
Technological advances in ligninolytic enzymes for the biological valorization of lignin
Ning
Fu
abc,
Ruo-Ying
Liu
abc,
Ya
Zhou
abc,
Bing-Zhi
Li
 *abc,
Ying-Jin
Yuan
abc and
Zhi-Hua
Liu
*abc
*abc,
Ying-Jin
Yuan
abc and
Zhi-Hua
Liu
*abc
aSchool of Chemical Engineering and Technology, Tianjin University, Tianjin 300072, China. E-mail: bzli@tju.edu.cn; zhliu@tju.edu.cn
bState Key Laboratory of Synthetic Biology and Frontiers Science Center for Synthetic Biology, Tianjin 300072, China
cFrontiers Research Institute for Synthetic Biology, Tianjin University, 301799, China
First published on 6th March 2025
Abstract
Lignin is a promising renewable aromatic resource with significant potential for conversion into high-value products, making it a key component in advancing biorefinery processes and supporting the bioeconomy. However, its structural heterogeneity and macromolecular complexity pose major challenges to its biological valorization. This work focuses on advancing the bio-depolymerization of lignin into highly bioavailable derivatives for downstream bioconversion by exploring cutting-edge technologies for the screening and modification of ligninolytic enzymes. Key enzymes involved in both extracellular depolymerization and intracellular transformation of lignin serve as critical targets for advancing its biological valorization. A range of state-of-the-art technologies can be employed for high-throughput screening of efficient ligninolytic enzymes or bacteria, thereby enriching the pool of available biocatalysts. Protein engineering offers a powerful approach for developing artificial enzymes with industrial production advantages. Additionally, artificial intelligence provides valuable strategies for designing and modifying ligninolytic enzymes. Overall, the interdisciplinary application of these advanced technologies is instrumental in propelling lignin biological valorization to a more sophisticated stage of green development.
Green foundation1. This work highlights recent advancements in biological lignin valorization using ligninolytic enzymes and cutting-edge technologies in the field. The interdisciplinary application of these innovations plays a crucial role in advancing lignin biological valorization, driving it toward a more sustainable stage of green chemistry development.2. Lignin is the largest renewable source of aromatics in nature. A thorough review of cutting-edge technologies could advance the biological valorization of lignin, potentially transforming its utilization and contributing to biorefining and the bioeconomy. 3. Future work will focus on scaling the practical applications of advanced technologies for discovering and optimizing ligninolytic enzymes and microbial strains. Enhancements in the robustness, efficiency, and specificity of these biological systems will unlock new research opportunities and establish a foundation for developing greener, more sustainable methods for industrial lignin valorization. |
1. Introduction
Lignin, one of the three major components of lignocellulosic biomass (LCB), is the most abundant terrestrial aromatic biopolymer.1 Lignin is an underutilized renewable energy source and holds great promise as an alternative aromatic material for value-added products in biological manufacturing.2–4 Its utilization can significantly reduce the dependence on nonrenewable fossil fuels, such as petroleum derived aromatic hydrocarbons.5,6 Despite this potential, lignin is a highly heterogeneous biopolymer.2 Its proportion varies among different lignocellulosic biomasses and is closely related to the proportions of cellulose and hemicellulose.7 Lignin is typically composed of three subunits: guaiacyl (G), syringyl (S), and p-hydroxyphenyl (H), which polymerize via C–C or C–O linkages.8 Therefore, the structural heterogeneity and macromolecular characteristics of lignin polymers, composed of different subunits, are major obstacles to their biological valorization.5 To overcome the obstacles posed by these inherent features, it is crucial to develop a wide range of deconstruction strategies and innovative transformation pathways.Depolymerization and fractionation of lignin are key steps in its utilization.8,9 Currently, various chemical, physical, and biological depolymerization methods have been developed.9 In the industrial application, chemical depolymerization of lignin faces significant challenges, such as high energy consumption, reaggregation tendencies, increased pollution levels, etc.10 Meanwhile, physical methods often serve primarily as auxiliary pretreatment steps to extract lignin suitable for industrial purposes rather than directly degrading lignin itself.11
Interestingly, through natural evolution, microorganisms have developed the ability to depolymerize lignin and utilize the aromatic molecules derived from it as carbon sources, establishing a natural pathway for lignin depolymerization. Investigating the enzymes and metabolic pathways of ligninolytic microorganisms has significantly improved the efficiency of lignin biotransformation and utilization.10,11 The biotransformation of lignin begins with the depolymerization and separation of lignin, followed by the metabolism of oligomers through the “biological funnel” route of aromatic compounds (Fig. 1).12 The upper pathway serves as a “biological funnel”, converting isomeric substrates into crucial aromatic product intermediates, such as protocatechuic acid and catechol.12,13 These intermediates undergo further cleavage via the aromatic ring cleavage pathway, opening the ring to produce acetyl coenzyme A (CoA) and succinyl CoA. Following the β-ketoadipic acid pathway, these compounds are then integrated into central carbon metabolism.13 Biological lignin utilization can be conducted under mild conditions and it is often considered environmentally friendly, aligning with global sustainability goals.14
 | ||
| Fig. 1 The main pathways and research progress of lignin biotransformation. (A) The biological valorization routes from lignin to valuable products. (B) Cutting-edge technologies applied in the progress of lignin biotransformation. (C) Papers published in the field of lignin biotransformation in the past 20 years. (D) Current research work with lignin as the core topic. | ||
However, the full exploitation and utilization of the microbial conversion of lignin have not yet been fully actualized. It is necessary to identify effective ligninolytic enzymes and pathways for lignin depolymerization and conversion.8,10,14 Recent efforts in lignin utilization have led to the introduction of cutting-edge technologies aimed at addressing these challenges.15,16 For example, high-throughput screening technologies harness the power of evolution to identify the desired properties of enzymes and strains.17–19 Laboratory evolution through high-throughput screening can tailor the properties and enhance the performance of targeted ligninolytic strains and enzymes.20,21 Protein engineering surpasses natural protein libraries for its potential to customize enzyme and strain characteristics.22 When necessary enzymes for specific target products are absent from metabolic pathways, new enzymes can be created through protein engineering.23 Additionally, the integration of biological big data with bioinformatics tools enables effective analysis and screening.24–27 Data-driven approaches using artificial intelligence (AI) facilitate advanced strain and enzyme design, guiding directed evolution and reducing experimental iterations.28–31 Incorporating AI concepts enhances the efficiency of obtaining target strains and enzymes with desired functions and properties.22,30–33 Overall, these pivotal technologies provide significant opportunities for improving the efficiency of ligninolytic strains and enzymes, thereby advancing the valorization of lignin.
This work aims to advance biological lignin valorization with ligninolytic enzymes by summarizing cutting-edge technologies in the field. First, the biological conversion processes of lignin are reviewed, including macromolecular depolymerization, oligomer degradation, intracellular biological funneling, and ring-opening pathways. Next, advanced technologies for the screening and modification of ligninolytic enzymes are summarized. Powerful tools such as protein engineering and artificial intelligence are highlighted to enable the design and optimization of artificial enzymes with advantages for industrial applications. The cross-application of these multidisciplinary technologies has the potential to drive lignin biological valorization toward a more advanced and intelligent stage of green development.
2. Key pathways and enzymes of biological lignin valorization
The microbial valorization of lignin requires a cascade bioconversion route, which mainly includes lignin depolymerization, aromatic metabolism, and product biosynthesis.22 Preliminary depolymerization of lignin aims to break ether bonds in the structure, resulting in oligomers or monomers for subsequent utilization. This depolymerization generates various heterogeneous lignin-derived aromatic hydrocarbons, which can be further decomposed through the “biological funnel” route. These compounds are metabolized into key intermediates through reactions such as O-demethylation, hydroxylation, and decarboxylation.34 Subsequently, the aromatic intermediates undergo aromatic ring cleavage mediated by dioxygenase. The opened lignin aromatic derivatives then enter the metabolic pathways of microorganisms, such as glycolysis and the tricarboxylic acid cycle, to enable the utilization of lignin-derived aromatic compounds and provide precursors for product biosynthesis.132.1 Depolymerization pathways and enzymes of lignin macromolecules
The initial step of lignin bioconversion involves the depolymerization of lignin by breaking ether bonds to acquire oligomers or monomers for downstream utilization. Various methods exist for the depolymerization of lignin macromolecules, which can be broadly categorized into chemical, biological, physical and physicochemical methods.8,22The structure and properties of natural lignin are influenced by its source and the methods used for fractionation.34 Softwood lignin is primarily composed of guaiacyl (G) units and is characterized by a higher molecular weight. Hardwood lignin consists of syringyl (S) and guaiacyl (G) units, with S units being more susceptible to cleavage during acid pretreatment. Herbaceous lignin is primarily composed of G and S units, with a minor contribution from p-hydroxyphenyl (H) units.35,36 In the chemical depolymerization methods of lignin, acid pretreatment is regarded as one of the most effective technologies for cleaving β-O-4 linkages of lignin; however, it leads to lignin's repolymerization, significantly altering the molecular weight and exhibiting S unit reactivity. Alkaline pretreatment primarily cleaves β-O-4 linkages through nucleophilic cleavage, impacting on the solubility and molecular weight based on alkali concentration and temperature.37,38 Organosolv pretreatment dissolves lignin in organic solvents, with the molecular weight and structure varying depending on solvent type, temperature, and pretreatment severity.39,40 This extraction can also be performed using several processes, including organocell, alcell, milox, and ionic liquid lignin extraction.41 Chemical fractionation methods demonstrated distinct characteristics in modifying lignin chemistry.42 Chemical depolymerization of lignin also can be performed using methanol, ethanol and peroxyformic acid, as well as various organic cations and inorganic anions (called green solvents).43 Binary solvent systems consisting of biomass-derived γ-valerolactone and one cosolvent, such as water, ionic liquids, dimethyl sulfoxide and dimethylformamide, were developed as highly efficient systems for the dissolution of various types of lignin.44 Stirring the sample at 40 and 70 °C induces lignin solubility higher than 200 g kg−1 and 300 g kg−1, respectively. Among various solvent systems, γ-valerolactone/water was the most efficient one. The solubility of lignin could reach 381 g kg−1 even at 313 K with the content of water being 50 wt%.44 Although these depolymerization approaches successfully yield aromatic compounds, the bioconversion efficiency of these depolymerized aromatics still requires further evaluation.45
Biological depolymerization methods can catalyze the linkages of lignin polymers with powerful ligninolytic enzyme systems. Fungi and bacteria serve as effective ligninolytic microorganisms for lignin depolymerization. Ligninolytic fungi are classified as soft rot fungi, white rot fungi, and brown rot fungi. Ligninolytic bacteria include Actinobacteria, Firmicutes, Proteobacteria, and Pasteurella, encompassing Actinomyces such as Streptomyces and Rhodococcus, as well as Proteobacteria such as Pseudomonas and Sphingomonas.46 Microorganisms achieve the depolymerization of lignin macromolecules predominantly through their potent oxidase system in lignin-decomposing microorganisms. Ligninolytic enzymes catalyzing lignin depolymerization include laccase, manganese peroxidase, lignin peroxidase, dye-decolorizing peroxidase, glutathione-dependent peroxidase, β-etherases, and auxiliary enzymes that produce hydrogen peroxide (Table 1).47
| Enzymes | Gene sources | Expression strains | Substrates | Enzyme activity | Catalysis reactions | Products | Ref. |
|---|---|---|---|---|---|---|---|
| SL, Spartina alterniflora Loisel; CotA, coating protein A; ABTS, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid); DMP, 2,6-dimethoxy phenol; SBR, Similipal Biosphere Reserve; GGE, guaiacylglycerol-β-guaiacyl ether; LMCs, lignin model compounds; DHP, dehydrogenative polymer; MWL, milled wood lignin; DMBQ, 2,6-dimethoxy-1,4-benzoquinone; VGE, veratrylglycerol-β-guaiacyl ether; ZEN, zearalenone; GUA, guaiacol; VA, veratryl alcohol; DCP, 2,4-dichlorophenol; RB5, reactive black 5 (a high-redox potential dye). | |||||||
| Laccase | Trametes villosa | — | SL | — | Redoxidation | H2 | 149 |
| CotA lac | Escherichia coli BL21 | — | Lignin phenol derivatives | — | Oxidation | — | 150 |
| LacBl | Bacillus sp. PCH94 | E. coli BL21 | ABTS, DMP and guaiacol | 40.64 IU mg−1 for ABTS | Depolymerization | Ferulic acid, acetovanillone | 151 |
| Lac | Protein Data Bank | — | Lignin polymer, glyphosate, isoproturon and parathion | 99.3% degradation for dihydroconiferyl | Detoxification | — | 152 |
| Lac | Bacillus licheniformis | E. coli BL21 | Methylene blue | 99.28% decolorization | Decoloration; oxidation | — | 153 |
| CotA lac | Bacillus subtilis | E. coli | Kraft lignin | — | Oxidation | Lower molecules | 154 |
| Lac | Streptomyces coelicolor A3 | Pseudomonas putida A514 | ABTS | 300 U mL−1 | — | — | 155 |
| Manganese peroxidase | SBR | Pseudoduganella violacea | Maillard products | 6.12 U mL−1 min−1 | Demethylation | — | 156 |
| MnP | Ceriporiopsis subvermispora | Pichia pastoris X-33 | GGE and KL | — | Polymerization | Biphenyl GGE, heptamer length compounds | 56 |
| MnP | B. aryabhattai | B. aryabhattai | Kraft lignin | 9.2 IU mL−1 | Demethylation; cleavage of ether linkages; oxidation | KL degradation metabolites | 157 |
| MnP | Phanerochaete chrysosporium | E. coli | Lignin and other phenolic compounds | — | Oxidation | — | 158 |
| PcLiP01 | P. chrysosporium | P. chrysosporium | LMCs, DHP and MWL | — | Caryl–Cα bond cleavage | DMBQ | 159 |
| PcLiPs | P. chrysosporium | E. coli BL21 | Melanin | Interrupting π–π stacking and/or hydrogen bonds | — | 160 | |
| LiP | P. chrysosporium | E. coli | Veratryl alcohol, GGE and VGE | — | Oxidation of nonphenolic aromatics | Dimer phenoxy radicals | 161 |
| Dye-decolorizing peroxidase | Bacillus sp. BL5 | E. coli BL21 | ABTS and alkali lignin | — | Partial oxidation of coniferyl and sinapyl groups of lignin | Low-molecular-weight compounds, vanillin and methoxyphenol | 162 |
| PoDyP4 | Pleurotus ostreatus | E. coli Rosetta | Zearalenone | 9.5 U mg−1 | Hydroxylation and polymerization | ZEN dimer and four other products | 163 |
| CsDyP | Comamonas serinivorans | E. coli BL21 | ABTS, GUA, VA, and DMP | 317.87 U mg−1 | Depolymerization and oxidation | Aromatic monomers, low-molecular-weight lignin fractions | 164 |
| BsDyP | B. subtilis | E. coli BL21 | Lignin model compounds, DMP, guaiacol and VA | — | Oxidation | — | 165 |
| DypB | Comamonas testosterone | E. coli BL21 | ABTS, DCP and guaiacol | — | Cleavage of Cα–Cβ linkages | Aldehyde and ketone | 58 |
| Versatile peroxidase | Pleurotus eryngii | Pichia pastoris | Rice straw | 101 U L−1 for DMP | Delignification and saccharification | Bioethanol | 166 |
| VPs | P. eryngii, P. ostreatus | Saccharomyces cerevisiae | ABTS, DMP, RB5, VA and Mn2+ | — | Oxidation | — | 134 |
| VP | Bjerkandera adusta | — | Lignin from Casuarina equisetifolia | 1 U mL−1 for co-immobilized system | Oxidation of nonphenolic rings | Vanillin | 167 |
| VP | Physisporinus sp. strain P18 | E. coli | Kraft lignin and natural lignin, dimeric lignin model compounds | 19.8 U mg−1 | Degradation of β-O-4 and 5–5′ types of lignin dimer | Monomeric products | 168 |
The first type of ligninolytic enzyme in bacteria is laccase, initially characterized in fungi.47 Bacterial laccase is known to enhance the operational stability of lignin and oxidize a wide range of lignin-derived aromatics.48 As a multicopper oxidase, it catalyzes the four-proton reduction of O2 to H2O accompanied by the one-electron oxidation of four substrate molecules.49 The depolymerization of lignin by laccase is initiated via abstracting a single electron from the phenylpropanoid unit in lignin.50 Laccase can directly oxidize the phenolic subunits of lignin in the presence of a suitable medium and can also oxidize the non-phenolic units of lignin or high redox potential compounds.51 The phenolic subunits of lignin are catalyzed to phenoxy radicals by laccase, and the non-phenolic units are oxidized to β-aryl radicals and benzylic radicals by the laccase mediator system.49 The laccase mediator system can degrade 80–90% of the lignin structure.52 Peroxidases represent another essential type of ligninolytic enzyme in bacteria, working in conjunction with the co-substrate hydrogen peroxide (H2O2) to degrade lignin.53 Based on the type of reaction, peroxidases are classified as manganese peroxidase (MnP), lignin peroxidase (LiP), dye-decolorizing peroxidase (DyP), and versatile peroxidase (VP)53,54 MnP is a heme-containing glycoprotein and it catalyzes the oxidation of manganese ions (Mn2+) to Mn3+ in the presence of H2O2.55 When using guaiacylglycerol-β-guaiacyl ether (GGE) as the substrate, the manganese peroxidase from Ceriporiopsis subvermispora (CsMnP) catalyzed the formation of biphenyl GGE linked at the 5–5′ position of the phenol ring and generated a heptamer compound.56 Additionally, CsMnP demonstrated a remarkable ability to polymerize kraft lignin, which increased the molecular weight of the reaction by 360% and significantly reduced the phenol content by 37% in the study using kraft lignin as the substrate.56 These experiments proved that MnP was an effective biocatalyzer for lignin phenol subunit polymerization. Additionally, the strategy using the enzyme complex of laccase, LiP and MnP was explored to improve the depolymerization efficiency of lignin.
β-Etherases have also demonstrated promising potential in the process of lignin degradation.57 The strains of Sphingobium, Novosphingobium and other α-Proteobacteria produce intracellular β-etherases that can utilize glutathione to attack the β-ether bond in lignin fragments.58 They are also able to cleave lignin dimer compounds through the β-aryl ether degradation pathway to generate vanillin, and then be funneled into the downstream pathway.59 Therefore, diverse ligninolytic enzymes in bacteria exhibited high substrate specificity, holding the potential to yield suitable lignin-derived aromatics for downstream biotransformation.
Chemical depolymerization methods enable the effective breakdown of lignin, resulting in more versatile aromatic products. However, these methods require high energy inputs and rely on chemical reagents, leading to increased costs.60 In contrast, biological depolymerization methods, which utilize mild ligninolytic enzyme systems, are more cost-effective and environmentally friendly, offering a viable solution to lignin's challenging degradability.61 Despite their advantages, biological methods still face challenges related to yield and efficiency, requiring further optimization to achieve more effective depolymerization and support subsequent biotransformation processes. Recently, significant progress has been made in combining chemical and biological depolymerization techniques, demonstrating substantial potential for improving the separation efficiency of lignin components.
2.2 Metabolic pathways and enzymes of depolymerized lignin oligomers
After preliminary depolymerization, the depolymerized lignin oligomers can be further metabolized by ligninolytic strains.62 The lignin-derived oligomers were first catabolized into vanillin and butyric acid, which underwent O-demethylation to form catechol derivatives. Catechol derivatives undergo aromatic ring cleavage to produce tricarboxylic acid cycle intermediates.63 In bacteria, various lignin-derived biaryl and monoaryl groups, along with guaiacyl and syringyl nucleotides, are converted into vanillin and syringic acid through specific ligninolytic enzymes. Vanillin and syringic acid are subsequently demethylated to yield aromatic compounds with catechol structures.64Sphingomonas sp. SYK-6 is regarded as an exemplary organism for the decomposition and metabolism of lignin-derived aromatic compounds (Fig. 2).14 It can absorb various lignin-derived diaryls, such as biphenyls, β-aryl ether, phenylcoumarin, diarylpropane, as well as monoaryls like ferulic acid, vanillin, syringal, and syringal acid.63 In SYK-6, protocatechuic acid ester 4,5-dioxygenase (LigAB) and gallic acid dioxygenase (DesB) play key roles in the downstream catabolism of lignin derived aromatics with guaiacol and eugenol nuclei, respectively.65 Due to the abundance of β-aromatic ether bonds in lignin, the study of β-aryl ether cleavage is a vital aspect of lignin degradation. Enzymes involved in β-aryl ether catabolism have been identified, with X-ray crystal structures determined for enzymes such as LigD, LigL, LigF, LigE, LigG, and LigO.66 The characterized β-aromatic ether catabolism system in bacteria closely resembles that of SYK-6, underscoring the importance of elucidating its role in the natural degradation of β-aromatic ethers.
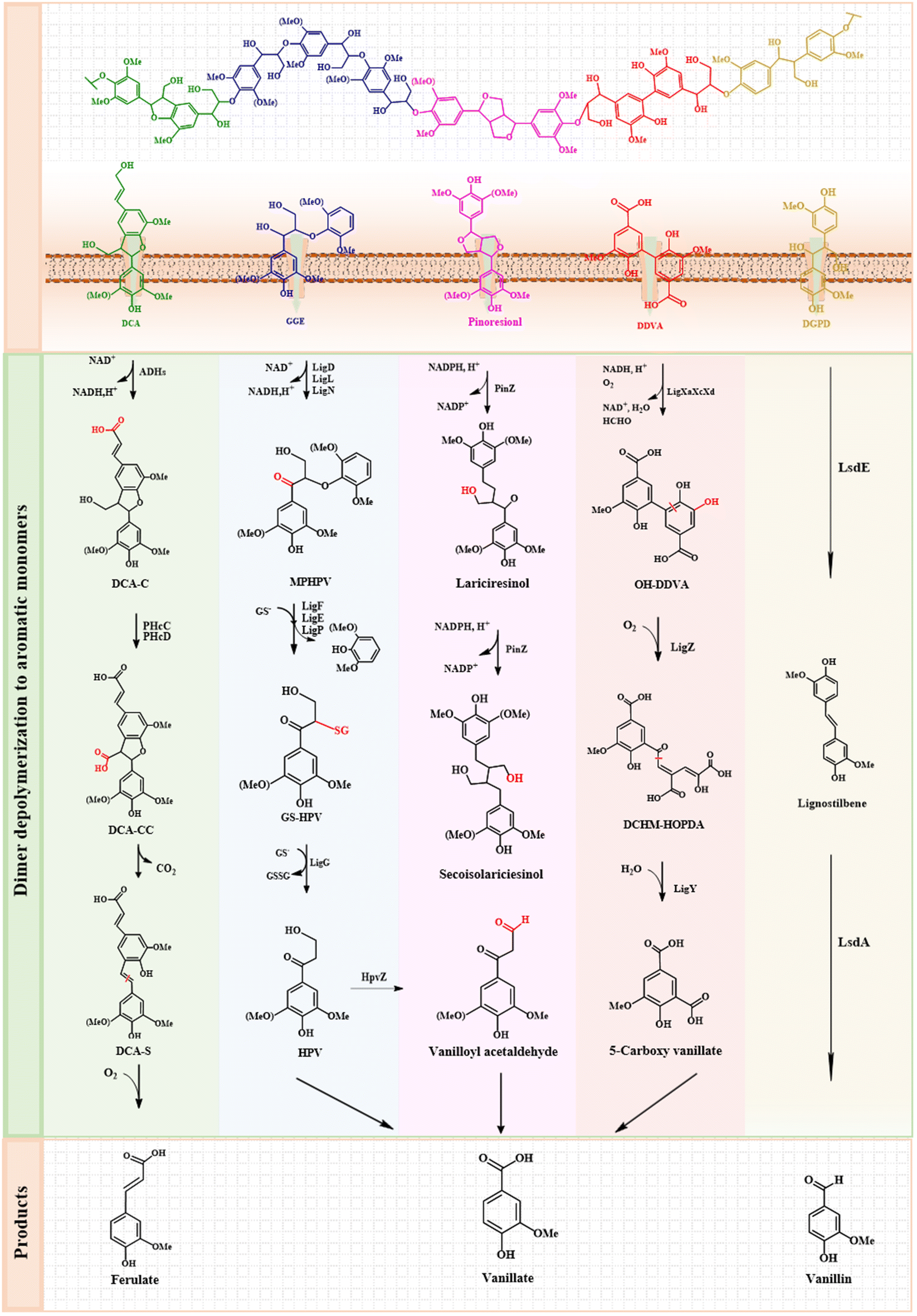 | ||
| Fig. 2 Main catabolic pathways of lignin-derived compounds. The lignin polymer model is shown at the top. Lignin polymer is initially depolymerized into oligomers and transported across the membrane into the cell, the oligomer compounds with guaiacyl and syringyl nuclei are then funnelled into vanillate and syringate, respectively. DCA, dehydrodiconiferyl alcohol; GGE, guaiacylglycerol-β-guaiacyl ether; DDVA, 5,5′-dehydrodivanillate; DCA-C, 3-(2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-7-methoxy-2,3-dihydrobenzofuran-5-yl)acrylate; DCA-CC, 5-(2-carboxyvinyl)-2-(4-hydroxy-3-methoxyphenyl)-7-methoxy-2,3-dihydrobenzofuran-3-carboxylate; DCA-S, 3-(4-hydroxy-3-(4-hydroxy-3-methoxystyryl)-5-methoxyphenyl)acrylate; MPHPV, α-(2-methoxyphenoxy)-β-hydroxypropiovanillone; GS-HPV, α-glutathionyl-β-hydroxypropiovanillone; HPV, β-hydroxypropiovanillone; OH-DDVA, 2,2′,3-trihydroxy-3′-methoxy-5,5′-dicarboxybiphenyl; DCHM-HOPDA, 4,11-dicarboxy-8-hydroxy-9-methoxy-2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate. | ||
A bacterial catabolic pathway to phenylcoumarin-type biaryl dehydrodiconiferyl alcohol (DCA) was proposed in S. paucimobilis TMY1009 and SYK-6.63 In the catabolic pathway to dehydrodiconiferyl alcohol identified in SYK-6, the alcohol group of the B-ring side chain of dehydrodiconiferyl alcohol was first oxidized to a carboxyl group and 3-(2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-7-methoxy-2,3-dihydrobenzofuran-5-yl) acrylate (DCA-C) was generated from 3-(2-(4-hydroxy-3-methoxyphenyl)-3-(hydroxymethyl)-7-methoxy-2,3-dihydrobenzofuran-5yl) acrylaldehyde (DCA-L). The alcohol group of the A-ring side chain of DCA-C was converted into 5-(2-carboxyvinyl)-2-(4-hydroxy-3-methoxyphenyl)-7-methoxy-2,3-dihydrobenzofuran-3-carboxylate (DCA-CC) by two-step oxidation. The Cγ-carboxyl group of the A-ring side chain of DCA-CC was decarboxylated to produce 3-(4-hydroxy-3-(4-hydroxy-3-methoxystyryl)-5-methoxyphenyl)acrylate (DCA-S), which was further cleaved between Cα and Cβ of the A-ring side linkage to form vanillin and 5-formylferulate. Finally, ferulic acid was prepared by removing the formyl group of 5-formylferulic acid.63 In this metabolic pathway, multiple quinone protein alcohols and aryl alcohol dehydrogenases in SYK-6 participate as dehydrodiconiferyl alcohol convertases. Aryl alcohol dehydrogenases play a major role in the oxidation of dehydrodiconiferyl alcohol, while aldehyde dehydrogenases participate in the transformation of DCA-L.67 Cytoplasmic and membrane-located SLG_09480 (phcC) and SLG_09500 (phcD) act as enantioselective oxidases responsible for the oxidation of Cγ alcohols in the A-ring side chain of DCA-C.68 Additionally, the guaiacylglycerol-β-guaiacyl ether oxidoreductase family in SYK-6 plays an important role in the oxidation of alcohols at Cγ of C6–C3 lignin-derived compounds.68
The β–β bond typically represents secondary linkages that need to be metabolized in other lignin-derived diaryl groups. Pinol and syringoresinol are typical β–β dimers, also known as lignans. (±)-Pinoresinol cannot be utilized as the only carbon source and energy material by SYK-6, whereas (±)-secoisolariciresinol can be produced through the two-step reductive ether cleavage of (±)-lariciresinol.69 The presence of the pinZ gene in SYK-6 is crucial, as the atypical short-chain dehydrogenase/reductase encoded by this gene enables the transformation of (±)-pinoresinol. This enzyme also exhibited activity against syringoresinol. Therefore, the accumulation of the secondary isoleucine glucoside in pinZ transgenic plants can be achieved by reducing the levels of abiesterol and its glucoside.67 As another secondary component of lignin, the β-1 structure mainly exists in the form of a spirodienone structure.70S. paucimobilis TMY1009 can degrade a β-1 compound such as 1,2-bis(4-hydroxy-3-methoxyphehyl)-1,3-propanediol (HMPPD). 1,2-Bis(4-hydroxy-3-methoxyphehyl)-1,3-propanediol initially underwent Cγ deformylation while dehydroxylation at Cα produced a stilbene-type compound. TMY1009 possessed four lignostilbene α,β-dioxygenase (LSD) isoenzymes to convert β-1 lignin stilbenes into two equivalents of vanillin. However, the actual involvement of LSD-I has not been elucidated in the catabolism of 1,2-bis(4-hydroxy-3-methoxyphehyl)-1,3-propanediol.
Overall, lignin-derived oligomers undergo further assimilation under the catalysis of various enzymes. This process not only reveals the potential biological conversion pathways for lignin but also lays the foundation for the subsequent development of “biological funnel” pathways. This step is a crucial component in the conversion of lignin into high-value products. At the same time, continuing to optimize enzyme catalytic efficiency, improve reaction selectivity, and develop new biological conversion pathways remains the key focus of this process.
2.3 Biological funnel pathways and enzymes of lignin-derived aromatic monomers
Representative monomers of lignin include p-coumaric acid (H-type lignin), ferulic acid (G-type lignin), and syringic acid (S-type lignin). The heterogeneous structure of lignin continues to pose significant challenges, necessitating extensive research efforts to advance its valorization.71 Some ligninolytic microorganisms such as Pseudomonas putida KT2440 have evolved a pathway that converges lignin-derived aromatics into central intermediates, such as protocatechuic acid (PCA).72 This metabolic pathway, termed “biological funneling”, comprises cascade enzymatic reactions like side-chain cleavage, decarboxylation and oxidation.72 This strategy enables microbes to funnel diverse aromatic compounds into a single product, preserving the energy stored in the aromatic ring and streamlining lignin metabolism.The metabolic pathways of H-type aromatics in bacteria can be divided into three categories: coenzyme A (CoA) dependent β-oxidation pathway, coenzyme A dependent non β-oxidation pathway, and coenzyme A independent decarboxylation pathway.73 The intermediate of p-hydroxybenzoic acid was synthesized through these three pathways, and then converted into protocatechuic acid through the 4-hydroxybenzoate-3-monooxygenase PobA. By integrating the beneficial genes for p-hydroxybenzoate hydroxylase (PobA) and vanillate O-demethylase oxygenase (VanAB) and implementing the fed-batch strategy, engineered P. putida exhibited remarkable PCA production from lignin-derived monomers, achieving a titer of 113.6 mM (17.5 g L−1).74 The metabolic pathways of G-type aromatics in bacteria include coenzyme A-dependent β-oxidation, coenzyme A-dependent non-β-oxidation, non-oxidative decarboxylation and side-chain reduction.75 These pathways enable the conversion of ferulic acid into vanillic acid intermediates, which are converted into vanillic acid by vanillic acid O-demethylase VanAB. The involvement of class IA oxygenases VanA and VanB reductase in the O-demethylation of vanillin is reported in Pseudomonas. A biological funnel pathway was successfully constructed in S. cerevisiae by deleting the ADH6, ADH7, BDH2, and FDC1 genes and other optimization strategies.76 The study successfully obtained protocatechuic acid titers up to 810 mg L−1 from lignin streams of p-coumaric acid and ferulic acid, highlighting that it was a sustainable approach for lignin valorization.
S-Type aromatics possess two methoxy groups in the aromatic ring. Due to a poor demethylation performance in most bacteria, their overall metabolic efficiency remains unsatisfactory.14Sphingosinomonas SYK-6 is the best bacterium for the metabolism of S-type aromatics. Degradation through the syringic acid pathway is initiated by tetrahydrofolate dependent O-methyltransferase LigM and DesA, and representative products mainly include protocatechuate, gallic acid and 3-O-methylgallic acid.77 Heterologous engineering of the O-demethylation system in P. putida KT2440 allowed it to metabolize S-type lignin. On this basis, the biosynthesis pathway for cis,cis-muconic acid (CCMA) was engineered to simultaneously process three types of lignin-derived aromatics, achieving a remarkable titer and yield of 13.1 mM and 99.5%, respectively.77
Other aromatic compounds like phenol, benzene, benzoate, toluene, and naphthalene can be directed to the catechol pathway for further metabolism.78 Catechol degradation is primarily catalyzed by dioxygenase through ortho- or meta-cleavage pathways.76 During the degradation process, catechol 1,2-dioxygenase (catA) and cis,cis-muconate cycloisomerase (catB) play important regulatory roles.79 With the overexpression of catA and inactivation of catB, the engineered strain produced 13.5 g L−1cis,cis-muconic acid from p-CA in a fed-batch bioreactor.80 Afterward, further research focused on modifying the metabolically engineered strain P. putida KT2440.81 A synthetic pathway module was designed under the control of the Pcat promoter, comprising both native catechol 1,2-dioxygenases catA and catA2. This modification aimed to enhance catechol tolerance, increase the levels of catechol 1,2-dioxygenase, and improve catechol conversion rates. The engineered MA-6 strain achieved an cis,cis-muconic acid titer of 64.2 g L−1 from catechol in a fed-batch process.81
The biological funnel pathway, along with the associated enzymatic activities, demonstrates the versatility of ligninolytic bacteria in breaking down various lignin-derived aromatic compounds into central intermediates for further metabolism and energy production. Understanding these pathways is crucial for optimizing lignin biotransformation.
2.4 Ring-opening metabolic pathways and enzymes for lignin transformation
Following the biological funnel pathway, key aromatic intermediates further undergo ring-opening pathways to generate various metabolites that can enter central metabolism. Protocatechuic acid (PCA) follows three distinct ring-opening pathways, namely, the 3,4-cleavage pathway (β-ketoadipic acid pathway), the 4,5-cleavage pathway, and the 2,3-cleavage pathway.P. putida KT2440 harnesses the 3,4-cleavage pathway of protocatechuic acid catalyzed by protocatechuic acid 3,4-dioxygenase and a series of enzymes, namely PcaB, PcaC, PcaD, and PcaI/J.12Paenibacillus sp. harnessed the 2,3-cleavage pathway of protocatechuic acid catalyzed by protocatechuic acid 2,3-oxygenase, with PraH, PraB, PraC, PraD and PraE, to yield 4-hydroxy-2-oxyvalerate. The 4,5-cleavage pathway of protocatechuic acid is found in Sphingobium sp. SYK-6 and Novosphingobium aromaticivorans, and protocatechuic acid can be converted into 4-carboxyl-2-hydroxy-mucilate-6-hemialdehyde (CHMS) by protocatechuic acid 4,5-dioxygenase LigAB.70 4-Carboxyl-2-hydroxy-mucilate-6-hemialdehyde can be automatically converted into the intramolecular hemiacetal form and then oxidized by 4-carboxyl-2-hydroxy-mucilate-6-hemialdehyde dehydrogenase LigC to obtain 2-pyrone-4,6-dicarboxylicacid.82 2-Pyrone-4,6-dicarboxylate can be further catalyzed by LigI/J to produce 4-carboxyl-4-hydroxy-2-oxadipate (CHA), which is then converted into pyruvate and oxaloacetate under the action of 4-carboxyl-4-hydroxy-2-oxadipate aldolase LigK.
Lignin-derived aromatic monomers can be converted into catechol by the catalytic action of protocatechuic acid decarboxylase. Catechol is then further degraded in bacteria through the ortho-cleavage and meta-cleavage pathways. P. putida KT2440 was engineered to replace the natural protocatechuic acid 3,4-oxygenase with protocatechuic acid decarboxylase, enabling the conversion of protocatechuic acid into catechol.83 In Pseudomonas CF600, the meso-cleavage pathway of catechol was initially catalyzed by catecho-2,3-dioxygenase (C2,3D) to produce 2-hydroxymyxohemialdehyde. Additionally, the inter-cleavage pathway of catechol was found in both Gram-negative and Gram-positive strains.84 Sphingosinomonas SYK-6 harbored DesZ or LigAB to catalyze 3-O-methylgallic acid to produce 2-pyrone-4,6-dicarboxylate, which could be integrated into the 4,5-cleavage pathway of protocatechuic acid and catalyzed by LigI to produce 4-oxychloroadone alcohol. Gallic acid was converted into 4-chlorinated salicylic acid by DesB or LigAB, and then to 4-carboxyl-4-hydroxy-2-oxadipate by LigJ. 4-Carboxyl-4-hydroxy-2-oxadipate can further be cleaved by LigK to form pyruvate and oxaloacetate.
The aforementioned information provides a concise summary of the ring-opening metabolic pathways associated with several key intermediates derived from lignin. It is important to note that different ligninolytic strains may exhibit distinct catabolic preferences for aromatic compounds, reflecting their unique enzymatic machinery and metabolic adaptations. These variations highlight the need for strain-specific optimization to maximize lignin valorization efficiency.
In conclusion, exploring and identifying more comprehensive and potent biocatalytic systems within microorganisms is crucial for advancing lignin utilization. Recent advances in multi-omics approaches have enabled the discovery of novel enzymes and pathways, which expand the toolbox for lignin depolymerization and bioconversion (Table 2). Additionally, the application of synthetic biology and protein engineering techniques enables the design of tailored microbial strains with enhanced lignin-degrading capabilities. Furthermore, understanding the regulatory mechanisms governing aromatic catabolism in ligninolytic microbes can provide valuable insights into improving their performance under industrial conditions. For instance, elucidating the role of transcriptional regulators and stress response systems in these organisms could lead to the development of more robust biocatalysts capable of withstanding inhibitory compounds and harsh processing environments. Therefore, the exploration of microbial biocatalytic systems could not only deepen our understanding of lignin metabolism but also pave the way for more efficient and sustainable lignin valorization.
| Screening methods | Evolution methods/library design | Library size | Expression strains | Enzymes | Detection substrates | Outcomes | Ref. |
|---|---|---|---|---|---|---|---|
| 2,6-DMP, 2,6-dimethoxy phenol; 4-MUC, 4-methylumbelliferyl-β-D-cellobioside; 2,5-DABSA, 2,5-diaminobenzenesulfonic acid; FCB, fluorescein di-β-D-cellobioside; ARTP, atmospheric and room temperature plasma; ABTS, 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid); vi, initial reaction rates; KD, equilibrium dissociation constants; kcat, Km, apparent steady-state kinetics; GcoAB enzyme system, a new architectural class of P450 with a cytochrome P450 monooxygenase (GcoA) and a three domain reductase (GcoB); DCP, 2,4-dichlorophenol; SEM, scanning electron microscopy; XRD, X-ray diffraction; RSM, response surface methodology; ANN-GA, artificial neural networks technique coupled to the genetic algorithm. | |||||||
| Two-step high-throughput screening | — | — | 82 bacterial and 46 fungal strains | Laccases | 2,6-DMP | Identified ten bacterial and five fungal strains with high laccase activities and the highest laccase activity achieved 133.44 U L−1 | 94 |
| InVitroFlow technology platform | Multisite saturation mutagenesis | 3.6 × 106 | Escherichia coli BL21 | CelA2 cellulase | 4-MUC | The cellulase variant CelA2-M3 showed an 8-fold increase in activity and a 41-fold increase in activity against 4-MUC | 98 |
| High-throughput colorimetric assay | Directed and focused molecular evolution | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
Saccharomyces cerevisiae | Laccases | Catechol and 2,5-DABSA | The laccase mutant was secreted at 37 mg L−1, and its catalytic efficiency for catechol and 2,5-DABSA was improved by 3.5 times | 99 |
| Droplet-based microfluidic platform based on a cellulase-catalyzed reaction | Atmospheric and room temperature plasma (ARTP) mutagenesis | — | Pichia pastoris | Heterogeneous cellulase | FCB | The cellulase mutant showed a 7-fold increase in enzyme activity to 11110 U mL−1 |
106 |
| Diverse functional profile screening experiments | PROSS stability design calculations | 43 | Yeast | Versatile peroxidases | ABTS | The activities of the two versatile peroxidase mutants were increased 11-fold and 14-fold, respectively | 122 |
| Measured vi, KD and kcat, Km parameters | Structure-guided protein engineering and molecular dynamics | — | P. putida KT2440 | GcoAB enzyme system | O- and p-vanillin | Demonstrated GcoAB and its single amino acid variants could catalyze a wide range of aromatic O-demethylations | 126 |
| High-throughput screening monitored for ABTS and DMP oxidation in 96-well plates | Three rounds of mutagenesis by error-prone PCR | 6000 | E. coli | Dyp | H2O2, ABTS and 2,6-DMP | The Dyp variant 6E10 achieved 100-fold increase in catalytic efficiency for 2,6-DMP, and it also demonstrated resistance to hydrogen peroxide inactivation and was produced in 2-fold higher yields | 130 |
| The libraries were screened for activity towards DCP and alkali Kraft lignin | Combinatorial active site saturation (CAST) method | 546 | Pseudomonas fluorescens Pf-5 | Dyp1B | DCP and alkali Kraft lignin | The H169L mutant showed an eightfold increase in catalytic efficiency | 119 |
| SEM and XRD monitored the changes of rice straw | Artificial intelligence modeling and molecular docking verification | 1000 | C. testosteroni FJ17 | Laccases | Rice straw | The degumming mechanisms of laccase and lignin compounds were revealed and the laccase activity reached 2016.7 U L−1 | 143 |
| Statistical analysis by RSM | Experimental modeling through the ANN-GA | 135 | Pleurotus sajor-caju | Laccase and MnP | Pulp wash (PW) | Laccase and manganese peroxidase could decolorize the dye up to 89.4% and 75%, respectively | 144 |
3. High-throughput screening identified ligninolytic enzymes and strains
3.1 Fluorescence-activated cell sorting (FACS) for ligninolytic enzymes
Fluorescence-activated cell sorting (FACS) is a sorting type of flow cytometry that enables the simultaneous analysis of multiple parameters of single cells from a mixed population and the rapid sorting of target cells (Fig. 3).85 FACS, which relies on fluorescence as a signal source, is the most widely used screening strategy. This method can be further classified based on the type of fluorescence used, including fluorescent substrates, fluorescent proteins, and DNA fluorescent probes. Fluorescent substrates, in particular, are commonly employed in enzyme engineering, as enzymes can cleave these substrates, releasing fluorescent products. Common fluorescent substrates include catechol, coumarin, fluorescein, and resorufin.86,87 The core of this technology is the coupling of genotype and phenotype, with the key being the conversion of phenotypic signals into detectable indicators for large-scale screening.88,89 | ||
| Fig. 3 The workflow for high-throughput screening identified ligninolytic enzymes and strains. FCB, fluorescein di-β-D-cellobioside. | ||
Lignin contains various conjugated structural units that emit a bluish-green glow when exposed to ultraviolet light, making it a key contributor to the fluorescence observed in plant cell walls.90 Lignin exhibited a peak fluorescence at approximately 400 nm, and demonstrated a broad emission spectrum that extends from the near-ultraviolet to the entire range of visible light wavelengths. However, the exact fluorophoric elements responsible for lignin's fluorescence are still not fully understood. Its fluorescent characteristics, nevertheless, resemble those of certain model compounds, particularly those with anionic, stilbene, and phenylcoumarone structures, including pinol.90,91 The variation in emission wavelengths of lignin's fluorescence can be attributed to the combined effects of clustering and the spatial conjugation of its phenylpropane components and benzene rings, at shorter and longer wavelengths, respectively. These characteristics of lignin enabled the synthesis and application of lignin-based fluorescence resonance energy transfer pH fluorescent probes. A significant technology for determining lignin content emerged by correlating its concentration with the level of fluorescence. Nevertheless, lignin's photoluminescent properties in solution can be influenced by various types of aggregation, including the dimming of fluorescence due to aggregation and the enhancement of brightness due to luminescent aggregation.92 Similarly, the complex and heterogeneous macromolecular structure of lignin complicates the design of fluorescent substrates. Environmental factors such as solution concentration, temperature, solvent polarity, pH, viscosity, ion type, ionic strength, and pressure also significantly affect lignin's fluorescence behavior. Bridging this knowledge gap is essential for advancing the use of lignin as a fluorescence tool in high-throughput screening technologies.
High-throughput screening technologies enable the rapid isolation of new ligninolytic microorganisms from a large number of natural biomasses.93 Advanced technologies in microbiological culture enrichment and enzyme-activity-based screening facilitated a comprehensive study conducted on decaying wood samples collected from the Ottawa region in Canada.94 A total of 50 samples were meticulously collected and subjected to a battery of tests to identify and isolate microorganisms pivotal in the decomposition process. This rigorous scientific inquiry successfully led to the isolation of a diverse microbial population, comprising 82 distinct bacterial strains and 46 unique fungal strains.94 The isolation of such a high number of strains underscored the richness of microbial diversity in decaying wood ecosystems and provided a valuable resource for further ecological and applied research. This methodology established a new standard for the isolation of microorganisms from natural environments, especially those involved in the degradation of lignocellulosic biomass. Additionally, a heat- and alkali-tolerant enterobacterium was isolated from sediment samples collected at the mouth of the Mandovi River on the west coast of India.95 The strain was identified as a highly efficient producer of xylanase, along with ligninolytic bacteria and fungi exhibiting high laccase activity. Notably, some of these strains also demonstrated additional enzymatic activities, including xylanase and β-glucanase.94 This method was applicable not only to screening ligninolytic microorganisms but also to identifying new microbial strains and enzymes from various natural samples, thereby increasing opportunities to isolate desired strains based on specific enzyme activities.95
Critical and efficient technologies have been developed for acquiring lignin hydrolase mutants. The combined active site-saturation test (CAST) was sophisticated technology employed to pinpoint critical amino acid residues near an enzyme's active site. This technology leveraged the enzyme's three-dimensional structural data to systematically identify and evaluate the importance of side chain locations crucial for its catalytic function. By saturating the active site with various amino acid substitutions, CAST enabled the determination of the functional significance of individual residues and their interactions within the active site environment. This approach not only enhanced our understanding of enzyme mechanics but also facilitated the rational design of enzymes with enhanced or novel functionalities.4 The OmniChange mutagenesis method represented a revolutionary tool for site-specific mutagenesis technologies. Unlike traditional methods, OmniChange could simultaneously saturate up to five codons, significantly accelerating the mutagenesis process and enabling a more comprehensive exploration of the sequence space. Moreover, it did not require a minimum distance between target codons, allowing tightly clustered sites to be mutated in a single experiment. This greatly simplified the process and reduced the time and resources required for repeated mutations.96 Following the generation of lignin hydrolase mutants, high-throughput screening technologies could be employed to identify mutants with optimal secretion levels, activity, stability, and pH applicability.
Among various high-throughput screening technologies, FACS, which uses cells as detection chambers, has emerged as the preferred choice due to its superior screening efficiency and greater versatility.97 The most common strategy involved converting non-fluorescent substrates into fluorescent products through enzyme-catalyzed reactions. This approach was typically applied to hydrolases such as cellulase, lipase, and esterase.11 In this strategy, the substrate must be able to cross the cell membrane or be transported into the cell, where it can generate a fluorescent signal under the catalysis of intracellular enzymes. The InVitroFlow technology screened mutant libraries by first generating them using plasmid DNA templates. Mutant cellulase libraries were then expressed cell-free within water-in-oil (w/o) single emulsions. The activity variations were classified using flow cytometry in water-in-oil-in-water (w/o/w) double emulsions. DNA was subsequently extracted from the (w/o/w) double emulsions, followed by PCR gene amplification, cloning into an expression host, and transformation to express beneficial clones. The plasmid DNA from the best variants was isolated and used as template DNA for the next round of directed evolution and screening.98 The InVitroFlow method enabled the screening of the multi-site saturated mutagenic library and OmniChange library, targeting four saturated sites at CelA2 cellulase active site. After screening over 36 million events, a significantly improved cellulase variant, CelA2-M3 (H288F/H524Q), was successfully obtained.86 The specific activity of the cellulase variant reached 83.9 U mg−1, which was 8 times higher than that of the wild-type CelA2. Additionally, the specific activity for the substrate 4-methylumbelliferyl-β-D-cellobioside was 41 times higher than that of the wild-type CelA2.98 These results clearly demonstrated the outstanding advantages of the InVitroFlow technology implemented to accelerate screening efforts and revealed the significant potential for improving enzyme activity through natural variation.
Additionally, enzymes such as proteases, which can break special functional bonds, can also be screened using surface display technologies, with yeast commonly used as a display carrier. By combining high-throughput screening technology and directed evolution, a strongly expressed alkaline laccase variant was identified in Saccharomyces cerevisiae.99 This high-throughput screening experiment was based on the oxidative cross-coupling between 2,5-diaminobenzenesulfonic acid (2,5-DABSA) and catechol, catalyzed by laccase at pH 8.0. After the enzyme-catalyzed oxidation reaction, a colorimetric reaction occurs between the originally colorless catechol and 2,5-DABSA. This colorimetric reaction was highly sensitive to pH and could serve as an indicator for evaluating the activity of the laccase variant enzymes. After conducting rigorous benchmark tests on laccase mutants for the oxidation of catechol and 2,5-DABSA, a mutation library of approximately 1700 mutants was constructed through further evolution. The mutants were then expressed in S. cerevisiae and screened using high-throughput colorimetric methods. The final laccase mutant was secreted at 37 mg L−1, and its catalytic oxidation efficiency for catechol and 2,5-DABSA at pH 8.0 was improved by 3.5 times compared to the wild type.99 The resulting laccase mutant was easily secreted, providing a highly significant platform for advancing work related to organic synthesis. Autonomous hypermutation yeast surface display technology (AHEAD) was developed.21 This synthetic recombinant antibody generation technology mimicked somatic hypermutations in engineered yeast. By applying this screening technology, potent nanobodies targeting SARS-CoV-2 S glycoproteins, G-protein-coupled receptors, and other targets were successfully generated.21 Additionally, the combination of substrate and fluorescence resonance energy transfer (FRET) probe allowed for the detection of enzyme activity or the screening of active mutants by observing the significant change in substrate fluorescence wavelength before and after the enzyme reaction. A novel cationic probe1 (Mito-H2O2) targeting mitochondria was sensitive to the presence of H2O2 and could be used to detect H2O2 quickly.100 The probe featured a fast response time, high specificity, and high sensitivity. This unique organelle targeting fluorescent probe is expected to become a practical tool for H2O2-related ligninolytic enzymes and to stimulate the production of new organelle specific fluorescent sensor devices.100
The sorting method primarily relied on highly sensitive fluorescent probes of hydrogen peroxide. It was also widely applicable to the directed evolution of other flavin adenine dinucleotide-dependent oxidases in E. coli. Fungi that transformed lignin in nature did not function well in neutral/alkaline pH ranges and were strongly inactivated at moderate concentrations of OH−. Due to the release of organic acids during the metabolism of white rot fungi, lignin decomposition occurred at acidic pH. However, the evolution of oxidation stability in versatile peroxidases can be achieved by designing a high-throughput screening method based on the molar ratio of H2O2 to enzyme. This allowed for the screening of clones with improved activity at alkaline pH while maintaining the activity at acidic pH, thereby altering the pH activity profile of these enzymes. In this way, variants that functioned under both neutral/alkaline and acidic conditions could be obtained, providing significant versatility for various industrial and environmental applications.101
As the most promising high-throughput screening method for enzymes, the introduction of FACS technology holds great potential in the field of directed evolution of ligninolytic enzymes. However, practical applications of this screening technology face several challenges. Variability among individual cells and inconsistencies in the transport of target products across cell membranes can lead to a high rate of false positive outcomes during large-scale screenings. To improve FACS technology, it is crucial to align the genotype with phenotype complexities, integrate both internal and external fluorescent signals, and mitigate the effects of erroneous positive results that may skew the information received. Furthermore, FACS technology is limited to detecting fluorescent signals linked with desired lignin derived products inside cells or fluorescent signals of membrane-associated metabolites. Its inability to screen for secreted metabolites and extracellular enzymes in high-producing cells restricts its broader applicability. Therefore, further research is needed to overcome these limitations and advance high-throughput screening instrumentation.
3.2 Droplet microfluidic screening of ligninolytic enzymes
Droplet microfluidic screening, a powerful approach for high-throughput screening, relied on a series of complex operations performed on microfluidic chips. These operations included DNA fragment assembly, host cell transformation, cell culture, recombinant protein expression and sophisticated droplet manipulation techniques coupled with ultrafast separation methods.17 The development of droplet microfluidic screening overcame limitations inherent to traditional liquid-based screening environments.102 This screening technology was advantageous for enhancing the biosynthesis of chemical substances or improving the properties of extracellular enzymes.103 It provided robust methodological support for discovering rare cells in bio-directed evolution experiments.104Microfluidic tools for high-throughput functional screening of enzyme libraries have enabled the routine screening of millions of enzyme variants, streamlining the identification of active variants in directed evolution and functional metagenomic projects. A universal high-throughput screening platform for droplet-based microfluidics was proposed and implemented in 2010.105 Compared to state-of-the-art robotic screening, this system was 1000 times faster, required 10 million times fewer test doses, and offered cost savings estimated to be about 10 million times greater. The system was successfully implemented in directed evolution, leading to the identification of a new mutant of horseradish peroxidase (HRP).105 Droplet microfluidic screening technology has been reported in enzyme and cell factory engineering. A droplet-based microfluidic high-throughput screening platform was successfully used for whole-cell directed evolution of P. pastoris and further improved heterogeneous cellulase production (Fig. 3).106 The fluorogenic substrate fluorescein di-β-D-cellobioside (FCB) was used to label heterogeneous cellulase, and methanol served as the organic solvent to form fluorescently labeled water-in-oil droplets. After off-chip incubation for cell growth and cellulase expression for 40 h at 30 °C, droplets were analyzed and sorted based on the fluorescence signal, and finally cells were recovered from the sorted droplets. At a sorting rate of 300 droplets per second, the sorting efficiency of the platform reached 94.4%. Finally, after five rounds of iterative atmospheric and room temperature plasma (ARTP) mutagenesis and microfluidic screening, the best mutant strain was obtained, exhibiting cellulase activity of 11110 U mL−1, nearly twofold higher than the starting strain.106 This high-throughput screening system can also be applied to engineer other protein-producing filamentous fungi and will provide valuable insights for accelerating whole-cell directed evolution of host strains and enzymes of high industrial interest. A microfluidic screening platform could enhance the efficiency of enzyme kinetics data collection by adjusting the flow rates of the droplets.107 This method was successfully applied to study the complex dynamics and thermodynamics of engineered haloalkane dehalogenase variants, offering insights into substrate specificity and hydration-related entropy effects. By combining global data analysis with molecular dynamics simulations, this approach guided the design of novel biocatalysts of lignin and provided direction for developing new biocatalysts.108
The practical value of high-throughput screening technology using droplet microfluidics is well established. Since the initiation of the Tox21 project, this approach has evaluated approximately 8500 unique chemicals, accumulating a vast repository of over 120 million data points using high-throughput screening technologies.19 The project's achievements highlighted the transformative power of technological advancements in tackling complex scientific challenges.19 This strategy was employed for the screening and evolution of ligninolytic enzymes. A genetically programmed vanilla-sensing bacterium was successfully developed and utilized for high-throughput screening of ligninolytic enzyme libraries.109 Vanilla-sensing bacteria were developed through RNA sequencing analysis, which identified a vanilla-induced promoter. The green fluorescent protein gene was then placed under the control of that promoter. The fluorescence of this biosensing cell was significantly enhanced in the presence of vanillin and could be easily observed using a fluorescence microscope. This biosensor was highly specific to vanillin and could detect vanillin concentrations as low as 200 μM.109 These engineered E. coli cells can be used as host cells to screen for enzymes capable of converting lignin into vanillin, thereby significantly enhancing the efficiency of screening ligninolytic enzymes.
Glycosidases, as prominent representatives of industrial hydrolase centers, are extensively used in the degradation of lignocellulosic biomass.110 A novel screening process was designed with three main steps: liquid droplets, colonies, and lysates, with plasmid transfer to E. coli enabling co-encapsulation of single bacteria, lysates, and fluorescent substrates.111 In a single experiment, absorbance measurements of droplets at multiple adjustable time points were designed, with the flow direction reversed to enable multiple detection of products generated for determining enzyme kinetic parameters.112 Twelve high-resolution and high-precision datasets of Michaelis–Menten kinetics were obtained, showcasing the substrate's characterization of the glycosidase potential across seven orders of magnitude of kcat/Km enzymatic hydrolysis.113 Glucosidase β-glucuronidase, a member of the glycoside hydrolase 3 family with few homologous enzymes, was successfully identified using the high-throughput screening platform.111 This system demonstrated high throughput, excellent data quality, and a wide dynamic range, making it suitable for detailed analysis of clones in large-scale combinatorial experiments.
Overall, high-throughput screening based on droplet microfluidics offered various advantages. Each droplet acted as an independent reactor, ensuring mutual independence of the culture environment between cells and minimizing cross-contamination. In vitro expression within droplets also reduced the loss of polymorphism and mitigated the toxic effects on host cells. The microfluidic system could process microdroplets at speeds of up to 108 per day, significantly enhancing the analytical capacity for large-scale samples. Additionally, the small volume of each microdrop, in the picoliter range, drastically reduced reagent consumption, which was particularly advantageous for analyzing expensive samples. However, current droplet microfluidic screening technology has limitations for the identification of ligninolytic enzymes. The entire process relies heavily on manual operation, making it complex and prone to errors. Moreover, issues such as leakage of fluorescent substrates and the need to further improve signal sensitivity and detection range remain for ligninolytic enzymes. Advances in integrating droplet microfluidic screening with new analytical technologies, such as fluorescence resonance energy transfer, fluorescence polarization, mid-infrared spectroscopy, or mass spectrometry, are crucial for enhancing its capabilities. As technology matures and progress is made, droplet microfluidic screening is poised to become a vital tool in synthetic biology and a powerful method for screening biobanks in directed evolution toward lignin valorization.
4. Protein engineering promoted the evolution of ligninolytic enzymes
For biological lignin valorization, it is of great significance to improve the catalysis performance of ligninolytic enzymes.23 Protein engineering technology enables the transformation of low-yield, promiscuous enzymatic activities into powerful and efficient biocatalysts, providing a promising tool for obtaining ligninolytic enzymes toward lignin valorization.114,115Fig. 4 presents the main process of using protein engineering to promote the evolution of ligninolytic enzymes.116–119 | ||
| Fig. 4 Protein engineering promoted the evolution of ligninolytic enzymes. (A) The main protein engineering process to modify ligninolytic enzymes. (B) Key steps in the design of versatile peroxidases with Rosetta modelling and PROSS stability design. (C) The main workflow for using the combinatorial active site saturation (CAST) method to enhance the activity of dye-decolorizing peroxidase Dyp1B enzyme for phenolic and polymeric lignin substrates. | ||
4.1 Protein engineering assists in designing new ligninolytic enzymes
Fungi deploy a suite of powerful oxidoreductase enzymes, with versatile peroxidases standing out due to their robust redox potential.116 Versatile peroxidases have garnered significant attention in industry due to their capability to break down environmental aromatic pollutants and process lignocellulosic biomass.117 Despite the promising attributes of versatile peroxidases, producing these enzymes through recombinant technologies poses substantial difficulties. These challenges primarily stem from the necessity to maintain enzyme stability, activity and functionality outside of their native context.118 So far, the scientific community has successfully characterized the structure of only one versatile peroxidase enzyme in detail.119 The optimization process aims to enhance the enzyme's expression levels, stability under various conditions, and catalytic activity. This achievement has paved the way for its optimization for recombinant expression, which is essential for practical applications.A new generation of ab initio structure prediction methods based on deep learning have emerged, with the latest methods achieving crystal structure accuracy.120 The de novo deep learning structure prediction method was seamlessly combined with the PROSS protein stability design strategy to obtain new versatile peroxidases with desired properties (Fig. 4).121 The stability design of versatile peroxidases was further refined through PROSS to automatically improve the expression and stability of the enzyme, reducing the need for screening thousands of variants.122 In research, the Rosetta structure prediction algorithm was firstly used to model 11 diverse versatile peroxidase sequences extracted from public databases and PROSS stability design calculations were run on each sequence.122 After the design was completed, the versatile peroxidase-encoded sequence was placed downstream of the α factor leader sequence of the yeast cell and transformed into the yeast cell for functional expression. Based on four diverse versatile peroxidases, a versatile peroxidase from Pleurotus eryngii (VPL), two paralogs from Pleurotus ostreatus (VP5 and VP11) and a versatile peroxidase from Ganoderma sp. 10597_SS1 (VP8), four functional versatile peroxidases were successfully obtained and denoted as VPLH, 5H, 11H and 8H. The activity of 8H and VPLH increased 11-fold and 14-fold compared to their wild-type enzymes, respectively. Remarkably, the 5H and 11H variants maintained their initial activity levels even after a week of incubation at pH 9. Of all the versatile peroxidase variants, 11H exhibited the highest stability to hydrogen peroxide.122 The reliability of this new structure prediction and design methodology expanded the scale and scope of computational enzyme optimization for versatile peroxidases, adding significant diversity to the lignin oxidoreductase library characterized to date.
O-Aryl-demethylation is an essential step in the aerobic degradation of lignin and its derivatives.123O-Demethoxylation facilitates the conversion of lignin derivatives into aromatic diols such as catechol, protocatechuic acid and gallic acid. Nearly all lignin-derived aromatics require this crucial step for their conversion into central intermediates.124 A cytochrome P450 system, GcoAB, was found to demethylate guaiacol (2-methoxyphenol), produced from coniferol-derived lignin, to form catechol.125 The naturally occurring GcoAB enzyme exhibited limited ability to process syringol (2,6-dimethoxyphenol). Fortunately, the evolution of GcoAB could be achieved through structure-guided protein engineering strategies.125,126 In the GcoA component of the GcoAB system, the F169 phenylalanine residue was found to obstruct syringol binding. This hindrance arose from a spatial conflict between the side chain of F169 and an additional methoxy group on syringol, which impeded effective binding of eugenol in the active site. Replacing the F169 residue with a smaller amino acid in the mutant GcoA effectively resolved this issue.125 This substitution mitigated spatial conflict with syringol, allowing syringol to bind more efficiently to the active site of the enzyme. Crystallographic studies and molecular dynamics simulations demonstrated that the mutant GcoAF169A, which involved replacing phenylalanine with alanine, performed exceptionally well. Consequently, syringol was efficiently demethylated, and its transformation was successfully achieved in P. putida KT2440.113
Subsequent studies successfully converted GcoA into a more effective catalyst through single amino acid substitutions.126 Combining X-ray crystallography and molecular dynamics simulations explained the structural basis for the enhanced activity of the GcoA variant. The active site of GcoA was described as a closely fitting hydrophobic pocket, where a series of hydrophobic amino acids were responsible for positioning the aromatic ring of the substrate. The aromatic ring of the substrate adopted the planar conformation and relative rotation observed in the wild-type enzyme, thereby providing an optimal presentation of the active methoxy group for the catalysis of the heme. Replacing the smaller alanine residue at position F169 allowed for the binding and demethylation of 2,6-dimethoxyphenol (eugenol). Substituting the primary alcohol (S296) with a secondary alcohol (T296) restored a hydrogen bonding pattern like the wild type around the vanillin aldehyde, enabling a stable binding mode for catalysis. These two amino acid changes reduced spatial conflicts with the aldehyde and provided hydrogen bond donors, making GcoAB an effective catalyst for vanillin isomers. GcoAF169S and GcoAT296S exhibited strong selectivity for both substrate entry and demethylation of guaiacol or its preferred vanillin isomer.126 This precise protein engineering not only improved the demethylation efficiency of syringol, but also highlighted the plasticity and potential of the cytochrome P450 system in the biotransformation of lignin-derived aromatics.
Cytochrome P450 systems are among the most versatile enzymes, renowned for their exceptional catalytic versatility.126 They serve as valuable scaffolds for biotransformation, handling a wide range of substrates and catalyzing numerous chemical reactions.127 It was demonstrated that the P450 system had an O-demethylation effect on guaiacol (2-methoxyphenol).128 Protein engineering of cytochrome P450 enzymes is evolving into a robust strategy for enhancing the conversion efficiency of lignin components, which is set to improve our understanding of lignin's complex structure and drive the development of innovative transformation technologies.
Overall, the strategic application of protein engineering to design ligninolytic enzymes offered a fresh perspective on harnessing this plentiful resource, potentially redefining the possibilities in lignin valorization and marking a significant step toward more sustainable processing.
4.2 Directed evolution assists in mining new ligninolytic enzymes
Dye-decolorizing peroxidases possess significant potential in lignocellulosic biorefineries due to their ability to oxidize lignin-related compounds.117 The first bacterial dye-decolorizing peroxidase identified to act on polymeric lignin was dye-decolorizing peroxidase B from R. jostii RHA1, a property previously attributed to fungal lignin peroxidase.129 Bacterial dyp-type peroxidases have demonstrated significant potential in converting lignin from industrial processes such as the pulp and paper and the biorefinery industries into value-added products.130Dye-decolorizing peroxidase from P. putida MET94 was successfully engineered using error-prone PCR.130 In the evolution of the PpDyP enzyme, a random mutagenesis approach was initially employed to construct a library, and first-generation screening was conducted based on the oxidation capacity of the variants to the substrate 2,2′-azino bis(3-ethylbenzthiazoline-6-sulfonic acid) (ABTS). Then the best-performing variants were selected as the parent for the next round of random mutagenesis. Each generation's screening combined random mutagenesis and used a color change as a key indicator. Following three rounds of random mutagenesis via error-prone PCR on the PpDyP gene, the 6E10 variant was subsequently identified through additional high-throughput screening, achieving a remarkable 100-fold increase in catalytic efficiency (kcat/Km) for 2,6-dimethoxyphenol (DMP). Furthermore, variant 6E10 also demonstrated resistance to hydrogen peroxide inactivation and was produced at 2-fold higher yields.130 The catalytic efficiency of the evolved variant of dye-decolorizing peroxidase from P. putida MET94 was significantly improved on phenolic and aromatic amine substrates in alkaline pH environments. These substrates included Kraft lignin and the model lignin dimer guaiacylglycerol-β-guaiacyl, which represented the main chain linkage type in lignin. These results opened numerous opportunities for custom applications utilizing immobilized PpDyP variants and studying protein structure within the DyP-type peroxidase family. More importantly, this report was the first to robustly demonstrate the laboratory evolution of bacterial dye-decolorizing peroxidase, highlighting the value of directed evolution in mining new ligninolytic enzymes.
The combinatorial active-site saturation test (CAST) method was developed to obtain effective ligninolytic enzymes.4 CAST offered an effective alternative to traditional epPCR as the initial step in directed evolution, addressing the long-standing challenge of broad substrate acceptance by enzymes. CAST also enabled the directional evolution of the dye-decolorizing peroxidase Dyp1B from P. fluorescens Pf-5 (Fig. 4).131 A focused library was established at seven active site residues near the heme auxiliary factor relying on saturation mutagenesis, and this library was initially screened using the high redox potential substrate 2,6-dichlorophenol. Then, alkaline Kraft lignin was used for secondary screening, monitoring for increases in absorbance at 465 nm. Analysis of the DyP peroxidase family suggested the presence of residues near the active site and on the protein surface that assisted in catalytic function. Through in vitro biotransformation or gene overexpression of ligninolytic strains, the activity of the mutant DyP enzyme on lignin substrate was significantly enhanced. The H169L mutant, which replaced His169 with Leu, exhibited the highest activity for the substrate 2,6-dichlorophenol. The kcat value of the H169L mutant was threefold higher than that of wild-type Dyp1B, resulting in an eightfold increase in catalytic efficiency.131 The discovery offered valuable insights and potential applications for using these more active mutant enzymes to convert lignin into high-value chemicals.
Although the physiological substrate and mechanism of action of dye-decolorizing peroxidases have not yet been clearly defined, these enzymes have garnered increasing attention and study due to their biotechnological potential in lignin valorization. Simultaneously, protein engineering and directed evolution provide promising new approaches for the development of recombinant enzymes and the transformation of lignin substrates. However, engineering robust ligninolytic enzymes remains a challenging task. It is crucial to develop computational modeling and structural analysis in protein engineering applications to understand the structure–function relationships of ligninolytic enzymes. After elucidating how these enzymes interact with the substrates at the molecular level, it is necessary to design effective mutations that enhance substrate binding, enzyme catalysis, and product release to accelerate the discovery and optimization of ligninolytic enzymes. To further improve the stable ligninolytic enzymes, computational engineering of these enzymes should be explored by building on the success of individual stable mutation designs and incorporating information from several complementary approaches based on evolutionary function or energetic function. Additionally, positive mutant libraries often fail to achieve the desired function due to the prevailing epistasis effect. Addressing these issues is essential for realizing the widespread and efficient application of protein engineering in the high-value conversion of lignin. Overall, protein engineering strategies are crucial for screening ligninolytic enzymes to unlock the potential of lignin as a renewable resource for valuable products.
5. Artificial intelligence (AI) boosted the identification of ligninolytic enzymes
Artificial intelligence encompasses the creation of intelligent systems designed to perform tasks that typically require human intelligence, such as learning, problem-solving, and decision-making.132 AI-driven tools can significantly accelerate the process of ligninolytic enzyme discovery. By integrating data from genomic databases and leveraging predictive models, AI can identify potential ligninolytic enzymes for novel applications without the need for exhaustive laboratory experiments.133 For instance, machine learning and big data have assisted in ligninolytic enzyme identification and design, thereby enabling sustainable lignin bioconversion toward valuable products.1345.1 Artificial intelligence elucidated the mechanism of ligninolytic enzymes
Ligninolytic enzymes exhibit unique molecular and chemical functionalities essential for the comprehensive modification and depolymerization of lignin polymers. These enzymes, pivotal as biocatalysts, boast diverse redox potentials, enabling efficient enzymatic reactions that enhance yields, lower process costs, and mitigate environmental waste.135 Understanding the intricate mechanisms underlying ligninolytic enzyme action is crucial for advancing lignin valorization.136,137 Integrating ligninolytic enzymes with artificial intelligence-driven computational frameworks holds promise for potentially achieving breakthroughs to meet escalating industrial demands of ligninolytic enzymes.31Artificial intelligence technology boosts the application of computational frameworks, including docking, molecular dynamics simulation and chemical cracking prediction, in predicting the bioconversion of lignin.138–141 A computational framework was employed as the theoretical basis for understanding the degradation mechanism and to enhance the ability to predict ligninolytic enzymes.142 The Protein Data Bank was utilized to retrieve crystal structures of ligninolytic enzymes including peroxidase, laccase, versatile peroxidase and dye type peroxidase. The ExPasy-ProtParam web server was employed to compute various parameters for each member of the ligninolytic enzyme family. The “SAS-Sequence Annotated by Structure” online server was accessed to predict the sequence annotation of ligninolytic enzymes. A mature modeling tool, CABS-Flex, starting with the CABS input structure, was employed to simulate protein flexibility modeling of ligninolytic enzymes. The final molecular docking between each member of the ligninolytic enzyme families and specific substrates was conducted using AutoDock Vina software (v.1.2.0.). For the final docked complexes, the BIOVIA Discovery Studio Visualizer and PyMOL software were used to capture the binding interactions of the ligand with each ligninolytic enzyme member.142 The data obtained from the final docking analysis elucidated that versatile peroxidase showed the lowest docking affinity of −9.2 kcal mol−1 for the 2,3,7,8-tetrachlorodibenzo-p-dioxin docked complex, indicating that it might effectively catalyze similar pollutants.142 These results highlighted the binding mode and potential catalysis mechanism of ligninolytic enzymes and further illustrated their potential in the field of ecological environmental remediation.
Artificial intelligence was also successfully employed to elucidate specific functional characteristics of laccases on the bioconversion of lignin-derived compounds. C. testosteroni FJ17 employed laccases for the delignification of rice straw (RS), modifying the apparent structure and reduced lignin content in rice straw (Fig. 5).143 Artificial intelligence modeling and molecular docking revealed the specific functional properties associated with the interaction between laccases and lignin compounds, achieving a peak laccase activity of 2016.7 U L−1 after 24 h. Scanning electron microscopy and X-ray diffraction analysis confirmed that laccases induced fractures and pores on the surface of rice straw, with crystallinity decreasing from 45.3% to 39.9%, and lignin content decreasing from 19.0% to 10.3%. Gas chromatography–mass spectrometry (GC-MS) and liquid chromatography–mass spectrometry (LC-MS) analysis indicated that the primary delignification mechanism of laccases involved the cleavage of β-O-4 and α-aryl ether cleavage, resulting in the formation of several small molecular products. The laccase genes were successfully cloned and bioinformatics analysis revealed a sequence comprising 317 amino acids with a predicted molecular weight of 33.13 kDa. Finally, laccase proteins were found to exhibit low binding energies with all tested lignin compounds, and lignin was oxidized by laccases through hydrogen-bonding interactions with the amino acid residues.143 In conclusion, docking verification, as a reliable method to evaluate the binding affinity between ligninolytic enzymes and substrates, can be used to clarify the mechanism of ligninolytic enzymes and help us predict the theoretical basis of ligninolytic enzymes.
 | ||
| Fig. 5 Artificial intelligence strategies and their applications in designing ligninolytic enzymes. | ||
The trained neural network models facilitated the predicted application of lignin-modifying enzymes. The artificial neural network models could optimize the biological decolorization of lignin-modifying enzymes for the azo dye Reactive Black 5 (RB5).144 Crude RB5 enzyme was obtained from Pleurotus sajor-caju as the source of ligninolytic enzymes. An artificial neural network was combined with a genetic algorithm and the response surface method to forecast the decolorization of Reactive Black 5. The results showed that the fungi producing ligninolytic enzymes showed higher laccase activity when growing in pulp washing solution. When immobilized, the fungi showed higher manganese peroxidase activity. Laccase and manganese peroxidase could decolorize the dye up to 89.4% and 75%, respectively.
Overall, artificial intelligence has demonstrated superior performance to tackle the constraints inherent to traditional simulation technologies for ligninolytic enzymes, including issues related to uncertainty, dependence on knowledge-driven models, and substantial computational expenses. To foster a more robust and advanced evolution of artificial intelligence for lignin variolization, there are several areas that warrant ongoing attention and effort. It is crucial to enhance the training of data samples through more extensive case studies of ligninolytic enzymes to evaluate the efficacy of diverse algorithms, and establish more stringent criteria for algorithm selection. It is also necessary to reinforce the processes of data collection, documentation, and sharing within pertinent domains of ligninolytic enzymes. Therefore, the effectiveness of artificial intelligence technologies would be significantly improved, which would guide the identification and engineering of ligninolytic enzymes with the improved accessibility and performance.
5.2 Artificial intelligence discovered new ligninolytic enzymes
As biological big data continue to expand, data-driven methodologies leveraging artificial intelligence technologies revolutionize protein and pathway design to tackle the persistent bottleneck in lignin utilization.28 Enter deep neural networks, which harness amino acid sequence information to forecast the spatial distance between amino acid residues within three-dimensional structures of ligninolytic enzymes. Through rigorous training on a vast array of diverse natural protein structures, deep neural networks have acquired the ability to encode specific protein structural properties. Building upon these advancements, a deep residual network has been devised, enabling the prediction of residue direction and distance swiftly and accurately. This network facilitated the generation of structural models guided by the Rosetta constrained energy minimization protocol, shedding light on the role of each amino acid in determining protein folding sequences. This breakthrough held promise for addressing current hurdles in the de novo design of ligninolytic enzyme proteins and was poised to be instrumental in a spectrum of protein structure prediction and design challenges.145 Furthermore, deep neural networks have been fine-tuned solely on natural protein sequences and structures, culminating in the creation of a network capable of generating novel proteins that transcend existing natural sequences, folding into stable structures independently. This innovative approach not only allowed for the reverse engineering of new ligninolytic enzymes but also complemented traditional physics-based models, ushering in a new era of protein design with enhanced functionalities and capabilities.146Linear discriminant analysis and random forest (RF) machine learning algorithms enabled the elucidation of the complex relationship between genetic information and wood rotting fungi decay patterns. This approach enabled the classification of white rot and brown rot fungi with an accuracy exceeding 98%.147 The machine learning algorithm primarily relied on the count of carbohydrate active enzyme genes for the differentiation of white rot and brown rot fungi. Within the random forest algorithm, specific genes engaged in cellulose and lignin degradation, such as polysaccharide monooxygenase and cellulose biohydrolase, emerged as pivotal factors in distinguishing between white rot and brown rot fungi.147 Leveraging algorithms presented a novel avenue for discovering previously unidentified ligninolytic enzymes in nature. These tools not only aided in the categorization of ligninolytic enzymes and microbial strains but also facilitated a deeper exploration of their identities.
The biological degradation of aromatic waste presented a green and sustainable approach. For example, exploring assumed enzyme sequences to unveil new aromatic plastic-degrading enzymes may offer a promising avenue. Enter the plastic enzyme degradation (PED) framework, a cutting-edge development rooted in machine learning technologies. This innovative framework predicted enzyme capabilities in degrading specific aromatic plastics by uncovering latent patterns within protein sequences.148 It excelled at pinpointing and comprehensively interpreting crucial enzymatic characteristics essential for aromatic plastic degradation. The novel context-aware enzyme sequence representation (CESR) method embedded within the framework excelled at capturing intricate context information within enzyme sequences, extracting enzyme features at both the amino acid and global sequence levels. With an impressive overall accuracy rate of 90.2%, surpassing existing sequence-based protein classification models, the PED framework emerged as a powerful tool for predicting and discovering aromatic plastic-degrading enzymes, paving the way for a more sustainable future.148
With the emergence and advancement of artificial intelligence, the integration of computer technology and experimental screening has enhanced the efficiency and intelligence of screening strategies for ligninolytic enzymes and bacteria. The utilization of big data research, driven by the fusion of computer science and artificial intelligence, along with advancements in machine learning, enables the rapid analysis of vast datasets, the identification of patterns, and the provision of accurate results. This significantly aids in the engineering and sustainable development of high-value ligninolytic enzymes. However, it is important to acknowledge that the development of artificial intelligence models poses unique challenges in terms of reproducibility. The widespread adoption of artificial intelligence technology heavily relies on data, necessitating both enough data and high data quality especially for biological lignin valorization. It needs to address the limitations of data quality in the future for achieving the widespread implementation of artificial intelligence.
6. Conclusions
Microorganisms have developed the ability to depolymerize lignin and utilize aromatic molecules derived from it as carbon sources, establishing a natural pathway for lignin depolymerization. However, several major challenges remain in the bioconversion of lignin. Biological lignin valorization requires effective depolymerization approaches for lignin and feasible bioconversion routes of lignin-derived aromatics. The efficient depolymerization and bioconversion of lignin have not been fully realized due to the lack of efficient ligninolytic enzymes. The synergistic advancement of synthetic biology alongside other scientific disciplines has paved the way for the discovery of effective ligninolytic enzymes, which serve as promising biocatalysts for depolymerizing and converting lignin into valuable derivatives that engineered microorganisms can further process. Advanced technologies—such as high-throughput screening, protein engineering, and artificial intelligence—could enhance the identification of efficient ligninolytic enzymes, thereby improving the overall efficiency of lignin depolymerization and bioconversion.First, high-throughput techniques can be employed to screen for efficient biocatalysts with specific functions from a vast array of enzymes or microorganisms. When combined with laboratory evolution, this approach allows for the tailoring of desired biological properties. However, high-throughput screening technologies still face practical challenges. Variability in the levels of single cells and the transport of target products between cells can lead to false positive rates in screenings such as FACS. To effectively screen ligninolytic bacteria and enzymes, it is essential to integrate genotype and phenotype, as well as intracellular and extracellular fluorescent signals, to mitigate misleading information caused by false positives. Second, protein engineering can refine the evolutionary strategies applied to ligninolytic enzymes. This field goes beyond natural protein libraries, aiming to enhance the functionality of modified enzymes with customized properties. This strategy is crucial for overcoming challenges related to missing biological pathways and catalytic inefficiencies. However, efficiently sampling across a broad design space remains a challenge in the quest to design new and more efficient ligninolytic enzyme pathways. Third, data-driven approaches that leverage artificial intelligence technology enable the design of more advanced target strains and enzymes. Integrating AI into the development of ligninolytic strains and enzymes can guide directed evolution, minimizing the number of experimental iterations required to efficiently obtain target strains and enzymes with specific functions and properties. Machine learning-guided directed evolution has proved successful in accelerating optimization cycles and alleviating experimental burdens. It is worth noting that when designing artificial intelligence strategies to assist in the screening and transformation of aromatic polymer degradation enzymes, it is crucial to meet the quantitative requirements of artificial intelligence while ensuring the authenticity and availability of data.
Overall, the integration of the cutting-edge technologies discussed above into lignin high-value research is set to yield groundbreaking advancements. This progress is expected to address the current challenges related to the limited variety and low efficiency of ligninolytic enzymes. However, it is important to recognize that the application of these technologies for lignin valorization is still in the early stages of research and development. Significant efforts remain necessary to achieve the widespread application of these innovative approaches, highlighting the need for continued dedication and progress in this field.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare that they have no competing interests in the article.Acknowledgements
This work was supported by the National Key Research and Development Program of China (2023YFC3403500) and the Key Research and Development Program of Ningxia Hui Autonomous Region (2024BEE02005).References
- E. T. C. Vogt and B. M. Weckhuysen, Nature, 2024, 629, 295–306 CrossRef CAS PubMed.
- M. Zuk, Science, 2020, 367, 1304–1305 CrossRef CAS PubMed.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann, P. T. Pienkos and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013–12018 CrossRef CAS PubMed.
- R. Y. Liu, Z. H. Liu, B. Z. Li and Y. J. Yuan, Green Chem., 2024, 26, 11378 RSC.
- Z. H. Liu, N. Hao, Y. Y. Wang, C. Dou, F. Lin, R. Shen, R. Bura, D. B. Hodge, B. E. Dale, A. J. Ragauskas, B. Yang and J. S. Yuan, Nat. Commun., 2021, 12, 3912 CrossRef CAS PubMed.
- D. Salvachúa, T. Rydzak, R. Auwae, A. De Capite, B. A. Black, J. T. Bouvier, N. S. Cleveland, J. R. Elmore, A. Furches, J. D. Huenemann, R. Katahira, W. E. Michener, D. J. Peterson, H. Rohrer, D. R. Vardon, G. T. Beckham and A. M. Guss, Microb. Biotechnol., 2019, 13, 290–298 CrossRef PubMed.
- J. R. Elmore, G. N. Dexter, D. Salvachúa, M. O'Brien, D. M. Klingeman, K. Gorday, J. K. Michener, D. J. Peterson, G. T. Beckham and A. M. Guss, Metab. Eng., 2020, 62, 62–71 CAS.
- E. Rosini, F. Molinari, D. Miani and L. Pollegioni, Catalysts, 2023, 13, 555 CAS.
- Z. H. Liu, H. Liu, T. Xu, Z. M. Zhao, A. J. Ragauskas, B. Z. Li, J. S. Yuan and Y. J. Yuan, Renewable Sustainable Energy Rev., 2025, 211, 115296 CAS.
- E. Erickson, A. Bleem, E. Kuatsjah, A. Z. Werner, J. L. DuBois, J. E. McGeehan, L. D. Eltis and G. T. Beckham, Nat. Catal., 2022, 5, 86–98 CAS.
- Y. Suzuki, Y. O. Abe, Y. Otsuka, T. Araki, M. Nojiri, N. Kamimura, E. Masai and M. Nakamura, Bioresour. Technol., 2022, 363, 127836 CAS.
- A. Z. Werner, W. T. Cordell, C. W. Lahive, C. A. Singer, B. C. Klein, E. C. D. Tan, M. A. Ingraham and K. J. Ramirez, Sci. Adv., 2023, 9, eadj0053 CAS.
- C. W. Johnson, D. Salvachúa, N. A. Rorrer, B. A. Black, D. R. Vardon, P. C. St. John, N. S. Cleveland, G. Dominick, J. R. Elmore, N. Grundl, P. Khanna, C. R. Martinez, W. E. Michener, D. J. Peterson, K. J. Ramirez, P. Singh, T. A. VanderWall, A. N. Wilson, X. Yi, M. J. Biddy, Y. J. Bomble, A. M. Guss and G. T. Beckham, Joule, 2019, 3, 1523–1537 CAS.
- J. Becker and C. Wittmann, Biotechnol. Adv., 2019, 37, 107360 CAS.
- Z. Liu, S. Chen and J. Wu, Trends Biotechnol., 2023, 41, 1168–1181 CAS.
- Y. Liu, H. Yuan, D. Ding, H. Dong, Q. Wang and D. Zhang, ACS Synth. Biol., 2021, 10, 1373–1383 CAS.
- J. Yang, R. Tu, H. Yuan, Q. Wang and L. Zhu, Crit. Rev. Biotechnol., 2021, 41, 1023–1045 CAS.
- S. Chityala, V. Nandana and D. Jayachandran, Adv. Yeast Biotechnol. Biof. Sustain, 2023, pp. 249–275 Search PubMed.
- N. Perera and R. S. H. Koralege, Encyclopedia of Toxicology, 2023, pp. 297–301 Search PubMed.
- M. Yang, Y. Yang, C. Xie, M. Ni, J. Liu, H. Yang, F. Mu and J. Wang, Nat. Mach. Intell., 2022, 4, 696–709 Search PubMed.
- A. Wellner, C. McMahon, M. S. A. Gilman, J. R. Clements, S. Clark, K. M. Nguyen, M. H. Ho, V. J. Hu, J.-E. Shin, J. Feldman, B. M. Hauser, T. M. Caradonna, L. M. Wingler, A. G. Schmidt, D. S. Marks, J. Abraham, A. C. Kruse and C. C. Liu, Nat. Chem. Biol., 2021, 17, 1057–1064 CAS.
- M. Paul, N. K. Pandey, A. Banerjee, G. K. Shroti, P. Tomer, R. K. Gazara, H. Thatoi, T. Bhaskar, S. Hazra and D. Ghosh, Bioresour. Technol., 2023, 379, 129045 CrossRef CAS PubMed.
- Y. Wang, H. Tang, L. Huang, L. Pan, L. Yang, H. Yang, F. Mu and M. Yang, Nat. Mach. Intell., 2023, 5, 845–860 CrossRef.
- D. D. Silva and D. Alahakoon, Patterns, 2022, 3, 100489 CrossRef PubMed.
- A. Holzinger, K. Keiblinger, P. Holub, K. Zatloukal and H. Müller, New Biotechnol., 2023, 74, 16–24 CrossRef CAS PubMed.
- M. Hutson, Science, 2022, 375, 129–129 CAS.
- T. H. Yu, H. Y. Cui, J. C. Li, Y. N. Luo, G. Jiang and H. M. Zhao, Science, 2023, 379, 1358–1363 CrossRef CAS PubMed.
- W. D. Jang, G. B. Kim, Y. Kim and S. Y. Lee, Curr. Opin. Biotechnol., 2022, 73, 101–107 CAS.
- R. L. Sokolik, O. Khersonsky, S. P. Schröder, C. de Boer, S. Y. Hoch and G. J. Davies, Science, 2023, 379, 195–201 Search PubMed.
- N. Konno and W. Iwasaki, Sci. Adv., 2023, 9, eadc9130 Search PubMed.
- A. K. Singh, H. M. N. Iqbal, N. Cardullo, V. Muccilli, J. Fernández-Lucas, J. E. Schmidt, T. Jesionowski and M. Bilal, Int. J. Biol. Macromol., 2023, 242, 124968 CAS.
- S. K. Jayasekara, H. D. Joni, B. Jayantha, L. Dissanayake, C. Mandrell, M. M. S. Sinharage, R. Molitor, T. Jayasekara, P. Sivakumar and L. N. Jayakody, Comput. Struct. Biotechnol. J., 2023, 21, 3513–3521 CAS.
- I. D. Lutz, S. Z. Wang, C. Norn, A. Courbet, A. J. Borst, Y. T. Zhao, A. Dosey, L. X. Cao, J. W. Xu, E. M. Leaf, C. Treichel, P. Litvicov, Z. Li, A. D. Goodson, P. R. Sánchez, A. M. Bratovianu, M. Baek, N. P. King, H. R. Baker and D. Baker, Science, 2023, 380, 266–273 CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 1246843 Search PubMed.
- Z. H. Liu, M. L. Olson, S. Shinde, X. Wang, N. Hao, C. G. Yoo, S. Bhagia, J. R. Dunlap, Y. Pu, K. C. Kao, A. J. Ragauskas, M. Jin and J. S. Yuan, Green Chem., 2017, 19, 4939–4955 CAS.
- Z. H. Liu, S. Shinde, S. Xie, N. Hao, F. Lin, M. Li, C. G. Yoo, A. J. Ragauskas and J. S. Yuan, Sustainable Energy Fuels, 2019, 8, 2024–2037 Search PubMed.
- J. S. Kim, Y. Y. Lee and T. H. Kim, Bioresour. Technol., 2016, 199, 42–48 CAS.
- H. Liu, Z. Chen, J. Q. Cui, S. Ntakirutimana, T. Xu, Z. H. Liu, B. Z. Li and Y. J. Yuan, Ind. Crops Prod., 2014, 218, 118956 Search PubMed.
- N. L. Radhika, S. Sachdeva and M. Kumar, Fuel, 2022, 312, 122935 CAS.
- N. Zhou, W. P. D. W. Thilakarathna, Q. S. He and H. P. V. Rupasinghe, Front. Energy Res., 2022, 9, 758744 Search PubMed.
- J. G. Linger, D. R. Vardon, M. T. Guarnieri, E. M. Karp, G. B. Hunsinger, M. A. Franden, C. W. Johnson, G. Chupka, T. J. Strathmann, P. T. Pienkos and G. T. Beckham, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 12013–12018 CAS.
- A. Tribot, G. Amer, M. Abdou Alio, H. de Baynast, C. Delattre, A. Pons, J. D. Mathias, J. M. Callois, C. Vial, P. Michaud and C. G. Dussap, Eur. Polym. J., 2019, 112, 228–240 CAS.
- N. Muhammad, Z. Man and M. A. Bustam Khalil, Eur. J. Wood Wood Prod., 2011, 70, 125–133 Search PubMed.
- Z. Xue, X. Zhao, R. C. Sun and T. Mu, ACS Sustainable Chem. Eng., 2016, 4, 3864–3870 CrossRef CAS.
- C. Mukesh, G. Huang, H. Qin, Y. Liu and X. Y. Ji, Biomass Bioenergy, 2024, 188, 107305 CrossRef CAS.
- D. P. Brink, K. Ravi, G. Lidén and M. F. G. Grauslund, Appl. Microbiol. Biotechnol., 2019, 103, 3979–4002 CrossRef CAS PubMed.
- G. W. Park, G. Gong, J. C. Joo, J. Song, J. Lee, J. P. Lee, H. T. Kim, M. H. Ryu, R. Sirohi, X. Zhuang and K. Min, Renewable Sustainable Energy Rev., 2022, 157, 112025 CrossRef CAS.
- S. T. Zhang, Z. J. Dong, J. Shi, C. R. Yang, Y. Fang, G. Chen, H. Chen and C. J. Tian, Bioresour. Technol., 2022, 361, 127699 CrossRef CAS PubMed.
- M. Zhou, O. A. Fakayode, M. Ren, H. X. Li, J. K. Liang, A. E. A. Yagoub, Z. Fan and C. S. Zhou, Bioresour. Bioprocess., 2023, 10, 21 CrossRef PubMed.
- M. Zhou, L. Z. Tao, P. Russell, R. D. Britt, T. Kasuga, X. Lü and Z. L. Fan, Ind. Crops Prod., 2022, 188, 115650 CrossRef CAS.
- Z. Li, J. Zhang, L. Qin and Y. Ge, ACS Sustainable Chem. Eng., 2018, 6, 2591–2595 CrossRef CAS.
- C. Chio, M. Sain and W. Qin, Renewable Sustainable Energy Rev., 2019, 107, 232–249 CrossRef CAS.
- O. D. V. Biko, M. V. Bloom and W. H. V. Zyl, Enzyme Microb. Technol., 2020, 141, 109669 CrossRef CAS PubMed.
- A. O. Falade, U. U. Nwodo, B. C. Iweriebor, E. Green, L. V. Mabinya and A. I. Okoh, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2017, 6, e00394 Search PubMed.
- A. Kumar and P. K. Arora, Front. Environ. Sci., 2022, 10, 875157 CrossRef.
- K. S. K. Teo, K. Kondo, T. Watanabe, T. Nagata and M. Katahira, ACS Sustainable Chem. Eng., 2024, 12, 2172–2182 CrossRef CAS.
- J. M. Guo, D. Liuc and Y. Xu, Sustainable Energy Fuels, 2024, 8, 1153–1184 RSC.
- G. M. M. Rashid and T. D. H. Bugg, Catal. Sci. Technol., 2021, 11, 3568–3577 RSC.
- P. D. Sainsbury, E. M. Hardiman, M. Ahmad, H. Otani, N. Seghezzi, L. D. Eltis and T. D. H. Bugg, ACS Chem. Biol., 2013, 8, 2151–2156 CrossRef CAS PubMed.
- N. L. Radhika, S. Sachdeva and M. Kumar, Fuel, 2022, 312, 122935 Search PubMed.
- Z. Chen and C. Wan, Renewable Sustainable Energy Rev., 2017, 73, 610–621 CrossRef CAS.
- E. Masai, Y. Katayama and M. Fukuda, Biosci., Biotechnol., Biochem., 2007, 71, 1–15 Search PubMed.
- N. Kamimura, K. Takahashi, K. Mori, T. Araki, M. Fujita, Y. Higuchi and E. Masai, Environ. Microbiol. Rep., 2017, 9, 679–705 Search PubMed.
- R. Y. Liu, H. N. Lan, Z. H. Liu, B. Z. Li and Y. J. Yuan, Renewable Sustainable Energy Rev., 2024, 191, 114205 CrossRef CAS.
- K. Mori, N. Kamimura and E. Masai, Appl. Microbiol. Biotechnol., 2018, 102, 4807–4816 CAS.
- J. H. Pereira, R. A. Heins, D. L. Gall, R. P. McAndrew, K. Deng, K. C. Holland, T. J. Donohue, D. R. Noguera, B. A. Simmons, K. L. Sale, J. Ralph and P. D. Adams, J. Biol. Chem., 2016, 291, 10228–10238 CAS.
- M. Tamura, Y. Tsuji, T. Kusunose, A. Okazawa, N. Kamimura, T. Mori, R. Nakabayashi, S. Hishiyama, Y. Fukuhara, H. Hara, K. Sato-Izawa, T. Muranaka, K. Saito, Y. Katayama, M. Fukuda, E. Masai and S. Kajita, Appl. Microbiol. Biotechnol., 2014, 98, 8165–8177 CAS.
- L. Kirmair and A. Skerra, Appl. Environ. Microbiol., 2014, 80, 2468–2477 Search PubMed.
- Y. Fukuhara, N. Kamimura, M. Nakajima, S. Hishiyama, H. Hara, D. Kasai, Y. Tsuji, S. Narita-Yamada, S. Nakamura, Y. Katano, N. Fujita, Y. Katayama, M. Fukuda, S. Kajita and E. Masai, Enzyme Microb. Technol., 2013, 52, 38–43 CAS.
- Y. Matsushita, S. Yagami, A. Kato, H. Mitsuda, D. Aoki and K. Fukushima, J. Agric. Food Chem., 2020, 68, 9245–9251 CAS.
- H. N. Lan, R. Y. Liu, Z. H. Liu, X. Li, B. Z. Li and Y. J. Yuan, Biotechnol. Adv., 2023, 64, 108107 CAS.
- H. Liu, Z. H. Liu, R. K. Zhang, J. S. Yuan, B. Z. Li and Y. J. Yuan, Biotechnol. Adv., 2022, 60, 108000 Search PubMed.
- D. H. Jung, E. J. Kim, E. Jung, R. J. Kazlauskas, K. Y. Choi and B. G. Kim, Biotechnol. Bioeng., 2015, 113, 1493–1503 Search PubMed.
- Z. Chen, H. Liu, Q. J. Zong, T. X. Liang, J. Sun, T. Xu, Z. H. Liu, J. P. Wu, B. Z. Li and Y. J. Yuan, ACS Sustainable Chem. Eng., 2024, 49, 17726–17738 Search PubMed.
- S. Notonier, A. Z. Werner, E. Kuatsjah, L. Dumalo, P. E. Abraham, E. A. Hatmaker, C. B. Hoyt, A. Amore, K. J. Ramirez, S. P. Woodworth, D. M. Klingeman, R. J. Giannone, A. M. Guss, R. L. Hettich, L. D. Eltis, C. W. Johnson and G. T. Beckham, Metab. Eng., 2021, 65, 111–122 Search PubMed.
- R. K. Zhang, Y. S. Tan, Y. Z. Cui, X. Xin, Z. H. Liu, B. Z. Li and Y. J. Yuan, Green Chem., 2021, 23, 6515 CAS.
- H. Liu, X. Tao, S. Ntakirutimana, Z. H. Liu, B. Z. Li and Y. J. Yuan, Chem. Eng. J., 2024, 495, 153375 Search PubMed.
- C. W. Johnson and G. T. Beckham, Metab. Eng., 2015, 28, 240–247 CAS.
- L. G. Zou, Y. T. Yao, F. F. Wen, X. Zhang, B. T. Liu, D. W. Li, Y. F. Yang, W. D. Yang, S. Balamurugan and H. Y. Li, J. Agric. Food Chem., 2023, 71, 10065–10074 CAS.
- D. R. Vardon, M. A. Franden, C. W. Johnson, E. M. Karp, M. T. Guarnieri, J. G. Linger, M. J. Salm, T. J. Strathmann and G. T. Beckham, Energy Environ. Sci., 2015, 8, 617–628 CAS.
- M. Kohlstedt, S. Starck, N. Barton, J. Stolzenberger, M. Selzer, K. Mehlmann, R. Schneider, D. Pleissner, J. Rinkel, J. S. Dickschat, J. Venus, J. B. J. H. V. Duuren and C. Wittmann, Metab. Eng., 2018, 47, 279–293 CrossRef CAS PubMed.
- J. M. Perez, W. S. Kontur, M. Alherech, J. Coplien, S. D. Karlen, S. S. Stahl, T. J. Donohue and D. R. Noguera, Green Chem., 2019, 21, 1340–1350 Search PubMed.
- C. W. Johnson, D. Salvachúa, P. Khanna, H. Smith, D. J. Peterson and G. T. Beckham, Metab. Eng. Commun., 2016, 3, 111–119 Search PubMed.
- R. Zhang, J. Wang, S. Milligan and Y. Yan, Green Chem., 2021, 23, 8238–8250 RSC.
- W. Zeng, L. Guo, S. Xu, J. Chen and J. Zhou, Trends Biotechnol., 2020, 38, 888–906 Search PubMed.
- A. Nakamura, N. Honma, Y. Tanaka, Y. Suzuki, Y. Shida, Y. Tsuda, K. Hidaka and W. Ogasawara, Anal. Chem., 2022, 94, 2416–2424 CrossRef CAS PubMed.
- M. Klaus, P. J. Zurek, T. S. Kaminski, A. Pushpanath, K. Neufeld and F. Hollfelder, ChemBioChem, 2021, 22, 3292–3299 Search PubMed.
- B. Kintses, C. Hein, M. F. Mohamed, M. Fischlechner, F. Courtois, C. Lainé and F. Hollfelder, Chem. Biol., 2012, 19, 1001–1009 Search PubMed.
- J. Panwar, A. Autour and C. A. Merten, Nat. Protoc., 2023, 18, 1090–1136 CAS.
- Q. C. Xu, Z. O. Sebastian, G. Lindquist, H. Jiang, Ch. Z. Li and W. B. Shannon, ACS Energy Lett., 2021, 6, 305–312 CAS.
- K. Radotić, A. Kalauzi, D. Djikanović, M. Jeremić, R. M. Leblanc and Z. G. Cerović, J. Photochem. Photobiol., B, 2006, 83, 1–10 CrossRef PubMed.
- G. Paës, Molecules, 2014, 19, 9380–9402 Search PubMed.
- J. Arnthong, C. Siamphan, C. Chuaseeharonnachai, N. Boonyuen and S. Suwannarangsee, J. Microbiol. Biotechnol., 2020, 30, 1670–1679 CAS.
- N. S. Ali, F. Huang, W. Qin and T. C. Yang, Biotechnol. Rep., 2023, 39, e00809 CAS.
- R. Khandeparkar and N. B. Bhosle, Res. Microbiol., 2006, 157, 315–325 CAS.
- Y. Ensari, G. V. Dhoke, M. D. Davari, A. J. Ruff and U. Schwaneberg, ChemBioChem, 2018, 19, 1563–1569 Search PubMed.
- G. Yang and S. G. Withers, ChemBioChem, 2009, 10, 2704–2715 CAS.
- G. Körfer, V. Besirlioglu, M. D. Davari, R. Martinez, L. Vojcic and U. Schwaneberg, Biotechnol. Bioeng., 2022, 119, 2076–2087 Search PubMed.
- A. I. Vicente, J. V. Gonzalez, P. S. Moriano, C. M. Alvarez, A. O. Ballesteros and M. Alcalde, J. Mol. Catal. B: Enzym., 2016, 134, 323–330 Search PubMed.
- J. Xu, Y. Zhang, H. Yu, X. Gao and S. Shao, Anal. Chem., 2015, 88, 1455–1461 Search PubMed.
- L. G. Wang, M. He, X. W. Liu, L. J. Zhai, L. X. Niu, Z. L. Xue and H. T. Wu, Green Chem., 2023, 25, 550–553 CAS.
- G. Sun, L. Qu, F. Azi, Y. Liu, J. Li, X. Lv, G. Du, J. Chen, C. H. Chen and L. Liu, Biosens. Bioelectron., 2023, 225, 115107 CAS.
- R. He, R. Ding, J. A. Heyman, D. Zhang and R. Tu, J. Ind. Microbiol. Biotechnol., 2019, 46, 1603–1610 CAS.
- B. Kintses, L. D. van Vliet, S. R. A. Devenish and F. Hollfelder, Curr. Opin. Chem. Biol., 2010, 14, 548–555 CAS.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahna, A. C. Rowat, J. C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6550 Search PubMed.
- H. Yuan, Y. Zhou, Y. Lin, R. Tu, Y. Guo, Y. Zhang and Q. Wang, Biotechnol. Biofuels Bioprod., 2022, 15, 50 CAS.
- I. Swyer, R. Fobel and A. R. Wheeler, Langmuir, 2019, 35, 5342–5352 CrossRef CAS PubMed.
- D. Hess, V. Dockalova, P. Kokkonen, D. Bednar, J. Damborsky, A. deMello, Z. Prokop and S. Stavrakis, Chem, 2021, 7, 1066–1079 CAS.
- B. Sana, K. H. B. Chia, S. S. Raghavan, B. Ramalingam, N. Nagarajan, J. Seayad and F. J. Ghadessy, Biotechnol. Biofuels, 2017, 10, 32 CrossRef PubMed.
- S. Neun, P. J. Zurek, T. S. Kaminski and F. Hollfelder, Methods Enzymol., 2020, 643, 317–343 CAS.
- S. Neun, P. Brear, E. Campbell, T. Tryfona, K. El Omari, A. Wagner, P. Dupree, M. Hyvonen and F. Hollfelder, Nat. Chem. Biol., 2022, 18, 1096–1103 CrossRef CAS PubMed.
- S. Neun, L. V. Vliet, F. Hollfelder and F. Gielen, Anal. Chem., 2022, 94, 16701–16710 CrossRef CAS PubMed.
- F. Gielen, T. Buryska, L. V. Vliet, M. Butz, J. Damborsky, Z. Prokop and F. Hollfelder, Anal. Chem., 2015, 87, 624–632 CrossRef CAS PubMed.
- Y. Cui, Y. Chen, X. Liu, S. Dong, Y. E. Tian, Y. Qiao, R. Mitra, J. Han, C. Li, X. Han, W. Liu, Q. Chen, W. Wei, X. Wang, W. Du, S. Tang, H. Xiang, H. Liu, Y. Liang, K. N. Houk and B. Wu, ACS Catal., 2021, 11, 1340–1350 CrossRef CAS.
- D. C. Miller, S. V. Athavale and F. H. Arnold, Nat. Synth., 2022, 1, 18–23 CrossRef CAS PubMed.
- C. Del Cerro, E. Erickson, T. Dong, A. R. Wong, E. K. Eder, S. O. Purvine, H. D. Mitchell, K. K. Weitz, L. M. Markillie, M. C. Burnet, D. W. Hoyt, R. K. Chu, J. F. Cheng, K. J. Ramirez, R. Katahira, W. Xiong, M. E. Himmel, V. Subramanian, J. G. Linger and D. Salvachua, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017381118 Search PubMed.
- C. C. Chen, L. Dai, L. Ma and R. T. Guo, Nat. Rev. Chem., 2020, 4, 114–126 Search PubMed.
- F. J. R. Duenas, M. Morales, E. Garcia, Y. Miki, M. J. Martinez and A. T. Martinez, J. Exp. Bot., 2009, 60, 441–452 CrossRef PubMed.
- M. P. Boada, F. J. R. Dueñas, R. Pogni, R. Basosi, T. Choinowski, M. Jesús Martínez, K. Piontek and A. T. Martínez, J. Mol. Biol., 2005, 354, 385–402 Search PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek, A. Potapenko, A. Bridgl, C. Meyer, S. A. A. Kohl, A. J. Ballard, A. Cowie, B. R. Paredes, S. Nikolov, R. Jain, J. Adler, T. Back, S. Petersen, D. Reiman, E. Clancy, M. Zielinski, M. Steinegger, M. Pacholska, T. Berghammer, S. Bodenstein, D. Silver, O. Vinyals, A. W. Senior, K. Kavukcuoglu, P. Kohli and D. Hassabis, Nature, 2021, 596, 583–589 CrossRef CAS PubMed.
- A. Goldenzweig, M. Goldsmith, S. E. Hill, O. Gertman, P. Laurino, Y. Ashani, O. Dym, T. Unger, S. Albeck, J. Prilusky, R. L. Lieberman, A. Aharoni, I. Silman, J. L. Sussman, D. S. Tawfik and S. J. Fleishman, Mol. Cell, 2016, 63, 337–346 CAS.
- S. B. Zucker, V. Mindel, E. G. Ruiz, J. J. Weinstein, M. Alcalde and S. J. Fleishman, J. Am. Chem. Soc., 2022, 144, 3564–3571 Search PubMed.
- D. Salvachúa, C. W. Johnson, C. A. Singer, H. Rohrer, D. J. Peterson, B. A. Black, A. Knapp and G. T. Beckham, Green Chem., 2018, 20, 5007–5019 Search PubMed.
- G. T. Beckham, C. W. Johnson, E. M. Karp, D. Salvachúa and D. R. Vardon, Curr. Opin. Biotechnol., 2016, 42, 40–53 CAS.
- M. M. Machovina, S. J. B. Mallinson, B. C. Knott, A. W. Meyers, M. G. Borràs, L. Bu, J. E. Gado, A. Oliver, G. P. Schmidt, D. J. Hinchen, M. F. Crowley, C. W. Johnson, E. L. Neidle, C. M. Payne, K. N. Houk, G. T. Beckham, J. E. McGeehan and J. L. DuBois, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13970–13976 CAS.
- E. S. Ellis, D. J. Hinchen, A. Bleem, L. Bu, S. J. B. Mallinson, M. D. Allen, B. R. Streit, M. M. Machovina, Q. V. Doolin, W. E. Michener, C. W. Johnson, B. C. Knott, G. T. Beckham, J. E. McGeehan and J. L. DuBois, JACS Au, 2021, 1, 252–261 CAS.
- Z. Li, Y. Jiang, F. P. Guengerich, L. Ma, S. Li and W. Zhang, J. Biol. Chem., 2020, 295, 833–849 Search PubMed.
- S. J. B. Mallinson, M. M. Machovina, R. L. Silveira, M. G. Borras, N. Gallup, C. W. Johnson, M. D. Allen, M. S. Skaf, M. F. Crowley, E. L. Neidle, K. N. Houk, G. T. Beckham, J. L. DuBois and J. E. McGeehan, Nat. Commun., 2018, 9, 2487 Search PubMed.
- M. Ahmad, J. N. Roberts, E. M. Hardiman, R. Singh, L. D. Eltis and T. D. H. Bugg, Biochemistry, 2011, 50, 5096–5107 CAS.
- V. Brissos, D. Tavares, A. C. Sousa, M. P. Robalo and L. O. Martins, ACS Catal., 2017, 7, 3454–3465 CAS.
- R. R. Pour, A. Ehibhatiomhan, Y. Huang, B. Ashley, G. M. Rashid, S. M. Williams and T. D. H. Bugg, Enzyme Microb. Technol., 2019, 123, 21–29 Search PubMed.
- A. Arias, G. Feijoo and M. T. Moreira, Environ. Technol. Innov., 2023, 32, 103277 Search PubMed.
- M. Liao and Y. Yao, GCB Bioenergy, 2021, 13, 774–802 Search PubMed.
- N. Prioux, R. Ouaret, G. Hetreux and J. P. Belaud, Clean Technol. Environ., 2023, 25, 689–702 Search PubMed.
- D. S. Vilar, M. Bilal, R. N. Bharagava, A. Kumar, A. Kumar Nadda, G. R. S. Banda, K. I. B. Eguiluz and L. F. R. Ferreira, J. Chem. Technol. Biotechnol., 2021, 97, 327–342 Search PubMed.
- A. K. Singh, M. Bilal, H. M. N. Iqbal and A. Raj, Int. J. Biol. Macromol., 2021, 177, 58–82 CAS.
- M. Bilal and H. M. N. Iqbal, Catal. Lett., 2020, 150, 524–543 CAS.
- S. S. Hernández, C. V. Ayala, P. L. I. Flores, M. J. R. Alanis, R. P. Saldivar, H. M. N. Iqbal and D. C. Nieves, Int. J. Biol. Macromol., 2020, 161, 1099–1116 Search PubMed.
- A. K. Singh, M. Bilal, H. M. N. Iqbal and A. Raj, Sci. Total Environ., 2021, 770, 144561 CAS.
- A. K. Singh, S. K. Katari, A. Umamaheswari and A. Raj, RSC Adv., 2021, 11, 14632–14653 CAS.
- A. K. Singh, M. Bilal, H. M. N. Iqbal and A. Raj, Chemosphere, 2022, 292, 133250 CAS.
- A. K. Singh, M. Bilal, T. Jesionowski and H. M. N. Iqbal, Chemosphere, 2023, 313, 137546 CAS.
- L. J. Wang, C. Xue, G. Owens and Z. L. Chen, Bioresour. Technol., 2022, 345, 126565 CAS.
- C. D. Fernandes, V. R. S. Nascimento, D. B. Meneses, D. S. Vilar, N. H. Torres, M. S. Leite, J. R. V. Baudrit, M. Bilal, H. M. N. Iqbal, R. N. Bharagava, S. M. Egues and L. F. R. Ferreira, J. Hazard. Mater., 2020, 399, 153–181 Search PubMed.
- J. Yang, I. Anishchenko, H. Park, Z. Peng, S. Ovchinnikov and D. Baker, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 1496–1503 CrossRef CAS PubMed.
- T. H. Tsui, M. C. M. V. Loosdrecht, Y. Dai and Y. W. Tong, Bioresour. Technol., 2023, 369, 128445 CrossRef CAS PubMed.
- N. Hasegawa, M. Sugiyama and K. Igarashi, BioRxiv, 2024, 15, 585254.
- R. Jiang, L. Shang, R. Wang, D. Wang and N. Wei, Environ. Sci. Technol. Lett., 2023, 10, 557–564 CrossRef CAS.
- Y. L. Xiang, Y. B. Zhang, J. Q. Wu, J. Zhu, B. W. Cao and C. Y. Xiong, Renewable Energy, 2024, 235, 121289 CrossRef CAS.
- J. Li, Z. Liu, J. Zhao, G. Wang and T. Xie, Int. J. Biol. Macromol., 2024, 256, 128487 CrossRef CAS PubMed.
- A. Manglesh, V. Kumar, D. Chandra, V. Thakur, U. Sharma and D. Singh, Int. J. Biol. Macromol., 2023, 234, 123601 CrossRef PubMed.
- P. Bhatt, K. Bhatt, W. J. Chen, Y. Huang, Y. Xiao, S. Wu, Q. Lei, J. Zhong, X. Zhu and S. Chen, J. Hazard. Mater., 2023, 443, 130319 CAS.
- N. K. Chopra and S. Sondhi, Int. J. Biol. Macromol., 2022, 206, 1003–1011 CrossRef CAS PubMed.
- S. A. Mayr, R. Subagia, R. Weiss, N. Schwaiger, H. K. Weber, J. Leitner, D. Ribitsch, G. S. Nyanhongo and G. M. Guebitz, Int. J. Mol. Sci., 2021, 22, 13161 CAS.
- L. Cao, L. Lin, H. Sui, H. Wang, Z. Zhang, N. Jiao and J. Zhou, Green Chem., 2021, 23, 2079–2094 RSC.
- N. K. Kheti, S. Rath and H. Thatoi, Sustainable Chem. Environ., 2023, 2, 100009 Search PubMed.
- A. Singh, R. Kumar, A. Maurya, P. Chowdhary and A. Raj, Biotechnol. Rep., 2022, 35, e00755 Search PubMed.
- A. Alfi, B. Zhu, J. Damnjanovic, T. Kojima, Y. Iwasaki and H. Nakano, J. Biosci. Bioeng., 2019, 128, 290–295 Search PubMed.
- T. V. T. Nguyen, S. Kim, C. G. Yoo, J. W. Choi, G. Leem and Y. H. Kim, ACS Catal., 2024, 14, 11733–11740 CrossRef CAS.
- H. Gye, H. Baek, S. Han, H. Kwon, T. V. T. Nguyen, L. T. M. Pham, S. Kang, Y. H. Nho, D. W. Lee and Y. H. Kim, Biomacromolecules, 2023, 24, 2633–2642 CrossRef CAS PubMed.
- D. Linde, I. A. Fernandez, M. Laloux, J. E. Aguiar-Cervera, A. L. de Lacey, F. J. Ruiz-Duenas and A. T. Martinez, Int. J. Mol. Sci., 2021, 22, 2629 CrossRef CAS PubMed.
- S. I. Khan, N. S. Zada, M. Sahinkaya, D. N. Colak, S. Ahmed, F. Hasan, A. O. Belduz, S. Canakci, S. Khan, M. Badshah and A. A. Shah, Enzyme Microb. Technol., 2021, 151, 109917 CrossRef CAS PubMed.
- S. Ding, C. Lin, Q. Xiao, F. Feng, J. Wang, X. Zhang, S. Yang, L. Li and F. Li, Sci. Total Environ., 2024, 908, 168500 CrossRef CAS PubMed.
- S. Sethupathy, R. Xie, N. Liang, R. M. B. Shafreen, M. Y. Ali, Z. Zhuang, L. Zhe, Z. Zahoor, Y. C. Yong and D. Zhu, Int. J. Biol. Macromol., 2023, 253, 127117 CrossRef CAS PubMed.
- P. Dhankhar, V. Dalal, V. Singh, A. K. Sharma and P. Kumar, Int. J. Biol. Macromol., 2021, 193, 601–608 CrossRef CAS PubMed.
- K. S. K. Teo, K. Kondo, S. M. R. Khattab, T. Watanabe, T. Nagata and M. Katahira, J. Agric. Food Chem., 2024, 72, 2657–2666 CAS.
- K. Saikia, D. Vishnu, A. K. Rathankumar, B. Palanisamy Athiyaman, R. A. Batista-Garcia, J. L. Folch-Mallol, H. Cabana and V. V. Kumar, J. Air Waste Manage. Assoc., 2020, 70, 1252–1259 CAS.
- F. Li, F. Ma, H. Zhao, S. Zhang, L. Wang, X. Zhang and H. Yu, Appl. Environ. Microbiol., 2019, 85, e02803 CAS.
| This journal is © The Royal Society of Chemistry 2025 |