
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective synthesis of trifluoromethylated 1,3-dioxanes by intramolecular oxa-Michael reaction†
Liliana
Becerra-Figueroa
a,
Elodie
Brun
b,
Michael
Mathieson
b,
Louis J.
Farrugia
b,
Claire
Wilson
b,
Joëlle
Prunet
*b and
Diego
Gamba-Sánchez
 *a
*a
aLaboratory of Organic Synthesis, Bio and Organocatalysis, Chemistry Department, Universidad de los Andes, Cra 1 No. 18A-12 Q:305, Bogotá 111711, Colombia. E-mail: joelle.prunet@glasgow.ac.uk; da.gamba1361@uniandes.edu.co
bWestCHEM, School of Chemistry, University of Glasgow, Joseph Black Building, University Avenue, Glasgow G12 8QQ, UK
First published on 23rd November 2016
Abstract
A highly diastereoselective synthesis of trifluoromethylated 1,3-dioxanes is described. The reaction proceeds by an addition/oxa-Michael sequence and works efficiently under mild reaction conditions, with a good substrate scope and acceptable to good yields.
Trifluoromethyl-containing heterocycles and particularly oxygenated heterocyclic systems have attracted growing attention in the last few years.1 They are currently investigated because of their interesting applications in medicine, agriculture and materials science, among others.2 In spite of the continuous growth in this field, the synthesis of 1,3-dioxanes remains unexplored. Consequently, the development of new and efficient synthetic methods for this kind of heterocyclic system is at the same time a challenge and very desirable.
Since the seminal work published by Evans and Prunet in 1993,3 the addition/oxa-Michael cascade has demonstrated to be useful in the construction of protected syn 1,3-diols4 (Scheme 1). The reaction substrates are homoallylic alcohols functionalised with a Michael acceptor. The most usual reagent is benzaldehyde but there are several examples in the literature where p-methoxy benzaldehyde has been used successfully.5 Concerning the Michael acceptor and the substitution pattern the reaction has been explored with esters and amides,3 sulfones6 and sulfoxides,7 and in very few cases with substituents α to the Michael acceptor.8
 | ||
| Scheme 1 Difference between the closest similar report and our work. | ||
Inspired by those previous results and by the need for a simple method to synthesise CF3-containing dioxanes, we decided to explore the intramolecular oxa-Michael reaction using trifluoroacetophenone and a homoallylic alcohol functionalised with an ester as substrates (Scheme 1).
During our studies, a similar transformation on cyclic dienones in the presence of triethylamine was described by Wang et al.9 Even if the yields were good, the selectivity was moderate in most cases.
With our substrates, if the reaction is under thermodynamic control as it is the case with benzaldehyde,3 the major diastereomer should have all the large substituents at equatorial positions (the two alkyl chains and presumably the trifluoromethyl group).
We started our investigation with the simple known ester 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 and explored the reaction under the conditions described by Wang et al. (Table 1, entry 1). After 24 hours no reaction was observed, and the starting material was recovered quantitatively. The use of a larger quantity of a base and ketone or the use of another weak base such as potassium acetate did not afford better results (entries 2 and 3); therefore, we decided to turn our attention to bases and reaction conditions more familiar to us.11
10 and explored the reaction under the conditions described by Wang et al. (Table 1, entry 1). After 24 hours no reaction was observed, and the starting material was recovered quantitatively. The use of a larger quantity of a base and ketone or the use of another weak base such as potassium acetate did not afford better results (entries 2 and 3); therefore, we decided to turn our attention to bases and reaction conditions more familiar to us.11
|
|
|||||
|---|---|---|---|---|---|
| Entry | Base (equiv.) | PhCOCF3 | Time | Yield (%) | Selectivityb |
| a The reaction was performed making successive additions of the base and the ketone every 15 min. b Determined by 1H NMR of the crude product. c 80% brsm. | |||||
| 1 | Et3N (0.2) | 1.2 equiv. | 24 h | — | — |
| 2 | Et3N (0.4) | 2 equiv. | 12 h | — | — |
| 3 | AcOK (0.4) | 2 equiv. | 12 h | — | — |
| 4 | tBuOLi (0.4) | 2 equiv. | 12 h | 49 | 80:20 |
| 5 | tBuOK (0.4) | 2 equiv. | 6 h | 84 | 88:12 |
| 6a | tBuOK (0.3) | 3.3 equiv. | 2 h | 70c | >98:2 |
| 7a | tBuOK (0.3) | 3.3 equiv. | 6 h | 79 | >98:2 |

With tBuOLi and 2 equivalents of ketone (entry 4), the reaction proceeded and the desired dioxane 2a was isolated in 49% yield with a moderate selectivity, the major product has the same stereochemistry in all cases (see below). The change of the counterion to potassium provided much better results and in 6 hours the product was isolated in 84% yield with good selectivity (entry 5). Typically an intramolecular conjugate addition reaction of this kind is performed using successive addition of a base and carbonyl compound; it was demonstrated that this serves to improve the selectivity and to increase the reaction yield.3 Hence, we used three additions of 1.1 equivalents of ketone and 0.1 equivalents of base, but after 2 hours the reaction was not complete and the product was only isolated in 70% yield (80% brsm, entry 6); however after 6 hours the starting material was completely consumed and the product was isolated in 79% yield (entry 7). In both cases only one diastereomer was detected in the 1H NMR spectrum of the crude reaction mixture and after purification of the product.
The stereochemistry of the major diastereomer remained uncertain. We were confident about the cis stereochemistry of the substituents at positions 4 and 6, but for position 2 the configuration will be dependent on the relative steric hindrance of Ph and CF3. Fortunately, compound 2a was crystalline and X-ray crystallographic analysis provided its structure (Fig. 1), showing that CF3 is indeed at the equatorial position.
 | ||
| Fig. 1 X-ray structure of product 2a. | ||
With these promising results in hand, we started the study of the reaction scope. All the starting materials 1 were obtained by a cross metathesis reaction between the corresponding homoallylic alcohol and methyl acrylate using the Grubbs II catalyst (2.5% mol). These homoallylic alcohols were easily prepared in one step from commercially available aldehydes and allyl bromide using an aqueous Barbier reaction.12 Then we applied the optimised conditions to all substrates 1 and we found excellent diastereoselectivity in all cases (see Table 2).
|
|
|||
|---|---|---|---|
| Entry | R | Product | Yieldb (%) |
| a The reaction was performed making successive additions of the base (0.1 equiv.) and the ketone (1.1 equiv.) every 15 min. b Yields are reported for pure compounds; the 1H NMR spectra of the crude reaction mixtures showed in all cases only one diastereomer. c This product was obtained from enantiopure lactic acid and the anti diastereomer from the cross metathesis reaction was isolated (see the ESI). d 80% brsm. | |||
| 1 | PhCH2CH2 | 2a | 79 |
| 2 | C6H13 | 2b | 80 |
| 3 | Isopropyl | 2c | 81 |
| 4 | Chx | 2d | 82 |
| 5 | CH3CHOBnc | 2e | 70 |
| 6 | Ph | 2f | 67d |
| 7 | p-MeOPh | 2g | 70 |
| 8 | p-ClPh | 2h | 63 |

The use of simple aliphatic substrates afforded the best results (entries 1 to 4); there is no noticeable effect induced by the chain extension, or by the presence of aromatic rings in the aliphatic chain (compare entries 1 and 2). The use of branched R groups is also acceptable and there is no influence on the reaction diastereoselectivity; in fact, as these groups are at the equatorial position we indeed expected better selectivities with these bulkier R groups. The reaction is also useful with aromatic R substituents (entries 6 to 8); however, yields are somehow lower when electron-withdrawing groups are inserted (entry 8 vs. entry 7).
The reaction mechanism is shown in Scheme 2. We postulated that this reaction is under thermodynamic control as it is the case with benzaldehyde,3 with a reversible oxa-Michael reaction. So, it is to be expected that the use of a more electrophilic carbonyl compound (trifluoroacetophenone) will cause a better reaction with the alkoxide 3, favouring the formation of hemiketal alkoxide 4. We hypothesise that this intermediate is stabilised by the inductive effect of the CF3 group, facilitating the reverse oxa-Michael addition and in consequence improving the selectivity of the whole process.
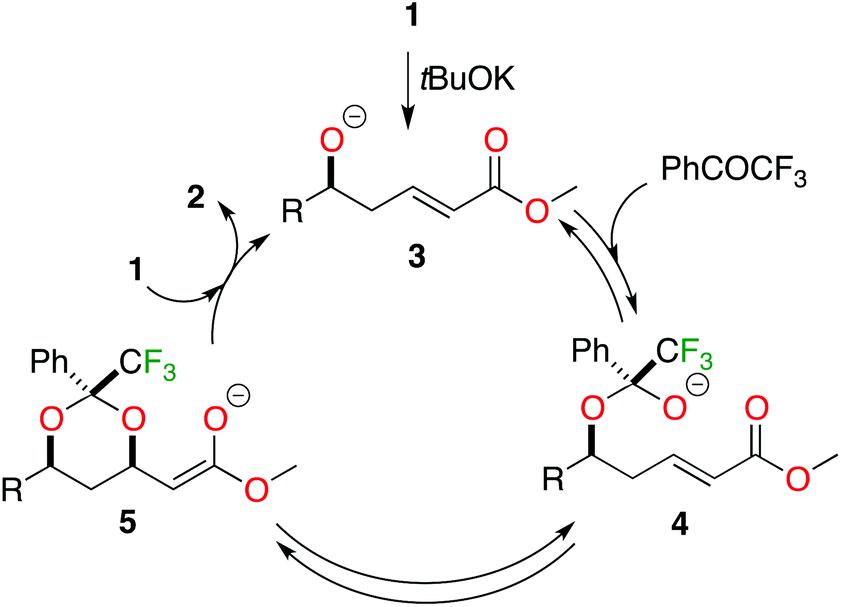 | ||
| Scheme 2 Reaction mechanism. | ||
The next step was the investigation of the effect of a substituent at the α position to the electron-withdrawing group8 (results in Table 3); for this purpose, we synthesised several α-substituted substrates 6 (see the ESI† for the reaction sequence). When the substituent at the α position was a methyl group (entry 1), the outcome of the reaction was disappointing. The product was observed in the 1H NMR spectrum along with the intermediate hemiketal and the elimination product. We were able to isolate the desired ketal with acceptable selectivity but in low yield. On the other hand, the reaction with product 6b works smoothly and the product was isolated with excellent selectivity and in acceptable yield (entry 2); curiously the reaction was not complete after 12 hours and some starting material was recovered. The introduction of a methyl group γ to the ester did not have a significant effect on the selectivity (entry 3); in this case the reaction was pushed to completion with 4 additions of a base and ketone to furnish 7c in 90% yield. It has to be noted that the reaction on a substrate similar to 6c (with R = iPr) with benzaldehyde as the electrophilic reagent furnished the corresponding product in 35% yield (53% brsm) with a 67:33 selectivity.8d
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | R1 | R2 | R3 | Product, yield | Selectivity |
| a This value was calculated by analysis of the 1H NMR spectrum; the crude mixture also contained the intermediate hemiketal and the elimination product; a sample was purified for analytical purpose. b Cb = CON(iPr)2. c 83% brsm. d The reaction was performed using 4 additions of a base and ketone. e The starting material was treated with 1.1 equivalents of base and ketone, and then consecutive additions of 0.1 equivalents of base and 1.1 equivalents of ketones were made. | ||||||
| 1 | PhCH2CH2 | H | Me | Et | 7a, <27%a | 82:18 |
| 2 | PhCH2CH2 | H | OCbb | Et | 7b, 54%c | >98:2 |
| 3d | PhCH2CH2 | Me | OCb | Et | 7c, 90% | >98:2 |
| 4e | PhCH2CH2 | H | NHCHO | Me | 7d, 85% | 82:18 |

We were also curious about the reactivity of a substrate with a nitrogen substituent at the α position. Depending on the substitution pattern of the nitrogen atom, the compound can have an enamine character or the N–H group can be acidic enough to be deprotonated before the alcohol.
We were able to synthesize compound 6d by a Horner–Wadsworth–Emmons reaction.13 As expected an extra equivalent of base was needed in order to completely deprotonate the NH group (entry 4). It is remarkable that this reaction proceeded giving very good yield and good selectivity, even if the deprotonation of the nitrogen can induce a nucleophilic character to the carbon at the β position to the ester. It is worth noting that in all the cases described in Table 3, the reported selectivity corresponds to the position of R2; the reaction proceeds with complete selectivity for the oxygen addition.
Fortunately, the stereochemistry of the substituent at the α position could be determined by X-ray crystallographic analysis of compound 7d as shown in Fig. 2. Comparison of NMR signals and coupling constants for the H-5 proton in 7d and 7b allowed us to define the configuration at C5 for compound 7b as shown in Scheme 3. The stereochemistry at C5 of 7c M was assumed to be the same as that of 7b.
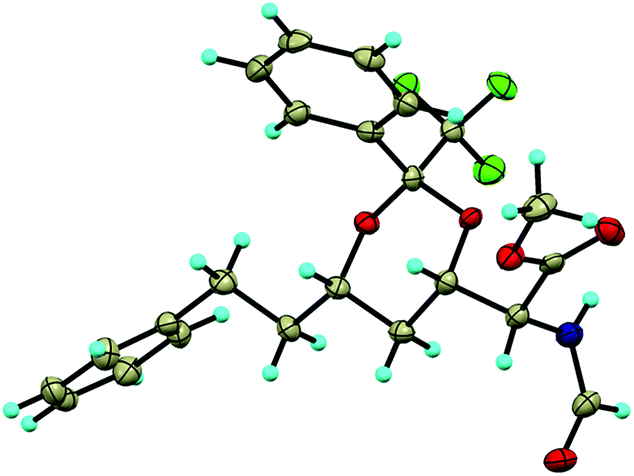 | ||
| Fig. 2 X-ray structure of product 7c. | ||
 | ||
| Scheme 3 Stereochemistry for major (M) and minor (m) diastereomers. | ||
Some additional experiments were carried out in order to illustrate the generality of our method. First, other Michael acceptors such as Weinreb amides or sulfones were explored. The cited literature3–8 shows that the reactivity of the Michael acceptors does not have a great influence on the yield of the oxa-Michael reaction. The main difference is the diastereoselectivity.
In consequence, we were curious to see if in our case Weinreb amides or sulfones are desirable substrates and if the selectivity is comparable to that obtained with esters.
As shown in Scheme 4 the results are very good, the reaction yields are comparable to those obtained with simple esters and in both cases, Scheme 4a and b, there was only one diastereoisomer visible by 1H NMR of the crude reaction mixture and isolated products 9 and 11. The stereochemistry of these compounds was assigned by comparison with that of the esters.
 | ||
| Scheme 4 Weinreb amide and sulfone as Michael acceptors. | ||
We also decided to use another trifluoromethyl ketone; we speculated that, if the ketone was more electrophilic than trifluoroacetophenone, the hemiketal alkoxide 4 would be less nucleophilic and so the reaction would be more difficult. Thus, if the reaction worked with a ketone substituted in the aromatic ring by a strong electron withdrawing group, then it would be predictable than the reaction could be extended to the use of almost any trifluoromethyl ketone. Scheme 5 shows the result of the reaction using m-nitro-trifluoroacetophenone as the electrophilic reagent. In this case the reaction proved to be more difficult; four additions of a base and 16 hours were needed to complete the reaction; however, product 13 was isolated in good yield and the reaction proceeded with complete diastereoselectivity.
 | ||
| Scheme 5 Reaction using m-nitrotrifluoroacetophenone. | ||
In summary, the additional experiments allow us to conclude that different Michael acceptors and other trifluoromethyl ketones can be used, thus making the method very general.
Finally, computational studies oriented to propose an explanation for the difference in reactivity and selectivity between the substrates, as well as additional experiments to increase even more the reaction scope are currently under investigation in our laboratory. The results will be reported in due course.
In conclusion, we have described a useful methodology for the synthesis of CF3-containing 1,3-dioxanes with excellent diastereoselectivity in most cases. We also reported our preliminary conclusions about the reaction mechanism based on literature reports and experimental observations. The stereochemistry of the products was established by X-ray crystallographic analysis and extrapolation.
Financial support for this work was provided by the University of Glasgow, the EPSRC (Doctoral Training Allocation EP/P50418X/1 for M. M.) and Universidad de los Andes. L. B.-F acknowledges the Universidad de los Andes and especially the Chemistry Department for a fellowship and the Faculty of Science for financial support for her internship. We also thank Mr David Sotelo for recording some of the NMR spectra.
Notes and references
- (a) F. M. D. Ismail, J. Fluorine Chem., 2002, 118, 27 CrossRef CAS; (b) J.-A. Ma and D. Cahard, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; (c) J. Nie, H.-C. Guo, D. Cahard and J.-A. Ma, Chem. Rev., 2011, 111, 455 CrossRef CAS PubMed; (d) Y.-J. Lin, L.-N. Du, T.-R. Kang, Q.-Z. Liu, Z.-Q. Chen and L. He, Chem. – Eur. J., 2015, 21, 11773 CrossRef CAS PubMed.
- (a) F. Li, J. Nie, L. Sun, Y. Zheng and J.-A. Ma, Angew. Chem., Int. Ed., 2013, 52, 6255 CrossRef CAS PubMed; (b) J. Wang, W.-G. Kong, F. Li, J. Liu, Q. Shen, L. Liu and W.-X. Zhao, Org. Biomol. Chem., 2015, 13, 5399 RSC; (c) Y. Zhu, Z. Dong, X. Cheng, X. Zhong, X. Liu, L. Lin, Z. Shen, P. Yang, Y. Li, H. Wang, W. Yan, K. Wang and R. Wang, Org. Lett., 2016, 18, 3546 CrossRef CAS PubMed.
- D. A. Evans and J. A. Gauchet-Prunet, J. Org. Chem., 1993, 58, 2446 CrossRef CAS.
- For a few selected examples, see: (a) D. T. Hung, J. B. Nerenberg and S. L. Schreiber, J. Am. Chem. Soc., 1996, 118, 11054 CrossRef CAS; (b) T. J. Hunter and G. A. O'Doherty, Org. Lett., 2001, 3, 2777 CrossRef CAS PubMed; (c) T. A. Dineen and W. R. Roush, Org. Lett., 2004, 6, 2043 CrossRef CAS PubMed; (d) A. Vincent and J. Prunet, Synlett, 2006, 2269 CAS; (e) E. de Lemos, F.-H. Porée, A. Bourin, J. Barbion, E. Agouridas, M.-I. Lannou, A. Commerçon, J.-F. Betzer, A. Pancrazi and J. Ardisson, Chem. – Eur. J., 2008, 14, 11092 CrossRef CAS PubMed; (f) S. S. Palimkar and J. Uenishi, Org. Lett., 2010, 12, 4160 CrossRef CAS PubMed; (g) A. M. M. Albury and M. P. Jennings, J. Org. Chem., 2012, 77, 6929 CrossRef CAS PubMed; (h) R. W. Bates and T. G. Lek, Synthesis, 2014, 1731 CrossRef; (i) T. J. Hunter, Y. Wang, J. Zheng and G. A. O'Doherty, Synthesis, 2016, 1700 CAS.
- D. A. Evans, P. Nagorny, D. J. Reynolds and K. J. McRae, Angew. Chem., Int. Ed., 2007, 46, 541 CrossRef CAS PubMed.
- (a) R. Oriez and J. Prunet, Tetrahedron Lett., 2010, 51, 256 CrossRef CAS; (b) L. Grimaud, D. Rotulo, R. Ros-Perez, L. Guitry-Azam and J. Prunet, Tetrahedron Lett., 2002, 43, 7477 CrossRef CAS.
- D. Gamba-Sanchez and J. Prunet, J. Org. Chem., 2010, 75, 3129 CrossRef CAS PubMed.
- (a) L. Grimaud, R. de Mesmay and J. Prunet, Org. Lett., 2002, 4, 419 CrossRef CAS PubMed; (b) D. Rotulo-Sims and J. Prunet, Org. Lett., 2007, 9, 4147 CrossRef CAS PubMed; (c) R. Aouzal and J. Prunet, Org. Biomol. Chem., 2009, 7, 3594 RSC; (d) R. Oriez, Master Thesis, Ecole Polytechnique (Palaiseau), 2006.
- F. Li, J. Wang, M. Xu, X. Zhao, X. Zhou, W.-X. Zhao and L. Liu, Org. Biomol. Chem., 2016, 14, 3981 CAS.
- J. A. Gazaille and T. Sammakia, Org. Lett., 2012, 14, 2678 CrossRef CAS PubMed.
- In our previous studies the typical base employed was tBuOK and we performed successive additions of a base and carbonyl compound.
- A. Chattopadhyay, J. Org. Chem., 1996, 61, 6104 CrossRef CAS PubMed.
- P. A. Alexander, S. P. Marsden, D. M. Muñoz Subtil and J. C. Reader, Org. Lett., 2005, 7, 5433 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data; experimental details and copies of NMR spectra. CCDC 1510756 and 1511482. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02333a |
| This journal is © The Royal Society of Chemistry 2017 |