
Alternating copolymerization of ethylenesulfonyl fluoride and styrene for the synthesis of SuFEx modifiable polymers
Chima
Anyaegbu
a,
Darsan
Haridas
a and
Joel F.
Hooper
 *ab
*ab
aSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: joel.hooper@monash.edu
bBioresource Processing Research Institute of Australia (BioPRIA), Monash University, Clayton, Victoria 3800, Australia
First published on 22nd April 2026
Abstract
This work investigates the copolymerization of ethenesulfonyl fluoride (ESF) and styrene, demonstrating this method as a practical approach to access highly functionalised sulfonyl fluoride-containing polymers. Instantaneous copolymer composition data were analyzed using the Mayo–Lewis equation, yielding reactivity ratios of r1 ≈ 0.062 (ESF) and r2 ≈ 0.896 (styrene), indicative of a strong tendency toward alternating copolymerisation. Kinetic studies demonstrated reversible deactivation radical polymerization in the presence of a RAFT agent with low dispersities. Post-polymerization modification of ESF-co-St copolymers was successfully achieved via reaction of the sulfonyl fluoride functionality, enabling versatile transformations for advanced material design. These findings provide a foundation for tailored copolymer architectures and open pathways for new functional material development.
Introduction
The design of functional polymers with controlled architectures is central to modern materials science, enabling post polymerization functionalization.1,2 Styrene, a widely used monomer in commodity and specialty polymers, has been extensively copolymerized with electron-deficient monomers such as maleic anhydride, N-substituted maleimides, and acrylonitrile, forming alternating copolymers. These copolymers exhibit unique thermal and mechanical properties and have been applied in compatibilizers, dispersants, and coatings. However, traditional electrophilic monomers often suffer from limited post-polymerization modification, hydrolytic instability, and restricted chemical diversity, limiting their utility in advanced functional systems.3,4To address these limitations, researchers have introduced activated esters such as pentafluorophenyl acrylate (PFPA) and p-nitrophenyl methacrylate into copolymers, enabling post-polymerization functionalization via nucleophilic substitution.5–7 While these strategies allow incorporation of diverse functionalities, they still present drawbacks including hydrolytic sensitivity, thus restricting their application in aqueous conditions.7
In contrast, sulfonyl fluorides have emerged as privileged functional groups due to their unique combination of stability and reactivity. Sulfur(VI) Fluoride Exchange (SuFEx) reactions proceed under mild, metal-free conditions and exhibit exceptional chemoselectivity, enabling efficient conversion of sulfonyl fluoride groups into sulfonamides, sulfonates, and sulfamides.8–11 SuFEx chemistry was first used in polymer synthesis by Dong, Wu and Sharpless in 2017 for the step-growth polymerisation of bisphenol A derivatives.12 Sulfonyl fluorides have also been introduced into vinyl-based polymers in reversible deactivation radical polymerizations by the use of functionalised RAFT agents,13 or through the use of functionalised monomers14 (Scheme 1) allowing for post-polymerisation functionalisation through SuFEx reactions.
 | ||
| Scheme 1 Synthesis of sulfonyl fluoride containing materials by radical polymerisation. | ||
Ethenesulfonyl fluoride (ESF), described as “the most perfect Michael acceptor”, is a cornerstone of SuFEx click chemistry, introduced by Sharpless and co-workers in 2014.15,16 Existing ESF-based polymers are limited to step-growth polysulfonates derived from ESF-amine adducts and bisphenols, which lack the architectural control and versatility offered by reversible deactivation radical polymerization.17,18 In 2003, Ueda and coworkers reported the homo- and co-polymerisation of ESF with a styrene-based monomer under free radical conditions for the production of photoresist materials.19
We have previously reported the use of ESF in C–H functionalisation reactions of small molecules,20 however, the copolymerisation kinetics of ESF-based polymers have not been reported nor have their application in SuFEx reactions been explored.
Herein, we report the first example of the radical copolymerisation of ESF under RAFT and conventional radical conditions. These copolymers serve as highly functionalizable platforms, enabling SuFEx-based derivatization to sulfonamides and sulfonates, as well as crosslinking with branched polyethyleneimine (bPEI) to produce a 3D network material (ESF-co-St-bPEI25K). Determination of the reactivity ratios of ESF and styrene using the Mayo–Lewis copolymer composition model provides critical insights into their tendency to undergo alternating copolymerisation.
Materials and methods
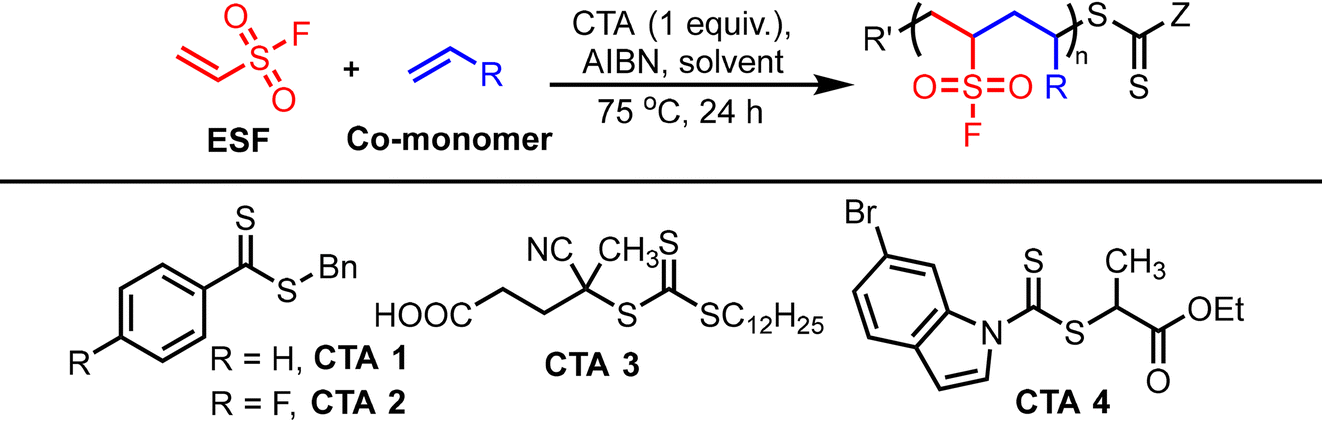
The following chemicals were purchased from Sigma Aldrich: vinyl acetate, methyl acrylate, 4-vinylbenzyl chloride, acetonitrile, toluene, dioxane, ethenesulfonyl fluoride, styrene, dimethylformamide, chloroform, polyethyleneimine, phenol, 4-methoxyphenol, azobisisobutyronitrile (AIBN), tetrahydrofuran, potassium carbonate. Benzyl benzodithioate (CTA 1) and 4-cyano-4-(((dodecylthio)carbonothioyl)thio) pentanoic acid (CTA 3), were purchased from Aaron Chem. Benzyl 4-fluorobenzodithioate (CTA 2)21 and ethyl 2-((6-bromo-1H-indole-1-carbonothioyl)thio)propanoate (CTA 4)22 were sythesised according to the previously reported methods.Gel permeation chromatography (GPC)/size exclusion chromatography (SEC) analysis was performed on an Agilent 1260 Infinity II GPC/SEC system with a refractive index detector (RID) and a PLgel MIXED-C column (7.5 mm × 300 mm, 5 μm). The samples, dissolved in THF at a concentration of 10 mg mL−1, were filtered by using a 0.22 μm syringe filter before analysis. Chromatography was carried out at 30 °C with a flow rate of 1 mL min−1 and a 50 μL injection volume. The system was calibrated against polystyrene standards. The molar masses were expressed as the number-average molar mass (Mn) and the weight-average molar mass (Mw).
NMR analysis: spectra were recorded in CDCl3 on a Bruker DRX400 NMR spectrometer operating at 400 MHz for proton and 100 MHz for carbon nuclei. Proton and 13C NMR spectra were calibrated using the residual solvent peak of CDCl3: δ = 7.26 ppm for 1H and δ = 77.16 ppm for 13C. 19F NMR spectra were recorded at 377 MHz in CDCl3, with chemical shifts externally referenced to CFCl3 (δ = 0.00 ppm).
Thermal gravimetry analysis: thermal properties of polymers and crosslinked material were evaluated using (Pyris 1, PerkinElmer, USA), a temperature program from 40 °C to 600 °C at a heating rate of 10 °C min−1 under a nitrogen atmosphere.
Rheological properties: measurements were done using a rotational rheometer (Anton Paar, MCR 703), plate diameter 40 mm, 1 mm gap. Gelation time was determined by performing a time-sweep test at a constant angular frequency and strain within the linear viscoelastic region. Gelation was defined as the point at which the storage modulus (G′) surpassed the loss modulus (G″). A frequency sweep was conducted at a frequency range (e.g., 0.1–100 rad s−1) at constant strain within the linear viscoelastic, at 25 °C.
FTIR analysis: spectra were collected using Agilent Cary 630 FTIR spectrometer in a wavenumber range of 400 to 4000 cm−1, by the accumulation of 32 scans, with 2 cm−1 resolution.
Typical polymerization procedure
A representative polymerization was carried out as follows: CTA-3 (8.1 mg, 0.02 mmol, 1 equiv.), AIBN (25 μL, 0.2 M solution in toluene, 0.005 mmol, 0.25 equiv.), ESF (220 mg, 2 mmol, 100 equiv.), and styrene (416 mg,4 mmol, 200 equiv.) were added to a reaction vial, followed by 1 mL of DMF. 4-Fluorobromobenzene was introduced as an internal standard. The mixture was purged with nitrogen for 15 min, and an aliquot was withdrawn prior to heating to determine the initial monomer ratios by 1H and 19F NMR spectroscopy. The sealed reaction mixture was then heated to 75 °C for the desired reaction time. At completion, the mixture was cooled to room temperature, and a second aliquot was taken for determination of monomer conversion by NMR analysis. The crude reaction mixture was precipitated into MeOH to isolate the polymer. The resulting solid was collected, redissolved in THF, and reprecipitated into MeOH. This dissolution-precipitation cycle was repeated two additional times to ensure thorough purification. The final polymer was dried under reduced pressure at 50 °C.Results and discussion
We began this study by investigating the copolymerization of ethylene sulfonyl fluoride (ESF) with styrene as co-monomer under RAFT polymerization conditions. The effect of different CTAs was investigated (Table 1, entries 1–5) beginning with dithiobenzoate CTA 1, which has been successfully applied in the alternating copolymerization of styrene with maleic anhydride.23 CTA 2 was also tested, as we anticipated that the fluorine atom could be used to quantify the ratio of CTA to sulfonyl fluoride in the resulting polymer. Dithiobenoate CTAs 1 and 2 showed 20 and 13% conversion of the ESF monomer, moderate dispersities (Đ = 1.50) and low Mn values of 3600 and 2800 respectively. CTA 3 (entry 3) was trialed next, as trithiocarbonates are compatible with both styrene and electron-deficient monomers.24 This showed better control, with ESF conversion of 32%, an Mn of 6900 that closely aligns with the theoretical value of 7000, a dispersity of 1.26 and an ESF mole fraction of 0.49, indicating a near 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of ESF and styrene in the isolated polymer. CTA 4 has recently been shown as an effective RAFT agent for acrylate monomers,22 which we hypothesized might be applicable to electron-deficient ESF. CTA 4 showed somewhat worse control than CTA 3, with 27% conversion of ESF and a dispersity of 1.39. Omitting the CTA entirely (Table 1, entry 5) gave an Mn of 6100 (compared with a theoretical value of 12600) and an increased Đ of 1.51, which is comparable to the use of CTA 1 or 2. The low Mn observed in the absence of a RAFT agent suggests that radical chain transfer may be occurring via reaction with the DMF solvent.25 To test this hypothesis, the reaction was next performed in MeCN (entry 6) as a less reactive solvent, resulting in an Mn of 39000 and a dispersity of 1.63.
:1 ratio of ESF and styrene in the isolated polymer. CTA 4 has recently been shown as an effective RAFT agent for acrylate monomers,22 which we hypothesized might be applicable to electron-deficient ESF. CTA 4 showed somewhat worse control than CTA 3, with 27% conversion of ESF and a dispersity of 1.39. Omitting the CTA entirely (Table 1, entry 5) gave an Mn of 6100 (compared with a theoretical value of 12600) and an increased Đ of 1.51, which is comparable to the use of CTA 1 or 2. The low Mn observed in the absence of a RAFT agent suggests that radical chain transfer may be occurring via reaction with the DMF solvent.25 To test this hypothesis, the reaction was next performed in MeCN (entry 6) as a less reactive solvent, resulting in an Mn of 39000 and a dispersity of 1.63.
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Ent. | ESF:co-monomer:CTA:AIBN |
Co-mono-mera | CTA | Solvent | ESF/co-monomer conv. (%) |
M
n
(theory) |
M
n
|
Đ | ESF mol. fract. |
| a St = styrene, VAc = vinyl acetate, MA = methyl acrylate, 4-Cl St = 4-chlorostyrene. b g mol−1. | |||||||||
| 1 | 100:50:1:0.25 |
St | 1 | DMF | 20/53 | 5000 | 3600 | 1.50 | 0.43 |
| 2 | 100:50:1:0.25 |
St | 2 | DMF | 13/34 | 3200 | 2800 | 1.50 | 0.43 |
| 3 | 100:50:1:0.25 |
St | 3 | DMF | 32/67 | 7000 | 6900 | 1.26 | 0.49 |
| 4 | 100:50:1:0.25 |
St | 4 | DMF | 27/70 | 6700 | 5800 | 1.39 | 0.44 |
| 5 | 100:50:1:0.25 |
St | None | DMF | 26/65 | 12600 |
6100 | 1.51 | 0.44 |
| 6 | 100:50:1:0.25 |
St | None | MeCN | 35/79 | 15900 |
39000 |
1.63 | 0.47 |
| 7 | 100:0:1:0.25 |
None | 3 | DMF | <2/n/a | — | — | — | — |
| 8 |
100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :50:1:0.25 :50:1:0.25
|
VAc | 3 | DMF | <1/29 | 1600 | — | — | — |
| 9 | 100:50:1:0.25 |
MA | 3 | DMF | <2/38 | 2100 | — | — | — |
| 10 | 100:50:1:0.25 |
4-Cl St | 3 | DMF | 44/82 | 9100 | 8400 | 1.37 | 0.52 |
| 11 |
50:25:1:0.25 |
St | 3 | DMF | 26/62 | 3100 | 4700 | 1.83 | 0.46 |
| 12 |
200:100:1:0.25 |
St | 3 | DMF | 21/60 | 10100 |
9100 | 1.40 | 0.41 |
| 13 |
100:200:1:0.25 |
St | 3 | DMF | 68/78 | 24300 |
21900 |
1.39 | 0.33 |
| 14 | 100:50:1:0.5 |
St | 3 | DMF | 37/88 | 8700 | 1900 | 2.96 | 0.46 |
| 15 | 100:50:1:0.125 |
St | 3 | DMF | 19/32 | 3700 | 5900 | 1.30 | 0.54 |
| 16 | 100:50:1:0.25 |
St | 3 | MeCN | 35/73 | 7700 | 7300 | 1.18 | 0.49 |
| 17 | 100:50:1:0.25 |
St | 3 | Toluene | 32/66 | 7000 | 6400 | 1.28 | 0.49 |
| 18 | 100:50:1:0.25 |
St | 3 | Dioxane | 31/70 | 7100 | 6100 | 1.28 | 0.47 |
| 19 | 100:50:1:0.25 |
St | 3 | None | 23/55 | 5400 | 5500 | 1.36 | 0.46 |
Chen and coworkers have recently reported low dispersity alternating radical copolymerisations of a number of fluorinated and non-fluorinated monomers.26 This study proposed that the absence of charge-transfer complexes (CTCs) between monomer pairs can increase propagation rates and reduce termination events, resulting in dispersities of 1.13–1.39. NMR analysis of a 1:1 mixture of ESF and styrene (Fig. S5) showed very little change in chemical shift compared with the pure monomers, suggesting that the formation of CTCs in this system is negligible.
When styrene was omitted (Table 1, entry 7), ESF conversion dropped to less than 2%, confirming that ESF does not homopolymerize efficiently under these conditions. Substituting styrene with vinyl acetate or methyl acrylate (Table 1, entries 8 and 9) resulted in similarly poor ESF incorporation (<2%) despite moderate conversions of the alternative monomers (29% and 38%, respectively). This suggests that ESF requires a co-monomer with complimentary reactivity for effective copolymerization. In contrast, replacing styrene with 4-chlorostyrene (Table 1, entry 10) significantly improves ESF conversion to 44% and comonomer conversion to 82%, yielding a polymer with Mn of 8400 and Đ of 1.37.
Variations in feed ratios and initiator loading were next investigated (entries 11–15). Reducing the total monomer feed ratio to 50 equivalents of ESF and 25 of styrene (entry 11) resulted in increased dispersity (Đ = 1.83) and a reduced ESF mole fraction of 0.46. Increasing the total monomer feed (entry 12) to 200:100 resulted in higher Mn (9100, compared with 10100 theoretical) while maintaining reasonable control (Đ = 1.40). Changing the ESF:St ratio to 100:200 gave high monomer conversion and higher molar mass (Mn = 21900), but gave a final ESF mole fraction of 0.33, indicating that an excess of ESF is required to achieve equimolar ratios in the final polymer. Doubling the amount of initiator (entry 14) caused severe loss of control, with dispersity soaring to 2.96 and Mn collapsing to 1900 despite high conversions (37% ESF, 88% styrene). Reducing the initiator concentration (entry 15) improves Đ to 1.30, though Mn remains moderate at 5900.
Solvent choice was also investigated in the ESF/styrene copolymerisation. MeCN (Table 1, entry 16) delivered the best control, with dispersity reduced to 1.18 and Mn increased to 7300, alongside conversions of 35% for ESF and 73% for styrene. Toluene and dioxane (entries 17 and 18) maintain similar conversion levels but exhibit slightly higher dispersities (1.28) and lower Mn values (6400 and 6100, respectively). Performing the reaction neat (entry 19) leads to the poorest outcome, with reduced conversions (23% ESF, 55% styrene), higher dispersity (Đ = 1.36), and Mn dropping to 5500. These results show that both polar and non-polar solvents are compatible with this reaction, with DMF giving the highest dispersity while solvent-free conditions show the lowest conversions and modest dispersity.
Overall, the data demonstrate that optimal conditions for balanced ESF–styrene incorporation and controlled polymerization involve polar solvents such as MeCN, CTA 3, and moderate initiator levels. 1H NMR analysis of the precipitated polymer (Table 1, entry 3) shows a 1:1 ratio of ESF and St, consistent with an alternating copolymerization (Fig. 1A). 19F NMR shows 2 broad fluorine environments between δ 40–50 ppm, with a relative integration of 1:3 (Fig. 1B). This ratio may indicate an atactic polymer, with rr, rm and mm stereochemical triads being present in a 1:2:1 ratio, and the signal at δ 50 ppm representing the rr or mm triads. Analysis of the polymer by 1H Diffusion Ordered Spectroscopy (DOSY) NMR showed a diffusion coefficient of 2.6–2.7 × 10−11 m2 s−1 for the signals associated with the polymer backbone and 3.2 × 10−11 m2 s−1 for the RAFT endgroup, compared with 1.7 × 10−9 m2 s−1 for the residual solvent signal (Fig. 1C). This is consistent with successful incorporation of the CTA into the polymer, where the diffusion coefficient for the polymer endgroups are slightly higher than for the polymer backbone.27 Similarly, 19F DOSY NMR shows the fluorine atoms diffusing at 2.7–2.8 × 10−11 m2 s−1.
 | ||
| Fig. 1 NMR spectra of polymer from Table 1, entry 3 (A) 1H NMR spectra and (B) 19F NMR spectra (C) 1H DOSY NMR (D) 19F DOSY NMR. | ||
The copolymerization kinetics were next investigated with an ESF:St feed ratio of 100:200 (conditions from Table 1, entry 13). The plot of ln([M0]/[MT]) vs. time (Fig. 2A) shows a linear correlation consistent with first-order kinetics, and no evidence of an induction period. The plot of molar mass (Mn) vs. total monomer conversion (Fig. 2A) is also linear, consistent with a reversible deactivation radical polymerization, with dispersity values below 1.4.
 | ||
| Fig. 2 (A) Plots of ln([M0]/[MT]) vs. time (B) Mn and dispersity vs. conversion. | ||
The polymerisation results from Table 1 show that ESF is not capable of homopolymerisation, and shows a tendency for a 1:1 ESF:St ratio in the final polymer, which may indicate an alternating copolymerisation. To investigate this, the reactivity ratios were estimated by nonlinear least squares (NLLS) fitting of the Mayo–Lewis equation using the Levenberg–Marquardt algorithm (Python, scipy.optimize.curve_fit). NLLS is preferred over linearized methods (Fineman-Ross, Kelen-Tüdös) as it preserves the error structure and avoids bias, especially in asymmetric systems where one monomer does not homopolymerise (r1 ≈ 0).28–31
The best-fit parameters are r1 (ESF) = 0.062 and r2 (styrene) = 0.896, with a product r1r2 ≈ 0.055. Fig. 3 shows the fitted composition curve alongside the experimental points and the ideal line F1 = f1. The experimentally determined copolymer compositions showed excellent agreement with values predicted by the terminal model using the calculated reactivity ratios (r1 = 0.062, r2 = 0.896), with residuals not exceeding ±0.03 over the entire composition range studied.
 | ||
| Fig. 3 Determination of reactivity ratios by fitting of the Mayo–Lewis equation. | ||
The fitted reactivity ratios indicate a pronounced preference for cross-propagation between ESF and styrene. Since r1 ≈ 0, ESF cannot homopolymerize and can therefore can only react with another monomer. Meanwhile r2 is moderately below unity, and the product r1r2 is well under one – a hallmark of alternating tendencies in radical copolymerization.
To demonstrate the synthetic utility of the sulfonyl fluoride containing polymers, we explored the post-polymerisation modification potential of the copolymer through SuFEx chemistry. This approach highlights the functionalizability of the backbone, enabling the introduction of new functional groups under mild conditions.
As shown in Fig. 4, the copolymer (from Table 1, entry 16) was treated with 4-methoxyphenol, phenol or 1-hexadecanamine in THF at 50 °C for 1 h. The SuFEx reactions successfully yielded the sulfonate and sulfonamide products, as observed from the proton and 19F NMR. In Fig. 4, the 1H NMR of the 4-methoxyphenol-containing polymer is shown, indicating the incorporation of four additional aromatic protons from the phenol, while the appearance of a distinct peak at 3.75 ppm suggests the introduction of a methoxy group. Notably, the broad fluorine signal from the sulfonyl fluoride group observed around 40–50 ppm in the starting polymer is completely absent in the product (Fig. S1), while DOSY NMR analysis of all three functionalised polymers (Fig. S1–3) showed consistent rates of diffusion between the polymer backbone signals and the functional groups introduced via SuFEx reaction, indicating successful functionalisation of the polymer by SuFEx click chemistry.
 | ||
| Fig. 4 Post-polymerisation functionalisation of ESF-co-St polymer. | ||
Building on these successful functionalisations, we hypothesized that the abundant amino groups in branched polyethyleneimine (bPEI) could be crosslinked with the sulfonyl fluoride moieties of ESF-co-St to form a 3-dimensional polymer network. To test this, equal masses of bPEI (25 kDa) and ESF-co-St were dissolved in THF and stirred at ambient temperature (Fig. 5A), resulting in rigid non-flowing gel within 30 minutes. Rheological analysis of this curing process (Fig. 5B) shows a rapid increase in the storage modulus (G′), with a gelation time of 12.7 minutes, as measured by the intercept of the G′ and G″ values.
 | ||
| Fig. 5 (A) Crosslinking reaction of bPEI25k and ESF-co-St. (B) Rheological analysis of crosslinking reaction. | ||
Crosslinked polyimines have shown application in chelation of heavy metals from aqueous media, addressing critical challenges in water purification. Compared to conventional adsorbents and previously reported functional copolymers, our ESF-based network may offer superior chemical stability, tuneable porosity, and modular functionality, positioning them as promising candidates for sustainable environmental technologies.32
Conclusion
We have demonstrated the novel copolymerization of ethenesulfonyl fluoride (ESF) and styrene, demonstrating a strong tendency toward alternating copolymerisation. Conversion profiles exhibited a near-linear increase of Mn with conversion, showing that this reaction proceeds via a reversible deactivation mechanism. Furthermore, we successfully showcased the post-polymerisation modification of ESF-co-St copolymers, leveraging the sulfonyl fluoride functionality for subsequent transformations, including small-molecule conjugation and the formation of a 3D polymer network. These post-modification strategies open pathways for creating advanced functional materials, and ongoing work in our group aims to expand this chemistry to achieve tailored architectures and properties for specialized applications.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): additional experimental protocols, characterisation data (NMR, GPC, FTIR) and polymerization kinetics. See DOI: https://doi.org/10.1039/d6py00136j.Data will be available upon request from the authors.
References
- S. Bag, S. Ghosh, S. Paul, M. E. H. Khan and P. De, Macromol. Rapid Commun., 2021, 42, 2100501 CrossRef CAS PubMed.
- D. J. T. Hill, J. H. O'Donnell and P. W. O'Sullivan, Macromolecules, 1982, 15, 960–966 CrossRef CAS.
- X. Zhang, Z. Yin, S. Xiang, H. Yan and H. Tian, Polymers, 2024, 16, 2807 CrossRef CAS PubMed.
- L. N. Woodard and M. A. Grunlan, ACS Macro Lett., 2018, 7, 976–982 CrossRef CAS PubMed.
- M. Eberhardt, R. Mruk, R. Zentel and P. Theato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS.
- J. G. Kim and H. G. Shin, Macromol. Res., 2022, 30, 757–765 Search PubMed.
- T. Tanaka, Polymers, 2024, 16, 1100 CrossRef CAS PubMed.
- T. A. Fattah, A. Saeed and F. Albericio, J. Fluor. Chem., 2018, 213, 87–112 CrossRef.
- S. N. Carneiro, S. R. Khasnavis, J. Lee, T. W. Butler, J. D. Majmudar, C. W. am Ende and N. D. Ball, Org. Biomol. Chem., 2023, 21, 1356–1372 RSC.
- Z. Liu, J. Li, S. Li, G. Li, K. B. Sharpless and P. Wu, J. Am. Chem. Soc., 2018, 140, 2919–2925 CrossRef CAS PubMed.
- J. A. Homer, L. Xu, N. Kayambu, Q. Zheng, E. J. Choi, B. M. Kim, K. B. Sharpless, H. Zuilhof, J. Dong and J. E. Moses, Nat. Rev. Methods Primers, 2023, 3, 58 CrossRef CAS PubMed.
- B. Gao, L. Zhang, Q. Zheng, F. Zhou, L. M. Klivansky, J. Lu, Y. Liu, J. Dong, P. Wu and K. B. Sharpless, Nat. Chem., 2017, 9, 1083–1088 CrossRef CAS PubMed.
- J. C. Brendel, L. Martin, J. Zhang and S. Perrier, Polym. Chem., 2017, 8, 7475–7485 RSC.
- M. Li, J.-A. Ma and S. Liao, Macromolecules, 2023, 56, 806–814 CrossRef CAS.
- Q. Chen, P. Mayer and H. Mayr, Angew. Chem., Int. Ed., 2016, 55, 12664–12667 CrossRef CAS PubMed.
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- H. Wang, F. Zhou, G. Ren, Q. Zheng, H. Chen, B. Gao, L. Klivansky, Y. Liu, B. Wu, Q. Xu, J. Lu, K. B. Sharpless and P. Wu, Angew. Chem., Int. Ed., 2017, 56, 11203–11208 CrossRef CAS PubMed.
- H. Kim, J. Zhao, J. Bae, L. M. Klivansky, E. A. Dailing, Y. Liu, J. R. Cappiello, K. B. Sharpless and P. Wu, ACS Cent. Sci., 2021, 7, 1919–1928 Search PubMed.
- T. Fujigaya, Y. Shibasaki, S. Ando, S. Kishimura, M. Endo, M. Sasago and M. Ueda, Chem. Mater., 2003, 15, 1512–1517 CrossRef CAS.
- C. E. Anyaegbu and J. F. Hooper, Org. Lett., 2025, 27, 5924–5929 CrossRef PubMed.
- A. Sudalai, S. Kanagasabapathy and B. C. Benicewicz, Org. Lett., 2000, 2, 3213–3216 CrossRef CAS PubMed.
- M. E. Driscoll, B. T. Nicholls and B. P. Fors, J. Am. Chem. Soc., 2025, 147, 16390–16395 CrossRef CAS PubMed.
- M. C. Davies, J. V. Dawkins and D. J. Hourston, Polymer, 2005, 46, 1739–1753 Search PubMed.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, J. Krstina, G. Moad, A. Postma and S. H. Thang, Macromolecules, 2000, 33, 243–245 CrossRef CAS.
- Y. Sugihara, P. O′connor, P. B. Zetterlund and F. Aldabbagh, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1856–1864 CrossRef CAS.
- Y. Gu, Z. Zhang, T. Gao, R. Gómez-Bombarelli and M. Chen, Angew. Chem., Int. Ed., 2024, 63, e202409744 CAS.
- E. B. Twum, E. F. McCord, D. F. Lyons, P. A. Fox and P. L. Rinaldi, Eur. Polym. J., 2014, 51, 136–150 CrossRef CAS.
- A. A. A. Autzen, S. Beuermann, M. Drache, C. M. Fellows, S. Harrisson, A. M. van Herk, R. A. Hutchinson, A. Kajiwara, D. J. Keddie, B. Klumperman and G. T. Russell, Polym. Chem., 2024, 15, 1851–1861 RSC.
- A. M. van Herk and B. Klumperman, Macromolecules, 2024, 57, 5121–5122 CrossRef CAS.
- A. J. Scott and A. Penlidis, Processes, 2018, 6, 8 CrossRef.
- F. R. Mayo and F. M. Lewis, J. Am. Chem. Soc., 1944, 66, 1594–1601 Search PubMed.
- A. Chandra, A. A. Singh, S. Prasad, M. R. Andersson and D. Gedefaw, Appl. Sci., 2025, 15, 4767 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |