
Mo-doped SnS2 with enriched S-vacancies for highly efficient electrocatalytic N2 reduction: the critical role of the Mo–Sn–Sn trimer†
Ke
Chu
 *,
Jing
Wang
,
Ya-ping
Liu
,
Qing-qing
Li
and
Ya-li
Guo
*,
Jing
Wang
,
Ya-ping
Liu
,
Qing-qing
Li
and
Ya-li
Guo
School of Materials Science and Engineering, Lanzhou Jiaotong University, Lanzhou 730070, China. E-mail: chukelut@163.com
First published on 24th March 2020
Abstract
Vacancy engineering and heteroatom doping are two effective approaches to tailor the electronic structures of catalysts for improved electrocatalytic activity. Herein, these two approaches were rationally combined to modulate the structure of SnS2 toward the N2 reduction reaction (NRR) by means of Mo-doping, which simultaneously induced the generation of enriched S-vacancies (Vs). The developed Mo-doped SnS2 nanosheets with enriched Vs presented a conspicuously enhanced NRR activity with an NH3 yield of 41.3 μg h−1 mg−1 (−0.5 V) and a faradaic efficiency of 20.8% (−0.4 V) and are among the best SnS2-based NRR catalysts to date. Mechanistic studies revealed that the co-presence of the Mo dopant and Vs enabled the creation of Mo–Sn–Sn trimer catalytic sites, capable of strongly activating N2 even for the cleavage of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond to the N
N triple bond to the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond at the N2 adsorption stage, consequently leading to a downhill process of the first hydrogenation step and a largely reduced energy barrier.
N double bond at the N2 adsorption stage, consequently leading to a downhill process of the first hydrogenation step and a largely reduced energy barrier.
Dinitrogen (N2) fixation to ammonia (NH3) is an essential chemical process for the modern industrial society because NH3 is a widely used feedstock for various chemical products and is also a promising carbon-free energy carrier.1 The electrochemical N2 reduction reaction (NRR) under ambient conditions holds great promise for green and sustainable NH3 production,2 in stark contrast to the industrial Haber–Bosch process which involves huge energy consumption and enormous CO2 emission. However, the NRR process is largely limited by the unsatisfactory NH3 production rate and low faradaic efficiency (FE), stemming from the extremely stable N
N bond, poor N2 adsorption/activation on the catalyst surface and the competing hydrogen evolution reaction (HER).3 Exploring highly active electrocatalysts may hold the key to mitigating these limitations and realizing high NRR efficiency. Over the past three years, an increasingly large number of noble metals,4–7 transition metal-based compounds,8–14 and metal-free materials15–19 have been designed as promising candidates toward the NRR.
SnS2-based materials have shown great potential in electrolysis applications, owing to their fascinating catalytic performance, good stability, tunable electronic structures, and 2D morphology with largely exposed active edges.20 Recently, SnS2 has been proved to be an attractive catalyst with favorable NRR activity as well.21,22 Nonetheless, the reported NRR performance of SnS2 materials is still far from satisfactory and competitive with that of the state-of-the-art NRR catalysts. The main bottlenecks are the poor NRR activation, i.e., the high energy barrier for the first hydrogenation step,22 and the low intrinsic electrical conductivity, i.e., the inefficient proton-coupled electron transfer process, even when employing approaches such as growth of amorphous Sn on crystalline SnS2 nanosheets (FE: 6.5%)21 and construction of well-aligned SnS2 nanoarrays on porous Ni foam (FE: 11.2%).22 Thus, more efficient methods are still needed to further enhance the intrinsic NRR activity of SnS2.
As an effective strategy for electronic structure modulation of the catalysts, vacancy engineering by introducing anion vacancies into the catalyst lattice,23 such as oxygen vacancies (TiO2,24 WO3 (ref. 25) and MnO2 (ref. 26)), and nitrogen vacancies (C3N4,27 VN28 and W2N3 (ref. 29)), provides unique active sites for the effective adsorption and activation of dinitrogen. In addition, heteroatom doping is another widely used method to optimize the adsorption of NRR intermediates on catalysts and promote the NRR reaction kinetics,30–33 which have been demonstrated in V-doped TiO2,34 Fe-doped Ni2P35 and Mo-doped MnO2.36 More importantly, heteroatom doping has been recently confirmed to facilitate the generation of vacancies, while the synergistic effect of dopants and vacancies leads to a conspicuously enhanced NRR performance.37,38 For instance, introducing Zr-dopants in TiO2 nanotubes has been reported to favorably generate both enriched oxygen vacancies and bi-Ti3+ pairs,38 and the bi-Ti3+ pairs serve as the dominant active sites in facilitating the catalytic performance of the NRR with an FE of 17.3%. Similar results and a more improved NRR performance (FE: 25.6%) have been presented in Fe-doped TiO2 nanoparticles,37 where the combined effect of bi-Ti3+ pairs and oxygen vacancies contributes to the substantially boosted NRR activity. Inspired by these studies, it is anticipated that the NRR performance of SnS2 may be greatly enhanced by the combination of vacancy engineering and heteroatom doping.
In this study, we rationally combined vacancy engineering and heteroatom doping to regulate the electronic structure of SnS2 by Mo-doping, which enabled the spontaneous generation of enriched S-vacancies (Vs) as well. The developed Mo-doped SnS2 nanosheets with enriched Vs exhibited a significantly enhanced NRR activity with an NH3 yield of 41.3 μg h−1 mg−1 (−0.5 V) and an FE of 20.8% (−0.4 V) and were among the best SnS2-based NRR catalysts to date. Density functional theory (DFT) calculations revealed that the co-presence of the Mo dopant and Vs enabled the creation of Mo–Sn–Sn trimer active sites that could strongly activate N2 and lower the reaction energy barrier.
Mo-doped SnS2 (Mo-SnS2) nanosheets were directly grown on CC via a facile hydrothermal method. Pristine SnS2/CC was also prepared as a reference. The synthesis details are provided in the ESI.† The obtained SnS2 and Mo-SnS2 nanosheets scraped from CC were first investigated by X-ray photoelectron spectroscopy (XPS). As shown in Fig. 1a, the deconvolution of the Mo3d spectrum results in three well-resolved peaks of S2s (226.4 eV), Mo4+3d3/2 (229.3 eV) and Mo4+3d1/2 (232.7 eV) for Mo-SnS2 nanosheets, which are absent in pristine SnS2 nanosheets, indicating the successful Mo-doping in the Mo-SnS2 nanosheets. Fig. 1b shows the XRD patterns of SnS2/CC and Mo-SnS2/CC. Excluding the peaks of CC, both samples display a pure hexagonal SnS2 phase (JCPDS no. 23-677) with no detectable impurities. Obviously, all the peaks of Mo-SnS2/CC are much weaker than those of SnS2/CC, indicating the lowered crystallinity of Mo-SnS2 caused by Mo-doping. The morphologies of SnS2 and Mo-SnS2 nanosheets were further investigated by scanning electron microscopy (SEM), transmission electron microscopy (TEM) and high-resolution TEM (HRTEM). The SEM images show that bushy nanosheets are densely and vertically anchored on the CC for both SnS2/CC (Fig. 1c) and Mo-SnS2/CC (Fig. 1d). The nanosheet features can be further revealed by TEM with the observation of clear wrinkles and corrugations on SnS2 (Fig. 1e) and Mo-SnS2 (Fig. 1f) nanosheets.39–41 The HRTEM image shows that the pristine SnS2 nanosheets (Fig. 1g) possess a high crystallinity as evidenced by the sharp lattice fringes with a spacing value of 0.59 nm, assigned to the (001) facet of SnS2. In contrast, blurred and discontinuous lattice fringes can be observed in Mo-SnS2 nanosheets (Fig. 1h), indicating that Mo-doping can remarkably reduce the crystallinity of Mo-SnS2 nanosheets, consistent with the XRD analysis (Fig. 1b). The selected areas in Fig. 1h (A and B squares), when applying an Inverse Fast Fourier Transform (IFFT) mask, display obvious distortions and dislocations in the lattice fringes,42–44 suggesting the existence of plentiful defects (i.e., vacancies) in Mo-SnS2 nanosheets, which rationalizes the reduced crystallinity of Mo-SnS2. The scanning TEM (STEM) elemental mapping images (Fig. 1i) reveal that considerable Mo dopants are uniformly distributed on the entire surface of Mo-SnS2 nanosheets.
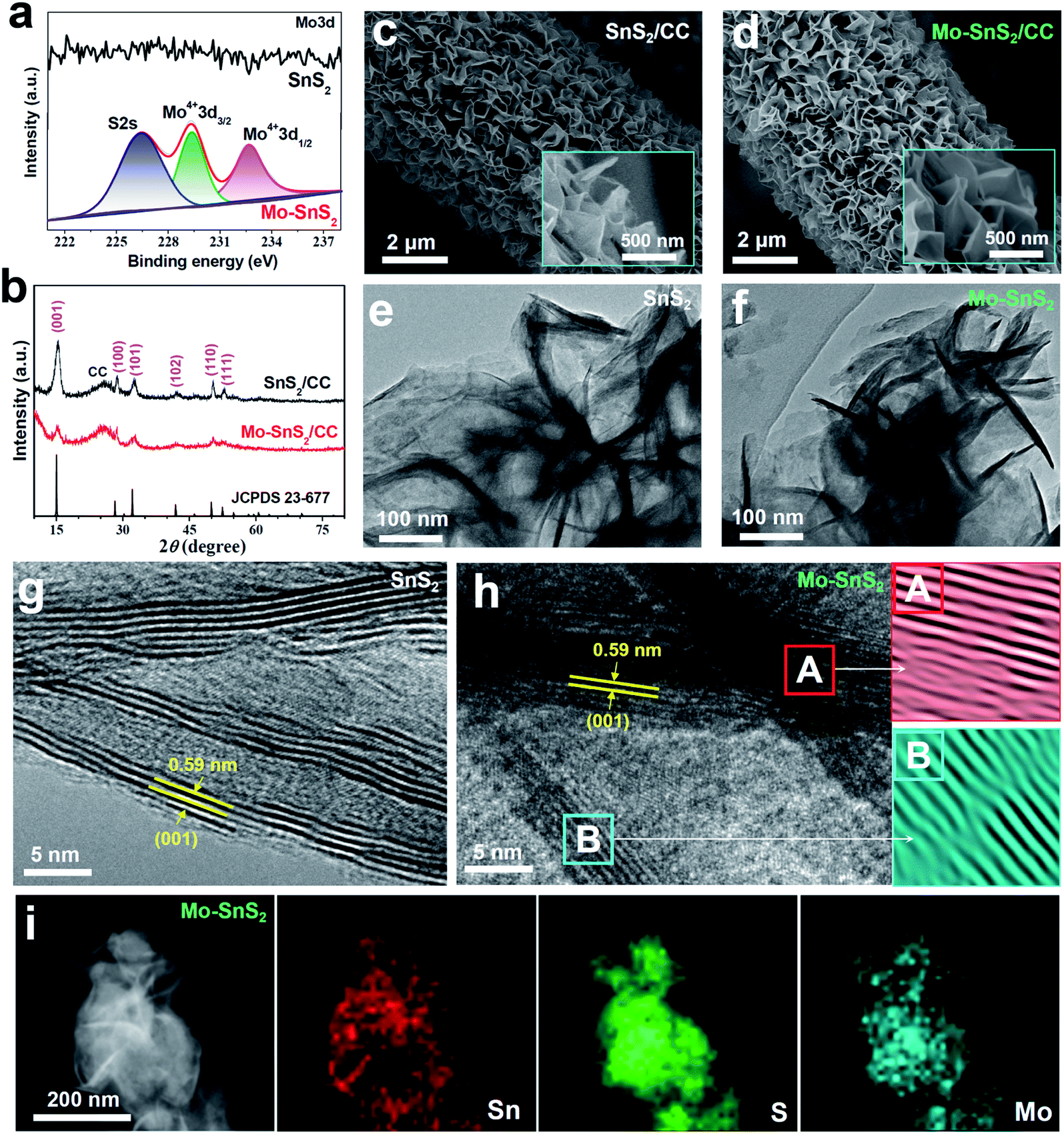 | ||
| Fig. 1 (a) XPS Mo3d spectrum of SnS2 and Mo-SnS2 nanosheets scraped from CC. (b) XRD patterns for SnS2/CC and Mo-SnS2/CC. (c and d) SEM images of (c) SnS2/CC and (d) Mo-SnS2/CC. (e and f) TEM images of (e) SnS2 and (f) Mo-SnS2 nanosheets. (g and h) HRTEM images of (g) SnS2 and (h) Mo-SnS2 nanosheets (A and B are the corresponding IFFT images recorded from regions A and B in (h)). (i) STEM element mapping images for Mo-SnS2 nanosheets. | ||
The vacancies existing in Mo-SnS2 nanosheets were further investigated by a combination of XPS, electron paramagnetic resonance (EPR) spectroscopy and DFT analysis. For both SnS2 and Mo-SnS2 nanosheets, the XPS Sn3d spectra (Fig. 2a) reveal Sn3d5/2 (∼486 eV) and Sn3d3/2 (∼495 eV) states of Sn4+, and the S2p spectra (Fig. 2b) reveal S2p3/2 (∼161 eV) and S2p1/2 (∼163 eV) states of S2−. With respect to those of the pristine SnS2 nanosheets, the Sn3d and S2p spectra of Mo-SnS2 nanosheets are negatively shifted by 0.4 and 0.3 eV, respectively, suggesting the decreased valence state and increased electrons in Mo-SnS2 nanosheets caused by the presence of Vs.45 The existence of enriched Vs in Mo-SnS2 nanosheets can also be directly verified from the remarkably reduced atomic ratio of S/Mo from 1.88 (SnS2) to 1.62 (Mo-SnS2) derived from the XPS element analysis. In the EPR spectra (Fig. 2c), the signal at g = 2.001 represents the electrons trapped in Vs. Compared to SnS2 nanosheets, Mo-SnS2 nanosheets exhibit a much stronger EPR signal intensity, suggesting a higher concentration of Vs,23 consistent with the XPS results. To gain further insight, DFT was applied to predict the Vs formation energy (Ef). As shown in Fig. 2d, the formation of isolated Vs in SnS2-Vs requires a largely positive Ef (5.25 eV) and thus is thermodynamically unfavorable. With the introduction of the Mo dopant (Fig. 2e), Vs can be favorably formed at the Mo-adjacent site of Mo-SnS2-Vs with a significantly reduced Ef (−0.78 eV). Therefore, the DFT results concur well with the XPS/EPR measurements and demonstrate that Mo-doping can considerably lower the Vs formation energy and enable the spontaneous generation of Vs in Mo-SnS2 nanosheets.
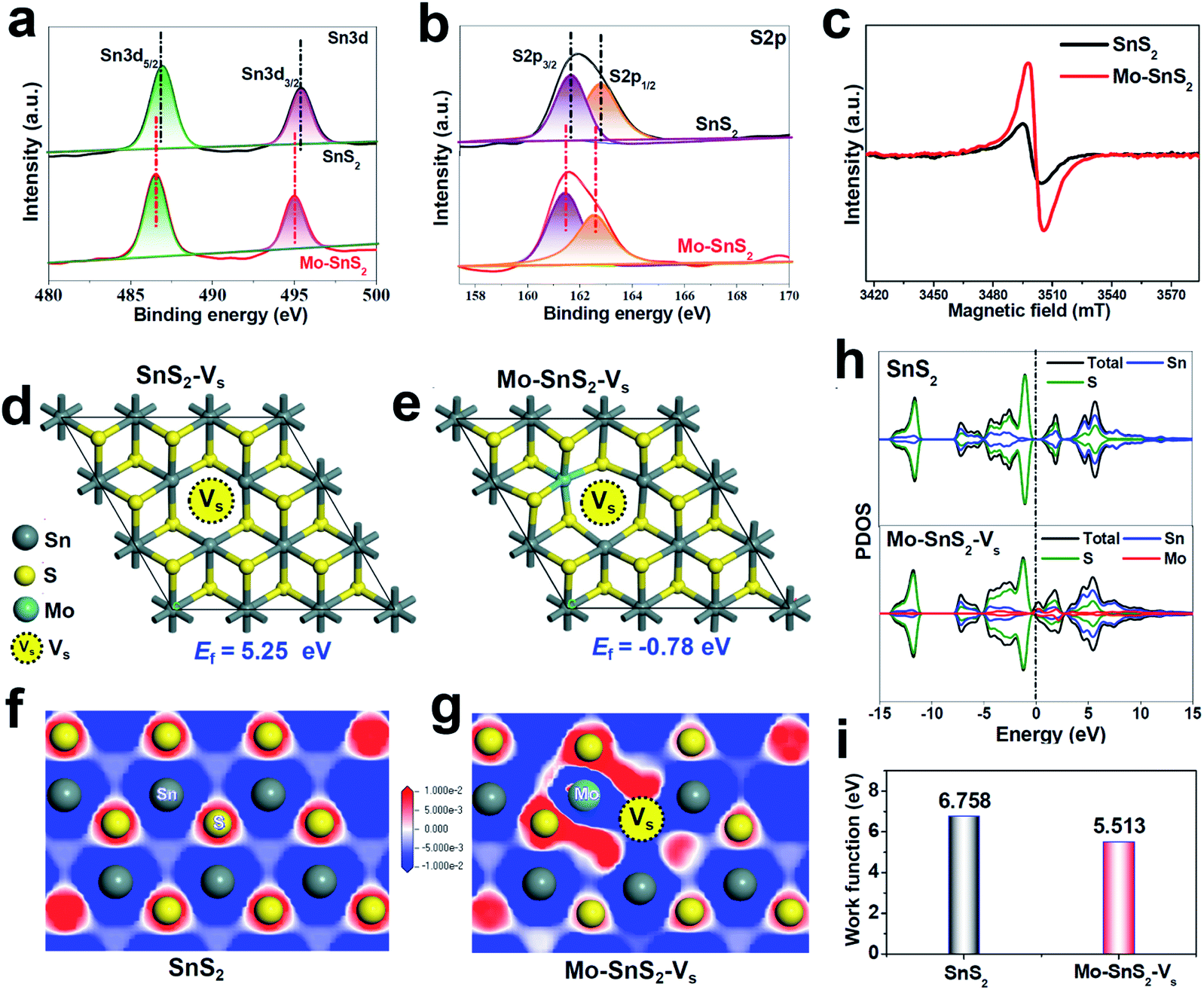 | ||
| Fig. 2 (a and b) XPS spectra of SnS2 and Mo-SnS2 nanosheets: (a) Sn3d and (b) S2p. (c) EPR spectra of SnS2 and Mo-SnS2 nanosheets. (d and e) Optimized SnS2 structures with isolated Vs (SnS2-Vs) and Mo-adjacent Vs (Mo-SnS2-Vs), and the corresponding formation energies (Ef). (f and g) Electron contour maps of the (001) slice for (f) SnS2 and (g) Mo-SnS2-Vs. The blue and red regions represent electron depletion and accumulation, respectively. (h) PDOS of SnS2 and Mo-SnS2-Vs. (i) Calculated work functions of SnS2 and Mo-SnS2-Vs (Fig. S2†). | ||
DFT calculations were used to further investigate the electronic structure of Mo-SnS2-Vs. From the electron contour maps sliced along the (001) plane, it can be seen that more electrons are accumulated in the Mo/Vs region in Mo-SnS2-Vs (Fig. 2g) as compared to SnS2 (Fig. 2f), which is also confirmed by the differential charge density (Fig. S1†). Upon N2 adsorption, it is believed that these Mo/Vs-induced abundant accumulated electrons can be easily transferred into the anti-bonding orbitals of N2 molecules for the weakening and dissociation of the NN bond.46 Projected density of states (PDOS, Fig. 2h) analysis indicates that SnS2 possesses a 0.64 eV bandgap indicative of its semiconducting character. In contrast, the co-presence of the Mo-dopant and Vs creates noticeable electronic states crossing the Fermi level, leading to the metallic characteristics of Mo-SnS2-Vs and thus higher conductivity relative to that of SnS2, which is favorable for the proton-coupled electron-transfer process to boost the NRR kinetics. Meanwhile, the calculated work function (Fig. 2i & S2†) indicates that Mo-SnS2-Vs (5.513 eV) possesses a lower work function than SnS2 (6.758 eV), which suggests the higher capability of Mo-SnS2-Vs for electron back-donation from its active sites to the absorbed N2 and NRR intermediates,47–49 thus facilitating N2 adsorption, activation, and hydrogenation.
The electrocatalytic NRR performance of Mo-SnS2/CC as a self-standing electrode was examined in N2-saturated 0.5 M LiClO4 using a gas-tight two-compartment cell, as displayed in Fig. S3.† An absorber was placed at the end of the cell to prevent the loss of produced NH3 by N2 flow during the NRR test.50 All potentials were converted into values versus the reversible hydrogen electrode (vs. RHE), and the standard RHE was experimentally calibrated using cyclic voltammetry curves in a high-purity hydrogen saturated solution (Fig. S4†).36 The concentration of generated NH3 was experimentally determined by the indophenol blue method,51 while the concentration of the possible N2H4 as a byproduct was measured by the Watt–Chrisp approach.52 Their standard calibration curves are shown in Fig. S5 and S6.† As displayed in Fig. S7,† N2H4 can hardly be detected, implying a high NRR selectivity for Mo-SnS2/CC for N2-to-NH3 conversion.
The polarization curves recorded by linear sweep voltammetry (LSV, Fig. 3a) show a higher current density in N2-saturated solution than in Ar-saturated solution, suggesting the feasibility of the NRR on Mo-SnS2/CC. Before the quantitative evaluation of the NRR performance, a series of control tests were preliminarily carried out to exclude the possible influences from any nitrogen contaminants.53 The isotopic labeling measurements based on 1H nuclear magnetic resonance (NMR) were first utilized to trace the origin of the N source, as shown in Fig. 3b. After NRR electrolysis using 14N2 or 15N2 as the feed gas, a triplet for 14NH4+ (1JN–H = 52 Hz) or a doublet for 15NH4+ (1JN–H = 72 Hz) can be distinguished, respectively, whereas no labeled 14NH4+ or 15NH4+ can be found when using Ar as the feed gas. In addition, UV-vis analysis (Fig. S8†) in an Ar-saturated solution, or at open circuit, or on pristine CC does not produce a detectable amount of NH3.54–56 Furthermore, the time-dependent test (Fig. S9†) shows that the produced NH3 increases linearly with the electrolysis time, indicating that NH3 can be continuously generated by NRR catalysis over Mo-SnS2/CC. We also employed the ion chromatography (IC) technique to quantitatively determine the concentration of NRR-derived NH3. As depicted in Fig. 3c and S10,† the IC measurement (1.27 μg mL−1) is very close to the 1.15 μg mL−1 attained by the indophenol blue method with a reasonable margin of experimental error. Therefore, all these control experiments convincingly demonstrate that the produced NH3 originates from the NRR.
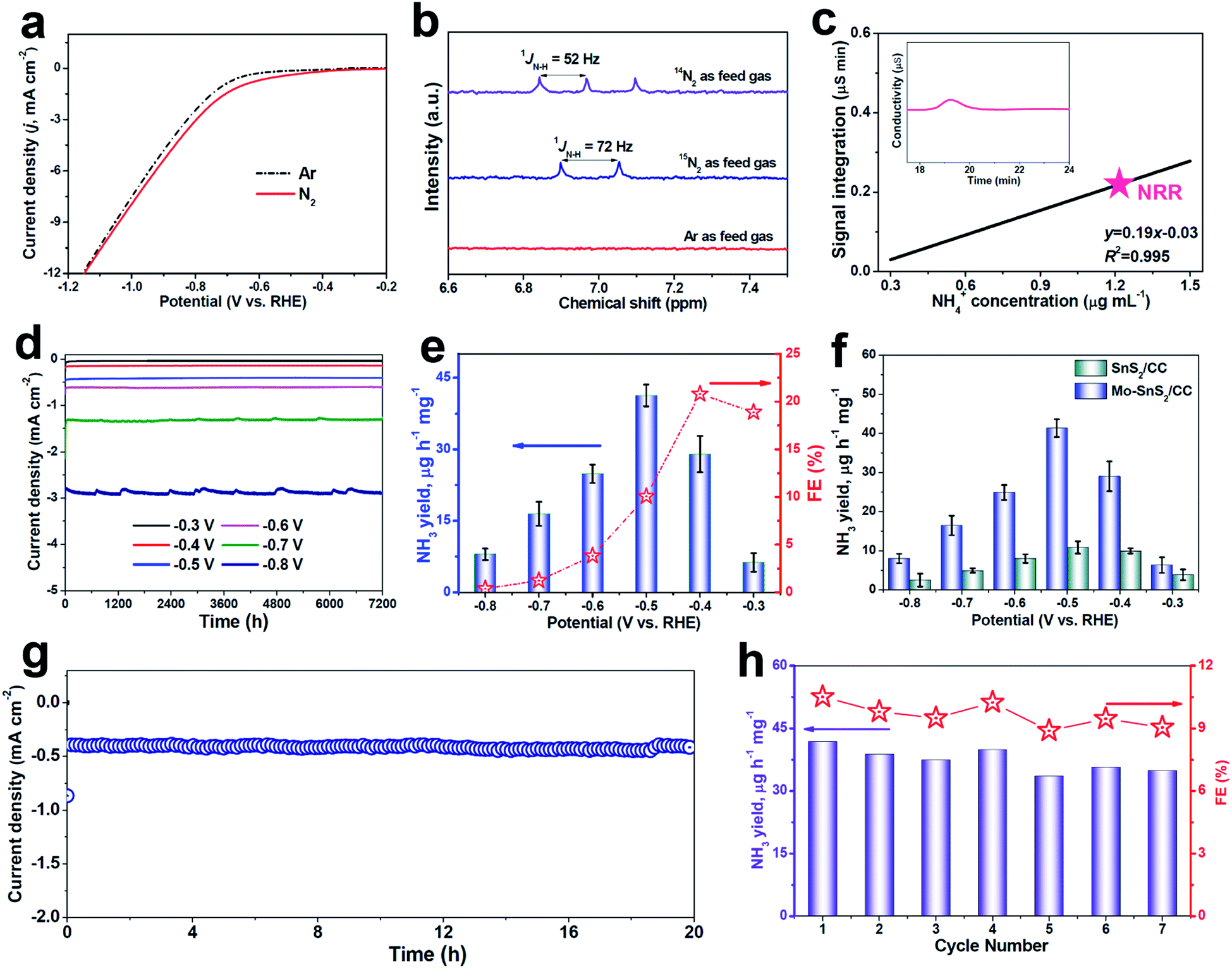 | ||
| Fig. 3 (a) LSV curves of Mo-SnS2/CC in Ar- and N2-saturated solutions. (b) 1H NMR spectra of electrolytes after NRR electrolysis for 2 h on Mo-SnS2/CC using 14N2, 15N2 or Ar as feed gases. (c) IC spectra of the electrolyte after NRR electrolysis on Mo-SnS2/CC for 2 h at −0.5 V (inset), and the determined NH4+ concentration of the electrolyte by referring to the calibration curve (Fig. S10†). (d) Chronoamperometry test results of Mo-SnS2/CC for 2 h of NRR electrolysis at various potentials, and (e) the corresponding NH3 yields and FEs. (f) NH3 yields of SnS2/CC and Mo-SnS2/CC at various potentials. (g) Chronoamperometry test results of Mo-SnS2/CC for 20 h of NRR electrolysis. (h) NH3 yields and FEs of Mo-SnS2/CC for seven cycles (each for 2 h of electrolysis at −0.5 V). | ||
The NRR performance of Mo-SnS2/CC was then quantitatively examined by the combination of chronoamperometry testing with each cycle for 2 h of electrolysis (Fig. 3d) and UV-vis analysis (average of three measurements) at various potentials. The corresponding data of NH3 yield and FE are shown in Fig. 3e. As observed, the NRR activity of Mo-SnS2/CC exhibits optimum NRR performance with an NH3 yield of 41.3 μg h−1 mg−1 at −0.5 V and a faradaic efficiency of 20.8% at −0.4 V. As shown in Table S1,† Mo-SnS2/CC is the best SnS2-based NRR catalyst reported to date and compares favorably to most state-of-the-art NRR catalysts. Nonetheless, the NRR performance shows a sharp decline at more negative potentials, which is attributed to the significantly enhanced HER.57
We also assessed the NRR performance of undoped SnS2/CC for comparison under identical conditions. As shown in Fig. 3f, SnS2/CC possesses a more inferior NRR activity with the highest NH3 yield of 10.6 μg h−1 mg−1 at −0.5 V, which is about one-quarter that of Mo-SnS2/CC (41.3 μg h−1 mg−1), demonstrating that Mo-doping is able to dramatically promote the NRR activity of SnS2 nanosheets. To elucidate the NRR enhancement observed in Mo-SnS2/CC, we determined the electrochemically active surface area (ECSA) by measuring the double-layer capacitance (Cdl). As shown in Fig. S11,† Mo-SnS2/CC exhibits only a 1.3 times higher Cdl, but a ∼3.9 times higher NH3 yield than SnS2/CC, suggesting that the ECSA is not the primary factor and Mo-SnS2/CC is intrinsically more active than SnS2/CC. In addition, as depicted in the electrochemical impedance spectra (EIS, Fig. S12†), Mo-SnS2/CC delivers a smaller charge-transport resistance than SnS2/CC, indicating faster electron-transfer and enhanced NRR reaction kinetics of Mo-SnS2/CC. This can be attributed to the synergistic role of the Mo-dopant and Vs in improving the conductivity of Mo-SnS2, as revealed by the DFT results (Fig. 2h). Therefore, the combined Mo-dopant and Vs can bring about an improved conductivity and elevated intrinsic NRR activity, resulting in significantly enhanced NRR performance of Mo-SnS2/CC.
We further evaluate the NRR stability which is another critical factor for practical applications.58,59 Chronopotentiometric response measurements (Fig. 3g) reveal that the current density over Mo-SnS2/CC presents a negligible degeneration for at least 20 h of continuous electrolysis, verifying the excellent long-term stability. Besides, when conducting seven chronoamperometric runs for Mo-SnS2/CC, there is no remarkable change in the UV-vis spectra of the resultant electrolytes (Fig. S13†), nor in the resulting NH3 yield and FE data (Fig. 3h), confirming the good cycling stability. Further, the morphology, crystal phase and chemical bonding states of Mo-SnS2 nanosheets can be well preserved after the stability test, as evidenced by SEM/TEM (Fig. S14†), XRD (Fig. S15†) and XPS (Fig. S16†) measurements. The outstanding stability of Mo-SnS2/CC is believed to originate from the strong atomic and electronic bonding of Mo-dopants, 2D confinement effect of the nanosheet structure,29 and direct nanosheet growth on CC with tight catalyst attachment.31 Therefore, Mo-SnS2/CC has great potential as a promising catalyst for electroreduction of N2 to NH3 with a favorable NH3 production rate and robust stability.
DFT calculations based on the energetically stable Mo-SnS2-Vs structure (Fig. 2e) were further performed to gain deep insights into the synergistic role of the Mo-dopant and Vs in facilitating the NRR. In view of N2 adsorption as the critical step to initialize the NRR, N2 adsorption behaviors over pristine SnS2 and Mo-SnS2-Vs were first analyzed. For pristine SnS2 (Fig. 4a and b), the N2 molecule barely adsorbs on either the central Sn site (Fig. 4a) or the edge Sn site (Fig. 4b), as evidenced by the negligible charge transfer and much less NN bond elongation (1.105 Å for the original N2 gas), suggesting that pristine SnS2 is almost inactive for the NRR. In sharp contrast, after N2 adsorption on the Vs site of Mo-SnS2-Vs (Fig. 4c), the NN bond is considerably elongated to 1.211 Å and 0.55|e| is injected into *N2, implying the greatly enhanced N2 adsorption on Mo-SnS2-Vs. The enlarged view (Fig. 4d) reveals a unique N2 adsorption mode on the Vs site of Mo-SnS2-Vs, that is, the N2 molecule is fixed to the Mo–Sn–Sn trimer center via a side-on configuration. Meanwhile, the NN triple bond is even cleaved to the NN double bond without hydrogenation, indicating that the Mo–Sn–Sn trimer can strongly activate N2 through an analogous dissociative pattern. As shown in the PDOS of the Mo–Sn–Sn trimer after N2 adsorption (Fig. 4e), the Mo4d and Sn5p orbitals are both considerably hybridized with the N2p orbitals both below and above the Fermi level, suggesting the efficient back-donation of electrons from the Mo–Sn–Sn trimer to *N2.60,61 The prominent back-donation of electrons from the Mo–Sn–Sn trimer to *N2 can be directly visualized from the differential charge density (Fig. S17†), showing the pronouncedly accumulated electrons on *N2. Mulliken charge analysis (Fig. S18†) reveals that the Mo–Sn–Sn trimer back-donates a total of 0.55|e| to *N2, with 0.37|e| from the Mo dopant and 0.09|e| from each of the two Sn atoms. Hence, the co-presence of the Mo dopant and Vs in Mo-SnS2-Vs enables the creation of Mo–Sn–Sn trimer catalytic sites that can strongly activate the N2 molecule even for the cleavage of the NN triple bond to the NN double bond at the N2 adsorption stage.
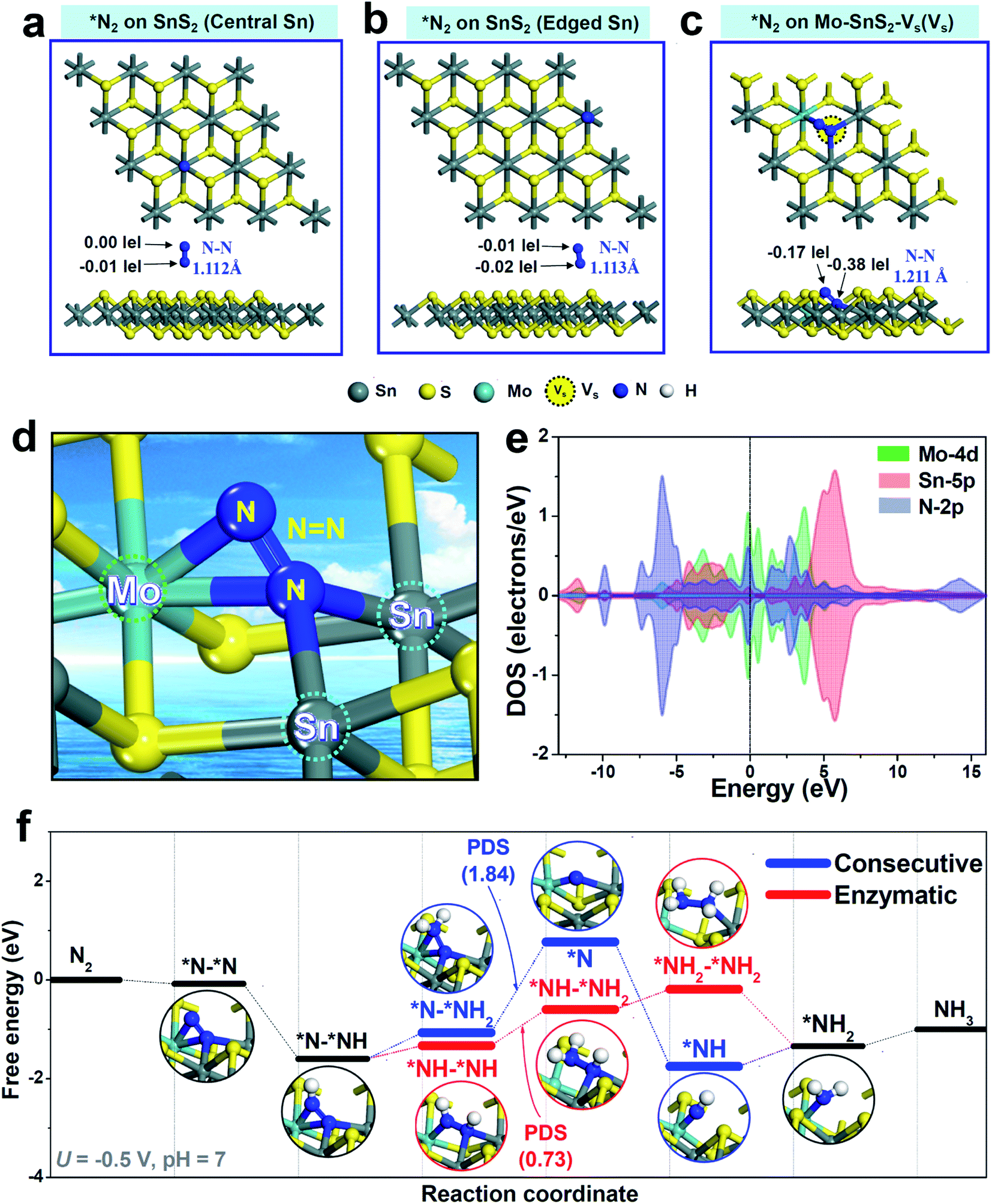 | ||
| Fig. 4 (a–c) Optimized structures of N2 adsorption on (a) SnS2 (central Sn site), (b) SnS2 (edge Sn site) and (c) Mo-SnS2-Vs (Vs site). (d) Enlarged view of the N2 adsorption mode on a Vs site of Mo-SnS2-Vs (Mo–Sn–Sn trimer). (e) PDOS of the Mo–Sn–Sn trimer after N2 adsorption. (f) Free energy diagrams of NRR consecutive/enzymatic reaction pathways over the Mo–Sn–Sn trimer at U = −0.5 V, pH = 7. | ||
The hydrogenation pathway of Mo-SnS2-Vs was then evaluated using Gibbs free energy profiles through the consecutive (analogous to the distal pathway but via side-on configuration62) and enzymatic associative mechanisms, as shown in Fig. 4f. Optimized structures of all the NRR intermediates are presented in Fig. S19† (consecutive) and Fig. S20† (enzymatic). Notably, with the strong N2 activation by the Mo–Sn–Sn trimer, the free energy of *N–*NH even becomes negative, making the first hydrogenation step (*N2 → *N–*NH) occur spontaneously. This is in stark contrast to the largely positive free energy of *N–NH (2.19 eV) observed in pristine SnS2,22 which has the first hydrogenation step (*N2 → *N–NH) as the potential determining step (PDS). After the first hydrogenation, *N–*NH can be further hydrogenated through a consecutive or enzymatic pathway. For the consecutive pathway, *N–*NH2 → *N is the PDS with a high energy barrier of 1.84 eV, while the PDS energy barrier (*NH–*NH → *NH–*NH2) is substantially reduced to 0.73 eV for the enzymatic pathway. Hence, the hydrogenation of Mo-SnS2-Vs prefers to proceed via the enzymatic pathway with an overpotential of 0.57 V,63 which is theoretically lower than that of most reported NRR catalysts.64–67 These results demonstrate that the highly active Mo–Sn–Sn trimer provides Mo-SnS2-Vs with the downhill process of the first hydrogenation step, leading to the significantly decreased energy barrier and largely enhanced NRR activity.
On the other hand, as reported in the literature,68,69 the surface charge and hydrogen bonding can affect the electrocatalytic calculations. As shown in Fig. S21,† after considering the effects of surface charge (adding one charge e− in the catalyst system) and hydrogen bonding (adding two H2O molecules on the catalyst surface), we find that these two factors can make the free energies of *N2 and *N–*NH become more negative but the downhill trend of the *N2 → *N–*NH process still remains. Therefore, our main conclusion is not affected by the effects of surface charge and hydrogen bonding.
In conclusion, through combined experimental and theoretical investigations, we demonstrated that the synergistic modulation of vacancy engineering and heteroatom doping has been successfully achieved in Mo-SnS2 nanosheets with enriched Vs, which showed greatly enhanced NRR performance with an NH3 yield of 41.3 μg h−1 mg−1 (−0.5 V) and an FE of 20.8% (−0.4 V). DFT calculations revealed the unique NRR mechanism of Mo-SnS2-Vs, in which the created Mo–Sn–Sn trimer active sites could strongly activate N2 for even the cleavage of the NN triple bond to the NN double bond at the N2 adsorption stage, consequently resulting in a downhill process of the first hydrogenation step and a largely reduced energy barrier. This work not only offers an efficient strategy towards the design of SnS2-based catalysts for highly efficient electrosynthesis of NH3, but also provides new insights into the synergistic role of vacancies and dopants in regulating the NRR activity.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (51761024), “Feitian Scholar” Program of Gansu Province, CAS “Light of West China” Program, and Foundation of A Hundred Youth Talents Training Program of Lanzhou Jiaotong University.References
- J. A. Brandes, N. Z. Boctor, G. D. Cody, B. A. Cooper, R. M. Hazen and H. S. Yoder Jr, Nature, 1998, 395, 365 CrossRef CAS PubMed.
- X. Zhu, S. Mou, Q. Peng, Q. Liu, Y. Luo, G. Chen, S. Gao and X. Sun, J. Mater. Chem. A, 2020, 8, 1545–1556 RSC.
- G. F. Chen, S. Y. Ren, L. L. Zhang, H. Cheng, Y. R. Luo, K. H. Zhu, L. X. Ding and H. H. Wang, Small Methods, 2019, 3, 1800337 CrossRef.
- S. J. Li, D. Bao, M. M. Shi, B. R. Wulan, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1700001 CrossRef PubMed.
- H. Xie, Q. Geng, X. Zhu, Y. Luo, L. Chang, X. Niu, X. Shi, A. M. Asiri, S. Gao, Z. Wang and X. Sun, J. Mater. Chem. A, 2019, 7, 24760–24764 RSC.
- R. Zhao, C. Liu, X. Zhang, X. Zhu, P. Wei, L. Ji, Y. Guo, S. Gao, Y. Luo, Z. Wang and X. Sun, J. Mater. Chem. A, 2020, 8, 77–81 RSC.
- G. Deng, T. Wang, A. A. Alshehri, K. A. Alzahrani, Y. Wang, H. Ye, Y. Luo and X. Sun, J. Mater. Chem. A, 2019, 7, 21674–21677 RSC.
- H. Xian, Q. Wang, G. Yu, H. Wang, Y. Li, Y. Wang and T. Li, Appl. Catal., A, 2019, 581, 116–122 CrossRef CAS.
- G. Yu, H. Guo, S. Liu, L. Chen, A. A. Alshehri, K. A. Alzahrani, F. Hao and T. Li, ACS Appl. Mater. Interfaces, 2019, 11, 35764–35769 CrossRef CAS PubMed.
- H. Xian, H. Guo, Z. Chen, G. Yu, A. A. Alshehri, K. A. Alzahrani, F. Hao, R. Song and T. Li, ACS Appl. Mater. Interfaces, 2020, 12, 2445–2451 CrossRef CAS PubMed.
- Y. R. Luo, G. F. Chen, L. Ding, X. Z. Chen, L. X. Ding and H. H. Wang, Joule, 2019, 3, 279–289 CrossRef CAS.
- H. Cheng, L. X. Ding, G. F. Chen, L. L. Zhang, J. Xue and H. H. Wang, Adv. Mater., 2018, 30, 1803694 CrossRef PubMed.
- K. Chu, Q. Q. Li, Y. H. Cheng and Y. P. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 11789–11796 CrossRef CAS PubMed.
- H. Cheng, P. X. Cui, F. R. Wang, L. X. Ding and H. H. Wang, Angew. Chem., Int. Ed., 2019, 58, 15541–15547 CrossRef CAS PubMed.
- X. Zhu, T. Wu, L. Ji, C. Li, T. Wang, S. Wen, S. Gao, X. Shi, Y. Luo, Q. Peng and X. Sun, J. Mater. Chem. A, 2019, 7, 16117–16121 RSC.
- L. Xia, X. Wu, Y. Wang, Z. Niu, Q. Liu, T. Li, X. Shi, A. M. Asiri and X. Sun, Small Methods, 2018, 3, 1800251 CrossRef.
- K. Chu, Q. Li, Y. Liu, J. Wang and Y. Cheng, Appl. Catal., B, 2020, 267, 118693 CrossRef CAS.
- L. L. Zhang, L. X. Ding, G. F. Chen, X. F. Yang and H. H. Wang, Angew. Chem., Int. Ed., 2019, 131, 2638–2642 CrossRef.
- L. Xia, J. Yang, H. Wang, R. Zhao, H. Chen, W. Fang, A. M. Asiri, F. Xie, G. Cui and X. Sun, Chem. Commun., 2019, 55, 3371–3374 RSC.
- F. Li, L. Chen, G. P. Knowles, D. R. MacFarlane and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 505–509 CrossRef CAS PubMed.
- P. Li, W. Fu, P. Zhuang, Y. Cao, C. Tang, A. B. Watson, P. Dong, J. Shen and M. Ye, Small, 2019, 15, 1902535 CrossRef PubMed.
- X. Chen, Y.-T. Liu, C. Ma, J. Yu and B. Ding, J. Mater. Chem. A, 2019, 7, 22235–22241 RSC.
- Y. B. Li, Y. P. Liu, J. Wang, Y. L. Guo and K. Chu, Inorg. Chem. Front., 2020, 7, 455–463 RSC.
- L. Yang, T. Wu, R. Zhang, H. Zhou, L. Xia, X. Shi, H. Zheng, Y. Zhang and X. Sun, Nanoscale, 2019, 11, 1555–1562 RSC.
- Z. Sun, R. Huo, C. Choi, S. Hong, T.-S. Wu, J. Qiu, C. Yan, Z. Han, Y. Liu, Y.-L. Soo and Y. Jung, Nano Energy, 2019, 62, 869–875 CrossRef CAS.
- L. Zhang, X.-Y. Xie, H. Wang, L. Ji, Y. Zhang, H. Chen, T. Li, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4627–4630 RSC.
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed.
- X. Yang, J. Nash, J. Anibal, M. Dunwell, S. Kattel, E. Stavitski, K. Attenkofer, J. G. Chen, Y. Yan and B. Xu, J. Am. Chem. Soc., 2018, 140, 13387–13391 CrossRef CAS PubMed.
- H. Jin, L. Li, X. Liu, C. Tang, W. Xu, S. Chen, L. Song, Y. Zheng and S.-Z. Qiao, Adv. Mater., 2019, 31, 1902709 CrossRef PubMed.
- X. H. Wang, J. Wang, Y. B. Li and K. Chu, ChemCatChem, 2019, 11, 4529–4536 CrossRef CAS.
- Y. P. Liu, Y. B. Li, H. Zhang and K. Chu, Inorg. Chem., 2019, 58, 10424–10431 CrossRef CAS PubMed.
- K. Chu, Y. Liu, Y. Chen and Q. Li, J. Mater. Chem. A, 2020, 8, 5200–5208 RSC.
- K. Chu, Y. H. Chen, Q. Q. Li, Y. P. Liu and Y. Tian, J. Mater. Chem. A, 2020, 8, 5865–5873 RSC.
- T. Wu, W. Kong, Y. Zhang, Z. Xing, J. Zhao, T. Wang, X. Shi, Y. Luo and X. Sun, Small Methods, 2019, 1900356 CrossRef CAS.
- C. Guo, X. Liu, L. Gao, X. Kuang, X. Ren, X. Ma, M. Zhao, H. Yang, X. Sun and Q. Wei, Appl. Catal., B, 2020, 263, 118296 CrossRef CAS.
- K. Chu, Y. P. Liu, Y. B. Li, Y. L. Guo, Y. Tian and H. Zhang, Appl. Catal., B, 2020, 264, 118525 CrossRef.
- T. Wu, Z. Xing, S. Mou, C. Li, Y. Qiao, Q. Liu, X. Zhu, Y. Luo, X. Shi, Y. Zhang and X. Sun, Angew. Chem., Int. Ed., 2019, 58, 18449–18453 CrossRef CAS PubMed.
- N. Cao, Z. Chen, K. Zang, J. Xu, J. Zhong, J. Luo, X. Xu and G. Zheng, Nat. Commun., 2019, 10, 1–12 CrossRef CAS PubMed.
- K. Chu, X. H. Wang, Y. B. Li, D. J. Huang, Z. R. Geng, X. L. Zhao, H. Liu and H. Zhang, Mater. Des., 2018, 140, 85–94 CrossRef CAS.
- K. Chu, J. Wang, Y. P. Liu and Z. R. Geng, Carbon, 2018, 140, 112–123 CrossRef CAS.
- K. Chu, X. H. Wang, F. Wang, Y. B. Li, D. J. Huang, H. Liu, W. L. Ma, F. X. Liu and H. Zhang, Carbon, 2018, 127, 102–112 CrossRef CAS.
- K. Chu, J. Wang, Y. P. Liu, Y. B. Li, C. C. Jia and H. Zhang, Carbon, 2019, 143, 85–96 CrossRef CAS.
- K. Chu, F. Wang, X. H. Wang, Y. B. Li, Z. R. Geng, D. J. Huang and H. Zhang, Mater. Des., 2018, 144, 290–303 CrossRef CAS.
- K. Chu, F. Wang, Y. B. Li, X. H. Wang, D. J. Huang and H. Zhang, Carbon, 2018, 133, 127–139 CrossRef CAS.
- L. Meng, S. Wang, F. Cao, W. Tian, R. Long and L. Li, Angew. Chem., Int. Ed., 2019, 58, 6761–6765 CrossRef CAS PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- J. Wang, Y. P. Liu, H. Zhang, D. J. Huang and K. Chu, Catal. Sci. Technol., 2019, 9, 4248–4254 RSC.
- Y. P. Liu, Y. B. Li, D. J. Huang, H. Zhang and K. Chu, Chem.–Eur. J., 2019, 25, 11933–11939 CrossRef CAS PubMed.
- K. Chu, Y. P. Liu, Y. B. Li, J. Wang and H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 31806–31815 CrossRef CAS PubMed.
- L. Li, C. Tang, B. Xia, H. Jin, Y. Zheng and S.-Z. Qiao, ACS Catal., 2019, 9, 2902–2908 CrossRef CAS.
- D. Zhu, L. Zhang, R. E. Ruther and R. J. Hamers, Nat. Mater., 2013, 12, 836 CrossRef CAS PubMed.
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS.
- K. Chu, Y. P. Liu, Y. B. Li, Y. L. Guo and Y. Tian, ACS Appl. Mater. Interfaces, 2020, 12, 7081–7090 CrossRef CAS PubMed.
- J. Yu, C. Li, B. Li, X. Zhu, R. Zhang, L. Ji, D. Tang, A. M. Asiri, X. Sun, Q. Li, S. Liu and Y. Luo, Chem. Commun., 2019, 55, 6401–6404 RSC.
- R. Zhang, L. Ji, W. Kong, H. Wang, R. Zhao, H. Chen, T. Li, B. Li, Y. Luo and X. Sun, Chem. Commun., 2019, 55, 5263–5266 RSC.
- C. Li, J. Yu, L. Yang, J. Zhao, W. Kong, T. Wang, A. M. Asiri, Q. Li and X. Sun, Inorg. Chem., 2019, 58, 9597–9601 CrossRef CAS PubMed.
- A. R. Singh, B. A. Rohr, J. A. Schwalbe, M. Cargnello, K. Chan, T. F. Jaramillo, I. Chorkendorff and J. K. Norskov, ACS Catal., 2016, 7, 706–709 CrossRef.
- P. Wang, Q. Q. Li, Y. H. Cheng and K. Chu, J. Mater. Sci., 2020, 55, 4624–4632 CrossRef CAS.
- F. Wang, Y. P. Liu, H. Zhang and K. Chu, ChemCatChem, 2019, 11, 1441–1447 CrossRef CAS.
- Q. Li, L. He, C. Sun and X. Zhang, J. Phys. Chem. C, 2017, 121, 27563–27568 CrossRef CAS.
- Y.-C. Hao, Y. Guo, L.-W. Chen, M. Shu, X.-Y. Wang, T.-A. Bu, W.-Y. Gao, N. Zhang, X. Su, X. Feng, J.-W. Zhou, B. Wang, C.-W. Hu, A.-X. Yin, R. Si, Y.-W. Zhang and C.-H. Yan, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- L. Shi, Q. Li, C. Ling, Y. Zhang, Y. Ouyang, X. Bai and J. Wang, J. Mater. Chem. A, 2019, 7, 4865–4871 RSC.
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- K. Chu, Y. Liu, J. Wang and H. Zhang, ACS Appl. Energy Mater., 2019, 2, 2288–2295 CrossRef CAS.
- K. Chu, Y. Liu, Y. Li, H. Zhang and Y. Tian, J. Mater. Chem. A, 2019, 7, 4389–4394 RSC.
- Z. Wang, F. Gong, L. Zhang, R. Wang, L. Ji, Q. Liu, Y. Luo, H. Guo, Y. Li, P. Gao, X. Shi, B. Li, B. Tang and X. Sun, Adv. Sci., 2018, 1801182 Search PubMed.
- X. Li, T. Li, Y. Ma, Q. Wei, W. Qiu, H. Guo, X. Shi, P. Zhang, A. M. Asiri, L. Chen, B. Tang and X. Sun, Adv. Energy Mater., 2018, 8, 1801357 CrossRef.
- X. Zhao and Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed.
- D. Kim, J. Shi and Y. Liu, J. Am. Chem. Soc., 2018, 140, 9127–9131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ta01688h |
| This journal is © The Royal Society of Chemistry 2020 |