
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conversion of oligo(ethyleneglycol)s into non-toxic highly selective biocompatible poly(ethyleneglycol)s: synthesis, antimicrobial and antibiofilm activity†
Sulbha
Kumari‡
a,
Arpita
Halder‡
b,
Aayush
Anand
a,
Oindrilla
Mukherjee
*b and
Subrata
Chattopadhyay
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Patna, Bihta, Patna 801106, Bihar, India. E-mail: sch@iitp.ac.in
bDepartment of Biotechnology, National Institute of Technology, Durgapur, Mahatma Gandhi Rd, A-Zone, Durgapur, West Bengal 713209, India. E-mail: omukherjee.bt@nitdgp.ac.in
First published on 26th February 2025
Abstract
Designing non-toxic, non-hemolytic, selective antimicrobials remains an important and challenging research problem. Herein, we report an affordable synthetic route to prepare a series of ten multifunctional polyethylene glycols (PEGs) via a cascade reaction approach involving aza-Michael polyaddition followed by post-polymerization modifications using triazolinedione-based click reactions. All polymers are characterized by NMR, IR, SEC, DSC and TG analyses. Antimicrobial and hemolytic studies reveal that structure plays a pivotal role in tuning the antimicrobial efficacy and selectivity (HC/MIC) of the functional PEGs. The selectivity (HC/MIC) reported for the best prototype (InPEG700-C12-TAD) is 129, 33 and 39 against P. aeruginosa, E. coli and S. aureus, respectively. Additionally, all the polymers are non-cytotoxic, as revealed by the MTT assay, and exhibit excellent antibiofilm activity.
1. Introduction
Infectious diseases are increasingly becoming a global problem in today's society, making the suitable design of antibiotics and antimicrobials a very important research topic.1–3 Since the first antibiotic ‘penicillin’ was reported by Alexander Fleming,4 several other antibiotics (low molecular weight and polymeric) have been reported.5 However, most conventional antibiotics are becoming increasingly ineffective because of antibiotic resistance, which results in multi-drug-resistant microbes. On the other hand, host defence peptides or antimicrobial peptides, which are an invariable part of many living organisms, defend host organisms against microbial infections effectively. Most of these antimicrobial peptides are composed of polymer chains of amino acids that include several cationic amino and hydrophobic functionalities. Due to their cationic nature, these peptides kill negatively charged bacteria via contact killing-membrane disruption mechanisms, which are explained via different models in the literature including the barrel-stave model, carpet-like model, toroidal-pore model and disordered toroidal-pore model.6–10 Due to the contact-active membrane disruption mechanism, such antimicrobial peptides are less able to develop antimicrobial resistance. However, it is extremely challenging to develop such antimicrobial peptides to combat infections as part of our day-to-day needs due to their lower synthetic upscalability, higher cost and the requirement to further improve their selectivity, indicated by the balance between antimicrobial and hemolytic activity.11–13 To address these issues, synthesis of contact-active antimicrobial polymers (or polymers showing intrinsic antimicrobial activity), which mimic the structural features of antimicrobial peptides, such as polymeric structure and the tuneable presence of cationic and hydrophobic groups within the backbone, has emerged as a very important research topic.14–16 In the literature, various antimicrobial polymers with diverse functional backbones have been reported, including polyacrylates,17–19 polyacrylamide,20,21 polystyrene,22 polyethyleneamine,23 polyvinylamine,24,25 chitosan,26–29 polytetrahydrofurans,30 polyoxazolines,31,32 polysulphoniums,33,34etc. Although antimicrobial properties can be tuned via adjusting the polymer's structure (hydrophobic/cationic balance), many of them suffer from higher hemolytic activity, resulting in lower selectivity (MIC/HC50), low biocompatibility and poor water solubility.Herein, we aim to synthesize polyethyleneglycol (PEG)-based contact-active antimicrobial polymers. PEGs are well known for their biocompatibility, non-immunogenicity and excellent water solubility. However, PEG, without any additional functionality, is not a potent antimicrobial, and functional PEGs have rarely been explored to develop contact-active intrinsic antimicrobials (Fig. 1). Recently, Kim et al. reported the first synthesis of PEG-based contact-active antimicrobials via ring-opening polymerization of various functional epoxides.35 In recent years, chitosan–PEG graft copolymers have shown potent antimicrobial activity due to grafting with the chitosan backbone; however, functional PEGs alone (without grafting) are not significantly antimicrobial.36
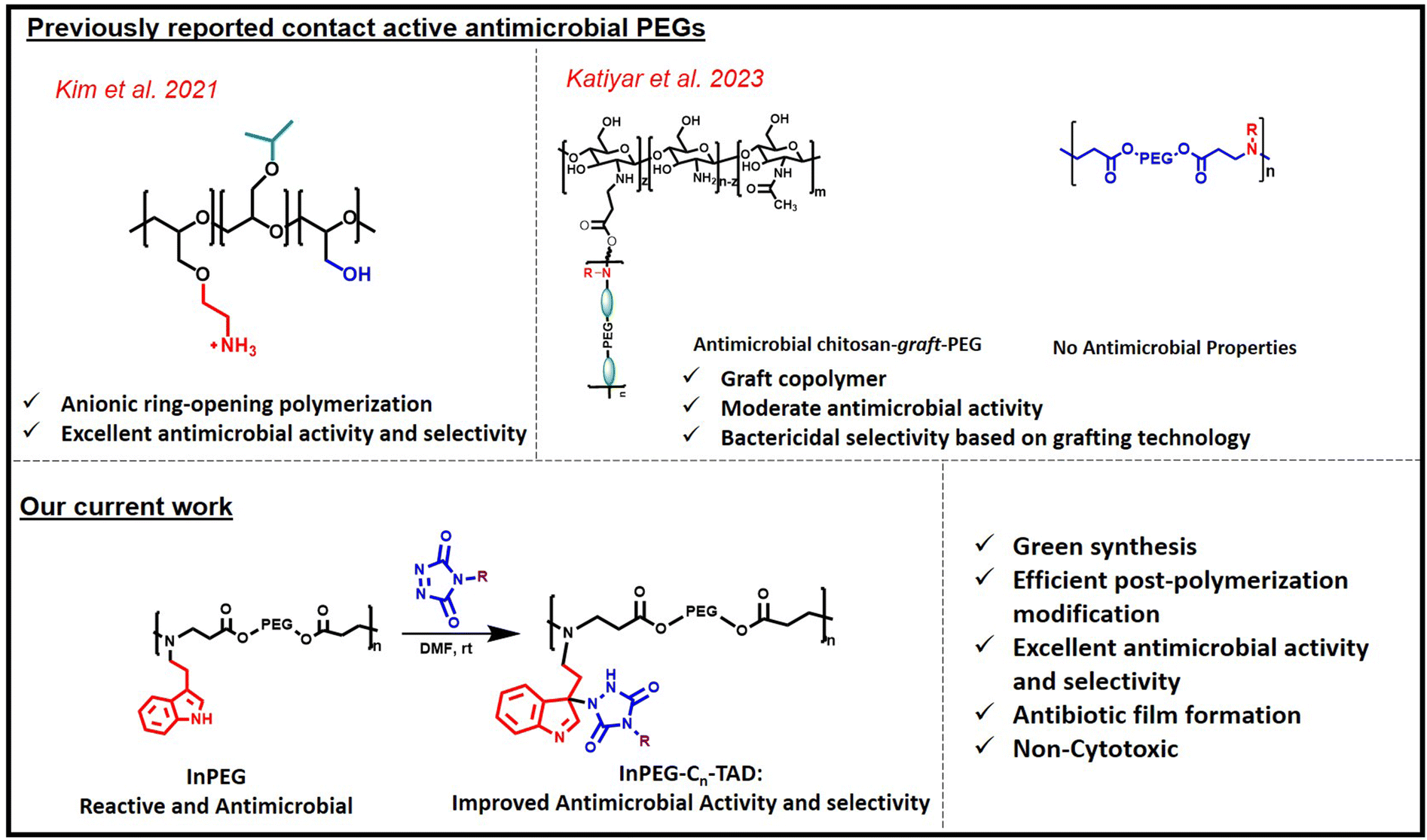 | ||
| Fig. 1 Scheme depicting earlier reported contact-active PEG-based copolymers and our current work on antimicrobials. | ||
In the current work, we prepared indole-decorated PEG periodic copolymers, where tryptamine functionality is periodically incorporated within the PEG backbone via a simple aza-Michael reaction between PEG diacrylates and tryptamine. In this approach, besides the periodic presence of amine groups in the backbone, the presence of indoles in the side chain serves two purposes. First, as reported in earlier studies, the presence of indole groups (due to their significant quaternisation in aqueous solution) is known to improve the antimicrobial efficacy of various non-cationic polymers, including poly(vinyl alcohol)37 and polyesters,38 and is therefore expected to further improve the antimicrobial properties in the current case. Additionally, indole acts as a reactive functional handle for TAD–indole reactions, enabling the further introduction of various alkyl chains to fine-tune both antimicrobial activity and selectivity (HC50/MIC).
2. Results and discussion
2.1 Synthesis of an indole-functionalized poly(ethylene glycol) polymer (InPEGx) and its post-polymerization modifications (InPEGx-Cn-TAD)
A versatile indole-functionalized polyethylene glycol (PEG) periodic copolymer, InPEGx, was designed and synthesized such that the biocompatible PEG backbone enhances its aqueous solubility and biocompatibility, while the indole and amine moieties confer antimicrobial activity. As illustrated in Scheme 1 (step 1), polymerization proceeds via an aza-Michael polyaddition reaction between commercially available PEG diacrylates (two analogues with Mn = 575 and 700 used as macromonomers) and tryptamine in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio using a 2:1 (v/v) mixture of water and DMF. The two PEG copolymers have different periodic nature, which was modulated by changing the PEG chain length (InPEGx; x = 575 and 700). The synthesized polymers were characterized using 1H- and 13C-NMR, HSQC and SEC analyses.
:1 molar ratio using a 2:1 (v/v) mixture of water and DMF. The two PEG copolymers have different periodic nature, which was modulated by changing the PEG chain length (InPEGx; x = 575 and 700). The synthesized polymers were characterized using 1H- and 13C-NMR, HSQC and SEC analyses.
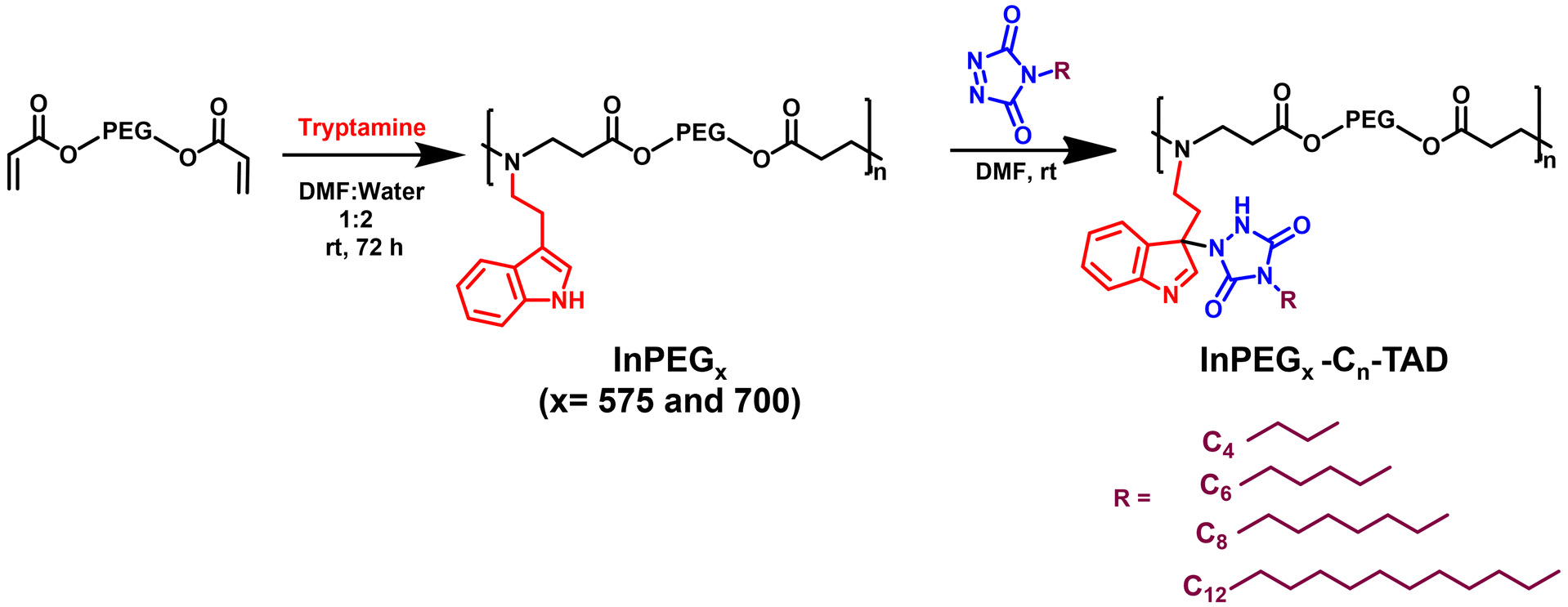 | ||
| Scheme 1 Synthesis of InPEGx and their post-polymerization modifications using TAD–indole reactions. | ||
The 1H-NMR spectrum of InPEG575 (Fig. 2A) clearly revealed the successful insertion of the indole functionality into the PEG copolymer, as evidenced by the appearance of characteristic aromatic and NH peaks of indole functionality between 6.9 and 7.6 ppm and at 10.6 ppm, respectively, along with the characteristic peaks for PEG. The 13C-NMR spectra (Fig. 2B) also provide strong evidence for the insertion of indole via an aza-Michael reaction. The characteristic methylene peaks at 23 ppm (–NH–CH2–![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) H2–Ar) and 49 ppm (–NH–H2–CH2–Ar) along with aromatic peaks at 111.8–127.7 ppm and 136.7 ppm are consistent with the indole moiety. Furthermore, the appearance of new methylene peaks at 32.5 ppm (OCO–H2–CH2–N) and 54.3 ppm (OCO–CH2–CH2–N) confirms the successful aza-Michael reaction between the acrylate (end) functionality and the amine group. HSQC spectra of InPEG575 (Fig. S1†) supported the same molecular details. Similar NMR evidence was observed for InPEG700 (Fig. S2†). The SEC analysis revealed the Mn of 5300 and 6100 g mol−1 for InPEG575 and InPEG700 copolymers, respectively.
H2–Ar) and 49 ppm (–NH–H2–CH2–Ar) along with aromatic peaks at 111.8–127.7 ppm and 136.7 ppm are consistent with the indole moiety. Furthermore, the appearance of new methylene peaks at 32.5 ppm (OCO–H2–CH2–N) and 54.3 ppm (OCO–CH2–CH2–N) confirms the successful aza-Michael reaction between the acrylate (end) functionality and the amine group. HSQC spectra of InPEG575 (Fig. S1†) supported the same molecular details. Similar NMR evidence was observed for InPEG700 (Fig. S2†). The SEC analysis revealed the Mn of 5300 and 6100 g mol−1 for InPEG575 and InPEG700 copolymers, respectively.
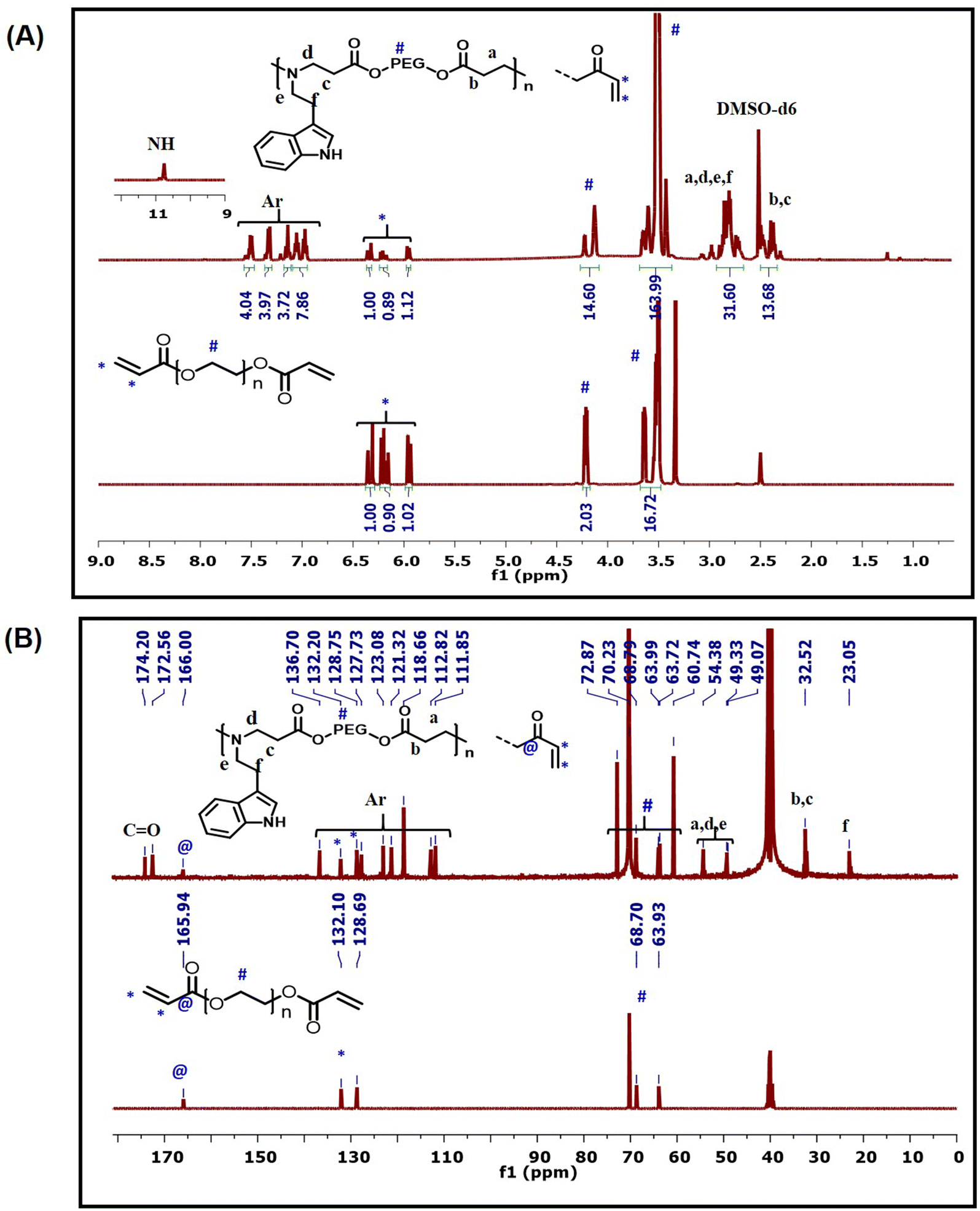 | ||
| Fig. 2 (A) 1H-NMR spectra and (B) 13C-NMR spectra of InPEG575 and PEG diacrylate (macromonomer). | ||
In the next stage, the indole group serves as an effective clickable site for triazolinedione (TAD) molecules via an ultrafast TAD–indole reaction under ambient reaction conditions.39–42 This facile approach offers a highly efficient method for introducing hydrophobic groups as pendant functionalities within polymer chains. For the purpose of post-polymerization modification, four different triazolinedione derivatives, having different alkyl chain lengths (butyl-, hexyl-, octyl- and dodecyl-TAD), were synthesized using a previously reported procedure and characterized using NMR, FTIR and mass spectroscopy.43,44
The four synthesized TAD derivatives with varying alkyl chain lengths (C4-TAD, C6-TAD, C8-TAD and C12-TAD) were subsequently employed for post-modification of InPEGxvia a TAD–indole click reaction. The TAD–indole click reaction was initiated by combining solutions of TAD and InPEGx in DMF at ambient temperature. After a 20 minute reaction period, the solvent was removed via lyophilization. Using this generalized approach, eight sets of hydrophobic post-modified PEG copolymers, with varying alkyl chains and PEG chain lengths, were prepared (InPEGx-Cn-TAD; x = 575 and 700, n = 4, 6, 8, 12), and all the modified copolymers were characterized using NMR, FTIR and SEC analyses. From the 1H-NMR spectra of the post-modified copolymer InPEG575-C4-TAD (Fig. 3A), the appearance of new characteristic peaks for the butyl group of the TAD moiety at 0.8, 1.0–1.55 and 3.4 ppm provided evidence of alkyl functionalization within the polymer backbone. Furthermore, the TAD–indole adduct was confirmed using 13C-NMR spectra (Fig. 3A). The presence of characteristic butyl peaks at 13.9, 19.7, 30.0 and 38.0 ppm, along with the appearance of a new peak at ∼77.9 ppm corresponding to the quaternary carbon after the TAD–indole linkage, a peak at ∼155.6 ppm associated with the carbonyl carbon of the TAD unit, and a peak at 163.8 ppm that corresponds to N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH of the indole unit supports the successful conjugation of the TAD moiety to the indole group of the copolymer. A similar observation was made in the NMR spectra of other hydrophobic TAD-modified InPEG575 and InPEG700 polymers (Fig. S3–S6†). Subsequently, SEC measurements were performed, and the resulting molecular weights, summarized in Table 1, support the successful post-polymerization modifications.
CH of the indole unit supports the successful conjugation of the TAD moiety to the indole group of the copolymer. A similar observation was made in the NMR spectra of other hydrophobic TAD-modified InPEG575 and InPEG700 polymers (Fig. S3–S6†). Subsequently, SEC measurements were performed, and the resulting molecular weights, summarized in Table 1, support the successful post-polymerization modifications.
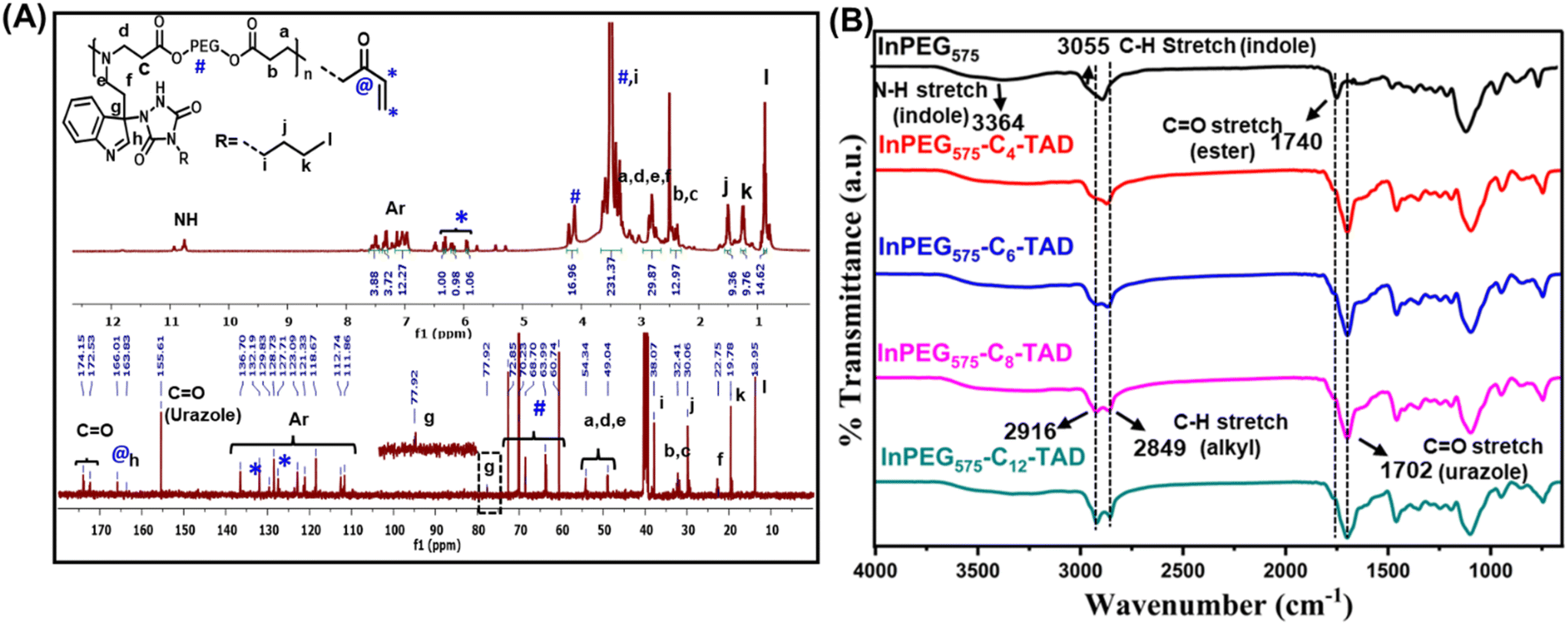 | ||
| Fig. 3 (A) 1H- and 13C-NMR spectra of InPEG575-C4-TAD and (B) FTIR spectra of InPEG575 and InPEG575-Cn-TAD (n = 4, 6, 8, 12). | ||
| Sample | M n (g mol−1) | Đ | Sample | M n (g mol−1) | Đ |
|---|---|---|---|---|---|
| InPEG575 | 5300 | 1.4 | InPEG700 | 6100 | 1.3 |
| InPEG575-C4-TAD | 5800 | 1.4 | InPEG700-C4TAD | 6200 | 1.4 |
| InPEG575-C6TAD | 5830 | 1.4 | InPEG700-C6TAD | 6350 | 1.4 |
| InPEG575-C8TAD | 6200 | 1.3 | InPEG700-C8TAD | 6630 | 1.4 |
| InPEG575-C12TAD | 6280 | 1.3 | In-PEG700-C12TAD | 6360 | 1.3 |
Furthermore, the synthesis of indolized PEGs and their post-polymer modifications were also evidenced by FTIR spectral analysis. Fig. 3B depicts the FTIR spectra of un-modified and modified InPEG575 with various mono-TADs. In the case of unmodified InPEG575, the appearance of bands at 3055 and 3364 cm−1, corresponding to the aromatic C–H and N–H stretching frequencies of the indole unit, supports the incorporation of the indole unit. After modification of the copolymer with hydrophobic TAD derivatives (Cn-TAD), enhanced bands at 2916 cm−1 and 2849 cm−1 that correspond to the C–H stretching frequency of the alkyl chain (of the TAD unit), along with the appearance of a band at 1702 cm−1, which is attributed to the CO stretching frequency of the urazole unit formed after the TAD–indole click reaction, support the modification.
To confirm the cationic nature of the current polymers in solution, their zeta potentials were measured in water (concentration = 0.5 mg mL−1). The zeta potential for the indole-functionalized PEG (InPEG575) was found to be +26 mV. After TAD modification (InPEG575-C8-TAD), the zeta potential decreased to +15 mV, confirming the cationic nature of all the polymers. The decrease of zeta potential after TAD modification can be ascribed to the formation of urazole linkages, which are significantly anionic at ambient pH, as their reported pKa is ∼5.3.45
The thermal properties of all ten synthesized PEG copolymers were analysed using differential scanning calorimetry (DSC) and thermo-gravimetric analysis (TGA). The TGA thermograms, as depicted in Fig. 4, reveal the thermal stability of the unmodified indole-functionalized PEG polymer (InPEGx) and hydrophobically modified polymers (InPEGx-Cn-TAD). A minor weight loss (∼5%) was observed around 100 °C (particularly for the more hydrophilic InPEGx polymers), due to the removal of adsorbed moisture. The thermograms revealed that both InPEG575 and InPEG700 polymers were stable up to 175 °C (Td10), which is atypical for the PEG backbone. Notably, the hydrophobically modified InPEGx-Cn-TAD polymers exhibited even greater thermal stability, with a slightly higher decomposition temperature (Td10) of ∼200 °C. Furthermore, DSC measurements (Fig. 5 and Fig. S8†) revealed that the glass transition temperature (Tg) values of unmodified InPEG575 and InPEG700 were −31 °C and −34 °C, respectively. The glass transition temperatures of the TAD derivatives are comparable to those of other similar PEG copolymers that have been reported.46 Modifying InPEGx with a hydrophobic TAD molecule leads to an increase in the Tg values (ranging from 0 to −5 °C) due to hydrogen bonding in the introduced urazole moieties.44,47
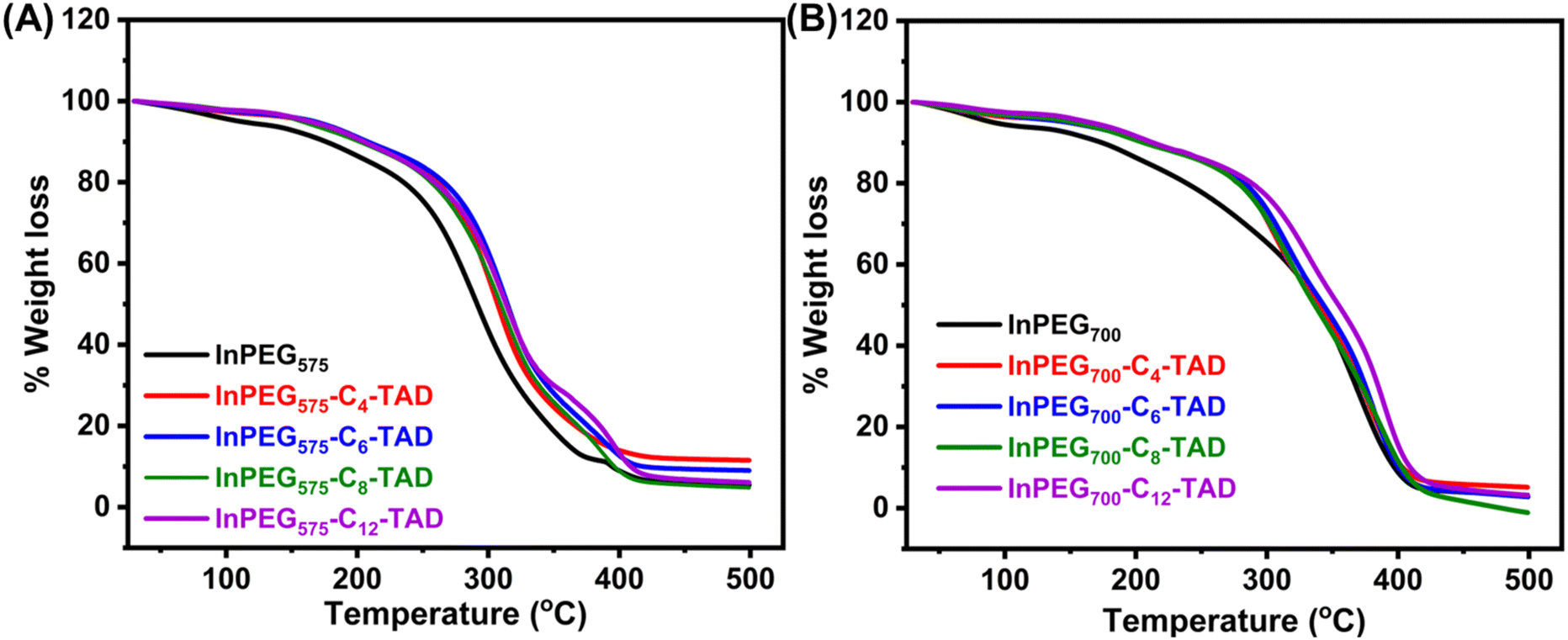 | ||
| Fig. 4 TGA thermograms of (A) InPEG575 and its four post-functional derivatives and (B) InPEG700 and its post-functional derivatives. | ||
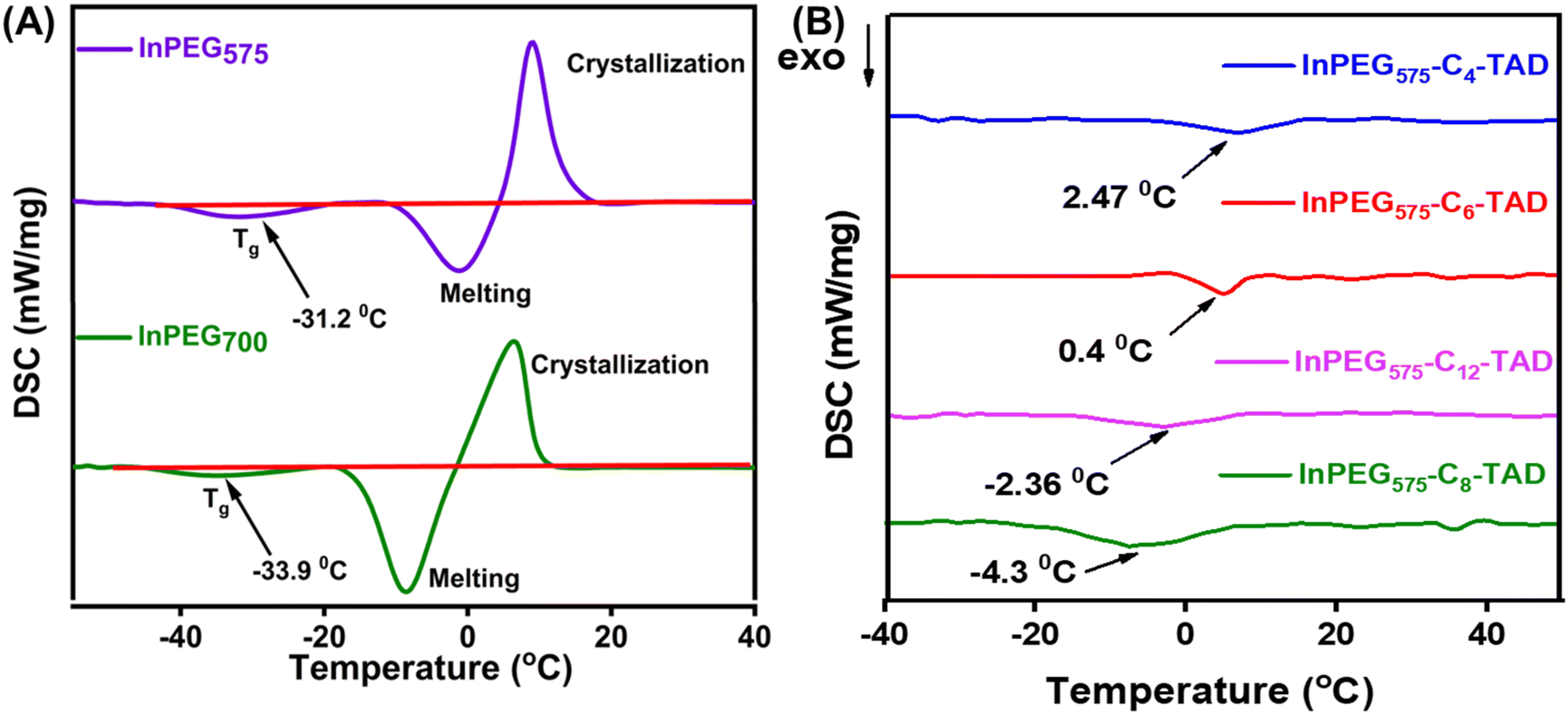 | ||
| Fig. 5 DSC thermograms of (A) InPEG575 and InPEG700 and (B) post-polymerization derivatives of InPEG575-Cn-TAD. | ||
2.2. Antimicrobial and hemolytic studies
The antimicrobial activity of all ten polymers was evaluated in solution against three different bacteria – Pseudomonas aeruginosa and Escherichia coli as model Gram-negative bacteria and Staphylococcus aureus as a model Gram-positive bacterium. For quantitative estimation and comparison, the minimum inhibitory concentration (MIC) was determined via the broth micro-dilution method, where the log (CFU mL−1) versus concentration curve was used to determine the MIC. Additionally, a hemolysis test was also performed to determine the HC50, which is the concentration required for 50% RBC lysis. A comparison between the HC and MIC was performed to assess the selectivity of the antimicrobial polymers. The results are reported in Table 2.| Polymer | MIC (μg mL−1) | HC50(μg mL−1) | Selectivity (HC/MIC) | ||||
|---|---|---|---|---|---|---|---|
| P. aeruginosa | E. coli | S. aureus | P. aeruginosa | E. coli | S. aureus | ||
| InPEG575 | 162 | 587 | 385 | 7130 | 44.13 | 12.14 | 18.50 |
| InPEG575-C4-TAD | 211 | 579 | 401 | 6582 | 31.19 | 11.36 | 16.43 |
| InPEG575-C6-TAD | 113 | 526 | 269 | 6760 | 59.93 | 12.84 | 25.15 |
| InPEG575-C8-TAD | 44 | 160 | 156 | 2022 | 46.42 | 12.66 | 13.00 |
| InPEG575-C12-TAD | 49 | 179 | 167 | 1855 | 37.85 | 10.37 | 11.12 |
| InPEG700 | 140 | 664 | 288 | 12008 |
86.06 | 18.07 | 41.69 |
| InPEG700-C4-TAD | 158 | 519 | 308 | 5772 | 36.60 | 11.13 | 18.75 |
| InPEG700-C6-TAD | 144 | 567 | 290 | 8934 | 62.14 | 15.77 | 30.84 |
| InPEG700-C8-TAD | 63 | 212 | 182 | 4809 | 76.86 | 22.69 | 26.40 |
| InPEG700-C12-TAD | 59 | 230 | 196 | 7601 | 129.47 | 33.03 | 38.81 |
For the structure–antimicrobial activity study, two structural variables were examined among the ten polymers (Fig. 6). For two individual sets – InPEG575-Cn-TAD and InPEG700-Cn-TAD – the PEG spacer length is different, which leads to different periodic structures. For each of these individual sets, 5 different polymers were synthesized with varying alkyl chain lengths (n) – unmodified InPEGx (n = 0), InPEGx-C4-TAD (n = 4), InPEGx-C6-TAD (n = 6), InPEGx-C8-TAD (n = 8), and InPEGx-C12-TAD (n = 12). It is evident that both InPEG575 and InPEG700 show good antimicrobial activity with excellent selectivity against all three bacteria, far surpassing that of unmodified PEG oligomers (which showed almost no antimicrobial activity). However, InPEG700 shows significantly lower hemolytic activity than InPEG575, resulting in higher selectivity of InPEG700. The higher selectivity can be ascribed to the higher PEG content in its periodic block copolymer structure. The next stage, which involved post-polymer modification of InPEGs, the antimicrobial efficacy initially decreases slightly in the case of C4-TAD; however, antimicrobial efficacy significantly improved when the length of alkyl chains was increased to C-8 and C-12. From the series of 10 polymers, InPEG700-C12-TAD was identified as the best antimicrobial agent considering both its MIC and selectivity. The MIC values of InPEG700-C12-TAD were 59, 230 and 196 μg mL−1 against P. aeruginosa, E. coli and S. aureus, respectively, with excellent selectivity (indicating very low hemolytic activity) of 129, 33 and 39. This further motivated us to calculate the actual hemolytic activity at its MIC. The calculated hemolysis values at 59, 230 and 196 μg mL−1 were 3.2, 4.3 and 4.1%, respectively. As all these values are below 5%, they are considered insignificant/null according to the American Society for Testing and Materials (ASTM).48,49
 | ||
| Fig. 6 Graphical representation of the structure–antimicrobial activity–hemolytic activity relationship of different PEG derivatives: (A), (C) and (E) represent the effect of different alkyl chain lengths on the antimicrobial activity and selectivity of the InPEG575-Cn-TAD polymer series against P. aeruginosa, S. aureus and E. coli, respectively; (B), (D) and (F) represent the effect of PEG chain length on antimicrobial activity and selectivity against P. aeruginosa, S. aureus and E. coli, respectively (the two best-performing polymers InPEG575-C8-TAD and InPEG700-C12-TAD were chosen for comparison). | ||
In the next stage, to validate the contact-active/membrane disruption antimicrobial mechanism, we conducted an N-phenyl-1-naphthylamine (NPN) uptake assay to determine bacterial membrane permeation by the polymer chains (Fig. 7A). In the literature, the outer membrane assay using 1-N-phenylnaphthylamine (NPN) is a well-established method to measure the permeability of bacterial outer membranes.50–52 This assay involves incubating bacteria with NPN, where the dye binds to the hydrophobic regions of the outer membrane. If membrane integrity is intact, NPN remains outside the cell. However, when the membrane is damaged, NPN enters the cell and exhibits increased fluorescence. Therefore, the intensity of fluorescence correlates with outer membrane permeability, allowing for the assessment of membrane integrity and the effectiveness of antimicrobial agents. The elevated fluorescence intensity following treatment with both InPEG700-C12-TAD and InPEG575-C8-TAD demonstrates that the polymers compromise the outer membrane integrity of the bacteria, thereby supporting membrane disruption as the mechanism underlying their antimicrobial effect.
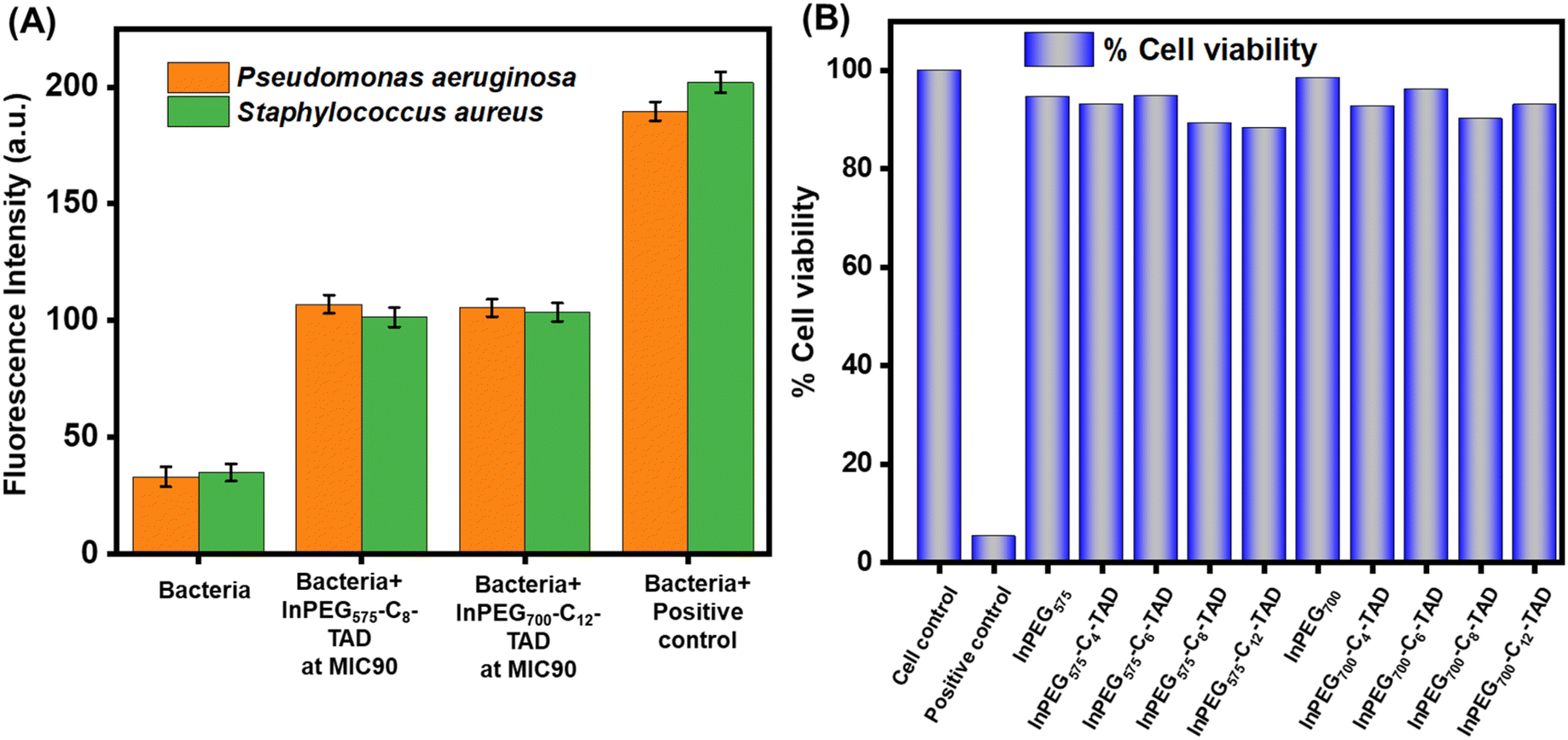 | ||
| Fig. 7 (A) NPN uptake assay of different bacteria in the presence of InPEG575-C8-TAD and InPEG700-C12-TAD. (B) MTT assay assessing the cytotoxicity of different PEG derivatives on HEK293 cells. | ||
Besides hemolytic activity, cell cytotoxicity is a crucial factor for the application of antimicrobials. To establish the biocompatibility and non-cytotoxic nature of the current polymers, an MTT assay was performed using HEK293 cells (Fig. 7B). Cell cytotoxicity was assessed at a polymer concentration of 1500 μg mL−1, which is significantly higher than the reported MIC of the polymers. The results clearly revealed that the polymers are non-toxic, as more than 90% cell viability was noted in all cases.
2.3. Biofilm inhibition assay
Biofilms can be described as communities of bacterial cells enclosed in a hydrated polyanionic exopolysaccharide complex that adheres to a biotic or abiotic surface. Biofilms are particularly important due to their role in persistent infections and antimicrobial tolerance.53 Bacteria can form biofilms on living surfaces such as host tissues and non-living surfaces such as medical devices and implants, and such cells have been proven to be significantly resistant to standard antibiotics and difficult to eliminate.54 Therefore, it is important to assess the biofilm inhibition ability of such polymers for their real-life applications.55 All ten polymers were assessed using the crystal violet assay (Fig. 8) against three of the most potent biofilm formers, P. aeruginosa, E. coli (Gram-negative bacteria), and S. aureus (Gram-positive bacteria), to determine their ability to inhibit biofilm formation. The results indicate that post-treatment, biofilm inhibition is observed with higher polymer concentrations (please note that in the absence of antimicrobial polymers, biofilm formation reaches 100%, as indicated in Fig. 8, concentration = 0). For most of the polymers, a concentration of 100–150 μg mL−1 is able to inhibit biofilm formation (95–100%) in all the bacteria. Although there seems to be no significant difference between the activity of the two individual sets – InPEG575-Cn-TAD and InPEG700-Cn-TAD – in terms of their structure, the biofilm inhibition activity increases as the length of the alkyl chain increases from C4 to C12. This trend aligns with the antimicrobial activity patterns observed for these polymers, suggesting that longer alkyl chains enhance both antimicrobial and anti-biofilm properties.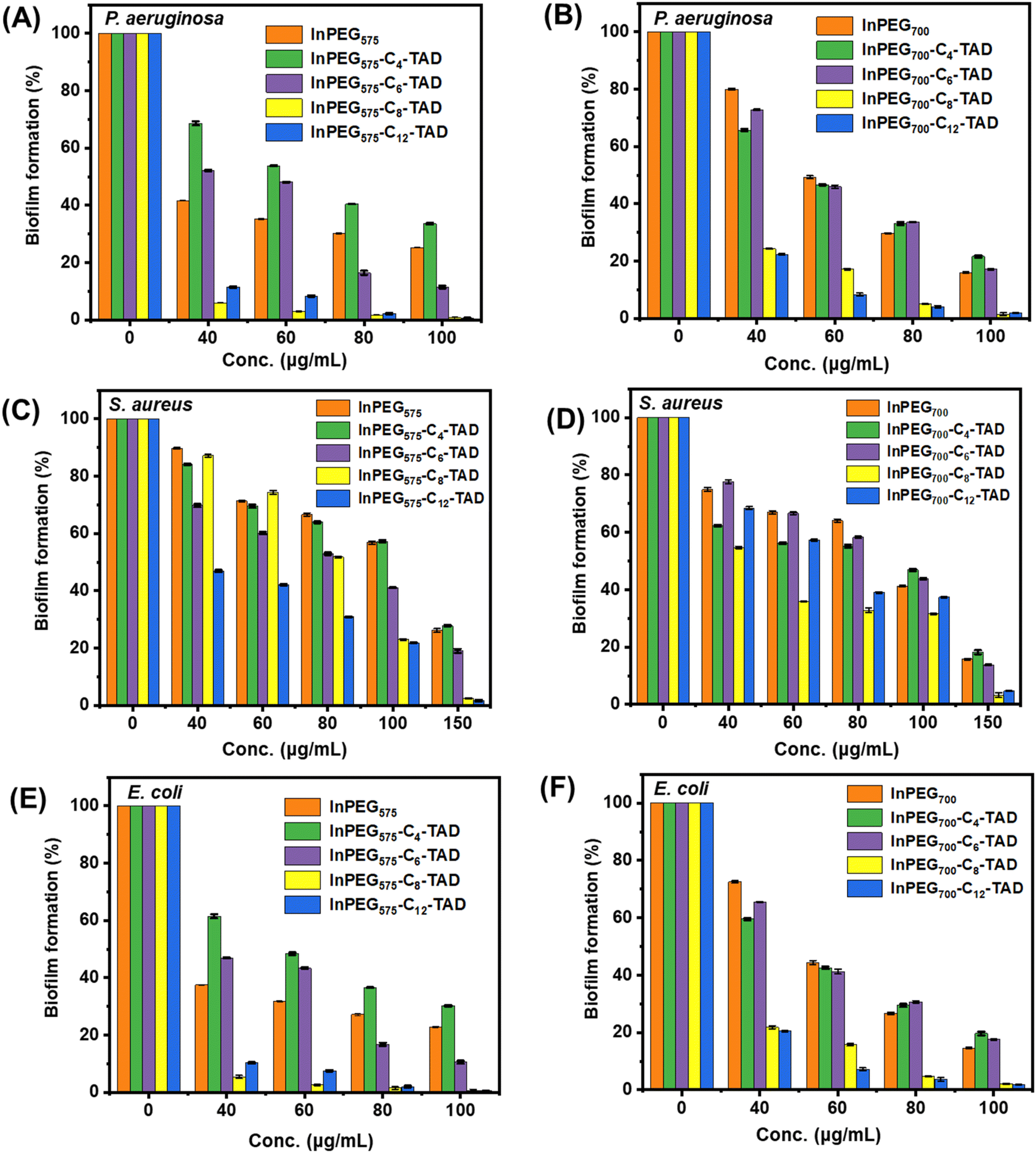 | ||
| Fig. 8 Crystal violet assay demonstrating the inhibition of biofilm formation in the presence of In-PEG575 and its derivatives when tested against P. aeruginosa: (A) for the InPEG575-Cn-TAD series and (B) for the InPEG700-Cn-TAD series; E. coli: (C) for the InPEG575-Cn-TAD series and (D) for the InPEG700-Cn-TAD series; and S. aureus: (E) for the InPEG575-Cn-TAD series and (F) for the InPEG700-Cn-TAD series. | ||
3. Conclusion
In summary, we demonstrated a green and affordable synthetic approach to prepare antimicrobial multifunctional polyethyleneglycols (PEGs) via cascade reactions. In the first step, indole-decorated periodic PEG copolymers (InPEG575 and InPEG700) were prepared via aza-Michael-based polyaddition reactions between tryptamine and commercially available PEG diacrylate macromonomers. Here, two different PEG chain lengths influence the polymer periodicity. In the second stage, both the polymers were further post-modified to functionalize them with different alkyl groups via TAD–indole reactions, resulting in a series of 10 different multifunctional PEGs. The molecular weight of all the polymers ranged between 5300 and 6600 Da, making them comparable analogues. The glass transition temperature (Tg) of InPEGs was reported to be around −30 °C, while post-polymerization modifications increased Tg to (−)4 to (+)5 °C, mainly due to hydrogen bonding between the incorporated urazole moieties. The antimicrobial activity of all the polymers was evaluated against three different bacteria, P. aeruginosa, E. coli and S. aureus. Antimicrobial and hemolytic studies revealed that structure plays a pivotal role in tuning the antimicrobial efficacy and selectivity (HC/MIC) of the polymers. The selectivity (HC/MIC) reported for the best prototype (InPEG700-C12-TAD) was 129, 33 and 39 against P. aeruginosa, E. coli and S. aureus, respectively. The NPN assay revealed bacterial membrane permeation by the polymers as the major bactericidal mechanism. All the polymers were non-cytotoxic, as revealed by the MTT assay. Furthermore, biofilm inhibition analysis of the polymers using the crystal violet assay revealed that both C8- and C12-functionalized analogues exhibited significant biofilm inhibition (>90%) above 60 μg mL−1, whereas complete inhibition was noted (∼99%) at 100 μg mL−1 for P. aeruginosa and E. coli and at 150 μg mL−1 for S. aureus.Author contributions
SK: synthesis, characterization, relevant data analysis (lead), and writing (lead); AH: biological experiments and analysis (lead) and writing (supporting); AA: synthesis, characterization (supporting), and writing (supporting); OM: supervision (biological experiments) and writing (supporting and review); and SC: conceptualization of the actual work and supervision (synthesis and relevant characterization) and writing (lead and review). All the authors have approved the final manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
SK and AA acknowledge IITP for the research fellowship. SC and OM acknowledge CSIR (02(0370)/19/EMR-II) and ICMR (Sanction letter no. 5/4/8-14/Obs./OM/2021-NCD-II) for funding support, respectively. SAIF, IITP is acknowledged for DSC, TGA and 2D NMR analyses.References
- R. Laxminarayan, A. Duse, C. Wattal, A. K. M. Zaidi, H. F. L. Wertheim, N. Sumpradit, E. Vlieghe, G. L. Hara, I. M. Gould, H. Goossens, C. Greko, A. D. So, M. Bigdeli, G. Tomson, W. Woodhouse, E. Ombaka, A. Q. Peralta, F. N. Qamar, F. Mir, S. Kariuki, Z. A. Bhutta, A. Coates, R. Bergstrom, G. D. Wright, E. D. Brown and O. Cars, Lancet Infect. Dis., 2013, 13, 1057–1098 CrossRef PubMed.
- M. A. Kohanski, D. J. Dwyer and J. J. Collins, Nat. Rev. Microbiol., 2010, 8, 423–435 CrossRef CAS PubMed.
- K. U. Jansen, C. Knirsch and A. S. Anderson, Nat. Med., 2018, 24, 10–19 CrossRef CAS PubMed.
- A. Fleming, Br. Med. Bull., 1944, 2, 4–5 CrossRef.
- A. Tomasz, Annu. Rev. Microbiol., 1979, 33, 113–137 Search PubMed.
- C. D. Fjell, J. A. Hiss, R. E. W. Hancock and G. Schneider, Nat. Rev. Drug Discovery, 2012, 11, 37–51 CrossRef CAS PubMed.
- K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 Search PubMed.
- M. Zasloff, Nature, 2002, 415, 389–395 CrossRef CAS PubMed.
- M. N. Melo, R. Ferre and M. A. R. B. Castanho, Nat. Rev. Microbiol., 2009, 7, 245–250 CrossRef CAS PubMed.
- H. Leontiadou, A. E. Mark and S. J. Marrink, J. Am. Chem. Soc., 2006, 128, 12156–12161 CrossRef CAS PubMed.
- R. E. W. Hancock and H.-G. Sahl, Nat. Biotechnol., 2006, 24, 1551–1557 CrossRef CAS PubMed.
- R. Spohn, L. Daruka, V. Lázár, A. Martins, F. Vidovics, G. Grézal, O. Méhi, B. Kintses, M. Számel, P. K. Jangir, B. Csörgő, Á. Györkei, Z. Bódi, A. Faragó, L. Bodai, I. Földesi, D. Kata, G. Maróti, B. Pap, R. Wirth, B. Papp and C. Pál, Nat. Commun., 2019, 10, 4538 CrossRef PubMed.
- E. A. Porter, B. Weisblum and S. H. Gellman, J. Am. Chem. Soc., 2002, 124, 7324–7330 CrossRef CAS PubMed.
- Y. Wu, G. Xia, W. Zhang, K. Chen, Y. Bi, S. Liu, W. Zhang and R. Liu, J. Mater. Chem. B, 2020, 8, 9173–9196 RSC.
- E.-R. Kenawy, S. D. Worley and R. Broughton, Biomacromolecules, 2007, 8, 1359–1384 CrossRef CAS PubMed.
- M. Haktaniyan and M. Bradley, Chem. Soc. Rev., 2022, 51, 8584–8611 RSC.
- Y. Ko, V. K. Truong, S. Y. Woo, M. D. Dickey, L. Hsiao and J. Genzer, Biomacromolecules, 2022, 23, 424–430 CrossRef CAS PubMed.
- A. Khlyustova, M. Kirsch, X. Ma, Y. Cheng and R. Yang, J. Mater. Chem. B, 2022, 10, 2728–2739 RSC.
- P. R. Judzewitsch, T.-K. Nguyen, S. Shanmugam, E. H. H. Wong and C. Boyer, Angew. Chem., Int. Ed., 2018, 57, 4559–4564 CrossRef CAS PubMed.
- P. Pham, S. Oliver, E. H. H. Wong and C. Boyer, Polym. Chem., 2021, 12, 5689–5703 RSC.
- P. T. Phuong, S. Oliver, J. He, E. H. H. Wong, R. T. Mathers and C. Boyer, Biomacromolecules, 2020, 21, 5241–5255 CrossRef CAS PubMed.
- A. O. Biying, M. J. H. Ong, W. Zhao, N. Li, H.-K. Luo and J. M. W. Chan, ACS Appl. Polym. Mater., 2024, 6, 8169–8177 Search PubMed.
- S. Chattopadhyay, E. Heine, A. Mourran, W. Richtering, H. Keul and M. Möller, Polym. Chem., 2016, 7, 364–369 Search PubMed.
- S. Chattopadhyay, E. T. Heine, H. Keul and M. Möller, Macromol. Biosci., 2014, 14, 1116–1124 CrossRef CAS PubMed.
- S. Chattopadhyay, H. Keul and M. Moeller, Green Chem., 2013, 15, 3135–3139 RSC.
- J. Zhang, Z. Yang, Y.-H. Li, S. Durrani, A.-P. Pang, Y. Gao, F.-G. Wu and F. Lin, ACS Appl. Polym. Mater., 2022, 4, 425–434 CrossRef CAS.
- P. M. Alves, R. F. Pereira, B. Costa, N. Tassi, C. Teixeira, V. Leiro, C. Monteiro, P. Gomes, F. Costa and M. C. L. Martins, ACS Appl. Polym. Mater., 2022, 4, 5012–5026 CrossRef CAS.
- M. Másson, Carbohydr. Polym., 2024, 337, 122159 CrossRef PubMed.
- B.-I. Andreica, L. Mititelu-Tartau, I. Rosca, I. M. Pelin, E. Nicol and L. Marin, Carbohydr. Polym., 2024, 342, 122389 Search PubMed.
- S. Chattopadhyay, E. Heine, H. Keul and M. Moeller, Polymers, 2014, 6, 1618–1630 CrossRef.
- C. Krumm, M. Hijazi, S. Trump, S. Saal, L. Richter, G. G. F. K. Noschmann, T.-D. Nguyen, K. Preslikoska, T. Moll and J. C. Tiller, Polymer, 2017, 118, 107–115 CrossRef CAS.
- M. Concilio, R. Garcia Maset, L. P. Lemonche, V. Kontrimas, J.-I. Song, S. K. Rajendrakumar, F. Harrison, C. R. Becer and S. Perrier, Adv. Healthcare Mater., 2023, 12, 2301961 CrossRef CAS PubMed.
- Y. Hu, J. Zhao, M. Yang, X. Wang, H. Zhang, J. Zhang, Z. Zhu and J. Rao, ACS Appl. Polym. Mater., 2022, 4, 4868–4875 CrossRef CAS.
- E. N. Şimşek, A. Akdağ and P. Z. Çulfaz-Emecen, Polymer, 2016, 106, 91–99 CrossRef.
- M. Kim, W. Mun, W. H. Jung, J. Lee, G. Cho, J. Kwon, D. J. Ahn, R. J. Mitchell and B.-S. Kim, ACS Nano, 2021, 15, 9143–9153 CrossRef CAS PubMed.
- J. D. Katiyar, A. Halder, M. Avais, H. Aidasani, O. Mukherjee and S. Chattopadhyay, ACS Appl. Polym. Mater., 2023, 5, 9742–9750 CrossRef CAS.
- X. Li, S. İlk, Y. Liu, D. B. Raina, D. Demircan and B. Zhang, Polym. Chem., 2022, 13, 2307–2319 RSC.
- X. Li, X. Wang, S. Subramaniyan, Y. Liu, J. Rao and B. Zhang, Biomacromolecules, 2021, 23, 150–162 CrossRef PubMed.
- S. Billiet, K. De Bruycker, F. Driessen, H. Goossens, V. Van Speybroeck, J. M. Winne and F. E. Du Prez, Nat. Chem., 2014, 6, 815–821 CrossRef CAS PubMed.
- S. Kumari, M. Avais and S. Chattopadhyay, Polymer, 2022, 256, 125219 CrossRef CAS.
- S. Kumari and S. Chattopadhyay, Surf. Interfaces, 2024, 46, 103884 CrossRef CAS.
- A. Anand, R. K. Bhagat, S. Ghosh and S. Chattopadhyay, ACS Appl. Polym. Mater., 2024, 6, 11487–11496 CrossRef CAS.
- L. Vlaminck, B. Van de Voorde and F. E. Du Prez, Green Chem., 2017, 19, 5659–5664 RSC.
- S. Kumari and S. Chattopadhyay, Mol. Syst. Des. Eng., 2024, 9, 490–499 RSC.
- S. Chattopadhyay and F. Du Prez, Eur. Polym. J., 2016, 81, 77–85 CrossRef CAS.
- M. Avais and S. Chattopadhyay, Macromol. Chem. Phys., 2022, 223, 2200109 CrossRef CAS.
- L. Vlaminck, K. De Bruycker, O. Türünç and F. E. Du Prez, Polym. Chem., 2016, 7, 5655–5663 RSC.
- U. Bernauer, L. Bodin, Q. Chaudhry, P. J. Coenraads, M. Dusinska, E. Gaffet, E. Panteri, V. Rogiers, C. Rousselle, M. Stepnik, T. Vanhaecke, S. Wijnhoven, N. von Goetz, W. H. de Jong and A. Simonnard, Regul. Toxicol. Pharmacol., 2020, 112, 104611 CrossRef CAS PubMed.
- R. Luna-Vázquez-Gómez, M. E. Arellano-García, J. C. García-Ramos, P. Radilla-Chávez, D. S. Salas-Vargas, F. Casillas-Figueroa, B. Ruiz-Ruiz, N. Bogdanchikova and A. Pestryakov, Materials, 2021, 14, 2792 CrossRef PubMed.
- F. Chen, Y. Tang, H. Zheng, Y. Xu, J. Wang and C. Wang, ChemMedChem, 2019, 14, 1457–1465 CrossRef CAS PubMed.
- J. Wang, S. Chou, L. Xu, X. Zhu, N. Dong, A. Shan and Z. Chen, Sci. Rep., 2015, 5, 15963 CrossRef CAS PubMed.
- I. M. Helander and T. Mattila-Sandholm, J. Appl. Microbiol., 2000, 88, 213–219 CrossRef CAS PubMed.
- T.-F. C. Mah and G. A. O'Toole, Trends Microbiol., 2001, 9, 34–39 CrossRef CAS PubMed.
- P. Keshvardoust, V. A. A. Huron, M. Clemson, F. Constancias, N. Barraud and S. A. Rice, npj Biofilms Microbiomes, 2019, 5, 22 CrossRef PubMed.
- Q. Li, L. Wang, L. Yu, C. Li, X. Xie, H. Yan, W. Zhou, C. Wang, Z. Liu, G. Hou and Y.-Q. Zhao, Biomacromolecules, 2024, 25, 1180–1190 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: All the experimental details and characterization details. See DOI: https://doi.org/10.1039/d4py01302f |
| ‡ These authors contributed equally to the current work. |
| This journal is © The Royal Society of Chemistry 2025 |