
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical semi-sacrificial growth of a self-supporting MOF-based electrode for urea electrooxidation-coupled water electrolysis†
Jiang
Ji
ab,
Yinsheng
Wang
ab,
Changsheng
Cao
 *b,
Xin-Tao
Wu
b and
Qi-Long
Zhu
*b
*b,
Xin-Tao
Wu
b and
Qi-Long
Zhu
*b
aCollege of Chemistry, Fuzhou University, Fuzhou 350002, China
bState Key Laboratory of Structural Chemistry and Fujian Provincial Key Laboratory of Materials and Techniques toward Hydrogen Energy, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou 350002, China. E-mail: cscao@fjirsm.ac.cn; qlzhu@fjirsm.ac.cn
First published on 26th April 2023
Abstract
Replacing the anodic oxygen evolution reaction in water electrolysis with thermodynamically more favorable oxidation reactions is appealing for reducing the energy consumption of hydrogen production but is limited by the lack of efficient yet cost-effective electrodes. Herein, a self-supporting NiFe-based Prussian blue analogue (PBA) electrode (NiFe-PBA-NF) was directly prepared via a facile semi-sacrificial anodic electrodeposition strategy, in which ultrasmall NiFe-PBA nanoparticles were grown uniformly and compactly on the nickel foam (NF) surface. Benefiting from its ingenious structure and the synergistic effect between Ni and Fe sites, the as-prepared NiFe-PBA-NF exhibited excellent electrochemical performance in the urea oxidation reaction (UOR) with a required potential of only 1.375 V to deliver a current density of 100 mA cm−2, outperforming the powdered NiFe-PBA and even the commercial RuO2 catalyst. Moreover, a Ru-NiFe-PBA-NF electrode assembled with ultrathin Ru-doped NiFe-PBA nanosheets was fabricated through further Ru-modification treatment, which exhibited a remarkable electrochemical performance in the hydrogen evolution reaction (HER), even better than that of the commercial Pt/C catalyst. Ultimately, a UOR-coupled energy-saving hybrid water electrolysis system was constructed by employing Ru-NiFe-PBA-NF and NiFe-PBA-NF as the electrodes for the cathodic HER and anodic UOR, respectively, which only requires a cell voltage of 1.36 V to deliver a current density of 10 mA cm−2, far superior to a conventional water electrolysis system. This work provides a novel way to design advanced organic–inorganic hybrid-based electrodes and innovative water electrolysis systems for efficient hydrogen production.
1. Introduction
Replacing traditional non-renewable fossil fuels with environmentally friendly and sustainable energy sources is expected to alleviate the increasingly serious environmental and energy dilemma, which is of great significance to achieve the sustainable development of human society.1–4 Among all alternatives, hydrogen is considered as the best choice due to its high energy density and zero carbon emission features.5–8 Different from traditional thermocatalytic strategies, electrocatalytic water splitting driven by sustainable energy including solar and wind is more in line with the modern concept of green hydrogen production.9It is well-known that a typical water electrolysis process involves two half-reactions, i.e., the hydrogen evolution reaction (HER) at the cathode and the oxygen evolution reaction (OER) at the anode.10,11 Generally, the sluggish four-electron transfer kinetics and large overpotentials of the OER make it a bottleneck reaction in water electrolysis.12,13 The most straightforward approach to address this issue is to develop efficient OER electrocatalysts.14,15 However, the high thermodynamic potential (i.e., 1.23 V vs. reversible hydrogen electrode (RHE)) of the OER makes it an insurmountable moat to substantially reduce the electric energy consumption of water electrolysis.16 Alternatively, replacing the anodic OER with the thermodynamically favorable urea oxidation reaction (UOR) in a water electrolysis system is expected to significantly reduce the energy input for H2 production while enabling the treatment of urea-rich wastewater, since the thermodynamic potential (0.37 V vs. RHE) of the UOR is much lower compared to the OER.17–21 Nevertheless, the key to improving the performance of a UOR-coupled water electrolysis system remains to be the development of efficient yet cost-effective electrocatalysts for the UOR.
Recently, metal–organic frameworks (MOFs) have been widely used in the field of electrochemistry because of their high surface areas, ordered porous structures, and easily tunable composition and morphology.22–26 However, the Achilles heel of MOFs as electrocatalysts is their poor conductivity and stability. Moreover, most MOFs prepared by conventional methods are in powder form, making organic binders indispensable during the fabrication of working electrodes, which will lead to partial coverage of active sites, and is thus detrimental to the electrocatalytic performance.27,28 Alternatively, direct growth of MOFs on conductive substrates with intimate contact not only facilitates charge transfer but also exposes more accessible active sites, thus leading to enhanced electrochemical performance.29 Although several effective strategies including solvothermal self-assembly have been reported to fabricate MOF-based self-supporting electrodes with significantly promoted electrocatalytic performance,30–32 the complex and tedious synthetic conditions with low controllability in most cases limit their large-scale application.33
Compared with conventional strategies, electrochemical deposition (ECD) techniques including anodic electrodeposition (AED) and cathodic electrodeposition (CED) have been proven to be environmentally friendly and time-saving, and show precise controllability and easy scalability, thus showing great potential in fabricating MOF-based self-supporting electrodes.34–37 More notably, the metal ion sources required for the construction of MOFs can be directly provided from their own metal substrate by the applied electric field due to electrochemical oxidization. Thereafter, the generated metal ions will further react with organic ligands to form MOFs and deposit on the surface of the substrate. For example, by precisely controlling the electrochemical conditions such as the applied potential and reaction time, Liu et al. realized the in situ growth of large-area Cu3(HHTP)2 (HHTP: 2,3,6,7,10,11-hexahydroxytriphenylene) MOF films on Cu (100) foil.38 Moreover, other Cu-MOF films using different ligands can also be synthesized through a similar growth strategy, confirming the effectiveness and universality of this strategy.39 On the other hand, it has been reported that Ni-based catalysts are the most attractive candidates for the UOR under alkaline conditions.40 Therefore, it is appealing to fabricate Ni-MOF-based self-supporting electrodes with excellent UOR performance via ECD techniques. Unfortunately, to the best of our knowledge, there are few reports on the synthesis of Ni-MOFs through electrochemical technologies so far.
Herein, a self-supporting NiFe-MOF-based nanocomposite electrode (NiFe-PBA-NF) composed of ultrasmall NiFe-based Prussian blue analogue (PBA) nanoparticles was prepared for the first time via a semi-sacrificial AED strategy, during which nickel foam (NF) was used as both the Ni source and the substrate, while K3[Fe(CN)6] was used as both the Fe source and the ligand. Notably, the composition and morphology of the NiFe-PBA-NF electrode and its corresponding electrochemical performance can be easily tuned by adjusting the AED conditions (e.g., applied potential and reaction time). When used directly as the working electrode for the UOR, the optimal one displayed excellent performance with a required applied potential of only 1.375 V to deliver a current density of 100 mA cm−2, which is superior to powdered NiFe-PBA and even commercial RuO2. Moreover, the Ru-NiFe-PBA-NF electrode with excellent HER activity was fabricated through further Ru-modification on the NiFe-PBA-NF electrode, in which ultrathin Ru-doped NiFe-PBA nanosheets were uniformly and densely distributed on the NF surface. Consequently, the UOR-coupled water electrolysis system assembled with Ru-NiFe-PBA-NF and NiFe-PBA-NF as the cathode and the anode, respectively, only needs a cell voltage of 1.36 V to deliver a current density of 10 mA cm−2, which is 102 mV lower compared with that for a conventional water electrolysis system.
2. Experimental section
2.1 Chemicals
Hydrochloric acid (HCl, ∼37 wt%), potassium ferricyanide(III) (K3[Fe(CN)6], ≥ 99.5%), KOH (85%), urea, and anhydrous ethanol were purchased from Sinopharm Chemical Reagent Co. Ltd. Ruthenium trichloride (RuCl3, 99%, 48.2% Ru) was bought from Energy Chemical Co. Ltd. Nickel foam (NF) was purchased from Kunshan Jiayisheng Electronics Co., Ltd. All chemicals were used directly without any further purification. Deionized water (DI) was used in all experiments.2.2 Fabrication of the NiFe-PBA-NF electrode
The self-supporting NiFe-PBA-NF electrode was fabricated by a semi-sacrificial electrodeposition (AED) strategy, where nickel foam (NF) was employed as both the Ni source and the substrate, while K3[Fe(CN)6] was used as both the Fe source and the ligand. Typically, a piece of NF with a size of 2.0 × 3.0 cm2 was first treated with HCl solution (1.0 m) and then washed with deionized water and ethanol several times. After vacuum drying at 60 °C, the pre-treated NF was collected for further use. The electrochemical fabrication of NiFe-PBA-NF was conducted in a three-electrode system with Ni foam (2.0 × 3.0 cm2) as the working electrode and a Pt mesh (1.0 × 1.0 cm2) and a saturated Ag/AgCl electrode as the counter and reference electrodes, respectively. 0.025 m K3[Fe(CN)6] solution was used as the electrolyte. The growth of NiFe-PBA-NF was achieved through a potentiostatic method with the appropriate potential and reaction time. Without a specific method, the sample named as NiFe-PBA-NF was fabricated at 3.0 V vs. Ag/AgCl for 3000 s. The mass loading of NiFe-PBA on NF was about 3.65 mg cm−2.2.3 Preparation of the Ru-NiFe-PBA-NF electrode
The Ru-NiFe-PBA-NF self-supporting electrode was prepared by a Ru-decorating treatment on the as-prepared NiFe-PBA-NF. Typically, 5.0 mg RuCl3 was first dissolved in a mixed solution containing 14.0 mL ethanol and 1.0 mL HCl solution (1.0 m). Then the above solution was transferred into a Teflon-lined autoclave (25 mL). After immersing a piece of the as-prepared NiFe-PBA-NF (2.0 × 3.0 cm2), the autoclave was sealed and heated at 120 °C for 12 h. After naturally cooling the sample to room temperature, it was taken out, washed with deionized water and ethanol several times, and finally dried at 60 °C overnight. Next, the pre-treated NiFe-PBA-NF was immersed in 20.0 mL methanol containing 160.0 mg NaBH4 under vigorous stirring for 30 min. Finally, the obtained Ru-NiFe-PBA-NF electrode was collected, washed with methanol several times, and dried under vacuum at room temperature.2.4 Preparation of NiFe-PBA@NF, RuO2@NF and Pt/C@NF electrodes
The NiFe-PBA@NF, RuO2@NF and Pt/C@NF electrodes were prepared as follows: 5.0 mg NiFe-PBA-p, commercial RuO2 and 20 wt% Pt/C were dispersed in a solution containing 0.45 mL H2O, 0.5 mL isopropyl alcohol, and 0.05 mL 5 wt% Nafion solution with ultrasonic treatment for 1 h. Then, 0.1 mL of the ink was dropped onto the surface of a piece of NF with a size of 1.0 × 1.0 cm2 and this was then dried at room temperature.2.5 Physical characterization
The powder X-ray diffraction (PXRD) patterns of the samples were recorded on a Rigaku MiniFlex 600 benchtop X-ray diffraction instrument with Cu Kα radiation. Scanning electron microscopy (SEM) images of the samples were collected using a Carl Zeiss Sigma 300 instrument. Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images of the samples were collected by using a FEI Tecnai G2 F30 instrument. X-ray photoelectron spectroscopy (XPS) spectra were obtained using a Thermo Fisher ESCALAB 250Xi X-ray photoelectron spectrometer with monochromatic Al Kα radiation (E = 1486.2 eV), and the binding energies were calibrated using C 1s to 284.8 eV. Raman spectra were recorded on a LabRAM HR Raman microscope with a 325 nm laser.2.6 Electrochemical characterization
The OER, UOR and HER measurements were performed in a three-electrode cell connected to an electrochemical workstation (CHI 760E), in which a saturated Ag/AgCl electrode and a Pt mesh (1.0 × 1.0 cm2) were used as the reference electrode and the counter electrode, respectively. The as-prepared electrodes with a size of 1.0 × 1.0 cm2 were directly used as the working electrodes. 1.0 m KOH with or without 0.33 m urea was used as the electrolyte. Linear sweep voltammetry (LSV) and cyclic voltammetry (CV) curves were collected at a scan rate of 5 mV s−1. All the measured potentials in this work were converted to reversible hydrogen electrodes (RHEs): ERHE = EAg/AgCl + 0.059 × pH + 0.197 (V). Without a specific description, all electrochemical data presented in this work were corrected with 90% iR-compensation. The electrochemical impedance spectra (EIS) in this work were recorded with the frequency ranging from 0.05 to 105 Hz at an AC amplitude of 5 mV.The electrochemical performances of UOR||HER and OER||HER were evaluated in a two-electrode cell, in which NiFe-PBA-NF and Ru-NiFe-PBA-NF were used as the anode and the cathode, respectively. 1.0 m KOH with or without 0.33 m urea was used as the electrolyte.
3. Results and discussion
As displayed in Scheme 1, the self-supporting NiFe-PBA-NF electrode was fabricated via a facile semi-sacrificial AED strategy, where NF was employed as both the Ni source and the substrate, while K3[Fe(CN)6] was used as both the Fe source and the ligand. The powder X-ray diffraction (PXRD) patterns (Fig. 1a) and Raman spectra (Fig. 1b) of the as-fabricated NiFe-PBA-NF electrode and powdered NiFe-PBA (NiFe-PBA-p) are all consistent with the typical NiFe-PBA (i.e., Ni2Fe(CN)6) reported in the literature,18 indicating the successful formation of Ni2Fe(CN)6 on the NF surface, which was further evidenced by the color change from the argenteus with metal luster to dull yellow (Fig. S1†). Moreover, scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were employed to reveal the microstructure of NiFe-PBA-NF. As can be observed in Fig. 1c and d and S1,† irregular cubic nanoparticles with a size of about 30–50 nm grew uniformly on the NF surface. Meanwhile, lattice fringes with a lattice spacing of 0.577 nm were observed in high-resolution TEM (HRTEM, inset in Fig. 1d), indicating the existence of Ni2Fe(CN)6 crystals, which is consistent with the PXRD patterns and Raman spectra. Additionally, the SEM energy-dispersive X-ray (EDX) spectra (Fig. S2†) illustrate that the Ni/Fe ratio is 8.27 in NiFe-PBA-NF. SEM–EDX elemental mapping images (Fig. S3†) show that C, O, N, Fe and Ni are uniformly distributed throughout the entire NiFe-PBA-NF, demonstrating the uniform growth of the Ni2Fe(CN)6 nanocrystals on the NF surface.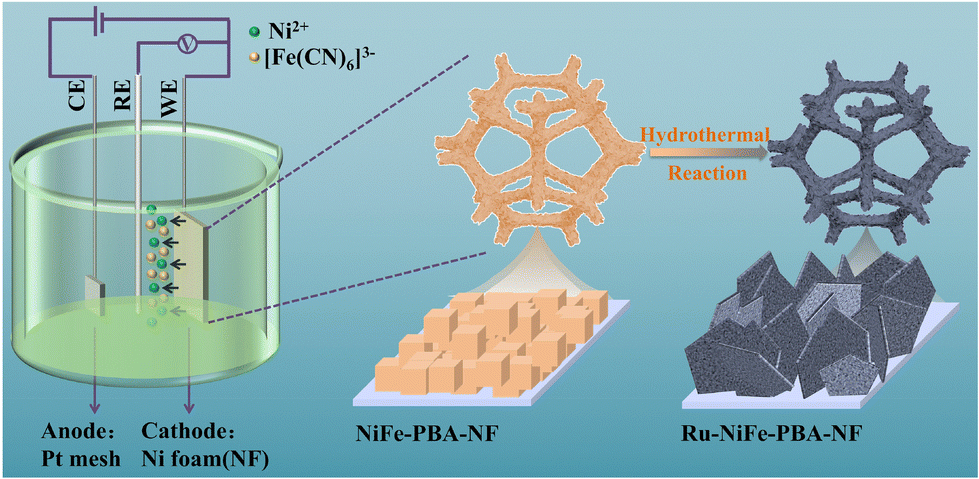 | ||
| Scheme 1 Schematic illustration of the fabrication of the NiFe-PBA-NF and Ru-NiFe-PBA-NF electrodes. | ||
 | ||
| Fig. 1 (a) PXRD patterns and (b) Raman spectra of NiFe-PBA-NF and NiFe-PBA-p. (c) SEM and (d) TEM images of NiFe-PBA-NF (inset in (d) is the HRTEM image). (e) N 1s and (f) Ni 2p spectra of NiFe-PBA-NF and NiFe-PBA-p. | ||
Besides its overall composition and morphology, the surface chemical composition and electronic states of NiFe-PBA-NF were further analyzed by X-ray photoelectron spectroscopy (XPS). As shown in the survey spectra (Fig. S16a†), both NiFe-PBA-NF and NiFe-PBA-p are composed of C, N, O, Fe and Ni. In the N 1s spectrum (Fig. 1e), the peak at 398.0 eV is assigned to the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond of the CN− species.41 Characteristic Ni 2p peaks in the Ni 2p spectrum (Fig. 1f) appeared at 856.3 eV (Ni 2p3/2) with a satellite peak at 861.5 eV, indicating their 2+ oxidation state. Meanwhile, another peak at 858.6 eV can be attributed to Ni3+ or the Ni–OH bond.29,42 Moreover, the two peaks at around 708.9 and 721.7 eV in the Fe 2p spectrum (Fig. S16b†) demonstrate their +2 oxidation state.43 Notably, the binding energies of the peaks corresponding to the CN bond, Ni2+ and Fe2+ in NiFe-PBA-NF and NiFe-PBA-p are almost the same, which further confirms the successful growth of NiFe-PBA on the NF surface. Since Ni2+ originates from the electrochemical oxidation of NF, the in situ formed Ni2Fe(CN)6 crystals can tightly adhere to the substrate, which is beneficial to enhance the electrochemical performance.
N bond of the CN− species.41 Characteristic Ni 2p peaks in the Ni 2p spectrum (Fig. 1f) appeared at 856.3 eV (Ni 2p3/2) with a satellite peak at 861.5 eV, indicating their 2+ oxidation state. Meanwhile, another peak at 858.6 eV can be attributed to Ni3+ or the Ni–OH bond.29,42 Moreover, the two peaks at around 708.9 and 721.7 eV in the Fe 2p spectrum (Fig. S16b†) demonstrate their +2 oxidation state.43 Notably, the binding energies of the peaks corresponding to the CN bond, Ni2+ and Fe2+ in NiFe-PBA-NF and NiFe-PBA-p are almost the same, which further confirms the successful growth of NiFe-PBA on the NF surface. Since Ni2+ originates from the electrochemical oxidation of NF, the in situ formed Ni2Fe(CN)6 crystals can tightly adhere to the substrate, which is beneficial to enhance the electrochemical performance.
According to previous studies, the growth of NiFe-PBA on the NF surface should be affected by AED conditions including the applied potential and reaction time.34 As shown in Fig. S4a and b,† at low applied potentials (e.g., 2.0 and 2.5 V), only a few nanoparticles grew randomly on the NF surface due to the low generated Ni2+concentration. Afterward, the Ni2+concentration gradually increased as the applied potential increased, thereby accelerating the nucleation and growth of NiFe-PBA nanoparticles. As a result, dense NiFe-PBA nanoparticles were tightly and uniformly grown on the NF surface (Fig. S4c and d†). However, overgrowth of NiFe-PBA nanoparticles at a too high applied potential (4.0 V) leads to the cracking of the uniform and dense MOF layer (Fig. S4e†), which may be detrimental to the electrocatalytic performance. Moreover, it is unsurprising that the reaction time has a similar effect on MOF growth to the applied voltage. As shown in Fig. S5b,† a too short reaction time (1000 s) results in incomplete formation of a dense MOF layer, while a too long reaction time (5000 s) leads to the destruction of the MOF layer (Fig. S5d†). Additionally, the SEM–EDX spectra and SEM–EDX elemental mapping images show that C, O, N, Fe, and Ni are all uniformly distributed throughout the as-prepared NiFe-PBA-NF electrodes with different applied potentials and reaction times (Fig. S2, S3 and S6–S13†). Meanwhile, the Ni/Fe ratio decreases with the increase of applied potential and reaction time (Fig. S14†), further demonstrating the gradual growth of NiFe-PBA nanoparticles, which corresponds to the result mentioned above. Furthermore, the electrochemical performance of the as-prepared NiFe-PBA-NF electrodes was determined in 1.0 m KOH with 0.33 M urea. From the results in Fig. S15,† the NiFe-PBA-NF electrode obtained by electrolysis at 3.0 V for 3000 s displayed the best UOR activity. Therefore, an appropriate applied potential and reaction time are critical for the fabrication of a uniform and robust self-supporting NiFe-PBA-NF electrode with optimal electrochemical performance.
The electrocatalytic OER performance of the as-prepared electrodes was first studied in 1.0 m KOH. As shown in Fig. 2a and S17 and 18a,† a pair of redox peaks corresponding to the Ni2+/Ni3+ couple was clearly observed between 1.0 and 1.5 V. Meanwhile, NiFe-PBA-NF displayed the best OER activity with required overpotentials of only 266 and 338 mV to deliver current density (j) values of 10 and 100 mA cm−2, respectively, outperforming NiFe-PBA@NF (281 and 392 mV) and even the benchmark RuO2@NF catalyst (302 and 427 mV). Further measurements revealed that NiFe-PBA-NF exhibits a lower Tafel slope compared with other electrodes (Fig. S18b†), indicating the improved reaction kinetics, which can greatly improve the OER performance. More notably, when 0.33 m urea was added, accompanied by the disappearance of the oxidation peak, the current densities for all electrodes increased significantly (Fig. 2b and S17†), demonstrating the occurrence of the UOR. Meanwhile, it is worth noting that the onset potentials of the UOR are near those of the oxidation peaks, implying that the UOR should be catalyzed by the in situ generated Ni3+ species, which is consistent with previous studies.18 Therefore, the larger peak area of the oxidation peak in NiFe-PBA-NF would contribute to better UOR activity. As a result, NiFe-PBA-NF exhibited the best UOR activity with required potentials of only 1.339 and 1.375 V to deliver j = 10 and 100 mA cm−2, respectively, which were much lower than those of NiFe-PBA@NF (1.373 and 1.464 V), RuO2@NF (1.369 and 1.623 V), and the most recently reported noble-metal free electrocatalysts (Table S1†), making the as-prepared NiFe-PBA-NF one of the most active MOF-based electrodes for the UOR.
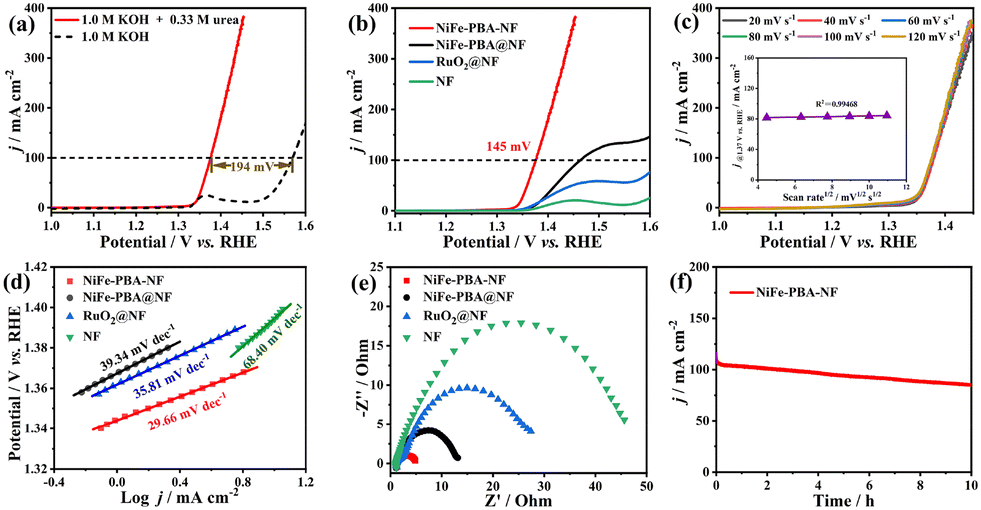 | ||
| Fig. 2 (a) LSV curves of NiFe-PBA-NF in 1.0 m KOH with and without 0.33 m urea. (b) LSV curves of NiFe-PBA-NF, NiFe-PBA@NF, RuO2@NF and NF in 1.0 m KOH with 0.33 m urea. (c) LSV curves of NiFe-PBA-NF in 1.0 m KOH with 0.33 m urea at scan rates from 20 to 120 mV s−1 (the inset shows the relationship between the current density and the square root of the scan rate at 1.37 V vs. RHE). (d) Tafel plots and (e) electrochemical impedance plots for NiFe-PBA-NF, NiFe-PBA@NF, RuO2@NF and NF in 1.0 m KOH with 0.33 m urea. (f) Long-term stability test for NiFe-PBA-NF at 1.48 V in 1.0 m KOH with 0.33 m urea. | ||
To gain more insight into the enhanced UOR activity, the reaction kinetics were first investigated. As shown in Fig. 2c, the nearly overlapping LSV curves collected at scanning rates from 20 to 120 mV s−1 imply that the UOR is a kinetic controlled process. Therefore, the corresponding Tafel plots were obtained to further analyze the reaction kinetics. As shown in Fig. 2d, NiFe-PBA-NF exhibited a lower Tafel slope (29.66 mV dec−1) compared with NiFe-PBA@NF (39.34 mV dec−1), RuO2@NF (35.81 mV dec−1), and NF (68.40 mV dec−1), suggesting its favorable UOR kinetics. Meanwhile, the charge-transfer kinetics were studied by electrochemical impedance spectroscopy (EIS), and the collected EIS plots are shown in Fig. 2e. Notably, the calculated charge-transfer resistance for NiFe-PBA-NF is much smaller than those of other electrodes, which would accelerate the electrocatalytic process. Furthermore, the measurement of electrochemically active surface areas (ECSAs) reveals that NiFe-PBA-NF possesses more exposed active sites with higher intrinsic activity compared with other electrodes (Fig. S19†). Consequently, the improved charge-transfer and reaction kinetics, as well as the adequate exposure of active sites with high intrinsic activity, synergistically endow NiFe-PBA-NF with excellent UOR performance. Additionally, control experiments show that the concentration of urea has an obvious effect on the UOR activity, where 0.33 M is the optimal value in this work (Fig. S20†).
Besides exhibiting excellent UOR activity, NiFe-PBA-NF also showed pretty good long-term stability. Specifically, it can deliver j = ∼100 mA cm−2 for more than 10 h (Fig. 2f). The gradually decreasing current density should be ascribed to the gradually decreasing urea concentration during continuous electrolysis. At the same time, the morphology and structure of NiFe-PBA-NF after a long-term stability test were further analyzed. As shown in Fig. S21,† the SEM and TEM images show inconspicuous changes after the stability test, implying a robust morphology. However, the drastically changed PXRD patterns and Raman and XPS spectra indicate the complete structural evolution of NiFe-PBA (Fig. S22 and S23). In particular, the signal of Fe 2p XPS sharply decreased, which may be due to the dissociation of ferricyanide ligands from NiFe-PBA during the UOR process. Therefore, we speculate that the in situ generated Fe-doped Ni(OH)2/NiOOH should be the real active species for the UOR, which has been extensively demonstrated previously.26,29
Apart from the OER and UOR in the anode, the electrocatalytic activity for the cathodic HER of NiFe-PBA-NF was also evaluated in alkaline electrolyte. Unfortunately, the HER performance of NiFe-PBA-NF is far behind that of commercial Pt/C (Fig. S24†). As a remedy, the Ru-decorated NiFe-PBA-NF electrode (Ru-NiFe-PBA-NF) was fabricated accordingly, since Ru has a comparable HER activity to Pt in alkaline electrolyte yet is much cheaper.42 Notably, the Ru content in Ru-NiFe-PBA-NF was only 0.79 wt% as determined by inductively coupled plasma atomic emission spectrometry (ICP-AES). The PXRD patterns (Fig. 3a) and Raman spectra (Fig. 3b) indicate that Ru-decorating has no effect on the structure of NiFe-PBA. Meanwhile, the diffraction peaks belonging to Ru-based species are not observed in the PXRD pattern, which may due to the low content and good dispersion of Ru species. Moreover, it is unexpectedly found that ultrathin Ru-decorated NiFe-PBA nanosheets are uniformly anchored on the NF surface, which is quite different from the parent nanoparticulate NiFe-PBA (Fig. 3c and S25†), demonstrating the occurrence of structural reorganization of NiFe-PBA during the Ru-decorating process. The SEM–EDX elemental mapping images show the even distribution of Ni, Ru and Fe throughout the entire Ru-NiFe-PBA-NF (Fig. S26†), illustrating the uniform dispersion of Ru species in Ru-NiFe-PBA-NF. The TEM and HRTEM images (Fig. 3d) show that the lattice spacing of Ru-NiFe-PBA-NF (0.577 nm) is the same as that of NiFe-PBA-NF, implying that the structure of NiFe-PBA remains unchanged after Ru decoration, corresponding to the result of the PXRD pattern. Meanwhile, no obvious metallic or oxidized Ru-based nanoparticles were observed (Fig. 3d and S27†), implying that Ru-based species may exist in Ru-NiFe-PBA-NF in the form of single atoms or ultrasmall clusters. Moreover, from the Ru 3p spectrum of Ru-NiFe-PBA-NF shown in Fig. 3e, the peaks at 462.3 and 484.5 eV are attributed to Ru0, while the peaks at 465.2 and 486.6 eV belong to the Rux+ species, indicating the co-existence of metallic and oxidized Ru species.44 Furthermore, the Ni 2p (Fig. 3f) and Fe 2p (Fig. S28b†) spectra of Ru-NiFe-PBA-NF were also collected. Compared with NiFe-PBA-NF, both Ni 2p and Fe 2p spectra in Ru-NiFe-PBA-NF shifted towards higher binding energy after Ru decoration, demonstrating that partial electrons were transferred from Ni and Fe sites to Ru sites, which will regulate the local electronic structure of the active sites, thereby enhancing the electrochemical performance.
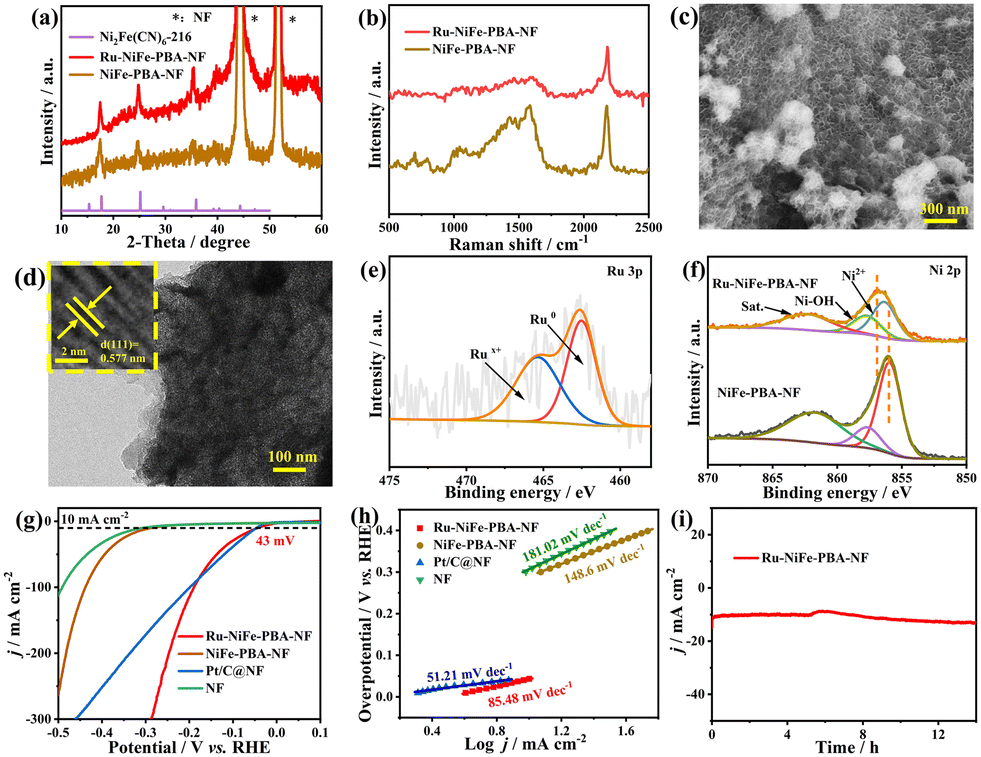 | ||
| Fig. 3 (a) PXRD patterns and (b) Raman spectra of NiFe-PBA-NF and Ru-NiFe-PBA-NF. (c) SEM and (d) TEM and HRTEM images of Ru-NiFe-PBA-NF. (e) Ru 3p spectra of Ru-NiFe-PBA-NF. (f) Ni 2p spectra of NiFe-PBA-NF and Ru-NiFe-PBA-NF. (g) LSV curves and the corresponding (h) Tafel plots of Ru-NiFe-PBA-NF, NiFe-PBA-NF, Pt/C@NF and NF in 1.0 m KOH with 0.33 m urea. (i) Long-term stability test for Ru-NiFe-PBA-NF at −0.12 V in 1.0 m KOH with 0.33 m urea. | ||
Accordingly, the HER performance of Ru-NiFe-PBA-NF was investigated in 1.0 m KOH with 0.33 m urea. Based on the preliminary experiment, the presence of urea in the electrolyte has little effect on the HER activity of Ru-NiFe-PBA-NF (Fig. S29†). As shown in Fig. 3g, Ru-NiFe-PBA-NF exhibited a boosted HER activity with an ultralow overpotential of 43 mV to deliver j = 10 mA cm−2, which is far superior to NiFe-PBA-NF (286 mV) and NF (306 mV) and comparable to commercial Pt/C (46 mV). More impressively, the needed overpotential to deliver j > 100 mA cm−2 for Ru-NiFe-PBA-NF is even much lower compared with Pt/C, indicating its excellent HER activity. Fig. 3h shows the Tafel plots for the as-prepared electrodes based on their corresponding LSV curves displayed in Fig. 3g. It can be clearly observed that the Tafel slope of Ru-NiFe-PBA-NF is much lower compared with NiFe-PBA-NF and NF within the whole test potential region (Fig. 3h and S30†), indicating the superior HER kinetics for Ru-NiFe-PBA-NF. Notably, the Tafel slope of Ru-NiFe-PBA-NF (85.48 mV dec−1) is slightly higher than that of Pt/C (51.21 mV dec−1) within a low-overpotential region of 0 to 0.05 V. However, within the high-overpotential region larger than 0.18 V (Fig. S30†), the derived Tafel slope for Ru-NiFe-PBA-NF (182.69 mV dec−1) is much lower than that for Pt/C (314.62 mV dec−1), illustrating that Pt/C has more excellent HER kinetics under low j values, while it is inferior to Ru-NiFe-PBA-NF at relatively high j values. Furthermore, as displayed in the EIS plots (Fig. S31a†), the semicircle diameter of Ru-NiFe-PBA-NF is close to that of Pt/C and is much smaller than those of NiFe-PBA-NF and NF, demonstrating the improved charge-transfer kinetics, which is helpful to promote HER performance. Moreover, the measurement of ESCAs shows that Ru-NiFe-PBA-NF is able to expose more accessible active sites with higher intrinsic activity than NiFe-PBA-NF and NF and even Pt/C (Fig. S31b and S32†), which may originate from the synergistic effect of Ru-decorating and the restructured nanosheet morphology. Besides having a notable activity, Ru-NiFe-PBA-NF also showed excellent long-term stability during the HER process. As shown in Fig. 3i, the value of j = ∼10 mA cm−2 can be maintained well without obvious decay during the long-term (>14 h) electrolysis.
Encouraged by the excellent UOR and HER performances of NiFe-PBA-NF and Ru-NiFe-PBA-NF, respectively, an undivided advanced hybrid water electrolysis system (UOR||HER) was constructed by replacing the anodic OER with the UOR, in which NiFe-PBA-NF and Ru-NiFe-PBA-NF were used as the anode and the cathode, respectively. Fig. 4a shows the electrochemical performance of UOR||HER and the conventional water electrolysis system (OER||HER) over the NiFe-PBA-NF||Ru-NiFe-PBA-NF pair. Impressively, it requires only 1.36 V to deliver a current density of 10 mA cm−2 for UOR||HER, which is 0.25 V lower than that for OER||HER (1.61 V), demonstrating the effectiveness of replacing the anodic OER with the UOR to facilitate H2 production in water electrolysis systems. Meanwhile, the UOR ||HER performance of the RuO2@NF || Pt/C@NF pair was measured for comparison. As displayed in Fig. 4b and c, the RuO2@NF||Pt/C@NF pair requires an undesirably higher energy input to output the same current density compared with the NiFe-PBA-NF||Ru-NiFe-PBA-NF pair, especially at high current density. For example, the cell voltages are 1.927 and 2.256 V to deliver current densities of 100 and 200 mA cm−2 for the NiFe-PBA-NF||Ru-NiFe-PBA-NF pair, respectively, which are 564 and 740 mV lower than those for the RuO2@NF||Pt/C@NF pair, respectively. To the best of our knowledge, this performance is superior to most recently reported hybrid water electrolysis systems (including UOR ||HER) by using noble-metal free electrocatalysts as electrodes (Table S2†). Besides having a gratifying activity, the NiFe-PBA-NF||Ru-NiFe-PBA-NF pair also presents fairly good long-term stability toward UOR||HER. As shown in Fig. 4d, it can maintain an output current density of 10 mA cm−2 for 24 h without significant decay, implying its potential application in hybrid water electrolysis devices.
 | ||
| Fig. 4 (a) LSV curves of the constructed OER||HER and UOR||HER cells based on the NiFe-PBA-NF||Ru-NiFe-PBA-NF pair. (b) LSV curves and (c) comparison of the needed cell voltages at different current densities of the NiFe-PBA-NF||Ru-NiFe-PBA-NF and RuO2@NF||Pt/C@NF pairs in the UOR||HER cell. (d) Long-term stability test for the NiFe-PBA-NF||Ru-NiFe-PBA-NF (1.36 V) and RuO2@NF||Pt/C@NF (1.45 V) pairs in the UOR||HER cell. | ||
Conclusions
In summary, a self-supporting NiFe-MOF-based nanocomposite electrode with a tunable composition and morphology was fabricated via an electrochemical strategy, in which ultrasmall NiFe-PBA nanoparticles are tightly anchored on the NF surface. Notably, the as-prepared NiFe-PBA-NF electrode exhibited an excellent UOR performance superior to the powdered NiFe-PBA and even commercial RuO2. Further coupling the NiFe-PBA-NF anode with the Ru-NiFe-PBA-NF cathode in a UOR-coupled water electrolysis system enables efficient hydrogen production while greatly reducing the energy input compared with a conventional water electrolysis system. This work provides an attractive way for the design of integrated MOF-based electrodes and novel water electrolysis systems for efficient hydrogen production.Author contributions
J. J., C. C. and Q.-L. Z. conceived the research and designed the experiments. Y. W. helped collect the electrochemical test data. J. J., C. C., X.-T. W., and Q.-L. Z. analyzed the data and drafted the manuscript. The manuscript was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (22105203 and 22175174), the Natural Science Foundation of Fujian Province (2020J01116, 2021J06033 and 2022L3092) and the China Postdoctoral Science Foundation (2021TQ0332 and 2021M703215).References
- M. S. Dresselhaus and I. L. Thomas, Nature, 2001, 414, 332–337 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7–7 CrossRef CAS PubMed.
- F. Luo, Q. Zhang, X. Yu, S. Xiao, Y. Ling, H. Hu, L. Guo, Z. Yang, L. Huang, W. Cai and H. Cheng, Angew. Chem., Int. Ed., 2018, 57, 14862–14867 CrossRef CAS PubMed.
- Y.-L. Wu, X. Li, Y.-S. Wei, Z. Fu, W. Wei, X.-T. Wu, Q.-L. Zhu and Q. Xu, Adv. Mater., 2021, 33, 2006965 CrossRef CAS PubMed.
- X. Meng, C. Ma, L. Jiang, R. Si, X. Meng, Y. Tu, L. Yu, X. Bao and D. Deng, Angew. Chem., 2020, 132, 10588–10593 CrossRef.
- K.-J. Jeon, H. R. Moon, A. M. Ruminski, B. Jiang, C. Kisielowski, R. Bardhan and J. J. Urban, Nat. Mater., 2011, 10, 286–290 CrossRef CAS PubMed.
- F.-H. Yuan, M.-R. Mohammadi, L.-L. Ma, Z.-D. Cui, S.-L. Zhu, Z.-Y. Li, S.-L. Wu, H. Jiang and Y.-Q. Liang, Rare Met., 2022, 41, 2624–2632 CrossRef CAS.
- C.-B. Hong, X. Li, W.-B. Wei, X.-T. Wu and Q.-L. Zhu, Appl. Catal., B, 2021, 294, 120230 CrossRef CAS.
- Y. Li, H. Wang, C. Priest, S. Li, P. Xu and G. Wu, Adv. Mater., 2021, 33, 2000381 CrossRef CAS PubMed.
- X. Cui, P. Ren, C. Ma, J. Zhao, R. Chen, S. Chen, N. P. Rajan, H. Li, L. Yu, Z. Tian and D. Deng, Adv. Mater., 2020, 32, 1908126 CrossRef CAS PubMed.
- G. Liu, B. Wang, L. Wang, W. Wei, Y. Quan, C. Wang, W. Zhu, H. Li and J. Xia, Green Energy Environ., 2022, 7, 423–431 CrossRef CAS.
- B. M. Hunter, H. B. Gray and A. M. Muller, Chem. Rev., 2016, 116, 14120–14136 CrossRef CAS PubMed.
- L. An, C. Wei, M. Lu, H. Liu, Y. Chen, G. G. Scherer, A. C. Fisher, P. Xi, Z. J. Xu and C.-H. Yan, Adv. Mater., 2021, 33, 2006328 CrossRef CAS PubMed.
- M. Yu, E. Budiyanto and H. Tuysuz, Angew. Chem., 2022, 61, e202103824 CAS.
- J. Abed, S. Ahmadi, L. Laverdure, A. Abdellah, C. P. O'Brien, K. Cole, P. Sobrinho, D. Sinton, D. Higgins, N. J. Mosey, S. J. Thorpe and E. H. Sargent, Adv. Mater., 2021, 33, 2103812 CrossRef CAS PubMed.
- J. Edgington, N. Schweitzer, S. Alayoglu and L. C. Seitz, J. Am. Chem. Soc., 2021, 143, 9961–9971 CrossRef CAS PubMed.
- S. J. Yao, S. K. Wolfson, B. K. Ahn and C. C. Liu, Nature, 1973, 241, 471–472 CrossRef CAS PubMed.
- S.-K. Geng, Y. Zheng, S.-Q. Li, H. Su, X. Zhao, J. Hu, H.-B. Shu, M. Jaroniec, P. Chen, Q.-H. Liu and S.-Z. Qiao, Nat. Energy, 2021, 6, 904–912 CrossRef CAS.
- L. Wang, Y. Zhu, Y. Wen, S. Li, C. Cui, F. Ni, Y. Liu, H. Lin, Y. Li, H. Peng and B. Zhang, Angew. Chem., 2021, 133, 10671–10676 CrossRef.
- Y. Zhang and C. Wang, Chin. Chem. Lett., 2021, 32, 2222–2228 CrossRef CAS.
- M. Yang, Y. Jiang, M. Qu, Y. Qin, Y. Wang, W. Shen, R. He, W. Su and M. Li, Appl. Catal., B, 2020, 269, 118803 CrossRef CAS.
- M. Li, H. Sun, J. Yang, M. Humayun, L. Li, X. Xu, X. Xue, A. Habibi-Yangjeh, K. Temst and C. Wang, Chem. Eng. J., 2022, 430, 132733 CrossRef CAS.
- S. Nangan, Y. Ding, A. Z. Alhakemy, Y. Liu and Z. Wen, Appl. Catal., B, 2021, 286, 119892 CrossRef CAS.
- C. Cao, D.-D. Ma, J. Jia, Q. Xu, X.-T. Wu and Q.-L. Zhu, Adv. Mater., 2021, 33, 2008631 CrossRef CAS PubMed.
- Y. Xu, M. Liu, M. Wang, T. Ren, K. Ren, Z. Wang, X. Li, L. Wang and H. Wang, Appl. Catal., B, 2022, 300, 120753 CrossRef CAS.
- M. Huang, C. Cao, L. Liu, W. Wei, Q.-L. Zhu and Z. Huang, eScience, 2023, 3, 100118 CrossRef.
- J. Nai, Y. Lu, L. Yu, X. Wang and X. W. Lou, Adv. Mater., 2017, 29, 1703870 CrossRef PubMed.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- C. Cao, D.-D. Ma, Q. Xu, X.-T. Wu and Q.-L. Zhu, Adv. Funct. Mater., 2019, 29, 1807418 CrossRef.
- W. Liu, R. Yin, X. Xu, L. Zhang, W. Shi and X. Cao, Adv. Sci., 2019, 6, 1802373 CrossRef PubMed.
- T. Rodenas, I. Luz, G. Prieto, B. Seoane, H. Miro, A. Corma, F. Kapteijn, F. X. Llabres i Xamena and J. Gascon, Nat. Mater., 2015, 14, 48–55 CrossRef CAS PubMed.
- F. Qiu, J. R. Edison, Z. Preisler, Y.-F. Zhang, G. Li, A. Pan, C.-H. Hsu, T. M. Mattox, P. Ercius, C. Song, K. Bustillo, M. A. Brady, E. W. Zaia, S. Jeong, J. B. Neaton, S. Du, S. Whitelam and J. J. Urban, Angew. Chem., 2018, 130, 13356–13360 CrossRef.
- S. Alizadeh and D. Nematollahi, J. Am. Chem. Soc., 2017, 139, 4753–4763 CrossRef CAS PubMed.
- X. Zhang, K. Wan, P. Subramanian, M. Xu, J. Luo and J. Fransaer, J. Mater. Chem. A, 2020, 8, 7569–7587 RSC.
- Q. Zhang, Z. Wu, Y. Lv, Y. Li, Y. Zhao, R. Zhang, Y. Xiao, X. Shi, D. Zhang, R. Hua, J. Yao, J. Guo, R. Huang, Y. Cui, Z. Kang, S. Goswami, L. Robison, K. Song, X. Li, Y. Han, L. Chi, O. K. Farha and G. Lu, Angew. Chem., Int. Ed., 2019, 58, 1123–1128 CrossRef CAS PubMed.
- X. Kang, K. Lyu, L. Li, J. Li, L. Kimberley, B. Wang, L. Liu, Y. Cheng, M. D. Frogley, S. Rudic, A. J. Ramirez-Cuesta, R. A. W. Dryfe, B. Han, S. Yang and M. Schroder, Nat. Commun., 2019, 10, 4466 CrossRef PubMed.
- X. Wang, P. She and Q. Zhang, SmartMat, 2021, 2, 299–325 CrossRef CAS.
- Y. Liu, Y. Wei, M. Liu, Y. Bai, X. Wang, S. Shang, J. Chen and Y. Liu, Angew. Chem., 2021, 60, 2887–2891 CrossRef CAS PubMed.
- X. Kang, L. Li, A. Sheveleva, X. Han, J. Li, L. Liu, F. Tuna, E. J. L. McInnes, B. Han, S. Yang and M. Schroder, Nat. Commun., 2020, 11, 5464 CrossRef CAS PubMed.
- R. M. A. Hameed and S. S. Medany, J. Colloid Interface Sci., 2018, 513, 536–548 CrossRef PubMed.
- X. Xiao, G. Zhang, Y. Xu, H. Zhang, X. Guo, Y. Liu and H. Pang, J. Mater. Chem. A, 2020, 7, 17266–17271 RSC.
- Y. Ma, Y. Ma, S. L. Dreyer, Q. Wang, K. Wang, D. Goonetilleke, A. Omar, D. Mikhailova, H. Hahn, B. Breitung and T. Brezesinski, Adv. Mater., 2021, 33, 2101342 CrossRef CAS PubMed.
- Z. Gao, Y. Li, C. Zhang, S. Zhang, Y. Jia and Y. Dong, Anal. Chim. Acta, 2020, 1097, 169–175 CrossRef CAS PubMed.
- P. Su, W. Pei, X. Wang, Y. Ma, Q. Jiang, J. Liang, S. Zhou, J. Zhao, J. Liu and G. Q. M. Lu, Angew. Chem., 2021, 133, 16180–16186 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ce00289f |
| This journal is © The Royal Society of Chemistry 2023 |