteins is indisputable and promising, further knowledge of the fundamentals, such as adsorption properties and the physical–chemical behavior of supercritical fluids under nonlinear and gradient conditions, should be further explored. Therefore, supplementary developments in the applications are needed for SFC to overcome HPLC performance.261
5. Conclusions
Peptides’ synthesis represents one of the most serious challenges in green chemistry. During the last 5 years, several new protocols have been developed by academic and industrial laboratories in order to increase the sustainability of the chemical iterative synthesis of peptide sequences. This systematic investigation allowed optimization of several pieces of the puzzle following an approach that can be extended to other fields of organic chemistry to decrease their environmental impact.Greening SPPS was one of the main subjects and, from all the overviewed data, it clearly emerges that, for the Fmoc/tBu-based approach, there is not a single combination of solvent(s)/base able to replace the DMF/piperidine one. As a result, in agreement with Albericio's observation, the reported data suggest that the specific substrate dictates the protocol of choice. From an industrial stand point, the use of greener solvents’ mixtures is hampered by the increased solvent costs and supply chain complexity. In addition, regulatory agencies apply stricter regulations to peptides with respect to small molecules, in terms of process change requirements. For all these reasons, the DMF/piperidine system continues to be applied in SPPS for the synthesis of API peptides, limiting the space for the introduction of the reported greener alternatives.
Moving to LPPS, Tamiaki's 2001 publication opened the way to the development of PA-LPPS, but despite several anchors that have been introduced, only recently has the protocol reached a good level of maturity. The developed technologies are characterized by the use of different coupling reagents, Fmoc deprotection systems and procedures to eliminate side-products, ranging from precipitation of the anchored peptides to the use of membranes or water washings. Unfortunately, in order to protect their technology, in addition to the patent filing, most of the companies kept their own data as trade secrets, describing the peptide synthesis only in general terms. The direct consequence is that the available information is not sufficient to calculate the upstream PMI, needed to make an unequivocal assessment on the greenness of the process. In addition, even if the solubility in organic solvents is increased by the presence of lipophilic anchors, the technology appears to be currently able to deliver only short peptide sequences.
An important innovation for both SPPS and PA-LPPS is the use of in-line analysis, that can decrease the amount of solvents, chemicals and process time. In upstream technology the main target is, indeed, to generate crude peptide with the highest possible purity, to facilitate the chromatographic purification. On the other hand, the downstream target is to decrease the number of chromatographic steps, introduce automation and increase the downstream yield. The use of semi-continuous chromatography, that immediately recycles the mixed fractions in the following column, allows one to decrease the amount of ACN/water that are still the main used eluents. Moving to cyclic peptides, the use of enzyme-catalysed macrocyclizations appears to be the technique of choice.
Considering all the overviewed literature, our impression is that the integration between different synthesis technologies, like SPPS, PA-LPPS, enzymatic catalysis, microwave-assisted synthesis, and continuous protocols, with innovative semi-continuous chromatographic separations is the way forward to decrease the PMI of chemical peptide synthesis. The stairway to ideal green peptide synthesis has probably still to be completed, but matching all the knowledge achieved to date in different areas may bring research closer to the goal.
Author contributions
LF, WC, AT, have been responsible for conceptualization, supervision, writing, review and editing the paper. AC, MC, GM, DC, PC, AM, TF, SF, CDL have been responsible for data collection and writing of the original draft. The manuscript was written in collaboration with all authors, who have given approval for the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We kindly acknowledge Alma Mater Studiorum University of Bologna for financial support. Fondazione CarisBo is gratefully acknowledged for the funding of the project #18668 “Tecnologie Avanzate per il Controllo e lo Sviluppo di Molecole Innovative per la Salute”. Fondazione del Monte di Bologna e Ravenna is kindly acknowledged for financial support (Prot. N°702bis/2019).Notes and references
- Pollution Prevention Act 1990.42 U.S.C., sections 1310113109.
- S. G. Koenig, D. K. Leahy and A. S. Wells, Org. Process Res. Dev., 2018, 22, 1344 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice. Oxford University Press, New York, 1998 Search PubMed.
- EPA-United States Environmental Protection Agency https://www.epa.gov/greenchemistry/information-about-green-chemistry-challenge.
- D. J. Constable, ACS Sustainable Chem. Eng., 2020, 8, 14675 Search PubMed.
- (a) K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31 RSC; (b) V. Martin, P. H. G. Egelund, H. Johansson, S. Thordal Le Quement, F. Wojcik and D. S. Pedersen, RSC Adv., 2020, 10, 42457 RSC.
- S. G. Koenig, C. Bee, A. Borovika, C. Briddell, J. Colberg, G. R. Humphrey, M. E. Kopach, I. Martinez, S. Nambiar, S. V. Plummer, S. D. Ribe, F. Roschangar, J. P. Scott and H. F. Sneddon, ACS Sustainable Chem. Eng., 2019, 7, 16937 CrossRef CAS.
- M. G. Braun, A. Diaz-Rodriguez, L. Diorazio, Z. Fei, K. Fraunhoffer, J. Hayler, M. Hickey, M. McLaws, P. Richardson, G. D. Roiban, A. T. Parsons, A. Steven, J. Terrett, T. White and J. Yin, Org. Process Res. Dev., 2019, 23, 1118 CrossRef CAS.
- (a) C. Jiménez-González, P. Poechlauer, Q. B. Broxterman, B. S. Yang, D. Am Ende, J. Baird, C. Bertsch, R. E. Hannah, P. Dell'Orco, H. Noorman, S. Yee, R. Reintjens, A. Wells, V. Massonneau and J. Manley, Org. Process Res. Dev., 2011, 15, 900 CrossRef; (b) K. Budzinski, M. Blewis, P. Dahlin, D. D'Aquila, J. Esparza, J. Gavin, S. V. Ho, C. Hutchens, D. Kahn, S. G. Koenig, R. Kottmeier, J. Millard, M. Snyder, B. Standard and L. Sun, New Biotechnol., 2019, 49, 37 CrossRef CAS PubMed; (c) A. Borovika, J. Albrecht, J. Li, A. S. Wells, C. Briddell, B. R. Dillon, L. J. Diorazio, J. R. Gage, F. Gallou, S. G. Koenig, M. E. Kopach, D. K. Leahy, I. Martinez, M. Olbrich, J. L. Piper, F. Roschangar, E. C. Sherer and M. D. Eastgate, Nat. Sustain., 2019, 2, 1034 CrossRef.
- (a) A. Tolomelli, L. Gentilucci, E. Mosconi, A. Viola, S. D. Dattoli, M. Baiula, S. Spampinato, L. Belvisi and M. Civera, ChemMedChem, 2011, 6, 2264 CrossRef CAS PubMed; (b) A. Tolomelli, M. Baiula, L. Belvisi, A. Viola, L. Gentilucci, S. Troisi, S. D. Dattoli, S. Spampinato, M. Civera, E. Juaristi and M. Escudero, Eur. J. Med. Chem., 2013, 66, 258 CrossRef CAS PubMed; (c) A. Tolomelli, M. Baiula, A. Viola, L. Ferrazzano, L. Gentilucci, S. D. Dattoli, S. Spampinato, E. Juaristi and M. Escudero, ACS Med. Chem. Lett., 2015, 6, 701 CrossRef CAS PubMed; (d) L. Ferrazzano, D. Corbisiero, E. Potenza, M. Baiula, S. D. Dattoli, S. Spampinato, L. Belvisi, M. Civera and A. Tolomelli, Sci. Rep., 2020, 10, 7410 CrossRef CAS PubMed.
- J. L. Lau and M. K. Dunn, Bioorg. Med. Chem., 2017, 26, 2700 CrossRef PubMed.
- https://www.fda.gov/drugs/development-approval-process-drugs/new-drugs-fda-cders-new-molecular-entities-and-new-therapeutic-biological-products (accessed on November 10th, 2021).
- (a) M. M. Goldenberg, Pharmaceutical approval update, P T, 2010, 35, 216 Search PubMed; (b) K. L. Bjerre and L. Jasper, Front. Endocrinol., 2019, 10, 155 CrossRef PubMed.
- Mordor Intelligence. Peptide Therapeutics Market - Growth, Trends, Covid-19 Impact, and Forecasts (2021–2026). https://www.mordorintelligence.com/industry-reports/peptide-therapeutics-market (accessed on November 10th, 2021).
- L. Wu, Peptide Therapeutics: Strategy and Tactics for Chemistry, Manufacturing, and Controls, ed. V. Srivastava, RSC publishing, Cambridge, 2013, p. 1 Search PubMed.
- Y. Gao, X. Yuan, Z. Zhu, D. Wang, Q. Liu and W. Gu, Exp. Ther. Med., 2020, 20, 234 CAS.
- M. Muttenthaler, G. F. King, D. J. Adams and P. F. Alewood, Nat. Rev. Drug Discovery, 2021, 20, 309 CrossRef CAS PubMed.
- (a) E. Valeur, S. M. Guért, H. Adihou, R. Gopalakrishnan, M. Lemurell, H. Waldmann, T. N. Grossmann and A. T. Plowright, Angew. Chem., Int. Ed., 2017, 56, 10294 CrossRef CAS PubMed; (b) A. M. Vargason, A. C. Anselmo and S. Mitragotri, Nat. Biomed. Eng., 2021, 5, 951 CrossRef PubMed.
- V. du Vigneaud, C. Ressler, J. M. Swan, C. W. Roberts and P. G. Katsoyannis, J. Am. Chem. Soc., 1954, 76, 3115 CrossRef CAS.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- J. Henninot, J. C. Collins and J. M. Nuss, J. Med. Chem., 2018, 61, 1382 CrossRef PubMed.
- (a) W. Cabri, P. Cantelmi, D. Corbisiero, T. Fantoni, L. Ferrazzano, G. Martelli, A. Mattellone and A. Tolomelli, Front. Mol. Biosci., 2021, 8, 697586 CrossRef CAS PubMed; (b) A. C. L. Lee, J. L. Harris, K. K. Khanna and J. H. Hong, Int. J. Mol. Sci., 2019, 20, 2383 CrossRef PubMed.
- Y. Huan, Q. Kong, H. Mou and H. Yi, Front. Microbiol., 2020, 11, 582559 Search PubMed.
- V. Khavinson, N. Linkova, A. Dyatlova, B. Kuznik and R. Umnov, Molecules, 2020, 25, 4389 CrossRef CAS PubMed; K. S. Bhullar, S. J. Drews and J. Wu, Eur. J. Pharmacol., 2021, 890, 173661 CrossRef PubMed.
- B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS PubMed.
- P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40 CrossRef CAS PubMed.
- T. Gain and P. S. Baran, J. Org. Chem., 2010, 75, 4657 CrossRef PubMed.
- (a) R. A. Sheldon, Green Chem., 2017, 19, 18 RSC; (b) F. Roschangar, A. Sheldon and C. H. Senanayake, Green Chem., 2015, 17, 752 RSC.
- (a) G. Martelli, P. Cantelmi, A. Tolomelli, D. Corbisiero, A. Mattellone, A. Ricci, T. Fantoni, W. Cabri, F. Vacondio, F. Ferlenghi, M. Mor and L. Ferrazzano, Green Chem., 2021, 23, 4095 RSC; (b) G. Martelli, P. Cantelmi, C. Palladino, A. Mattellone, D. Corbisiero, T. Fantoni, A. Tolomelli, M. Macis, A. Ricci, W. Cabri and L. Ferrazzano, Green Chem., 2021, 23, 8096 RSC; (c) J. Pawlas and J. H. Rasmussen, Green Chem., 2019, 21, 5990 RSC; (d) J. Yeo, L. Peeva, S. Chung, P. Gaffney, D. Kim, C. Luciani, S. Tsukanov, K. Seibert, M. Kopach, F. Albericio and A. Livingston, Angew. Chem., Int. Ed., 2021, 60, 7786 CrossRef CAS PubMed; (e) O. Maurin, P. Verdié, G. Subra, F. Lamaty, J. Martinez and T. X. Métro, Beilstein J. Org. Chem., 2017, 13, 2087 CrossRef CAS PubMed.
- R. Gani, Comput. Chem. Eng., 2004, 28, 2441 CrossRef CAS.
- D. K. Babi, K. Kulajanpeng, A. Tongrod, A. Kammafoo, K. Lourvanij and R. Gani, A Systematic Approach to Green Solvent Selection, Design, and Verification. The Application of Green Solvents in Separation Processes, Elsevier, 2017, p. 57 Search PubMed.
- Roots Analysis research report. Peptide Therapeutics: contract API manufacturing market, 2020–2030.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. R. McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 7 CrossRef.
- L. Cseri, M. Razali, P. Pogany and G. Szekely, Organic Solvents in Sustainable Synthesis and Engineering. Green Chemistry - An inclusive approach, Elsevier, 2018, 513 Search PubMed.
- (a) R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binksa and A. D. Curzons, Green Chem., 2011, 13, 854 RSC; (b) C. M. Alder, D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879 RSC; (c) C. Jimenéz-Gonzàlez, A. D. Curzons, D. J. C. Constable and V. L. Cunningham, Clean Technol. Environ. Policy, 2004, 7, 42 CrossRef.
- D. Prat, O. Pardigon, H. W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517 CrossRef CAS.
- (a) L. J. Diorazio, D. R. J. Hose and N. K. Adlington, Org. Process Res. Dev., 2016, 20, 760 CrossRef CAS; (b) D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. A. Shehada and P. J. Dunn, Green Chem., 2016, 18, 288 RSC.
- M. Tobiszewski, S. Tsakovski, V. Simeonov, J. Namiesnik and F. Pena-Pereira, Green Chem., 2015, 17, 4773 RSC.
- (a) J. Sherwood, M. De bruyn, A. Constantinou, L. Moity, C. R. McElroy, T. J. Farmer, T. Duncan, W. Raverty, A. J. Hunt and J. H. Clark, Chem. Commun., 2014, 50, 9650 RSC; (b) H. J. Salavagione, J. Sherwood, M. De bruyn, V. L. Budarin, G. J. Ellis, J. H. Clark and P. S. Shuttleworth, Green Chem., 2017, 19, 2550 RSC.
- (a) F. M. Kerton and R. Marriott, Alternative solvents for green chemistry, RSC publishing, Cambridge, 2nd edn, 2013 Search PubMed; (b) C. Reichardt and T. Welton, Solvents and solvent effects in organic chemistry, Wiley-VCH, Weinheim, 4th edn, 2011 Search PubMed; (c) A. S. Matlack, Introduction to green chemistry, Marcel Dekker, New York, 2001, p. 201 Search PubMed.
- (a) Y. Yang, Side reactions in peptide synthesis, Tsinghua University Press Limited, Published by Elsevier Inc, 2016 Search PubMed; (b) O. Al Musaimi, B. G. de la Torre and F. Albericio, Green Chem., 2020, 22, 996 RSC.
- J. M. Palomo, RSC Adv., 2014, 4, 32658 RSC.
- R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC.
- K. Hojo, A. Hara and H. Kitai, et al. , Chem. Cent. J., 2011, 5, 49 CrossRef CAS PubMed.
- V. Martin, S. Jadhav, P. H. G. Egelund, R. Liffert, H. J. Castro, T. Krüger, K. F. Haselmann, S. Thordal Le Quement, F. Albericio, F. Dettner, C. Lechner, R. Schönleber and D. S. Pedersen, Green Chem., 2021, 23, 3295 RSC.
- Data for each solvent retrieved at https://echa.europa.eu/it/candidate-list-table (accessed on November 10th, 2021).
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082 RSC.
- J. M. Collins, Microwaves in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 3rd edn, 2012 Search PubMed.
- http://www.cem.com (accessed on November 10th, 2021).
- http://www.csbio.com (accessed on November 10th, 2021).
- G. Sabatino, A. D'Ercole, L. Pacini, M. Zini, A. Ribecai, A. Paio, P. Rovero and A. M. Papini, Org. Process Res. Dev., 2021, 25, 552 CrossRef CAS.
- https://www.ambiopharm.com/news-press/cem-corporation-and-ambiopharm-form-u-s-partnership-for-gmp-peptide-production/ (accessed on November 10th, 2021).
- A. Kumar, Y. E. Jad, J. M. Collins, F. Albericio and B. G. de la Torre, ACS Sustainable Chem. Eng., 2018, 6, 8034 CrossRef CAS.
- A. Kumar, Y. E. Jad, A. El-Faham, B. G. de la Torre and F. Albericio, Tetrahedron Lett., 2017, 58, 2986 CrossRef CAS.
- G. A. Acosta, M. del Fresno, M. Paradis-Bas, M. Rigau-DeLlobet, S. Cote, M. Royo and F. Albericio, J. Pept. Sci., 2009, 15, 629 CrossRef CAS PubMed.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2015, 13, 2393 RSC.
- V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369 CrossRef CAS PubMed.
- Y. E. Jad, G. A. Acosta, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Amino Acids, 2016, 48, 419 CrossRef CAS PubMed.
- Y. E. Jad, G. A. Acosta, T. Govender, H. G. Kruger, A. El-Faham, B. G. de la Torre and F. Albericio, ACS Sustainable Chem. Eng., 2016, 4, 6809 CrossRef CAS.
- T. Govender, H. G. Kruger, A. El-Faham, B. de la Torre and F. Albericio, Org. Proc. Res. Dev., 2017, 21, 365 CrossRef.
- (a) A. Kumar, A. Sharma, B. G. de la Torre and F. Albericio, Molecules, 2019, 24, 4004 CrossRef CAS PubMed; (b) A. Kumar, A. Niyi, K. P. Nandhini, J. M. Collins, F. Albericio and B. G. de la Torre, Org. Process Res. Dev., 2019, 23, 1096 CrossRef CAS.
- https://www.solvay.com/en/brands/rhodiasolv-polarclean (accessed on November 10th, 2021).
- A. Kumar, A. Sharma, B. G. de la Torre and F. Albericio, Green Chem. Lett. Rev., 2021, 14, 544 Search PubMed.
- J. Pawlas and J. H. Rasmussen, ChemSusChem, 2021, 14, 3231 CrossRef CAS PubMed.
- S. Lawrenson, M. North, F. Peigneguy and A. Routledge, Green Chem., 2017, 19, 952 RSC.
- S. B. Lawrenson, R. Arav and M. North, Green Chem., 2017, 19, 1685 RSC.
- J. Lopez, S. Pletscher, A. Aemissegger, C. Bucher and F. Gallou, Org. Process Res. Dev., 2018, 22, 494 CrossRef CAS.
- (a) B. G. de la Torre, A. Kumar, M. Alhassan, C. Bucher, F. Albericio and J. Lopez, Green Chem., 2020, 22, 3162 RSC; (b) A. Kumar, M. Alhassan, J. Lopez, F. Albericio and B. G. de la Torre, ChemSusChem, 2020, 13, 5288 CrossRef CAS PubMed.
- (a) L. Ferrazzano, G. Martelli, T. Fantoni, A. Daka, D. Corbisiero, A. Viola, A. Ricci, W. Cabri and A. Tolomelli, Org. Lett., 2020, 22, 3969 CrossRef CAS PubMed; (b) T. Fantoni, S. Bernardoni, Al. Mattellone, G. Martelli, L. Ferrazzano, P. Cantelmi, D. Corbisiero, A. Tolomelli, W. Cabri, F. Vacondio, F. Ferlenghi, M. Mor and A. Ricci, ChemSusChem, 2021, 14, 2591 CrossRef CAS PubMed.
- P. Palladino and D. A. Stetsenko, Org. Lett., 2012, 14, 6346 CrossRef CAS PubMed.
- M. Alhassan, O. Al Musaimi, J. M. Collins, F. Albericio and B. G. de la Torre, Green Chem., 2020, 22, 2840 RSC.
- J. Pawlas, T. Svensson and J. H. Rasmussen, RSC Adv., 2019, 9, 38928 RSC.
- B. G. de La Torre and D. Andreu, J. Pept. Sci., 2008, 14, 360 CrossRef CAS PubMed.
- O. Al Musaimi, Y. E. Jad, A. Kumar, J. M. Collins, A. Basso, B. G. de la Torre and F. Albericio, Curr. Opin. Green Sustain. Chem., 2018, 11, 99 CrossRef.
- O. Al Musaimi, Y. E. Jad, A. Kumar, A. El-Faham, J. M. Collins, A. Basso, B. G. de la Torre and F. Albericio, Org. Process Res. Dev., 2018, 22, 1809 CrossRef CAS.
- K. Watanabe, N. Yamagiwa and Y. Torisawa, Org. Process Res. Dev., 2007, 11, 251 CrossRef CAS.
- J. Pawlas, B. Antonic, M. Lundqvist, T. Svensson, J. Finnman and J. H. Rasmussen, Green Chem., 2019, 21, 2594 RSC.
- Y. Ran, F. Byrne, I. D. V. Ingram and M. North, Chem. – Eur. J., 2019, 25, 4951 CrossRef CAS PubMed.
- L. Ferrazzano, D. Corbisiero, G. Martelli, A. Tolomelli, A. Viola, A. Ricci and W. Cabri, ACS Sustainable Chem. Eng., 2019, 7, 12867 CrossRef CAS.
- Y. Marcus, Solvent mixtures-Properties and selective solvation, Marcel Dekker, New York/Besel, 2002 Search PubMed.
- (a) C. Amadi-Kamalu, H. Clarke, M. McRobie, J. Mortimer, M. North, Y. Ran, A. Routledge, D. Sibbald, M. Tickias, K. Tse and H. Willway, ChemistryOpen, 2020, 9, 431 CrossRef CAS PubMed; (b) Data available at https://www.webofscience.com/wos/woscc/analyze-results/5a129d4c-2318-4366-9f89-f39db566beac-0d2e7594 using as key words “resin swelling green solvent” (accessed on November 10th, 2021).
- J. K. Magtaan, M. Devocelle and F. Kelleher, J. Pept. Sci., 2020, 26, e3250 CrossRef CAS PubMed.
- A. Randová, L. Bartovská, P. Morávek, P. Matějka, M. Novotná, S. Matějková, E. Drioli, A. Figoli, M. Lanč and K. Friess, J. Mol. Liq., 2016, 224, 1163 CrossRef.
- S. Knauer, N. Koch, C. Uth, R. Meusinger, O. Avrutina and H. Kolmar, Angew. Chem., Int. Ed., 2020, 59, 12984 CrossRef CAS PubMed.
- S. Jadhav, V. Martin, P. H. G. Egelund, H. Johansson Castro, T. Krüger, F. Richner, S. Thordal Le Quement, F. Albericio, F. Dettner, C. Lechner, R. Schönleber and D. S. Pedersen, Green Chem., 2021, 23, 3312 RSC.
- O. Al Musaimi, D. Al Shaer, B. G. de la Torre and F. Albericio, Chem. Today, 2021, 39, 18 Search PubMed.
- K. Kümmerer, J. H. Clark and V. G. Zuin, Science, 2020, 367, 369 CrossRef PubMed.
- B. G. de la Torre, S. Ramkisson, F. Albericio and J. Lopez, Org. Process Res. Dev., 2021, 25, 1047 CrossRef.
- O. Ludemann-Hombourger, I. Martinuzzi, C. Bobier and E. Francomme, Patent applicationWO2019/211531A1, 2019 Search PubMed.
- F. Guzmán, A. Gauna, O. Luna, T. Román, C. Álvarez, C. Pareja-Barrueto and L. Mercado, Molecules, 2021, 26, 5035 CrossRef PubMed.
- L. A. Carpino and G. Y. Han, J. Am. Chem. Soc., 1970, 92, 5748 CrossRef CAS.
- E. Atherton, H. Fox, D. Harkiss, C. J. Logan, R. C. Sheppard and B. J. Williams, J. Chem. Soc., Chem. Commun., 1978, 537 RSC.
- C.-D. Chang and J. Meienhofer, Int. J. Pept. Protein Res., 1978, 11, 246 CrossRef CAS PubMed.
- (a) G. B. Fields, Methods in Molecular Biology, in Peptide Synthesis Protocol, 1995, vol. 35, p. 17 Search PubMed; (b) W. Li, N. M. O'Brien-Simpson, M. A. Hossain and J. D. Wade, Aust. J. Chem., 2020, 73, 271 CrossRef CAS; (c) A. El-Faham and F. Albericio, Pept. Sci., 2020, 112, e24164 CAS.
- L. A. Carpino, Acc. Chem. Res., 1987, 20, 401 CrossRef CAS.
- M. S. Sonders, J. F. W. Keana and E. Weber, Trends Neurosci., 1988, 11, 37 CrossRef CAS.
- Red List of the International Narcotics Control Board available at https://www.incb.org/incb/en/precursors/Red_Forms/red-list.html, 18th edn, 2021 (accessed on November 10th, 2021).
- R. K. Henderson, A. P. Hill, A. M. Redman and H. F. Sneddon, Green Chem., 2015, 17, 945 RSC.
- J. D. Wade, M. N. Mathieu, M. Macris and G. W. Tregear, Lett. Pept. Sci., 2000, 7, 107 CrossRef CAS.
- O. Seitz, F. Bergmann and D. Heindl, Angew. Chem., Int. Ed., 1999, 38, 2203 CrossRef CAS PubMed.
- J. Hachmann and M. Lebl, J. Comb. Chem., 2006, 8, 149 CrossRef CAS PubMed.
- P. H. G. Egelund, S. Jadhav, V. Martin, H. Johansson Castro, F. Richner, S. T. Le Quement, F. Dettner, C. Lechner, R. Schoenleber and D. S. Pedersen, ACS Sustainable Chem. Eng., 2021, 9, 14202 CrossRef CAS.
- (a) A. K. Tickler, C. J. Barrow and J. D. Wade, J. Pept. Sci., 2001, 7, 488 CrossRef CAS PubMed; (b) J. D. Wade, J. Bedford, R. C. Sheppard and G. W. Tregear, Pept. Res., 1991, 4, 194 CAS.
- (a) I. Guryanov, A. Orlandin, A. Viola, B. Biondi, D. Badocco, F. Formaggio, A. Ricci and W. Cabri, Org. Process Res. Dev., 2019, 23, 2746 CrossRef CAS; (b) I. Guryanov, A. Orlandin, A. Viola, B. Biondi, F. Formaggio, A. Ricci and W. Cabri, Org. Process Res. Dev., 2020, 24, 274 CrossRef CAS.
- S. A. Kates, N. A. Sole, M. Beyermann, G. Barany and F. Albericio, Pept. Res., 1996, 9, 106 CAS.
- K. Ralhan, V. G. KrishnaKumar and S. Gupta, RSC Adv., 2015, 5, 104417 RSC.
- J. E. Sheppeck, H. Kar and H. Hong, Tetrahedron Lett., 2000, 41, 5329 CrossRef CAS.
- (a) C. F. Vergel Galeano, Z. J. Rivera Monroy, J. E. Rosas Pérez and J. E. Garcia Castañeda, J. Mex. Chem. Soc., 2014, 58, 386 Search PubMed; (b) O. F. Luna, J. Gomez, C. Cárdenas, F. Albericio, S. H. Marshall and F. Guzmán, Molecules, 2016, 21, 1542 CrossRef PubMed; (c) V. Rodríguez, H. Pineda, N. Ardila, D. Insuasty, K. Cárdenas, J. Román, M. Urrea, D. Ramírez, R. Fierro, Z. Rivera and J. García, Int. J. Pept. Res. Ther., 2020, 26, 585 CrossRef.
- (a) A. Přibylka, V. Krchňák and E. Schütznerová, Green Chem., 2019, 21, 775 RSC; (b) A. Přibylka, V. Krchňák and E. Schütznerová, J. Org. Chem., 2020, 85, 8798 CrossRef PubMed; (c) A. Přibylka, M. Pastorek, M. Grepl and E. Schütznerová, Tetrahedron, 2021, 99, 132452 CrossRef.
- The European Chemicals Agency (ECHA) Brief Profile for 4-methylpiperidine: https://echa.europa.eu/it/brief-profile/-/briefprofile/100.009.959 (accessed on November 10th, 2021).
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615 CrossRef CAS PubMed.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed.
- (a) S. R. Manne, B. G. de la Torre, A. El-Faham and F. Albericio, Synthesis, 2020, 3189 CAS; (b) R. Subirós-Funosas, S. N. Khattab, L. Nieto-Rodríguez, A. El-Faham and F. Albericio, Aldrichimica Acta, 2013, 46, 21 Search PubMed; (c) F. Albericio and A. El-Faham, Org. Process Res. Dev., 2018, 22, 760 CrossRef CAS.
- (a) W. König and R. Geiger, Chem. Ber., 1970, 103, 788 CrossRef PubMed; (b) L. A. Carpino, J. Am. Chem. Soc., 1993, 115, 4397 CrossRef CAS; (c) A. El-Faham and F. Albericio, Org. Lett., 2007, 9, 4475 CrossRef CAS PubMed.
- K. J. McKnelly, W. Sokol and J. S. J. Nowick, J. Org. Chem., 2020, 85, 1764 CrossRef CAS PubMed.
- (a) R. Knorr, A. Trzeciak, W. Bannwarth and D. Gillessen, Tetrahedron Lett., 1989, 30, 1927 CrossRef CAS; (b) V. Dourtoglou, J.-C. Ziegler and B. Gross, Tetrahedron Lett., 1978, 19, 1269 CrossRef; (c) L. A. Carpino, A. El-Faham and F. Albericio, Tetrahedron Lett., 1994, 35, 2279 CrossRef CAS; (d) L. A. Carpino, A. El-Faham, C. A. Minor and F. Albericio, J. Chem. Soc., Chem. Commun., 1994, 201 RSC; (e) O. Marder, Y. Shvo and F. Albericio, Chimica Oggi, 2002, 20, 37 CAS; (f) J. Coste, D. Le-Nguyen, G. Evin and B. Castro, Tetrahedron Lett., 1990, 31, 205 CrossRef CAS; (g) R. Subiròs-Funosas, J. A. Moreno, N. Bayo-Puxan, K. Abu-Rabeah, A. Ewenson, D. Atias, R. S. Marks and F. Albericio, Chimica Oggi, 2008, 26, 10 Search PubMed; (h) F. Albericio, M. Cases, J. Alsina, S. A. Triolo, L. A. Carpino and S. A. Kates, Tetrahedron Lett., 1997, 38, 4853 CrossRef CAS; (i) L. A. Carpino, A. El-Faham, C. A. Minor and F. Albericio, J. Chem. Soc., Chem. Commun., 1994, 201 RSC.
- A. El-Faham and F. Albericio, Eur. J. Org. Chem., 2009, 1499 CrossRef CAS.
- R. Subiròs-Funosas, R. Prohens, R. Barbas, A. El-Faham and F. Albericio, Chem. – Eur. J., 2009, 15, 9394 CrossRef PubMed.
- J. M. Collins, S. K. Singh and G. S. Vanier, Chem. Today, 2012, 30, 2 Search PubMed.
- P. Cherkupally, G. A. Acosta, L. Nieto-Rodriguez, J. Spengler, H. Rodriguez, S. N. Khattab, A. El-Faham, M. Shamis, Y. Luxembourg, R. Prohens, R. Subiros-Funosas and F. Albericio, Eur. J. Org. Chem., 2013, 6372 CrossRef CAS.
- (a) A. El-Faham, R. Subiròs-Funosas, R. Prohens and F. Albericio, Chem. – Eur. J., 2009, 15, 9404 CrossRef CAS PubMed; (b) A. El-Faham and F. Albericio, J. Pept. Sci., 2010, 16, 6 CrossRef CAS PubMed.
- R. Subirós-Funosas, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2010, 8, 3665 RSC.
- Y. E. Jad, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Org. Biomol. Chem., 2014, 12, 8379 RSC.
- Y. E. Jad, S. N. Khattab, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Molecules, 2014, 19, 18953 CrossRef PubMed.
- Y. E. Jad, B. G. de la Torre, T. Govender, H. G. Kruger, A. El-Faham and F. Albericio, Tetrahedron Lett., 2016, 57, 3523 CrossRef CAS.
- D. S. MacMillan, J. Murray, H. F. Sneddon, C. Jamieson and A. J. B. Watson, Green Chem., 2013, 15, 596 RSC.
- R. Subirós-Funosas, L. Nieto-Rodriguez, K. J. Jensen and F. Albericio, J. Pept. Sci., 2013, 19, 408 CrossRef PubMed.
- A. Kumar, Y. E. Jad, B. G. de la Torre, A. El-Faham and F. Albericio, J. Pept. Sci., 2017, 23, 763 CrossRef CAS PubMed.
- A. D. McFarland, J. Y. Buser, M. C. Embry, C. B. Held and S. P. Kolis, Org. Process Res. Dev., 2019, 23, 2099 CrossRef CAS.
- S. R. Manne, A. El-Faham, B. G. de la Torre and F. Albericio, Tetrahedron, 2021, 52, 3189 Search PubMed.
- M. Erny, M. Lundqvist, J. H. Rasmussen, O. Ludemann-Hombourger, F. Bihel and J. Pawlas, Org. Process Res. Dev., 2020, 24, 1341 CrossRef CAS.
- G. A. Rockwood, D. E. Thompson and I. Petrikovics, Toxicol. Ind. Health, 2016, 32, 2009 CrossRef CAS PubMed.
- S. R. Manne, O. Luna, G. A. Acosta, M. Royo, A. El-Faham, G. Orosz, B. G. de la Torre and F. Albericio, Org. Lett., 2021, 23, 6900 CrossRef CAS PubMed.
- (a) R. Dunetz, Y. Xiang, A. Baldwin and J. Ringling, Org. Lett., 2011, 13, 5048 CrossRef PubMed; (b) S. Zhang, L. M. De Leon Rodriguez, E. Lacey, A. M. Piggott, I. K. H. Leung and M. A. Brimble, Eur. J. Org. Chem., 2017, 149 CrossRef; (c) E. K. Davison, A. J. Cameron, P. W. R. Harris and M. A. Brimble, MedChemComm, 2019, 10, 693 RSC.
- O. Al Musaimi, R. Wisdom, P. Talbiersky, B. G. de la Torre and F. Albericio, ChemistrySelect, 2021, 6, 2649 CrossRef CAS.
- J. B. Sperry, J. B. Minteer, J. Tao, R. Johnson, R. Duzguner, M. Hawksworth, S. Oke, P. F. Richardson, R. Barnhart, D. R. Bill, R. A. Giusto and J. D. Weaver, Org. Process Res. Dev., 2018, 22, 1262 CrossRef CAS.
- (a) Handoko, S. Satishkumar, N. R. Panigrahi and P. S. Arora, J. Am. Chem. Soc., 2019, 141, 15977 CrossRef CAS PubMed; (b) H. Bukya, K. Nayani, P. Gangireddy and P. S. Mainkar, Eur. J. Org. Chem., 2020, 5358 CrossRef CAS.
- A. K. Mishra, S. K. Santra, A. Bazylevich, O. Dorfman, J. Rahamim and A. M. Szpilman, Angew. Chem., Int. Ed., 2021, 60, 2 CrossRef.
- J. Li, Y. Zhu, B. Liu, F. Tang, X. Zheng and W. Huang, Org. Lett., 2021, 23, 7571 CrossRef CAS PubMed.
- (a) A. A. Zompra, A. S. Galanis, O. Werbitzky and F. Albericio, Future Med. Chem., 2009, 1, 361 CrossRef CAS PubMed; (b) C. Meneses, S. L. Nicoll and L. Trembleau, J. Org. Chem., 2010, 75, 564 CrossRef CAS PubMed.
- (a) C. M. Gabriel, M. Keener, F. Gallou and B. H. Lipshutz, Org. Lett., 2015, 17, 3968 CrossRef CAS PubMed; (b) M. Cortes-Clerget, J.-Y. Berthon, I. Krolikiewicz-Renimel, L. Chaisemartin and B. H. Liphutz, Green Chem., 2017, 19, 2463 RSC; (c) M. Cortes-Clerget, S. E. Spink, G. P. Gallagher, L. Chaisemartin, E. Filaire, J.-Y. Berthon and B. H. Liphutz, Green Chem., 2019, 21, 2610 RSC.
- T. Bruckdorfer, O. Marder and F. Albericio, Curr. Pharm. Biotechnol., 2004, 5, 29 CAS.
- (a) PEG: V. N. R. Pillai, M. Mutter, E. Bayer and I. Gatfield, J. Org. Chem., 1980, 45, 5364 CrossRef CAS; (b) Soluble Styrene: M. Narita, Bull. Chem. Soc. Jpn., 1978, 51, 1477 CrossRef CAS; (c) Fluorinated compunds: M. Mizuno, K. Goto, T. Miura, D. Hosaka and T. Inazu, Chem. Commun., 2003, 972 RSC; (d) Aromatic phenol derivatives: H. Tamiaki, T. Obata, Y. Azefu and K. Toma, Bull. Chem. Soc. Jpn., 2001, 74, 733 CrossRef CAS.
- (a) K. Chiba, S. Kim and Y. Kono, US Patent8633298B2, 2014 Search PubMed; (b) K. Chiba, S. Kim and Y. Kono, US Patent8293948B2, 2012 Search PubMed.
- (a) D. Takahashi, US Patent9353148B2, 2016 Search PubMed; (b) D. Takahashi, US Patent9334302B2, 2016 Search PubMed; (c) D. Takahashi, US Patent2016060198A1, 2016 Search PubMed; (d) D. Takahashi, US Patent9206230B2, 2015 Search PubMed; (e) D. Takahashi, US Patent9029504B2, 2015 Search PubMed.
- (a) Technology – Exactmer, https://exactmer.com/technology/, (accessed on November 10th, 2021); (b) C. Noti, M. Cristau, J. Riegler, V. d. l. P. d. M. Castro Pinzon, H. M. Rodriguez Cabrera, F. Albericio Palomera, W. Chen and A. G. Livingston, WO Patent2016188835A1, 2016 Search PubMed; (c) A. G. Livingston, J. Kim, I. B. Valtcheva, P. R. J. Gaffney and M. Schaepertoens, WO Patent2016020708A1, 2016 Search PubMed; (d) A. G. Livingston, J. Kim, I. B. Valtcheva and P. R. J. Gaffney, WO Patent2016 024105A1, 2016 Search PubMed; (e) A. G. Livingston, S. Kumbharkar, L. Peeva and J. Da Silva Burgal, US Patent20170007963A1, 2017 Search PubMed; (f) M. Mechelhoff, P. Marchetti, A. Livingston and Z. Karina, US Patent10239024B2, 2019 Search PubMed; (g) M. Schaepertoens, P. R. J. Gaffney, G. Szekely and A. G. Livingston, US Patent10239996B2, 2019 Search PubMed; (h) M. Cook and A. Livingston, US Pat., 20200122094A1, 2020; (i) A. G. Livingston, P. Marchetti, P. Gaffney and R. Liu, US Patent10913033B2, 2021 Search PubMed; (j) A. G. Livingston and M. F. Jimenez Solomon, US Patent11117104B2, 2021 Search PubMed; (k) A. G. Livingston, Z. Jiang, R. Dong, J. Xu and S. Li, WO Patent2021205141A1, 2021 Search PubMed.
- (a) C. Seifert, US Patent20210079036A1, 2021 Search PubMed; (b) C. Seifert, WO Patent2020159837A1, 2020 Search PubMed; (c) C. Seifert, WO Patent2019217116A1, 2019 Search PubMed; (d) C. Seifert, WO Patent2019231760A1, 2019 Search PubMed; (e) C. Seifert and G. Li, US Patent10947267B2, 2021 Search PubMed.
- (a) G. Tana, S. Kitada, S. Fujita, Y. Okada, S. Kim and K. Chiba, Chem. Commun., 2010, 46, 8219 RSC; (b) Y. Okada, H. Suzuki, T. Nakae, S. Fujita, H. Abe, K. Nagano, T. Yamada, N. Ebata, S. Kim and K. Chiba, J. Org. Chem., 2013, 78, 320 CrossRef CAS PubMed; (c) H. Wakamatsu, Y. Okada, M. Sugai, S. R. Hussaini and K. Chiba, Asian J. Org. Chem., 2017, 6, 1584 CrossRef CAS.
- (a) K. Chiba, M. Sugihara, K. Yoshida, Y. Mikami and S. Kim, Tetrahedron, 2009, 65, 8014 CrossRef CAS; (b) S. Kitada, S. Fujita, Y. Okada, S. Kim and K. Chiba, Bioorg. Med. Chem. Lett., 2011, 21, 4476 CrossRef CAS PubMed; (c) S. Kitada, M. Takahashi, Y. Yamaguchi, Y. Okada and K. Chiba, Org. Lett., 2012, 14, 5960 CrossRef CAS PubMed; (d) Y. Fujita, S. Fujita, Y. Okada and K. Chiba, Org. Lett., 2013, 15, 1155 CrossRef CAS PubMed; (e) S. Kitada, S. Fujita, Y. Okada, S. Kim and K. Chiba, Tetrahedron, 2013, 69, 2555 CrossRef CAS; (f) Y. Okada, S. Hosoya, H. Suzuki and K. Chiba, Org. Lett., 2014, 16, 6448 CrossRef CAS PubMed; (g) E. Matsumoto, Y. Fujita, Y. Okada, E. I. Kauppinen, H. Kamiya and K. Chiba, J. Pept. Sci., 2015, 21, 691 CrossRef CAS PubMed; (h) Y. Okada, H. Wakamatsu, M. Sugai, E. I. Kauppinen and K. Chiba, Org. Lett., 2015, 17, 4264 CrossRef CAS PubMed; (i) Y. Okada, H. Asama, H. Wakamatsu, K. Chiba and H. Kamiya, Eur. J. Org. Chem., 2017, 5961 CrossRef CAS; (j) K. Ogami, Y. Okada and K. Chiba, Chem. Lett., 2018, 47, 138 CrossRef CAS; (k) S. Yamagami, Y. Okada, Y. Kitano and K. Chiba, Eur. J. Org. Chem., 2021, 31338 Search PubMed.
- Y. Okada, R. Takasawa, D. Kubo, N. Iwanaga, S. Fujita, K. Suzuki, H. Suzuki, H. Kamiya and K. Chiba, Org. Process Res. Dev., 2019, 23, 2576 CrossRef CAS.
- (a) BACHEM and JITSUBO enter into exclusive licensing agreement, https://www.bachem.com/news/bachem-and-jitsubo-enter-into-exclusive-licensing-agreement/, (accessed on November 2021); (b) Cost-efficient and green manufacturing of peptides thanks to Molecular HivingTM technology, https://www.smi-online.co.uk/pharmaceuticals/webinar/molecular-hiving-technology, (accessed on November 10th, 2021).
- https://www.ajibio-pharma.com/cdmo-services/oligo-peptide-synthesis/ajiphase-synthesis/ (accessed on November 10th, 2021).
- D. Takahashi and T. Yamamoto, Tetrahedron Lett., 2012, 53, 1936 CrossRef CAS.
- D. Takahashi, T. Yano and T. Fukui, Org. Lett., 2012, 14, 4514 CrossRef CAS.
- D. Takahashi, T. Inomata and T. Fukui, Angew. Chem., 2017, 129, 7911 CrossRef.
- (a) S. So, L. G. Peeva, E. W. Tate, R. J. Leatherbarrow and A. G. Livingston, Chem. Commun., 2010, 46, 2808 RSC; (b) S. So, L. G. Peeva, E. W. Tate, R. J. Leatherbarrow and A. G. Livingston, Org. Process Res. Dev., 2010, 14, 1313 CrossRef CAS; (c) V. Castro, C. Noti, W. Chen, M. Cristau, A. Livingston, H. Rodrìguez and F. Albericio, Macromolecules, 2017, 50, 1626 CrossRef CAS.
- I. B. Valtcheva, P. Marchetti and A. G. Livingston, J. Membr. Sci., 2015, 493, 568 CrossRef CAS.
- C. W. Seifert, A. Paniagua, G. A. White, L. Cai and G. Li, Eur. J. Org. Chem., 2016, 1714 CrossRef CAS PubMed.
- https://gappeptides.com/publications/ (accessed on November 10th, 2021).
- (a) H. Li, J. Chao, J. Hasan, G. Tian, Y. Jin, Z. Zhang and C. Qin, J. Org. Chem., 2020, 85, 6271 CrossRef CAS PubMed; (b) H. Li, J. Chao, J. Hasan, G. Tian, Y. Jin, N. Chang and C. Qin, Org. Lett., 2020, 22, 3323 CrossRef CAS PubMed; (c) H. Li, J. Chao, G. Tian, J. Hasan, Y. Jin, Z. Zhang and C. Qin, Org. Chem. Front., 2020, 7, 689 RSC; (d) H. Li, J. Ren, J. Li, Z. Zhang, M. Chang and C. Qin, Org. Biomol. Chem., 2020, 18, 8433 RSC.
- (a) I. F. Eggen, P. B. W. Ten Kortenaar and C. A. G. Haasnoot, US Patent20030018164A1, 2003 Search PubMed; (b) https://sb3000.tech/ (accessed on November 10th, 2021).
- I. F. Eggen and P. B. W. Ten Kortenaar, US Patent6864357B2, 2005 Search PubMed.
- I. F. Eggen, F. T. Bakelaar, A. Petersen, P. B. W. Ten Kortenaar, N. H. S. Ankone, H. E. J. M. Bijsterveld, G. H. L. Bours, F. El Bellaj, M. J. Hartsuiker, G. J. Kuiper and E. J. M. Ter Voert, J. Pept. Sci., 2005, 11, 633 CrossRef CAS PubMed.
- I. F. Eggen, F. T. Bakelaar, A. Petersen and P. B. W. Ten Kortenaar, Org. Process Res. Dev., 2005, 9, 98 CrossRef CAS.
- I. F. Eggen, F. T. Bakelaar, P. B. W. Ten Kortenaar, K. Adermann, W. G. Forssmann and A. Schulz, Rapid Solution-phase Synthesis of a 20-mer Peptide According to the DioRaSSP® Method, Understanding Biology Using Peptides. America Peptide Symposia, ed. S. E. Blondelle, Springer, New York, 2005 Search PubMed.
- https://www.aspenapi.com/api-catalogue/ (accessed on November 10th, 2021).
- R. A. Houghten, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 5131 CrossRef CAS PubMed.
- J. Rantanen, S. Mulvany, W. Lennard, R. G. Petersen, Y. Ermolovich and J. Bukrinski, Innovative Chemistry for Peptide Manufacturing and Analysis Meeting, March 2021, oral presentation essentially focused on the DOE of the piperidine deprotection.
- S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius and U. T. Bornscheuer, Angew. Chem., Int. Ed., 2021, 60, 88 CrossRef CAS PubMed.
- (a) A. M. Weeks and J. A. Wells, Chem. Rev., 2020, 120, 3127 CrossRef CAS PubMed; (b) T. Nuijens, A. Toplak, M. Schmidt, A. Ricci and W. Cabri, Front. Chem., 2019, 7, 829 CrossRef CAS PubMed.
- (a) S. K. Mazmanian, G. Liu, H. Ton-That and O. Schneewind, Science, 1999, 285, 760 CrossRef CAS PubMed; (b) H. Ton-Taht, G. Liu, S. K. Mazmanian, K. F. Faull and O. Schneewind, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12424 CrossRef PubMed; (c) U. Ilangovan, H. Ton-Tht, J. Iwahara, O. Scheewind and R. T. Clubb, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6056 CrossRef CAS PubMed; (d) H. Ton-That, S. K. Mazmanian, K. F. Faull and O. Schneewind, J. Biol. Chem., 2000, 13, 9876 CrossRef PubMed.
- R. L. Policarpo, H. Kang, X. Liao, A. E. Rabideau, M. D. Simon and B. L. Pentelute, Angew. Chem., Int. Ed., 2014, 53, 9203 CrossRef CAS PubMed.
- B. M. Dorr, H. O. Ham, C. An, E. L. Chaikof and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13343 CrossRef CAS PubMed.
- I. Chen, B. M. Dorr and D. R. Liu, Proc. Natl. Acad. Sci. U. S. A., 2011, 118, 11399 CrossRef PubMed.
- Z. Zou, D. M. Mate, M. Nöth, F. Jakob and U. Schwaneberg, Chem. – Eur. J., 2020, 26, 13568 CrossRef CAS PubMed.
- G. K. T. Nguyen, S. Wang, Y. Qiu, X. Hemu, Y. Lian and J. P. Tam, Nat. Chem. Biol., 2014, 10, 732 CrossRef CAS PubMed.
- D. J. Craik, N. L. Daly, T. Bond and C. Waine, J. Mol. Biol., 1999, 294, 1327 CrossRef CAS PubMed.
- G. K. T. Nguyen, Y. Cao, W. Wang, C. F. Liu and J. P. Tam, Angew. Chem., Int. Ed., 2015, 54, 15694 CrossRef CAS PubMed.
- X. Hemu, X. Zhang, G. K. T. Nguyen, J. To, A. Serra, S. Loo, S. Kwan Sze, C.-F. Liu and J. P. Tam, RSC Adv., 2021, 11, 23105 RSC.
- A. M. James, J. Haywood, J. Leroux, K. Ignasiak, A. G. Elliott, J. W. Schmidberger, M. F. Fisher, S. G. Nonis, R. Fenske, C. S. Bond and J. S. Mylne, Plant J., 2019, 98, 988 CAS.
- X. Hemu, A. El Sahili, S. Hu, X. Zhang, A. Serra, B. C. Goh, D. A. Darwis, M. W. Chen, S. K. Sze, C. Liu, J. Lescar and J. P. Tam, ACS Catal., 2020, 10, 8825 CrossRef CAS.
- (a) K. S. Harris, T. Durek, Q. Kaas, A. G. Poth, E. K. Gilding, B. F. Conlan, I. Saska, N. L. Daly, N. L. van der Weerden, D. J. Craik and M. A. Anderson, Nat. Commun., 2015, 6, 10199 CrossRef CAS PubMed; (b) K. S. Harris, R. F. Guarino, R. S. Dissanayake, P. Quimbar, O. C. McCorkelle, S. Poon, Q. Kaas, T. Durek, E. K. Gilding, M. A. Jackson, D. J. Craik, N. L. van der Weerden, R. F. Anders and M. A. Anderson, Sci. Rep., 2019, 9, 10820 CrossRef PubMed.
- M. A. Jackson, E. K. Gilding, T. Shafee, K. S. Harris, Q. Kaas, S. Poon, K. Yap, H. Jia, R. Guarino, L. Y. Chan, T. Durek, M. A. Anderson and D. J. Craik, Nat. Commun., 2018, 9, 2411 CrossRef CAS PubMed.
- X. Hemu, A. El Sahili, S. Hu, K. Wong, Y. Chen, Y. H. Wong, X. Zhang, A. Serra, B. C. Goh, D. A. Darwis, M. W. Chen, S. K. Sze, C.-F. Liu, J. Lescar and J. P. Tam, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11737 CAS.
- (a) R. Yang, Y. H. Wong, G. K. T. Nguyen, J. P. Tam, J. Lescar and B. Wu, J. Am. Chem. Soc., 2017, 139, 5351 CrossRef CAS PubMed; (b) J. Du, K. Yap, L. Y. Chan, F. B. H. Rehm, F. Y. Looi, A. G. Poth, E. K. Gilding, Q. Kaas, T. Durek and D. J. Craik, Nat. Commun., 2020, 11, 1575 CrossRef CAS PubMed.
- X. Hemu, J. To, X. Zhang and J. P. Tam, J. Org. Chem., 2020, 85, 1504 CrossRef CAS PubMed.
- F. B. H. Rehm, T. J. Tyler, J. Xie, K. Yap, T. Durek and D. J. Craik, ChemBioChem, 2021, 22, 2079 CrossRef CAS PubMed.
- (a) K. E. Neet and D. E. Koshland, Proc. Natl. Acad. Sci. U. S. A., 1996, 56(5), 1606 CrossRef PubMed; (b) L. Polgár and M. L. Beder, Biochemistry, 1967, 6, 610 CrossRef PubMed.
- (a) A. C. Braisted, J. K. Judice and J. A. Wells, Methods Enzymol., 1997, 289, 298 CAS; (b) L. Abrahmsen, J. Tom, J. Burnier, K. A. Butcher, A. Kossiakoff and J. A. Wells, Biochemistry, 1991, 30, 4151 CrossRef CAS PubMed.
- A. Toplak, T. Nuijens, P. J. L. M. Quaedflieg, B. Wu and D. B. Janssen, Adv. Synth. Catal., 2016, 358, 2140 CrossRef CAS.
- A. Toplak, E. F. T. de Oliveira, M. Schmidt, H. J. Rozeboom, H. J. Wijma, L. K. M. Meekels, R. de Visser, D. B. Janssen and T. Nuijens, Comput. Struct. Biotechnol. J., 2021, 19, 1277 CrossRef CAS PubMed.
- (a) J. Pawlas, T. Nuijens, J. Persson, T. Svensson, M. Schmidt, A. Toplak, M. Nilsson and J. H. Rasmussen, Green Chem., 2019, 21, 6451 RSC; (b) M. Schmidt and T. Nujiens, Chapter 4 - Chemoenzymatic Synthesis of Linear and Head-to-Tail Cyclic Peptides Using Omniligase-1, Enzyme-Mediated Ligation Methods, Humana, New York, 2019, vol. 2012 Search PubMed.
- M. Schmidt, A. Toplak, H. J. Rozeboom, H. J. Wijma, P. J. L. M. Quaedflieg, J. H. van Maarseveen, D. B. Janssen and T. Nuijens, Org. Biomol. Chem., 2018, 16, 609 RSC.
- M. Schmidt, A. Toplak, P. J. L. M. Quaedflieg, H. Ippel, G. J. J. Richelle, T. M. Hackeng, J. H. van Maarseveen and T. Nuijens, Adv. Synth. Catal., 2017, 359, 2050 CrossRef CAS.
- S. Liebscher, M. Schöpfel, T. Aumüller, A. Sharkhuukhen, A. Pech, E. Höss, C. Parthier, G. Jahreis, M. T. Stubbs and F. Bordusa, Angew. Chem., Int. Ed., 2014, 53, 3024 CrossRef CAS PubMed.
- S. Liebscher, S. Mathea, T. Aumüller, A. Pech and F. Bordusa, ChemBioChem, 2021, 22, 1201 CrossRef CAS PubMed.
- (a) J.-L. Do and T. Friscic, ACS Cent. Sci., 2017, 3, 13 CrossRef CAS PubMed; (b) S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friscic, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steedk and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413 RSC; (c) J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080 RSC.
- L. Takacs, Chem. Soc. Rev., 2013, 42, 7649 RSC.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334 RSC.
- A. Dicks and A. Hent, Green Chem. Metrics, 2015, 45 Search PubMed.
- J. G. Hernandez and C. Bolm, J. Org. Chem., 2017, 82, 4007 CrossRef CAS PubMed.
- D. Tan, L. Loots and T. Friscic, Chem. Commun., 2016, 52, 7760 RSC.
- V. Declerck, P. Nun, J. Martinez and F. Lamaty, Angew. Chem., Int. Ed., 2009, 48, 9318 CrossRef CAS PubMed.
- J. Bonnamour, T.-X. Métro, J. Martinez and F. Lamaty, Green Chem., 2013, 15, 1116 RSC.
- J. G. Herneandez and E. Juaristi, J. Org. Chem., 2010, 75, 7107 CrossRef PubMed.
- (a) V. Strukil, B. Bartolec, T. Portada, I. Dilovic, I. Halaszc and D. Margetic, Chem. Commun., 2012, 48, 12100 RSC; (b) C. Duangkamol, S. Jaita, S. Wangngae, W. Phakhodee and M. Pattarawarapan, RSC Adv., 2015, 5, 52624 RSC; (c) J. M. Landeros and E. Juaristi, Eur. J. Org. Chem., 2017, 687 CrossRef CAS; (d) Y. Yeboue, M. Jean, G. Subra, J. Martinez, F. Lamaty and T.-X. Métro, Org. Lett., 2021, 23, 631 CrossRef CAS PubMed.
- V. Porte, M. Thioloy, T. Pigoux, T.-X. Métro, J. Martinez and F. Lamaty, Eur. J. Org. Chem., 2016, 3505 CrossRef CAS.
- O. Maurin, P. Verdié, G. Subra, F. Lamaty, J. Martinez and T.-X. Métro, Beilstein J. Org. Chem., 2017, 13, 208 Search PubMed.
- L. Gonnet, T. Tintillier, N. Venturini, L. Konnert, J.-F. Hernandez, F. Lamaty, G. Laconde, J. Martinez and E. Colacino, ACS Sustainable Chem. Eng., 2017, 5, 2936 CrossRef CAS.
- M. Anselmi, P. Stavole, E. Boanini, A. Bigi, E. Juaristi and L. Gentilucci, Future Med. Chem., 2020, 12, 479 CrossRef CAS PubMed.
- K. Yazawa and K. Numata, Molecules, 2014, 19, 13755 CrossRef PubMed.
- J. G. Hernández, K. J. Ardila-Fierro, D. Crawford, S. L. James and C. Bolm, Green Chem., 2017, 19, 2620 RSC.
- M. O. Frederick, R. A. Boyse, T. M. Braden, J. R. Calvin, B. M. Campbell, S. M. Changi, S. R. Coffin, C. Condon, O. Gowran, J. McClary Groh, S. R. Groskreutz, Z. D. Harms, A. A. Humenik, N. J. Kallman, N. D. Klitzing, M. E. Kopach, J. K. Kretsinger, G. R. Lambertus, J. T. Lampert, L. M. Maguire, H. A. Moynihan, N. S. Mullane, J. D. Murphy, M. E. O'Mahony, R. N. Richey, K. D. Seibert, R. D. Spencer, M. A. Strege, N. Tandogan, F. L. Torres Torres, S. V. Tsukanov and H. Xia, Org. Process Res. Dev., 2021, 25, 1628 CrossRef CAS.
- C. de Luca, G. Lievore, D. Bozza, A. Buratti, A. Cavazzini, A. Ricci, M. Macis, W. Cabri, S. Felletti and M. Catani, Molecules, 2021, 26, 1 CrossRef PubMed.
- W. Zhang and B. W. Cue, Green Techniques for Organic Synthesis and Medicinal Chemistry, Wiley-VCH, 2012 Search PubMed.
- (a) H. Schmidt-Traub, Preparative chromatography of fine chemicals and pharmaceutical agents, Wiley-VCH, 2005 CrossRef; (b) P. J. Dunn, A. S. Wells and M. T. Williams, Green Chemistry in the Pharmaceutical Industry, Wiley-VCH, 2010 CrossRef; (c) C. J. Welch, N. Wu, M. Biba, R. Hartman, T. Brkovic, X. Gong, R. Helmy, W. Schafer, J. Cuff, Z. Pirzada and L. Zhou, Trends Anal. Chem., 2010, 29, 667 CrossRef CAS; (d) M. I. Shaik and N. M. Sarbon, Food Rev. Int., 2020, 1–21 CrossRef; (e) C. M. Grill and L. Miller, J. Chromatogr., A, 1998, 827, 359 CrossRef CAS.
- C. de Luca, S. Felletti, G. Lievore, A. Buratti, S. Vogg, M. Morbidelli, A. Cavazzini, M. Catani, M. Macis, A. Ricci and W. Cabri, J. Chromatogr. A, 2020, 1625, 461304 CrossRef PubMed.
- Y. Shen, B. Chen and T. A. van Beek, Green Chem., 2015, 17, 4073 RSC.
- A. Kremser and C. A. White, Chem. Today, 2019, 37, 12 CAS.
- B. Vanholme, T. Desmet, F. Ronsse, K. Rabaey, F. van Breusegem, M. de Mey, W. Soetaert and W. Boerjan, Front. Plant Sci., 2013, 4, 174 Search PubMed.
- D. Guo, C. T. Mant and R. S. Hodges, J. Chromatogr. A, 1987, 386, 205–222 CrossRef CAS PubMed.
- K. R. Solomon, G. J. M. Velders, S. R. Wilson, S. Madronich, J. Longstreth, P. J. Aucamp and J. F. Bornman, J. Toxicol. Environ. Health, 2016, 19, 289 CrossRef CAS PubMed.
- J. Cornish, K. E. Callon, C. Q.-X. Lin, C. L. Xiao, T. B. Mulvey, G. J. S. Cooper and I. R. Reid, Am. J. Physiol., 1999, 277, E779 CAS.
- K. Sikora, M. Jaśkiewicz, D. Neubauer, D. Migoń and W. Kamysz, Pharmaceuticals, 2020, 13, 442 CrossRef CAS PubMed.
- (a) J. E. Rivier, J. Liq. Chromatogr., 1978, 1, 1 CrossRef; (b) C. T. Mant, Y. Chen, Z. Yan, T. V. Popa, J. M. Kovacs, J. B. Mills, B. P. Tripet and R. S. Hodges, Methods Mol. Biol., 2007, 386, 3 CrossRef CAS PubMed.
- O. Reimann, O. Seitz, D. Sarma and R. Zitterbart, J. Pept. Sci., 2019, 25, e3136 CrossRef PubMed.
- The only commercial c&r method has been reported by D. W. Anderson, G. J. Cotton, M. H. Alastair, W. A. Paul and W. Ian, US20130211047, 2011.
- T. Müller-Späth, G. Ströhlein, O. Lyngberg and D. Maclean, Chem. Today, 2013, 31, 56 Search PubMed.
- S. Vogg, N. Ulmer, J. Souquet, H. Broly and M. Morbidelli, Biotechnol. J., 2019, 14, 1800732 CrossRef PubMed.
- (a) C. de Luca, S. Felletti, D. Bozza, G. Lievore, M. Morbidelli, M. Sponchioni, A. Cavazzini, M. Catani, W. Cabri, M. Macis and A. Ricci, Ind. Eng. Chem. Res., 2021, 60, 6826 CrossRef CAS; (b) D. Gétaz, G. Stroehlein, A. Butté and M. Morbidelli, J. Chromatogr. A, 2013, 1284, 69 CrossRef PubMed; (c) F. Steinebach, T. Müller-Späth and M. Morbidelli, Biotechnol. J., 2016, 11, 1126 CrossRef CAS PubMed.
- (a) C. M. Grill, J. Chromatogr. A, 1998, 796, 101 CrossRef CAS PubMed; (b) C. Grill, L. Miller and T. Yan, J. Chromatogr. A, 2004, 1026, 101 CrossRef CAS PubMed; (c) I. Quiñones, C. M. Grill, L. Miller and G. Guiochon, J. Chromatogr. A, 2000, 867, 1 CrossRef; (d) J. Dingenen and J. N. Kinkel, J. Chromatogr. A, 1994, 666, 627 CrossRef CAS.
- (a) D. W. Guest, J. Chromatogr. A, 1997, 760, 159 CrossRef CAS; (b) D. Broughton and C. Gerhold, PatentUS2985589A, 1961 Search PubMed.
- Y. Xie, S. Mun, J. Kim and N.-H. L. Wang, Biotechnol. Prog., 2002, 18, 1332 CrossRef CAS PubMed.
- D. Pfister, L. Nicoud and M. Morbidelli, Continuous Biopharmaceutical Processes: Chromatography, Bioconjugation, and Protein Stability, Cambridge University Press, 2018 Search PubMed.
- (a) E. R. Francotte and P. Richert, J. Chromatogr. A, 1997, 769, 101 CrossRef CAS; (b) V. P. Chernev, A. v. Wouwer and A. Kienle, Processes, 2016, 8, 1316 CrossRef.
- S. Imamoglu, Adv. Biochem. Eng./Biotechnol., 2002, 76, 211 CrossRef CAS PubMed.
- D. Pfister, L. Nicoud and M. Morbidelli, Continuous Biopharmaceutical Processes: Chromatography, Bioconjugation, and Protein Stability, Cambridge University Press, 2018. DOI:10.1017/9781108332897.
- (a) C. de Luca, S. Felletti, M. Macis, W. Cabri, G. Lievore, T. Chenet, L. Pasti, M. Morbidelli, A. Cavazzini, M. Catani and A. Ricci, J. Chromatogr. A, 2020, 1616, 460789 CrossRef CAS PubMed; (b) N. Marchetti, F. Dondi, A. Felinger, R. Guerrini, S. Salvadori and A. Cavazzini, J. Chromatogr. A, 2005, 1079, 162 CrossRef CAS PubMed; (c) L. Aumann and M. Morbidelli, Biotechnol. Bioeng., 2007, 98, 1043 CrossRef CAS PubMed.
- A. L. Zydney, Biotechnol. Bioeng., 2016, 113, 465 CrossRef CAS PubMed.
- Y. Xie, S. Mun, C. Y. Chin and N.-H. L. Wang, Front. Biomed. Eng., 2003, 507 Search PubMed.
- (a) L. Aumann and M. Morbidelli, PatentEP1877769B1, 2006 Search PubMed; (b) L. Aumann and M. Morbidelli, Biotechnol. Bioeng., 2008, 99, 728 CrossRef CAS PubMed; (c) G. Ströhlein, L. Aumann, M. Mazzotti and M. Morbidelli, J. Chromatogr. A, 2006, 1126, 338 CrossRef PubMed.
- (a) T. Müller-Späth, L. Aumann, L. Melter, G. Ströhlein and M. Morbidelli, Biotechnol. Bioeng., 2008, 100, 1166 CrossRef PubMed; (b) B. Schenkel, L. Aumann, G. Stroehlein and M. Morbidelli, BioPharm Int., 2009, 22, 46 Search PubMed; (c) C. de Luca, S. Felletti, G. Lievore, T. Chenet, M. Morbidelli, M. Sponchioni, A. Cavazzini and M. Catani, TrAC, Trends Anal. Chem., 2020, 132, 116051 CrossRef CAS PubMed; (d) G. Stroehlein, L. Aumann, T. Müller-Späth, A. Tarafder and M. Morbidelli, BioPharm Int., 2007, 22, 42 Search PubMed; (e) F. Steinebach, M. Krättli, G. Storti and M. Morbidelli, Ind. Eng. Chem. Res., 2017, 56, 13482 CrossRef CAS; (f) G. Subramanian, Continuous processing in pharmaceutical manufacturing, Wiley-VHC, 2005 Search PubMed.
- L. Aumann, G. Stroehlein and M. Morbidelli, Biotechnol. Bioeng., 2007, 98, 1029 CrossRef CAS PubMed.
- F. Steinebach, N. Ulmer, L. Decker, L. Aumann and M. Morbidelli, J. Chromatogr. A, 2017, 1492, 19 CrossRef CAS PubMed.
- M. Catani, C. de Luca, J. Medeiros Garcia Alcântara, N. Manfredini, D. Perrone, E. Marchesi, R. Weldon, T. Müller-Späth, A. Cavazzini, M. Morbidelli and M. Sponchioni, Biotechnol. J., 2020, 15, 1900226 CrossRef CAS PubMed.
- A. Tarafder, G. Ströhlein, L. Aumann and M. Morbidelli, J. Chromatogr. A, 2008, 1183, 87 CrossRef CAS PubMed.
- T. Müller-Späth and M. Bavand, Pharm. Eng., 2019, 39, 68 Search PubMed.
- https://www.bachem.com/event/continuous-chromatography-of-peptides/ .
- (a) G. Guiochon and A. Tarafder, J. Chromatogr. A, 2011, 1218, 1037 CrossRef CAS PubMed; (b) C. West, Anal. Bioanal. Chem., 2018, 410, 6441 CrossRef CAS PubMed.
- (a) K. Govender, T. Naicker, S. Baijnath, H. G. Kruger and T. Govender, J. Pharm. Biomed. Anal., 2020, 190, 113539 CrossRef CAS PubMed; (b) M. Saito, J. Biosci. Bioeng., 2015, 115, 590 CrossRef PubMed.
- M. Perrut, PatentUS4478720A, 1983 Search PubMed.
- S. Felletti, O. H. Ismail, C. de Luca, V. Costa, F. Gasparrini, L. Pasti, N. Marchetti, A. Cavazzini and M. Catani, Chromatographia, 2019, 82, 65 CrossRef CAS.
- https://www.americanpharmaceuticalreview.com/Featured-Articles/185891-A-Perspective-on-the-Application-of-Preparative-Supercritical-Fluid-Chromatography-Using-Achiral-Stationary-Phases-in-Pharmaceutical-Drug-Discovery-and-Development/ (accessed November 4, 2021).
- (a) O. H. Ismail, S. Felletti, C. de Luca, L. Pasti, N. Marchetti, V. Costa, F. Gasparrini, A. Cavazzini and M. Catani, Molecules, 2018, 23, 2709 CrossRef PubMed; (b) C. West, TrAC, Trends Anal. Chem., 2019, 120, 115648 CrossRef CAS; (c) J. Molineau, M. Hideux and C. West, J. Pharm. Biomed. Anal., 2021, 193, 113736 CrossRef CAS PubMed.
- (a) K. B. Thurbide and J. Zhang, Anal. Bioanal. Chem., 2005, 382, 1227 CrossRef CAS PubMed; (b) J. Zhang and K. B. Thurbide, J. Chromatogr. A, 2006, 1101, 286 CrossRef CAS PubMed; (c) J. Zheng, J. D. Pinkston, P. H. Zoutendam and L. T. Taylor, Anal. Chem., 2006, 78, 1535 CrossRef CAS PubMed; (d) Y. Shao, C. Wang, A. Apedo, O. Mcconnell, Y. Shao, C. Wang, A. Apedo and O. Mcconnell, J. Anal. Sci., Methods Instrum., 2016, 6, 23 CAS; (e) M. Enmark, E. Glenne, M. Leśko, A. Langborg Weinmann, T. Leek, K. Kaczmarski, M. Klarqvist, J. Samuelsson and T. Fornstedt, J. Chromatogr. A, 2018, 1568, 177 CrossRef CAS PubMed; (f) V. Spelling and M. Stefansson, J. Chromatogr. A, 2020, 1633, 461646 CrossRef CAS PubMed.
- N. M. Schiavone, R. Bennett, M. B. Hicks, G. F. Pirrone, E. L. Regalado, I. Mangion and A. A. Makarov, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1110–1111, 94 CrossRef CAS PubMed.
- M. Ventura, J. Pharm. Biomed. Anal., 2020, 185, 113227 CrossRef CAS PubMed.
- V. Kasche, R. Schlothauer and G. Brunner, Biotechnol. Lett., 1988, 10, 569 CrossRef CAS.
- J. Zagrobelny and F. V. Bright, Biotechnol. Prog., 1992, 8, 421 CrossRef CAS PubMed.
- M. Ventura, J. Pharm. Biomed. Anal., 2020, 185, 113227 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |