
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocatalytic asymmetric ring-opening of dihydroisoxazoles: a cyanide-free route to complementary enantiomers of β-hydroxy nitriles from olefins†‡
Daijun
Zheng
and
Yasuhisa
Asano
 *
*
Biotechnology Research Center and Department of Biotechnology, Toyama Prefectural University, 5180 Kurokawa, Imizu, Toyama 939-0398, Japan. E-mail: asano@pu-toyama.ac.jp
First published on 24th June 2020
Abstract
By combination of the cyanide-free synthesis of chiral nitriles and the Kemp elimination reaction catalyzed by aldoxime dehydratases, we herein report a new application of aldoxime dehydratase in the asymmetric ring-opening of 5-sub-4,5-dihydroisoxazoles to synthesize chiral β-hydroxy nitriles with broad substrate scope, excellent enantioselectivity (up to 99% ee), and good turnover number (up to 11 s−1). Upon simple isolation and treatment with an alkaline reagent, the remaining chiral 5-sub-4,5-dihydroisoxazoles can be easily transformed into their corresponding β-hydroxy nitriles. Using site-directed mutagenesis, a ferrous Heme-containing active site was confirmed and two possible deprotonation pathways were proposed. To the best of our knowledge, this is the first enzymatic reaction used to construct a chiral hydroxyl group and nitrile group in one-step starting from a simple alkene, which provides a novel and useful strategy for the synthesis of complementary enantiomers of β-hydroxy nitriles.
Introduction
Chiral β-hydroxy nitriles are versatile building blocks that can be easily transformed into chiral β-hydroxy acids, γ-hydroxy amines, lactones etc.,1–4 which are important motifs of pharmaceutical and biologically active molecules (Scheme 1), and can be used as potential monomers for the preparation of biodegradable plastics.5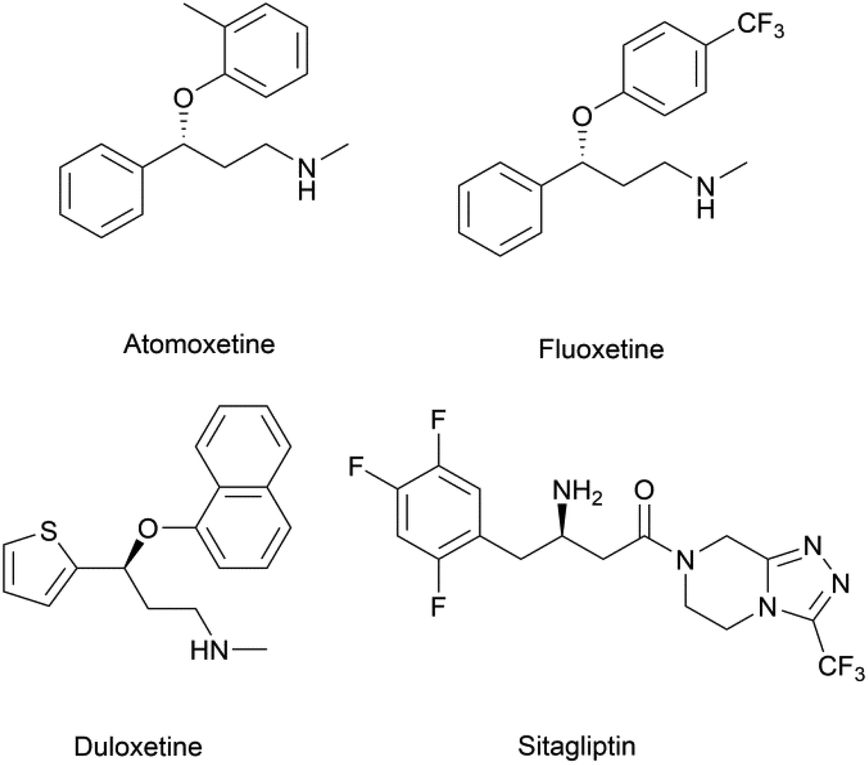 | ||
| Scheme 1 Biologically-active molecules containing the motifs derived from β-hydroxy nitriles. | ||
Due to the versatility of this family of compounds, the methods used for their efficient synthesis have been extensively explored over the last few decades. The most common strategy used in organic synthesis involves asymmetric transfer hydrogenation (ATH) to reduce keto-nitrile precursors into their corresponding hydroxyl nitrile compounds using transition-metal complexes such as ruthenium(II)-complex6–10 and iridium-complexes.11–13 In addition, alternative methods using the β-borylation of organoborane compounds with acceptor of α,β-unsaturated nitriles catalysed by Copper-complex following with oxidation to synthesize corresponding alcohols have been reported.14–16 What's more, the asymmetric cross aldol reaction of aldehydes with different nucleophilic addition donors and catalysts, such as acetonitrile/Cu alkoxide-chiral phosphine complex,17 TMSCH2CN/copper fluoride,18 organolithium reagents,19 cyanomethylzinc bromide/DPMPM20 have been well developed. Moreover, the region-selective ring-opening of epoxide with cyanide was also a classic synthetic method accessing to β-hydroxy nitriles.21 In general, all these synthetic routes inevitably use costly chiral transition-metal complexes as catalysts to construct the chiral centre. Nowadays, benefitting from the great development of biotechnology and green chemistry, a variety of synthetic strategies to β-hydroxy nitriles have been developed using purified enzymes or whole cell catalysts. Among them, lipases,2,22,23 reductases,1,24–26 dehydrogenases,27 and nitrilases28,29 have been mostly employed toward the synthesis of chiral β-hydroxy nitrile via the kinetic resolution of racemic hydroxyl nitriles or the asymmetric reduction of β-oxo-nitriles. In addition, few publications have reported the use of monooxygenase for the asymmetric hydroxylation of 3-phenylpropanenitrile30 or multi-enzymatic biosynthesis system of alcohol dehydrogenase/halohydrin dehalogenase treating on α-halo ketones and cyanide31 to form this family of chiral compounds. In part, these biocatalytic methods overcome the use of costly transition-metal catalysts, but are confined to a limited scope of precursors, including keto-nitriles and racemic hydroxyl nitriles, which are prepared using tedious synthetic processes or inevitably use poisonous cyanide as a starting material. By combination of cyanide-free synthesis of chiral nitriles (Scheme 2a)32 and the Kemp elimination reaction (Scheme 2b)33 catalysed by aldoxime dehydratases (EC 4.99.1.5, EC 4.99.1.6, EC 4.99.1.7), we herein report a novel cyanide-free enzymatic approach to construct a chiral hydroxyl group and nitrile group in one-step toward the efficient synthesis of the complementary enantiomers of β-hydroxy nitriles via the asymmetric ring-opening of 5-sub-4,5-dihydroisozazoles (Scheme 2c).
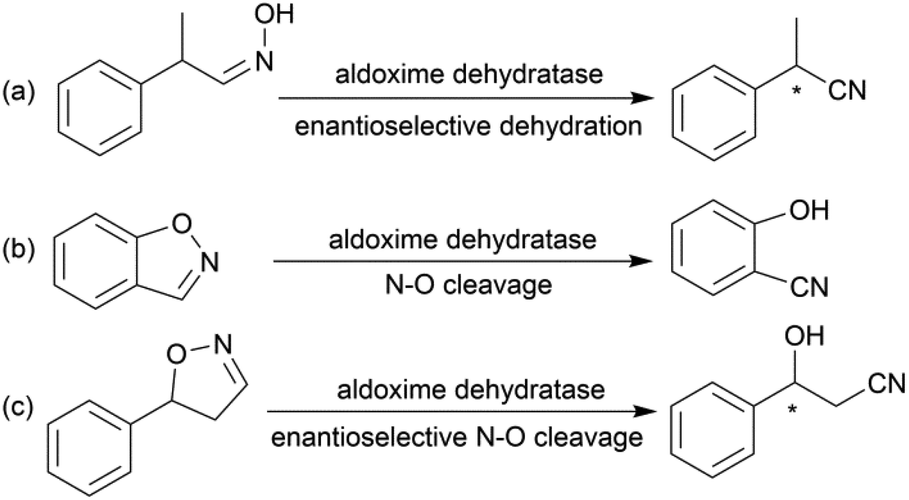 | ||
| Scheme 2 (a) Enantioselective dehydration catalysed by aldoxime dehydratase. (b) The Kemp elimination reaction catalysed by aldoxime dehydratase. (c) Asymmetric ring-opening of dihydroisoxazole catalysed by aldoxime dehydratase. | ||
Aldoxime dehydratase is a class of Heme-containing enzyme involved in the cyanogenesis pathway observed in nature, which catalyses the dehydration of aldoximes to their corresponding nitrile derivatives.34 Using the dehydration function of the enzyme, we established a cyanide-free platform for the synthesis of nitriles utilizing aldoxime precursors.35–38 Recently, we discovered the enzyme catalyses the Kemp elimination reaction via a –Fe–N– complex catalytic pathway,33 which is different from the classic acid–base mechanism.39,40 In this enzymatic elimination reaction, the N–O bond is initially opened with the action of the –Fe–N– complex (Heme Fe-proximal histidine) followed by deprotonation by a distal histidine residue to form the nitrile triple-bond. Upon investigation of the catalytic mechanism, it has been reported that the Kemp elimination reaction can also occur based on the redox catalysis of Heme-containing P450-BM3 via heterolysis to liberate an intermediate containing a nitrogen radical moiety (Fe(III)–N˙) and phenoxyl anion. The product is formed by bond rotation and proton-transfer to the resulting phenoxyl anion.41 It was also mentioned that the metabolism of leflunomide, an immunomodulatory therapeutic drug, could be transferred into teriflunomide via isoxazole ring-opening catalysed by human cytochrome P450. This provides the idea of synthesizing a chiral β-hydroxy nitrile product using a prochiral substrate containing the dihydroisoxazole motif. To verify this hypothesis, the substrate 5-phenyl-4,5-dihydroisoxazole was designed and tested in the reaction with aldoxime dehydratase Oxd-B obtained from Bacillus sp. OxB-1.42–44
Results and discussion
In the initial screening process, the reaction was carried out in the absence of a reductant, which showed excellent enantioselectivity on the cleavage of the N–O bond as expected, but the activity was barely satisfactory. In view of the important role of a reductant in the aldoxime dehydration45 and Kemp elimination reactions,33 which can significantly enhance the enzyme activity, different reductants were tested for the asymmetric ring-opening of 5-phenyl-4,5-dihydroisoxazole (Fig. S1‡). The enhanced effect on the enzymatic activity upon the addition of a reductant was observed, which showed the activity can be improved 1.9–6.0 fold using a variety of reductants. In addition, the enantioselectivity of the enzyme was not affected by the additives with the ee of the product being maintained at ≥99% in each case. The reductant effect implies a ferrous-Heme active state was involved in the reaction, which is consistent with the sensitivity of Oxd B to oxygen.42The classic Kemp elimination reaction can take place under alkaline conditions via an acid–base mechanism.40 Accordingly, this reaction was investigated in a range of different pH buffers (Fig. 1a). Although the yield varied significantly at different pH in a various buffer types, the enzyme could work at pH 5.0–10.0 with the desired product formed in 2–36% yield, and the optical purity of the product was maintained at ≥99% at pH 5.0–8.0. From the control results, spontaneous reactions were detected in pH 9.0–10.0 (Fig. 1b), which caused a sharp decrease in the product ee. Enlightened by these results, an useful method to prepare racemic β-hydroxy nitriles is described from alkenes via a 5-sub-4,5-dihydroisoxazole intermediate46,47 upon treatment with a methanolic NaOH solution at room temperature (Scheme 3, and detailed data are shown in the ESI, section 5‡). The result is consistent with the previous reports that β-hydroxy nitriles were formed by treating dihydroisoxazoles with bases (such as trimethylamine,48 1,8-diazabicyclo[5.4.0]undec-7-ene,49 NaOMe,50 ring-opening of 3-bromo-2-isoxazolines upon treatment with an alkanethiolate.51
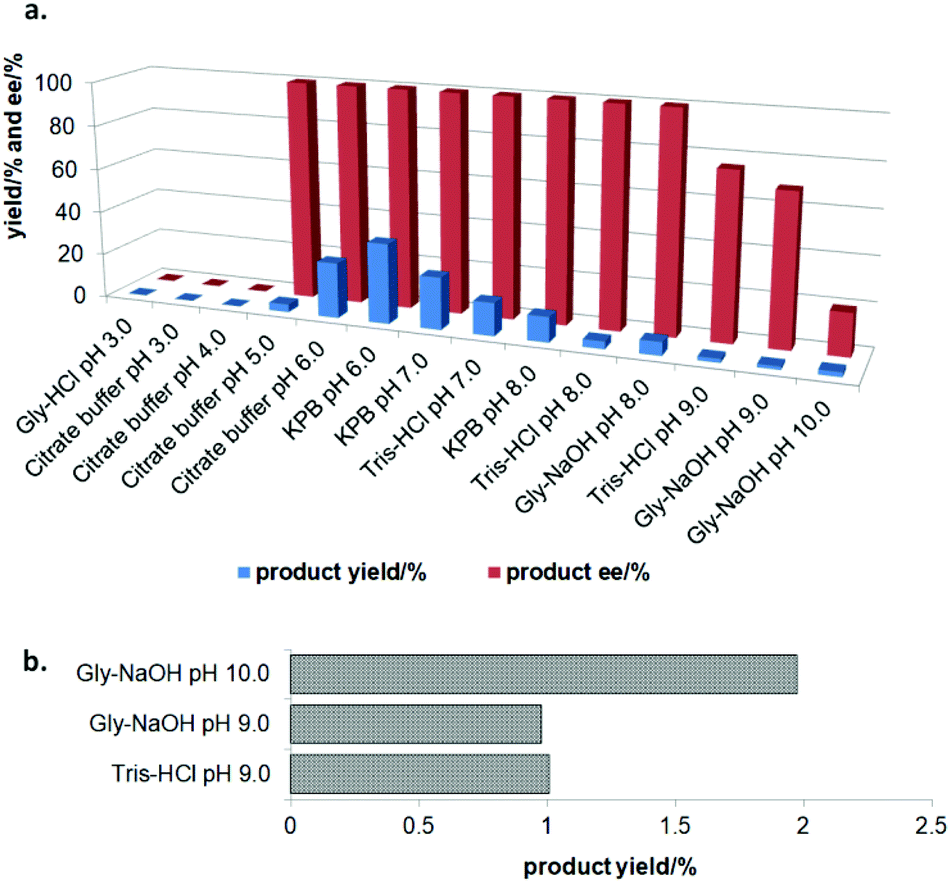 | ||
| Fig. 1 (a) The pH effect on the activity of Oxd B in the catalytic asymmetric ring-opening of 5-phenyl-4,5-dihydroisoxazole. The reactions were performed on a 500 μL scale using 100 mM buffer (pH 3.0–10.0), 1 U Oxd-B, 20 mM 5-phenyl-4,5-dihydroisoxazole, 0.05 mM FMN, and 1 mM Na2S2O5 at 30 °C for 30 min. The buffer solutions used were Gly-HCl (pH 3.0), Citrate-buffer (pH 3.0, 4.0, 5.0, and 6.0), KPB (pH 6.0, 7.0, and 8.0), Tris-HCl (pH 7.0, 8.0, and 9.0), and Gly-NaOH (pH 8.0, 9.0, and 10.0). (b) The control experiments were carried out without Oxd B at pH 9.0 and pH 10.0. The yield and ee were determined using chiral HPLC (n = 2). | ||
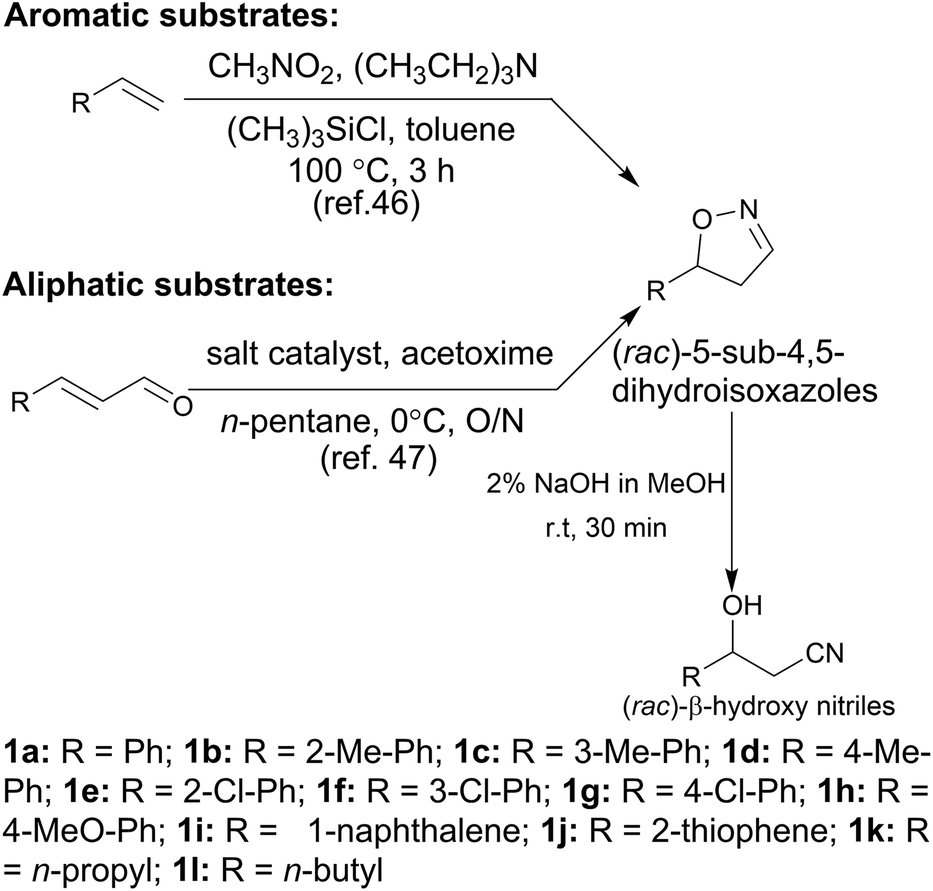 | ||
| Scheme 3 Synthesis of dihydroisoxazoles and (rac)-β-hydroxy nitriles from alkenes. | ||
To further improve the product yield, the reaction was monitored for a prolonged time of 3 h (Fig. 2). During the enantioselective transformation of the substrate, the ee of the substrate 5-phenyl-4,5-dihydroisoxazole (1a) increased to 99% over 2 h and the product (2a) ee was maintained at 99%, indicating the excellent catalytic enantioselectivity of Oxd B on this substrate. Additionally, it is often beneficial to agitate a reaction to improve the mass transfer process and as a result, enhance the reaction rate. However, a ∼5-fold decrease in the yield of the β-hydroxy nitrile product was observed upon shaking the reaction when compared to the static-state reaction over 1 h. As reported in the dehydration of aldoximes, the activity of Oxd B can be increased by 5-fold under anaerobic conditions.42 Therefore, the oxygen sensitivity of Oxd B seems to be the reason of the decrease in the enzymatic activity in a shaking mode. By shaking, the enzyme is much more susceptible to exposure to oxygen in the air, which leads to the oxidation of ferrous-Heme to ferric-Heme, the resting-state of Heme in the ring-opening reaction of dihydroisoxazole.52,53
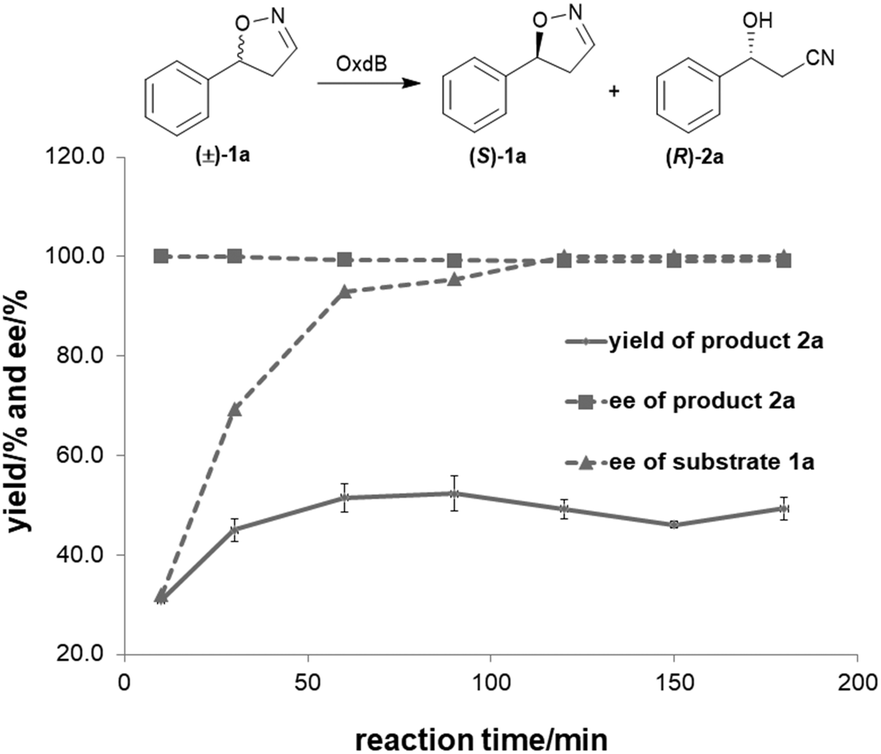 | ||
| Fig. 2 Time course of the asymmetric ring-opening of 5-phenyl-4,5-dihydroisoxazole using Oxd B. The reactions were performed on a 500 μL scale using 100 mM KPB buffer (pH 6.0), 1 U Oxd B, 20 mM 5-phenyl-4,5-dihydroisoxazole, 0.05 mM FMN, and 1 mM Na2S2O5 at 30 °C. The yield and ee were determined using chiral HPLC (n = 2). | ||
With the optimized reaction conditions, a series of substrates were investigated using Oxd B (Table 1). Oxd B exhibited good selectivity toward most aromatic substrates with desired product S/R-2 formed in 48–99% ee. Among them, substrates bearing an electron-donating group on the aryl ring showed relatively higher ee values than those bearing an electron-withdrawing group. In addition, the enzyme preferred substrates bearing a substituent at the para-position (Table 1, 1d, 1g, and 1h), rather than the ortho- (Table 1, 1b and 1e) or meta-position (Table 1, 1c and 1f). The selectivity toward heterocyclic aromatic substrates (Table 1, 1j) was also very excellent, but a slight decrease in activity was observed. However, increasing the size of the aromatic ring to a naphthalene group leads to the reaction conversion becoming negligible (Table 1, 1i), which may be caused by the lower solubility of the substrate in water. For aliphatic substrates (Table 1, 1k and 1l), Oxd B exhibited a deterioration in the reaction selectivity.
|
|
||||||
|---|---|---|---|---|---|---|
| Substrate (±)-1 | Enzyme | Conv.b [%] | ee of S/R-1 [%] | ee of S/R-2c [%] | Relative activityd [%] | |
| a For Oxd A, the reactions were performed on a 500 μL scale using 100 mM KPB buffer (pH 7.0), 10 U Oxd A, 20 mM substrate, and 4 mM Na2S2O4 at 30 °C for 3 h (n = 2); for Oxd B, the reactions were performed on a 500 μL scale using 100 mM KPB buffer (pH 6.0), 20 U Oxd-B, 20 mM substrate, 0.05 mM FMN, and 1 mM Na2S2O5 at 30 °C for 2 h (n = 2); and for Oxd RE, the reactions were performed on a 500 μL scale using 100 mM KPB buffer (pH 7.0), 1 U Oxd RE, 6 mM substrate, 1 mM Na2S at 30 °C for 2 h (n = 2). b The conversion was calculated using conv = ees/(ees + eep).57 c The ee was determined using chiral HPLC and chiral GC. d The activity for substrate 1a was taken to be 100%. The relative activity for Oxd B was determined on a 500 μL scale using 100 mM KPB buffer (pH 6.0), 1 U Oxd B, 20 mM substrate, 0.05 mM FMN, and 1 mM Na2S2O5 at 30 °C for 2 h (n = 2). | ||||||
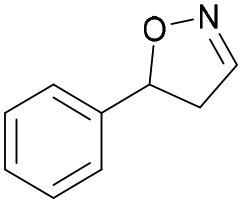
| 1a |
|
Oxd A | 93 | 24 (S) | 2 (R) | 100 |
| Oxd B | 51 | 99 (S) | 96 (R) | 100 | ||
| Oxd RE | 64 | 68 (S) | 38 (R) | 100 | ||
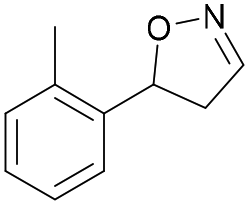
| 1b |
|
Oxd A | 70 | 25 (R) | 11 (S) | 75 |
| Oxd B | 31 | 43 (S) | 95 (R) | 8 | ||
| Oxd RE | 21 | 10 (R) | 36 (S) | 33 | ||
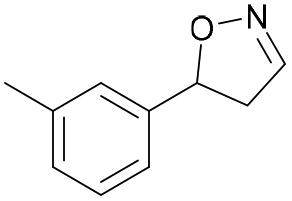
| 1c |
|
Oxd A | 70 | 77 (S) | 33 (R) | 75 |
| Oxd B | 47 | 76 (S) | 86 (R) | 42 | ||
| Oxd RE | 20 | 17 (S) | 66 (R) | 32 | ||
| 1d |
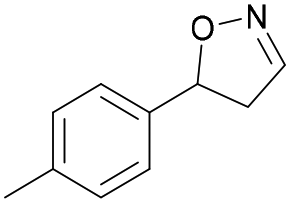
|
Oxd A | 95 | 21 (S) | 1 (R) | 102 |
| Oxd B | 32 | 45 (S) | 97 (R) | 8 | ||
| Oxd RE | 23 | 12 (S) | 41 (R) | 36 | ||
| 1e |
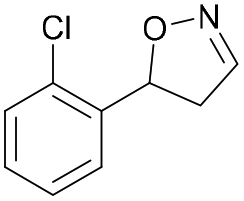
|
Oxd A | 67 | 25 (R) | 12 (S) | 74 |
| Oxd B | 47 | 47 (S) | 53 (R) | 55 | ||
| Oxd RE | 59 | 18 (R) | 13 (S) | 93 | ||
| 1f |
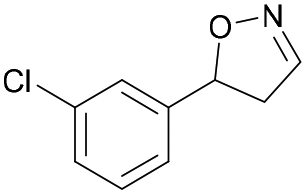
|
Oxd A | 52 | 19 (S) | 18 (R) | 55 |
| Oxd B | 53 | 64 (S) | 56 (R) | 38 | ||
| Oxd RE | 38 | 38 (S) | 62 (R) | 59 | ||
| 1g |
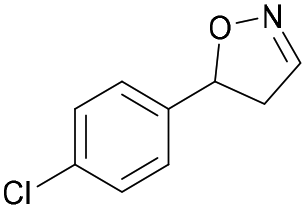
|
Oxd A | 94 | 99 (S) | 6 (R) | 101 |
| Oxd B | 40 | 63 (S) | 95 (R) | 28 | ||
| Oxd RE | 39 | 18 (S) | 28 (R) | 61 | ||
| 1h |
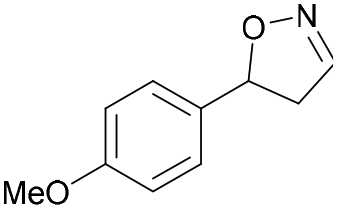
|
Oxd A | 74 | 77 (S) | 27 (R) | 79 |
| Oxd B | 47 | 87 (S) | 99 (R) | 25 | ||
| Oxd RE | 23 | 23 (S) | 77 (R) | 36 | ||
| 1i |
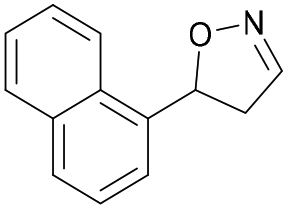
|
Oxd A | 0.5 | 0 (—) | 13 (—) | 0 |
| Oxd B | 8 | 6 (—) | 71 (—) | 7 | ||
| Oxd RE | 9 | 1 (—) | 6 (—) | 15 | ||
| 1j |
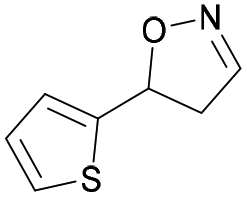
|
Oxd A | 77 | 9 (S) | 3 (R) | 83 |
| Oxd B | 49 | 96 (S) | 99 (R) | 79 | ||
| Oxd RE | 79 | 75 (S) | 20 (R) | 123 | ||
| 1k |
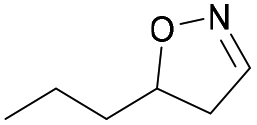
|
Oxd A | 83 | 84 (R) | 17 (S) | 89 |
| Oxd B | 67 | 99 (R) | 48 (S) | 12 | ||
| Oxd RE | 31 | 39 (R) | 87 (S) | 48 | ||
| 1l |
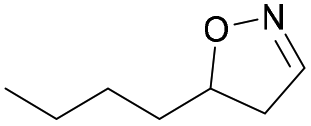
|
Oxd A | 88 | 87 (R) | 11 (S) | 95 |
| Oxd B | 59 | 94 (R) | 66 (S) | 80 | ||
| Oxd RE | 38 | 49 (R) | 82 (S) | 58 | ||
To establish an efficient catalytic system for aliphatic substrates, another two aldoxime dehydratases obtained from Pseudomonas chlororaphis B23 (Oxd A)54,55 and Rhodococcus sp. N-771 (Oxd RE),45,56 were selected and investigated using a range of different substrates. They were found to display higher activity toward aliphatic substrates when compared with aromatic substrates in the aldoxime dehydration reaction.
Prior to investigating the substrate scope, optimization of the reductant was also carried out. Oxd A showed the best activity with sodium dithionite (Na2S2O4), which was ∼79-fold higher than that observed in the control experiment performed in the absence of a reductant (Fig. S7‡). Oxd RE preferred sodium sulfide (Na2S), which exhibited a ∼8.2-fold higher activity than the control (Fig. S3‡). The optimal reductant concentration for Oxd A and Oxd RE were 4 mM (Fig. S8‡) and 1 mM (Fig. S4‡), respectively. Upon monitoring the asymmetric ring-opening reaction of 5-phenyl-4,5-dihydroisoxazole with time, Oxd A could catalyse the reaction in 83% yield over 3 h, but gave a nearly racemic product (Fig. S9‡). The same result was also obtained using Oxd RE, the product was formed in 28% yield and 53% ee at pH 6.0, and 48% yield and 55% ee at pH 7.0 over 2 h, respectively (Fig. S5‡). Our substrate specificity investigations show that Oxd A exhibited very poor enantioselectivity toward the substrates studied, though the conversion is high (Table 1). Oxd RE showed relatively better results toward aliphatic substrates when compared to aromatic substrates. In addition, it also exhibited slightly better enantioselectivity than Oxd B in the asymmetric ring-opening of aliphatic substrates (Table 1; 1k and 1l). Interestingly, Oxd A and Oxd RE exhibited different selectivity toward ortho-substituted aromatic substrates when compared with Oxd B (Table 1; 1b and 1e). What causes this difference in selectivity compared with other substrates and Oxd B is still not clear.
The kinetic parameters for both Oxd B and Oxd RE were determined under the optimized reaction conditions (Table S2‡). The turnover number of Oxd B reached 11 s−1 with a specific activity of 18 U mg−1, which is about one third of the activity observed for (Z)-phenylacetaldoxime dehydration (53 U mg−1) using the assay method containing Na2S2O5 (ESI, section 3‡). However, the Oxd RE activity was lower than Oxd B at 4 U mg−1 with a turnover number of 3 s−1, ∼15% of the activity observed for the (Z)-phenylacetaldoxime dehydration reaction (30 U mg−1). In addition, the catalytic efficiency of Oxd B (Kcat/Km = 3.7 × 104 M−1 s−1) is ∼9-fold higher than Oxd RE (Kcat/Km = 4.3 × 103 M−1 s−1). An obvious substrate inhibition effect was observed using 1 U of the enzyme upon increasing the substrate concentration to 30 mM for Oxd B and 6 mM for Oxd RE, but no any effect on the optical purity of the product (Fig. S2 and S6‡). Therefore, a scale-up reaction was performed based on a substrate concentration of 20 mM and 1 U Oxd B. Under the optimal conditions, the reaction concentration could reach 150 mM with the product formed with 50% conversion and 99% ee (Table S1‡).
To elucidate the reaction mechanism, a homology model of Oxd B was established based on the crystal structure of Oxd-RE.58 In addition, the corresponding residues located in the catalytic pocket of the Heme were selected for an alanine-scanning study (Fig. 3a). In this scanning study, L128A, T202A, Q204A, H282A, F289A, and H306A mutants were constructed via site-mutagenesis (ESI, section 7‡), which were purified as the wild type of Oxd B to obtain the pure enzyme (ESI, section 2.2‡) and used for activity comparison. To begin with, the activity of the mutants toward (Z)-phenylacetaldoxime was assayed. As reported in previous publication, no activity was detected for the T202A, H282A, and H306A mutants, which are corresponding to the S219, H299, and H320 residues in Oxd RE, the crucial residues in the dehydration of butyraldehyde oxime.33 These results reveal that the homology model of Oxd B is workable to some extent, although the protein sequence homology between Oxd B and Oxd RE is only 31%. After verifying the reliability of the homology model, the mutants were used in the 5-phenyl-4,5-dihydroisoxazole ring-opening reaction. As expected, the H282A mutant showed no activity toward the substrate because the proximal axial residue ligating to iron is thought to affect the redox potential of the Heme, thus leading to a great difference in the catalytic activity for bond cleavage.59–61 Surprisingly, the activity of the mutants was determined to be 20% for T202A and 5% for H306A when compared with wild-Oxd B (Fig. 3b), which is similar to that reported in the Kemp elimination reaction.33 To confirm the transformation in mutant of H306A, the amount of Oxd B-H306A was increased 10-fold, and the product could be obtained in 3.8% yield and 99% ee over 10 min, 21% yield and 95% ee in 1 h, respectively. This implied a discriminating catalytic mechanism between the ring-opening of dihydroisoxazole and aldoxime dehydration reactions. It seems that the ring-strain in dihydroisoxazole could facilitate the cleavage of the N–O bond via redox of ferrous-Heme without the assistance of the other residues (T202 and H306 in the (Z)-phenylacetaldoxime dehydration reaction), even though its catalytic efficiency was slower than that observed under the assistance of the other residues. In particular, weak activity was detected for H306A, which is thought to play an important role in the next deprotonation step to form the nitrile triple-bond (Scheme 4a). This suggests that in addition to the efficient deprotonation step by histidine (H306), it may have another route for deprotonation, which we refer to as the oxygen anion deprotonation pathway.41 After ring-opening upon attack of the ferrous-Heme, the resulting oxygen anion will approach the hydrogen atom of H–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– (via single bond rotation) followed by deprotonation and electron transfer back to ferric-Heme to produce the active state of ferrous-Heme, which undergoes the next catalytic cycle (Scheme 4b). We also noticed that the product ee did not change significantly in the presence of any mutants. This reveals the cooperative control of the enantioselectivity with the surrounding residues of the Heme, not only from certain residues in the catalytic pocket.
N– (via single bond rotation) followed by deprotonation and electron transfer back to ferric-Heme to produce the active state of ferrous-Heme, which undergoes the next catalytic cycle (Scheme 4b). We also noticed that the product ee did not change significantly in the presence of any mutants. This reveals the cooperative control of the enantioselectivity with the surrounding residues of the Heme, not only from certain residues in the catalytic pocket.
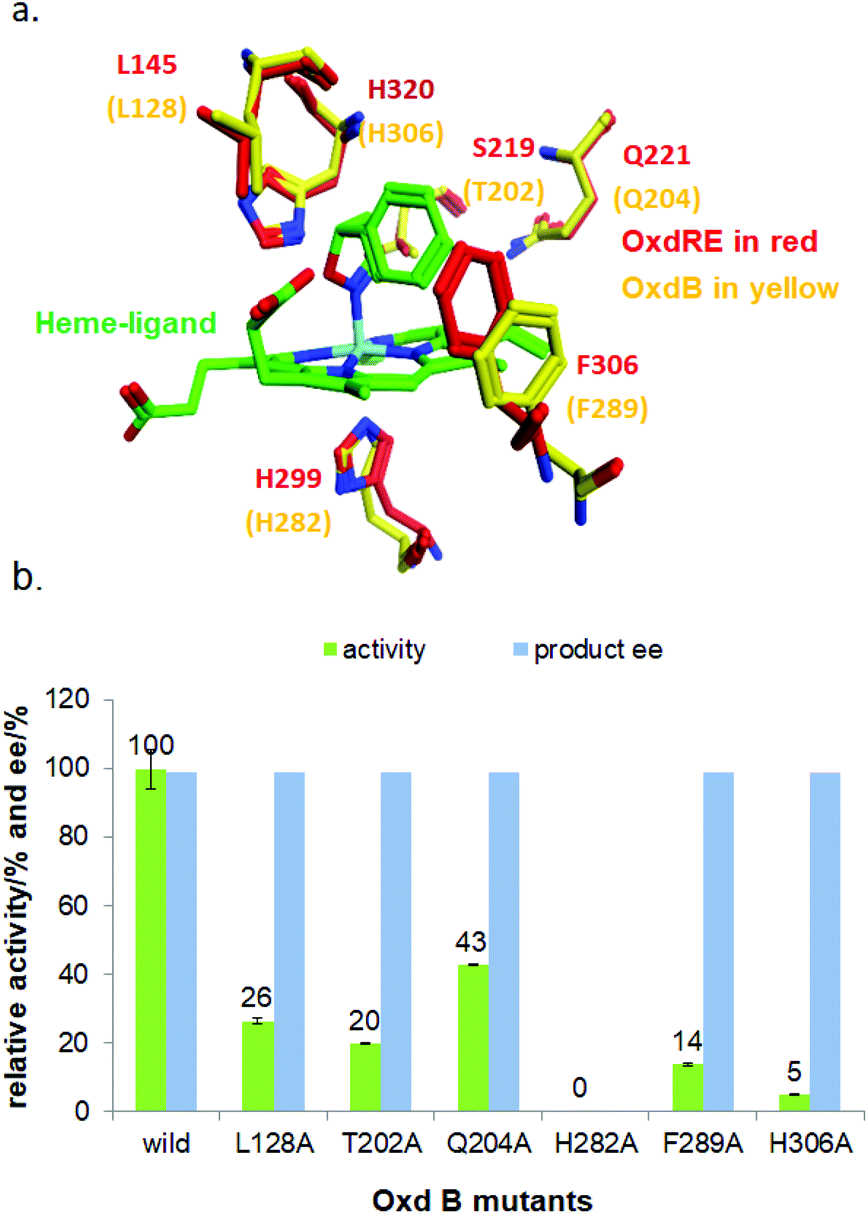 | ||
| Fig. 3 (a) Oxd B homology model and superposition to Oxd RE structure docking with substrate 1a; (b) Comparison of the Oxd B mutants’ activities for substrate 1a. The reactions were performed on a 500 μL scale using 100 mM KPB buffer (pH 6.0), 2 μg of Oxd-B mutants, 20 mM 5-phenyl-4,5-dihydroisoxazole, 0.05 mM FMN, and 1 mM Na2S2O5 at 30 °C for 10 min. The yield and ee were determined using chiral HPLC (n = 2). | ||
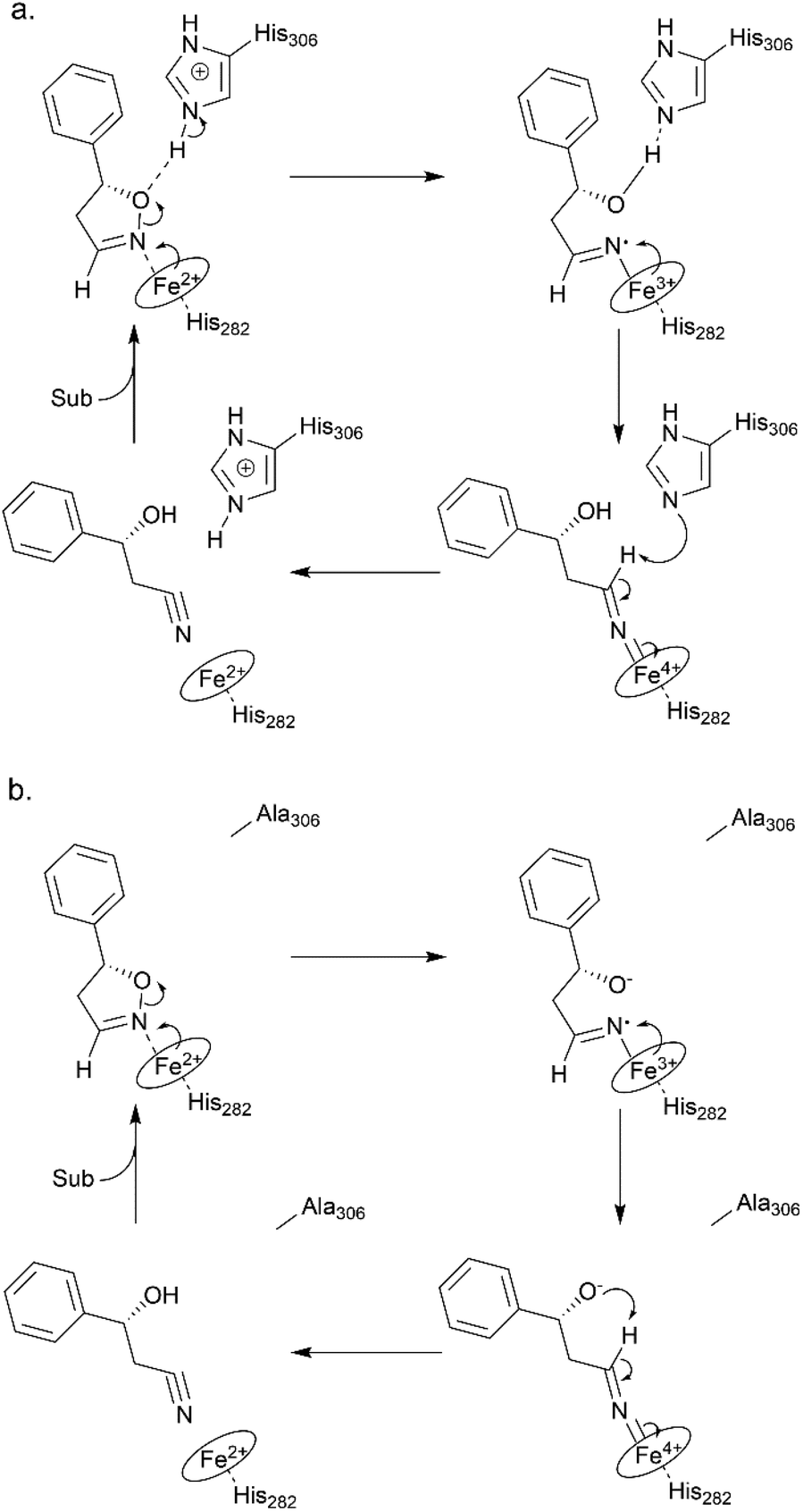 | ||
| Scheme 4 Proposed reaction mechanism for Oxd B-wild (a) and Oxd B-H306A (b). | ||
To verify the practicability of this novel method in organic synthesis, a preparative scale reaction of 5-phenyl-4,5-dihydroisoxazole (1a) was performed at 30 °C using 5 mmol of substrate, 0.25 mM FMN, 500 U Oxd B (see the activity assay method in ESI, section 3‡), and 5 mM Na2S2O5 in 50 mL KPB (pH 6.0, 100 mM) stirred slowly for 2 h. After purification by silica gel column chromatography, (R)-3-hydroxy-3-phenylpropanenitrile (R-2a) was isolated in 42% yield and 99% ee, and (S)-5-phenyl-4,5-dihydroisoxazole (S-1a) was isolated in 48% yield and 99% ee. The obtained (S)-5-phenyl-4,5-dihydroisoxazole (S-1a) was transformed into (S)-3-hydroxy-3-phenylpropanenitrile (S-2a) in 95% yield and 94% ee upon treatment with 2% NaOH methanol solution (10 mL) for 30 min (Scheme 5).
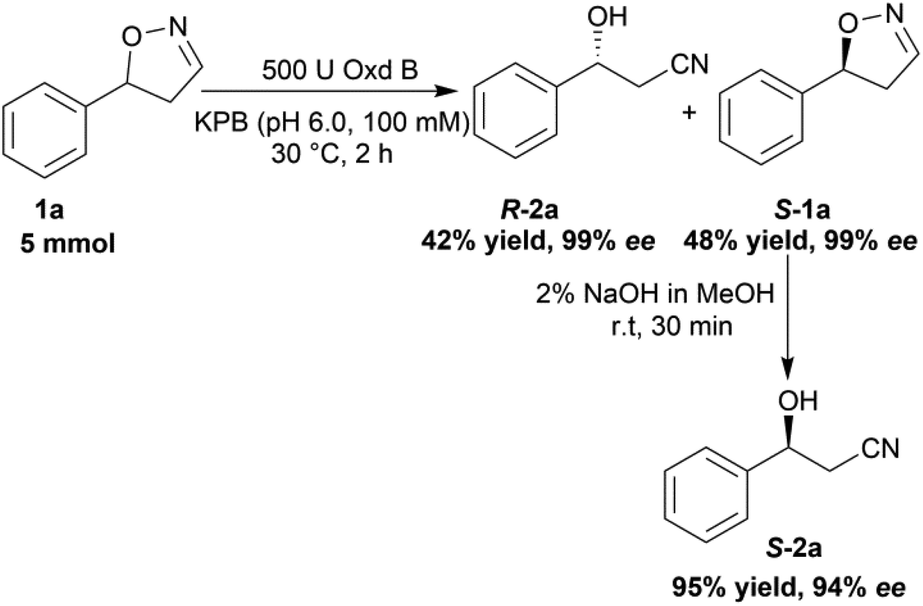 | ||
| Scheme 5 Preparative scale reaction using substrate 1a with Oxd B. The reaction was performed on a 50 mL scale using 100 mM KPB buffer (pH 6.0), 500 U Oxd B, 100 mM 5-phenyl-4,5-dihydroisoxazole, 0.25 mM FMN, and 5 mM Na2S2O5 at 30 °C for 2 h. The isolated yield was calculated after purification by silica gel column chromatography and the ee was determined by chiral HPLC. | ||
Conclusion
In summary, by combination of cyanide-free synthesis of chiral nitriles and the Kemp elimination reaction, we have achieved the first enzymatic reaction to synthesize chiral β-hydroxy nitriles from alkenes via the key step of asymmetric ring-opening of dihydroisoxazoles catalysed by aldoxime dehydratase with broad substrate scope and excellent enantioselectivity (up to 99% ee). The reaction can be carried out with a good turnover number (kcat up to 11 s−1) and high catalytic efficiency (kcat/km up to 3.7 × 104 M−1 s−1). Using site-directed mutagenesis analysis, the reaction was found to mostly occur in the Heme-containing catalytic pocket involving residues H282 and H306. The proximal histidine ligated to the Heme plays a very important role in improving the redox potential of ferrous-Heme, which ensures its reductive ability to open the N–O bond. Without the assistance of any other residues, the N–O bond can be also slowly opened via its redox reaction with ferrous-Heme. Two kinds of deprotonation pathways (histidine deprotonation and oxygen anion deprotonation) seem to exist in the catalytic process, which both contribute to the formation of the nitrile triple-bond.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We are grateful for the support from the JST ERATO Asano Active Enzyme Molecule Project, Japan (Grant Number JPMJRER1102) and Grant-in-Aid for Scientific Research (S) from the Japan Society for Promotion of Sciences (Grant No. 17H06169) awarded to Y. Asano.References
- H. Ankati, D. Zhu, Y. Yang, E. R. Biehl and L. Hua, J. Org. Chem., 2009, 74, 1658–1662 CrossRef CAS PubMed
.
- O. Pàmies and J.-E. Bäckvall, Adv. Synth. Catal., 2002, 344, 947–952 CrossRef
- S. D. Sharma, R. D. Anand and G. Kaur, Synth. Commun., 2004, 34, 1855–1862 CrossRef CAS
- J. M. Concellón and C. Concellón, J. Org. Chem., 2006, 71, 4428–4432 CrossRef PubMed
- E. Bugnicourt, P. Cinelli, A. Lazzeri and V. Alvarez, eXPRESS Polym. Lett., 2014, 8, 791–808 CrossRef
- T. Touge, T. Hakamata, H. Nara, T. Kobayashi, N. Sayo, T. Saito, Y. Kayaki and T. Ikariya, J. Am. Chem. Soc., 2011, 133, 14960–14963 CrossRef CAS PubMed
- A. Kišić, M. Stephan and B. Mohar, Adv. Synth. Catal., 2014, 356, 3193–3198 CrossRef
- Y. Li, Z. Li, F. Li, Q. Wang and F. Tao, Org. Biomol. Chem., 2005, 3, 2513–2518 RSC
- P. N. Liu, P. M. Gu, F. Wang and Y. Q. Tu, Org. Lett., 2004, 6, 169–172 CrossRef CAS PubMed
- W. Masahito, K. Murata and T. Ikariya, J. Org. Chem., 2002, 67, 1712–1715 CrossRef PubMed
- H. Vazquez-Villa, S. Reber, M. A. Ariger and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 8979–8981 CrossRef CAS PubMed
- C. Chen, L. Kong, T. Cheng, R. Jin and G. Liu, Chem. Commun., 2014, 50, 10891–10893 RSC
- O. Soltani, M. A. Ariger, H. Vázquez-Villa and E. M. Carreira, Org. Lett., 2010, 12, 2893–2895 CrossRef CAS PubMed
- J.-E. Lee and J. Yun, Angew. Chem., Int. Ed., 2008, 47, 145–147 CrossRef CAS PubMed
- L. Zhu, T. Kitanosono, P. Xu and S. Kobayashi, Chem. Commun., 2015, 51, 11685–11688 RSC
- S. Mun, J.-E. Lee and J. Yun, Org. Lett., 2006, 8, 4887–4889 CrossRef CAS PubMed
- Y. Suto, R. Tsuji, M. Kanai and M. Shibasaki, Org. Lett., 2005, 7, 3757–3760 CrossRef CAS PubMed
- Y. Suto, N. Kumagai, S. Matsunaga, M. Kanai and M. Shibasaki, Org. Lett., 2003, 5, 3147–3150 CrossRef CAS PubMed
- J. Granander, J. Eriksson and G. Hilmersson, Tetrahedron: Asymmetry, 2006, 17, 2021–2027 CrossRef CAS
- K. Soai, Y. Hirose and S. Sakata, Tetrahedron: Asymmetry, 1992, 3, 677–680 CrossRef CAS
- D. Mitchell and T. M. Koenig, Synth. Commun., 1995, 25, 1231–1238 CrossRef CAS
- B. Xia, J. Xu, Z. Xiang, Y. Cen, Y. Hu, X. Lin and Q. Wu, ACS Catal., 2017, 7, 4542–4549 CrossRef CAS
- E. García-Urdiales, F. Rebolledo and V. Gotor, Tetrahedron: Asymmetry, 2001, 12, 3047–3052 CrossRef
- M. L. Contente, I. Serra, F. Molinari, R. Gandolfi, A. Pinto and D. Romano, Tetrahedron, 2016, 72, 3974–3979 CrossRef CAS
- D. Zhu, H. Ankati, C. Mukherjee, Y. Yang, E. R. Biehl and L. Hua, Org. Lett., 2007, 9, 2561–2563 CrossRef CAS PubMed
- L.-J. Wang, C.-X. Li, Y. Ni, J. Zhang, X. Liu and J.-H. Xu, Bioresour. Technol., 2011, 102, 7023–7028 CrossRef CAS PubMed
- C. Rodriguez, W. Borzecka, J. H. Sattler, W. Kroutil, I. Lavandera and V. Gotor, Org. Biomol. Chem., 2014, 12, 673–681 RSC
- S. Kamila, D. Zhu, E. R. Biehl and L. Hua, Org. Lett., 2006, 8, 4429–4431 CrossRef CAS PubMed
- C. Mukherjee, D. Zhu, E. R. Biehl and L. Hua, Eur. J. Org. Chem., 2006, 5238–5242 CrossRef CAS
- Y. Chen, F. Lie and Z. Li, Adv. Synth. Catal., 2009, 351, 2107–2112 CrossRef CAS
- S.-Y. Chen, C.-X. Yang, J.-P. Wu, G. Xu and L.-R. Yang, Adv. Synth. Catal., 2013, 355, 3179–3190 CrossRef CAS
- R. Metzner, S. Okazaki, Y. Asano and H. Gröger, ChemCatChem, 2014, 6, 3105–3109 CrossRef CAS
- Y. Miao, R. Metzner and Y. Asano, ChemBioChem, 2017, 18, 451–454 CrossRef CAS PubMed
- M. Dadashipour and Y. Asano, ACS Catal., 2011, 1, 1121–1149 CrossRef CAS
- T. Betke, P. Rommelmann, K. Oike, Y. Asano and H. Gröger, Angew. Chem., Int. Ed., 2017, 56, 12361–12366 CrossRef CAS PubMed
- T. Betke, J. Higuchi, P. Rommelmann, K. Oike, T. Nomura, Y. Kato, Y. Asano and H. Gröger, ChemBioChem, 2018, 19, 768–779 CrossRef CAS PubMed
- C. Plass, A. Hinzmann, M. Terhorst, W. Brauer, K. Oike, H. Yavuzer, Y. Asano, A. J. Vorholt, T. Betke and H. Gröger, ACS Catal., 2019, 9, 5198–5203 CrossRef CAS
- A. Hinzmann, S. Glinski, M. Worm and H. Gröger, J. Org. Chem., 2019, 84, 4867–4872 CrossRef CAS PubMed
- A. N. Alexandrova, D. Röthlisberger, D. Baker and W. L. Jorgensen, J. Am. Chem. Soc., 2008, 130, 15907–15915 CrossRef CAS PubMed
- M. L. Casey, D. S. Kemp, K. G. Paul and D. D. Cox, J. Org. Chem., 1973, 38, 2294–2301 CrossRef CAS
- A. Li, B. Wang, A. Ilie, K. D. Dubey, G. Bange, I. V. Korendovych, S. Shaik and M. T. Reetz, Nat. Commun., 2017, 8, 14876 CrossRef CAS PubMed
- Y. Kato, K. Nakamura, H. Sakiyama, S. G. Mayhew and Y. Asano, Biochemistry, 2000, 39, 800–809 CrossRef CAS PubMed
- Y. Kato and Y. Asano, Protein Expression Purif., 2003, 28, 131–139 CrossRef CAS PubMed
- Y. Asano and Y. Kato, FEMS Microbiol. Lett., 1998, 158, 185–190 CrossRef CAS
- Y. Kato, S. Yoshida, S.-X. Xie and Y. Asano, J. Biosci. Bioeng., 2004, 97, 250–259 CrossRef CAS PubMed
- F. J. F. Castro, M. M. Vila, P. R. Jenkins, M. L. Sharma, G. Tustin, J. Fawcett and D. R. Russell, Synlett, 1999, 798–800 CrossRef CAS
- A. Pohjakallio, P. M. Pihko and U. M. Laitinen, Chem. – Eur. J., 2010, 16, 11325–11339 CrossRef CAS PubMed
- R. Huisgen and M. Christl, Chem. Ber., 1973, 106, 3291–3311 CrossRef CAS
- A. Pohjakallio, P. M. Pihko and J. Liu, J. Org. Chem., 2010, 75, 6712–6715 CrossRef CAS PubMed
- A. Yashiro, Y. Nishida, K. Kobayashi and M. Ohno, Synlett, 2000, 361–362 CAS
- M. H. Seo, Y. Y. Lee and Y. M. Goo, Synth. Commun., 1994, 24, 1433–1439 CrossRef CAS
- R.-Z. Liao and W. Thiel, J. Phys. Chem. B, 2012, 116, 9396–9408 CrossRef CAS PubMed
- E. Pinakoulaki, C. Koutsoupakis, H. Sawai, A. Pavlou, Y. Kato, Y. Asano and S. Aono, J. Phys. Chem. B, 2011, 115, 13012–13018 CrossRef CAS PubMed
- K. Oinuma, Y. Hashimoto, K. Konishi, M. Goda, T. Noguchi, H. Higashibata and M. Kobayashi, J. Biol. Chem., 2003, 278, 29600–29608 CrossRef CAS PubMed
- J. Nomura, H. Hashimoto, T. Ohta, Y. Hashimoto, K. Wada, Y. Naruta, K.-I. Oinuma and M. Kobayashi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2810–2815 CrossRef CAS PubMed
- Y. Kato, R. Ooi and Y. Asano, Arch. Microbiol., 1998, 170, 85–90 CrossRef CAS PubMed
- C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294–7299 CrossRef CAS
- H. Sawai, H. Sugimoto, Y. Kato, Y. Asano, Y. Shiro and S. Aono, J. Biol. Chem., 2009, 284, 32089–32096 CrossRef CAS PubMed
- Z. J. Wang, H. Renata, N. E. Peck, C. C. Farwell, P. S. Coelho and F. H. Arnold, Angew. Chem., Int. Ed., 2014, 53, 6810–6813 CrossRef CAS PubMed
- T. K. Hyster and F. H. Arnold, Isr. J. Chem., 2015, 55, 14–20 CrossRef CAS
- K. Kobayashi, M. Kubo, S. Yoshioka, T. Kitagawa, Y. Kato, Y. Asano and S. Aono, ChemBioChem, 2006, 7, 2004–2009 CrossRef CAS PubMed
Footnotes |
| † This work was supported by the Asano Active Enzyme Molecule Project, ERATO, JST. We are grateful to Prof. T. Sugai at Keio University for his useful suggestions on this research and to Dr. R. Metzner for his contribution to the preparation of this manuscript. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc01445a |
| This journal is © The Royal Society of Chemistry 2020 |