
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diborane heterolysis: breaking and making B–B bonds at magnesium†
Anne-Frédérique
Pécharman
,
Michael S.
Hill
 * and
Mary F.
Mahon
* and
Mary F.
Mahon
Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: msh27@bath.ac.uk
First published on 9th May 2018
Abstract
Reaction of the dimeric β-diketiminato magnesium hydride [(BDI)MgH]2 (BDI = HC{(Me)CN-2,6-i-Pr2C6H3}2) with bis-pinacolatodiborane (B2pin2) resulted in B–O bond activation and formation of a magnesium complex of an unusual borylborohydride anion. In contrast, similar treatment of the mononuclear organomagnesium [{BDI}Mg(n-Bu)] with 4,4,4′,4′,6,6′-hexamethyl-2,2′-bi(1,3,2-dioxaborinane) (B2hex2) provided a B(sp2)–B(sp3) diborane anion, [(hex)BB(n-Bu)(hex)]−, with a constitution which is analogous to that formed in the previously reported reaction with bis(pinacolato)diboron (B2pin2). Subsequent addition of 4-dimethylaminopyridine to a solution of this compound induced alkylborane displacement and provided a magnesium boryl derivative containing a terminal Mg–B(hex) interaction (Mg–B 2.319(3) Å), a result which reinforces the generality of this approach for the synthesis of boryl anions by B–B bond heterolysis. Further studies of the reactivity of the initially formed B(sp2)–B(sp3) anions with diborane small molecules also resulted in alkylborane displacement and the production of triboron anions, which are propagated by contiguous and electron precise (2c–2e) B–B–B interactions.
Introduction
The selective installation of boryl (–BX2) units into organic molecules is an important capability in contemporary chemical synthesis. The resultant C–B bonds provide a straightforward means to introduce further functionality, either through oxidation or their use in catalytic C–C bond formation through the application of Suzuki–Miyaura protocols.1–3 Boryl moieties are conventionally transferred to an organic substrate by a variety of routes (e.g. alkene hydroboration) which are reliant upon the Lewis acidic and electrophilic behaviour of the electropositive boron centre in reaction with an organic nucleophile. Although boryl ligands can be generated by oxidative addition of B–X (B = H, B or halide) bonds to transition metals,2,4 Yamashita and Nozaki's seminal report of the lithium boryl, [{HCN(Dipp)}2BLi(DME)] (1, Dipp = 2,6-di-iso-propylphenyl, DME = dimethoxyethane, Scheme 1) was widely recognised as the first true boron-centred nucleophile.5,6 The isolation of compound 1 requires strongly reducing and inconvenient reaction conditions, however, and its isolation is also crucially dependent on the high degree of kinetic stabilisation provided by sterically demanding substituents about the boron centre. While compound 1 has since been shown to display broad applicability in boron-to-element bond forming reactions with a palette of organic and metal-centred electrophiles,7–15 this latter structural feature dictates that some of these transformations display only limited specificity. Reactions of compound 1 with organohalides, RX (X = Cl, Br), for example, not only provide the expected products of nucleophilic substitution, [{HCN(Dipp)}2B-R], but also suffer from competitive halogen abstraction to give [{HCN(Dipp)}2B-X].16,17 | ||
| Scheme 1 Compound 1 and synthetic route to compounds 3–5. | ||
While a wide variety of alternative boron-centred nucleophiles have been described since the report of 1,18–36 a majority of routes to these species still require an alkali metal reduction step. With these issues in mind, we have recently reported that terminal magnesium boryl species may be easily generated by heterolysis of the B–B bond of commercially available bis(pincolato)diborane (B2pin2) within the coordination sphere of a β-diketiminato magnesium derivative (Scheme 1).37 Treatment of the magnesium n-butyl complex, [(BDI)Mg(n-Bu)] (2) (BDI = HC{(Me)CN-2,6-i-Pr2C6H3}2) with one equivalent of B2pin2 provided compound 3, which contains a diborane-derived anion in which one of the boron centres has been quaternised. This anion is strongly reminiscent of the variety of B(sp2)–B(sp3) adducts of B2pin2 with neutral or anionic donors that have been shown to act as viable sources of nucleophilic {Bpin} units.38–40 With this reactivity in mind, we have very recently shown that the {Bpin} moiety of compound 3 may be utilised to form electron-precise B–B bonds through reactions with the boron electrophiles Ph3B and borabicyclo[3.3.1]nonane (9-BBN) albeit, in common with the previously mentioned B(sp2)–B(sp3) adducts, these compounds are synthesised without the explicit generation of boryl anions.41 Reaction of compound 3 with an additional equivalent of B2pin2 also resulted in the displacement of n-BuBpin and the formation of an unusual derivative (4) of the catenated triboron [B3pin3]− anion, while treatment of either 3 or 4 with 4-dimethylaminopyridine (DMAP) provided a magnesium complex (5) which does contain the terminal [Bpin]− anion (Scheme 1). Initial assessment of the reactivity of compound 5 with both iodomethane and with non-halogenated organic electrophiles provided completely specific B–C bond formation and a definitive demonstration of its potential as a source of the [Bpin]− nucleophile.37 In this contribution we continue to explore the generality of these routes to magnesium boryl equivalents and catena-triborane anions.
Results and discussion
The molecular magnesium hydride [(BDI)MgH]2 (6) has played a central role in our development of a suite of catalytic bond activation processes.42,43 We, thus, carried out the reaction of compound 6 with B2pin2. Although the reaction performed in d8-toluene required brief heating to ensure the formation of a homogeneous solution, inspection of the resultant 1H and 13C{1H} NMR spectra revealed the emergence of two new sets of β-diketiminate ligand environments, which had been formed in a strict 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio by relative integration. Although the corresponding 11B{1H} NMR spectrum was broad and uninformative, the origin of these observations was resolved through the isolation of single crystals from the reaction solution. The resultant X-ray diffraction analysis revealed that compound 7 is the result of formal addition of both hydrides of the dimeric magnesium reagent (6) to a single boron centre within B2pin2 (Fig. 1). This process ensues with retention of the B–B bond and ring opening of one of the pinacolato boron heterocycles through rupture of the B2–O4 bond. The resultant diborane(5) anion bridges unsymmetrically between the two magnesium centres of the dimer, with consequent disruption of the C2v symmetry of the hydride precursor (6) that was apparent in the NMR spectra of 7. The new diborane anion coordinates to Mg1 through O1 and O3, which remain bound to B1 and B2, respectively, and to Mg2 via coordination by the now formal alkoxide donor O4 and μ2-BH2-Mg bridging interactions. Although the specific constitution of this diborane anion is unprecedented, the B1–B2 distance [1.713(2) Å] is closely comparable to the similar linkages within previously reported B(sp2)–B(sp3) bonded compounds.41,44
:1 ratio by relative integration. Although the corresponding 11B{1H} NMR spectrum was broad and uninformative, the origin of these observations was resolved through the isolation of single crystals from the reaction solution. The resultant X-ray diffraction analysis revealed that compound 7 is the result of formal addition of both hydrides of the dimeric magnesium reagent (6) to a single boron centre within B2pin2 (Fig. 1). This process ensues with retention of the B–B bond and ring opening of one of the pinacolato boron heterocycles through rupture of the B2–O4 bond. The resultant diborane(5) anion bridges unsymmetrically between the two magnesium centres of the dimer, with consequent disruption of the C2v symmetry of the hydride precursor (6) that was apparent in the NMR spectra of 7. The new diborane anion coordinates to Mg1 through O1 and O3, which remain bound to B1 and B2, respectively, and to Mg2 via coordination by the now formal alkoxide donor O4 and μ2-BH2-Mg bridging interactions. Although the specific constitution of this diborane anion is unprecedented, the B1–B2 distance [1.713(2) Å] is closely comparable to the similar linkages within previously reported B(sp2)–B(sp3) bonded compounds.41,44
 | ||
| Fig. 1 ORTEP representation of compound 7 (25% probability ellipsoids). Isopropyl methyl groups and carbon-bonded hydrogen atoms are removed for clarity. Selected bond lengths (Å) and angles (°): Mg1–O1 2.0899(11), Mg1–O3 1.9784(10), Mg1–N1 2.0560(13), Mg1–N2 2.0604(13), Mg2–O4 1.8527(11), Mg2–N3 2.0819(13), Mg2–N4 2.0892(13), B1–B2 1.713(2), O1–B1 1.4216(18), O2–C31 1.4563(17), O2–B1 1.356(2), O3–B2 1.5307(18), O3–Mg1–O1 90.07(4), O3–Mg1–N1 127.70(5), O3–Mg1–N2 124.49(5), N1–Mg1–O1 107.24(5), N1–Mg1–N2 93.72(5), O4–Mg2–N3 123.34(5), O4–Mg2–N4 117.15(5), O4–Mg2–B2 84.78(5), N3–Mg2–N4 90.49(5). | ||
We suggest that the B–O bond activation observed in the synthesis of compound 7 is a possible consequence of the initial dimeric structure of the hydride derivative 6. With this in mind, we returned our attention to the mononuclear magnesium n-butyl reagent, 2, to further investigate the generality of the synthetic approach illustrated for B2pin2 in Scheme 1. Reaction of an equimolar quantity of 2 with the alternative, but still commercially available, diborane reagent 4,4,4′,4′,6,6′-hexamethyl-2,2′-bi(1,3,2-dioxaborinane) (B2hex2) provided the colourless compound 8 after work-up in high (>90%) yield (Scheme 2). Although the 1H and 13C{1H} NMR spectra of 8 were complex, the corresponding 11B{1H} NMR spectrum was reminiscent of that observed for compound 3 in comprising two broad resonances at δ 30.5 and 4.0 ppm, consistent with the presence of three- and four-coordinate boron, respectively. This supposition was confirmed through the isolation of single crystals suitable for X-ray diffraction analysis, the results of which are illustrated in Fig. 2(a). The solid state structure confirmed that 8 is an analogue of compound 3 in which one of the boron centres (labelled as B2) has been quaternised by addition of an n-butyl group. The resultant anion coordinates to the 4-coordinate magnesium through O1 and O3, which are adjacent to the less sterically congested chiral monomethyl-substituted (C31 and C37) glycolato carbon atoms. The carbon centres of each glycolato chain were seen to exhibit disorder in a 55:45 ratio. Although the solution structure of compound 8 was not investigated any further, on this basis, we ascribe the complexity of the 1H and 13C NMR spectra of 8 to the persistence of several possible diastereomers in solution, arising from use of the racemic B2hex2 reagent.
 | ||
| Scheme 2 Synthesis of compounds 8 and 9. | ||
 | ||
| Fig. 2 ORTEP representations (25% probability ellipsoids) of (a) compound 8 and (b) compound 9. Isopropyl methyl groups are removed for clarity. Selected bond lengths (Å) and angles (°): (8) Mg1–O1 2.0466(14), Mg1–O3 1.9378(16), Mg1–N1 2.0550(16), Mg1–N2 2.0524(16), B1–B2 1.738(4), O1–Mg1–N1 118.56(7), O1–Mg1–N2 109.95(7), N2–Mg1–N1 94.34(7); (9) Mg1–N1 2.0832(19), Mg1–N2 2.0744(19), Mg1–N3 2.1422(18), Mg1–B1 2.319(3), N1–Mg1–N3 102.94(7), N1–Mg1–B1 133.12(8), N2–Mg1–N1 91.26(7), N2–Mg1–N3 106.23(7), N2–Mg1–B1 114.33(9), N3–Mg1–B1 106.09(8). | ||
Monitoring by 1H and 11B NMR spectroscopy of a sample of compound 8 treated with DMAP evidenced the generation of a single new β-diketiminato magnesium complex (9) and the elimination of the organoborane n-BuB(hex) (δ11B = 34.2 ppm). Although the 11B{1H} NMR spectrum was otherwise broad and uninformative, the constitution of compound 9 as a magnesium derivative of a terminal [(hex)B]− anion was confirmed through the isolation of single crystals from n-hexane solution at −35 °C. The structure of compound 9, shown in Fig. 2(b), is comparable to that of 5 and confirms the generality of the straightforward magnesium-centred B–B activation for the synthesis of terminally bound boryl anions. Like 5, compound 9 is a four-coordinate magnesium derivative with three of the magnesium to ligand contacts provided by the nitrogen atoms of the β-diketiminate ligand and a single unidentate DMAP ligand and with the final coordination site occupied by the sp2 boron donor. The Mg1–B1 distance [2.319(3) Å] of compound 9 is effectively identical to that determined for 5 [2.324(2) Å] and lies within the range observed in three reported magnesium derivatives synthesised by reactions of compound 1 with MgBr2 [2.281(6)–2.377(4) Å].7
Although a complete study of the behaviour of compounds 8 and 9 towards organic electrophiles will be described elsewhere, these structural and spectroscopic observations suggest the reactivity of both compounds should be comparable to those of the previously reported derivatives, 3 and 5. The facile reaction of compound 3 with B2pin2 to provide the triboron anion of compound 4 prompted us to assess the generality of this reaction to provide unusual homocatenated triboron species. Accordingly, toluene solutions of compound 3 were treated with equimolar quantities of B2hex2 and 5,5,5′,5′-tetramethyl-2,2′-bi(1,3,2-dioxaborinane) (B2neo2), respectively (Scheme 3). Examination of the resultant 1H NMR spectra after 2 hours at 40 °C evidenced, in both cases, the formation of two new β-diketiminato magnesium complexes, compounds 10 and 11. Both of the corresponding 11B{1H} NMR spectra were also observed to contain resonances at δ 34.2 ppm, consistent with elimination of n-BuBpin, which appeared alongside further broad signals ca. δ 30 and 1 ppm. These latter signals are reminiscent of the 11B resonances arising from compound 4 and, taken together, these data strongly imply the formation of similar homocatenated triborane anions.
 | ||
| Scheme 3 Synthesis of compounds 10 and 11. | ||
These deductions were corroborated by single crystal X-ray diffraction analysis, which was performed on crystals of compounds 10 and 11 isolated, in both cases, from concentrated n-hexane solutions. Fig. 3(a) and (b) show the structures of both compounds, which are derivatives of catenated triboron anions, analogous to that observed previously in compound 4. All of the B–B bond lengths [ca. 1.73 Å] across both structures are effectively identical and are consistent with their identification as electron precise (2c–2e) interactions. Although the triboron chain propagates via sequences of B(sp2)–B(sp3)–B(sp2) bonding interactions, these B–B bonds lie within the range established for various reported neutral and acyclic triborane molecules in which the catenate propagates wholly through trigonal boron centres.45–48
 | ||
| Fig. 3 ORTEP representation of (a) compound 10 and (b) compound 11 (25% probability ellipsoids). Isopropyl methyl groups are removed for clarity. Selected bond lengths (Å) and angles (°): (10) Mg1–O1 1.9384(14), Mg1–O3 2.0538(14), Mg1–N1 2.0465(17), Mg1–N2 2.0524(16), B1–B2 1.729(3), B1–B3 1.728(3), O1–Mg1–O3 92.53(6), O1–Mg1–N1 115.75(7), O1–Mg1–N2 129.90(7), B3–B1–B2 107.17(15); (11) Mg1–O1 2.0193(15), Mg1–O3 1.9153(15), B1–B2 1.733(3), B2 –B3 1.732(3), Mg1–N1 2.0406(18), Mg1–N2 2.0262(19), O1–Mg1–N1 105.39(7), O1–Mg1–N2 112.28(7), O3–Mg1–O1 90.59(7), O3–Mg1–N1 134.77(8), O3–Mg1–N2 117.36(8), N2–Mg1–N1 95.54(8), B3–B2–B1 106.59(18). | ||
The isolation of both compounds 10 and 11 apparently ensues with the simultaneous elimination of n-BuBpin from compound 3. Although this observation implies that the diborane insertion reactions take place with exclusive displacement of the pre-quaternised boron centre and retention of the B–B bond of the subsequent diborane reagent employed, a further reaction indicated that this reactivity is also prone to redistribution of the σ-bonded boron centres. A reaction of compound 8 with B2pin2 was performed under identical conditions to those employed in the synthesis of compounds 10 and 11. Initial inspection of the resultant 1H and 11B NMR spectra indicated the generation of a single reaction product (12), which was anticipated to be the result of formal n-BuB(hex) elimination to form a magnesium complex of the catenated [pinB(Bpin)B(hex)]− anion. Work up and crystallisation of compound 12, however, revealed that this process had produced an isomeric form of the anticipated triborane anion (Scheme 4). The resultant single crystal X-ray analysis (Fig. 4) identified a triboron anion in which the central sp3 boron centre is provided by a {B(hex)} moiety rather than through retention of the entire {B2pin2} unit. Although it has not been possible to determine whether this apparent cross metathesis process occurs during or subsequent to the displacement of the n-Bu(hex) by-product, we suggest that the ultimate structure adopted by 12 may be dictated by kinetic factors and the lower steric demands of the monomethyl substitution of C31, which is adjacent to the {B(hex)} oxygen donor (O1) to the magnesium centre.
 | ||
| Scheme 4 Synthesis of compound 12. | ||
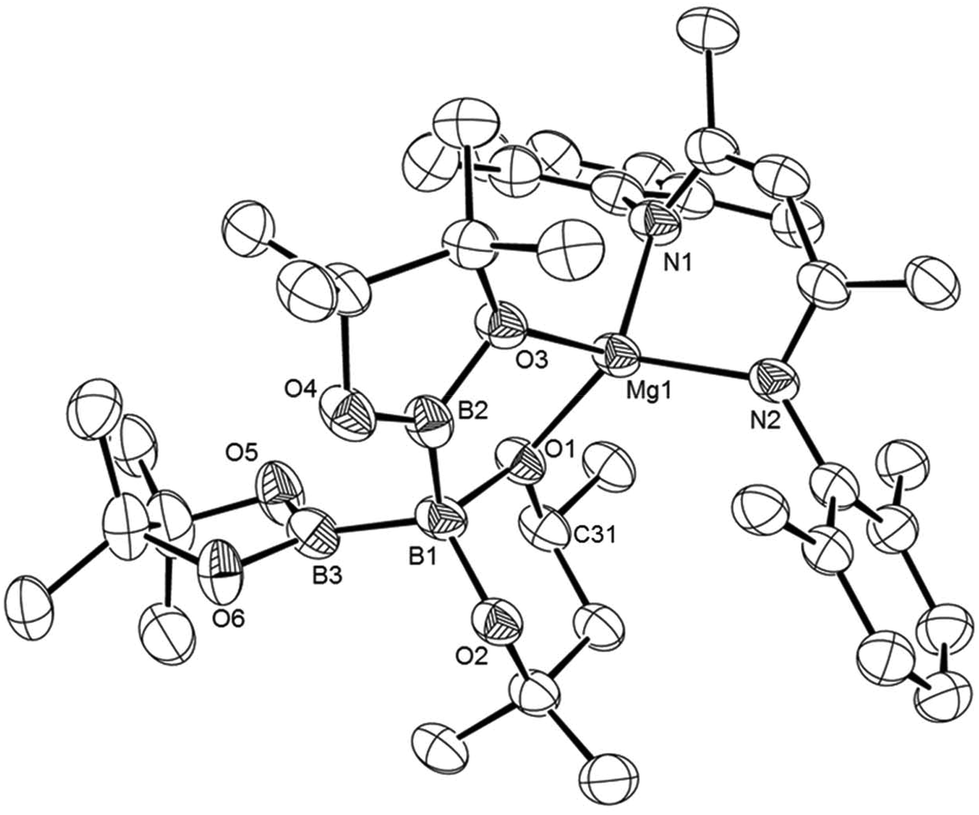 | ||
| Fig. 4 ORTEP representation of compound 12 (25% probability ellipsoids). Isopropyl methyl groups are removed for clarity. Selected bond lengths (Å) and angles (°): Mg1–O1 1.943(2), Mg1–O3 2.043(2), Mg1–N1 2.031(3), Mg1–N2 2.042(3), B1–B2 1.724(6), B1–B3 1.722(6), O1–Mg1–O3 90.30(10), O1–Mg1–N1 119.14(12), O1–Mg1–N2 127.47(12), N1–Mg1–O3 113.70(11), N1–Mg1–N2 95.23(12), N2–Mg1–O3 112.00(11), B3–B1–B2 106.9(3). | ||
Conclusions
The synthesis of compound 7, in which a B–O bond of B2pin2 is cleaved with retention of the B–B bond, implies that the nuclearity of the [(BDI)MgH]2 reagent has a direct influence over the mode of activation of the diborane. In contrast, the reactivity of [(BDI)Mg(n-Bu)] with B2hex2 follows an analogous path to that observed with B2pin2, as does the outcome of treatment of the resultant mono-quaternised diborane(5) anion with DMAP. This latter process provides a terminal magnesium boryl species through displacement of the alkylborane and confirms the generality of this strategy for the facile heterolytic activation of the B–B single bonds of commercially available diboranes. In a similar manner, reaction of the previously reported pinacolatodiborane(5) derivative (3) with B2hex2 and B2neo2 also induces displacement of n-BuBpin and formation of unusual and unsymmetrical catena-triborane(6) anions in which the B–B bond of the externally added diborane reagents are retained. In contrast, although treatment of compound 8, derived by quaternisation of B2hex2, prompts the anticipated n-BuB(hex) elimination, the resultant product comprises the triborane(5) anion in which the two pinacolatoboron units are bonded to the four-coordinate boron of the remaining {B(hex)} moiety. Although no experimental corroboration could be obtained, this observation is tentatively ascribed to the relative steric demands of the boron-bound pinacolato and glycolato chelates and suggests that these species are prone to B–B exchange equilibria in which the ultimate product is determined by kinetic factors. The significance of these observations will become apparent during the future elaboration of this chemistry and our further studies of the onward reactivity of these readily available sources of nucleophilic boron with a range of organic and inorganic electrophiles.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank the EPSRC (UK) for funding of this research (EP/N014456/1).References
- A. Suzuki, Angew. Chem., Int. Ed., 2011, 50, 6722–6737 CrossRef PubMed.
- L. Dang, Z. Y. Lin and T. B. Marder, Chem. Commun., 2009, 3987–3995 RSC.
- J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef PubMed.
- (a) G. J. Irvine, M. J. G. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, E. G. Robins, W. R. Roper, G. R. Whittell and L. J. Wright, Chem. Rev., 1998, 98, 2685–2722 CrossRef PubMed; (b) D. L. Kays and S. Aldridge, in Contemporary Metal Boron Chemistry I: Borylenes, Boryls, Borane, ed. T. B. Marder and Z. Lin, 2008, vol. 130, pp. 29–122 Search PubMed.
- (a) Y. Segawa, M. Yamashita and K. Nozaki, Science, 2006, 314, 113–115 CrossRef PubMed; (b) T. B. Marder, Science, 2006, 314, 69–70 CrossRef PubMed; (c) H. Braunschweig, Angew. Chem., Int. Ed., 2007, 46, 1946–1948 CrossRef PubMed.
- Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 CrossRef PubMed.
- M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, J. Am. Chem. Soc., 2007, 129, 9570 CrossRef PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef PubMed.
- T. Terabayashi, T. Kajiwara, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14162 CrossRef PubMed.
- T. Arnold, H. Braunschweig, W. C. Ewing, T. Kramer, J. Mies and J. K. Schuster, Chem. Commun., 2015, 51, 737–740 RSC.
- L. M. A. Saleh, K. H. Birjkumar, A. V. Protchenko, A. D. Schwarz, S. Aldridge, C. Jones, N. Kaltsoyannis and P. Mountford, J. Am. Chem. Soc., 2011, 133, 3836–3839 CrossRef PubMed.
- A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef PubMed.
- A. V. Protchenko, D. Dange, M. P. Blake, A. D. Schwarz, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2014, 136, 10902–10905 CrossRef PubMed.
- D. Dange, A. Davey, J. A. B. Abdalla, S. Aldridge and C. Jones, Chem. Commun., 2015, 51, 7128–7131 RSC.
- J. Campos and S. Aldridge, Angew. Chem., Int. Ed., 2015, 54, 14159–14163 CrossRef PubMed.
- M. Yamashita, Y. Suzuki, Y. Segawa and K. Nozaki, Chem. Lett., 2008, 37, 802–803 CrossRef.
- M. S. Cheung, T. B. Marder and Z. Y. Lin, Organometallics, 2011, 30, 3018–3028 CrossRef.
- Y. Okuno, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2011, 50, 920–923 CrossRef PubMed.
- B. Wang, Y. Li, R. Ganguly, H. Hirao and R. Kinjo, Nat. Commun., 2016, 7, 11871 CrossRef PubMed.
- W. Lu, H. Hu, Y. Li, R. Ganguly and R. Kinjo, J. Am. Chem. Soc., 2016, 138, 6650–6661 CrossRef PubMed.
- H. Braunschweig, C.-W. Chiu, K. Radacki and T. Kupfer, Angew. Chem., Int. Ed., 2010, 49, 2041–2044 CrossRef PubMed.
- H. Braunschweig, M. Burzler, R. D. Dewhurst and K. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5650–5653 CrossRef PubMed.
- H. Braunschweig, R. D. Dewhurst, L. Pentecost, K. Radacki, A. Vargas and Q. Ye, Angew. Chem., Int. Ed., 2016, 55, 436–440 CrossRef PubMed.
- R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef PubMed.
- D. A. Ruiz, M. Melaimi and G. Bertrand, Chem. Commun., 2014, 50, 7837–7839 RSC.
- L. Kong, Y. Li, R. Ganguly, D. Vidovic and R. Kinjo, Angew. Chem., Int. Ed., 2014, 53, 9280–9283 CrossRef PubMed.
- L. Kong, R. Ganguly, Y. Li and R. Kinjo, Chem. Sci., 2015, 6, 2893–2902 RSC.
- D. Wu, L. Kong, Y. Li, R. Ganguly and R. Kinjo, Nat. Commun., 2015, 6, 7340 CrossRef PubMed.
- T. Imamoto and T. Hikosaka, J. Org. Chem., 1994, 59, 6753–6759 CrossRef.
- L. Kong, W. Lu, L. Yongxin, R. Ganguly and R. Kinjo, Inorg. Chem., 2017, 56, 5586–5593 CrossRef PubMed.
- J. Monot, A. Solovyev, H. Bonin-Dubarle, E. Derat, D. P. Curran, M. Robert, L. Fensterbank, M. Malacria and E. Lacôte, Angew. Chem., Int. Ed., 2010, 49, 9166–9169 CrossRef PubMed.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590–7592 CrossRef PubMed.
- E. Bernhardt, V. Bernhardt-Pitchougina, H. Willner and N. V. Ignatiev, Angew. Chem., Int. Ed., 2011, 50, 12085–12088 CrossRef PubMed.
- J. Landmann, J. A. P. Sprenger, R. Bertermann, N. Ignat'iv, V. Bernhardt-Pitchougina, E. Bernhardt, H. Willner and M. Finze, Chem. Commun., 2015, 51, 4989–4992 RSC.
- J. Landmann, F. Keppner, D. B. Hofmann, J. A. P. Sprenger, M. Häring, S. H. Zottnick, K. Müller-Buschbaum, N. V. Ignat'ev and M. Finze, Angew. Chem., Int. Ed., 2017, 56, 2795–2799 CrossRef PubMed.
- J. Landmann, P. T. Hennig, N. V. Ignat'ev and M. Finze, Chem. Sci., 2017, 8, 5962–5968 RSC.
- (a) A. F. Pécharman, A. L. Colebatch, M. S. Hill, C. L. McMullin, M. F. Mahon and C. Weetman, Nat. Commun., 2017, 8, 15022 CrossRef PubMed. For recent reviews of the chemistry of diboron (4) compounds and transition metal-free diboration reactions, see: (b) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef PubMed; (c) A. B. Cuenca, R. Shishido, H. Ito and E. Fernández, Chem. Soc. Rev., 2017, 46, 415–430 RSC.
- (a) M. Gao, S. B. Thorpe and W. L. Santos, Org. Lett., 2009, 11, 3478 CrossRef PubMed; (b) S. B. Thorpe, X. Guo and W. L. Santos, Chem. Commun., 2011, 47, 424 RSC; (c) M. Gao, S. B. Thorpe, C. Kleeberg, C. Slebodnick, T. B. Marder and W. Santos, J. Org. Chem., 2011, 76, 3997–4007 CrossRef PubMed.
- (a) H. Gulyas, A. Bonet, C. Pubill-Ulldemolins, C. Sole, J. Cid and E. Fernandez, Pure Appl. Chem., 2012, 84, 2219–2231 CrossRef; (b) J. Cid, H. Gulyas, J. J. Carbo and E. Fernandez, Chem. Soc. Rev., 2012, 41, 3558–3570 RSC.
- R. D. Dewhurst, E. C. Neeve, H. Braunschweig and T. B. Marder, Chem. Commun., 2015, 51, 9594–9607 RSC.
- M. S. Hill, M. F. Mahon, C. L. McMullin and A.-F. Pecharman, Angew. Chem., Int. Ed., 2017, 56, 16363–16366 CrossRef PubMed.
- S. J. Bonyhady, C. Jones, S. Nembenna, A. Stasch, A. J. Edwards and G. J. McIntyre, Chem. – Eur. J., 2010, 16, 938–955 CrossRef PubMed.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- S. Pietsch, E. C. Neeve, D. C. Apperley, R. Bertermann, F. Y. Mo, D. Qiu, M. S. Cheung, L. Dang, J. B. Wang, U. Radius, Z. Y. Lin, C. Kleeberg and T. B. Marder, Chem. – Eur. J., 2015, 21, 7082–7098 CrossRef PubMed.
- M. Arrowsmith, H. Braunschweig and T. E. Stennett, Angew. Chem., Int. Ed., 2017, 56, 96–115 CrossRef PubMed.
- K. Nozaki, Y. Aramaki, M. Yamashita, S. H. Ueng, M. Malacria, E. Lacote and D. P. Curran, J. Am. Chem. Soc., 2010, 132, 11449–11451 CrossRef PubMed.
- Y. Hayashi, Y. Segawa, M. Yamashita and K. Nozaki, Chem. Commun., 2011, 47, 5888–5890 RSC.
- G. Linti, D. Loderer, H. Nöth, K. Polborn and W. Rattay, Chem. Ber., 1994, 127, 1909–1922 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterisation data of compounds 7–12, along with NMR spectra. CCDC 1824654–1824659 for compounds 7–12 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8dt01451e |
| This journal is © The Royal Society of Chemistry 2018 |