
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Concurrent ultrafast twisting and proton transfer photoreactions in new pyrano[2,3-c]pyrazole derivatives†
Marius
Navickas
 *a,
Karolis
Gineitis
a,
Arminas
Urbonavičius
b,
Sonata
Krikštolė
c,
Vytas
Martynaitis
c,
Eglė
Arbačiauskienė
c,
Miglė
Dagilienė
b,
Algirdas
Šačkus
bc and
Mikas
Vengris
a
*a,
Karolis
Gineitis
a,
Arminas
Urbonavičius
b,
Sonata
Krikštolė
c,
Vytas
Martynaitis
c,
Eglė
Arbačiauskienė
c,
Miglė
Dagilienė
b,
Algirdas
Šačkus
bc and
Mikas
Vengris
a
aLaser Research Center, Vilnius University, Saulėtekio av. 10, LT-10223, Lithuania. E-mail: marius.navickas@ff.vu.lt
bInstitute of Synthetic Chemistry, Kaunas University of Technology, K. Baršausko str. 59, Kaunas, LT-51423, Lithuania
cDepartment of Organic Chemistry, Kaunas University of Technology, Radvilėnų rd. 19, Kaunas, LT-50254, Lithuania
First published on 20th December 2024
Abstract
Pyrano[2,3-c]pyrazole derivatives are a class of compounds exhibiting dual solvent-dependent fluorescence. This interesting and potentially useful optical property is attributed to the excited state intramolecular proton transfer (ESIPT). We have investigated excited state dynamics of these molecules in detail using femtosecond time-resolved fluorescence and transient absorption spectroscopy. We found that when the compounds containing methoxy groups in a phenyl ring are dissolved in a polar protic solvent (methanol), they undergo excited state twisting that competes with the ESIPT reaction. Additionally, the dumping of the tautomer stimulated emission allowed us to populate a short-lived ground-state tautomer and track a ground-state proton transfer (GSIPT) back reaction. We found that the GSIPT decays on the sub-picosecond to picosecond time scale, and a fast process is more pronounced in less polar solvents.
1 Introduction
Excited state intramolecular proton transfer (ESIPT) is a ubiquitous photoreaction that takes place in both synthetic and natural molecular systems.1–3 It is also an important mechanism underlying the phenomena of multiple fluorescence.4,5 As a result, ESIPT has received considerable attention and a number of investigations have been devoted to it in recent decades.6,7 Even though the number of known molecules exhibiting ESIPT is vast, synthesis and characterization of new compounds with desirable and interesting properties remain an important scientific task to improve and extend the applications of such materials. One of the strategies of looking for such new compounds is the creative variations of the structure of known molecules undergoing ESIPT. One such class is 3-hydroxyflavones (3-HF). They are members of a wider avonoid family, a group of naturally occurring substances with variable phenolic structures, found in fruits, vegetables, flowers, etc.8–10 3-HF have been investigated as therapeutic imaging agents, including as fluorescence sensors and probes for the detection of the microenvironment, metal ions, and structures of proteins and DNA.11 3-HF exhibit fluorescence spectra with two emission bands, the intensities of which are strongly affected by the solvent polarity. The presence of these bands is attributed to the ESIPT reaction. A number of studies of ESIPT in 3-HF revealed that proton transfer dynamics is influenced by different factors, with solute/solvent complexes playing a significant role. Namely, the impact of hydrogen bonding with solvent protons, altering the ESIPT reaction rate in 3-HF derivatives, has been a topic of interest. The presence and strength of hydrogen bonds have been found to change the relative intensities of normal and phototautomer emissive species.12 It was also shown that the presence of electron-donating substitutes, such as hydroxy or amino groups, could be used to tune the position of normal and tautomer emission bands and make them more or less sensitive to the polarity of the solvent.13–15Recently, a series of new pyrano[2,3-c]pyrazole derivatives analogous to 3-HF have been synthesized and characterized.16 Pyrazoles are known to exhibit anti-inflammatory, analgesic, anticancer, antimicrobial, anti-infective and other activities.17–20 The newly synthesized pyrano[2,3-c]pyrazoles exhibit dual emission behaviour that was attributed to the ESIPT reaction based on the similarity to 3-HF.16 The most pronounced dual emission behaviour of pyrano[2,3-c]pyrazoles was demonstrated in methanol solution for the compounds containing methoxy substituents. It must be noted that the picture of the photocycle in pyrano[2,3-c]pyrazoles was proposed only from steady-state experiments and the analogies with structurally similar compounds. Some observations suggest that this picture may not be complete. From the steady-state emission experiments on compounds with methoxy groups, it is obvious that the Stokes shift of the blue emission band is larger than that expected from a locally excited molecular state. Therefore, its origin requires explanation. The rates of ESIPT reactions in various solvents and their dependencies on different side groups have not been addressed in detail. The full photocycle of the newly synthesized molecules also includes ground-state proton back transfer, the dynamics of which is unknown. Therefore, full understanding of photoinduced dynamics in these compounds requires a more extensive study using time-resolved spectroscopic methods.
Only a few such studies on 3-HF derivatives have been published, and the lifetime of the ground-state tautomer (T) was found to be spread over a large range of values, depending on the specific molecule and its environment. For example, in 4′-N,N-diethylamino-3-hydroxyflavone in toluene and tetrahydrofuran, the ESIPT rate varies from 16 ps to 30 ps; meanwhile, the proton transfer in the ground state is faster – its rate was obtained to be from 1.7 ps to 10 ps.21
This paper is our attempt at a detailed study of ultrafast excited and ground state proton transfer in pyrano[2,3-c]pyrazole derivatives, containing methoxy groups that are effective electron donors. For this study, we performed steady state absorption, fluorescence and kinetic fluorescence measurements together with ultrafast pump–probe and pump–dump–probe experiments to monitor the proton transfer reaction in the excited and ground states.
2 Materials and methods
The synthesis of 6-(3,4-dimethoxyphenyl)-5-hydroxy-2-phenylpyrano[2,3-c]pyrazol-4(2H)-one (M1) and 5-hydroxy-6-(4-methoxyphenyl)-2-phenylpyrano[2,3-c]pyrazol-4(2H)-one (M2) was carried out as described in ref. 16 and the structures of these molecules are presented in the insets of Fig. 1(a) and (b). The steady-state absorption and fluorescence spectra of pyrano[2,3-c]pyrazole derivatives in methanol (MeOH) and chloroform (CHCl3) were recorded using a scanning spectrophotometer (Shimadzu UV-3101PC) and a fluorimeter (PerkinElmer LS55), respectively. For steady-state fluorescence measurements, the sample solutions were diluted to an optical density of ca. 0.1 O.D. in a 1 cm optical path length Hellma fused quartz cell. Femtosecond-to-nanosecond fluorescence dynamics of sample solutions were measured using a Harpia-TF time-resolved fluorescence spectrometer (Light Conversion, Ltd) combining optical Kerr shutter and time-correlated single photon counting (TCSPC) techniques. Fluorescence excitation pulse for Kerr gating and TCSPC measurements was generated as the third harmonic (344 nm) from the fundamental wavelength of the Yb:KGW laser (PHAROS, Light-Conversion), emitting 230 fs pulses and operating at 10 kHz and 100 kHz repetition rates, respectively. | ||
| Fig. 1 Steady-state absorption and fluorescence spectra of (a) M1 and (b) M2 compounds, dissolved in methanol and chloroform. | ||
Two and three-pulse ultrafast transient absorption (TA) spectra and kinetic traces were recorded using a Harpia-TA (Light Conversion, Ltd) transient absorption spectrometer. The pump pulse was generated using an optical parametric amplifier (TOPAS-800, Light Conversion) which was pumped by the fundamental harmonic of a commercial Ti:Sapphire femtosecond laser (800 nm, 50 fs, 2.5 W, 1 kHz, Coherent, Libra). For ultrafast pump–probe experiments, the different sample solutions were excited with 344 nm, 70 fs laser pulses and the diameter of the pump beam at a sample plane was 120 μm. Excitation pulse energy was set to less than 1 μJ. To probe the population dynamics, we used a white light continuum generated in a CaF2 plate. The polarization of the pump beam was aligned parallel to one another and at a 54.7° (“magic”) angle with respect to the polarization of the probe beam. In all ultrafast measurements, the sample was continuously moved in a grid-pattern to avoid degradation. Both time-resolved fluorescence and TA data were globally fitted to construct the model of the entire photocycle in investigated compounds. The target analysis of TA data was performed using CarpetView software (Light Conversion, Ltd).
3 Results
3.1 Steady-state spectroscopy
Steady-state absorption and fluorescence spectra of pyrano[2,3-c]pyrazole derivatives dissolved in methanol (MeOH) (ε = 32.70) and chloroform (CHCl3) (ε = 4.80) are shown in Fig. 1. MeOH is regarded as a protic and polar solvent, whereas CHCl3 is significantly less polar and aprotic. As presented in Fig. 1(a), the absorption line shapes M1 in both solvents are different, especially in the UV region. When the molecule is dissolved in CHCl3, the relative intensity of absorption in 250–300 nm is noticeably lower than in MeOH.In MeOH solution, the dual emission is seen in the fluorescence spectrum, around 480 nm and 580 nm. The fluorescence band, centred at 480 nm according to A. Urbonavičius et al., was attributed as N* emission; meanwhile, the 580 nm band was attributed to T* emission.16 It is noted that the same dual behaviour was found in 4′-N,N-diethylamino-3-hydroxyflavone dissolved in tetrahydrofuran and toluene where the respective emission bands were attributed to the excited normal molecule and excited tautomer emission.21 For comparison, the steady-state emission of the N* state in CHCl3 sample solution is not detectable. The same results were obtained for M2 compounds (Fig. 1(b)). A very important feature of the dual fluorescence deserving attention is a relatively large Stokes shift of the blue emission bands for both M1 and M2 compounds. For M1, it is ca. 6410 cm−1, while that of M2 MeOH solution is ca. 5747 cm−1. Such values are not typical for molecules that do not undergo either a major structural change, or charge redistribution upon excitation. For example, in rhodamine 6G, the Stokes shift is just 800 cm−1.22
3.2 Time-resolved fluorescence spectroscopy
To follow the appearance and evolution of N* form emission with a high temporal resolution, we combined the Kerr gating fluorescence technique together with TCSPC. The observed results of M1 in MeOH and CHCl3 solutions are presented in Fig. 2(a) and (b). The initially detected spectral peak at around 420 nm in both solutions exhibits rough mirror symmetry with the lowest lying absorption band. The apparent structure in the spectra is probably due to experimental noise. On a time scale of 50 ps, the blue fluorescence of M1 in MeOH continuously decreases and shifts to the red. After 50 ps, the fluorescence achieves its plateau centered around 480 nm. Here, the red shift stops and the band at 480 nm further decays in time. The observed red shift of the fluorescence on a 50 ps time scale has been observed by earlier works on similar molecules by Chou et al.23 and Kimura et al.24 They attributed such dynamics to the solvent relaxation that occurred in response to the large redistribution of the electronic charge upon excitation. However, in our case, we observe significant decay of the signal in addition to the red shift. While solvent relaxation can decrease the excited-state energy, which explains the red-shift, it is not obvious why it should also change the magnitude of the transition dipole moment. Therefore, the observed decay on the picosecond timescale is more likely due to a structural change. | ||
| Fig. 2 Time-resolved fluorescence spectra of M1 dissolved in (a) methanol and (b) chloroform. Steady-state fluorescence spectra are shown by filled-in graphs for comparison. The panels (c) and (d) show the TCSPC fluorescence decay traces in both solvents. | ||
The shape of the fluorescence spectra, recorded at longer detection times, coincides with the blue part of the steady-state dual emission. Furthermore, as presented in Fig. 2(c), the decrease of blue fluorescence is accompanied by the appearance of 580 nm T* form fluorescence. The lifetimes of both fluorescent states were estimated during the exponential fitting of the kinetics, taken at 480 nm and 580 nm. Fitting of the 580 nm decay trace with a double rising and decaying exponent gives satisfactory results when τ1 = 270 ps (k = 37 × 108 s−1) and τ2 = 3.0 ns (k = 3.3 × 108 s−1). The value of the rise time τ1 closely matches the decay time of 480 nm emission. Interestingly, the decay of 480 nm emission also contains a weak long component of 1.2 ns, the amplitude of which is ca. 200 times smaller than that of the main 280 ps component. The physical origin of the long decay component is probably related to the ESIPT barrier, created by polar protic MeOH. In some molecules, if the proton does not transfer before the hydrogen bonds trap it, these molecules may retain their original configuration. In this state, blue fluorescence becomes the only means of returning to the ground state. However, the strength of this signal is too weak to justify further detailed analysis.
In CHCl3 solution, the fluorescence maximum at the initial gate time is centered around 440 nm and shifts slightly to 450 nm and decays rapidly. The dynamic Stokes shift is less apparent than in MeOH. In CHCl3, the decay time of N* form emission was estimated to be less than 30 ps, whereas tautomer emission in chloroform solution decays with a lifetime of 4 ns (Fig. 2(d)), 1.3 times longer than in MeOH. The observed dynamic red shift in MeOH solution, along with an unusually large Stokes shift of the blue fluorescence band, becomes a strong experimental finding suggesting that the ESIPT is not the only photophysical process that occurs in the M1 molecule.
3.3 TA spectroscopy
TA spectroscopy allows monitoring not only fluorescent but also dark states; they are observable via their induced absorption and ground state bleach bands. We used this technique to get more detailed information about the photocycle in pyrano[2,3-c]pyrazole derivative molecules. The broadband TA data of M1 in MeOH and CHCl3 are shown in Fig. 3. In these spectra, the excited state absorption (ESA), ground state bleaching (GSB) and stimulated emission (SE) can be identified clearly. The spectra in MeOH (Fig. 3(b)) show clear and distinct dual emission. At a probe time of 0.3 ps, the TA spectrum shows the ESA across most of the visible spectral range. The negative signal in UV is assigned to the GSB in accordance with the steady-state absorption spectrum. Furthermore, a dip of the ESA around 420 nm represents the stimulated N* emission that undergoes a slight red shift (black → red → green) and transforms into a shoulder around 530 nm (green spectrum). The same red shift was obtained from time-resolved fluorescence experiments in MeOH (Fig. 2(a)), where the fluorescence shifts into the red over 50 ps. Furthermore, the same TA spectrum undergoes a second transformation whereby the ESA in the range of 400–550 nm is featuring the shoulder around 530 nm transforms into a broader ESA (dashed spectra). The latter ESA reaches its maximum plateau at 550 ps, and in subsequent probe times, it maintains the same spectral form, which is governed by the decrease in amplitude. Additionally, the changes of the ESA are followed by a concurrent rise of a new SE band around 580 nm, which is in line with the steady-state T* emission due to the ESIPT reaction. The observed spectral transformation suggests the involvement of at least three different transient states; two of them can be expected to be N* and T* forms, emitting near 420 and 600 nm. The origin of the transient, appearing in a time scale of 50 ps, is not obvious; however, it is clearly visible due to its spectrotemporal signature.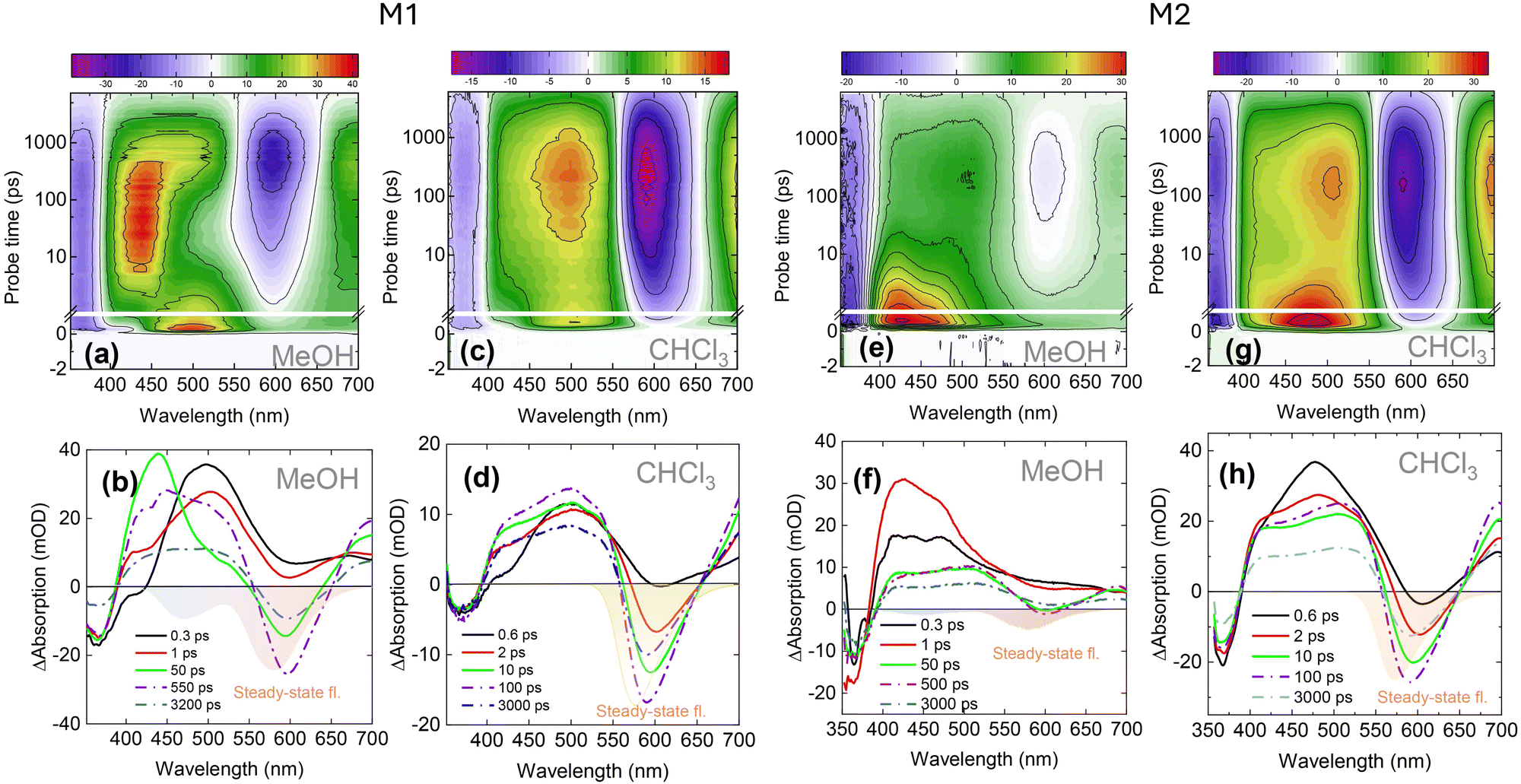 | ||
| Fig. 3 TA dynamics spectra of M1 dissolved in (a) methanol and (d) chloroform. The panels (c) and (d) show the TA spectra at selected probe times in both solvents. The TA data of the M2 compound are depicted in (e)–(h) panels. The color-filled area represents the steady-state fluorescence spectra. | ||
Comparatively, the ESIPT dynamics of M1 in CHCl3 exhibits similar spectral shapes but proceeds significantly faster than in MeOH (Fig. 3(d)). Firstly, a small negative dip of around 420 nm appears, attributing the SE of the N* state. This SE over several picoseconds is replaced by ESA, ranging from ca. 400 to 550 nm. Subsequently, in sub-hundreds of picoseconds, the ESA grows up and after 100 ps reaches its plateau which further remains virtually unchanged and only decreases in magnitude. Simultaneously, with the evolution of ESA, the second SE band around 570 nm appears at several picoseconds and grows until it reaches its maximum amplitude at a 100 ps probe time. After this, no major spectral changes are observed in TA spectra – the bleach, ESA and SE decrease uniformly. In summary, the main difference between two solvents lies in the intermediate 50 ps transition observed in MeOH that cannot be discerned in CHCl3. Additionally, the SE attributed to the tautomer develops slower in MeOH taking ca. 550 ps to reach its peak, whereas in CHCl3 this process only takes 100 ps.
To quantitatively examine the effect of the methoxy groups, we conducted the same experiment on the compound containing a single methoxy group (M2). The absorption and fluorescence spectra of M2, together with its structure, are shown in Fig. 1(b) and the TA dynamics are summarised in Fig. 3(e)–(h). In the TA spectra of MeOH sample solution (Fig. 3(f)), the ESA is dominant; therefore, the SE of the N* state is not visible. Furthermore, the ESA demonstrates a slight transformation over the time. Firstly, after 345 nm excitation, the ESA covers almost all visible spectral ranges. After a 5 ps delay time, a small dip of around 600 nm, representing the SE of T*, appears and peaks at approximately 300 ps. During the same 300 ps, the ESA in the 400–450 nm region decreases while the signal at 450–500 nm remains constant. In contrast to M1 in MeOH, M2 has a very weak SE of the T* form. However, in CHCl3, the emission of both compounds is very strong (Fig. 3(c), (d), and (g)–(h)) and develops quickly. The kinetic traces of the M2 molecule dissolved in MeOH and CHCl3, along with the global analysis results, are shown in Fig. S1 and S2 (ESI†).
The totality of experimental observations in pyrano[2,3-c]pyrazole with methoxy groups (large initial Stokes shift, spectral transformations on 50 ps time scales) suggests that an additional dynamic pathway is involved in excited state dynamics. From the molecular structure,16 the most obvious candidate for such a mechanism would be the twisting of the molecule around the C–C bond (the bond between 3,4-dimethoxyphenyl and position 6 on the pyrano[2,3-c]pyrazol fragment) that occurs together with the ESIPT reaction. To check whether the observed dynamics is related to large scale conformational changes in compound M1, we conducted the pump–probe measurements in a sample solution containing glycerol (60 proc. of MeOH and 40 proc. of glycerol, the resultant viscosity of the mixture is ca. 40 mPa s). It should be noted that the dielectric constant of glycerol (ε = 43) is similar to the dielectric constant of MeOH (ε = 32.7) but the viscosity of glycerol is significantly higher (0.545 mPa s of MeOH and 1412 mPa s of glycerol at a 20 °C temperature). The measured kinetics of SE in MeOH and glycerol solution versus pure MeOH sample solution are presented in Fig. S3 (ESI†), and the global fitting of the data is shown in Fig. S4 (ESI†). The result in glycerol shows that the dynamics indeed slows down. On the other hand, without the slowdown of conformational dynamics, solvent relaxation also slows down at a similar rate, leaving uncertainty about which process is more affected by the increased viscosity.
3.4 Three-pulse TA spectroscopy
Three-pulse TA spectroscopy is a variation of conventional pump–probe (PP) spectroscopy involving an additional pulse that can either transfer the population to the ground-state (pump–dump–probe, i.e. PDP) or re-pump the molecule to a higher excited state (pump–repump–probe, i.e. PRpP). In both cases, the three-pulse TA allows opening new reaction pathways or observing cleaner dynamics in complex kinetic schemes. We employed this technique to unveil the origin of TA spectra detected in tens of picoseconds in MeOH and to study the ground-state proton transfer in M1. First, we consider the TA spectral form of M1 at 50 ps (Fig. 3(b), green curve). The induced absorption of this TA spectrum observed in the 400–550 nm spectral range significantly differs from the equilibrated T* spectrum – it has a strong peak at 440 nm and only a small shoulder around 500 nm. It is reasonable to assume that such an intermediate spectrum may be a superposition of N* and some other states. This other state may feature an SE band which is masked by the dominating ESA of N*. The contribution of this SE to the overall TA signal would then manifest itself as a dip in the ESA. To test this hypothesis and disentangle the overlapping spectral bands, we applied the re-pumping laser pulse that was in resonance with the 480 nm ESA band. The re-pump pulse was timed at 13 ps after the pump, and its effect was probed at 18 ps. If there is SE overlapping with ESA, such a pulse should produce two different outcomes: (a) re-pump some of the excited molecules to a higher excited state via the ESA, and (b) demote some of them to the ground state via SE. The first channel will be dominant if the overall TA spectrum is positive. However, the higher excited state created by the re-pump will relax back to the lowest excited state over several picoseconds, and only the effects of dumped emission will remain visible. The obtained result is summarised in Fig. 4(a) and shows that ca. 5 ps after the re-pump pulse action, in the double TA spectrum (PRpP-PP), there is a loss of the GSB signal together with the decrease of ESA around 450 nm. The re-pump pulse also promotes some gain of ESA around 480 nm. The disappearance of the GSB and ESA around 450 nm arises due to the demotion of the molecules back into the ground state. However, the fact that the ESA signal around 480 nm increases instead of decreasing points towards the loss of SE that is masked by the ESA of a state unaffected by the repump pulse. In our case, this ESA is due to the tautomer state. The discussed changes are better illustrated by the fractional change of the TA signal (PRpP-PP/PP) depicted in Fig. 4(b). Here, we clearly recognise the decrease of GSB and ESA signals together with a small gain of 480 nm ESA where we expect to obtain SE of the separate state. It should be noted that the observed gain around 480 nm closely matches the spectral position of the blue band in the steady-state fluorescence spectrum. This gives further credence that the main effect of an additional pulse observed at this delay time is the depletion of the blue-emitting state via SE. In contrast, the emission of the tautomer around 610 nm remains virtually unaffected by the re-pump.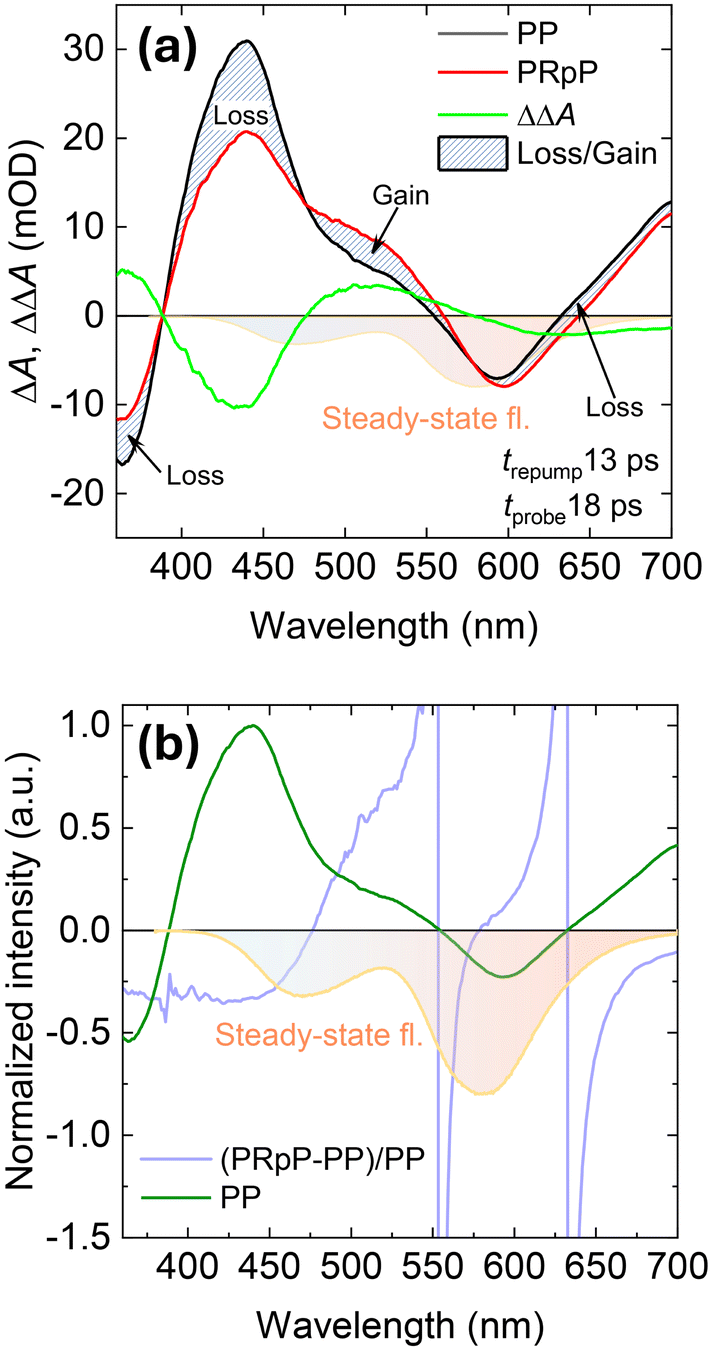 | ||
| Fig. 4 (a) The femtosecond TA spectrum of M1 in MeOH at a 18 ps delay time in the presence (black line, PDP) and absence (red line, PP) of a repump pulse arriving at 13 ps after the initial 345 nm excitation. The green curve shows the double difference absorption i.e. PRpP-PP. The blue curve in (b) shows the fractional loss and gain behavior of the TA after repump, i.e. PRpP-PP/PP. | ||
After the ESIPT reaction and the relaxation of the excited state, the proton must come back to the negatively charged oxygen and re-form the N state, i.e. the ground state proton transfer (GSIPT) takes place. Under normal pump–probe conditions, this reaction can be difficult to detect, when the rate-limiting step is the radiative relaxation of the tautomer. In this case, population of the ground-state tautomer is very low because GSIPT depletes it faster than it can be created. To circumvent this, the ground state of the tautomer can be filled by appropriately timed and wavelength-tuned dump pulse.25–27 We applied a 610 nm dump pulse with an energy of 1 μJ that arrives at the probe time when SE of T* achieves the maximum (τprobe = 550 ps). To illustrate the spectral change induced by the dump pulse, the TA spectra with the absence and presence of the dump pulse are presented in Fig. 5(a) and (b). It becomes evident that comparison of both TA spectra at 550 ps shows a significant depletion of T* SE at 600 nm (Fig. 5(a)). Moreover, in the double TA spectrum shown in panel (b), (the difference between dumped and normal PP spectra), there is a clearly pronounced new ESA at the blue edge of the steady-state fluorescence (Fig. 5(b)). This is the expected absorption position of the ground-state tautomer species populated by the dump pulse. After 30 ps of dumping (τprobe = 580 ps), the TA spectra with and without dumping pulses display similar spectral forms, suggesting a very fast relaxation of the ground-state tautomer.
 | ||
| Fig. 5 Femtosecond TA spectra of M1 in MeOH at the selected delay times in the presence (filled area, PDP) and absence (solid line, PP) of a dump pulse arriving at 550 ps after the initial 345 nm excitation. Panel (b) represents the double difference absorption spectrum 2.2 × PDP-PP which denote the pure difference spectrum corresponding to the ground state. The kinetic trace with the presence and absence of a dumping pulse is presented in panel (c), while panel (d) shows the relaxation of the ground-state tautomer. | ||
The relaxation of the ground-state tautomer is better illustrated by the kinetic traces taken at the blue-edge of M1 tautomer SE and is presented in Fig. 5(c) and (d). The blue-filled circles represent the kinetic trace of the conventional pump–probe without the dumping pulse, while the empty circles illustrate the TA dynamics with a dumping pulse, arriving at 550 ps. Up to a 550 ps probe time, both traces overlap, but at 550 ps the dumped trace switches from a negative (SE) to a positive (ESA) signal. This is a feature of the ground-state tautomer featuring the induced absorption at 556 nm. Fig. 5(d) presents the decay of the ground-state tautomer in more detail. The fitting according to the global analysis procedure shows a bi-exponential decay fashion of the ground-state tautomer with 0.7 ps and 8 ps lifetimes.
To compare the ground-state dynamics in polar and less-polar solvents, we conducted the same experiment on M1 in CHCl3 solution because in the excited state, it seems that ESIPT proceeds significantly faster in this solvent. The effect of the same dump pulse, which is timed at 250 ps after the excitation (time, when the SE reaches the maximum in CHCl3 solution), is presented in Fig. 6(a) and (b). After dumping, the ESA appears around 550 nm but it is less pronounced than in MeOH, indicating a lower absorption cross-section for the ground-state tautomer T. Another difference in CHCl3vs. MeOH is a faster relaxation of the ground-state tautomer – in MeOH, and this reaction takes 8 ps, while in CHCl3 most of the reaction occurs in less than a picosecond (Fig. 6(d)). In the case of M2 MeOH sample solution, we could not investigate the ground-state dynamics due to a very weak T* SE preventing us from the efficient dumping of the T* state.
 | ||
| Fig. 6 (a) TA spectra of M1 in CHCl3 with the presence (green) and absence (black) of the SE dumping pulse, arriving at a 250 ps probe time and panel (b) shows the double difference absorption spectrum 1.7 × PDP-PP which denotes the pure difference spectrum corresponding to the ground state. The kinetic trace in the presence and absence of a dumping pulse is presented in panel (c), while panel (d) shows the kinetic traces representing the decay of dump-induced transients in MeOH and CHCl3 solutions. | ||
4 Discussion
4.1 The kinetic model
To examine the intramolecular processes, we employed target analysis using the kinetic model shown in Fig. 7. This model was selected based on the plausibility of transient spectra obtained from the global fitting of the pump–dump–probe data. An important element of the model is the excited-state ESIPT reaction, discussed in the literature as the primary mechanism of dual emission.16 This proton transfer occurs between the hydroxy group at position 5 and the carboxy group at position 4, as depicted by the N* → T* transition in Fig. 7. In MeOH, time-resolved fluorescence of M1 reveals a dynamic Stokes shift, which is absent in CHCl3 solution. Moreover, two and three-pulse pump–probe measurements revealed an additional transient that affects the ESIPT reaction. The polar moieties, picosecond fluorescence decay and drastic changes of TA spectra in MeOH indicate the involvement of conformational changes in one of the reactions. These observations together suggest a physical state influencing the ESIPT rate, leading us to postulate an additional transition from the normal excited state N* to a twisted intramolecular charge transfer (TICT*) state.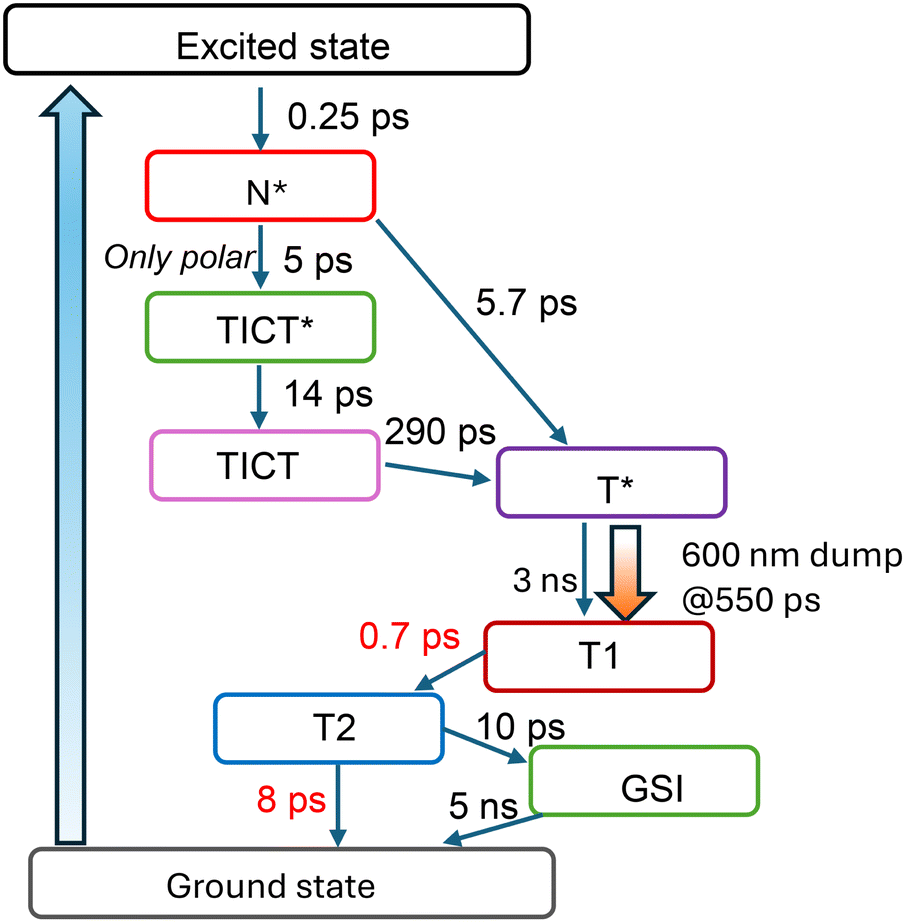 | ||
| Fig. 7 Target evolution model of M1 used in global analysis. | ||
TCSPC measurements of M1 in MeOH indicate tautomer emission appearing after 290 ps, while pump–probe data exhibit a minor tautomer SE signal at initial probe times, indicating a direct ESIPT reaction and the N* → T* transition in the model. Thus, there are two channels for tautomer formation: direct ESIPT and ESIPT via the TICT state. To fully describe the photocycle, ground-state dynamics are also considered. Following radiative relaxation, the ground-state tautomer T is produced, which subsequently reverts to its original state N. During this relaxation, T undergoes solvation and proton back transfer in the ground state, thus involving the T → T1 → T2 transition. We avoid assigning specific time constants to each process because both of them can proceed in a non-exponential fashion: different solvation shells will react to the ground state population at different rates, and the proton transfer will depend on the particular configuration of the hydrogen bond network around the molecule. Two exponents with 0.7 ps and 8 ps lifetimes provided an adequate description of this complex transition. In addition to proton back transfer, the data suggest the presence of a long-lasting dump effect on the ground state. It was accounted for by including an additional ground state intermediate, GSI, in the model. The possible origin of this component will be discussed below.
The species-associated difference absorption spectra (SADS) representing the TA of each component are shown in Fig. 8(a), and the quality of the fits can be evaluated from the comparison of experimental and fitted curves shown in Fig. S5(d) of the ESI.† The general idea of the proposed model goes as follows: the 345 nm laser pulse creates an initial excited Franck–Condon (FC) state which within ca. 0.5 ps evolves into a more stable N* state. After this, the molecules evolve either directly into the excited tautomer T* state or into the TICT* state, with a branching ratio close to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The twisting is accompanied by solvation and the TICT* state relaxes within 14 ps. The corresponding change in SADS results in a significant red shift of an emission maximum and lesser compensation between SE and ESA (see green and blue curves in Fig. 8(a)) It should be noted that the spectral shape of TICT SADS fairly coincides with the inverse double difference absorption spectrum, obtained from the re-pumping experiment on M1 in MeOH (see the dashed line in Fig. 8(a)). This provides an additional indication that the suggested target analysis model is correct. Finally, the relaxed conformer transforms into the excited tautomer T* within 290 ps, resulting in the SADS depicted by a yellow line in Fig. 8(a). The latter then relaxes to the ground state with a 3.0 ns time constant. In summary, there are two distinct excited state pathways of T* formation: directly from the ‘normal’ excited state and via the TICT state undergoing several relaxation steps in between.
:1. The twisting is accompanied by solvation and the TICT* state relaxes within 14 ps. The corresponding change in SADS results in a significant red shift of an emission maximum and lesser compensation between SE and ESA (see green and blue curves in Fig. 8(a)) It should be noted that the spectral shape of TICT SADS fairly coincides with the inverse double difference absorption spectrum, obtained from the re-pumping experiment on M1 in MeOH (see the dashed line in Fig. 8(a)). This provides an additional indication that the suggested target analysis model is correct. Finally, the relaxed conformer transforms into the excited tautomer T* within 290 ps, resulting in the SADS depicted by a yellow line in Fig. 8(a). The latter then relaxes to the ground state with a 3.0 ns time constant. In summary, there are two distinct excited state pathways of T* formation: directly from the ‘normal’ excited state and via the TICT state undergoing several relaxation steps in between.
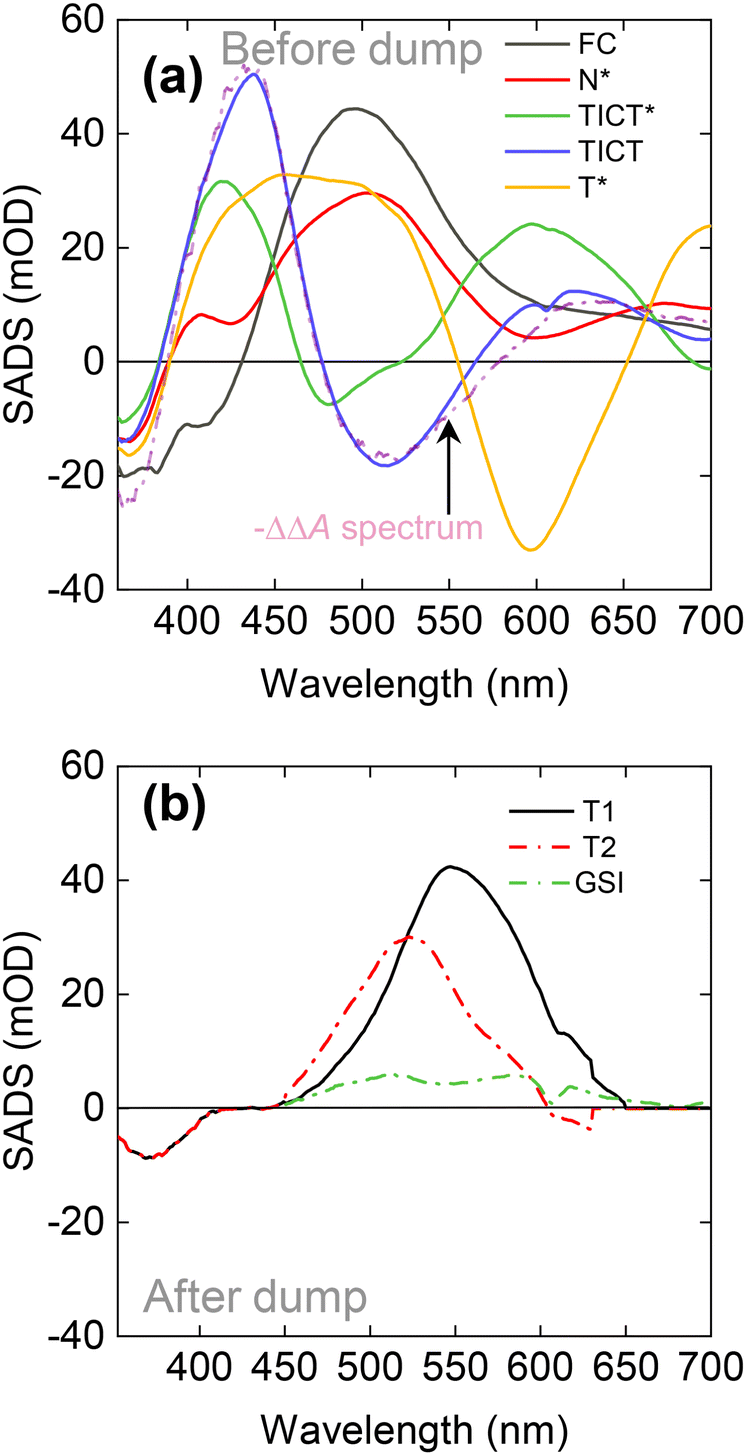 | ||
| Fig. 8 SADS of M1, retrieved from global analysis. Panel (a) shows the spectra of excited-state intermediates, while the spectra assigned to ground states revealed by the dump are shown in panel (b). | ||
The model is branched; therefore, several of its compartments are populated at any given time. This makes it more difficult to compare SADS to measured TA spectra directly. However, the first transition from the excited state to N* is clearly discernible in the experimental data. The SADS of the early components fully correspond to the earliest experimental TA spectra. Such verifications become harder when N* branches into TICT* and T*. The TA at 50 ps is a superposition of TICT and T* states. The ESA of T* SADS is very broad and dominates the experimental TA spectrum, obscuring the SE of the TICT state at 500 nm. The global analysis allows us to distinguish the SADS of TICT and T* states. The situation again becomes less complicated after the full formation of T*. The TA spectra after ca. 1 ns represent the relaxation of the T* state.
TICT occurring alongside ESIPT is not a new idea. S. M. Ormson et al. suggested the presence of such states to explain the dual emission in 3-HF.28 Furthermore, the calculations based on density functional theory of the DMAF compound (a derivative of 3-HF) confirmed the conformational changes.29 Our spectroscopic data i.e. fluorescence decay in tens of picoseconds, the presence of electron-donating moieties in both M1 and M2 molecules points towards a conformational change concurrent with the proton transfer.
The reaction scheme, shown in Fig. 7, allows more than one physical interpretation. The dynamics observed in both compounds share many similarities with those reported for amino-hydroxyflavones.23,24 These studies found that the N* state possesses a significant charge-transfer (CT) character, which is stabilized more in polar solvents, leading to a higher barrier for ESIPT. As a result, ESIPT slows down with increased solvent polarity. The initial ESIPT reaction can occur directly from the non-relaxed N* and proceeds faster, but it slows down as solvent relaxes. The 'fast' protons account for a minor T* SE at very early times in pyrano[2,3-c]pyrazole dissolved in MeOH. Assuming this approach, the N* state of the pyrazole molecule would have the ICT/TICT character itself. This interpretation is also consistent with the observations in the weakly polar solvent CHCl3 where conversion to T* is faster than that in methanol. Then, the N* → TICT* → TICT depicted in Fig. 7 would simply account for different stages of solvent relaxation of the N*(ICT/TICT) state. They would also cause the dynamic fluorescence Stokes shift seen in Fig. 2. On the other hand, the pump–repump–probe experiment revealed a definite separate spectrum of the ICT/TICT state. Initial changes of TA spectra in Fig. 3 (black → red → green curve) seem to be too drastic to be caused by just solvation. The fluorescence decay of M1 on the timescale of tens of picoseconds is most readily explained by the change of the molecular structure that causes the decrease of the transition dipole moment. Both molecules contain polar moieties on a ring separated from the backbone of the molecule by a single bond. The presence of these structural features allows to expect both charge transfer and twisting. Considering all these arguments, we lean towards the interpretation that N* and ICT/TICT states are separate and twisting plays a role in the observed dynamics.
Using time-resolved fluorescence data as the reference, it becomes possible to decide whether the dips in the induced absorption bands observed in pump–probe experiments are actually smaller ESA or overlapping SE contribution. The observed dynamic Stokes shift, occurring alongside with the picosecond decay of the M1 fluorescence, here can be attributed to the formation of TICT from the N* state. The red shift of the blue fluorescence takes about 50 ps and is in a good agreement with the population time of the TICT* state retrieved from global analysis on TA data. After 50 ps, the amplitude of the fluorescence spectrum further decreases in time and it means that the decay, measured during TCSPC experiments, corresponds to the relaxation of TICT and formation of the T* state. The decay time of TICT fluorescence in M1 is 280 ps and is close to the time constant of TICT → T* transition, retrieved from the global analysis of TA data (transition from blue to red SADS in Fig. 8(a)). It also matches the time constant of the appearance of the T* emission at 580 nm (270 ps). In the case of M2, we did not show the temporal evolution of the blue fluorescence because the signal was under the detection threshold. It also exhibits a smaller Stokes shift. This is consistent with previous results by A. S. Klymchenko et al. on 3-HF, where they proposed that electron donor groups cause a more significant red shift of the blue emission than of the T*.30
We observed the solvent effect by comparing the experimental TA results of M1 dissolved in MeOH and CHCl3. The TA dynamics in both solvents are different. In CHCl3 solution, the TA representing the TICT state is absent, and the ESA together with SE of the T* form appears faster than in MeOH, indicating that the proton transfer rate dominates over the twisting rate. Therefore, the TA dynamics in CHCl3 were fitted using a simple sequential model, skipping the TICT branch, as presented in Fig. S6(a) (ESI†). The obtained SADS together with the fits of the kinetic traces, showing the fitting quality, are presented in Fig. S6(b) and (c) of the ESI,† respectively. The TA spectra at 200 ps and later are virtually identical. Given the large Stokes shift of the fluorescence in CHCl3 (Fig. 1(a)) and its similarity to the SE in the TA spectra, we conclude that the TA signals at 200 ps and later correspond to the tautomer TA. From the literature on ESIPT in 3-HF, it is known that a decrease of the solvent polarity leads to a faster proton transfer.31 On the other hand, an increase of H-bond donating ability in polar solvents selectively stabilizes N* and hinders the ESIPT reaction.14 Changing the MeOH to CHCl3, the polarity of the solvent significantly decreases (from ε = 32.7 to ε = 4.8), as well as it reduces its H-bonding ability. Based on previous observations in 3-HF, it is assumed that the ESIPT reaction also becomes faster in CHCl3.
Femtosecond time-resolved fluorescence data in CHCl3 lack the dynamic Stokes shift observed in MeOH; only fast decay is observed. The emission at 575 nm decays slightly slower in CHCl3 (4.2 ns) than in MeOH (3.0 ns) indicating a slowdown of the radiative relaxation rate. Similar behavior was previously obtained for the same 3-HF by Klymchenko14 and Ormson,28 where they proposed that in less polar solvents the emission lifetime increases along with the fluorescence quantum yield.
4.2 Ground-state dynamics
The evolution of the ground-state reaction is a very different process than that of the excited state. According to the Franck–Condon principle, the nuclear configuration of the molecule immediately after the dump should be the same to that of the excited tautomer T*. The T generated by the deactivation of T* subsequently rearranges its nuclei and the surrounding solvent from the T* configuration back to the equilibrated ground-state configuration. To describe the proton shuttling in the ground state (T → N), we involve the stages T1 → T2 → GSI in the model, as presented in Fig. 7 (SADS presented in Fig. 8(b)). In the case of M1 in MeOH, we observed a very fast GSIPT featuring two time constants, 0.7 ps and 8 ps. Such a fast relaxation of the ground state transient has been observed before in 3-HF, where it also lasts only a few picoseconds.21,32 It should be noted that both the magnitude and the direction of the dipolar moments of T* and T states are different; therefore, the ground state transient should also undergo solvation which could be responsible for a fast decay component. It is tempting to attribute the 8 ps component to the main GSIPT rate, and the faster component to the solvation. Indeed, the observed blue-shift of induced absorption produced by the dump (see Fig. 8(b)) looks like the mirror image of solvation dynamics usually observed in SE signals in the pump–probe. However, in reality, the solvent interactions are probably an integral part of the proton transfer process, resulting in the observed multiexponential relaxation. This is supported by the pump–dump–probe measurements in CHCl3, as shown in Fig. 6(c). In this case, the dumping of the 500 nJ pulse arriving at 250 ps after excitation also results in a bi-exponential decay of the ground-state intermediate but the fast component is even faster than in MeOH and is more dominant in the kinetics.In order to fit the data adequately, we needed to include a ground state intermediate (GSI) component in the model that relaxes on a 5 ns timescale. Its physical origin is unclear. One hypothesis for this long-lived state is that in some cases the ground-sate proton transfer could be hindered by particularly strong H-bonding with surrounding protons. After dumping, the ground state contains a number of tautomers T, and most of them return to the original N form within 8 ps. However, a small fraction of tautomers may find themselves in an H-bond configuration that traps the proton in its tautomer position and forms a ground state barrier for proton back transfer. Such explanation would predict that the long-lived ground state would only be observed in MeOH that is both highly polar and protic. Indeed, in CHCl3, the dumped kinetics decays fully during the 10 ps after the dump, while in MeOH there is a small fraction of the induced TA signal that remains for several nanoseconds.
5 Conclusions
In conclusion, we have investigated the excited and ground state dynamics of a new set of compounds exhibiting intramolecular proton transfer, pyrano[2,3-c]pyrazoles possessing methoxy substituents. To understand the photophysics of these compounds, we conducted time-resolved fluorescence, conventional pump–probe, and pump–dump/repump–probe spectroscopies. Femtosecond time-resolved fluorescence spectroscopy of M1 in MeOH revealed a dynamic Stokes shift that was attributed to the formation of a twisted intramolecular charge transfer state competing with the proton transfer process. Both conventional transient absorption (TA) and pump–repump–probe spectroscopies confirmed this association, allowing us to construct a kinetic model to describe the photocycle in M1.Using stimulated emission dumping, we were also able to observe the ground-state proton transfer reaction. The ground-state intramolecular proton transfer (GSIPT) reaction was shown to be very fast and demonstrated a bi-exponential decay fashion. We also investigated the substitution effect and found that the ESIPT rate differs in compounds with one and two methoxy substituents. For the compound with one methoxy group (M2) in MeOH, the stimulated emission (SE) of T* was practically invisible; meanwhile, for M1, it was perfectly resolved.
Data availability
Quality of the fits provided by the kinetic model has been included in the ESI.† Other data are available upon reasonable request from the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. U., S. K., V. M., E. A., M. D. and A. S. acknowledge funding from the Research Council of Lithuania (LMTLT), grant no. S-MIP-23-51. M. N. and M. V. acknowledge the “Universities’ Excellence Initiative” programme by the Ministry of Education, Science and Sports of the Republic of Lithuania under the agreement with the Research Council of Lithuania (project no. S-A-UEI-23-6).Notes and references
- A. Douhal, S. Kim and A. Zewail, Nature, 1995, 378, 260–263 CrossRef CAS PubMed.
- A. Douhal, F. Lahmani and A. H. Zewail, Chem. Phys., 1996, 207, 477–498 CrossRef CAS.
- M. Rini, B.-Z. Magnes, E. Pines and E. T. Nibbering, Science, 2003, 301, 349–352 CrossRef CAS PubMed.
- X. Zhang, L. Guo, F.-Y. Wu and Y.-B. Jiang, Org. Lett., 2003, 5, 2667–2670 CrossRef CAS PubMed.
- X. Peng, Y. Wu, J. Fan, M. Tian and K. Han, J. Org. Chem., 2005, 70, 10524–10531 CrossRef CAS PubMed.
- H. C. Joshi and L. Antonov, Molecules, 2021, 26, 1475 CrossRef CAS PubMed.
- S. Ameer-Beg, S. M. Ormson, R. G. Brown, P. Matousek, M. Towrie, E. T. Nibbering, P. Foggi and F. V. Neuwahl, J. Phys. Chem. A, 2001, 105, 3709–3718 CrossRef CAS.
- J. M. Al-Khayri, G. R. Sahana, P. Nagella, B. V. Joseph, F. M. Alessa and M. Q. Al-Mssallem, Molecules, 2022, 27, 2901 CrossRef CAS PubMed.
- A. N. Panche, A. D. Diwan and S. R. Chandra, J. Nutr. Sci., 2016, 5, e47 CrossRef CAS PubMed.
- A. Ullah, S. Munir, S. L. Badshah, N. Khan, L. Ghani, B. G. Poulson, A.-H. Emwas and M. Jaremko, Molecules, 2020, 25, 5243 CrossRef CAS PubMed.
- M. Sarkar, J. G. Ray and P. K. Sengupta, Spectrochim. Acta, Part A, 1996, 52, 275–278 CrossRef.
- A. S. Klymchenko, V. G. Pivovarenko and A. P. Demchenko, J. Phys. Chem. A, 2003, 107, 4211–4216 CrossRef CAS.
- A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC.
- A. S. Klymchenko and A. P. Demchenko, Phys. Chem. Chem. Phys., 2003, 5, 461–468 RSC.
- D. McMorrow and M. Kasha, J. Phys. Chem., 1984, 88, 2235–2243 CrossRef CAS.
- A. Urbonavičius, S. Krikštolaitytė, A. Bieliauskas, V. Martynaitis, J. Solovjova, A. Žukauskaitė, E. Arbačiauskienė and A. Šačkus, Molecules, 2023, 28, 6599 CrossRef PubMed.
- J. V. Faria, P. F. Vegi, A. G. C. Miguita, M. S. Dos Santos, N. Boechat and A. M. R. Bernardino, Bioorg. Med. Chem., 2017, 25, 5891–5903 CrossRef CAS PubMed.
- F. E. Bennani, L. Doudach, Y. Cherrah, Y. Ramli, K. Karrouchi and M. E. A. Faouzi, et al. , Bioorg. Chem., 2020, 97, 103470 CrossRef CAS PubMed.
- A. Ansari, A. Ali and M. Asif, et al. , New J. Chem., 2017, 41, 16–41 RSC.
- M. Ramadan, A. A. Aly, L. E. A. El-Haleem, M. B. Alshammari and S. Bräse, Molecules, 2021, 26, 4995 CrossRef CAS PubMed.
- Z. Kuang, Q. Guo, X. Wang, H. Song, M. Maroncelli and A. Xia, J. Phys. Chem. Lett., 2018, 9, 4174–4181 CrossRef CAS PubMed.
- T. Govindaunny and B. Sivaram, Appl. Phys., 1980, 23, 253–258 Search PubMed.
- P.-T. Chou, S.-C. Pu, Y.-M. Cheng, W.-S. Yu, Y.-C. Yu, F.-T. Hung and W.-P. Hu, J. Phys. Chem. A, 2005, 109, 3777–3787 CrossRef CAS PubMed.
- Y. Kimura, M. Fukuda, K. Suda and M. Terazima, J. Phys. Chem. B, 2010, 114, 11847–11858 CrossRef CAS PubMed.
- J. T. Kennis, D. S. Larsen, I. H. van Stokkum, M. Vengris, J. J. van Thor and R. van Grondelle, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17988–17993 CrossRef CAS PubMed.
- E. Papagiannakis, M. Vengris, D. S. Larsen, I. H. Van Stokkum, R. G. Hiller and R. Van Grondelle, J. Phys. Chem. B, 2006, 110, 512–521 CrossRef CAS PubMed.
- D. S. Larsen, I. H. van Stokkum, M. Vengris, M. A. van Der Horst, F. L. de Weerd, K. J. Hellingwerf and R. van Grondelle, Biophys. J., 2004, 87, 1858–1872 CrossRef CAS PubMed.
- S. M. Ormson, R. G. Brown, F. Vollmer and W. Rettig, J. Photochem. Photobiol., A, 1994, 81, 65–72 CrossRef CAS.
- Y. Wang, Y. Shi, L. Cong and H. Li, Spectrochim. Acta, Part A, 2015, 137, 913–918 CrossRef CAS PubMed.
- A. S. Klymchenko, V. G. Pivovarenko, T. Ozturk and A. P. Demchenko, New J. Chem., 2003, 27, 1336–1343 RSC.
- V. V. Shynkar, Y. Mely, G. Duportail, E. Piemont, A. S. Klymchenko and A. P. Demchenko, J. Phys. Chem. A, 2003, 107, 9522–9529 CrossRef CAS.
- A. N. Bader, V. G. Pivovarenko, A. P. Demchenko, F. Ariese and C. Gooijer, J. Phys. Chem. B, 2004, 108, 10589–10595 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cp03805c |
| This journal is © the Owner Societies 2025 |