
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceJanus head type lone pair–π–lone pair and S⋯F⋯S interactions in retaining hexafluorobenzene†
Sonam
Mehrotra
and
Raja
Angamuthu
*
Laboratory of Inorganic Synthesis and Bioinspired Catalysis (LISBIC), Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur 208016, India. E-mail: Raja@iitk.ac.in
First published on 5th May 2016
Abstract
A series of eight tris-arylthiotriazines were synthesized to study the lone pair–π interaction between the triazine ring centroid of these molecules and halogenated solvents. All the eight compounds were characterized using 1H and 13C NMR spectroscopy and single crystal X-ray diffraction techniques. All these compounds show interesting structural properties in the solid state. Unprecedented Janus head type lp⋯π⋯lp and S⋯F⋯S interactions were observed between one of the tris-arylthiotriazines and hexafluorobenzene.
1. Introduction
It is apparent from the recent literature that lone pair-π (lp⋯π) interactions are weak yet important for the stabilization of three-dimensional structures of biomolecules1 in addition to other well-established weak interactions such as hydrogen bonding, anion–π interactions, and cation–π interactions. Some experimental evidence has been reported very recently to establish the applications of anion–π interactions2–6 and cation–π interactions7–10 while the lp⋯π interactions are yet to be proved useful in small molecular systems. To the best of our knowledge, all the reported synthetic molecular systems that exhibit lp⋯π interactions were discovered serendipitously. We hypothesised to devise molecular systems that will have lone pair–π interactions with small molecules of environmental and industrial interest (CFCs, CO2, SO2) as these interactions may be strong enough to sequester the small molecules and weak enough to release them with minimal energy input such as slight elevation of temperature and/or depression in pressure. This prompted us to investigate host molecules comprising electron deficient ring systems in order to demonstrate the functional relevance of lp⋯π interactions. With this motivation, we have synthesized a number of tris-arylthiotriazine molecules (TArTTz), as the valley of electron deficiency present at the centre of the C3N3 ring of these molecules6,11–13 is known to attract lone pairs of electronegative atoms of neighbouring molecules.14–16 We have anticipated that this will allow us to park small molecules in between C3N3 rings of adjacent molecules through lp⋯π interactions. For this, we wished to take advantage of the well-known “Piedfort Diad” formation (Chart 1).17–23 According to literature reports17,18sym-triaryloxytriazines (TArOTz) have an interesting structural feature of peripheral aryl rings oriented almost perpendicular to the central C3N3 ring and form stacked diads known as Piedfort units21 which eventually are the synthons for higher level supramolecular assemblies.17–19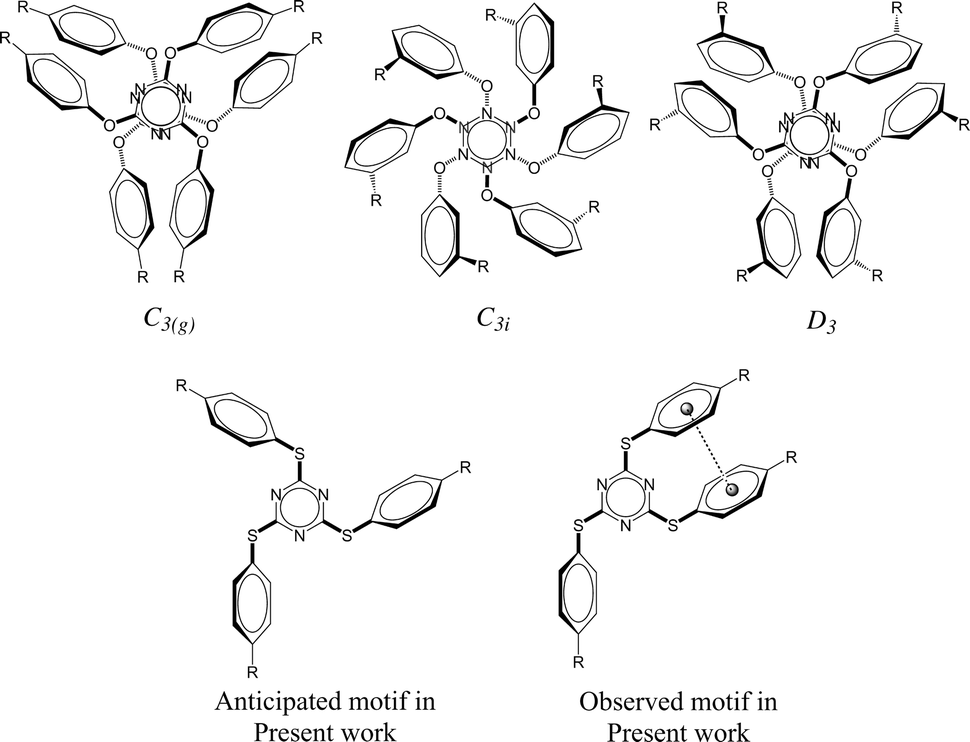 | ||
| Chart 1 Piedfort units of tris-aryloxotriazines (TArOTz) with 3-fold symmetry (C3(g), C3i, and D3)17–21,23–25 and the motif anticipated and observed in the present work for tris-arylthiotriazines (TArTTz). | ||
One of the inherent disadvantages of the known ‘Piedfort Diads’ is that the distance DTz···Tz between the C3N3 rings are in the range of 3 and 4 Å whereas to park a small molecule in between two sym-triaryloxytriazines through lp⋯π interaction(s), a minimum of ∼8 Å distance might be required.17–19 To achieve this we have intended to utilize arylthio substitutions instead of aryloxy units where the aryl rings comprise varying substitutions in order to tune the distance between adjacent C3N3 rings and the type of interactions between neighbouring molecules.
Herein we report eight sym-triazine based molecules that are decorated with aryl rings possessing 4-methyl (2), 4-chloro (3), 3-nitro (4), 2-methyl-5-nitro (5), 2,6-dimethyl (6), pentamethyl (7) and 2,3,5,6-tetrafluoro (8) substitutions (Scheme 1). All the eight compounds were characterized using 1H and 13C NMR spectroscopy. The solid state structures of all the molecules were studied by employing single crystal X-ray diffraction (SCXRD) and were found to comprise a number of inter and intramolecular interactions such as π⋯π and lp⋯π including Janus head type lp⋯π⋯lp and S⋯F⋯S interactions.
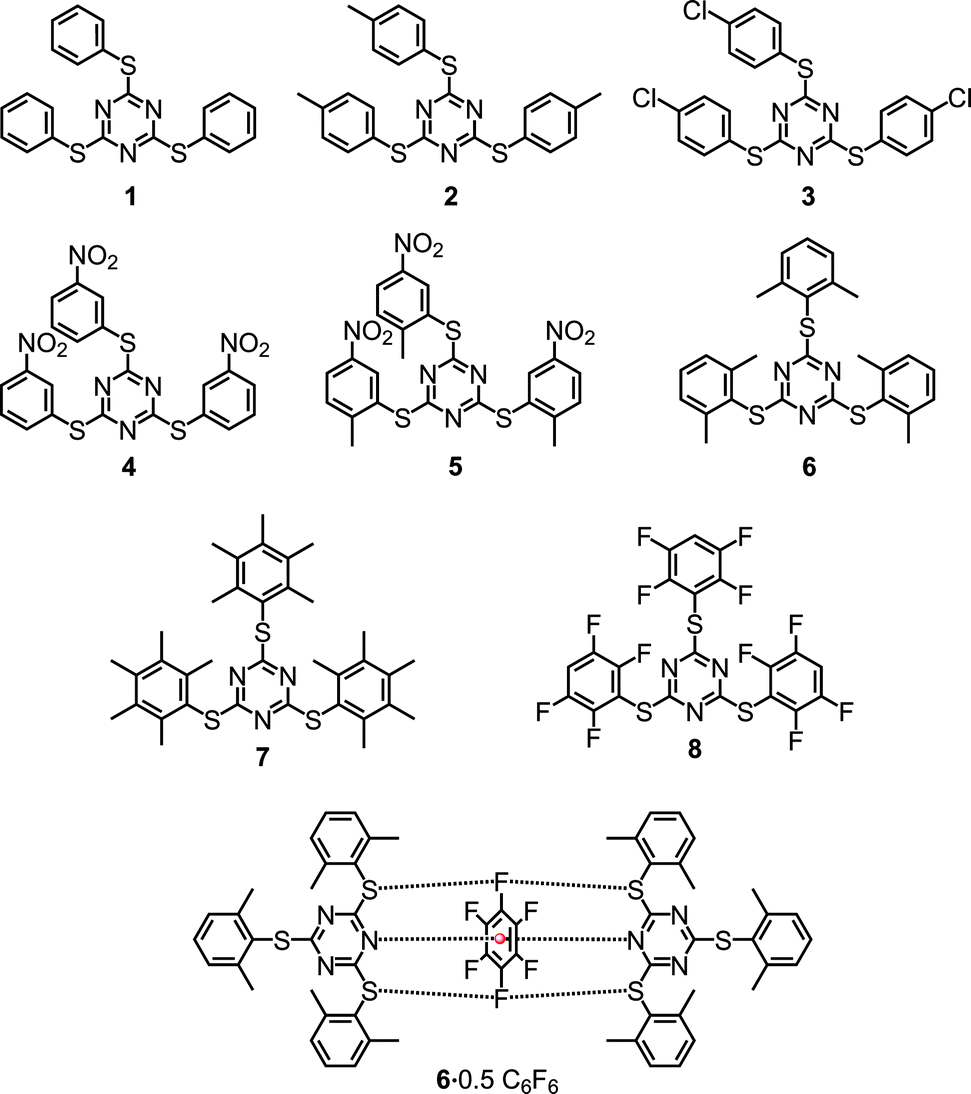 | ||
| Scheme 1 Tris-1,3,5-arylthiotriazines (TArTTz) synthesised for the present study and the schematic diagram of the observed synergism of fluorine bonding and lone pair–π interactions. | ||
2. Results and discussion
2.1. Syntheses of tris-arylthiotriazines 1–8
An equivalent of cyanuric chloride was reacted with three equivalents of thiophenol, 4-methylthiophenol, 4-chlorothiophenol, 3-nitrobenzenethiol, 2-methyl-5-nitrobenzenethiol, 2,6-dimethylthiophenol, 2,3,4,5,6-pentamethylthiophenol and 2,3,5,6-tetrafluorothiophenol in the presence of a suitable base to obtain 2,4,6-tris(phenylthio)-1,3,5-triazine (1), 2,4,6-tris(p-tolylthio)-1,3,5-triazine (2),26 2,4,6-tris((4-chlorophenyl)thio)-1,3,5-triazine (3), 2,4,6-tris((3-nitrophenyl)thio)-1,3,5-triazine (4), 2,4,6-tris((2-methyl-5-nitrophenyl)thio)-1,3,5-triazine (5), 2,4,6-tris(2,6-dimethylphenylthio)-1,3,5-triazine (6), 2,4,6-tris(2,6-dimethylphenylthio)-1,3,5-triazine (7) and 2,4,6-tris((2,3,5,6-tetrafluorophenyl)thio)-1,3,5-triazine (8), respectively (Scheme 1). All the eight compounds were characterized using 1H and 13C NMR spectroscopy and the single-crystal X-ray diffraction technique. The thermal stability of these molecules was studied by thermogravimetric analysis.2.2. NMR spectroscopy
The NMR spectra of all the eight TArTTz compounds indicate that these molecules are C3 symmetric in solution at room temperature in NMR time scale though it is not the case in the solid state as seen in the SCXRD structures of these TArTTz compounds. As 1H NMR spectroscopy does not differentiate the mono and disubstituted triazines from TArTTz, 13C NMR spectroscopy was used as the finite preliminary analysis where all the TArTTz exhibit a signal around 180 ppm indicating the presence of three similar tertiary carbons in the triazine ring system.2.3. Description of the structures
Single crystals of TArTTz 1–8 suitable for SCXRD studies were grown by slow evaporation or diffusion methods as reported in the experimental section in detail. Unfortunately, none of the TArTTz molecules crystallised in any of the three required symmetries (C3(g), C3i or D3),17–19 which would have allowed the formation of the anticipated Piedfort units. To the best of our knowledge, many known free tris-aryloxotriazines form Piedfort units in their solid states.17–23 However, TArOTz are also known to attain lower symmetry when they are coordinated to metal ions.27 One possible reason for the lowering of symmetry on going from TArOTz to TArTTz could be the possibility of intramolecular π⋯π interaction in TArTTZ molecules owing to the bigger size of sulphur. All the TArTTz molecules exhibit weak intramolecular π⋯π interactions of distances between 3.94 and 4.38 Å, which were not retained in solution at room temperature as observed from their NMR spectra.The TArTTz 1, where the phenyl rings do not have any substitution, possesses an intermolecular lone pair⋯π interaction between the lone pair of thioether sulphur and the triazine ring (S(lp)⋯π) with a distance (DS(lp)⋯π) of 3.207 Å which is shorter than the sum of van der Waals radii (C + S = 3.5 Å; N + S = 3.35 Å).13 The S3 A⋯ring distances are 3.471 Å (S3 A⋯N1B), 3.472 Å (S3 A⋯N2B), 3.527 Å (S3 A⋯N3B), 3.391 Å (S3 A⋯C1B), 3.453 Å (S3 A⋯C2B), and 3.525 Å (S3 A⋯C3B). This may be the first observed S(lp)⋯π involving a 1,3,5-triazine ring system whereas few other known S(lp)⋯π involve 1,2,3-triazine and 1,3,4-triazine rings (Fig. 1).13 The 4-methyl substituted TArTTz 2 does not have any significant lp⋯π interactions whereas the 4-chloro substituted TArTTz 3 exhibits an intermolecular Cl(lp)⋯π interaction (DCl(lp)⋯π = 3.542 Å). The sums of the van der Waals radii are 3.45 Å (C + Cl) and 3.30 Å (N + Cl);13 the Cl2⋯ring distances are 3.648 Å (Cl2⋯N1), 4.181 Å (Cl2⋯N2), 3.550 Å (Cl2⋯N3), 3.981 Å (Cl2⋯C1), 3.931 Å (Cl2⋯C2), and 3.353 Å (Cl2⋯C3). According to Mooibroek et al., the observed mean DCl(lp)⋯π is 3.346 Å.12 Hence, the Cl(lp)⋯π interaction observed in TArTTz 3 may be considered as a weak one.
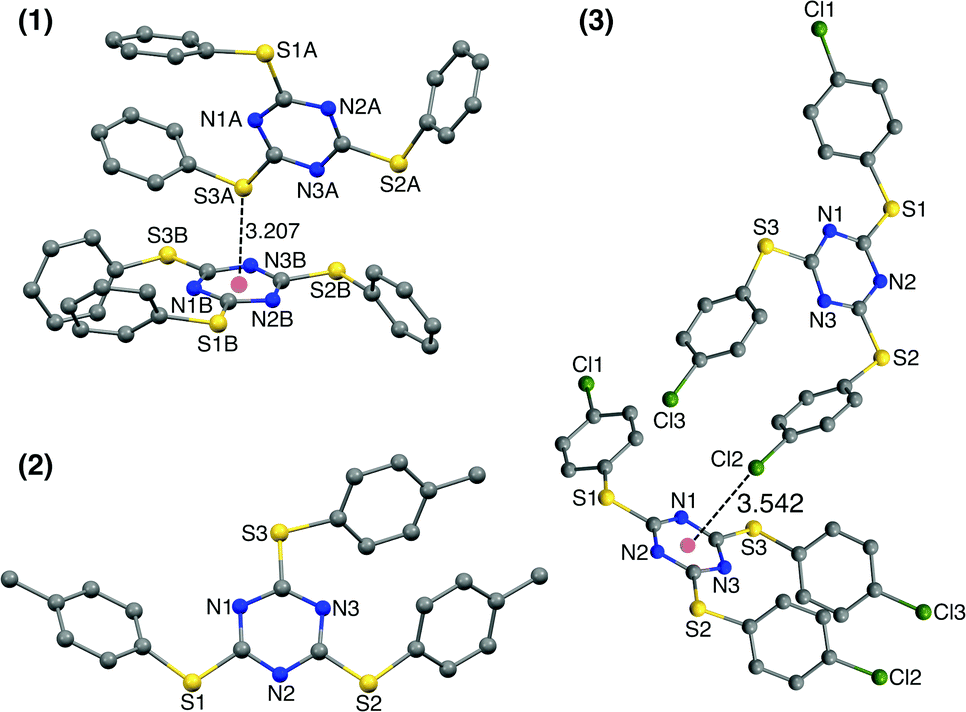 | ||
| Fig. 1 X-ray structures of 2,4,6-tris(phenylthio)-1,3,5-triazine (1), 2,4,6-tris(p-tolylthio)-1,3,5-triazine (2), and 2,4,6-tris((4-chlorophenyl)thio)-1,3,5-triazine (3). The hydrogen atoms are not shown for clarity. Intramolecular π⋯π interactions: (1) 4.287 and 4.350; (2) 4.384; (3) 4.158 Å. | ||
The 3-nitro substituted TArTTz 4 has all three nitro groups facing the same side of the triazine ring while the 2-methyl-5-nitro substituted TArTTz 5 has two nitro groups in one side and the other one facing the opposite side of the triazine ring. This indicates that the 2-methyl-5-nitrophenyl rings have free rotation in solution as TArTTz 5 has C3 symmetry in solution. Apart from the intramolecular π⋯π interactions, TArTTz 4 has an interesting intermolecular π⋯π interaction (DCg⋯Cg = 3.616 Å) between the triazine ring of one molecule with the 3-nitrophenyl ring of another molecule. The TArTTz 5 possesses an intermolecular π⋯π interaction (DCg⋯Cg = 3.652 Å), which presents a ladder of four aryl rings (Fig. 2). The TArTTz 6 and 7 also present the same ladder-type structure of four dimethylphenyl rings and four pentamethylphenyl rings with intermolecular (DCg⋯Cg = 3.868 Å in 6; 3.913 Å in 7) and intramolecular π⋯π stacking (DCg⋯Cg = 3.965 Å in 6; 4.114 Å in 7).
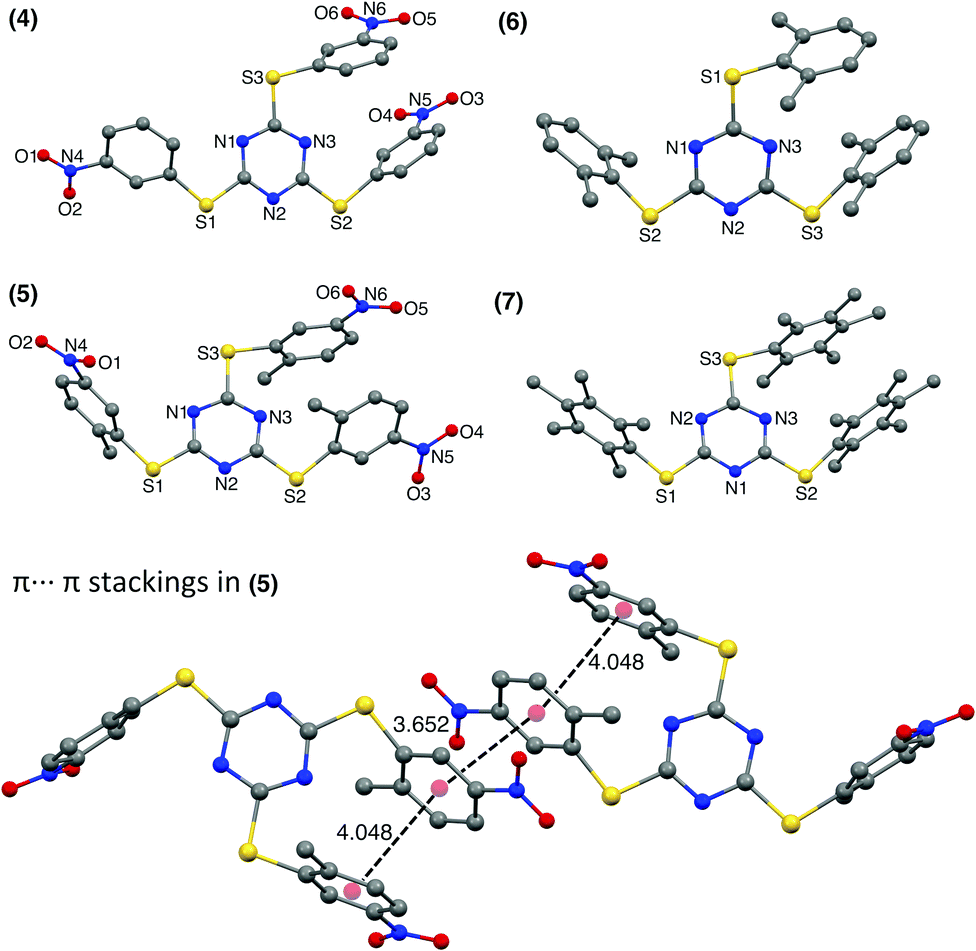 | ||
| Fig. 2 X-ray structures of 2,4,6-tris((3-nitrophenyl)thio)-1,3,5-triazine (4), 2,4,6-tris((2-methyl-5-nitrophenyl)thio)-1,3,5-triazine (5), 2,4,6-tris(2,6-dimethylphenylthio)-1,3,5-triazine (6), and 2,4,6-tris(2,6-dimethylphenylthio)-1,3,5-triazine (7). The hydrogen atoms are not shown for clarity. Intramolecular π⋯π interactions: (4), 4.041; (5), 4.048; (6), 3.965; (7), 4.114 Å. Intermolecular π⋯π interactions: (4), 3.616; (5), 3.652 (6); 3.868; (7), 3.913 Å. | ||
The SCXRD structure of TArTTz 8 (Fig. 3) presents an interesting dimeric motif exhibiting many intermolecular interactions such as S⋯F fluorine bonds (S3⋯F8, 2.991 Å; S1···F1, 3.251 Å; S2⋯F2, 3.498 Å), an intermolecular F(lp)···π interaction (DF(lp)⋯π = 2.967 Å) and an intramolecular π⋯π interaction (DCg⋯Cg = 3.944 Å). The F(lp)⋯π interaction observed in TArTTz 8 is a strong one as the DF(lp)⋯π of 2.967 Å is well below the sums of the van der Waals radii (C + F = 3.17 Å; N + F = 3.02 Å).13 The F4···ring distances are 3.273 Å (F4⋯N1), 3.111 Å (F4⋯N2), 3.414 Å (F4⋯N3), 3.404 Å (F4⋯C1), 3.077 Å (F4⋯C2), and 3.212 Å (F4⋯C3) which are not too far from the sum of the van der Waals radii.
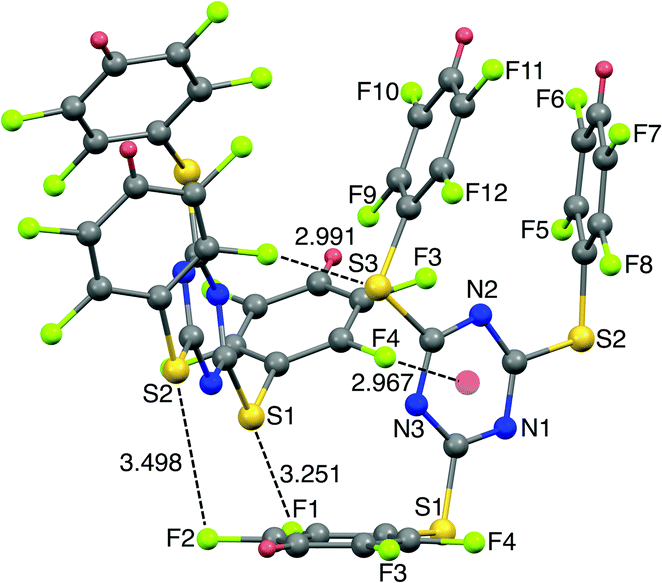 | ||
| Fig. 3 X-ray structure of 2,4,6-tris((2,3,5,6-tetrafluorophenyl)thio)-1,3,5-triazine (8). Intramolecular π⋯π interaction distance: 3.944 Å. | ||
As we have observed the intermolecular lp⋯π interactions in TArTTz 1, 3 and 8, we set out to investigate the interactions with fluorinated molecules as solvents. The halogenated aromatic molecules, especially C6F6, are known to be involved in lp⋯π interactions as they can participate through the halogen lone pair and through the electron deficient centre of the aromatic ring. We have observed a peculiar crystal structure where the C6F6 molecule involves in Janus head type of lp⋯π⋯lp interaction on both sides of the ring (DN(lp)⋯π = 3.51 Å) where the nitrogen lone pairs of two TArTTz 6 molecules interact with the ring centre of C6F6 (Fig. 4). Furthermore, the fluorine atoms at the 1,4-positions of the C6F6 ring also take part in fluorine bonding28–31 with the thioether sulphur atoms of the TArTTz 6 molecules with interesting S⋯F⋯S motifs (DS⋯F = 3.365 and 3.442 Å). Apart from the lp⋯π⋯lp and S⋯F⋯S interactions, the structure of 6·0.5C6F6 possesses extensive inter and intramolecular π⋯π stacking networks.
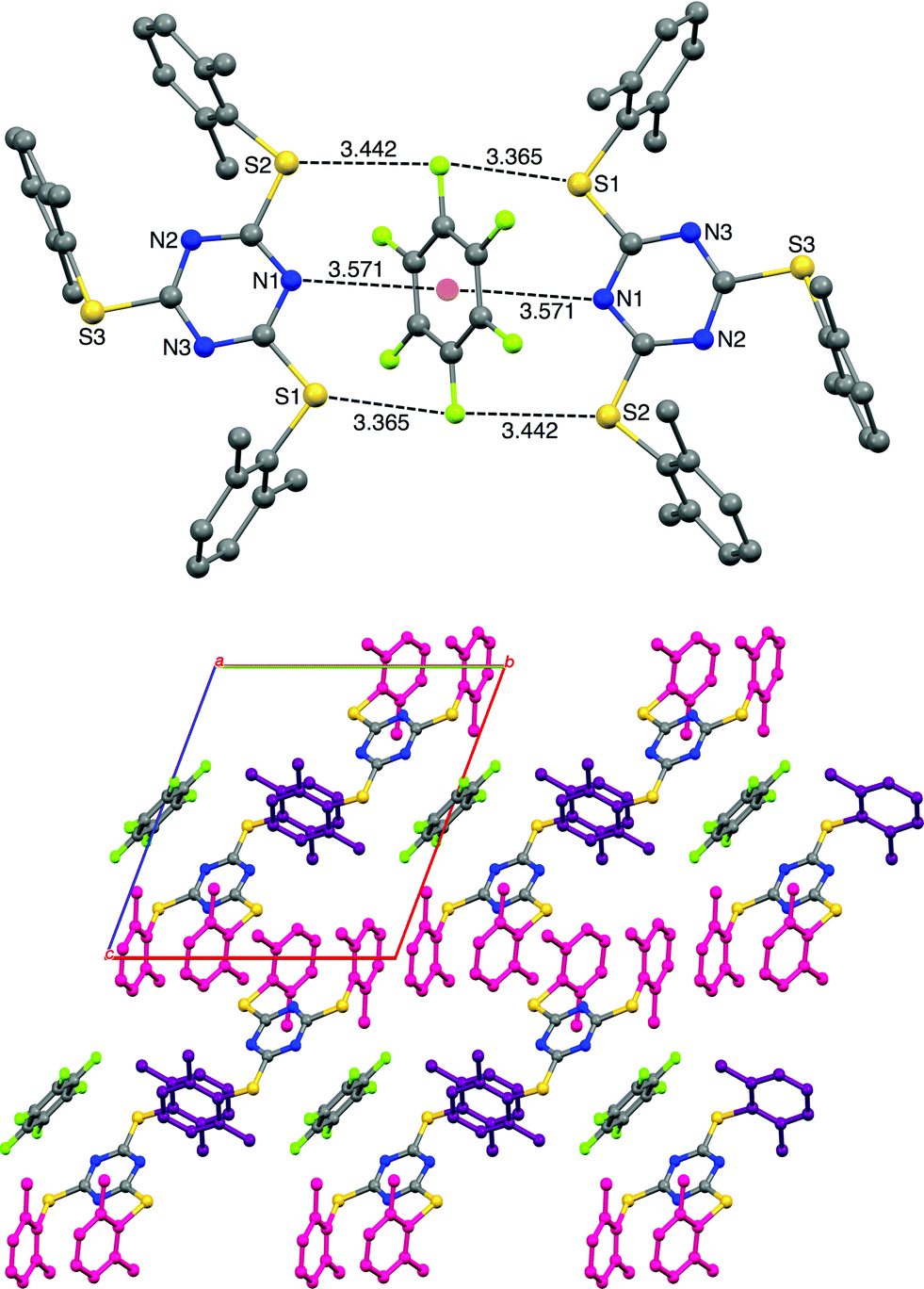 | ||
| Fig. 4 X-ray structure of 2,4,6-tris(2,6-dimethylphenylthio)-1,3,5-triazine (6) crystallized in hexafluorobenzene (6·0.5C6F6). The hydrogen atoms are not shown for clarity. The packing diagram of 6·0.5C6F6 shows the presence of extensive inter and intramolecular π⋯π stacking. Pink coloured dimethylphenyl rings are involved in both inter and intramolecular stacking and the purple coloured rings are involved in the intermolecular stacking. π⋯π interactions: 4.100 Å (intra, pink); 4.160 Å (inter, pink); 4.280 Å (inter, purple). | ||
2.4. Thermal stability of TArTTz molecules
The thermal stability of TArTTz compounds 1–8 was studied by means of thermogravimetric analysis. Compounds 1–3, 6 and 7 were found to be stable until ∼280 °C, 4 was stable until ∼260 °C, whereas compounds 5 and 8 began to decompose at ∼230 °C. Interestingly, adduct compound 6·0.5C6F6 lost the hexafluorobenzene molecule at 140 °C (Fig. 5) which is 60 °C above the boiling point of C6F6 which indicates the synergism of the lp⋯π⋯lp and S⋯F⋯S interactions in retaining the C6F6 molecule in the lattice.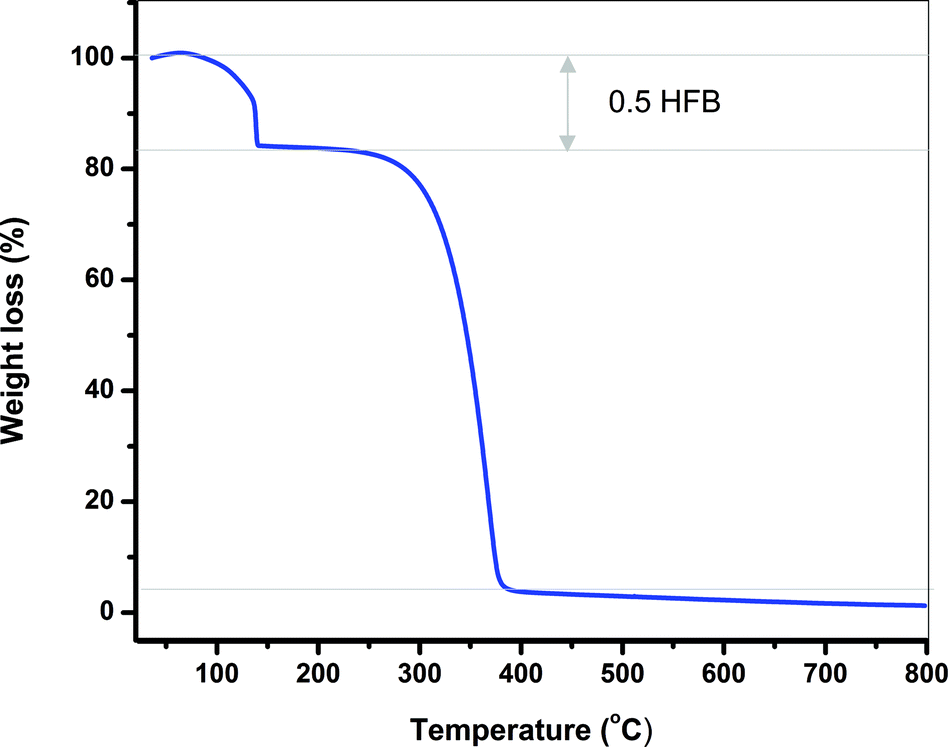 | ||
| Fig. 5 The thermogravimetric diagram of 6·0.5C6F6 showing the loss of the C6F6 solvate at 140 °C and the decomposition of 6 at 280 °C. | ||
3. Conclusions
The tris-arylthiotriazines reported herein show that the lone pair–π interactions can indeed be engineered by design and be used for applications such as capturing or retaining small molecules which posses at least one lone pair (halocarbons, CO2, or SO2). The observed feature of the synergistic Janus head lp⋯π⋯lp and S⋯F⋯S motif is interesting in the context of fluorine bonding and warrants further experimental and computational studies as the fluorine bonding is a unique and emerging section of weak interactions which might be relevant to pharmacologically important fluorinated drug molecules.294. Experimental
4.1. Materials
Cyanuric chloride (Avra Synthesis), thiophenol (SDFCL), 4-methylthiophenol (Avra Synthesis), 4-chlorothiophenol (Avra Synthesis), nitrobenzene (Merck), chlorosulfonic acid (Spectrochem), triphenylphosphine (Avra synthesis), 4-nitrotoluene (Avra), 2,6-dimethylthiophenol (Alfa Aesar), 2,3,4,5,6-pentamethylbenzene-1-sulfonylchloride (Alfa Aesar) and 2,3,5,6-tetrafluorothiophenol (Alfa Aesar) were used as received from commercial sources. Solvents were distilled under dry nitrogen atmosphere using conventional methods. Heavy-walled high-pressure round bottom flasks were acquired from Ace Glass Inc.4.2. Methods
Elemental analyses were carried out on a Perkin-Elmer CHNS/O analyser. The NMR spectra were recorded on JEOL 500 MHz and JEOL 400 MHz spectrometers. The temperature was kept constant using a variable temperature unit within the error limit of ±1 K. The software MestReNova was used for the processing of the NMR spectra.32 Tetramethylsilane (TMS) or the deuterated solvent residual peaks were used for calibration. Mass spectrometry experiments were performed on a Waters-Q-ToF-Premier-HAB213 equipped with an electrospray interface. The spectra were collected by constant infusion of the sample dissolved in methanol or acetonitrile with 0.1% formic acid. Thermogravimetric analyses were carried out in dry nitrogen atmosphere at a heating rate of 10 °C min−1 on a Mettler Toledo TGA/DSC1 Stare System.Warning: Reactions using heavy-walled high-pressure round bottom flask with screw cap must be carried cautiously. The danger of explosion can be avoided by performing the reactions in smaller scales. The scales presented here are safe with a 250 ml heavy-walled high-pressure round bottom flask.
4.3. Synthetic procedures
4.4. X-ray crystallography
Single-crystal X-ray data were collected on a Bruker SMART APEX CCD diffractometer using graphite-monochromated Mo Kα radiation (λ = 0.71069 Å). The linear absorption coefficients, the scattering factors for the atoms, and the anomalous dispersion corrections were taken from International Tables for X-ray Crystallography. Data integration and reduction were conducted with SAINT. An empirical absorption correction was applied to the collected reflections with SADABS using XPREP. The structures were determined by direct methods using SHELXTL and refined on F2 by a full-matrix least-squares technique using the SHELXL-97 program package. The lattice parameters and structural data are listed at the end of the ESI.Acknowledgements
This work was supported by the Ministry of Earth Sciences, India and generous funding from the Indian Institute of Technology Kanpur (IITK, India) through the “New Faculty Initiation Grant” (IITK/CHM/20120078). SM acknowledges the University Grants Commission (UGC, India) for the Senior Research Fellowship. RA acknowledges Dr. Tiddo J. Mooibroek for inspiration. This paper is dedicated to Professor Ray J. Butcher on the occasion of his 70th birthday.Notes and references
- A. Jain, V. Ramanathan and R. Sankararamakrishnan, Protein Sci., 2009, 18, 595 CAS
.
- R. E. Dawson, A. Hennig, D. P. Weimann, D. Emery, V. Ravikumar, J. Montenegro, T. Takeuchi, S. Gabutti, M. Mayor, J. Mareda, C. A. Schalley and S. Matile, Nat. Chem., 2010, 2, 533 CrossRef CAS PubMed
- Q. He, Y.-F. Ao, Z.-T. Huang and D.-X. Wang, Angew. Chem., Int. Ed., 2015, 54, 11785 CrossRef CAS PubMed
- M. Giese, M. Albrecht and K. Rissanen, Chem. Commun., 2016, 52, 1778 RSC
- N. Busschaert, C. Caltagirone, W. Van Rossom and P. A. Gale, Chem. Rev., 2015, 115, 8038 CrossRef CAS PubMed
- A. Bauza, T. J. Mooibroek and A. Frontera, CrystEngComm, 2016, 18, 10 RSC
- E. Nagy, E. St Germain, P. Cosme, P. Maity, A. C. Terentis and S. D. Lepore, Chem. Commun., 2016, 52, 2311 RSC
- A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2013, 113, 2100 CrossRef CAS PubMed
- S. Gao, G. Shi and H. Fang, Nanoscale, 2016, 8, 1451 RSC
- I. D. Mucic, M. R. Nikolic and S. D. Stojanovic, Protoplasma, 2015, 252, 947 CrossRef CAS PubMed
- N. Mohan, C. H. Suresh, A. Kumar and S. R. Gadre, Phys. Chem. Chem. Phys., 2013, 15, 18401 RSC
- T. J. Mooibroek and P. Gamez, CrystEngComm, 2012, 14, 3902 RSC
- T. J. Mooibroek, P. Gamez and J. Reedijk, CrystEngComm, 2008, 10, 1501 RSC
- A. Kumar, S. R. Gadre, N. Mohan and C. H. Suresh, J. Phys. Chem. A, 2014, 118, 526 CrossRef CAS PubMed
- R. J. Gotz, A. Robertazzi, I. Mutikainen, U. Turpeinen, P. Gamez and J. Reedijk, Chem. Commun., 2008, 3384 RSC
- Z. L. Lu, P. Gamez, I. Mutikainen, U. Turpeinen and J. Reedijk, Cryst. Growth Des., 2007, 7, 1669 CAS
- V. R. Thalladi, R. Boese, S. Brasselet, I. Ledoux, J. Zyss, R. K. R. Jetti and G. R. Desiraju, Chem. Commun., 1999, 1639 RSC
- V. R. Thalladi, S. Brasselet, H. C. Weiss, D. Blaser, A. K. Katz, H. L. Carrell, R. Boese, J. Zyss, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 2563 CrossRef CAS
- P. Bombicz and A. Kalman, Cryst. Growth Des., 2008, 8, 2821 CAS
- R. Boese, G. R. Desiraju, R. K. R. Jetti, M. T. Kirchner, I. Ledoux, V. R. Thalladi and J. Zyss, Struct. Chem., 2002, 13, 321 CrossRef CAS
- A. S. Jessiman, D. D. Macnicol, P. R. Mallinson and I. Vallance, J. Chem. Soc., Chem. Commun., 1990, 1619 RSC
- L. Fabian, P. Bombicz, M. Czugler, A. Kalman, E. Weber and M. Hecker, Supramol. Chem., 1999, 11, 151 CrossRef CAS
- K. Henderson, D. D. Macnicol, P. R. Mallinson and I. Vallance, Supramol. Chem., 1995, 5, 301 CrossRef CAS
- M. Czugler, E. Weber, L. Parkanyi, P. P. Korkas and P. Bombicz, Chem. – Eur. J., 2003, 9, 3741 CrossRef CAS PubMed
- Y. X. Ke, D. J. Collins, D. F. Sun and H. C. Zhou, Inorg. Chem., 2006, 45, 1897 CrossRef CAS PubMed
- A. L. Isfahani, I. Mohammadpoor-Baltork, V. Mirkhani, M. Moghadam, A. R. Khosropour, S. Tangestaninejad, M. Nasr-Esfahani and H. A. Rudbari, Synlett, 2014, 25, 645 CrossRef CAS
- M. Munakata, M. Wen, Y. Suenaga, T. Kuroda-Sowa, M. Maekawa and M. Anahata, Polyhedron, 2001, 20, 2321 CrossRef CAS
- P. Metrangolo, J. S. Murray, T. Pilati, P. Politzer, G. Resnati and G. Terraneo, CrystEngComm, 2011, 13, 6593 RSC
- P. Zhou, J. Zou, F. Tian and Z. Shang, J. Chem. Inf. Model., 2009, 49, 2344 CrossRef CAS PubMed
- K. Reichenbacher, H. I. Suss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC
- S. Kawai, A. Sadeghi, F. Xu, L. Peng, A. Orita, J. Otera, S. Goedecker and E. Meyer, ACS Nano, 2015, 9, 2574 CrossRef CAS PubMed
- MestReNova, Mestrelab Research S.L., Santiago de Compostela, Spain, http://www.mestrelab.com, 2014
- A. L. Isfahani, I. Mohammadpoor-Baltork, V. Mirkhani, M. Moghadam, A. R. Khosropour, S. Tangestaninejad, M. Nasr-Esfahani and H. A. Rudbari, Synlett, 2014, 25, 645 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of all the compounds. X-ray crystallographic data in CIF format have been deposited with the Cambridge Structural Database. CCDC 1403546–1403550 (1–5), 1436791–1436793 (6·0.5C6F6, 6, 7) and 1452878 (8) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00496b |
| This journal is © The Royal Society of Chemistry 2016 |