In situ growth of gold nanoparticles on magnetic γ-Fe2O3@cellulose nanocomposites: a highly active and recyclable catalyst for reduction of 4-nitrophenol†
Rui Xiong,
Yaru Wang,
Xinxing Zhang,
Canhui Lu* and
Lidan Lan
State Key Laboratory of Polymer Materials Engineering, Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: canhuilu@scu.edu.cn; Fax: +86-28-85402465; Tel: +86-28-85460607
First published on 24th December 2013
Abstract
In this study, a facile synthetic method has been developed for in situ growth of Au nanoparticles (NPs) on magnetic γ-Fe2O3@carboxylated cellulose nanospheres using 2,2,6,6-tetramethylpiperidinyl-1-oxyl radical (TEMPO) oxide cellulose as linkage and reducing agent. The size of the as-prepared Au NPs can be tuned from 14 to 38 nm by the initial concentration of the gold salt used in the reaction mixture. Only one step was needed to synthesize the magnetic γ-Fe2O3@cellulose nanospheres. The formed γ-Fe2O3@carboxylated cellulose@Au is highly dispersible in aqueous solution and its potential as a magnetic catalyst is proved by the reduction reaction of 4-nitrophenol to 4-aminophenol. In particular, there was no visible decrease in the catalytic activity of the reused catalysts even after being recycled five times, which is preferred in terms of cost and environmental protection. Due to the absence of any other reducing agent during the proposed process, both the synthesis steps and the reaction cost were remarkably decreased, which makes it very suitable for industrial-scale production of recyclable catalysts.
Introduction
Au nanoparticles (NPs) have attracted considerable attention owing to their unique physical and chemical properties as well as their broad and important applications in catalysis,1 sensing,2 therapy,3 delivery4 and surface enhance Raman scattering (SERS).5 In particular, their large specific surface area made them highly catalytically active and find wide applications in catalysis.6 From a practical point of view, however, there are two primary drawbacks to the application of Au NPs, including their aggregation and the difficulty in complete recovery after use. Pure Au NPs are easy to aggregate into large particles via the interparticle dipolar force,7,8 leading to the loss of size effect and the decrease of specific surface area, which would result in a remarkable reduction in their catalytic activity.9 Moreover, it is necessary to separate and recover the Au NPs from the reaction mixture solution due to their high cost and limited supply. However, it is difficult to separate Au NPs completely because of their small size. In order to solve these problems, immobilizing Au NPs on the surface of solid supports is regarded as a conventional way.10–12 Nanosized solid supports such as ZrO2,12 TiO2 (ref. 13) and SiO2 (ref. 14) have attracted much attention due to their large surface area, inherent adsorptive properties, and active sites.15Iron oxide magnetic NPs (MNPs), especially Fe3O4 and γ-Fe2O3, have been considered as an ideal oxide support because they are easy to prepare, have a very active surface for adsorptions or immobilization of metals, and can be separated by magnetic field after the reaction.16,17 However, pure MNPs are poorly dispersed in aqueous medium due to their hydrophobic surface.18 To enhance its dispersity in aqueous medium, MNPs have to be coated with inorganic shells or hydrophilic polymers. Among these, coating with hydrophilic polymers through both physical adsorption and in situ polymerizations, such as PEG,19 chitosan20 and polypeptide,21 has attracted great attention in recent years. Because the polymers not only protected the magnetic core from the environment, rendering stable and water dispersible MNPs, but also provided the desired functionality for further modifications. As the emphasis of science and technology is to shift more towards environmentally friendly and sustainable resources and processes, it is necessary to develop renewable and green coating materials for the MNPs.
Cellulose has been considered as an interesting alternative to coat the MNPs owing to the fact that cellulose is one of the most abundant and renewable biopolymers on earth. It is built up of β (1, 4)-linked D-glucose units and consists of three hydroxyl groups per anhydroglucose unit (AGU). The electron-rich feature of hydroxyl and ether groups in these components makes them suitable for the preparation and immobility of Au NPs.22–25 However, the highly ordered crystalline nature of cellulose along with strong inter- and intramolecular hydrogen bonding makes it tend to precipitate in water.26 To increase the dispersibility of cellulose particles or cellulose chains in an aqueous system, typically chemical modification of cellulose is carried out. Chemical modification of its natural structure (e.g., –OH group modification leading to –NH2 and –COOH) can provide electrostatic repulsion within and between the cellulose chains, which leads to good dispersibility.27
In this work, we report a facile and environmentally friendly method to prepare well dispersed and stable magnetic Au NPs in aqueous solution (Fig. 1). After the γ-Fe2O3@cellulose NPs were first prepared via an ionic liquid assisted co-precipitation process, 2,2,6,6-tetramethylpiperidinyl-1-oxyl radical (TEMPO) mediated oxidation of cellulose was carried out to introduce carboxyl groups on the surface of MNPs. The carboxyl groups can provide electrostatic repulsion between the MNPs, which leads to water soluble MNPs. Furthermore, Au NPS could be formed through the in situ reduction of Au3+ by cellulose and subsequently deposit highly dispersed Au NPs directly onto the surface of γ-Fe2O3@carboxylated cellulose MNPs. This process did not employ any other reductants, capping or dispersing agents. To the best of our knowledge, the approach of using TEMPO-oxide cellulose coating on MNPs to synthesis the Au NPs has not been explored. The resulted magnetic biopolymer-metal nanohybrids are used as recyclable catalyst in the reduction process of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP). The γ-Fe2O3@carboxylated cellulose@Au MNPs showed excellent catalytic performance towards the reduction of 4-NP to 4-AP in aqueous solution, which would be very useful in various catalytic reductions.
 | ||
| Fig. 1 Schematic illustration for the preparation of the iron oxide@carboxylated cellulose@Au NPs. | ||
Experimental section
Materials
Microcrystalline cellulose (MCC), ferric chloride hexahydrate (FeCl3·6H2O), ferrous chloride tetrahydrate (FeCl2·4H2O), ammonia (NH3·H2O) and 4-nitrophenol (4-NP) were provided by Kelong Co. Ltd. (Chengdu, China). 1-butyl-3-methylimidazolium chloride [BmimCl] was obtained from Chengjie Co. Ltd. (Shanghai, China). Hydrogen tetrachloroaurate(III) (HAuCl4) and 2,2,6,6-tetramethylpiperidinyl-1-oxyl radical (TEMPO) were obtained from J&K Co. Ltd. All the reagents were used without further purification.Synthesis of iron oxide@cellulose (IC)
MCC (150 mg), FeCl3·6H2O (0.5 g, 1.85 mmol), and FeCl2·4H2O (0.184 g, 0.925 mmol) were dissolved in 30 g [BmimCl] at 80 °C. To this solution was added 7.5 mL of NH3·H2O (28% in water) while stirring vigorously. At this time, the color of the solution changed to dark black, indicating the formation of iron oxide nanoparticles. Then, an external magnetic field (Mext) was applied to the solution using a rare-earth magnet. Within minutes all the black particles sank down toward the magnet, and the supernatant was discarded. The black precipitate was stirred gently in 30 mL of distilled water to redisperse, and Mext was applied again to remove the supernatant. The resultant was washed repeatedly twice more, and finally, the remaining particles were sonicated at 200 W in 30 mL of distilled water for 15 min. Afterwards, Mext was applied overnight to precipitate and collect the IC particles.Synthesis of iron oxide@carboxylated cellulose (ICC)
About 510 mg of iron oxide@cellulose were suspended in water (200 mL) and sonicated for 30 min. TEMPO (30 mg) and NaBr (300 mg) were added to the suspension. 200 wt% (compared to mass of cellulose in solution) of NaClO (chloride content 4%) was then added, and the mixture was stirred for 3 h; The pH of the mixture was maintained at 11 at room temperature by adding 0.5 M NaOH while stirring the suspension. After 3 h, the oxidation was terminated by adding methanol (ca. 1 mL) and the pH was adjusted to 7 with 0.5 M HCl. The water insoluble fraction was recovered by a magnet and washed thoroughly with water. The iron oxide@carboxylated cellulose were then dialyzed overnight against DI water (pH 7) and lyophilized to dry (yield: 0.471 g, 92%). The concentration of functional groups on the surface of the iron oxide@cellulose was determined via conductometric analysis. A solution of 0.05 wt% iron oxide@carboxylated cellulose was dispersed in water via overnight sonication. Hydrochloric acid (12 M) was added in 10 μL increments until the pH of the solution reached 2–3. Titrations were then performed with a 0.1 M sodium hydroxide solution in multiples of three, yielding an average carboxylate concentration of 980 mmol kg−1.Synthesis of iron oxide@carboxylated cellulose@Au NPs (ICC@Au)
0.1, 0.2 and 0.3 mL of HAuCl4 solution (10 mM) was added into 10 mL of iron oxide@carboxylated cellulose (0.1%) solution and the mixture was stirred for 1 h to absorb the Au3+ on the surface of iron oxide@carboxylated cellulose. Then the mixture was transferred into a 50 mL Teflon-lined autoclave. The autoclave was sealed and maintained at 150 °C for 8 h to reduce the Au3+ to Au NPs, and then allowed to cool to room temperature naturally. Iron oxide@carboxylated cellulose MNPs were coded as ICC@Au0.1, ICC@Au0.2 and ICC@Au0.3, according to the concentration of HAuCl4 solution.Catalytic reduction of 4-NP
In a typical reaction, 3 × 10−5 mmol of catalyst (ICC@Au0.1, ICC@Au0.2 and ICC@Au0.3) was added to a solution consisting of 48.6 mg NaBH4 in 10 mL (0.1 mM) 4-NP to catalyze the reduction of 4-NP. Reactions were performed in room temperature with continuous stirring. The color of the solutions was changed gradually from yellow to colorless as the reaction proceeded. The conversion of 4-NP was monitored by measuring the absorbance peak at 400 nm for 4-nitrophenolate ion.Recycle catalysis experiments were conducted in order to study the reusability of the magnetic catalysts. Similar to the above reduction process, a given amount of the as-prepared ICC@Au0.1, 3 × 10−7 M were used to catalyze 4-NP (c(4-NP) = 1 × 10−4 M, c(NaBH4) = 0.128 M). After reacted for 5 min, the catalysts were separated by a magnet, and the supernatant was measured using UV-vis spectroscopy. The same procedures were repeated four times. No significant loss in the catalytic activity of ICC@Au0.1 was detected.
Characterization
The morphology of the sample was determined by transmission electron microscope (TEM, JEOL JEM-100CX, Japan) at 80 kV. The UV-vis spectra were recorded on a MAPADA UV-1800 spectrophotometer. Particle size and zeta potential of the nanoparticles were characterized using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, UK). The Fourier transform infrared (FTIR) spectra of all the samples were recoreded on a Nicolet 560 spectrophotometer (USA) over the frequency range of 4000–400 cm−1 at a resolution of 4 cm−1. The KBr disk method was carried out, and the samples were dried in the vacuum oven for 24 h before test. XRD patterns were recorded on a Philips Analytical X'Pert X-diffractometer (Philips Co., Netherlands) with Cu Ka radiation (λ= 1.5406 Å) at 40 kV and 30 mA in the range of 10–70° at room temperature. X-ray photoelectron spectroscopy (XPS) was performed on an ESCALab220i-XL electron spectrometer from VG Scientific using 300 W Al Kα radiation. Magnetization measurement of the sample was performed with a vibrating sample magnetometer (VSM, Lake Shore, 7304, USA) at room temperature under ambient atmosphere.Results and discussion
Preparation and characterization of ICC MNPs
The IC MNPs prepared via an ionic liquid assisted one-step co-precipitation method. This method is based on the ability of ionic liquids to co-dissolve cellulose and iron salt. The addition of ammonia into the ionic liquid was shown to significantly decrease the solubility of cellulose, resulting in the formation of the regenerated cellulose from the cellulose solution.28 Because of the addition of ammonia in the cellulose solution, chemical co-precipitation of iron ions can occur, and iron oxide and regenerated cellulose will be precipitated simultaneously to form cellulose coated iron oxide MNPs. Then, TEMPO-mediated oxidation of cellulose was carried out to introduce carboxyl groups on the surface of MNPs. Fig. 2 depicts the structure of the obtained IC and ICC MNPs. In order to evaluate the stability and dispersibility of the ICC in aqueous solution, analysis of the particle size and surface charge were carried out. Dynamic light-scattering (DLS) measurements revealed that ICC MNPs has a much smaller size than IC MNPs, indicating the increase of dispersibility of the MNPs. Moreover, there was also a remarkable decrease in zeta potential from −8.5 to −48.1 mV, indicating that the hydroxyl was converted to a negatively charged carboxylic acid surface. On the other hand, ICC MNPs dispersed well in water and were stable for up to a month under ambient conditions without any sign of aggregation (Fig. 2), while the IC aggregated and precipitated immediately. This high stability is attributed to the existence of a charged and firm polymeric coating layer on MNPs.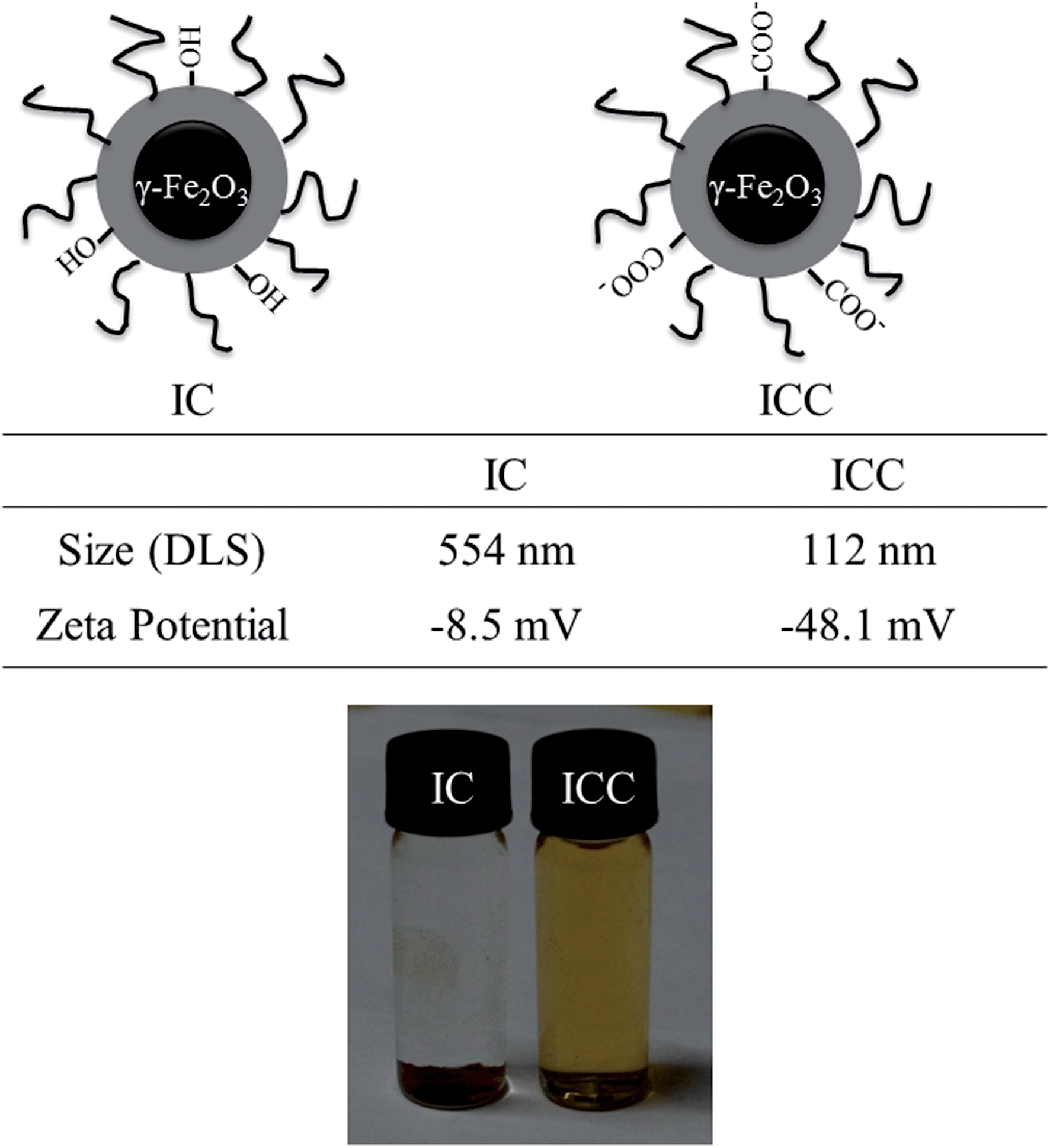 | ||
| Fig. 2 The schematic illustration of the obtained IC and ICC MNPs with size and zeta potential changes and photographs of IC and ICC dispersed in aqueous solution. | ||
As revealed by TEM images in Fig. 3a and b, it can be observed that the magnetic cores are monodispersed and ultrasmall with a particle size of 5 nm. However, through TEMPO oxidation process, ICC MNPs still shows some aggregations in the TEM image (Fig. 3b), which is probably due to the water evaporation. The inserted image in Fig. 3b is a HRTEM image of the as-prepared nanoparticles. The atomic lattice fringes indicate the γ-Fe2O3 to be single crystalline. The interfringe distance is measured to be 0.296 nm, which is close to the lattice spacing of the (220) plane. However, only the core of γ-Fe2O3 nanospheres can be observed, and the cellulose shell is not discernible due to the lack of contrast.
 | ||
| Fig. 3 TEM images of IC (a) and ICC (b). | ||
FTIR is a useful tool for obtaining rapid information about the structure changes of cellulose due to TEMPO oxidation treatment. The FTIR transmission spectra of IC and ICC NPs were recorded and shown in Fig. 4. The spectra are normalized at 1160 cm−1 which assigned to ν–C–O– within the anhydroglucose ring. The FTIR transmission spectra of both samples reveal characteristic bands of both species, i.e., γ-Fe2O3 NPs (∼560 cm−1),29 as well as cellulose (CH, CO, and OH stretching and deformation vibrations). Through the TEMPO oxidation process, a new band at 1732 cm−1 (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) for the carboxylate group of cellulose appears in the spectra of ICC, indicating the successful introduction of carboxylate group. Additionally, in the spectra of ICC the peak at 3410 cm−1 (ν–OH) become narrow and shifted towards lower wavelength, which also indicate the introduction of carboxylate group.
O) for the carboxylate group of cellulose appears in the spectra of ICC, indicating the successful introduction of carboxylate group. Additionally, in the spectra of ICC the peak at 3410 cm−1 (ν–OH) become narrow and shifted towards lower wavelength, which also indicate the introduction of carboxylate group.
 | ||
| Fig. 4 FTIR spectra of IC and ICC. | ||
In situ growth and characterization of Au NPs
The environmentally friendly ICC was used for the reduction and immobilization of Au NPs. The reduction of Au3+ ions to Au NPs was achieved by cellulose directly on the surface of ICC MNPs. Moreover, the size of the obtained Au NPs can be controlled simply by adjusting the concentration of HAuCl4. The morphology and size distribution of ICC@Aux NPs colloids (x = 0.1, 0.2 and 0.3) are determined by TEM and shown in Fig. 5. Fig. 5a and b shows the TEM image of ICC@Au0.1 NPs and the corresponding histogram of particle size distribution, respectively. The mean particle diameter of ICC@Au0.1 is 14.3 ± 5.3 nm, and no agglomeration of Au NPs is observed. The appearance of fringes with different spacing and directions in the corresponding HRTEM micrograph of ICC@Au0.1 NPs suggests that ICC@Au0.1 NPs are polycrystalline. Moreover, selected area electron diffraction (SAED) patterns exhibited spotty diffraction rings, further confirming the polycrystalline feature. Furthermore, the particle size distribution of ICC@Au0.1 is in a narrow range, which suggests that cellulose is highly efficient for the stabilization and/or dispersion of Au NPs. The results indicated that ICC MNPs was able to reduce Au3+ to form Au NPs and served as a support to prevent agglomeration of Au NPs. Larger Au NPs with slightly wider size distribution (24.1 ± 6.1 nm) are formed if the concentration of HAuCl4 increases to 0.2 mM, as shown in Fig. 5c and d. With further increasing the concentration of HAuCl4, a similar trend can also be observed in Fig. 5e and f. The particle size distribution of ICC@Au0.3 is 38.3 ± 8.8 nm. | ||
| Fig. 5 TEM image and size distribution of ICC@Au0.1 (a and b), ICC@Au0.2 (c and d) and ICC@Au0.3 (d and f). The insert images are HRTEM and SEAD of Au NPs in ICC@Au0.1. | ||
The size changes involved in the reduction reaction lead to changes in the surface plasmon resonance (SPR) of the Au NPs, so that the reaction could be monitored by UV-vis spectroscopy. Fig. 6 shows the photograph of the dispersions of ICC MNPs in aqueous solution, before and after reduction with different amounts of HAuCl4, along with their UV-vis spectra. The color of the dispersions changes with increasing concentrations of HAuCl4 and such color changes are reflected in the SPR redshifts. Moreover, the band width of the extinction spectra broadens with increasing concentrations of HAuCl4, indicating that particle size distributions become wider, as evidenced by TEM.
 | ||
| Fig. 6 Photographs of the dispersions of ICC MNPs in aqueous solution, before and after reduction with different amounts of HAuCl4 (0.1, 0.2 and 0.3 mM), along with their UV-vis spectra. | ||
Fig. 7 shows the XRD patterns of ICC and ICC@Au0.1 to further verify the formation of the Au NPs. In the pattern of ICC, two peaks at 20.1° and 21.2° are attributed to (110) and (200) diffractions of cellulose II.30 Furthermore, the iron oxide phase in magnetic NPs is verified from diffraction peaks at 30.0°, 35.3°, 42.8°, 56.9° and 62.5° of (220), (311), (400), (511) and (440) planes of γ-Fe2O3 (JCPDS Card no. 39–1346), indicating the successful synthesis of iron oxide NPs. Moreover, Au NPs deposited onto the surface of ICC can be confirmed from the corresponding XRD data of ICC@Au0.1. Excepting the characteristic diffraction peaks of ICC, there are four other diffraction peaks in the figure. The four peaks positioned at 38.2°, 44.4°, 64.5° and 77.4° could be attributed to the reflections of the (111), (200), (220), and (311) crystalline planes of cubic Au, respectively.
 | ||
| Fig. 7 XRD patterns of ICC and ICC@Au0.1. | ||
As the standard XRD patterns of Fe3O4 and γ-Fe2O3 are almost identical, XPS were used to distinguish them. In Fig. 8a, XPS data show peaks at 710.9 and 724.6 eV which are in good agreement with the known values of Fe 2p3/2 and Fe 2p1/2 of γ-Fe2O3, respectively.31 The peak positions of Fe 2p1/2 and Fe 2p3/2 for Fe3O4 are comparatively lower, located at 724.1 and 709.6 eV.32 Furthermore, there is a characteristic satellite peak of γ-Fe2O3 obtained at 718.8 eV, confirming that these magnetic NPs are indeed γ-Fe2O3.33 Meanwhile, the Au 4f XPS spectrum of the ICC@Au0.1 NPs (Fig. 8b) displays an Au 4f7/2 and Au 4f5/2 binding energy of 83.5 and 87.1 eV, respectively, which are the typical characteristics of Au0.34 On the other hand, no characteristic binding energy of Au3+ and Au+ is observed, indicating the completely reduction of Au3+.
 | ||
| Fig. 8 Fe 2p (a) and Au 4f (b) XPS spectra of ICC@Au0.1. | ||
Since the magnetic properties of the magnetic materials are critical to ensure their application, the magnetic properties of ICC and ICC@Au0.1 were studied. Fig. 9 shows the magnetic hysteresis loops of the ICC and ICC@Au0.1 at room temperature. They exhibited super-paramagnetic behavior and little hysteresis, remanence and coercivity due to the fact that the particles were composed of ultrafine γ-Fe2O3 nanocrystals. This result indicates that no residual magnetism for γ-Fe2O3 NPs is retained without external magnetic field; otherwise these NPs may aggregate irreversibly. The magnetic saturation (Ms) values are 30 and 18 emu g−1, respectively. Comparing to the Ms value of ICC, that of ICC@Au0.1 is lower, which might be due to the decrease in the density of γ-Fe2O3 after immobilization of the Au NPs. It should be noted that the ICC@Au0.1 still exhibited strong magnetization, which ensured their suitability for magnetic separation and targeting. The inserted image in Fig. 9 illustrates its strong magnetic response.
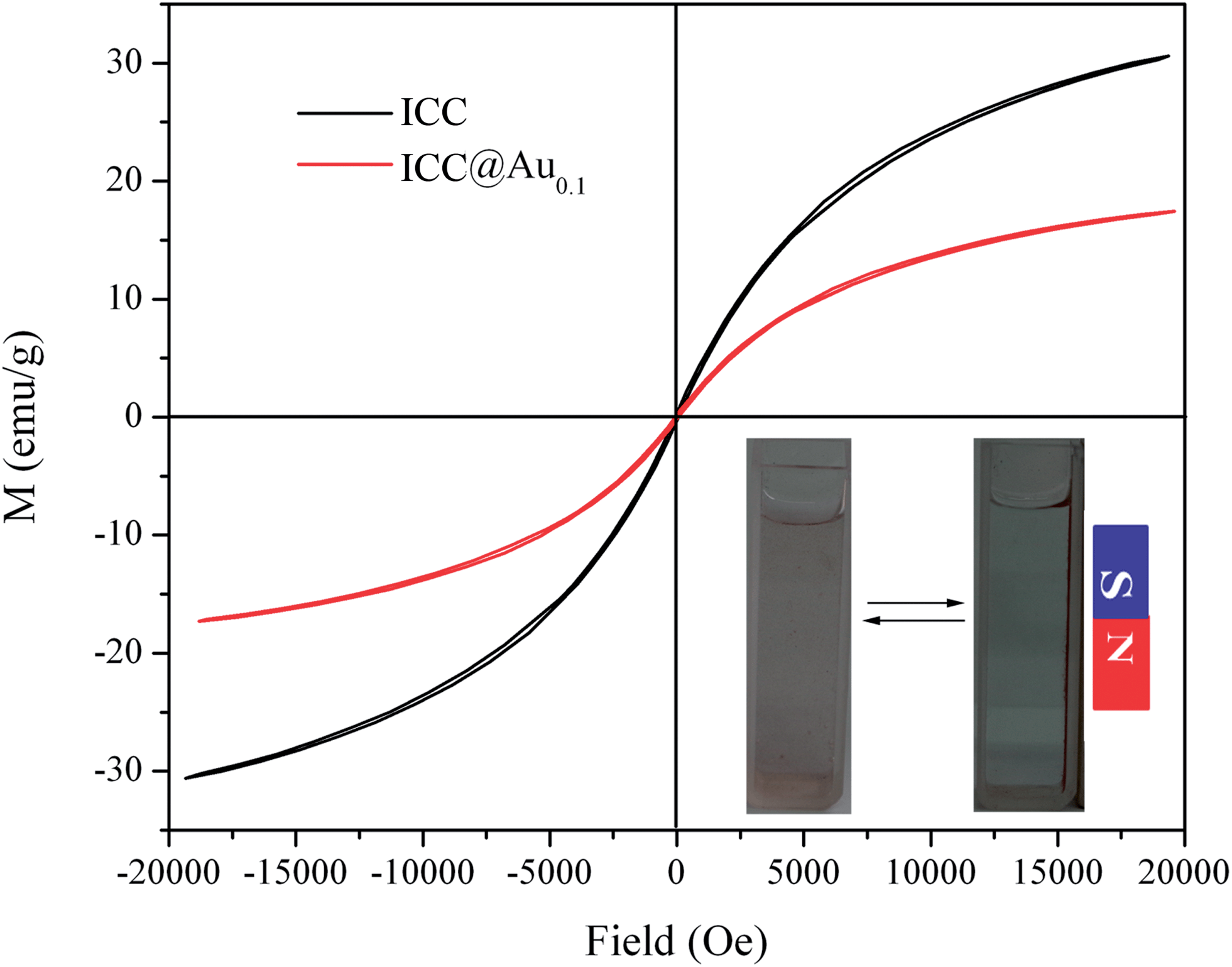 | ||
| Fig. 9 Magnetic hysteresis loops of the ICC and ICC@Au0.1 at room temperature. The inset pattern is a photograph of the magnetic separation. | ||
Catalytic activity
It has been demonstrated that metallic Au NPs are excellent catalysts with high activity and selectivity.1,35 Immobilized on a solid support, Au NPs may serve as a practical recyclable nanocatalyst towards many reactions. 4-NP is one of the most common organic pollutants in industrial and agricultural wastewaters, while 4-AP is very useful and important in many applications including analgesic and antipyretic drugs, photographic development, corrosion inhibition, anticorrosion lubrication, etc.36–38 Here, the as-prepared ICC@Au catalyst was used to catalyze the reduction of 4-NP in the presence of NaBH4. As shown in Fig. S1,† the pure 4-NP solution exhibited an absorption maximum at 317 nm, and the absorption peak shifted to 400 nm immediately after the addition of the NaBH4 solution, which corresponded to a color change of light yellow to bright yellow due to the formation of 4-NP ion in alkaline condition.39,40 In the absence of catalyst, virtually no reduction of 4-NP was observed (Fig. S2†). Before investigating the catalytic activity of the ICC@Au NPs, the catalytic ability of the equal amount of ICC was studied. The UV-vis absorption spectrum also remains unaltered with time in the presence of ICC as shown in Fig. S3,† which means that the ICC does not catalyze this reaction. However, by adding a small amount of ICC@Au0.1 NPs, the color of 4-NP solution faded in only several minutes, indicating the reduction of 4-NP by NaBH4 occurs very fast in the presence of ICC@Au0.1 NPs.Fig. 10a shows the successive UV-vis spectra of 4-NP in the presence of NaBH4 and ICC@Au0.1 NPs in aqueous solution. The color of the 4-NP solution vanishes gradually in several minutes, with the gradual decrease in absorbance value at 400 nm. Meanwhile, a new peak centered at 300 nm appears and increases with reaction time. This peak could be indexed to the characteristic absorbance peak of 4-AP, which further confirmed the reduction of 4-NP to 4-AP.41 The progress of the catalytic reduction of 4-NP can be easily followed by the decrease in absorbance at max of 4-NP with time. It should be noted that the reduction of 4-NP by NaBH4 was finished within 5 minutes with the addition of ICC@Au0.1 (Fig. 10a). However, a longer reaction time was required to achieve the full reduction of 4-NP by NaBH4 using ICC@Au0.2 and ICC@Au0.3 as catalysts (Fig. 10b and c). Owing to the great excess amount of NaBH4, it remained essentially constant throughout the reaction. The reduction rate is assumed to be independent of the NaBH4 concentration. Therefore, a pseudo first-order kinetic equation can be applied to evaluate the catalytic rate. The linear relationship between ln(Ct/C0) (C0 and Ct is the absorption peak at 400 nm initially and at time t.) and reaction time is displayed in Fig. 10d, from which the reaction rate constant is calculated to be 5.6 × 10−1 min−1, 2.5 × 10−1 min−1 and 4.0 × 10−2 min−1 for ICC@Au0.1, ICC@Au0.2 and ICC@Au0.3, respectively. These results clearly indicate the catalytic activity of the obtained nanocomposites for the reduction of 4-NP followed this order: ICC@Au0.1 > ICC@Au0.2 > ICC@Au0.3. This is due to the increasing size of ICC@Au0.1, ICC@Au0.2 and ICC@Au0.3, resulting in the decrease of their active surface areas.42 It is worth to mention that the reaction constant of ICC@Au0.1 is higher than many reported for the same catalytic conversion using catalysts based on Au NPs with smaller sizes.42–46
 | ||
| Fig. 10 (a)The successive UV-vis spectra of 4-NP in the presence of NaBH4 and ICC@Au0.1 (a), ICC@Au0.2 (b) and ICC@Au0.3 (c) in aqueous solution. (d) The ln(Ct/C0) versus the reaction time for the reduction of 4-NP over ICC@Au0.1, ICC@Au0.2 and ICC@Au0.3 at 25 °C. C0 and Ct is the absorption peak at 400 nm initially and at time t. | ||
The recyclability of catalysts is an important factor for a more economical process. Therefore, we investigated the efficiency of the prepared ICC@Au0.1 catalyst in repeated cycles of reaction. At the end of the reaction, the catalyst was easily separated from the reaction medium using a magnet. Then the catalyst was washed thoroughly with distilled water and reused for the next reduction reaction. After this procedure has been repeated four times, ICC@Au0.1 catalyst was still stable and exhibited a high catalytic activity (the conversion was up to 99% as shown in Fig. 11), indicating the excellent recyclability of the prepared ICC@Au0.1 as a catalyst for reduction reaction.
 | ||
| Fig. 11 The reusability of ICC@Au0.1 as a catalyst for the reduction of 4-NP with NaBH4. | ||
Conclusion
In conclusion, a facile in situ reduction route for immobilizing Au NPs on the surface of γ-Fe2O3@carboxylated cellulose has been successfully developed. Cellulose not only acted as reducing agent for the gold nucleation and growth, but also acts as adhesive to the metallic Au surface. The as-prepared γ-Fe2O3@carboxylated cellulose@Au can be used as catalysts for the reduction of 4-NP with NaBH4. The catalysts can be easily recovered for reuse via a magnetic separation technique, which is preferred in terms of cost and environmental protection. The biodegradable, biocompatibility and good dispersion properties of the developed nanohybrids can be readily applied in the areas of biotechnology/biomedicine, health care, magnetic resonance imaging, sensors, as well as inorganic carriers for enzyme immobilization and controlled drug delivery, etc.Acknowledgements
The authors would like to thank the National Science Foundation of China (51203105) and National High Technology Research and Development Program (863 Program, SS2012AA062613) for financial support.References
- Y. Choi, H. S. Bae, E. Seo, S. Jang, K. H. Park and B. S. Kim, J. Mater. Chem., 2011, 21, 15431–15436 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- A. Llevot and D. Astruc, Chem. Soc. Rev., 2012, 41, 242–257 RSC.
- K. Niikura, N. Iyo, Y. Matsuo, H. Mitomo and K. Ijiro, ACS Appl. Mater. Interfaces, 2013, 5, 3900–3907 CAS.
- S. Kundu and M. Jayachandran, RSC Adv., 2013, 3, 16486–16498 RSC.
- G. Marcelo, A. Muñoz-Bonilla and M. Fernández-García, J. Phys. Chem. C, 2012, 116, 24717–24725 CAS.
- J. Zhang, D. Han, H. Zhang, M. Chaker, Y. Zhao and D. Ma, Chem. Commun., 2012, 48, 11510–11512 RSC.
- L. B. Devi and A. B. Mandal, RSC Adv., 2013, 3, 5238–5253 RSC.
- C. Zhu, L. Han, P. Hu and S. Dong, Nanoscale, 2012, 4, 1641–1646 RSC.
- M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 3371–3393 RSC.
- N. T. S. Phan, C. S. Gill, J. V. Nguyen, Z. J. Zhang and C. W. Jones, Angew. Chem., Int. Ed., 2006, 45, 2209–2212 CrossRef CAS PubMed.
- R. Güttel, M. Paul, C. Galeano and F. Schüth, J. Catal., 2012, 289, 100–104 CrossRef PubMed.
- J. Zhao, S. Sallard, B. M. Smarsly, S. Gross, M. Bertino, C. Boissière, H. Chen and J. Shi, J. Mater. Chem., 2010, 20, 2831–2839 RSC.
- M. C. Ferrara, L. Mirenghi, A. Mevoli and L. Tapfer, Nanotechnology, 2008, 19, 365706 CrossRef CAS PubMed.
- M. B. Gawande, R. K. Pandey and R. V. Jayaram, Catal. Sci. Technol., 2012, 2, 1113–1125 CAS.
- K. V. S. Ranganath and F. Glorius, Catal. Sci. Technol., 2011, 1, 13–22 CAS.
- V. Polshettiwar, R. Luque, A. Fihri, H. B. Zhu, M. Bouhrara and J. M. Bassett, Chem. Rev., 2011, 111, 3036–3075 CrossRef CAS PubMed.
- D. Kim, N. Lee, M. Park, B. Y. Kim, K. An and T. Hyeon, J. Am. Chem. Soc., 2009, 131, 454–455 CrossRef CAS PubMed.
- H. Lee, E. Lee, D. K. Kim, N. K. Jang, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2006, 128, 7383–7389 CrossRef CAS PubMed.
- Z. Marková, K. Šišková, J. Filip, K. Šafářová, R. Prucek, A. Panáček, M. Kolář and R. Zbořil, Green Chem., 2012, 14, 2550–2558 RSC.
- G. Marcelo, A. Munoz-Bonilla, J. Rodríguez-Hernández and M. Fernández-García, Polym. Chem., 2013, 4, 558–567 RSC.
- B. Liu, X. Li, C. Zheng, X. Wang and R. Sun, Nanotechnology, 2013, 24, 235601 CrossRef CAS PubMed.
- S. Sharma, P. Sanpui, A. Chattopadhyay and S. Sankar Ghosh, RSC Adv., 2012, 2, 5837–5843 RSC.
- X. Lin, M. Wu, D. Wu, S. Kuga, T. Endo and Y. Huang, Green Chem., 2011, 13, 283–287 RSC.
- R. Xiong, C. Lu, W. Zhang, Z. Zhou and X. Zhang, Carbohydr. Polym., 2013, 95, 214–219 CrossRef CAS PubMed.
- M. Adsul, S. K. Soni, S. K. Bhargava and V. Bansal, Biomacromolecules, 2012, 13, 2890–2895 CrossRef CAS PubMed.
- D. Klemm, F. Kramer, S. Moritz, T. Lindstrom, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- X. Yang, X. Zhang, Y. Ma, Y. Huang, Y. Wang and Y. Chen, J. Mater. Chem., 2009, 19, 2710–2714 RSC.
- J. Cai, S. Kimura, M. Wada and S. Kuga, Biomacromolecules, 2009, 10, 87–94 CrossRef CAS PubMed.
- L. S. Zhong, J. S. Hu, H. P. Liang, A. M. Cao, W. G. Song and L. J. Wan, Adv. Mater., 2006, 18, 2426–2431 CrossRef CAS.
- E. Smit, M. M. Schooneveld, F. Cinquini, H. Bluhm, P. Sautet, F. M. F. Groot and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2011, 123, 1622–1626 CrossRef.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441–2449 CrossRef CAS PubMed.
- H. Wei, Z. Wang, L. Yang, S. Tian, C. Hou and Y. Lu, Analyst, 2010, 135, 1406–1410 RSC.
- J. Han, Y. Liu and R. Guo, J. Am. Chem. Soc., 2009, 131, 2060–2061 CrossRef CAS PubMed.
- Y. Du, H. Chen, R. Chen and N. Xu, Appl. Catal., A, 2004, 277, 259–264 CrossRef CAS PubMed.
- J. F. Corbett, Dyes Pigm., 1999, 41, 127–136 CrossRef CAS.
- C. V. Rode, M. J. Vaidya and R. V. Chaudhari, Org. Process Res. Dev., 1999, 3, 465–470 CrossRef CAS.
- K. Hayakawa, T. Yoshimura and K. Esumi, Langmuir, 2003, 19, 5517–5521 CrossRef CAS.
- S. Praharaj, S. Nath, S. K. Ghosh, S. Kundu and T. Pal, Langmuir, 2004, 20, 9889–9892 CrossRef CAS PubMed.
- M. Zhu, C. Wang, D. Meng and G. Diao, J. Mater. Chem. A, 2013, 1, 2118–2125 CAS.
- Y. Zhang, S. Liu, W. Lu, L. Wang, J. Tian and X. Sun, Catal. Sci. Technol., 2011, 1, 1142–1144 CAS.
- H. Wu, X. Huang, M. Gao, X. Liao and B. Shi, Green Chem., 2011, 13, 651–658 RSC.
- P. Zhang, C. Shao, X. Li, M. Zhang, X. Zhang, C. Su, N. Lu, K. Wang and Y. Liu, Phys. Chem. Chem. Phys., 2013, 15, 10453–10458 RSC.
- T. Huang, F. Meng and L. Qi, J. Phys. Chem. C, 2009, 113, 13636–13642 CAS.
- Y. Zhu, J. Shen, K. Zhou, C. Chen, X. Yang and C. Li, J. Phys. Chem. C, 2011, 115, 1614–1619 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra46761a |
| This journal is © The Royal Society of Chemistry 2014 |