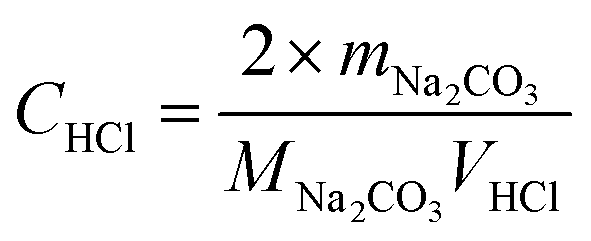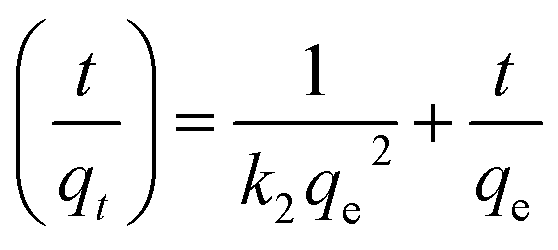DOI:
10.1039/C5RA09761D
(Paper)
RSC Adv., 2015,
5, 60208-60219
Magnetic amine-functionalized polyacrylic acid-nanomagnetite for hexavalent chromium removal from polluted water
Received
24th May 2015
, Accepted 16th June 2015
First published on 16th June 2015
Abstract
A novel magnetic amine-functionalized polyacrylic acid-nanomagnetite (Fe3O4-PAA-NH2) adsorbent prepared using a facile surface-initiated polymerization (SIP) method has delivered a great Cr(VI) removal performance compared to as-received Fe3O4 nanoparticles. The maximum amine group (–NH2) concentration grafted onto Fe3O4-PAA is determined to be 3.925 mg g−1 based on acid–base titrimetric analysis. The optimal pH value for Cr(VI) adsorption is around 2.0 with a Fe3O4-PAA-NH2 dose of 30 mg and contact time of 10 min at room temperature. A multilayer adsorption for the Freundlich isotherm model is well-fitted and fits better than the monolayer adsorption of the Langmuir isotherm model. The kinetics of Cr(VI) removal by the Fe3O4-PAA-NH2 nanoadsorbent is found to follow pseudo-second-order behavior with a calculated room temperature rate constant of 1.23 g mg−1 min−1 for a solution with an initial Cr(VI) concentration of 7.0 mg L−1 and pH value of 2.5. The competition adsorption tests show that the presence of other metals in polluted water, including Cu(II), Zn(II), Cd(II), K(I), Ca(II), Na(I), and Mg(II), favors the Cr(VI) adsorption by the fabricated Fe3O4-PAA-NH2 nanoadsorbent due to the affinity of the chemical potential and electronegativity of each metallic element. Moreover, the prepared Fe3O4-PAA-NH2 nanoadsorbent exhibits a good reusability and retains around 85% of its Cr(VI) adsorption capacity even after 5 cycles.
1. Introduction
Currently, increasing contamination in wastewater systems is a critical problem to solve.1,2 In particular, the heavy metals, such as chromium (Cr), cadmium (Cd), mercury (Hg), lead (Pb), and arsenic (As), are highly toxic water pollutants resulting from industries including metal plating facilities, mining operations, fertilizer industries, tanneries, batteries, paper industries, pesticides, etc.,3,4 which can cause severe public health problems to animals and human beings since they can be stored, accumulated and transferred by organisms.5 Among these heavy metal ions, hexavalent chromium (Cr(VI)) is a common contaminant in polluted water and exhibits extreme toxicity and notorious mobility.6 The U.S. Environmental Protection Agency (EPA) has set a maximum contaminant level for total Cr in drinking water as 0.1 mg L−1 according to the national primary drinking water regulations.7,8 Therefore, rapid, efficient and economical technologies are required to be used to stringently treat industrial wastewater before discharge in order to meet the limitations.9 Adsorption is a traditional, favorable and feasible technique10 to eliminate heavy metal ions for environmental remediation due to its low cost and high efficiency.11 In addition, adsorption can effectively remove heavy metals present in the wastewater system at lower concentrations compared with other methods, such as chemical precipitation and electrochemical methods.12,13
Recently, nanostructured adsorbents have shown promise for application in water decontamination owing to their high specific surface area and having many more active sites than bulk materials for trapping heavy metal ions.14,15 However, the recycling of these nanosized adsorbents after treatment of polluted water still remains a challenge.16 Generally, the introduction of magnetism into nanoadsorbents can help with the recycling of adsorbents from the wastewater system via direct application of an external magnetic field.1 These recycled magnetic nanoadsorbents can also be regenerated and reused for economic and practical applications. Therefore, researchers have started to pay attention to the design of magnetic nanoadsorbents for heavy metal remediation. For example, Kim et al.16 prepared a hierarchical structured MnO2-coated magnetite nanocomposite (Fe3O4/MnO2) using a mild hydrothermal process with a maximum adsorption capacity toward Cd(II) of 53.2 mg g−1. Liu et al.9 fabricated magnetic biopolymer hybrid hydrogels consisting of cellulose and chitosan as coating polymers and Fe3O4 as the core nanoparticles from ionic liquid as the solvent, which exhibited high adsorption capacities for different heavy metals (Cu(II), Fe(II), and Pb(II)). Gu et al.2 reported that magnetic Fe3O4–polyaniline (PANI) nanocomposites showed a unique capability to remove Cr(VI) from polluted water with a wide pH range and could be easily regenerated and reused without decreasing the Cr(VI) removal performance.
Meanwhile, it has been found that the surface functionality of adsorbents could greatly affect the adsorption of heavy metals at the interface.17 The introduced functional groups, such as carboxylate, hydroxyl, sulfate and amino groups, on the adsorbent surface are responsible for the heavy metal adsorption due to their affinity for heavy metals, forming metal complexes or chelates.18–20 Zhang et al.21 reported that thiol-functionalized multiwalled carbon nanotube/magnetite nanocomposites (CNT/Fe3O4) showed maximum adsorption capacities for Hg(II) and Pb(II) of 65.52 and 65.40 mg g−1, respectively. Xin et al.22 noted that amine-functionalized mesoporous Fe3O4 nanoparticles obtained using a hydrothermal method possessed maximum adsorption capacities for Pb(II), Cd(II), and Cu(II) of 369.0 to 523.6 mg g−1. Qiu et al.23 prepared ethyl cellulose (EC) composites modified with 20.0 wt% polyethylenimine (PEI) (PEI/ECs), which showed effective Cr(VI) removal from solutions with a wide pH range.
Based on the aforementioned advantages, this work combines magnetism with surface functionality to design a novel magnetic amine-functionalized polyacrylic acid (PAA)-nanomagnetite (Fe3O4-PAA-NH2) nanoadsorbent, which shows efficient Cr(VI) removal from polluted water. The PAA is introduced on the surface of Fe3O4 using a facile surface-initiated polymerization (SIP) method in order to link the nanomagnetite with the amine groups. The presence of PAA on the surface of Fe3O4 can also protect Fe3O4 from acid etching. The effects of initial Cr(VI) concentration, solution pH values and adsorbent doses on the Cr(VI) removal have been systematically investigated. The room temperature adsorption kinetics are explored by studying the Cr(VI) concentration change with different contact times. The effect of the presence of other heavy metals, including Cu(II), Zn(II), and Cd(II), on the adsorption of Cr(VI) and the stability of the nanoadsorbent is studied as well. The synthesized Fe3O4-PAA-NH2 nanoadsorbent can satisfy the limitation of the US EPA requirement.
2. Experimental
2.1. Materials
Acrylic acid (≥99.0%) was provided by Shanghai RichJoint Chemical Reagents Co., Ltd. Triethylene tetramine (TETA), potassium dichromate (K2Cr2O7), nitric acid (HNO3, 65–68 wt%), potassium nitrate (KNO3, ≥99.0%, density: 2.109 g cm−3 (16 °C)), magnesium nitrate (hexahydrate) (Mg(NO3)2·6H2O, ≥99.0%, density: 1.464 g cm−3), sodium nitrate (NaNO3, ≥99.0%, density, 2257 g cm−3), calcium nitrate (tetrahydrate) (Ca(NO3)2·4H2O, ≥99.0%, density: 1.896 g cm−3), zinc nitrate (hexahydrate) (Zn(NO3)·6H2O, ≥99.0%, density: 2.065 g cm−3), and cupric nitrate (trihydrate) (Cu(NO3)·3H2O, 99.0–102.0%, density, 2.32 g cm−3) were purchased from Sinopharm Chemical Reagent Co., Ltd. Cadmium (99.98%) was obtained from Aladdin. Ammonium persulfate (APS, (NH4)2S2O8, ≥98.0%) was from Chinasun Specialty Products Co. Ltd. Fe3O4 nanoparticles with an average size of 12 nm were obtained from Nanjing Emperor Nano Material Co., Ltd. All the chemicals were used as-received without any further treatment.
2.2. Preparation of PAA-modified Fe3O4 nanoparticles
The PAA-modified Fe3O4 nanoparticles (Fe3O4-PAA) were prepared using a SIP method. First, the Fe3O4 nanoparticles (2.0 g) and oxidant APS (1.0 g) were mixed in a 250 mL beaker with 100 mL deionized water for 20 minutes with sonication. Then, the dispersed solution was transferred into a 250 mL three-neck flask and heated to 70 °C in an oil bath. After that, the acrylic acid was dropped into the above solution under mechanical stirring (300 rpm) and heated at reflux for an additional 4 hours in the oil bath at 70 °C for polymerization of the acrylic acid to form PAA. Finally, the product (Fe3O4-PAA) was vacuum filtered and washed with deionized water to remove the remaining oxidant and any oligomers before amine-functionalization.
2.3. Fabrication of Fe3O4-PAA-NH2 nanoparticles
The obtained Fe3O4-PAA nanoparticles were transferred into a 250 mL three-neck flask and mechanically stirred in the oil bath under reflux at 70 °C. TETA, with a weight ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to the added acrylic acid, was dropped into the above solution for a further reaction time of 3 hours. The obtained powder was vacuum filtered and washed several times with deionized water and ethanol. The final amine-functionalized Fe3O4-PAA-NH2 nanocomposite was dried at 60 °C in an oven overnight. The preparation procedure of Fe3O4-PAA-NH2 is shown in Scheme 1.
:1 to the added acrylic acid, was dropped into the above solution for a further reaction time of 3 hours. The obtained powder was vacuum filtered and washed several times with deionized water and ethanol. The final amine-functionalized Fe3O4-PAA-NH2 nanocomposite was dried at 60 °C in an oven overnight. The preparation procedure of Fe3O4-PAA-NH2 is shown in Scheme 1.
 |
| | Scheme 1 Preparation procedure of Fe3O4-PAA-NH2. | |
2.4. Determination of amine group content in the Fe3O4-PAA-NH2 nanoadsorbent
The amine group content in Fe3O4-PAA-NH2 was determined using acid–base titrimetric analysis. Around 0.1 g of Fe3O4-PAA-NH2 was weighed precisely and put into a 250 mL glass conical flask containing 50 mL deionized water and mixed with a bromocresol green–methyl red indicator. A standard hydrochloric acid (HCl) solution was used to titrate the above solution until the color of the above solution turned from green to colorless or red. The amine content (Camine: mg g−1) was evaluated from eqn (1):| |
 | (1) |
where CHCl (mol L−1) is the concentration of the standard HCl solution, VHCl (L) is the volume of the consumed standard HCl solution, MNH2 (g mol−1) is the molecular weight of the amine group, and mamine (g) is the weight of the used Fe3O4-PAA-NH2.
The concentration of the standard HCl solution was obtained using following procedure. 0.0052 g of anhydrous sodium carbonate (Na2CO3) was dissolved in 50 mL of deionized water in a 250 mL glass conical flask with bromocresol green–methyl red as a mixed indicator. The standard HCl solution was employed to titrate the Na2CO3 solution. As the color of the solution changed from green to red, the solution was boiled for 2 min and cooled down to room temperature. Then, the solution was further titrated with the standard HCl solution until the solution became colorless or red. Thus, the concentration of the standard HCl solution was calculated using eqn (2):
| |
 | (2) |
where
mNa2CO3 (g) is the weight of the anhydrous Na
2CO
3,
MNa2CO3 (g mol
−1) is the molecular weight of the anhydrous Na
2CO
3, and
VHCl (L) is the volume of the consumed standard HCl solution.
2.5. Characterization of Fe3O4-PAA-NH2 nanoadsorbent
The chemical structure of the as-prepared nanoadsorbent was analyzed using Fourier transform infrared spectroscopy (FT-IR, Thermo Nicolet NEXUS, Thermo Scientific) in the range of 500 to 4000 cm−1 with a resolution of 4 cm−1. The morphologies of the synthesized nanocomposites were observed using field emission scanning electron microscopy (FE-SEM, Hitachi S-4800 system). The samples were prepared by adhering the powders onto an aluminum plate. Elemental mapping was carried out with an electron-probe micro-analyser (JXA-8100, JEOL). Thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) were performed from ambient temperature to 700 °C in air at a heating rate of 5 °C min−1 using a SDT thermal-microbalance apparatus. X-ray photoelectron spectroscopy (XPS) was carried out with a Kratos AXIS Ultra DLD spectrometer using Al Kα (hν = 1486.6 eV) radiation as the excitation source under an anode voltage of 12 kV and an emission current of 10 mA. The C1s peaks were deconvoluted into the components consisting of a Gaussian line shape Lorentzian function (Gaussian = 80%, Lorentzian = 20%) on Shirley background. The Brunauer–Emmett–Teller (BET) method was used to measure the specific surface area of the as-prepared nanoadsorbent. BET adsorption and desorption isotherms were obtained using a surface area analyzer (TriStar 3000, Micromeritics Instrument Corp.). The samples (around 0.1 g) were weighed and placed inside the sample holder cell. The refrigerant used was liquid nitrogen placed in a vacuum Dewar at about 77 K and the carrier gas was nitrogen. The magnetic properties of the as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2 were measured on a magnetic property measurement system (Lakeshore 735 VSM Controller 7300 Series Magnetometer).
2.6. Cr(VI) removal tests
All the Cr(VI) removal tests were conducted in a 50 mL glass conical flask at room temperature. The concentration of heavy metals was determined using atomic absorption spectroscopy (AAS, Agilent 3510, USA). The reported values were the average of three measurements of each sample with a standard deviation of ±5%.
2.6.1 Effect of pH value. The effect of pH value on the Cr(VI) removal by the synthesized Fe3O4-PAA-NH2 was investigated by selecting solutions with pH values of 1.0, 2.0, 3.0, 5.0, 7.0, 9.0, 10.0 and 11.0. The initial pH value of the Cr(VI) solutions was adjusted using NaOH (1.0 mol L−1) and HCl (1 mol L−1) with a pH meter (model: PHS-25C). The Fe3O4-PAA-NH2 (30.0 mg) was ultrasonically dispersed (model: KQ-800KDE) for 10 min in 20.0 mL solutions with an initial Cr(VI) concentration of 5.4 mg L−1. Then, the solution was taken out and centrifuged (model: TDL-80-2B) for Cr(VI) concentration determination. Meanwhile, the Fe3O4-PAA-NH2 could also be separated from the solutions by using a permanent magnet to give similar results.
2.6.2 Effect of initial Cr(VI) concentration. The effect of initial Cr(VI) concentration on the Cr(VI) removal was investigated by using Fe3O4-PAA-NH2 (30.0 mg) to treat Cr(VI) solutions (20.0 mL) with initial Cr(VI) concentration varying from 1.3 to 7.5 mg L−1 for 10 min.
2.6.3 Effect of Fe3O4-PAA-NH2 dose. The effect of the dose of synthesized Fe3O4-PAA-NH2 on the Cr(VI) removal was studied by using Fe3O4-PAA-NH2 with loadings from 0.25 to 4.0 g L−1 to treat 20.0 mL Cr(VI) solutions with an initial Cr(VI) concentration of 7.5 mg L−1 and an initial pH of 3.0 for 10 min. For comparison, as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2 (10.0 mg) were used to treat a neutral solution of 20.0 mL Cr(VI) with an initial Cr(VI) concentration of 1.0 mg L−1 for 5 min.
2.6.4 Isotherm study. For the isotherm study, the synthesized Fe3O4-PAA-NH2 (20.0 mg) nanoparticles were used to treat different initial Cr(VI) concentrations varying from 0.5 to 4.5 mg L−1 with a contact time of 15 min at room temperature.
2.6.5 Kinetics study. For the kinetics study, the synthesized Fe3O4-PAA-NH2 (30.0 mg) was used to treat a 20.0 mL solution with an initial Cr(VI) concentration of 7.0 mg L−1 and pH of 3.0 for different contact times.
2.6.6 Effect of multiple metals on the adsorption of Cr(VI) by Fe3O4-PAA-NH2. To investigate the effect of other heavy metals, including Cu(II), Zn(II), and Cd(II), and other metal ions, such as K(I), Na(I), Ca(II), Mg(II) (which are normally present in real water systems), in the solution on the adsorption of Cr(VI) by Fe3O4-PAA-NH2, Fe3O4-PAA-NH2 (30.0 mg) was added into the mixed-metal solution with an initial heavy metal concentration of 5.0 mg L−1 and pH of 3.0 for 10 min treatment.
2.6.7 Regeneration of Fe3O4-PAA-NH2. The Fe3O4-PAA-NH2 (30.0 mg) was used to treat a 20.0 mL solution with an initial Cr(VI) concentration of 5.0 mg L−1 for 15 min. After adsorption, the used Fe3O4-PAA-NH2 nanoparticles were separated from the solution using a permanent magnet. The separated Fe3O4-PAA-NH2 nanoparticles were placed into a conical flask containing 20 mL of deionized water and sonicated for 15 min for desorption, and then separated from the solution using a permanent magnet. Finally, the regenerated Fe3O4-PAA-NH2 nanoparticles were dried at 60 °C for 3 h for reuse.The Cr(VI) removal percentage (R%) is obtained using eqn (3):
| |
 | (3) |
where
C0 (mg L
−1) is the initial Cr(
VI) concentration, and
Ce (mg L
−1) is the final Cr(
VI) concentration in the solution after treatment. The Cr(
VI) removal capacity (
q, mg g
−1) is quantified by
eqn (4):
| |
 | (4) |
where
V (L) represents the volume of the Cr(
VI) solution, and
m (g) stands for the mass of the used Fe
3O
4-PAA-NH
2 nanoadsorbent.
3. Results and discussion
3.1. Structural characterization of amine-functionalized Fe3O4 nanoadsorbent
Fig. 1 illustrates the SEM microstructures of the as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2. The as-received Fe3O4 nanoparticles are ball-like in morphology with an average diameter of 13.3 nm, determined using the Nano measure software (Fig. 1(a)), which is consistent with the information obtained from the supplier. However, Fe3O4-PAA exhibits a different morphology from the as-received Fe3O4 nanoparticles. The Fe3O4-PAA is observed to be stuck together due to the polymerized PAA on the surface of the Fe3O4 nanoparticles (Fig. 1(b)). After amino-functionalization, the Fe3O4-PAA-NH2 becomes particulate again with an average diameter of 20.1 nm obtained from the Nano measure software (Fig. 1(c) and (d)). The increased nanoparticle diameter of Fe3O4-PAA-NH2 arises from the formed PAA and amino-functionalization process.
 |
| | Fig. 1 SEM micrographs of (a) as-received Fe3O4 nanoparticles, (b) Fe3O4-PAA, and (c) Fe3O4-PAA-NH2, and (d) enlarged picture of Fe3O4-PAA-NH2 nanoadsorbent. | |
Elemental mapping of microstructures by SEM with energy dispersive X-ray spectroscopy (EDS) is widely applied in science, engineering, and technology fields.24 An element map is an image to display the 2-dimensional elements distributions in a material. In this work, the elemental mapping is conducted on the synthesized Fe3O4-PAA-NH2 to further clarify the specific components of Fe3O4-PAA-NH2. Fig. 2 shows the zero loss image (a), elemental maps of (b) O, (c) C, (d) Fe, (e) N and (f) the summation of the O, C, Fe, N elements. The different colors are used to represent the different elements in order to identify their compositions in the synthesized Fe3O4-PAA-NH2. The presence of N element confirms the successful preparation of amino-functionalized Fe3O4-PAA.
 |
| | Fig. 2 SEM elemental mapping photographs of (a) Fe3O4-PAA-NH2 nanoparticles, (b) O map, (c) C map, (d) Fe map, (e) N map, and (f) C + O + Fe + N map. | |
Fig. 3(A) shows the FT-IR spectra of the as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2. The absorption peak at around 533 cm−1 in the as-received Fe3O4 nanoparticles, Fig. 3(A)-a, is attributed to the vibration of the Fe–O band,25 which shifts to 567 and 581 cm−1 for Fe3O4-PAA (Fig. 3(A)-b) and Fe3O4-PAA-NH2 (Fig. 3(A)-c), respectively. In the Fe3O4-PAA, Fig. 3(A)-b, the strong absorption peaks at 1552 and 1710 cm−1 correspond to the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the carboxylic groups, respectively.13 The peak at around 2909 cm−1 is related to the C–H stretching vibration of the PAA polymer backbone.26 The peak at 3354 cm−1 is assigned to the O–H stretching vibration in the carboxylic groups of PAA. For the Fe3O4-PAA-NH2, Fig. 3(A)-c, the peaks at around 3419 and 1320 cm−1 are due to the stretching and bending vibrations of N–H, respectively.27 The peak at 2924 cm−1 is related to the C–H stretching vibration. The band at 1563 cm−1 (C–N stretching vibration) together with the small band at 1642 cm−1 (CO stretching vibration) are the characteristic absorption peaks for the formation of –CONH– groups,13 which confirms the linkage of TETA onto the PAA polymer backbone.
O stretching vibrations of the carboxylic groups, respectively.13 The peak at around 2909 cm−1 is related to the C–H stretching vibration of the PAA polymer backbone.26 The peak at 3354 cm−1 is assigned to the O–H stretching vibration in the carboxylic groups of PAA. For the Fe3O4-PAA-NH2, Fig. 3(A)-c, the peaks at around 3419 and 1320 cm−1 are due to the stretching and bending vibrations of N–H, respectively.27 The peak at 2924 cm−1 is related to the C–H stretching vibration. The band at 1563 cm−1 (C–N stretching vibration) together with the small band at 1642 cm−1 (CO stretching vibration) are the characteristic absorption peaks for the formation of –CONH– groups,13 which confirms the linkage of TETA onto the PAA polymer backbone.
 |
| | Fig. 3 (A) FT-IR spectra of (a) as-received Fe3O4 nanoparticles, (b) Fe3O4-PAA, (c) Fe3O4-PAA-NH2; (B) pore size distribution of as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2; (C) TGA curves, (D) DSC curves of as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2; high resolution C1s XPS spectra of (E) Fe3O4-PAA, and (F) Fe3O4-PAA-NH2. | |
Fig. 3(B) depicts the pore size distribution of the as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2. It is observed that the pore diameter of these three samples is widely distributed within the range of 1.5–40 nm. For the as-received Fe3O4 nanoparticles, the highest pore volume of 0.05297 cm3 g−1 corresponds to the pore diameter of 15.1 nm. However, for Fe3O4-PAA, the highest pore volume of 0.03254 cm3 g−1 appears at pore diameters of 15.5 and 5.3 nm, and for Fe3O4-PAA-NH2, the highest pore volume of 0.03653 cm3 g−1 corresponds to the pore diameter of 8.7 nm. These results show that the pore volume of the Fe3O4 nanoparticles is reduced after polymerization of PAA and amino functionalization on the surface of the Fe3O4 nanoparticles. The BET specific area of the as-received Fe3O4 nanoparticles is measured to be 81.4216 m2 g−1, which is reduced to 61.6277 and 69.9001 m2 g−1 for Fe3O4-PAA and Fe3O4-PAA-NH2, respectively.
Fig. 3(C)&(D) show the TGA and DSC curves, respectively, of the as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2 in air. In the TGA curve of the as-received Fe3O4 nanoparticles, the slight weight loss before 150 °C is attributed to the loss of moisture and there is no weight change after a further increase in temperature. This is consistent with the DSC curve of the as-received Fe3O4 nanoparticles, in which no endothermic peak is observed during the whole procedure (Fig. 3(D)). However, for Fe3O4-PAA, there is a significant weight loss observed around the temperature range of 250–300 °C, which corresponds to the thermal degradation of the PAA polymer chains. In the DSC curve (Fig. 3(D)) the thermal degradation of the PAA polymer chain appears as a sharp endothermic peak (the temperature is around 260 °C). Normally, the Fe3O4 nanoparticles will become oxidized to form hematite (α-Fe2O3) at high temperature.25 The weight residue of Fe3O4-PAA in the TGA curve is determined to be 89.8% (weight percentage of α-Fe2O3) at 700 °C, which corresponds to 86.8 wt% of Fe3O4 nanoparticles. This means that the Fe3O4-PAA contains 13.2 wt% of PAA polymer. Meanwhile, the TGA profile of Fe3O4-PAA-NH2 is distinct from that of Fe3O4-PAA, in which two weight loss regions are observed. The weight loss from 100 to 200 °C is due to the elimination of the functionalized TETA small molecules on the PAA polymer backbone. The major weight loss around the temperature range of 250–300 °C is assigned to the thermal degradation of the PAA polymer backbone, which is similar to that of Fe3O4-PAA. Both the decomposition of the TETA small molecules and PAA polymer contributes to the broad endothermic peak in the DSC curve of Fe3O4-PAA-NH2 (Fig. 3(D)).
Fig. 3(E)&(F) show the deconvolution of the high-resolution C1s XPS spectra of Fe3O4-PAA and Fe3O4-PAA-NH2, respectively. The C1s peak from Fe3O4-PAA is deconvoluted into three major components with peaks at 284.9, 285.6 and 289.0 eV, which are attributed to the –CH2–, ≡CH, and CO of the PAA polymer backbone, respectively, as shown in Fig. 3(E).28 However, the C1s spectrum of Fe3O4-PAA-NH2 is distinct from that of Fe3O4-PAA, in which four curves is properly fitted (Fig. 3(F)). The first peak, located at 285.0 eV, is attributed to the aliphatic carbon of –CH2–. The higher binding energy of 286.1 arises from the C–C of PAA and TETA. The binding energy peak located at 289.1 eV is due to the carboxylate carbon (–COOH). Most importantly, the characteristic peak of amide (–CONH–) appears at 287.0 eV.29 These results indicate the successful fabrication of amino-functionalized Fe3O4-PAA nanocomposites. The amine content is determined to be 3.925 mg g−1 from the acid–base titrimetric analysis.
Fig. 4 shows the magnetization curves of the as-received Fe3O4 nanoparticles and Fe3O4-PAA-NH2 at room temperature. Both samples show no hysteresis loop in their magnetization curve. This means that the coercivity (Hc) is zero Oe, which indicates a superparamagnetic behavior.30 The magnetization of neither sample reaches saturation in the measured magnetic field range. Therefore, the saturation magnetization (Ms) is determined from the extrapolated Ms obtained from the intercept of M ∼ H−1 at high magnetic field.31 The Ms of the as-received Fe3O4 NPs is 54.47 emu g−1, which is smaller than that of the bulk Fe3O4 (92 emu g−1).32 The obtained Ms value of Fe3O4-PAA-NH2 is 47.71 emu g−1. This value is sufficient for the immediate recycling of Fe3O4-PAA-NH2 from a solution using a permanent magnet, as shown in the inset of Fig. 4. This is an essential factor in the separation and reuse of magnetic Fe3O4-PAA-NH2 for the purification of water.33 The weight percentage of Fe3O4 in the synthesized Fe3O4-PAA-NH2, estimated from the Ms value, is found to be around 87.0%, which is consistent with the TGA result (86.8%).
 |
| | Fig. 4 Magnetization curves of as-received Fe3O4 nanoparticles and Fe3O4-PAA-NH2. The inset shows that Fe3O4-PAA-NH2 can be attracted by a permanent magnet. | |
3.2. Evaluation of Cr(VI) removal by Fe3O4-PAA-NH2 nanoadsorbent
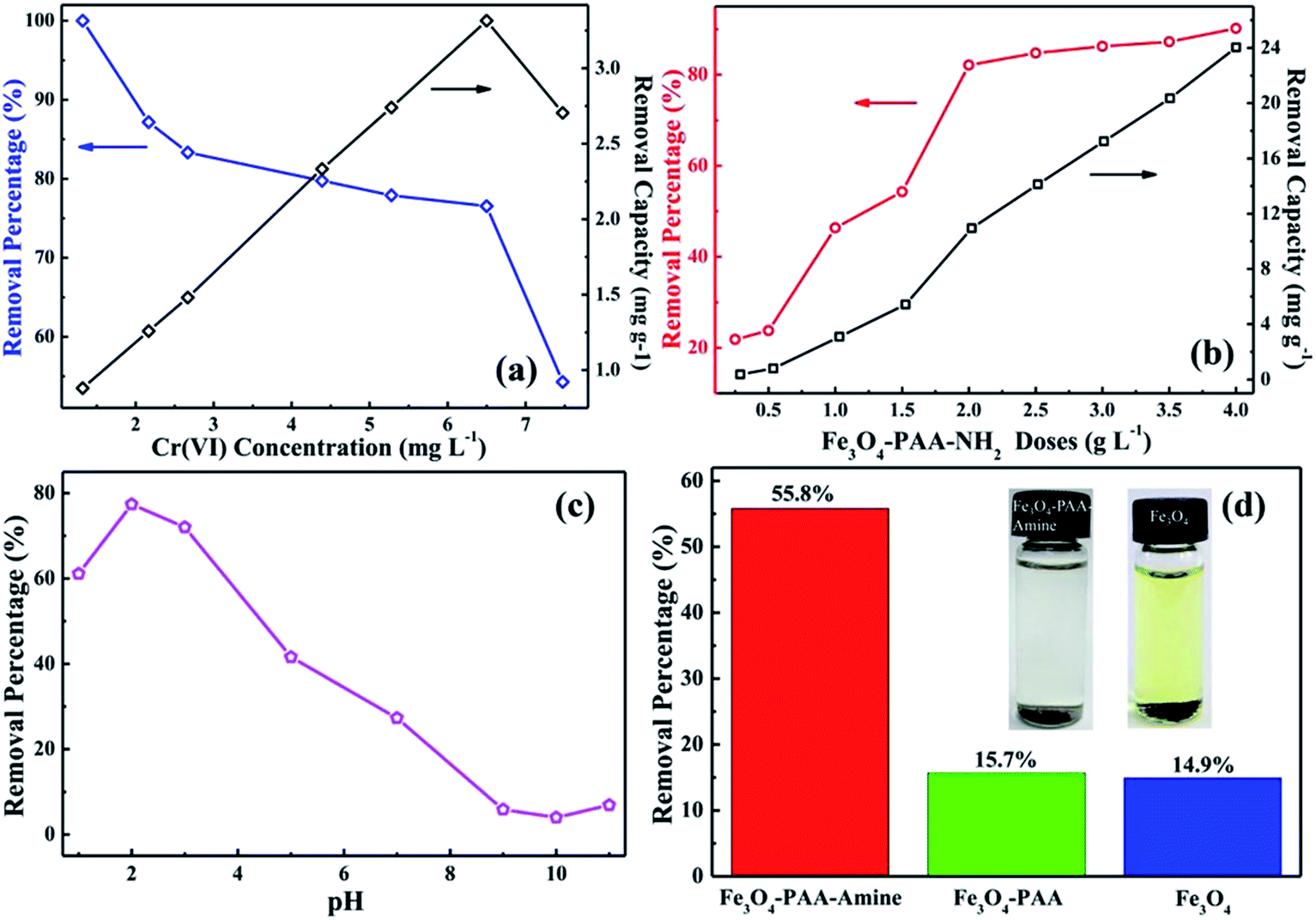
Fig. 5(a) shows the Cr(VI) removal percentage and removal capacity from the solutions with different initial Cr(VI) concentrations at pH of 3.0 after treatment with Fe3O4-PAA-NH2 for 10 min. The synthesized Fe3O4-PAA-NH2 with a weight of 30.0 mg is observed to be able to treat a 20.0 mL solution with an initial Cr(VI) concentration of 1.3 mg L−1 for 100% Cr(VI) removal, which achieves the limitation of the US EPA requirement. After further increasing the initial Cr(VI) concentration, the Cr(VI) removal percentage of Fe3O4-PAA-NH2 decreases to 54.3% for a solution with an initial Cr(VI) concentration of 7.5 mg L−1. In contrast, the Cr(VI) removal capacity increases from 0.88 to 3.31 mg g−1 for solutions with an initial Cr(VI) concentration changing from 1.3 to 6.5 mg L−1, and then decreases to 2.70 mg g−1 for solutions with an initial Cr(VI) concentration of 7.5 mg L−1. Normally, the active sites of the amine-functionalized nanoadsorbent result from the chelation between the heavy metals and the amine functional groups.34 As the gradual saturation of these active sites occurs at high Cr(VI) concentrations, the Fe3O4-PAA-NH2 cannot accommodate the excess Cr(VI), leading to the decreased Cr(VI) removal percentage and removal capacity as the initial Cr(VI) concentration increases to 7.5 mg L−1. A similar result is also observed with carboxyl group-functionalized multi-walled carbon nanotubes (MWNTs).4
 |
| | Fig. 5 (a) Removal percentage and removal capacity of 30.0 mg Fe3O4-PAA-NH2 for 20.0 mL Cr(VI) solutions with different initial Cr(VI) concentrations after 10 min contact time at pH of 3.0 at room temperature; (b) removal percentage and removal capacity of different Fe3O4-PAA-NH2 doses for 20.0 mL Cr(VI) solution with an initial concentration of 7.5 mg L−1 and pH of 3.0 after 10 min contact time at room temperature; (c) removal percentage of 30 mg Fe3O4-PAA-NH2 at different pH values of 20.0 mL Cr(VI) solutions with an initial concentration of 5.4 mg L−1 after 10 min contact time at room temperature; (d) removal percentage comparison of 10.0 mg Fe3O4-PAA-NH2, Fe3O4-PAA, and as-received Fe3O4 nanoparticles treated with 20.0 mL Cr(VI) solution with initial Cr(VI) concentration of 1.0 mg L−1 after 5 min contact time. The inset shows the Fe3O4-PAA-NH2 nanoadsorbent and as-received Fe3O4 nanoparticles after immersion in 1.0 mol L−1 HCl for one hour. | |
Fig. 5(b) shows the Cr(VI) removal percentage and removal capacity with different Fe3O4-PAA-NH2 doses in a 20.0 mL solution with initial Cr(VI) concentration of 7.5 mg L−1 and pH of 3.0 after 10 min treatment at room temperature. Both the Cr(VI) removal percentage and removal capacity are observed to increase with increasing Fe3O4-PAA-NH2 dose due to the increased active sites and amine groups for trapping Cr(VI).35 The Cr(VI) removal percentage increases from 21.8% for a Fe3O4-PAA-NH2 dose of 0.25 g L−1 to 90.2% for a Fe3O4-PAA-NH2 dose of 4 g L−1. Meanwhile, the Cr(VI) removal capacity increases from 0.36 mg g−1 for a Fe3O4-PAA-NH2 dose of 0.25 g L−1 to 24.0 mg g−1 for a Fe3O4-PAA-NH2 dose of 4 g L−1.
Fig. 5(c) shows the Cr(VI) removal percentage for the solutions with an initial Cr(VI) concentration of 5.4 mg L−1 and different pH values after treatment with the Fe3O4-PAA-NH2 nanoadsorbent for 10 min at room temperature. The Cr(VI) removal percentage by Fe3O4-PAA-NH2 is observed to be strongly dependent on the pH value of the solution. The Cr(VI) removal percentage increases from 61.1% for pH = 1.0 solution to 77.4% for pH = 2.0 solution and sharply decreases with increasing solution pH. The Cr(VI) removal percentages for pH = 3.0, 5.0, and 7.0 are 72.0, 41.6 and 27.3%, respectively. As solution pH increases to 9.0, 10.0, and 11.0, the Cr(VI) removal percentages are only around 5.9, 4.0, and 6.8%, respectively. These results demonstrate that the amine-functionalized Fe3O4 nanoadsorbent is not sufficient for the Cr(VI) removal if the pH of the solution is higher than 5, especially for the basic solution. Normally, the pH-dependent heavy metal removal performance is associated with both the metal chemistry in the solution and the type of adsorbent.36 As one of the heavy metals, the existence of Cr(VI) in solution is complicated and there are several species of Cr(VI) present in solutions with the different pH values. Generally, H2CrO4 is a strong acid, which can be ionized in aqueous solution as shown in eqn (5)&(6).37
| | |
H2CrO4 ⇌ H+ + HCrO4−, K7 = 10−0.75
| (5) |
| | |
HCrO4− ⇌ H+ + CrO42−, K8 = 10−6.45
| (6) |
In the pH < 2 solution, both H2CrO4 and HCrO4− are present in the solution; in the pH range from 2 to 4, HCrO4− is the predominant form in the solution; when the pH of the solution is higher than 4, CrO42− starts to appear and its content increases with increasing solution pH; when the pH of the solution is above 9, HCrO4− disappears, leaving only CrO42− in the solution.38 The Cr(VI) removal performance of the amine-functionalized nanoadsorbent is strongly related to the existence of these different forms of Cr(VI). The obtained results from Fig. 5(c) indicate that the amine group prefers to chelate with HCrO4− ions in the solution rather than to chelate with H2CrO4 and CrO42−. Therefore, Fe3O4-PAA-NH2 delivers a good Cr(VI) removal performance within the pH range of 2–3 and a poor performance at pH > 5. The proposed Cr(VI) removal mechanism is illustrated in Scheme 2. These results are consistent with the mechanism reported by Mohan et al.18 They confirmed that the Cr(VI) adsorption on aminated polyacrylonitrile fibers was affected by both the electrostatic attraction and surface complexation at low solution pH values (around pH of 3.0). Mayer-Gall et al.39 also reported that the amino groups are easily protonated to form the ammonium salts in the acidic solution, which could adsorb the negatively charged chromate ion.
 |
| | Scheme 2 Proposed adsorption of Cr(VI) species on Fe3O4-PAA-NH2 in the pH 2–4 solution. | |
Fig. 5(d) illustrates the Cr(VI) removal percentage in a solution with an initial Cr(VI) concentration of 1.0 mg L−1 after treatment with the as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2 (10.0 mg) for 5 min for comparison. The as-received Fe3O4 nanoparticles only exhibit Cr(VI) removal from the aqueous solution with a low removal percentage of 14.9%, which is lower than that of a previous report (around 22%).40 Fe3O4-PAA also shows a poor Cr(VI) removal percentage of 15.7%. In contrast, the synthesized Fe3O4-PAA-NH2 exhibits a Cr(VI) removal percentage of 55.8%, which is almost 4 times higher than the as-received Fe3O4 nanoparticles and the prepared Fe3O4-PAA. As previously mentioned, the BET results show that the average specific surface areas for the as-received Fe3O4 nanoparticles, Fe3O4-PAA, and Fe3O4-PAA-NH2 are 81.4216, 61.6277, and 69.9001 m2 g−1, respectively. Even though the specific surface area decreases for the Fe3O4 nanoparticles after amino functionalization, the Cr(VI) removal percentage is increased. These results demonstrate that the amine group plays an important role in the Cr(VI) removal from the polluted water system. This further confirms the significance of the functional groups on the adsorbent for water treatment. A 1 mol L−1 HCl solution is used to measure the stability of the as-received Fe3O4 nanoparticles and synthesized Fe3O4-PAA-NH2 in acidic solution. The obtained results are shown in the inset of Fig. 5(d). After immersion in the acidic solution for 1 h, the as-received Fe3O4 nanoparticles are observed to be dissolved in the acid solution with the formation of a light yellow-green color. However, the Fe3O4-PAA-NH2 is still at the bottom of the solution and the color of the solution is still clear without any changes. These results confirm that the amine-functionalized Fe3O4-PAA nanoadsorbent is more stable in acid solution than the as-received Fe3O4 nanoparticles.
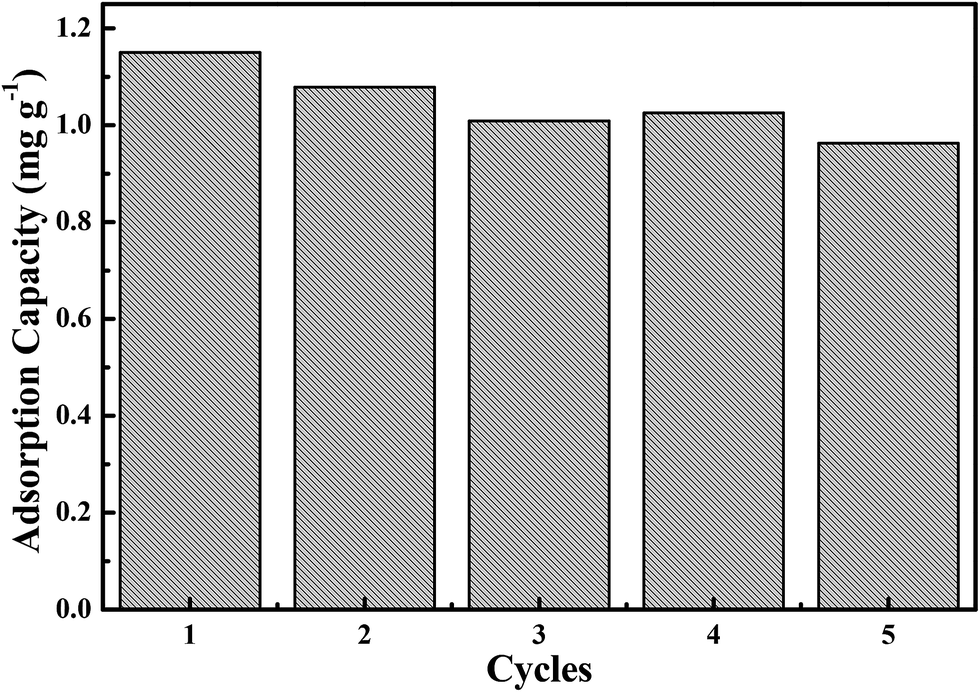
From an economic and practical point of view, the regeneration and reusability was investigated in this work. After treating a Cr(VI) solution, the Fe3O4-PAA-NH2 was placed into deionized water and sonicated for 15 min for desorption at room temperature. The regenerated Fe3O4-PAA-NH2 was used to treat an initial Cr(VI) concentration of 5.0 mg L−1 (20 mL) for 15 min. The adsorption–desorption process was conducted for 5 cycles and the obtained adsorption capacity is depicted in Fig. 6. It is observed that the adsorption capacity of Fe3O4-PAA-NH2 still remains around 85% after 5 cycles, exhibiting relatively good reusability and stability. This indicates that the synthesized Fe3O4-PAA-NH2 can be used as a reversible efficient nanoadsorbent for practical wastewater treatment.
 |
| | Fig. 6 Cr(VI) adsorption capacity of the regenerated Fe3O4-PAA-NH2 in a 20 mL solution with an initial Cr(VI) concentration of 5.0 mg L−1 and a contact time of 15 min. | |
3.3. Cr(VI) adsorption isotherm of Fe3O4-PAA-NH2 nanoadsorbent
The adsorption isotherm can describe the adsorption equilibrium of adsorbate at the surface of an adsorbent. The adsorption equilibrium models can provide some insights into the adsorption mechanism, the surface properties and the affinity of the adsorbent.41 Two commonly used isotherm models, namely the Langmuir and Freundlich isotherm models, can explain solid–liquid adsorption systems. The Langmuir model is often used to describe monomolecular layer adsorption, which assumes uniform distribution of adsorption energy on the surface and no migration of adsorbate in the plane of surface.42 The Langmuir model is expressed as eqn (7):| |
 | (7) |
where Ce is the equilibrium concentration (mg L−1) of Cr(VI), qe is the amount of adsorbed Cr(VI) at equilibrium (mg g−1), and a (mg g−1) and b (L mg−1) are Langmuir isotherm parameters. The Freundlich model is an empirical equation, which focuses on the adsorption on an energetically heterogeneous surface. The expression of the Freundlich model is given as eqn (8), which provides the relationship between the adsorbed concentration and the solute concentration:where kf is the Freundlich equilibrium constant, indicating the extent of adsorption, and η is the power term of the Freundlich isotherm and the heterogeneity factor, illustrating the intensity of the adsorption.43
In order to study the adsorption equilibrium of Cr(VI) at the surface of the nanoadsorbent, the synthesized Fe3O4-PAA-NH2 (20.0 mg) was used to treat different initial Cr(VI) concentrations varying from 0.5 to 4.5 mg L−1 with a contact time of 15 min at room temperature. Both Langmuir and Freundlich isotherm models are used to fit the Cr(VI) adsorption equilibrium process of Fe3O4-PAA-NH2. The obtained results are shown in Fig. 7 and the fitting parameters are listed in Table 1. According to the correlation coefficient values, the Freundlich isotherm model (R2 = 0.976) is observed to be well-fitted for the Cr(VI) adsorption by Fe3O4-PAA-NH2, demonstrating a multilayer adsorption mechanism. However, the correlation coefficient values for these two models only show a little difference for the Cr(VI) removal adsorption (R2 = 0.960 for the Langmuir model). The maximum Cr(VI) adsorption capacity by Fe3O4-PAA-NH2 obtained from parameter a of the Langmuir model is 9.791 mg g−1, which is higher than those of activated carbon (around 1.1–9.5 mg g−1)18 and chitosan flakes (around 5.246–7.943 mg g−1)44 and lower than those of magnetic carbon nanocomposites prepared from Fe(NO3)3/cellulose (15.3 mg g−1),45 chitosan-coated gauze containing amino groups (12.4 mg g−1),46 and chitosan/poly(vinyl alcohol) containing cerium(III) (52.8 mg g−1).47
 |
| | Fig. 7 Adsorption isotherm of 20.0 mL Cr(VI) solution (5 mg L−1) on Fe3O4-PAA-NH2 (20 mg) with 15 min contact time at room temperature. | |
Table 1 Langmuir and Freundlich adsorption isotherm parameters
| Parameter |
Langmuir |
Freundlich |
| a (mg g−1) |
b (L mg−1) |
R2 |
kf (mg1−η Lη g−1) |
η |
R2 |
| Value |
9.791 |
0.074 |
0.960 |
0.731 |
0.799 |
0.976 |
3.4. Cr(VI) removal kinetics of Fe3O4-PAA-NH2 nanoadsorbent
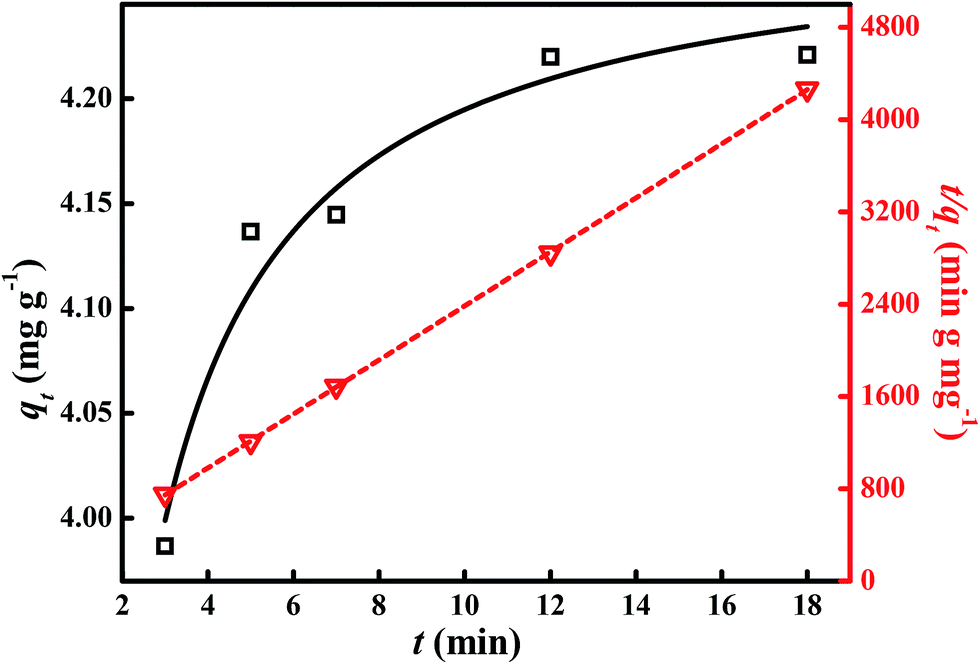
The adsorption kinetics are one of the important parameters to describe the Cr(VI) uptake rate and control of the residue time of the adsorbent at the solid–solution interface, from which the efficiency of the Cr(VI) adsorption by the nanoadsorbent can be determined.48 Therefore, the Cr(VI) removal kinetics are investigated to understand the adsorption behavior of Cr(VI) by the prepared Fe3O4-PAA-NH2 nanoadsorbent. The amount of adsorbed Cr(VI) (qt, mg g−1) at time t is calculated using eqn (9):| |
 | (9) |
where C0 is the initial Cr(VI) concentration (mg L−1) and Ct represents the Cr(VI) concentration in mg L−1 at time t (min). The calculated qt in solution with pH = 2.5 after treatment with Fe3O4-PAA-NH2 nanoadsorbent for different contact times (3, 5, 7, 12, and 18 min) is depicted in Fig. 8. It is observed that the qt increases until contact time reaches to 12 min and then become flat after the contact time increases further to 18 min. This indicates that the adsorption of Cr(VI) by the Fe3O4-PAA-NH2 nanoadsorbent gradually becomes saturated as time increases to 18 min.49 An appropriate kinetic model is required to quantify the changes in adsorbed Cr(VI) with time. Hence, the pseudo-second-order adsorption kinetic model is introduced to describe the Cr(VI) adsorption behavior by the Fe3O4-PAA-NH2 nanoadsorbent, which is expressed by eqn (10):50| |
 | (10) |
where k2 is the pseudo-second-order adsorption rate constant (g mg−1 min−1), and qe stands for the adsorption capacity at equilibrium time (mg g−1). If the pseudo-second-order adsorption kinetic model is suitable for this system, the plot of t/qt ∼ t is supposed to give a linear line, and the qe and k2 can be calculated from the slope coefficient (1/qe) and intercept (1/k2qe2) of the plot. The obtained corresponding kinetic plot is also shown in Fig. 8. A good linear fit of the t/qt ∼ t plot is observed with a fitting correlation coefficient R2 of 0.9996. This means that the Cr(VI) adsorption kinetics of the Fe3O4-PAA-NH2 nanoadsorbent obey pseudo-second-order adsorption behavior. The obtained equilibrium adsorption capacity of qe is 4.27 mg g−1 at the initial Cr(VI) concentration of 7.0 mg L−1, which is consistent with the calculated qt at 18 min of 4.22 mg g−1 (Fig. 8). The adsorption rate constant k2 is calculated to be 1.23 g mg−1 min−1, which is higher than that of chitosan/nanoclay bionanocomposites (8.763 × 10−4 g mg−1 min−1).51 The initial adsorption rate h (mg g−1 min−1) at time t approaching zero is defined as eqn (11):52
 |
| | Fig. 8 Cr(VI) removal rate qt vs. t for 30.0 mg Fe3O4-PAA-NH2 after treatment with 20.0 mL Cr(VI) solution (7.0 mg L−1) under different contact times at room temperature (square line), and corresponding kinetic plot (triangle line). | |
The calculated results show that the prepared Fe3O4-PAA-NH2 nanoadsorbent exhibits an initial adsorption rate of 22.51 mg g−1 min−1, which is much higher than those of magnetic graphene material (0.28 mg g−1 min−1)6 and magnetic polypropylene nanocomposites (0.229 mg g−1 min−1).8
3.5. Effect of multiple metals on the adsorption of Cr(VI)
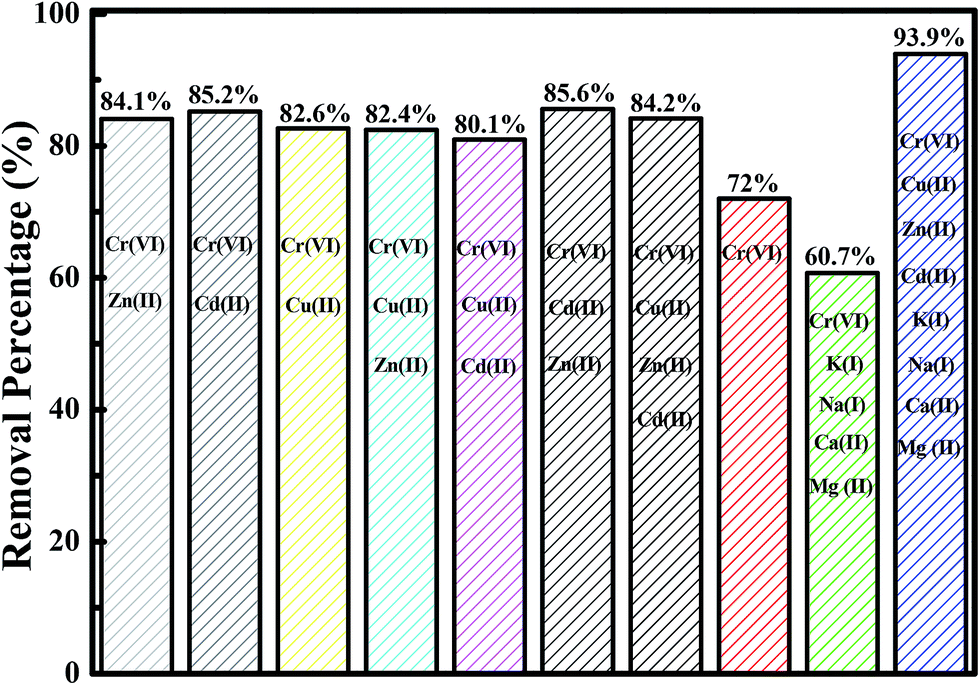
Normally, many different kinds of toxic heavy metals coexist in polluted water systems53 and the investigation of the effect of these multiple metal ions on the adsorption of Cr(VI) is most important since the other heavy metal ions may possess the active sites of the adsorbent and compete with each other for the adsorption sites.54 Meanwhile, the presence of other metals, such as K, Ca, Na, and Mg, may also influence the adsorption process.55 In this work, the competition adsorption of multiple heavy metals, including Zn(II), Cd(II), and Cu(II), in the solution with equal initial heavy metal concentration is explored and the effect of the presence of K, Ca, Na and Mg is also investigated. The obtained results are shown in Fig. 9. For the solution with Cr(VI), the Cr(VI) removal percentage is around 72%. However, for the bi-metal solutions consisting of Cr(VI) with Zn(II), Cr(VI) with Cd(II), and Cr(VI) with Cu(II), the Cr(VI) removal percentages are 84.1, 85.2 and 82.6%, respectively. For the tri-metal solutions composed of Cr(VI) with Cu(II)&Zn(II), Cr(VI) with Cu(II)&Cd(II), and Cr(VI) with Cd(II)&Zn(II), the Cr(VI) removal percentages are 82.4, 80.1, and 85.6%, respectively. For the tetra-metal solution of Cr(VI), Cu(II), Zn(II), and Cd(II), the Cr(VI) removal percentage is found to be 84.2%. These results indicate that after introduction of multiple heavy metal ions, such as Cu(II), Zn(II), and Cd(II), into the solution, the Cr(VI) removal performance is increased. The presence of Cu(II), Zn(II), and Cd(II) favors the Cr(VI) adsorption by the fabricated Fe3O4-PAA-NH2. The heavy metal Cd(II) is suggested to have greater effect on the Cr(VI) removal performance than the heavy metals Zn(II) and Cu(II). Similar results are also reported by Ghorbel-Abid et al.56 They found that Cd(II) could significantly increase the adsorption capacity for Cr(VI) arising from the affinity of the chemical properties of each element (such as chemical potential, electronegativity, etc.). For the solution consisting of Cr(VI), K(I), Ca(II), Na(I), and Mg(II), the Cr(VI) removal percentage is 60.7%. However, the Cr(VI) removal percentage is increased to 93.9% for the solution containing eight metal ions.
 |
| | Fig. 9 Cr(VI) removal percentage in the multi-metal solutions including Cr(VI), Cu(II), Zn(II), Cd(II), K(I), Ca(II), Na(II), and Mg(II) with an equal initial concentration of 5.0 mg L−1 and pH of 3.0 after treatment for 10 min with 30 mg Fe3O4-PAA-NH2. | |
4. Conclusions
In this study, the ability of a magnetic amine-functionalized Fe3O4-PAA-NH2 nanoadsorbent synthesized using the SIP method to remove Cr(VI) from polluted water is evaluated. Even though the average specific surface area of Fe3O4-PAA-NH2 is lower than that of as-received Fe3O4 nanoparticles, as shown by the BET results, the Cr(VI) removal percentage of Fe3O4-PAA-NH2 is almost 4 times higher than that of the as-received Fe3O4 nanoparticles due to the presence of amine groups on the surface of Fe3O4-PAA-NH2. 30 mg of the Fe3O4-PAA-NH2 nanoadsorbent is able to completely remove Cr(VI) from a 20.0 mL solution with initial Cr(VI) concentration of 1.32 mg L−1 after 10 min contact time, which can satisfy the limitation of the US EPA requirement. The Fe3O4-PAA-NH2 dose of 4 g L−1 could remove 90.2% of Cr(VI) from a 20.0 mL 7.5 mg L−1 Cr(VI) solution with pH of 3.0. The Cr(VI) removal percentage of the Fe3O4-PAA-NH2 nanoadsorbent is observed to highly depend on the pH value of the solution and the optimal pH value for Cr(VI) removal is found in the solution with pH of 2.0. The Cr(VI) adsorption isotherm is fitted to the Freundlich isotherm model. The Cr(VI) removal kinetics of the Fe3O4-PAA-NH2 nanoadsorbent obeys pseudo-second-order behavior and the calculated room temperature rate constant is 1.23 g mg−1 min−1 for the solution with an initial Cr(VI) concentration of 7.0 mg L−1 and pH value of 2.5. The presence of other metals, including Cu(II), Zn(II), Cd(II), K(I), Ca(II), Na(I), and Mg(II), in polluted water is beneficial for the Cr(VI) adsorption by the Fe3O4-PAA-NH2 nanoadsorbent. Meanwhile, the prepared Fe3O4-PAA-NH2 nanoadsorbent can prevent acid etching of the Fe3O4 nanoparticles and be easily recycled from water using a permanent magnet for reuse. The prepared Fe3O4-PAA-NH2 nanoadsorbent still retains around 85% of Cr(VI) adsorption performance even after 5 cycles.
Acknowledgements
The authors thank the financial support from the Science and Technology Commission of Shanghai Municipality (no. 15YF1412700) and Tongji University (no. 2014KJ028).
References
- N. Wu, H. Wei and L. Zhang, Environ. Sci. Technol., 2011, 46, 419–425 CrossRef PubMed.
- H. Gu, S. Rapole, J. Sharma, Y. Huang, D. Cao, H. A. Colorado, Z. Luo, N. Haldolaarachchige, D. P. Young, S. Wei and Z. Guo, RSC Adv., 2012, 2, 11007–11018 RSC.
- Y. Ni, K. Mi, C. Cheng, J. Xia, X. Ma and J. Hong, Chem. Commun., 2011, 47, 5891–5893 RSC.
- H. Gu, S. B. Rapole, Y. Huang, D. Cao, Z. Luo, S. Wei and Z. Guo, J. Mater. Chem. A, 2013, 1, 2011–2021 CAS.
- J. Gong, T. Liu, X. Wang, X. Hu and L. Zhang, Environ. Sci. Technol., 2011, 45, 6181–6187 CrossRef CAS PubMed.
- J. Zhu, S. Wei, H. Gu, S. B. Rapole, Q. Wang, Z. Luo, N. Haldolaarachchige, D. P. Young and Z. Guo, Environ. Sci. Technol., 2012, 46, 977–985 CrossRef CAS PubMed.
- D. Zhang, S. Wei, C. Kaila, X. Su, J. Wu, A. B. Karki, D. P. Young and Z. Guo, Nanoscale, 2010, 2, 917–919 RSC.
- J. Zhu, H. Gu, S. B. Rapole, Z. Luo, S. Pallavkar, N. Haldolaarachchige, T. J. Benson, T. C. Ho, J. Hopper, D. P. Young, S. Wei and Z. Guo, RSC Adv., 2012, 2, 4844–4856 RSC.
- Z. Liu, H. Wang, C. Liu, Y. Jiang, G. Yu, X. Mu and X. Wang, Chem. Commun., 2012, 48, 7350–7352 RSC.
- G. Zhao, J. Li, X. Ren, C. Chen and X. Wang, Environ. Sci. Technol., 2011, 45, 10454–10462 CrossRef CAS PubMed.
- K. Kadirvelu, C. Faur-Brasquet and P. L. Cloirec, Langmuir, 2000, 16, 8404–8409 CrossRef CAS.
- M. A. A. Zaini, R. Okayama and M. Machida, J. Hazard. Mater., 2009, 170, 1119–1124 CrossRef CAS PubMed.
- J.-K. Yang, H.-J. Park, H.-D. Lee and S.-M. Lee, Colloids Surf., A, 2009, 337, 154–158 CrossRef CAS PubMed.
- M. Fernandez-Garcia, A. Martinez-Arias, J. Hanson and J. Rodriguez, Chem. Rev., 2004, 104, 4063–4104 CrossRef CAS PubMed.
- W. Zhang, X. Shi, Y. Zhang, W. Gu, B. Li and Y. Xian, J. Mater. Chem. A, 2013, 1, 1745–1753 CAS.
- E.-J. Kim, C.-S. Lee, Y.-Y. Chang and Y.-S. Chang, ACS Appl. Mater. Interfaces, 2013, 5, 9628–9634 CAS.
- S.-J. Park and Y.-S. Jang, J. Colloid Interface Sci., 2002, 249, 458–463 CrossRef CAS PubMed.
- D. Mohan and C. U. Pittman Jr, J. Hazard. Mater., 2006, 137, 762–811 CrossRef CAS PubMed.
- D. Sud, G. Mahajan and M. Kaur, Bioresour. Technol., 2008, 99, 6017–6027 CrossRef CAS PubMed.
- B. Gao, J. Lu, R. Zhuang and G. Zhang, J. Appl. Polym. Sci., 2009, 114, 3487–3494 CrossRef CAS PubMed.
- C. Zhang, J. Sui, J. Li, Y. Tang and W. Cai, Chem. Eng. J., 2012, 210, 45–52 CrossRef CAS PubMed.
- X. Xin, Q. Wei, J. Yang, L. Yan, R. Feng, G. Chen, B. Du and H. Li, Chem. Eng. J., 2012, 184, 132–140 CrossRef CAS PubMed.
- B. Qiu, J. Guo, X. Zhang, D. Sun, H. Gu, Q. Wang, H. Wang, X. Wang, X. Zhang and B. L. Weeks, ACS Appl. Mater. Interfaces, 2014, 6, 19816–19824 CAS.
- D. E. Newbury and N. W. M. Ritchie, J. Anal. At. Spectrom., 2013, 28, 973–988 RSC.
- J. Guo, H. Gu, H. Wei, Q. Zhang, N. S. Haldolaarachchige, Y. Li, D. P. Young, S. Wei and Z. Guo, J. Phys. Chem. C, 2013, 117, 10191–10202 CAS.
- M. Imamoglu and O. Tekir, Desalination, 2008, 228, 108–113 CrossRef CAS PubMed.
- A.-N. A. El-Hendawy, J. Hazard. Mater., 2009, 167, 260–267 CrossRef CAS PubMed.
- B. C. Beard and P. Spellane, Chem. Mater., 1997, 9, 1949–1953 CrossRef CAS.
- H. Hasar, J. Hazard. Mater., 2003, 97, 49–57 CrossRef CAS.
- H. Gu, S. Tadakamalla, Y. Huang, H. A. Colorado, Z. Luo, N. Haldolaarachchige, D. P. Young, S. Wei and Z. Guo, ACS Appl. Mater. Interfaces, 2012, 4, 5613–5624 CAS.
- Y. Li, H. Zhu, H. Gu, H. Dai, Z. Fang, N. J. Weadock, Z. Guo and L. Hu, J. Mater. Chem. A, 2013, 1, 15278–15283 CAS.
- H. Gu, Y. Huang, X. Zhang, Q. Wang, J. Zhu, L. Shao, N. Haldolaarachchige, D. P. Young, S. Wei and Z. Guo, Polymer, 2012, 53, 801–809 CrossRef CAS PubMed.
- S. Li, Y. Gong, Y. Yang, C. He, L. Hu, L. Zhu, L. Sun and D. Shu, Chem. Eng. J., 2015, 260, 231–239 CrossRef CAS PubMed.
- L. Shao, X. Chang, Y. Zhang, Y. Huang, Y. Yao and Z. Guo, Appl. Surf. Sci., 2013, 280, 989–992 CrossRef CAS PubMed.
- J. Hu, C. Chen, X. Zhu and X. Wang, J. Hazard. Mater., 2009, 162, 1542–1550 CrossRef CAS PubMed.
- S. Mor, K. Ravindra and N. R. Bishnoi, Bioresour. Technol., 2007, 98, 954–957 CrossRef CAS PubMed.
- P. Mishra and R. Patel, J. Hazard. Mater., 2009, 168, 319–325 CrossRef CAS PubMed.
- K. Kadirvelu, C. Faur-Brasquet and P. L. Cloirec, Langmuir, 2000, 16, 8404–8409 CrossRef CAS.
- T. Mayer-Gall, K. Opwis and J. S. Gutmann, J. Mater. Chem. A, 2015, 3, 386–394 CAS.
- P. Yuan, D. Liu, M. Fan, D. Yang, R. Zhu, F. Ge, J. Zhu and H. He, J. Hazard. Mater., 2010, 173, 614–621 CrossRef CAS PubMed.
- M. Brdar, M. Šćiban, A. Takači and T. Došenović, Chem. Eng. J., 2012, 183, 108–111 CrossRef CAS PubMed.
- R. Cheng, S. Ou, M. Li, Y. Li and B. Xiang, J. Hazard. Mater., 2009, 172, 1665–1670 CrossRef CAS PubMed.
- S. Sharma and G. P. Agarwal, J. Colloid Interface Sci., 2001, 243, 61–72 CrossRef CAS.
- Y. A. Aydin and N. D. Aksoy, Chem. Eng. J., 2009, 151, 188–194 CrossRef CAS PubMed.
- B. Qiu, H. Gu, X. Yan, J. Guo, Y. Wang, D. Sun, Q. Wang, M. Khan, X. Zhang, B. L. Weeks, D. P. Young, Z. Guo and S. Wei, J. Mater. Chem. A, 2014, 2, 17454–17462 CAS.
- F. Ferrero, C. Tonetti and M. Periolatto, Carbohydr. Polym., 2014, 110, 367–373 CrossRef CAS PubMed.
- F. Wang and M. Ge, Text. Res. J., 2012, 83, 628–637 CrossRef PubMed.
- E. Demirbas, M. Kobya, E. Senturk and T. Ozkan, Water, 2004, 30, 533–539 CAS.
- P. A. Kumar, M. Ray and S. Chakraborty, Chem. Eng. J., 2009, 149, 340–347 CrossRef CAS PubMed.
- C. Gerente, V. K. C. Lee, P. L. Cloirec and G. McKay, Crit. Rev. Environ. Sci. Technol., 2007, 37, 41–127 CrossRef CAS PubMed.
- S. Pandey and S. B. Mishra, J. Colloid Interface Sci., 2011, 361, 509–520 CrossRef CAS PubMed.
- A. Gürses, Ç. Doğar, M. Yalçın, M. Açıkyıldız, R. Bayrak and S. Karaca, J. Hazard. Mater., 2006, 131, 217–228 CrossRef PubMed.
- S.-Y. Wang, M.-H. Tsai, S.-F. Lo and M.-J. Tsai, Bioresour. Technol., 2008, 99, 7027–7033 CrossRef CAS PubMed.
- F. Lu, L. Gu, M. J. Meziani, X. Wang, P. G. Luo, L. M. Veca, L. Cao and Y. P. Sun, Adv. Mater., 2009, 21, 139–152 CrossRef CAS PubMed.
- S. Yalçin and R. Apak, Anal. Chim. Acta, 2004, 505, 25–35 CrossRef.
- I. Ghorbel-Abid and M. Trabelsi-Ayadi, Arabian J. Chem., 2015, 8, 25–31 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.