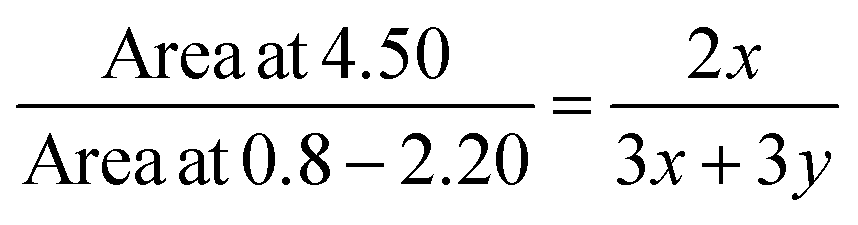DOI:
10.1039/C4RA00864B
(Paper)
RSC Adv., 2014,
4, 28653-28663
Synthesis of conductive polyaniline-modified polymers via a combination of nitroxide-mediated polymerization and “click chemistry”
Received
30th January 2014
, Accepted 19th June 2014
First published on 19th June 2014
Abstract
A new strategy for graft copolymerization of aniline onto polystyrene by a multi step process is suggested. For this purpose, firstly poly(styrene-co-4-chloromethyl styrene) [P(St-co-CMSt)] was synthesized via nitroxide-mediated polymerization (NMP) and then the chlorine groups of copolymer were converted to azide groups by a substitution nucleophilic reaction in the presence of a solvent composed of sodium azide (NaN3), and dry N,N-dimethylformamide (DMF). Afterwards, “click” cycloaddition reaction was employed for the introduction of phenylamine groups into the polystyrenic backbone by reaction between the azido copolymer and 4-aminobenzonitrile in the presence of anhydrous ZnCl2 as the catalyst. The graft copolymerization of aniline monomers onto phenylamine-functionalized polystyrene was initiated by oxidized phenylamine groups after addition of ammonium peroxydisulfate (APS), and p-toluenesulfonic acid-doped polyaniline was chemically grafted onto the polystyrenic backbone via oxidation polymerization. The obtained graft copolymer was characterized by 1H nuclear magnetic resonance (NMR) and Fourier transform infrared (FTIR) spectroscopy, and its electroactivity behavior was verified under cyclic voltammetric conditions. Moreover, the thermal behaviors of the obtained polymers were examined by differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA).
1. Introduction
In recent decades a great deal of research effort has focused on the development of new organic materials for both optical and electronic applications. Among the wide range of materials that may be suitable for these applications intrinsically conductive polymers (ICPs) are of particular interest.1–5 On the other hand, since the discovery of doped polyacetylene as a conductive polymer in 1977 (ref. 6) it has been found that only a few polymers are stable enough under normal processing conditions to be incorporated in practical applications. Among leading candidates are polythiophene (PTh), polypyrrole (PPy), and polyaniline (PANI).7–12 In these important conductive polymers, polyaniline has many advantages over other conducting polymers such as polythiophene, and polypyrrole. Polyaniline is thermally stable up to 250 °C and can be easily synthesized chemically, and electrochemically via oxidative polymerization in various organic solvents and/or in aqueous media.5,13 Moreover, polyaniline has stimulated great interest due to its low cost, good environmental stability,14,15 adequate level of electrical conductivity,16,17 and wide range of commercial and technological applications such as secondary batteries,18,19 electromagnetic interference (EMI) shielding,20,21 solar cells,22,23 bio/chemical sensors,24,25 corrosion devices,26,27 and many more.
However, the applications rang of polyaniline, similar to other π-conjugated polymers, is limited due to its insolubility and infusibility. Lack of solubility of PANI may be resulted from the stiffness of its main chain and the existence of a strongly conjugated π electron system. Therefore, in order to improve the solubility and induce fusibility of the stiff chain of this polymer, various procedures have been adapted. For example, self-doped PANI with sulfonic acid groups substituted onto the polymer has been synthesized.28,29 Several rings and N-substituted PANI (soluble in common organic solvents) have been prepared directly from the polymerization of the corresponding aniline monomers.30,31 Other effective way to overcome the certain deficiencies of polyaniline is synthesis of its copolymers with processable thermoplastics. Currently, to the best of our knowledge, controlled radical polymerization (CRP) methods are the most widely used approaches to prepare PANI copolymers.5,7,8,32
Controlled radical polymerization has received significant attention in recent years, as it allows production of polymers with controlled molecular weight, narrow dispersity index, and complex macromolecular architectures.33–35 Based on this technique, three main types of controlled radical polymerizations have been investigated: nitroxide-mediated polymerization (NMP),36,37 atom transfer radical polymerization (ATRP),38,39 and reversible addition-fragmentation chain transfer polymerization (RAFT).40,41
Graft copolymers are generally synthesized using macromonomer techniques,42–44 by the linkage between two different polymers through a coupling reaction,8,45 or by the polymerization of a monomer from active sites on a polymer backbone.32,46 Macromonomers are usually referred to as reactive oligomers or polymers in which a polymerizable functional group is incorporated into the chain end(s). Macromonomers can be synthesized using various methods, including anionic, cationic, and radical polymerization, as well as the chemical modification of polymer ends.42,47 The development of new polymeric materials usually requires the use of highly selective and efficient functionalization reactions. In this respect, “click chemistry” has been emerged as one of the most powerful tool for functionalization of polymeric materials.48 The copper catalyzed reaction of azides with alkynes to produce 1,2,3-triazoles regioselectively (the 1,3-dipolar Huisgen cycloaddition) has become the most widely used “click” reaction.49,50 Moreover, the 1,3-dipolar cycloaddition of azide to organic nitriles in the presence of a protic or Lewis acid at higher temperature leading to 5-substituted tetrazoles, is an example of a “click chemistry” reaction.51,52
For the first time, graft copolymerization of aniline onto polystyrene via a combination of nitroxide-mediated polymerization (NMP) and “click chemistry” reaction is reported. For this purpose, at first poly(styrene-co-4-chloromethyl styrene) was synthesized via nitroxide-mediated polymerization and then the phenylamine-functionalized polystyrene macromonomer (PhAPStM) was synthesized by “click” cycloaddition reaction between azide-functionalized polystyrene and 4-aminobenzonitrile in the presence of anhydrous ZnCl2 as the catalyst. The resultant macromonomer subsequently copolymerized with aniline momomers via oxidation polymerization method to produce a polystyrene-graft-polyaniline graft copolymer.
2. Experimental
2.1. Materials
Styrene, 4-chloromethyl styrene, and aniline monomers from Merck (Darmstadt, Germany) were distilled under a reduced pressure before use. 2,2,6,6-Tetramethyl-1-piperidinyloxy (TEMPO) was prepared in our laboratory.7,36,46 Tetrahydrofuran (THF) from Merck was dried by refluxing over sodium and distilled under argon prior to use. Ammonium peroxydisulfate (APS) from Merck was recrystallized at room temperature from ethanol–water. Sodium azide, 4-aminobenzonitrile, and zinc chloride (ZnCl2) were provided from Merck and were used as received. All other reagents were purchased from Merck and purified according to the standard methods.
2.2. Synthesis of P(St-co-CMSt) via NMP
Styrene (3.5 ml, 30 mmol), 4-chloromethyl styrene (1.1 ml, 8 mmol), benzyl peroxide (0.05 g, 0.20 mmol), and TEMPO (0.05 g, 0.30 mmol) were placed in an ampoule degassed with several freeze–pump–thaw cycles, sealed off under vacuum and placed in an oil bath at 95 °C for 4 hours. At the end of this period, the temperature was increased to 130 °C and the mixture stirred for another 12 hours. The product was cooled, diluted with THF and precipitated into excess methanol. The solid was dried overnight in vacuum at room temperature (Scheme 1). The 4-chloromethyl styrene content in the copolymer was 19.8% (by mole) based on peak at 4.50 ppm (–CH2Cl) in the 1H NMR spectrum (yield: 3.52 g, 80.5%).
 |
| | Scheme 1 Synthesis of poly(styrene-co-4-chloromethyl styrene) via NMP. | |
2.3. Synthesis of azide-functionalized polystyrene (PSt-N3)
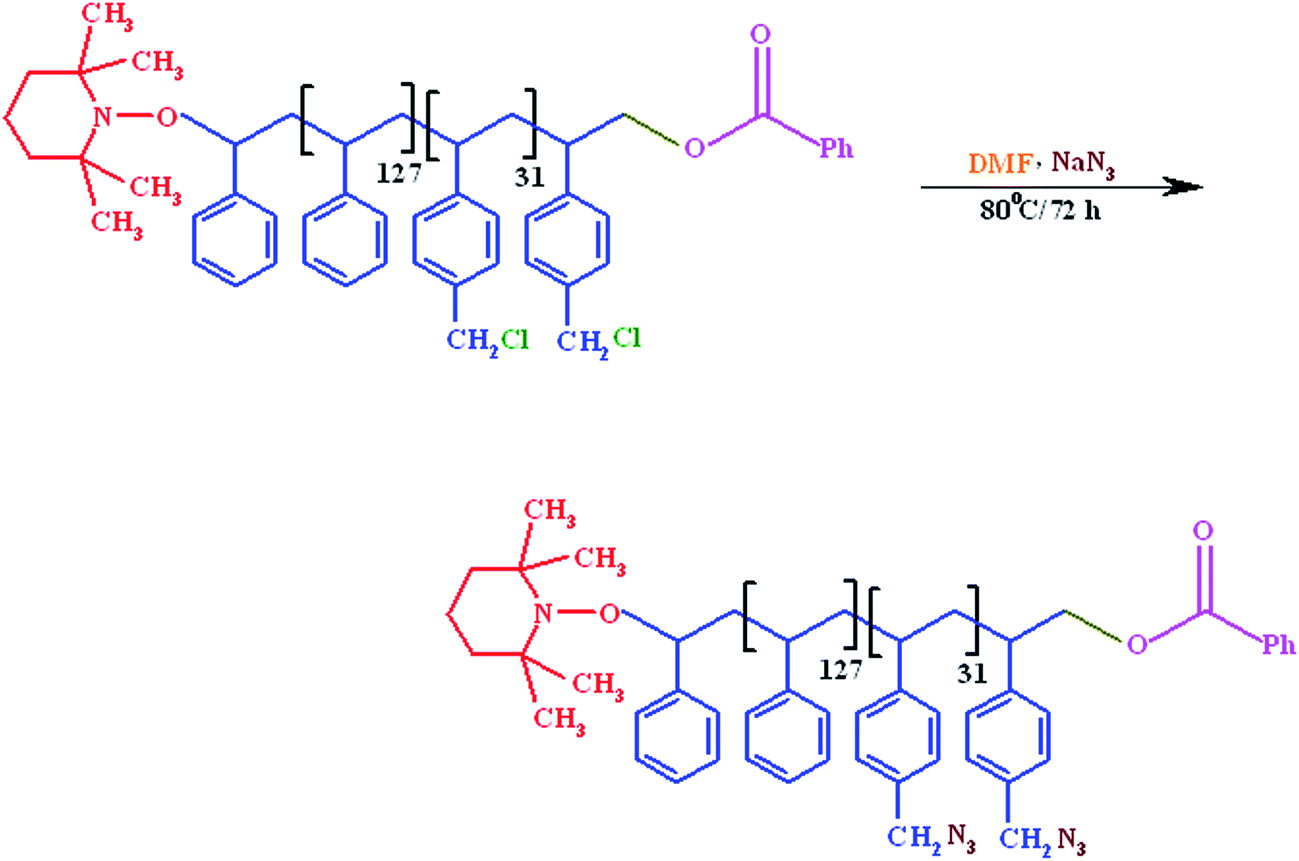
In a three-neck round-bottom flask equipped with a condenser, gas inlet/outlet and a magnetic stirrer, 2 g (17.5 mmol) of P(St-co-CMSt) was dissolved in anhydrous N,N-dimethylformamide (50 ml) and added to 0.45 g (7 mmol) of sodium azide. The solution was de-aerated by bubbling highly pure argon for 20 minutes and the mixture was refluxed at 80 °C under argon atmosphere for about 72 hours. The reaction was terminated by pouring the contents of the flask into a large amount of ethyl ether. The obtained solid was filtered, washed several times with cold water and dried under vacuum at room temperature (Scheme 2).
 |
| | Scheme 2 Synthesis of azide-functionalized polystyrene (PSt-N3). | |
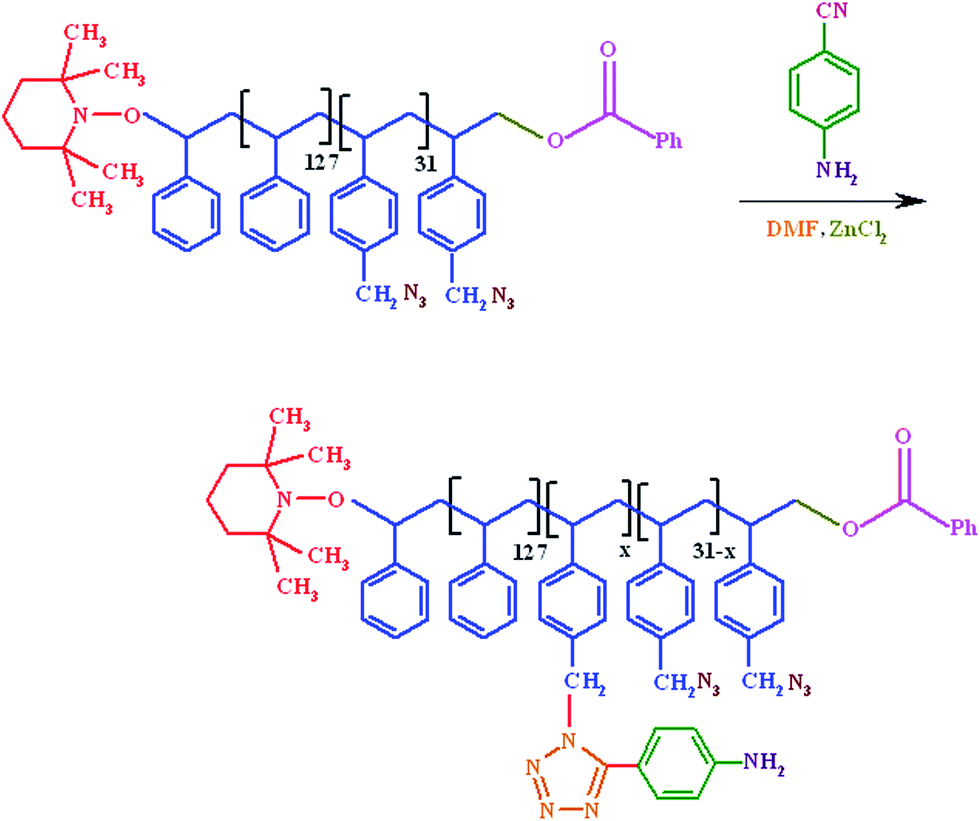
2.4. Synthesis of phenylamine-functionalized polystyrene macromonomer (PhAPStM)
In a three-neck round-bottom flask equipped with condenser, gas inlet/outlet, and a magnetic stirrer, 1 g of azide-functionalized polystyrene (PSt-N3), was dissolved in 50 ml of dried N,N-dimethylformamide (DMF). Anhydrous ZnCl2 (2.85 g, 21 mmol), and 4-aminobenzonitrile (1.2 g, 10 mmol) were added, and the reaction mixture was de-aerated by bubbling highly pure argon for 20 minutes. The reaction mixture was stirred at 130 °C for about 48 hours under argon atmosphere. At the end of this period, after cooling to 65 °C, 15 ml of diluted HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 by volume in water) was added, and the reaction mixture was kept at this temperature for another 6 hours. The phenylamine-functionalized polystyrene macromonomer (PhAPStM) was separated by precipitation of the contents of the flask in large amount of ethyl ether, filtrated, washed several times with cold water and dried in vacuum at room temperature (Scheme 3).
:10 by volume in water) was added, and the reaction mixture was kept at this temperature for another 6 hours. The phenylamine-functionalized polystyrene macromonomer (PhAPStM) was separated by precipitation of the contents of the flask in large amount of ethyl ether, filtrated, washed several times with cold water and dried in vacuum at room temperature (Scheme 3).
 |
| | Scheme 3 Synthesis of phenylamine-functionalized polystyrene macromonomer (PhAPStM). | |
2.5. Graft copolymerization of aniline onto phenylamine-functionalized polystyrene
A 250 ml round-bottom flask containing 60 ml tetrahydrofuran (THF), and phenylamine-functionalized polystyrene macromonomer (PhAPStM) (1 g) was equipped with a mechanical stirrer. Afterwards, 1 g (10.7 mmol) of aniline and 1 g (5.15 mmol) of p-toluenesulfonic acid were added to the solution. The mixture was vigorously stirred and temperature was reduced to 0 °C. In a separate container, 2 g (8.76 mmol) of ammonium peroxydisulfate (APS) was dissolved in 60 ml of p-toluenesulfonic acid solution (1 M). The oxidant solution was slowly added at a rate of 5 ml min−1 to the mixture. The mixture was stirred for about 8 hours at 0 °C, and then the reaction was terminated by pouring the content of the flask into a large amount of methanol. The crude product was filtered, washed several times with methanol (Scheme 4). The crude product was extracted with cyclohexane in a Soxhlet apparatus for 24 hours, in order to remove any residual ungrafted polystyrenic chains. The synthesized polystyrene-graft-polyaniline is partially soluble in cyclohexane, while ungrafted polystyrenic chains are completely soluble in cyclohexane. Moreover, the product was extracted with tetrahydrofuran (THF) for three times, in order to remove pure polyaniline. Polyaniline is partially soluble in THF (high molecular weight polyaniline is not soluble in THF); while the PSt-g-PANI graft copolymer is completely soluble in THF (Table 2). The polymer solution was filtered, precipitated into excess methanol, and dried in vacuum at room temperature7,8 (yield (after purification): 1.37 g, 37%).
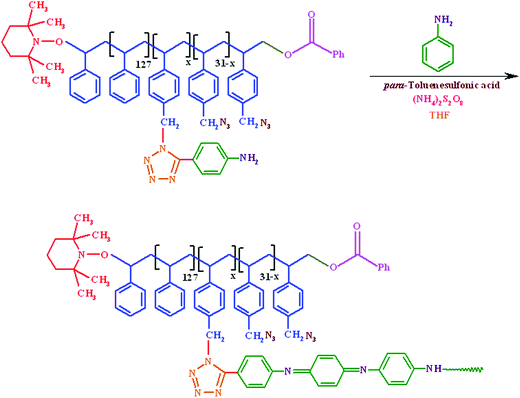 |
| | Scheme 4 Graft copolymerization of aniline onto polystyrene. | |
2.6. Synthesis of polyaniline
The pure polyaniline was synthesized by a chemical oxidation polymerization method. A typical synthesis was as follows: a 250 ml round-bottom flask was charged with THF (60 ml), aniline (1 g, 10.7 mmol), and p-toluenesulfonic (1 g, 5.15 mmol). The mixture was vigorously stirred and temperature was reduced to 0 °C. In a separate container, 2 g (8.76 mmol) of ammonium peroxydisulfate (APS) was dissolved in 60 ml of p-toluenesulfonic acid solution (1 M) as dopant. The oxidant solution was slowly added at a rate of 5 ml min−1 to the mixture. The mixture was stirred for about 8 hours at 0 °C, and then the polymer solution was filtered and by-products or oligomers were removed by excess water and methanol. The final dark green PANI powder (doped PANI) was obtained after drying at room temperature for 24 hours under reduced pressure (Scheme 5) (yield (undoped): 0.76 g, 76%).
 |
| | Scheme 5 Synthesis of polyaniline. | |
2.7. Electrochemical system
The electrochemical measurements were carried out using Auto-Lab equipment (ECO Chemie, Utrecht, The Netherlands) equipped with a three-electrode cell assembly. Glassy carbon (GC) microelectrode (with a surface area of 0.03 cm2), platinum rod, and Ag/AgCl were used as working, counter, and reference electrodes, respectively. The surface of the working electrode was polished with emery paper followed by 1.0 and 0.5 μm alumina. To prepare glassy carbon microelectrode coated by synthesized samples, the polymers were dissolved in THF to form a 1.0 mg ml−1 solution and ultrasonically treated for 15 minutes. The solution was dropped onto the GC microelectrode surface and allowed to dry under ambient conditions. The electrochemical measurements were accomplished in the aqueous solution of p-toluenesulfonic acid (1 M), by applying a sequential linear potential scan rate of 25–200 mV s−1 between −0.3 and +1.0 V vs. Ag/AgCl electrode. All experimental solutions were de-aerated by bubbling highly pure argon for 15 minutes, and an argon atmosphere was kept over the solutions during the measurements.
2.8. Characterization
Fourier transform infrared (FTIR) spectra of the samples were obtained on a Shimadzu 8101M FTIR (Shimadzu, Kyoto, Japan). The samples were prepared by grinding the dry powders with potassium bromide (KBr) and compressing the mixture into disks. The disks were stored in a desiccator to avoid moisture absorption. The spectra were recorded at room temperature. 1H nuclear magnetic resonance (NMR) spectra were obtained at 25 °C using an FT-NMR (400 MHz) Bruker spectrometer (Bruker, Ettlingen, Germany). The sample for NMR spectroscopy was prepared by dissolving about 10 mg of products in 1 ml of deuterated chloroform and chemical shifts were reported in ppm units with tetramethylsilane as internal standard. The molecular weights of the P(St-co-CMSt), and PSt-g-PANI were measured with a gel permeation chromatograph (GPC) (Maxima 820 GPC Analysis Report, Ventura, California, USA) using polystyrene (106, 105 and 104 Å) calibration standard with a THF mobile phase at a flow rate of 1 ml min−1 and column temperature at 25 °C. Thermal properties of the samples were examined with a TGA-PL STA 1640 equipment (Polymer Laboratories, Shropshire, U. K.). About 10 mg of the sample was heated between 25 and 700 °C at a rate of 10 °C min−1 under flowing nitrogen. Differential scanning calorimetric analyses were performed with a NETZSCH (Selb, Germany), DSC 200 F3 Maia. The sample was first heated to 200 °C, and then, allowed to cool for 5 minutes to eliminate the thermal history. Thereafter, the sample was reheated to 200 °C at a rate of 10 °C min−1. The entire test was performed under nitrogen purging at a flow rate of 50 ml min−1. Ultraviolet-visible (UV-vis) absorption spectra of the samples were measured using a Shimadzu 1601 PC UV-vis spectrophotometer (Shimadzu, Kyoto, Japan) in the wavelength range of 300–1000 nm. Electrochemical experiments were conducted using Auto-Lab PGSTA T302N. The electrochemical cell contained five openings: three of them were used for the electrodes and two for argon bubbling in the solutions during all experiments. The four probe technique (Azar Electric, Urmia, Iran) was used to measure the conductivity of the PSt-g-PANI and pure PANI samples at room temperature.
3. Results and discussion
The synthesis of PANI copolymers with conventional thermoplastics provides an effective way to overcome its certain deficiencies such as solubility and processability. It has been a scientific challenge and industrially interesting subject to prepare graft or block copolymers having low dispersity index (Đ = ![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) w/n), and stereoregularity on the main chain. In this respect, graft or block copolymerization of aniline onto various polymers synthesized via controlled radical polymerization (CRP) methods have been proposed. As illustrated in Scheme 4, in this study the phenylamine-functionalized polystyrene macromonomer (PhAPStM) used as the multicenter macromonomer for the aniline monomer grafts via the oxidation polymerization method.
w/n), and stereoregularity on the main chain. In this respect, graft or block copolymerization of aniline onto various polymers synthesized via controlled radical polymerization (CRP) methods have been proposed. As illustrated in Scheme 4, in this study the phenylamine-functionalized polystyrene macromonomer (PhAPStM) used as the multicenter macromonomer for the aniline monomer grafts via the oxidation polymerization method.
3.1. Synthesis of azide-functionalized polystyrene (PSt-N3)
Fig. 1 shows the 1H nuclear magnetic resonance (NMR) spectra of the P(St-co-CMSt), and azide-functionalized polystyrene (PSt-N3). The copolymer compositions were calculated from the 1H NMR spectra data. In the past few decades 1H NMR spectroscopic analysis has been established as a powerful tool for the determination of copolymer compositions because of its simplicity, rapidity, and sensitivity.8,9,42 The molar compositions of styrene and 4-chloromethyl styrene in the synthesized copolymer were calculated from the ratio integrated intensities of the peaks around 4.50 ppm, corresponding to two methylene protons of benzyl chloride in 4-chloromethyl styrene units to the total area between 0.80 and 2.20 ppm, which were attributed to six aliphatic protons in 4-chloromethyl styrene and styrene units. The 4-chloromethyl styrene and styrene contents in the synthesized copolymer were calculated from eqn (1) and (2), where x and y are the mole fractions of 4-chloromethyl styrene and styrene, respectively.| |
 | (1) |
 |
| | Fig. 1 1H nuclear magnetic resonance (NMR) spectra of P(St-co-CMSt), and azide-functionalized polystyrene. | |
In 1H NMR spectrum of P(St-co-CMSt) solving the simultaneous equations with the integrated areas, it can be seen that the molar compositions of styrene and 4-chloromethyl styrene in the synthesized copolymer are 80.2% and 19.8%, respectively.
The 1H NMR spectrum of the azide-functionalized polystyrene show similar peaks with minor differences. The most distinctive feature in the 1H NMR spectra of PSt-N3 and P(St-co-CMSt) is the presence of a new peak at 4.15 ppm, corresponding to CH2–N3 protons. Moreover, the 1H NMR spectrum of PSt-N3 indicates that all of the chlorine groups in the P(St-co-CMSt), were substituted with azide groups, because the chemical shift of –CH2Cl groups at 4.50 ppm is completely disappears, and the new peak for –CH2N3 groups is observed at 4.15 ppm.
3.2. Synthesis of phenylamine-functionalized polystyrene macromonomer (PhAPStM)
As aforementioned, the phenylamine-functionalized polystyrene macromonomer (PhAPStM) was synthesized via “click” cycloaddition reaction between the PSt-N3 and 4-aminobenzonitrile in the presence of anhydrous ZnCl2 as the catalyst. The 1H NMR spectrum of the PhAPStM is shown in Fig. 2. The most distinctive features after introduction of phenylamine groups into PSt are the appearance of new peaks at 3.72 and 4.61 ppm corresponding to –NH2 groups (g), and tetrazole–CH2 groups (h), respectively. Moreover, the peak at 4.15 ppm (c) indicates that some of the azide group's does not converted to the phenylamine groups.
 |
| | Fig. 2 1H nuclear magnetic resonance (NMR) spectrum of phenylamine-functionalized polystyrene macromonomer (PhAPStM). | |
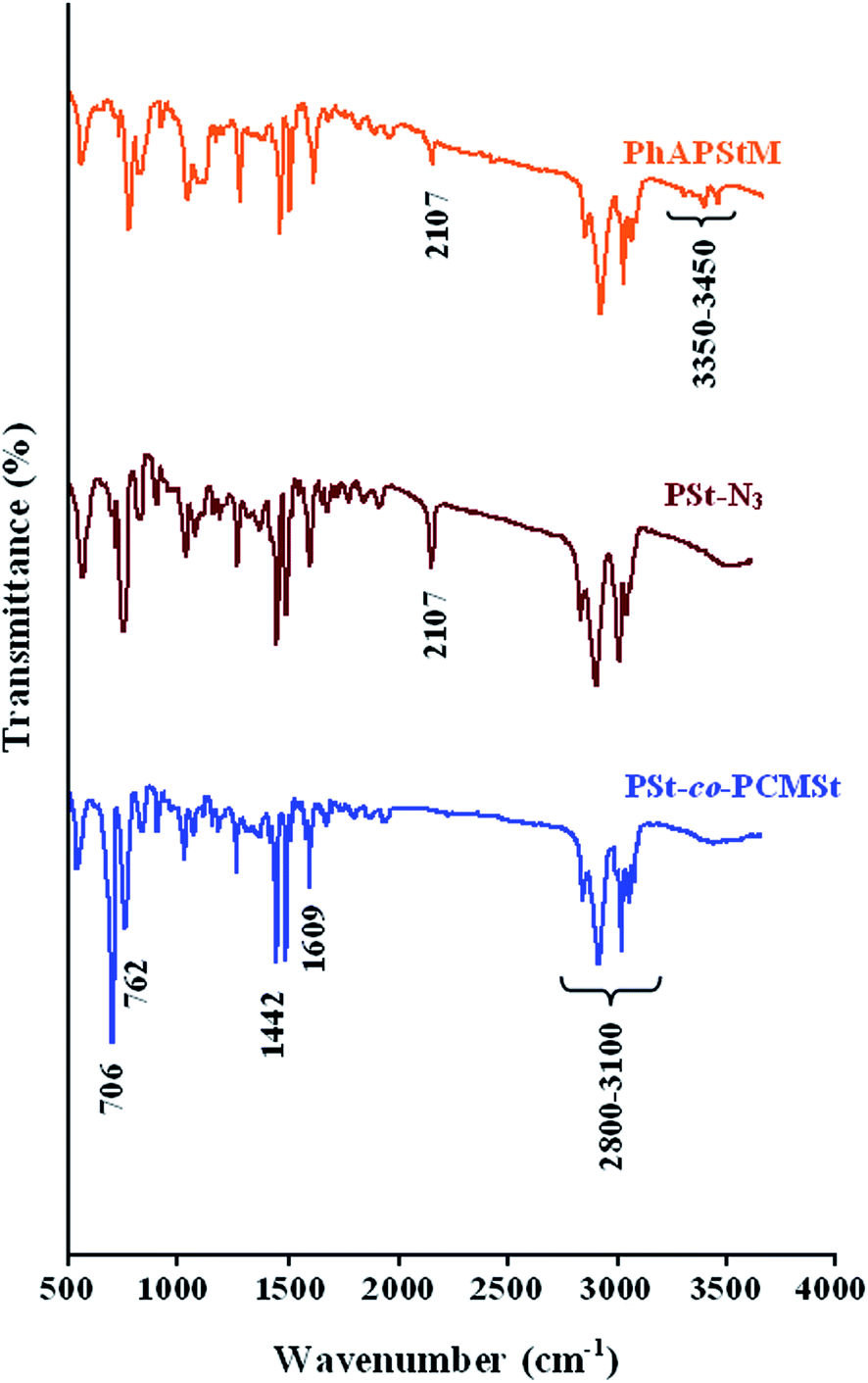
The FTIR spectra of the P(St-co-CMSt), PSt-N3, and PhAPStM macromonomer are shown in Fig. 3. The FTIR spectrum of the P(St-co-CMSt) copolymer shows the characteristic absorption bands due to the stretching vibrations of C–H (3100–2800 cm−1), weak aromatic overtone and combination bands in the 1900–1650 cm−1 region, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations (1609 and 1486 cm−1), CH2 bending vibrations (1442 and 1375 cm−1), and γ(C–H) in the aromatic ring (849 and 762 cm−1). Moreover, the strong peak at 706 cm−1 is attributed to the C–Cl group in the FTIR spectrum of the P(St-co-CMSt) copolymer. The FTIR spectrum of the azide-functionalized polystyrene shows similar peaks with minor differences. The most distinctive features of PSt-N3 in comparison with P(St-co-CMSt), in the FTIR spectra are the appearance of a new band at 2107 cm−1 corresponding to azide groups, and disappearance of the strong peak at 706 cm−1 (related to the C–Cl group). After introduction of phenylamine groups into PSt backbone, it can be clearly seen that the characteristic absorption bands due to the stretching vibration of amine groups (–NH2) is appeared at 3450–3350 cm−1 region. Moreover, the intensity of the azide group at 2107 cm−1 significantly decreased, because most of the azide groups were converted to the phenylamine groups, according to the 1H NMR spectrum. These FTIR and 1H NMR spectra assignments verify that the phenylamine groups were introduced into the PSt backbone, and thus the PhAPStM was successfully synthesized.
C stretching vibrations (1609 and 1486 cm−1), CH2 bending vibrations (1442 and 1375 cm−1), and γ(C–H) in the aromatic ring (849 and 762 cm−1). Moreover, the strong peak at 706 cm−1 is attributed to the C–Cl group in the FTIR spectrum of the P(St-co-CMSt) copolymer. The FTIR spectrum of the azide-functionalized polystyrene shows similar peaks with minor differences. The most distinctive features of PSt-N3 in comparison with P(St-co-CMSt), in the FTIR spectra are the appearance of a new band at 2107 cm−1 corresponding to azide groups, and disappearance of the strong peak at 706 cm−1 (related to the C–Cl group). After introduction of phenylamine groups into PSt backbone, it can be clearly seen that the characteristic absorption bands due to the stretching vibration of amine groups (–NH2) is appeared at 3450–3350 cm−1 region. Moreover, the intensity of the azide group at 2107 cm−1 significantly decreased, because most of the azide groups were converted to the phenylamine groups, according to the 1H NMR spectrum. These FTIR and 1H NMR spectra assignments verify that the phenylamine groups were introduced into the PSt backbone, and thus the PhAPStM was successfully synthesized.
 |
| | Fig. 3 Fourier transform infrared spectra of P(St-co-CMSt), PSt-N3, and PhAPStM. | |
3.3. Synthesis of PSt-g-PANI
Two kinds of cation radicals initiate aniline polymerization after addition of APS. One is oxidized phenylamine groups coupled to the polystyrene backbone; the other is oxidized aniline cation radicals. As reaction time increasing, more aniline monomers join in the polymerization. Some are entrapped into the polymer chains initiated by oxidized aniline groups coupled to the polystyrenic chains; others incorporate the polymer chains initiated by oxidized aniline cation radicals. These polymer chains could not be linked chemically to the polystyrene backbone. To remove the ungrafted polystyrene chains and pure polyaniline, the crude product was purified as given in the experimental section.
Living polymerization is characterized by a linear increase of the molecular weight with conversion and reaction time, and a narrow molecular weight distribution as evidenced by a dispersity index approaching 1. Table 1 represents the molecular weights analysis data of P(St-co-CMSt), and PSt-g-PANI copolymers by GPC. The molecular weight distributions of P(St-co-CMSt), and PSt-g-PANI copolymers are 1.18 and 1.36, respectively. The GPC chromatograms of P(St-co-CMSt), and PSt-g-PANI copolymers are shown in Fig. 4. The GPC chromatograms verify that PANI was chemically grafted onto PSt backbone because the chromatogram of the PSt-g-PANI is shifted to higher molecular weight compared with the chromatogram of P(St-co-CMSt). Moreover, the single peak of the chromatogram of the PSt-g-PANI graft copolymer indicates that the product cannot be a blend of P(St-co-CMSt) and PANI; if so, the chromatogram should appear as two peaks, one for P(St-co-CMSt) and the other for PANI. Thus the PSt-g-PANI graft copolymer was successfully synthesized.
Table 1 Molecular weights' analysis data of P(St-co-CMSt), and PSt-g-PANI copolymers by GPC
| Samples |
n |
w |
Đ = w/n |
P(St-co-CMSt)a [%] |
PANIa [%] |
| Composition calculated from the n values of the starting polystyrenic sample and the resulting graft copolymer. |
| P(St-co-CMSt) |
18462 |
21853 |
1.18 |
100 |
— |
| PSt-g-PANI |
25017 |
34157 |
1.36 |
64.5 |
35.5 |
 |
| | Fig. 4 GPC chromatograms of P(St-co-CMSt), and PSt-g-PANI copolymers. | |
The FTIR spectra of the pure PANI and PSt-g-PANI are shown in Fig. 5. The FTIR spectrum of the pure PANI shows the characteristic absorption bands due to stretching vibration of the CN in the quinoidal units at 1605 cm−1, the benzenoid stretches at 1496 cm−1, Caromatic–N stretching at 1314 cm−1, and the N–H stretches at 3427 cm−1. The FTIR spectrum of the PSt-g-PANI graft copolymer shows the characteristic absorption band of polyaniline, namely, CN in the quinoidal units, appearing at 1605 cm−1, the benzenoid stretches at 1496 cm−1, and N–H stretches at 3427 cm−1. The Caromatic–N stretching band of an aromatic amine appears at 1314 cm−1. The absorption band at 1153 cm−1 is characteristic of electron-like absorption of the NQN vibration (where Q denotes the quinoid ring), and the band at 825 cm−1 is attributed to the 1,4-disubstituted benzene ring (out-of-plane). Moreover, the characteristic absorption bands due to stretching vibration of the C–H (aliphatic and aromatic) at 3100–2800 cm−1 region, is verify the presence of polystyrene in the sample. These FTIR spectra assignments verify that the aniline has grown onto polystyrenic backbone and then the PSt-g-PANI graft copolymer was successfully synthesized.
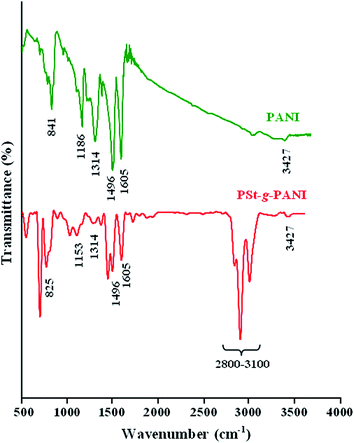 |
| | Fig. 5 Fourier transform infrared spectra of the pure PANI and PSt-g-PANI graft copolymer. | |
Additional evidence for the synthesis of the PSt-g-PANI was also obtained from 1H NMR data (Fig. 6). To calculate the extent of the polystyrenic chains and PANI chains in the graft copolymer the following method was adopted. The ratio of aliphatic hydrogens to aromatic hydrogens in the P(St-co-CMSt), is 3:(0.198 × 4) + (0.802 × 5) ∼ 6.88:10.39; after graft copolymerization of aniline onto polystyrenic chains, this ratio changed to 3:(0.198 × 4) + (0.802 × 5) + 4X ∼ 0.2927:0.6072, where X is the percent of PANI in the graft copolymer, 0.198 and 4 are the percent of PCMSt and number of its aromatic hydrogens in the P(St-co-CMSt) part, 0.802 and 5 are the percent of PSt and number of its aromatic hydrogens in the P(St-co-CMSt) part, and 4 is the number of aromatic hydrogens in the PANI segments. In Fig. 6, by solving the equation, it can be seen that X = 35.6%.
 |
| | Fig. 6 1H nuclear magnetic resonance (NMR) spectrum of PSt-g-PANI. | |
The solubility of the PSt-g-PANI graft copolymer and pure PANI in common organic solvents are shown in Table 2. Polystyrenic chains have excellent solubility in non-polar solvents. The solubility of PSt-g-PANI in common organic solvents improved compared to pure PANI, because aniline has grown onto polystyrenic backbone.
Table 2 Solubility of PSt-g-PANI and pure PANI in common organic solventsa
| Solvent |
DMSO |
NMP |
THF |
CHCl3 |
Xylene |
DMF |
| + + +: soluble; + +: sparingly soluble; +: slightly soluble; –: insoluble; the concentration used in the solubility test was 10 mg of each polymer in 1 ml of solvents (DMSO, dimethylsulfoxide; NMP, N-methylpyrrolidone; THF, tetrahydrofuran; DMF, N,N-dimethylformamide). |
| PANI |
+ + |
+ + |
+ |
− |
− |
+ + |
| PSt-g-PANI |
+ + + |
+ + + |
+ + + |
+ + |
+ + |
+ + + |
3.4. Electrical conductivity and electroactivity measurements
The most important property of PANI and its copolymers which make them suitable for various practical and technological applications is their electrical conductivity. The electrical resistivity (R) of the synthesized samples, was measured at room temperature and then converted to volume specific resistivity (ρ) using the equation R = ρL/A. In this equation, A is the test sample cross-section area and L the sample thickness. Finally, the electrical conductivity (σ) is the reciprocal of ρ(σ = 1/ρ). The electrical conductivity values (σ) of the pure PANI and PSt-g-PANI were obtained 2.45 and 0.73 S cm−1, respectively.
As illustrated in Fig. 7 the effect of the potential scanning rate (V) on the peak current for the P(St-co-CMSt), pure PANI, and PSt-g-PANI modified electrodes were studied in the range of 25–200 mV s−1 scan rate, in a aqueous solution of p-toluenesulfonic acid (1 M). The polymer films were prepared on glassy carbon (GC) microelectrode by casting. Typical cyclic voltammograms (CVs) of the pure PANI film are shown in Fig. 7b. When the CVs of pure PANI are recorded between −0.3 and +1.0 V vs. Ag/AgCl at scan rate of 25–200 mV s−1, two typical redox couples with anodic peaks at 0.22 and 0.61 V (vs. Ag/AgCl) are observed. Moreover, the anodic peaks shifts in the direction of positive potential with increasing scan rate, which indicates the electrochemical oxidation/reduction (doping/dedoping) of the casted PANI film was chemically reversible. In the cyclic voltammograms of the pure polystyrenic film no detectable redox peak was observed. Fig. 7a shows that the P(St-co-CMSt) sample is a non-electroactive polymer.
 |
| | Fig. 7 Cyclic voltammetry curves of (a) pure P(St-co-CMSt), (b) neat PANI and (c) PSt-g-PANI, and (d) linear relationship between current and scan rate in the (1) PANI and (2) PSt-g-PANI. | |
As shown in Fig. 7c, cyclic voltammograms of the PSt-g-PANI graft copolymer exhibit some qualitative similarities with those of pure PANI. Similar to cyclic voltammograms of pure PANI for PSt-g-PANI two typical redox couples with anodic peaks at 0.3 and 0.55 V (vs. Ag/AgCl) are observed and the anodic peaks current are linearly increased with increasing of scan rate. The cyclic voltammograms of the PSt-g-PANI indicated that the grafted PANI onto polystyrene backbone still remained a good redox activity in the resulting graft copolymer, and the resulting graft copolymer was high stable. To evaluate the electroactivity further, it was determined the relationship between the peak current size vs. scan rate. Fig. 7d shows linear relationships between the current and scan rate between 25–200 mV s−1 in the PSt-g-PANI graft copolymer and pure PANI. This linear relationship is typical of a redox active polymer attached to the electrodes and also exemplifies the stability of PSt-g-PANI and PANI films toward doping/dedoping.
On the bases of the evidence from electrical conductivity and electroactivity measurements the conclusion could be drawn that the PSt-g-PANI exhibit lowers electrical conductivity and electroactivity than those of the pure PANI due to the decreased conjugation length distribution. However, the lowers electrical conductivity and electroactivity levels in the modified polyaniline can be improved at the price of solubility and processability.
3.5. Thermal property study
Thermal behaviors of the obtained polymers were investigated by differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA). Fig. 8 shows the DSC traces of the P(St-co-CMSt), and PSt-g-PANI graft copolymer. The P(St-co-CMSt) copolymer is a non-crystalline polymer and therefore does not exhibit any crystallization or melting transitions. The DSC curve of P(St-co-CMSt) exhibits an endothermic peak approximately at 86 °C, corresponding to the glass transition temperature (Tg) of the copolymer. In DSC curve of the PSt-g-PANI the transition observed at 93 °C, can be designed as the glass transition temperature of the graft copolymer. In addition, the Tg slightly increased in the presence of PANI segments by impeding chain flexibility.7,8
 |
| | Fig. 8 Differential scanning calorimeter traces of the P(St-co-CMSt), and PSt-g-PANI. | |
Characteristic TGA curves of the P(St-co-CMSt), and PSt-g-PANI graft copolymer are shown in Fig. 9. It is evident that the PSt-g-PANI graft copolymer undergoes a two-step decomposition; the first step corresponds to the decomposition of the PSt chain (290–370 °C), whereas the second step is associated with PANI chain scission (380–480 °C), after which the loss rate slows down. However, decomposition of the P(St-co-CMSt) copolymer was occurring in one step around 270–410 °C, after which the loss rate slows down. According to the Fig. 9, we can draw the conclusion that the final weight residue at 650 °C, for the P(St-co-CMSt), and PSt-g-PANI were 4 and 25 wt%, respectively.
 |
| | Fig. 9 Thermogravimetric analysis of the P(St-co-CMSt), and PSt-g-PANI. | |
3.6. UV-vis spectroscopy
The optical properties of the synthesized samples were studied using ultraviolet-visible (UV-vis) spectroscopy. The samples for UV-vis spectroscopy were prepared by dissolving the same amount of the obtained polymers in dimethylsulfoxide (DMSO) followed by ultrasonic treatment for 10 minutes. The UV-vis spectra of the pure PANI and PSt-g-PANI graft copolymer are shown in Fig. 10. The UV-vis spectrum of the pure PANI was characterized by two electronic transitions respectively at about 326 and 438 nm. The first absorption band corresponds to the overlap of the π–π* transition of the benzenoid rings of PANI, while the latter one is assigned as exciton absorption of the quinoid (quinonimine) rings of PANI.8 However, the UV-vis spectrum of the PSt-g-PANI graft copolymer also was characterized by two electronic transitions at about 413 and 835 nm, respectively.
 |
| | Fig. 10 Electronic spectra of pure PANI and PSt-g-PANI in DMSO solution. | |
4. Conclusion
This work has shown an efficient and novel approach for the synthesis of conductive polyaniline-modified polymers via a combination of nitroxide-mediated polymerization (NMP) and “click chemistry”. The growth of aniline onto functionalized polystyrene enhanced its solubility and processability compared with pure polyaniline. The synthesized PSt-g-PANI showed the polaronic band, at lower wavelength (higher energy) than pure PANI in the UV-vis spectra due to lower concentration of conjugated units in the case of PSt-g-PANI. The cyclic voltammograms (CVs) of the PSt-g-PANI exhibited some qualitative similarities with those of pure PANI. The CVs of PSt-g-PANI indicated that the grafted polyaniline onto polystyrenic chains still remained a good redox activity in the resulting graft copolymer, and the synthesized graft copolymer was high stable. The PSt-g-PANI exhibited lowers electrical conductivity and electroactivity than those of the pure PANI due to the decreased conjugation length distribution. However, the lowers electrical conductivity and electroactivity levels in the PSt-g-PANI can be improved at the price of solubility and processability. Investigation of thermal behaviors of the synthesized graft copolymer showed the increased flexibility compared with neat PANI due to the grafting process.
Abbreviations
| ICPs: | Intrinsically conductive polymers |
| CRP: | Controlled radical polymerization |
| NMP: | Nitroxide-mediated polymerization |
| TEMPO: | 2,2,6,6-Tetramethyl-1-piperidinyloxy |
| PCMSt: | Poly(4-chloromethyl styrene) |
| PSt: | Polystyrene |
| PANI: | Polyaniline |
| THF: | Tetrahydrofuran |
| Tg: | Glass transition temperature |
| DSC: | Differential scanning calorimetry |
| TGA: | Thermogravimetric analysis |
Acknowledgements
We express our gratitude to the Research Center for Pharmaceutical Nanotechnology, Tabriz University of Medical Sciences and Payame Noor University for supporting this project.
References
- B. M. Schulze, N. T. Shewmon, J. Zhang, D. L. Watkins, J. P. Mudrick, W. Cao, R. B. Zerdan, A. J. Quartararo, I. Ghiviriga, J. Xue and R. K. Castellano, J. Mater. Chem. A, 2014, 2, 1541–1549 CAS
 .
. - X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS PubMed .
- H. S. Abdulla and A. I. Abbo, Int. J. Electrochem. Sci., 2012, 7, 10666–10678 CAS .
- K. H. Ong, S. L. Lim, J. Li, H. K. Wong, H. S. Tan, T. T. Lin, L. C. H. Moh, J. C. De-Mello and Z. K. Chen, Polym. Chem., 2013, 4, 1863–1873 RSC .
- M. Jaymand, Prog. Polym. Sci., 2013, 38, 1287–1306 CrossRef CAS PubMed .
- H. Shirakawa, E. J. Louis, A. G. Mac-Diarmid, C. K. Chiang and A. J. Heeger, Chem. Commun., 1977, 578–580 RSC .
- M. Jaymand, Polymer (Korea), 2010, 34, 553–559 CAS .
- M. Hatamzadeh, A. Mahyar and M. Jaymand, J. Braz. Chem. Soc., 2012, 23, 1008–1017 CrossRef CAS PubMed .
- M. Hatamzadeh, M. Jaymand and B. Massoumi, Polym. Int., 2014, 63, 402–412 CrossRef CAS .
- M. Jaymand, Des. Monomers Polym., 2011, 14, 433–444 CrossRef CAS PubMed .
- A. M. Youssef, RSC Adv., 2014, 4, 6811–6820 RSC .
- H. J. Wang, Y. P. Chen, Y. C. Chen, C. P. Chen, R. H. Lee, L. H. Chan and R. J. Jeng, Polymer, 2012, 53, 4091–4103 CrossRef CAS PubMed .
- M. Abbasian, M. Jaymand and S. E. S. Bonab, J. Appl. Polym. Sci., 2012, 125, E131–E140 CrossRef CAS .
- Y. Wang, H. D. Tran, L. Liao, X. Duan and R. B. Kaner, J. Am. Chem. Soc., 2010, 132, 10365–10373 CrossRef CAS PubMed .
- H. Kawashima and H. Goto, Soft Nanosci. Lett., 2011, 1, 71–75 CrossRef CAS .
- P. Saini, R. Jalan and S. K. Dhawan, J. Appl. Polym. Sci., 2008, 108, 1437–1446 CrossRef CAS .
- J. Niziol, M. Sniechowski, A. Podraza-Guba and J. Pielichowski, Polym. Bull., 2011, 66, 761–770 CrossRef CAS .
- L. Li, E. Liu, Y. Yang, H. Shen, Z. Huang and X. Xiang, Mater. Lett., 2012, 64, 2115–2117 CrossRef PubMed .
- A. Guerfi, J. Trottier, I. Boyano, I. De-Meatza, J. A. Blazquez, S. Brewer, K. S. Ryder, A. Vijh and K. Zaghib, J. Power Sources, 2014, 248, 1099–1104 CrossRef CAS PubMed .
- J. Yun and H. I. Kim, Polym. Bull., 2012, 68, 561–573 CrossRef CAS .
- P. Saini, V. Choudhary, N. Vijayan and R. K. Kotnala, J. Phys. Chem. C, 2012, 116, 13403–13412 CAS .
- Y. Qiao, C. M. Li, S. J. Bao and Q. L. Bao, J. Power Sources, 2007, 170, 79–84 CrossRef CAS PubMed .
- J. Chen, B. Li, J. Zheng, J. Zhao, H. Jing and Z. Zhu, Electrochim. Acta, 2011, 56, 4624–4630 CrossRef CAS PubMed .
- Q. Lin, Y. Li and M. Yang, Anal. Chim. Acta, 2012, 748, 73–80 CrossRef CAS PubMed .
- C. H. A. Esteves, B. A. Iglesias, R. W. C. Lia, T. Ogawa, K. Araki and J. Gruber, Sens. Actuators, B, 2014, 193, 136–141 CrossRef CAS PubMed .
- N. Plesu, G. Ilia, A. Pascariu and G. Vlase, Synth. Met., 2006, 156, 230–238 CrossRef CAS PubMed .
- W. K. Lu, R. L. Elsenbaumer and B. Wessling, Synth. Met., 1995, 71, 2163–2166 CrossRef CAS .
- H. R. Ghenaatian, M. F. Mousavi and M. S. Rahmanifar, Electrochim. Acta, 2012, 78, 212–222 CrossRef CAS PubMed .
- G. Ramya, C. Renugadevi, C. R. K. Rao and D. C. Trivedi, React. Funct. Polym., 2008, 68, 701–709 CrossRef CAS PubMed .
- M. Grigoras, A. M. Catargiu, F. Tudorache and M. Dobromir, Iran. Polym. J., 2012, 21, 131–141 CrossRef CAS .
- Z. F. Li and E. Ruckenstein, J. Colloid Interface Sci., 2004, 269, 62–71 CrossRef CAS .
- M. Ghorbani, H. Gheybi and A. A. Entezami, J. Appl. Polym. Sci., 2012, 123, 2299–2308 CrossRef CAS .
- J. Ran, L. Wu, Z. Zhang and T. Xu, Prog. Polym. Sci., 2014, 39, 124–144 CrossRef CAS PubMed .
- M. J. Monteiro and M. F. Cunningham, Macromolecules, 2012, 45, 4939–4957 CrossRef CAS .
- C. Detrembleur, C. Jérôme, J. D. Winter, P. Gerbaux, J. L. Clément, Y. Guillaneuf and D. Gigmes, Polym. Chem., 2014, 5, 335–340 RSC .
- M. Jaymand, Polymer, 2011, 52, 4760–4769 CrossRef CAS PubMed .
- J. Nicolasa, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef PubMed .
- M. Jaymand, J. Polym. Res., 2011, 18, 1617–1624 CrossRef CAS PubMed .
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4428 CrossRef CAS .
- M. Ahmed and R. Narain, Prog. Polym. Sci., 2013, 38, 767–790 CrossRef CAS PubMed .
- H. Willcock, A. Lu, C. F. Hansell, E. Chapman, I. R. Collins and R. K. O'Reilly, Polym. Chem., 2014, 5, 1023–1030 RSC .
- M. Jaymand, Polym. J., 2011, 43, 901–908 CrossRef CAS .
- Z. Vazifehasl, S. Hemmati, M. Zamanloo and M. Jaymand, Macromol. Res., 2013, 21, 427–434 CrossRef CAS PubMed .
- A. Thomas, K. Niederer, F. Wurm and H. Frey, Polym. Chem., 2014, 5, 899–909 RSC .
- S. H. Lin, S. J. Wu, C. C. Ho and W. F. Su, Macromolecules, 2013, 46, 2725–2732 CrossRef CAS .
- M. Jaymand, Polym. J., 2011, 43, 186–193 CrossRef CAS .
- K. O. Kim and T. L. Choi, Macromolecules, 2013, 46, 5905–5914 CrossRef CAS .
- P. L. Golas and K. Matyjaszewski, QSAR Comb. Sci., 2007, 26, 1116–1134 CAS .
- A. M. Eissa and E. Khosravi, Eur. Polym. J., 2011, 47, 61–69 CrossRef CAS PubMed .
- H. Nulwala, D. J. Burke, A. Khan, A. Serrano and C. J. Hawker, Macromolecules, 2010, 43, 5474–5477 CrossRef CAS .
- V. Aureggi and G. Sedelmeier, Angew. Chem., Int. Ed., 2007, 46, 8440–8444 CrossRef CAS PubMed .
- N. V. Tsarevsky, K. V. Bernaerts, B. Dufour, F. E. Du-Prez and K. Matyjaszewski, Macromolecules, 2004, 37, 9308–9313 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.