
Manipulation of photoresponsive liquid-crystalline polymers and their applications: from nanoscale to macroscale
Yufan
Ji
and
Haifeng
Yu
 *
*
School of Material Science and Engineering, Key Laboratory of Polymer Chemistry and Physics of Ministry of Education, Peking University, Beijing, 100871, China. E-mail: yuhaifeng@pku.edu.cn
First published on 26th June 2024
Abstract
As a smart material, photoresponsive liquid-crystalline polymers exhibit interestingly tunable properties from the nanoscale to the macroscale, which are attributed to their combination of liquid crystal ordering and photoswitchable features. An azobenzene-containing liquid-crystalline polymer is a typical representative, which has been studied from many different aspects. However, the lack of research on the structure–performance relationship and multi-scale manipulation has limited its applications in a broader field. Over the past few years, great efforts have been devoted to achieving the control at multiple scales using light, which has promoted their applications in the fields of energy storage, advanced anticounterfeiting and switchable adhesion. Here, we summarize recent progress in photoresponsive liquid-crystalline polymers, including molecular design, fabrication techniques and the manipulation mechanism from the nanoscale to the micrometre scale and then the macroscale. Their various applications based on abundant photoresponsive properties are systematically summarized. Finally, the existing challenges and opportunities in this field are discussed.
 Yufan Ji | Yufan Ji is currently a PhD candidate at Peking University. His current research interests focus on fabricating micro/nanostructures using azobenzene-containing photoresponsive materials. |
 Haifeng Yu | Haifeng Yu received his PhD degree from the Tsinghua University in 2003. He then worked as a researcher at JST-CREST, and then as a postdoc in JSPS at the Tokyo Institute of Technology and Kyodo University. In 2008, he started his independent research as an associate professor by special appointment at the Nagaoka University of Technology. In 2012, he joined Peking University as a research professor as the “2011 National Thousand Young Talents Program” recipient. In 2013, he obtained the “NSFC Award” for Excellent Young Scholar. In 2023, he was selected as one of the National high-level leading talents. His research interests focus on photoresponsive soft matter, supramolecular self-assembled materials and polymer-based composite materials. |
1. Introduction
With the development of information technology, human society has gradually become more diversified and intelligent. Simultaneously, the development and utilization of functional materials are continuously expanding. Generally, most of them are smart materials, that is, materials that can respond to external environments, including metallic,1,2 inorganic non-metallic,3,4 polymeric and their composite materials.5,6 Among them, polymer materials have incomparable advantages over other materials due to their excellent processability and designability. Therefore, combining polymer materials with stimuli-responsive behaviours often brings about obvious variations in properties, such as luminescence,7–9 discoloration,10–12 deformation,13–15 actuation,16–18 and their combinations.19–21 In particular, as an external stimulus, light has attracted much attention since it can achieve remote control, high speed and precise manipulation.22–24 If the physiochemical properties of polymers can be modulated by light, they are called photoresponsive polymers. Photoresponsive polymers can undergo isomerization,25 crosslinking,26 or degradation27 upon light irradiation, and these photochemically induced changes in molecular configuration provide an opportunity for altering their macroscopic characteristics, such as colour, phase state, solubility, refractive index, glass-transition temperature (Tg), and surface energy. Taking advantage of these intriguing properties, they have broad prospects in the development of photo-actuation devices, fabrication of optical components, chip manufacturing, and information storage.Typically, an azobenzene (AZ)-containing polymer (or azopolymer) is one of the most studied photoresponsive polymers,28–32 since its photoisomerization is a simple and fast process, which is more suitable for the construction of photoresponsive systems. Additionally, AZ in its trans form can be used as a mesogen due to its rod-like geometry, while its cis state with a bent shape is induced upon UV illumination (Fig. 1), which is often isotropic at room temperature.15 Moreover, trans-AZ groups have unique properties of liquid crystals (LCs): mesogenic self-assembly, molecular cooperative motion, optical anisotropy and birefringent behaviours. The LC phase is a special state of matter, where the most important feature is that the mesogenic molecule has a director, so it will expand perpendicularly to the director and contract along the director upon phase transition at the clearing point (Ti) upon heating.33 For trans-AZ mesogens or AZ-containing LC systems, another way to achieve the above-mentioned phenomenon is photoisomerization upon UV light irradiation since the order parameters can be decreased by the photoisomerization and the following molecular cooperative motion.34–36 Furthermore, cis-AZ is thermally unstable, and it often recovers to its trans state spontaneously. Also, suitable-wavelength visible-light irradiation or thermal treatment can achieve the cis-to-trans back-isomerization. This reversible isomerization process can be completed in a timescale from femtoseconds to several seconds depending on the kind of AZ moities,36–38 offering the possibility of applications in various fields.
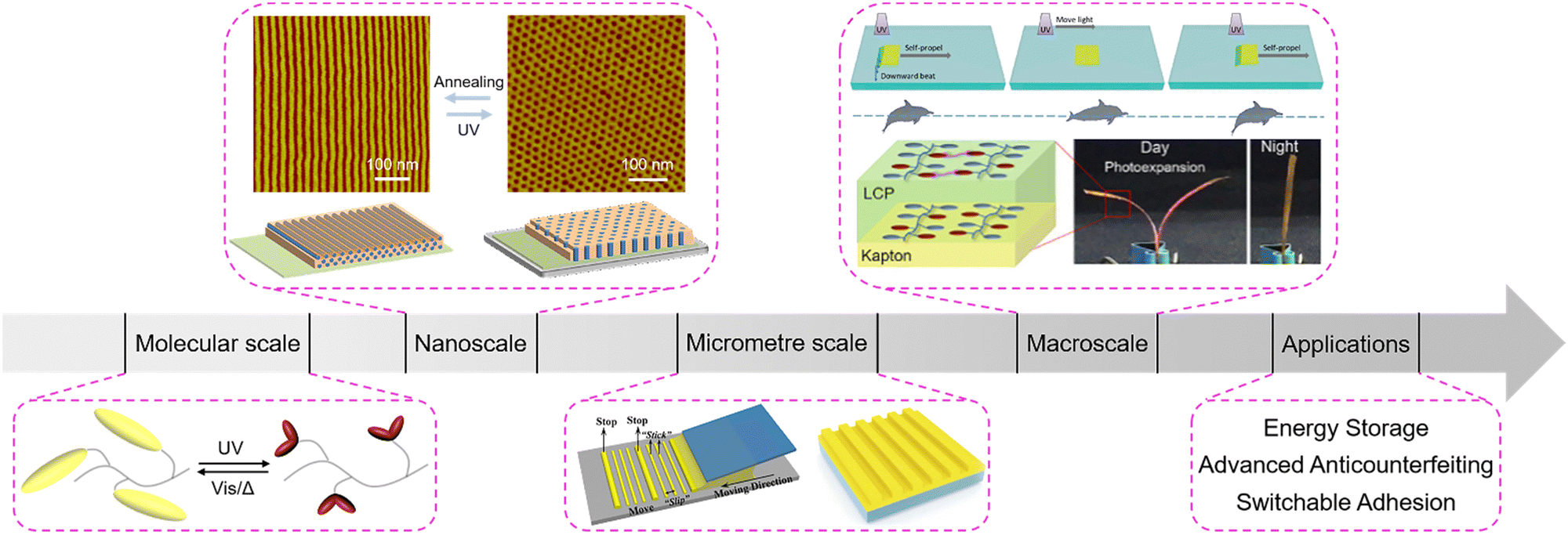 | ||
| Fig. 1 Schematic illustration of the manipulation of photoresponsive LCPs at multiple scales: the molecular scale, nanoscale, micrometre scale, and macroscale. Reproduced from ref. 39; copyright 2006, Wiley-VCH; reproduced from ref. 40; copyright 2007, American Chemical Society; reproduced from ref. 41; copyright 2017, American Chemical Society; reproduced from ref. 42; copyright 2018, American Chemical Society; reproduced from ref. 43; copyright 2020, Wiley-VCH; reproduced from ref. 44; copyright 2019, Wiley-VCH; reproduced from ref. 45; copyright 2019, The Royal Society of Chemistry. | ||
In this review, we discuss recent progress in the manipulation of photoresponsive liquid-crystalline polymers (LCPs) at multiple scales (Fig. 1). Considering that the switchable topographical deformation and numerous applications of LCPs have been reviewed previously,15,46–48 here we focus on multi-scale manipulation of LCPs from the nanoscale, and the micrometre scale to the macroscale. Firstly, we focus on the construction method of different material systems. Then, we discuss the manipulation of nanostructures and hierarchically supramolecular self-assembly in liquid-crystalline block copolymers (LCBCs). Subsequently, we highlight the fabrication techniques of microstructures in different LCPs. Moreover, the development of photo-actuation devices and their manipulation mechanisms have been systematically reviewed. Then, we present some potential applications in energy storage, advanced anticounterfeiting and switchable adhesion. Finally, we summarize this review and provide some promising prospects in this field.
2. Construction of different material systems
2.1. Supramolecular systems
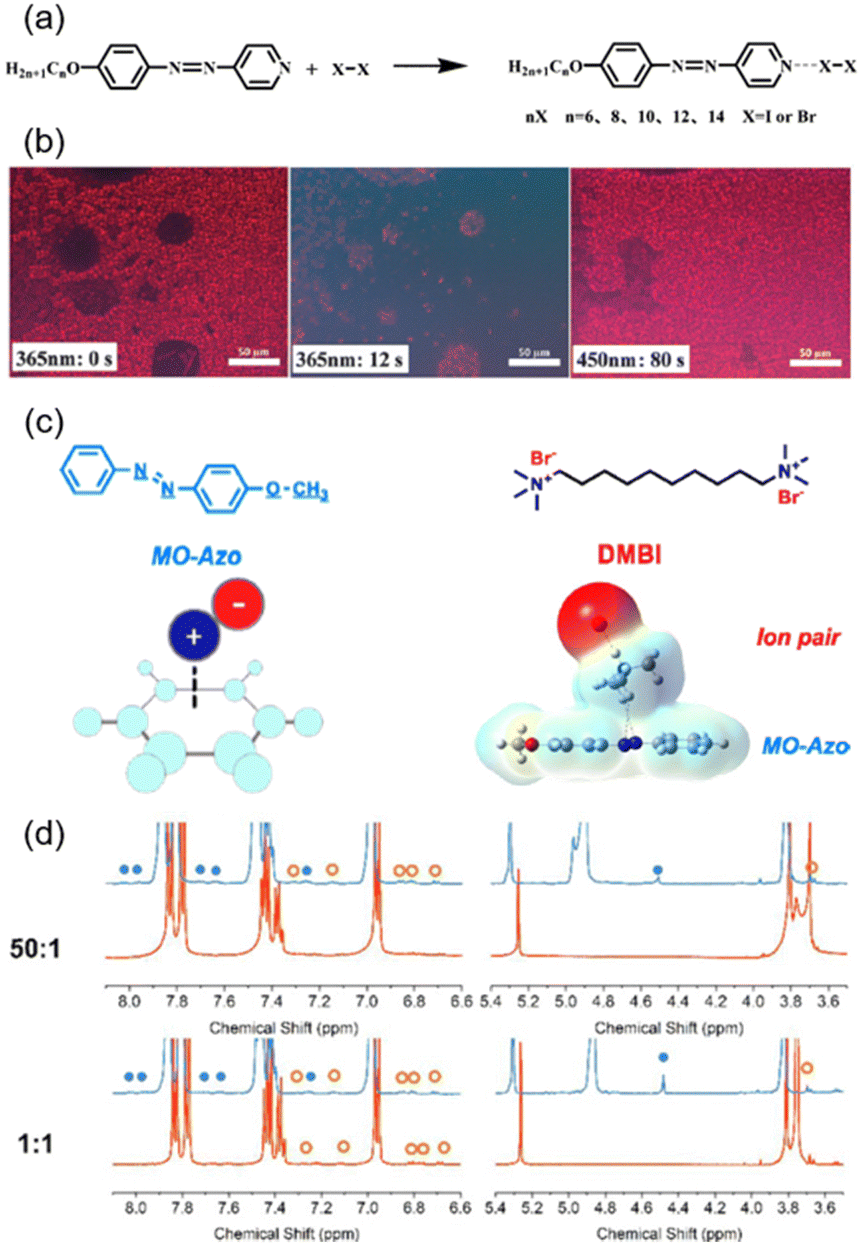
Supramolecular interaction including hydrogen bonds (HBs), halogen bonds (XBs), and cation–π interactions is a kind of noncovalent bonding interaction formed by self-organization.49 In particular, self-assembly via HBs is a simple but efficient way to fabricate supramolecular systems. The carboxylic AZ-containing homopolymer (PM6AzCOOH) is a typical example with response to light and pH value. As shown in Fig. 2a and b, our group reported the emergence of vesicle-like structures resulting from the self-assembly of PM6AzCOOH in a mixed solvent of water and N,N-dimethyl formamide (DMF), and these structures could be manipulated upon changing the pH value of the solution.50,51 Under acidic conditions, a HB was easily formed between the carboxylic acids and the morphology of the PM6AzCOOH aggregates could be observed clearly, while in a basic environment, the HB was destroyed due to the formation of carboxylates and then the micelle-like structures became much smaller (Fig. 2b). Another typical example of forming HBs is between an azopyridine-containing polymer (PM11AzPy) and oleic acid (OA) (Fig. 2c).52–54 The obtained PM11AzPy-OA gel with different feed molar ratios had distinct mesogenic characteristics (Fig. 2d) and morphologies of three-dimensional (3D) networks (Fig. 2e). Based on these unique features, an organogel with multi-responsive behaviours has been developed. | ||
Fig. 2 (a) Schematic illustration of the influence of pH value on the HB formation of PM6AzCOOH. (b) Transmission electron microscopy (TEM) images of PM6AzCOOH/DMF + water at pH = 3 (left), pH = 7 (middle), and pH = 13 (right). Reproduced from ref. 50; copyright 2015, The Royal Society of Chemistry. (c) Chemical structures of the azopyridine-containing polymer (PM11AzPy), oleic acid (OA), and the compound PM11AzPy-OA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) gel. (d) POM images of the PM11AzPy-OA gel with different molar ratios. (e) Field emission scanning electron microscopy (FESEM) images of the PM11AzPy-OA gel with different molar ratios. Reproduced from ref. 52; copyright 2019, American Chemical Society. :1) gel. (d) POM images of the PM11AzPy-OA gel with different molar ratios. (e) Field emission scanning electron microscopy (FESEM) images of the PM11AzPy-OA gel with different molar ratios. Reproduced from ref. 52; copyright 2019, American Chemical Society. | ||
XB is an emerging topic in recent years, which can be described in general as D⋯X–Y, where X is an electrophilic halogen atom, D is the donor of electron density, and Y is a carbon, nitrogen or halogen atom.55 A series of photoresponsive XB complexes were successfully obtained using azopyridine-containing derivatives with molecular iodine or bromine (Fig. 3a).56 According to polarized optical microscopy (POM) results, photoinduced phase transition from the LC to the isotropic phase occurred in the iodine-bonded complex upon UV illumination, and the LC texture can be recovered under visible-light irradiation (Fig. 3b), just like other photoresponsive LC materials.57–59 However, there is no photochemical phase transition in bromine-bonded LC materials, exhibiting high mesophase stability in this system.
 | ||
| Fig. 3 (a) Molecular structures of the photoresponsive XB complexes. (b) POM images of an iodine-bonded complex upon UV illumination and then visible-light irradiation. Reproduced from ref. 56; copyright 2014, The Royal Society of Chemistry. (c) Chemical structures, schemes, and simulation results of the supramolecular system. (d) 1H NMR of the compounds at 25 °C (orange line) and −30 °C (blue line), where the characteristic peaks of the cation–π interaction are marked as blue solid circles and those of the cis-isomer are marked as orange hollow circles. Reproduced from ref. 60; copyright 2022, American Chemical Society. | ||
The cation–π interaction is the “youngest” supramolecular interaction compared to the above-mentioned HB and XB, which was first named by Dougherty in 1996.61 The mechanism of this special interaction is different from the HB.62 Based on these properties, our group originally proposed a method to fabricate a supramolecular system by introducing the cation–π interaction into AZ-containing materials (Fig. 3c).60 By comparing 1H nuclear magnetic resonance (NMR) spectra at room temperature and −30 °C, the increased chemical shift could be observed, which is different from the peak of the cis-isomer (Fig. 3d), demonstrating the existence of the cation–π interaction. This interaction is of great importance to applications in biological and medical fields.63–65 Besides, amphiphilic and ionic interactions can be used to fabricate self-assembled nanofibers using low-molecular-weight azopyridinium salts with different additional components.66–68
2.2. Homopolymers
A homopolymer is the most common polymer but there are still many issues that need to be addressed. One of them is that polymers with a high Tg are difficult to process at room temperature, so polymers with a switchable Tg and a reversible solid-to-liquid transition have been extensively studied.69–71 Recently, we investigated the phototunable mechanics of an AZ-containing LCP poly(6-(4-((4-butylphenyl) diazenyl)-phenoxy) hexyl methacrylate) (PM6AZC4),43 as shown in Fig. 4a. PM6AZC4 in the trans-rich state has a Tg of 76 °C and a Ti of 94 °C, while the Tg of its cis-rich state is only 3 °C. Upon exposure to UV light, the homopolymer can undergo phase transition from a smectic LC phase to an isotropic phase (Fig. 4a). In addition, the loss tangent (tanδ), a ratio of loss modulus to storage modulus, can be used to characterize the state of the materials. The tan δ value of trans-rich PM6AZC4 is below 0.2 in the whole frequency range (Fig. 4b), while cis-rich PM6AZC4 has a tanδ value larger than 2.0 (Fig. 4c), indicating that trans-rich PM6AZC4 is a glassy plastic at room temperature, but cis-rich PM6AZC4 is a flowing fluid under the same conditions. Moreover, Tg of trans-rich and cis-rich PM6AZC4 can be derived from the temperature sweep curves, respectively, as shown in Fig. 4d and e.
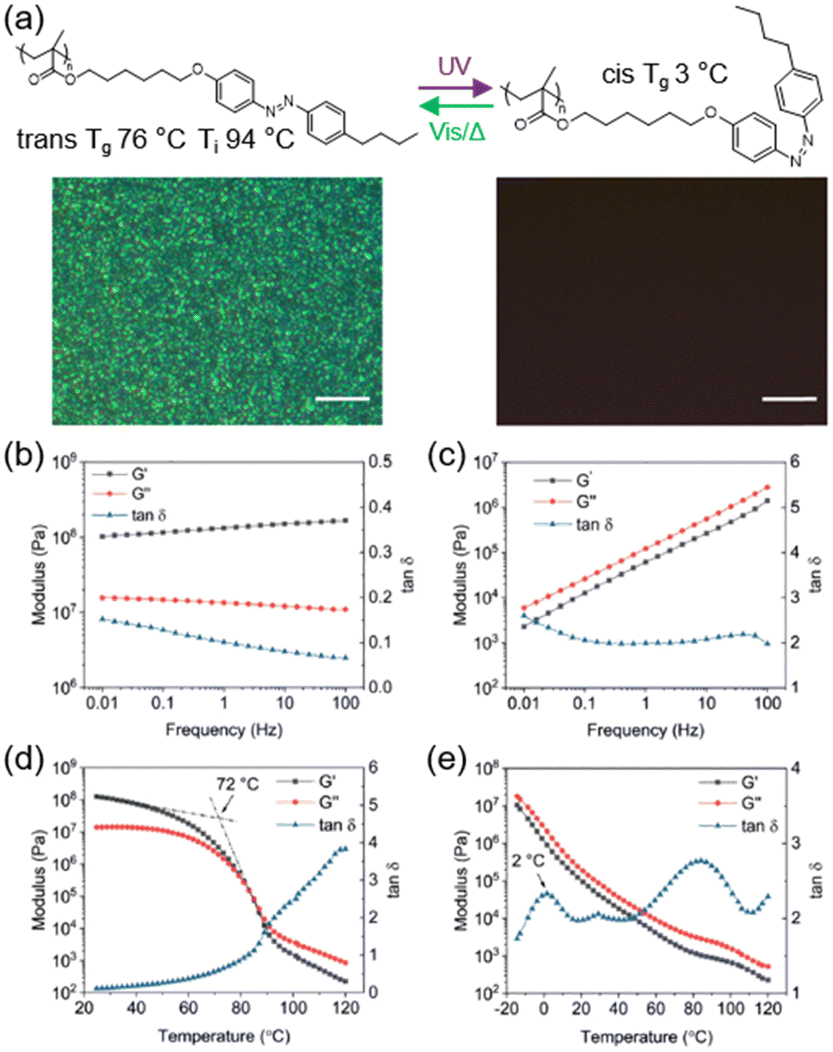 | ||
| Fig. 4 (a) Molecular structures and POM images of the azopolymer with a photo-switchable Tg. Scale bars are all 10 μm. (b) Frequency sweep of PM6AZC4 in the trans state. (c) Frequency sweep of PM6AZC4 in the cis state. (d) Temperature sweep of PM6AZC4 in the trans state. (e) Temperature sweep of PM6AZC4 in the cis state. Reproduced from ref. 43; copyright 2020, Wiley-VCH. | ||
These results are consistent with the differential scanning calorimetry (DSC) results, demonstrating the successful construction of a homopolymer system with a photo-switchable Tg.
2.3. Block copolymers
AZ-containing LCBC systems with supramolecular cooperative motion are competitive candidates for fabricating microphase-separated nanostructures in macroscopic ordering. Therefore, an amphiphilic LCBC consisting of poly(ethylene oxide) (PEO) and AZ-containing LC polymethacrylate was synthesized to explore its unique properties. As shown in Fig. 5a, one mechanical rubbing method was used to achieve homogeneous LC alignment along the rubbing direction after annealing.39 This simple technique provides an opportunity for manufacturing various nanostructures since it is convenient and low-cost compared to other methods.58 Additionally, PEO nanocylinders in the LCBC film can be aligned perpendicular to the polarization direction of linearly polarized light (LPL) due to the supramolecular cooperative motion between AZ moieties and PEO nanocylinders (Fig. 5b).72 This remote and precise photocontrol method offers the possibility to fabricate parallel array of nanocylinders in any region, even curved surfaces.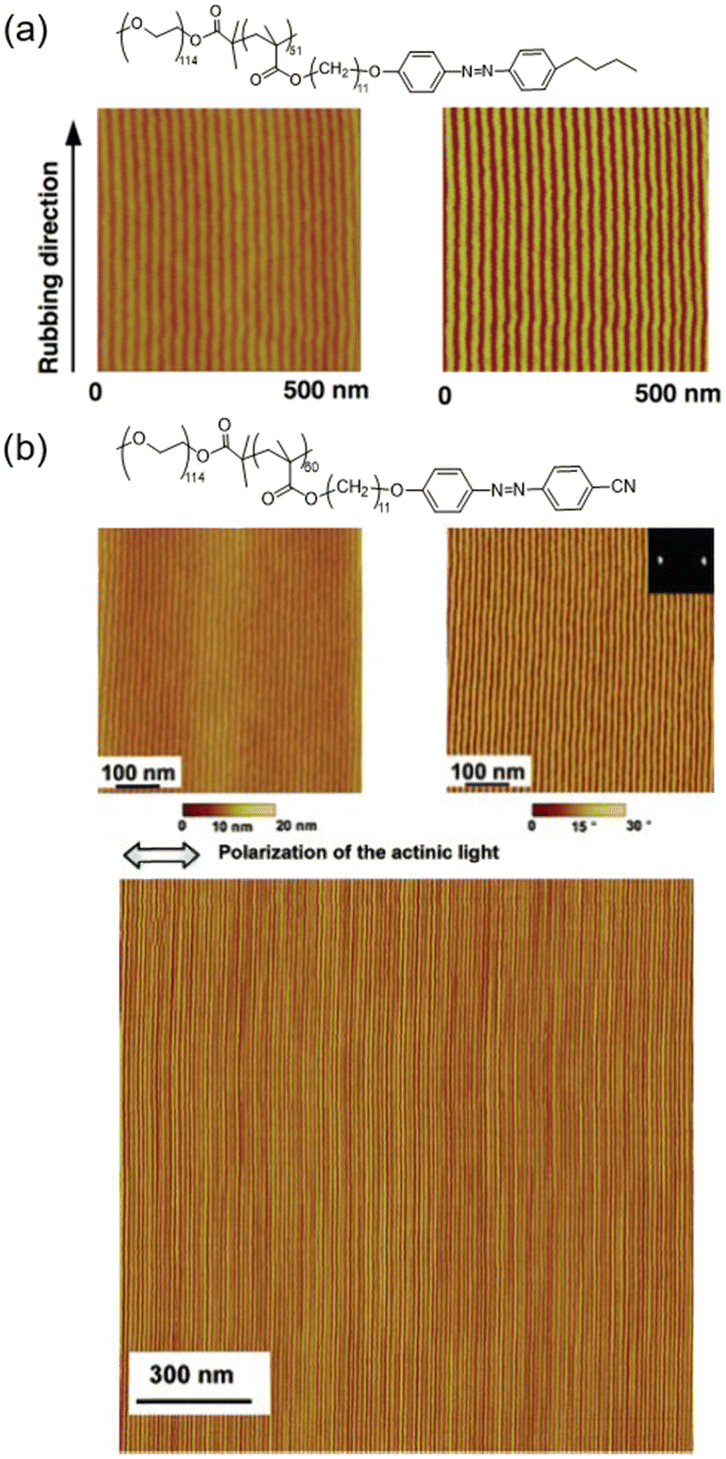 | ||
| Fig. 5 (a) Molecular structure and atomic force microscopy (AFM) height (left) and phase (right) images of the LCBC film after rubbing and annealing treatment. Reproduced from ref. 39; copyright 2006, Wiley-VCH. (b) Chemical structure and AFM height (left) and phase (right) images of the LCBC film upon irradiation. The bottom image is the AFM phase image in a larger area. Reproduced from ref. 72; copyright 2006, American Chemical Society. | ||
Another interesting LCBC system is an ABA-type triblock copolymer (TBC). It is well-known that there is physical cross-linking in polystyrene-b-polybutadiene-b-polystyrene (SBS),73,74 where the hard segment with a high Tg acts as a “physical cross-linker” and the soft segment with a low Tg functions as an elastic strand in the network.75 To mimic this phase behaviour, a well-defined SBS-like structure including an azopolymer as the middle block and polystyrene (PS) as the end blocks was designed, as shown in Fig. 6a.76 The Tg of this azopolymer in the middle block is higher than room temperature, so its trans state exhibits stiffness at room temperature (Fig. 6b). After UV irradiation, the Tg of the azopolymer drops below 0 °C, which can act as soft domains, while the PS block with a Tg higher than room temperature serves as a “physical cross-linker” (Fig. 6b), allowing this TBC to behave as an elastomer.
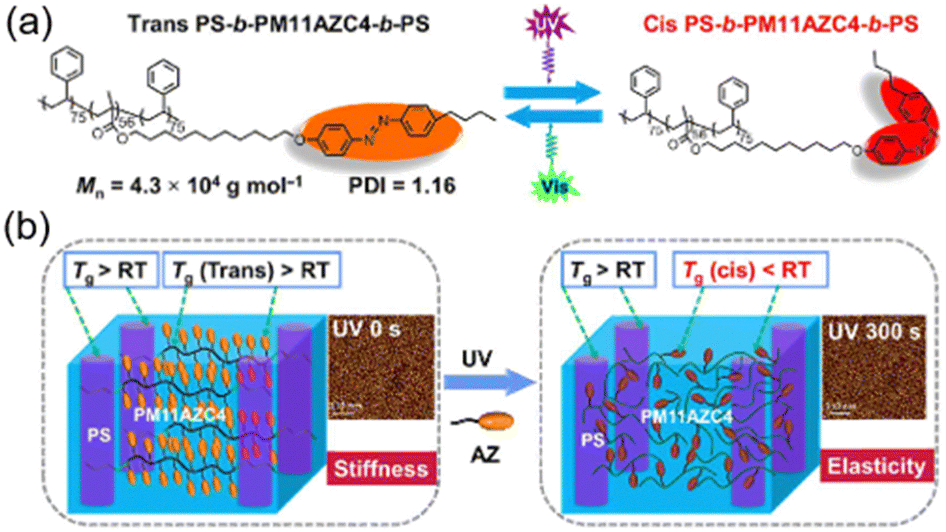 | ||
| Fig. 6 (a) Molecular structures of the AZ-containing TBC. (b) Schematic illustration of the AZ-containing TBC and the nanostructures of this TBC film before and after UV irradiation. Reproduced from ref. 76; copyright 2022, American Association for the Advancement of Science. | ||
2.4. Composite systems
AZ-containing composites can be divided into many categories, such as polymer-dispersed LCs (PDLCs)/graphene oxide (GO) nanocomposites, AZ derivatives/low-density polyethylene (LDPE) films, photoresponsive LCPs/Kapton (commercial polyimide) films and so on.To take full advantage of sunlight for the photo-actuation of composite films, homodispersed GO, a nematic LC molecule (5CB) and a photoresponsive AZ molecule (5CAZ) shown in Fig. 7a were introduced into the poly(vinyl alcohol) (PVA)-dispersed LC system.77 Then, the stretched PDLC/GO composite film could respond to near-infrared (NIR) light or visible-light at room temperature (Fig. 7a). Upon irradiation, the film bent toward the light source along the stretching direction when its LC-rich layer faced the light source, while the bending direction is opposite if the other face of the film was irradiated.77 The reason for this interesting photomechanical deformation is that the photothermal effect of GO leads to the temperature rise of the film. When the temperature was higher than the Ti of the LC mixture, the phase transition from the LC to the isotropic occurred. Therefore, molecular contraction along the stretching direction in the LC-rich layer is much more than that in the LC-poor layer, resulting in a different bending direction. Moreover, phase transition from the nematic LC to the isotropic phase could occur upon exposure to UV light, since the photoisomerization of 5CAZ would bring about the photochemical phase transition. Hence, the composite film bent upon UV illumination, similarly to that actuated by NIR or visible-light (Fig. 7a). In addition, the results remained the same when the PVA matrix was replaced with shape-memory polyurethane (SMPU).78 Similarly, the SMPU-based composite system consisting of 5CAZ and upconversion nanoparticles (UCNPs) could respond to UV-Vis-NIR light.79 This design prevents the temperature rise of the film since the UCNPs can convert NIR light into green fluorescence, causing 5CAZ to undergo trans–cis isomerization, which is different from the photothermal mechanism of the PDLC/GO nanocomposite system.
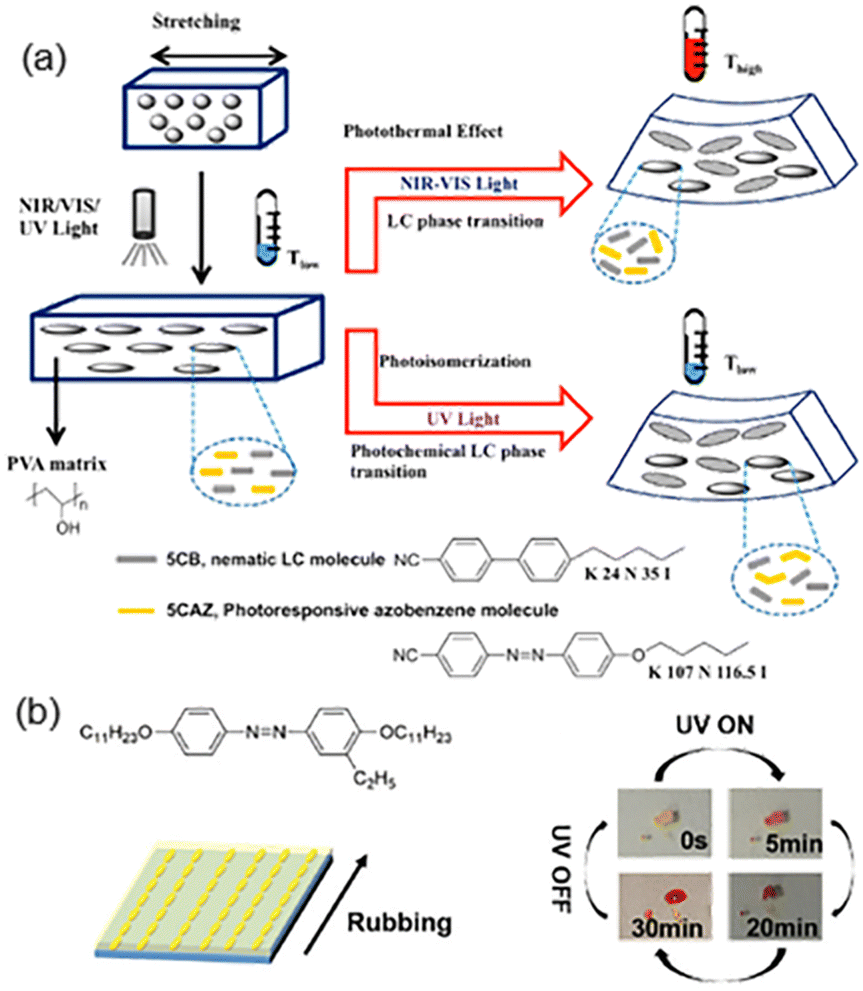 | ||
| Fig. 7 (a) Schematic illustration of the NIR-Vis-UV light-responsive composite film composed of GO and the LC mixture. Reproduced from ref. 77; copyright 2015, American Chemical Society. (b) Molecular structures of the AZ derivative and its reversible photo-liquefaction from a crystal to an isotropic liquid. A schematic illustration of the bilayer actuator composed of the AZ derivative and the LDPE film is also provided. Reproduced from ref. 80; copyright 2018, The Royal Society of Chemistry. | ||
Another composite system was fabricated by dropwise coating a tetrahydrofuran (THF) solution of an AZ derivative on the mechanically-rubbed LDPE film and then annealing at a certain temperature.80 The composite film bent towards the AZ layer when exposed to UV light, which is in agreement with other systems.77,81 The AZ derivative used in this system could undergo a photoinduced transition directly from a crystal to an isotropic liquid upon UV irradiation due to photoisomerization and the decreased melting point, as shown in Fig. 7b. Moreover, the rubbing-induced microgrooves on the LDPE film surface and the good fluidity of the AZ derivative during the thermal annealing process could facilitate the AZ orientation along the microgroove (Fig. 7b). Then, the photoinduced phase transition of the aligned AZ derivative would drive the whole film to bend towards the AZ layer.
Furthermore, a bilayer composite film composed of the AZ-containing LCP and the Kapton substrate was fabricated using a facile drop casting method.45 One homopolymer PM6ABOC2 and two random copolymers P(M6ABOC2x-r-M6AzPy1−x) were synthesized (Fig. 8a), where introduction of the pyridine group offered a cross-linking site. After drop casting the LCP solution on the Kapton film, the composite film was then annealed at the LC temperature to obtain an oriented nematic phase (Fig. 8b). Subsequently, the annealed film was cross-linked by the quaternization reaction using 1,4-diidonetetra-butane vapor (Fig. 8b). So far, the bilayer composite film exhibiting photoinduced bending towards the Kapton layer upon UV illumination has been prepared successfully.
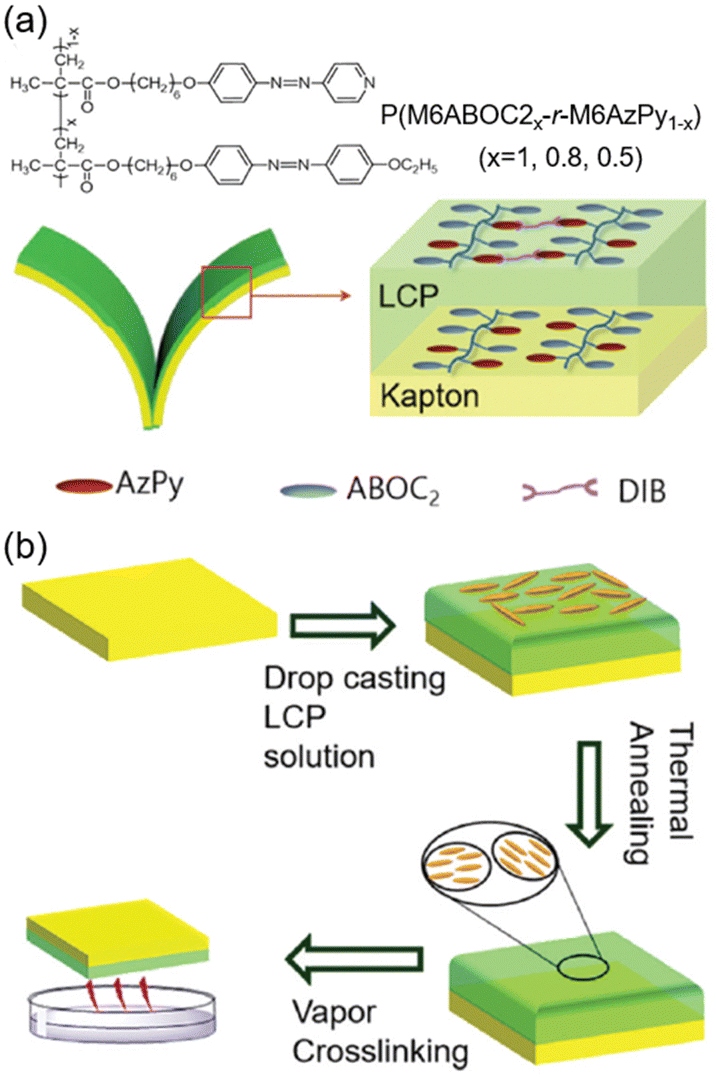 | ||
| Fig. 8 (a) Molecular structures and schematic illustration of the bilayer film. (b) Scheme of the drop casting method to prepare the bilayer composite film. Reproduced from ref. 45; copyright 2019, The Royal Society of Chemistry. | ||
3. Manipulation at the nanoscale
3.1. Microphase-separated nanostructures in LCBCs
AZ-containing LCBCs with delicate molecular structures and narrow molecular-weight distributions possess unique features in that microphase-separated (MPS) nanostructures occur spontaneously and the supramolecular self-assembled nanostructures can interact with the ordered mesogenic groups under certain conditions.39,72 As shown in Fig. 9a–e, the LC to isotropic phase transition induced by UV illumination can bring about significant effects on the MPS nanostructures in the LCBC film.41 As the irradiation time increased, in-plane nanocylinders began to fracture into dot-like nanostructures and then became more ordered until hexagonal packing was obtained. The whole process of the photocontrol of the nanocylinder orientation from in-plane to out-of-plane was completed in several seconds, and after reannealing at 145 °C, the initial state reappeared (Fig. 9f). In addition, after the hexagonal nanocylinder array was fabricated, thermal stabilization could be greatly improved by coating water-soluble PSSNa onto the film surface.82 Then, even if the film was reannealed at a higher temperature, the orientation of PEO nanocylinders remained unchanged. Therefore, the photo-patterned film with PSSNa coating has two different nanostructures after reannealing at 125 °C: dot-like nanostructures in the UV-irradiated area and linear nanostructures in the non-irradiated area (Fig. 9g). Moreover, UV-Vis absorption spectra reveal that thermal stabilization of the nanostructures was independent of the orientation of AZ moieties, and just depended on the PEO nanocylinder domains (Fig. 9h).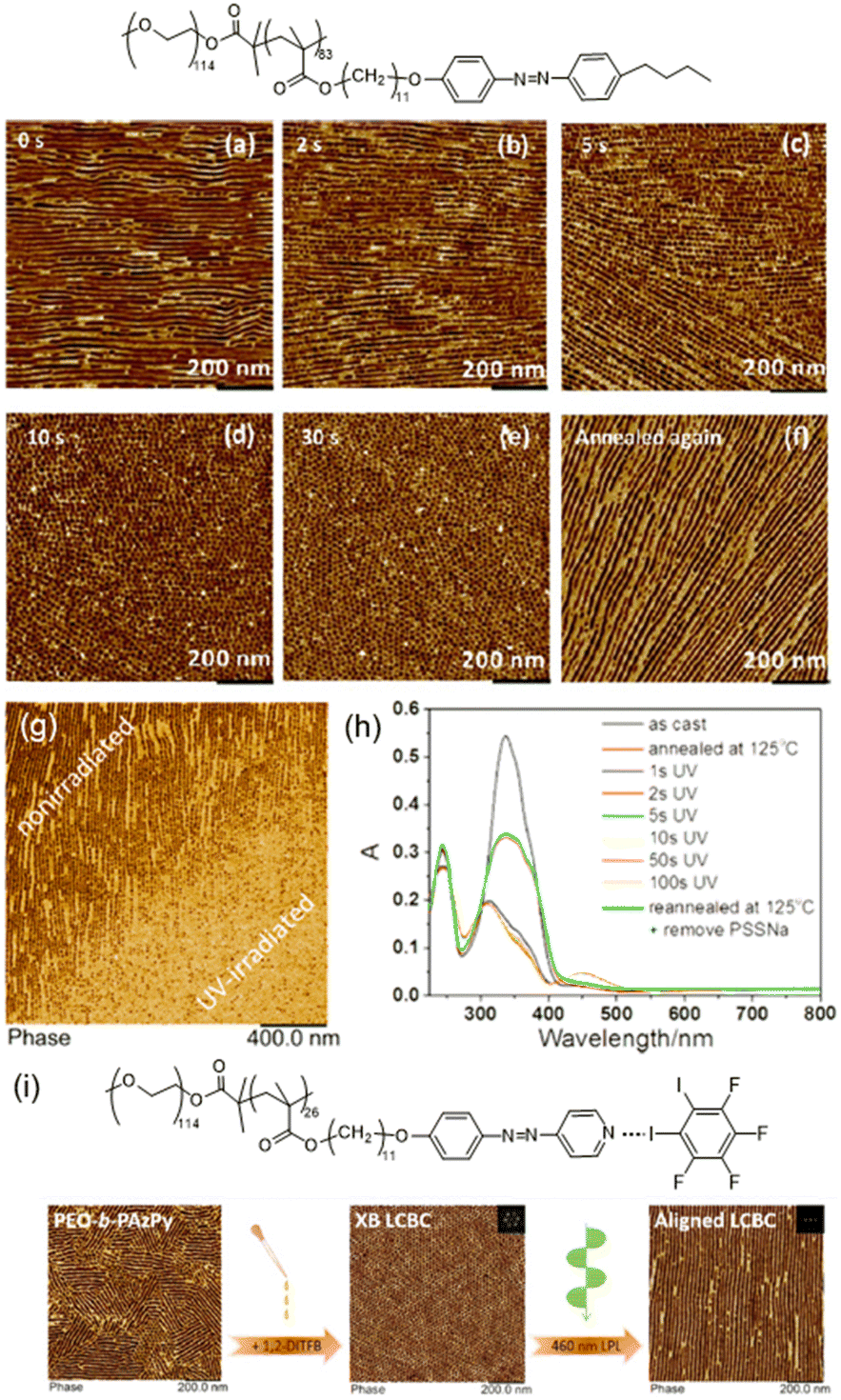 | ||
| Fig. 9 (a)–(e) The variation of the MPS nanostructures in the LCBC film after UV illumination. (f) MPS nanostructures after reannealing at 145 °C. Reproduced from ref. 41; copyright 2017, American Chemical Society. (g) AFM phase image of the photopatterned film with PSSNa coating after reannealing at 125 °C. (h) UV-Vis absorption spectra during the whole process. Reproduced from ref. 82; copyright 2019, American Chemical Society. (i) The variation in the AFM phase image after the introduction of the XB and 460 nm linearly polarized light (LPL) irradiation. Reproduced from ref. 83; copyright 2020, American Chemical Society. | ||
Another more sophisticated system is based on the supramolecular XB interaction between 1,2-diiodo-3,4,5,6-tetrafluorobenzene and one non-mesogenic block copolymer composed of PEO and azopyridine-containing polymethacrylate,83 as shown in Fig. 9i. Generally, a XB is more directional than a HB, which results in the formation of highly ordered supramolecular mesogenic ordering. So the introduction of XB-driven supramolecular self-assembly enhanced the ordering of the MPS nanostructures in the polymer film.83 Furthermore, upon actinic LPL irradiation, the PEO nanocylinders were aligned consistent with the orientation of the supramolecular mesogens, demonstrating that this XB-involved LCBC system is promising for applications in optics.
3.2. Hierarchical self-assembly in LCBCs
Fabrication of hierarchically self-assembled sub-10 nm structures has attracted much attention in science and engineering fields in recent years.84,85 An elegant method to obtain sub-10 nm nanostructures through the self-assembly of LCBCs by introducing urethane groups in the side chain of the hydrophobic block of the LCBC has been proposed (Fig. 10a).86 In the LCBC film, the HB should exist among urethanes (Fig. 10b) or between urethane and oxygen in the PEO block (Fig. 10c). Upon the MPS process, the in-plane orientation of PEO nanocylinders in the LCBC film was observed, as shown in Fig. 10d. Surprisingly, hierarchically ordered structures inside the PEO nanodomains appeared along the axis direction of nanocylinders, indicating that supramolecular self-assembly occurred inside the PEO nanodomains (Fig. 10d). This is attributed to the MPS nanoconfinement effect and the formation of the HB between urethane and PEO, exerting a great impact on the PEO crystallization process. Then, the insufficient crystallization of PEO segments allows crystallized and amorphous PEO to coexist in the MPS nanodomains, enabling the formation of lamellar structures (about 4 nm) inside the PEO nanocylinders.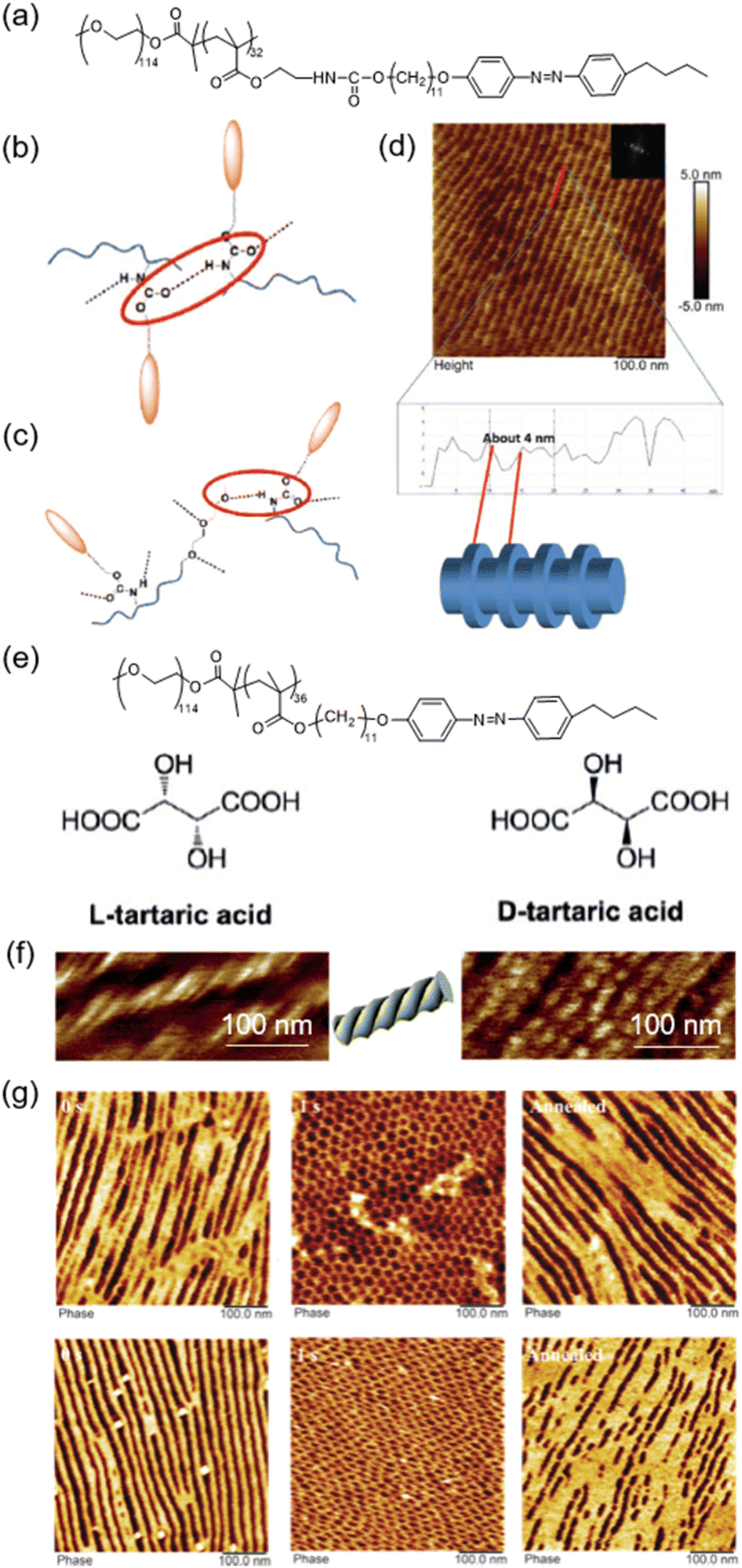 | ||
| Fig. 10 (a) Chemical structure of the urethane-containing LCBC system. (b) and (c) Structural models of two kinds of HBs. (d) AFM height image and cross-sectional profile of the annealed urethane-containing LCBC film. Reproduced from ref. 86; copyright 2018, Wiley-VCH. (e) Chemical structures of the LCBC and chiral additives. (f) AFM images of the LCBC composite films after being annealed for 1200 min. The dopant was L-TA (left) or D-TA (right). (g) AFM images of annealed films doped with L-TA (top) or D-TA (bottom) upon UV illumination for 1 s and then being annealed for 1200 min. Reproduced from ref. 87; copyright 2018, Wiley-VCH. | ||
In addition, helical nanostructures can be prepared by using LCBC doped with functional chiral additives, like enantiopure tartaric acid (TA) (Fig. 10e).87,88 The chirality transferred from a small molecular chiral dopant to non-chiral polymer phase-domains leads to the hierarchical self-assembly in the composite system.89–91 Therefore, helical nanostructures can be observed in the LCBC doped with L-TA or D-TA (Fig. 10f). It is worth noting that the induced aggregation chirality in the mesogenic segments was not determined by the handedness of the dopants, which is the reason why the result was the same whether the dopant was L-TA or D-TA. The HB introduced by TA enhanced the interactions between the two blocks of the LCBC,92,93 providing an opportunity for the emergence of aggregation chirality in the continuous phase and the formation of helical nanostructures in the film.
Furthermore, the manipulation of aggregation chirality to achieve the photocontrol of MPS helical nanostructures was successfully realized. Upon UV irradiation, photoisomerization and phase transition occurred simultaneously, and the in-plane orientation of chiral nanostructures in the film became out-of-plane correspondingly (Fig. 10g).41,82 Then, after thermal annealing for a long time (1200 min), the aggregation chirality reappeared (Fig. 10g), showing a reversible photocontrol process. This tunable hierarchical self-assembly offers a facile method to functionalize chiral materials with controllable properties and opens a door for countless applications in chiral recognition, enantiomer separation, enantioselective permeation, and chiral sensory systems for bio- and nanotechnology.87
4. Manipulation at the micrometre scale
4.1 Hierarchical stripes formed via flow-enabled self-assembly
Evaporation-induced self-assembly in a droplet is a competitive candidate for fabricating sophisticated patterns.94,95 For example, hierarchical stripes can be generated by flow-enabled self-assembly (FESA) of one photoresponsive AZ-containing LCP,42 as shown in Fig. 11. First, one solution of PM6ABOC2 was placed in two parallel plates, where the upper one was fixed and the lower one was in a “stop-and-move” manner (Fig. 11a). When the lower substrate was stopped, the solvent evaporation process would allow the polymer to move to the contact line and form the primary stripe. If the substrate moved at a certain speed, the meniscus was stretched and the contact angle would decrease to a critical value, then the depinning force became larger than the pinning force,96 leading to a change in the position of the contact line and the formation of the secondary stripe. This periodic variation in the contact line would permit the emergence of regular hierarchical stripes (Fig. 11b). Additionally, the formation of secondary stripes was independent of the existence of the primary stripes.42 If the substrate moved at an appropriate speed continuously, no primary stripes would appear but secondary stripes still existed (Fig. 11c). Furthermore, whether it can produce regular secondary stripes depends on the concentration of the solution (c) and the moving speed of the lower substrate (v). As shown in Fig. 11d, regular secondary stripes could be generated when c and v were in the shadow area. If c is too low or v is too fast, there is no sufficient polymer or time to be pinned at the contact line to form stripes. In contrast, when v is too slow, the pinning time is enough but the distance between two adjacent secondary stripes is short, resulting in the generation of irregular stripes. Therefore, this method with exquisite control paves a way for applications in the field of energy and biology.97–100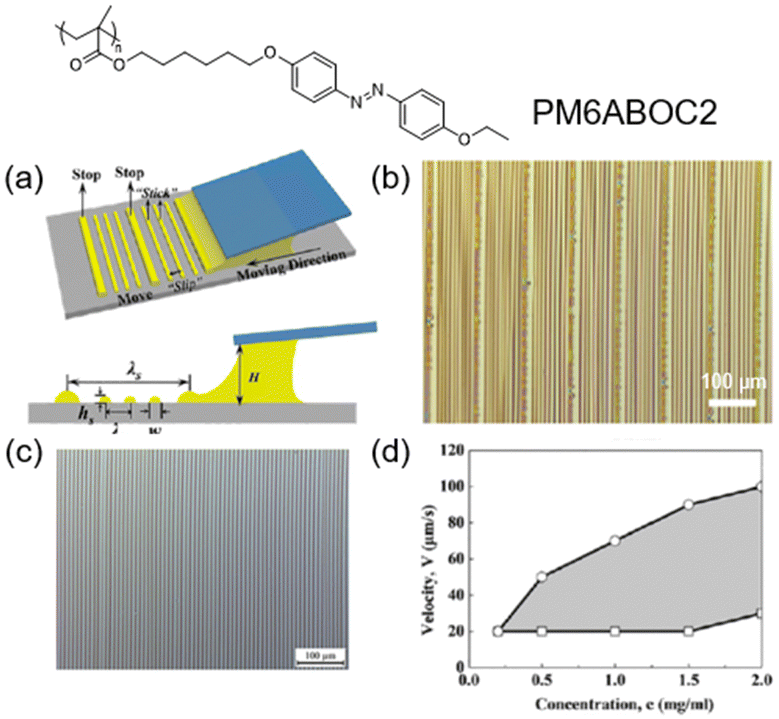 | ||
| Fig. 11 (a) Schematic illustration of the formation of hierarchical stripes via FESA. (b) Optical microscopic photograph of the hierarchical stripes produced by FESA. (c) Secondary stripes generated by moving the lower substrate at an appropriate speed continuously. (d) Two-dimensional (2D) map describing how the concentration of the solution and the moving speed of the lower substrate influence the formation of secondary stripes. If the parameters are in the shadow area, regular secondary stripes will exist. Reproduced from ref. 42; copyright 2018, American Chemical Society. | ||
4.2. Fabrication of gratings and micro-patterns
Gratings with switchable performance are of great importance to academic research and industrial applications. Recently, a facile method was reported to fabricate diffraction gratings in a supramolecular system,52 and the chemical structures of the system are shown in Fig. 2c. Upon irradiation of one interference pattern, holographic gratings were successfully recorded in the PM11AzPy-OA gel (Fig. 12a–c). As expected, the obtained diffraction gratings could respond to multiple external stimuli based on the intriguing properties of this supramolecular LC organogel. When increasing the temperature, the grating structures first became more obvious due to the enhancement of birefringence at the mesogenic phase, and then gradually disappeared because of the weakened anisotropic properties (Fig. 12a). As a result, the bright field under POM turned dark at Ti, and the grating structures disappeared completely. After cooling down to room temperature, gratings could be inscribed again. In addition, UV illumination could achieve the manipulation of the gratings. Since the photoisomerization of azopyridine would break the organogel, the birefringence became weaker until it completely disappeared with the increasing irradiation time (Fig. 12b). After thermal annealing, it went back to the initial state and gratings can be re-recorded. Moreover, the grating structure can be affected by the ethanol solution of organic silver. Due to the strong coordination interaction between azopyridine and Ag+,52 grating structures collapsed and birefringence gradually disappeared as the time passed (Fig. 12c). Even after thermal annealing, the disappeared birefringence would not recover. This phenomenon provides an opportunity for applications in detecting the presence of Ag+.101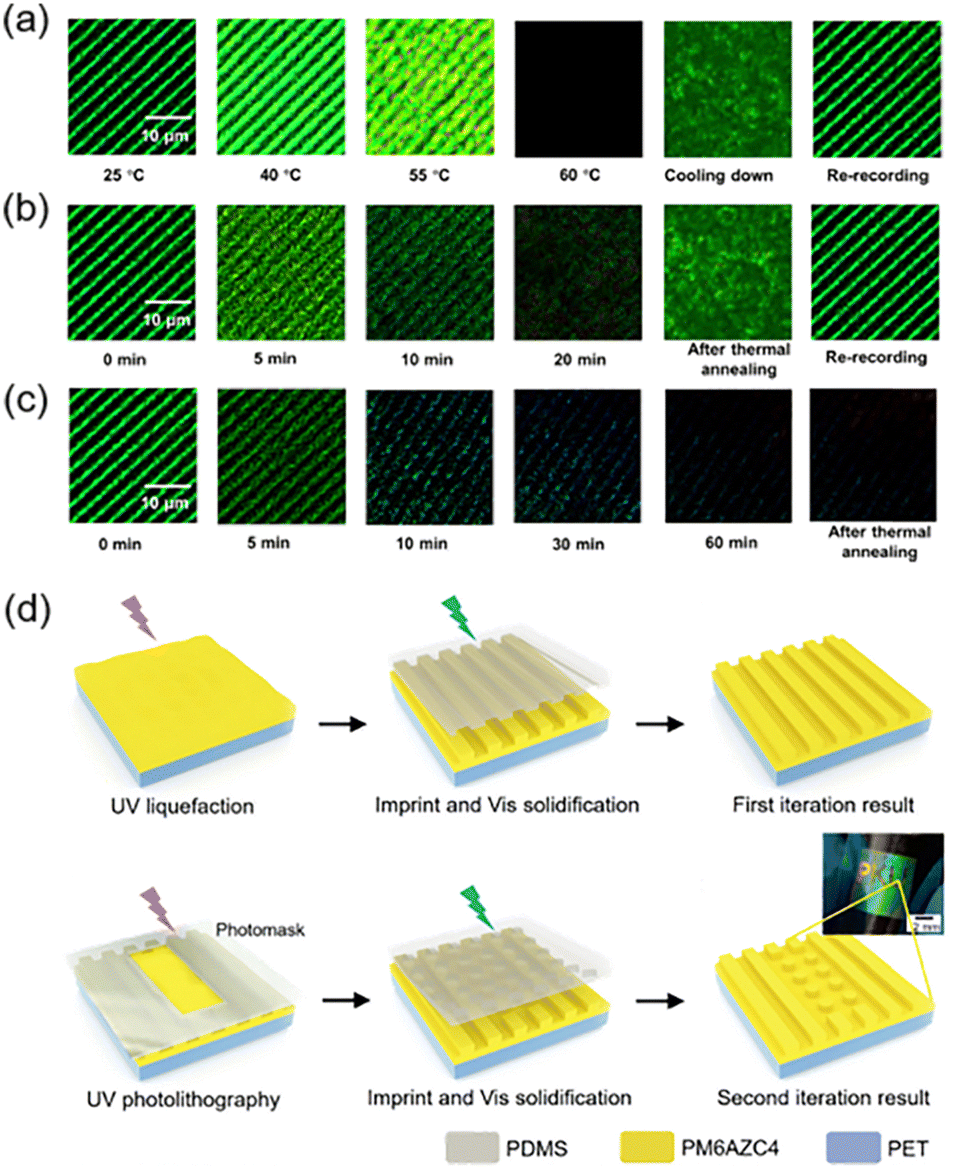 | ||
| Fig. 12 (a) POM images of the gratings in response to heating. (b) POM images of the gratings in response to UV irradiation. (c) POM images of the gratings in response to Ag+ ions. Reproduced from ref. 52; copyright 2019, American Chemical Society. (d) Schematic illustration of the procedures to fabricate micro-patterns. Reproduced from ref. 43; copyright 2020, Wiley-VCH. | ||
Another method to fabricate gratings and other micro-patterns is combining athermal nanoimprint lithography (AT-NIL) with photo-patterning.43 First, one THF solution of the azopolymer (PM6AZC4, half-life of the cis isomer: 14.6 h, Fig. 4a) was spin-coated on the poly(ethylene terephthalate) (PET) substrate and global UV illumination was provided (Fig. 12d). Then, a pre-designed poly(dimethyl siloxane) (PDMS) mold was placed on the film with an external mechanical stress and cis-rich PM6AZC4 would flow into the microarray of the mold. Subsequently, exposure to green light to cause cis–trans back-isomerization could achieve solidification of the azopolymer, and the first iteration result was obtained after removal of the mold. Furthermore, multiple selective photoisomerization in local areas was feasible by using a photomask, since the solid-to-liquid transition of PM6AZC4 was completely reversible. As shown in Fig. 12d, three letters of “P”, “K” and “U” were successfully imprinted on the flexible PET substrate.
Recently, microstructures fabricated by using the mass transport method have become popular, especially Marangoni flow.102–106 Marangoni flow is a kind of interfacial agitation driven by local variations of interfacial tension,107,108 which can be used in the photoresponsive AZ-containing system. Our group has developed a two-step method to fabricate microstructures on the film surface of PM6AZC4 (half-life: 14.6h, Fig. 4a): selective photoisomerization using a photomask and then immersion in a solvent (Fig. 13a).109 After UV-patterning the azopolymer film, the periodic distributions of trans and cis isomers were acquired. Since the surface energy of the trans-azopolymer and cis-azopolymer alternately distributed in the film was different, mass transport occurred when the pre-patterned film was immersed in a solvent such as ethanol (Fig. 13a). It is notable that the flow direction of the azopolymer in the solvent is opposite to the traditional Marangoni flow of the polymer itself,102,109 indicating that the interaction between the solvent and the film surface with wettability gradient exists, allowing the solvent to drag the underlying polymer to transport, and then surface morphing is formed (Fig. 13a). Essentially, this flow from the flowing fluid (cis-rich PM6AZC4) to the glassy plastic (trans-rich PM6AZC4) is the capillary flow induced by the flow of the solvent on the film surface with wettability gradients.110 Therefore, combining this unique phenomenon with the traditional Marangoni flow, a steerable mass transport direction was successfully realized.110 To obtain well-controlled bidirectional mass transport, one commercial small-molecule nematic LC, 4-cyano-4′-pentylbiphenyl (5CB), was introduced into the azopolymer as a functional additive to build a host–guest system (half-life: 7.8 h).110 Upon optimizing the host–guest ratio (the doping amount of 5CB) and the width of the photoisomerized areas, the polymer flow in both directions was obtained simultaneously (Fig. 13b, the middle scheme). As shown in Fig. 13b, when the width of the photoisomerized area was 5 μm, polymer flow occurred from the cis-rich region to the trans-rich region, while the direction was opposite if the width reached or exceeded 20 μm.
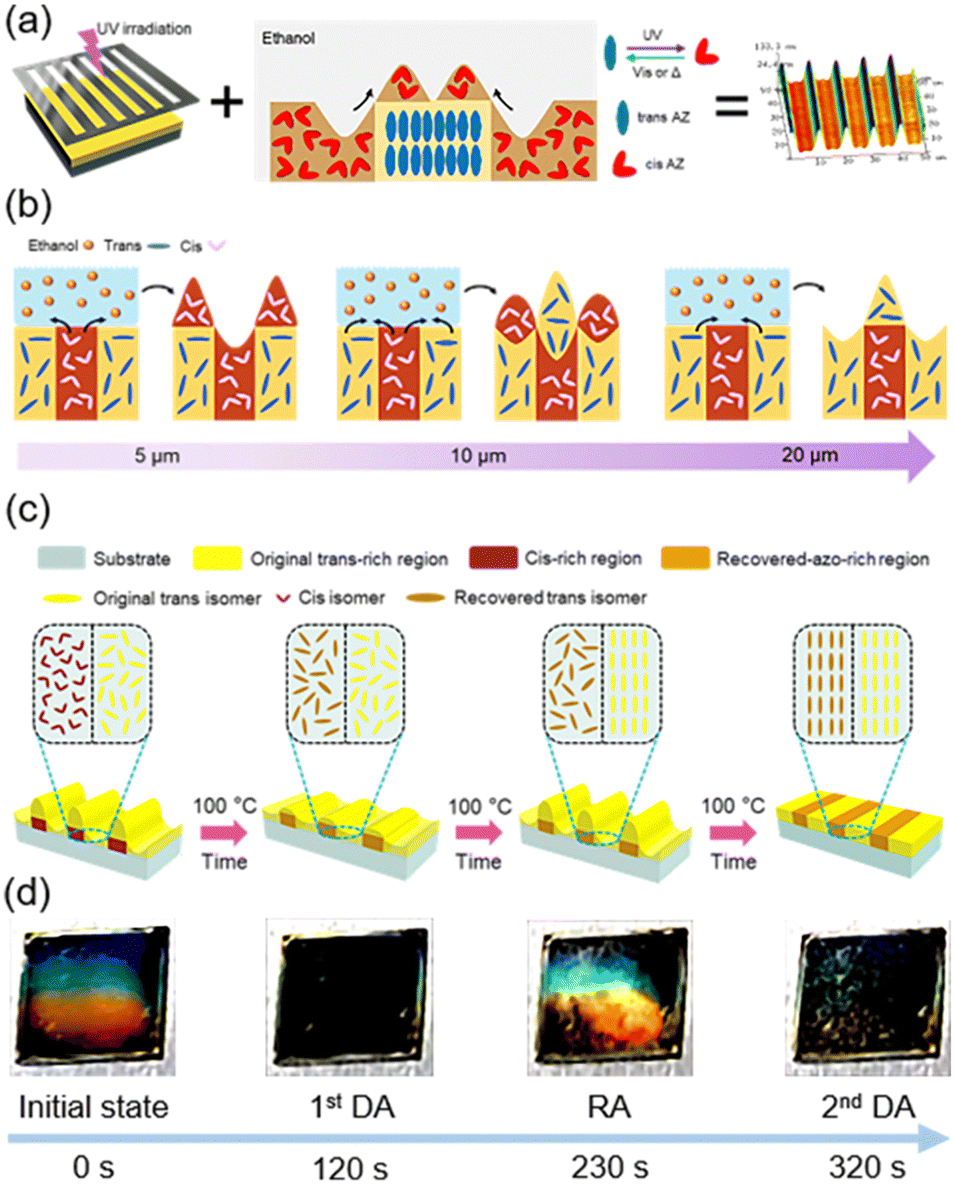 | ||
| Fig. 13 (a) Schematic illustration of the two-step method. The first step is UV-patterning, and the second step is immersion in a solvent. Reproduced from ref. 109; copyright 2023, American Chemical Society. (b) Schematic illustration of the variation in the mass transport direction of the host–guest system as the width of the photoisomerized area increases. Reproduced from ref. 110; copyright 2024, Cell Press. (c) Schematic illustration of the dynamic process upon heating at 100 °C. (d) Photographs of the variation in structural color during the whole process. The film was spin-coated on the silicon wafer with a size of 1 cm × 1 cm. Reproduced from ref. 111; copyright 2024, Wiley-VCH. | ||
Furthermore, the microstructures fabricated using the host–guest system (half-life: 7.8 h) at room temperature were responsive to thermal treatment. It is well-known that cis-AZ groups will undergo cis–trans back-isomerization upon heating and the recovered trans-AZ moieties may self-organize into the mesogenic phase.111,112 Therefore, surface morphing has been conveniently manipulated by just heating the film with periodic microstructures, exhibiting a vivid structural color (Fig. 13c and d).111 When heating the host–guest film at 100 °C, cis–trans back-isomerization occurred first, leading to the first disappearance of the structural color (1st DA) and then mesogenic assembly of the recovered AZ groups occurred due to the inherent properties at the LC phase. Since there was a difference in the time required for accomplishing the assembly between recovered AZ moieties and original trans-AZ groups, the surface-energy gradient could be reproduced and Marangoni flow would occur, resulting in the reappearance of structural color (RA). Finally, the assembly of the AZ moieties in all regions was completed, so the structural color disappeared again (2nd DA).
5. Manipulation at the macroscale
5.1. Photoinduced bending deformation
Photoinduced bending is the most frequently reported photomechanical deformation among the numerous photo-actuation devices.113 In the cross-linked LCP (CLCP) fibre system, it was reported that if the AZ group was in the cross-linker, it would bend toward the light source, as previously reported;114–118 however, if the AZ moiety was only linked in the side chain, it bent away from the light source (Fig. 14a).119 Although trans–cis isomerization occurred when exposed to UV light, no phase transition from the LC to the isotropic phase occurred upon UV irradiation. It is speculated that the photochemical reaction of AZ in the side chain was decoupled from the cross-linked network. A possible explanation for this unusual bending behaviour is the free volume theory. Photoisomerization of the AZ group usually requires more free space to occur,31,32,120–123 so the irradiated area will expand along the horizontal direction or the fibre axis direction, while the non-irradiated side remains unchangeable since there is no photoisomerization. As a result, the fibre bent away from the light source (Fig. 14a). As for the bilayer composite system shown in Fig. 8a, the result was the same: it always bent towards the Kapton layer independent of the incident direction of UV light,45 since the photoisomerization of AZ in the side chain led to a volume expansion in the LCP layer (Fig. 14b). Similarly, small-molecule crystals of AZ derivatives could bend in three different ways at different temperatures: away from the light source, towards the light source, and bidirectional motions.124 Also, polymorphic crystals with different molecular packings exhibit remarkable different photoinduced behaviours.125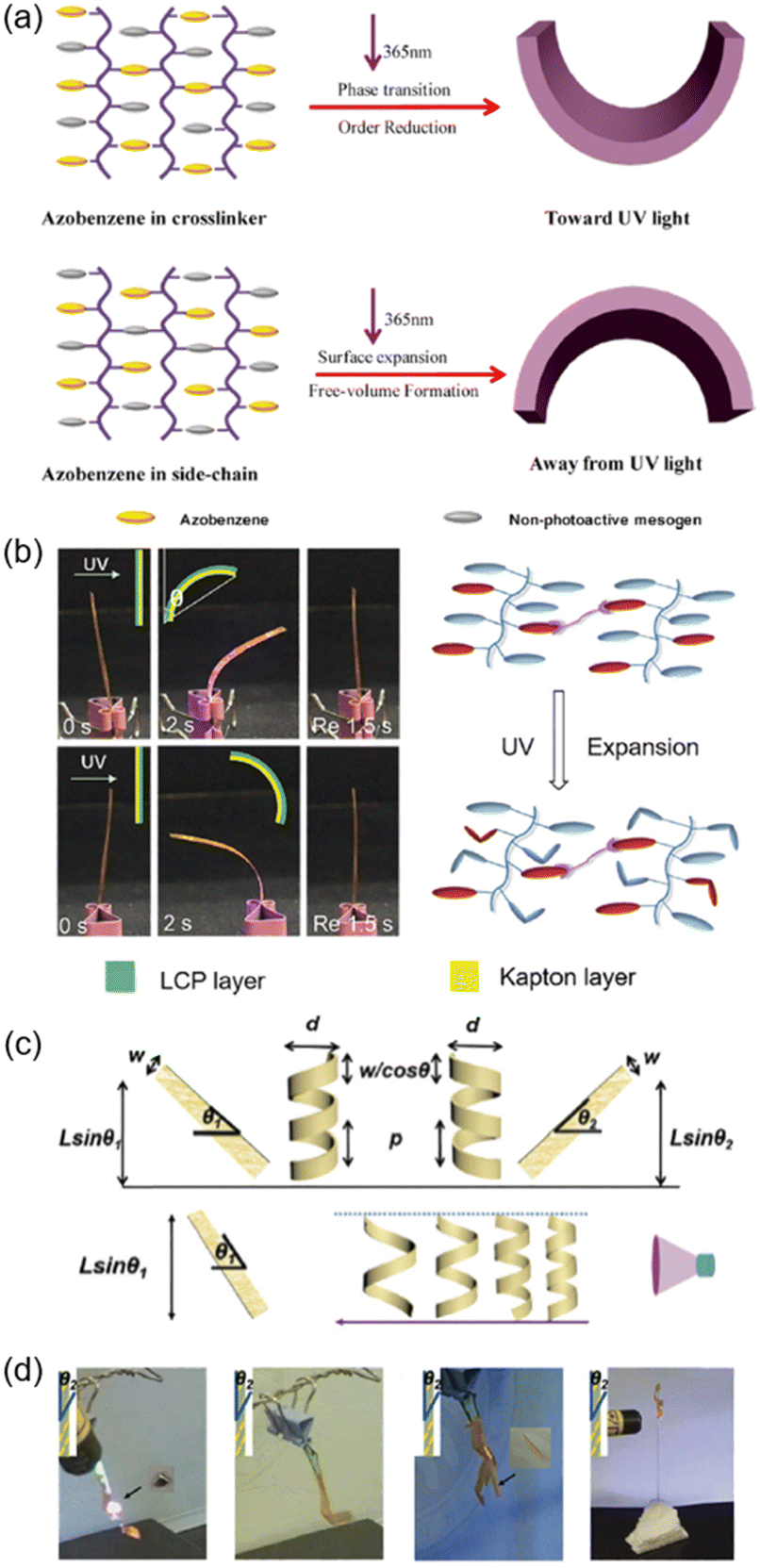 | ||
| Fig. 14 (a) Schematic illustration of different kinds of photoinduced bending that occur in the CLCP system. Reproduced from ref. 119; copyright 2017, American Chemical Society. (b) Photographs of the photoinduced bending of the LCP-Kapton bilayer composite system and the schematic illustration of its mechanism. Reproduced from ref. 45; copyright 2019, The Royal Society of Chemistry. (c) Schematic diagram of the photoinduced helices of the photo-actuators. (d) Photographs of the photo-actuators mimicking the predation of pythons. Reproduced from ref. 80; Copyright 2018, The Royal Society of Chemistry. | ||
Furthermore, if the photo-actuator was obtained by cutting from the AZ derivative/LDPE bilayer film (Fig. 7b) and there is an angle between its long axis (L) and rubbing-caused microgroove direction (M), helically twisted motions would occur (Fig. 14c and d).80 In addition, the size of the actuator and the light intensity could influence the photomechanical performance. As shown in Fig. 14c, there is a positive connection between the deformation amplitude of the actuator and the light intensity. When the amplitudes of these photoinduced helices were large enough, it could grasp objects, just like the predation of pythons. Specifically, objects with irregular shapes and different sizes such as oblate metal plates or slender wood sticks can be captured using this photo-actuator (Fig. 14d). The metal plate or wood stick was released upon removal of UV light due to the reversible deformation behaviours. Moreover, the actuator could enwind around the slender rod helically and the winding tightness could be manipulated by tuning the light intensity. This biomimetic actuator paves the way for applications in miniaturized robotics.
5.2. Light-powered self-oscillation
Oscillation is a common phenomenon in the world, such as in the heartbeat of animals, flapping of wings, waves on the water and so on. So, a photoresponsive self-oscillator can be prepared in many ways.126–128 As shown in Fig. 15a, one random copolymer containing the supramolecular HB was used as a photoresponsive layer, and the commercial Kapton was the substrate layer.129 After thermal annealing, this bilayer composite film exhibited chaotic self-oscillation when exposed to UV light (Fig. 15b). The introduction of M6BCOOH (in Fig. 15a) brought about three advantages: the acceleration of the trans–cis isomerization rate of AZs, the enhancement of the driving force for photomechanical deformation since the HB can function as the physical crosslinking sites, and the extension of the timescale of the self-oscillation behaviour due to the spatial confinement effect of acidic dimers.129 Under continuous UV illumination, the bilayer film bent away from the light source due to the photoinduced volume expansion of the LCP layer when the driving force (D) was larger than the resistance force (R) (Fig. 15c). Then, it passed the equilibrium position and reached its maximum deflection. At this time, R was larger than D, so it would recover and passed the equilibrium position again. This led to a result that D became larger than R and photoinduced bending could occur again (Fig. 15c). This continuous process would stop until the driving force provided by the LCP layer was insufficient to allow the bilayer film to bend.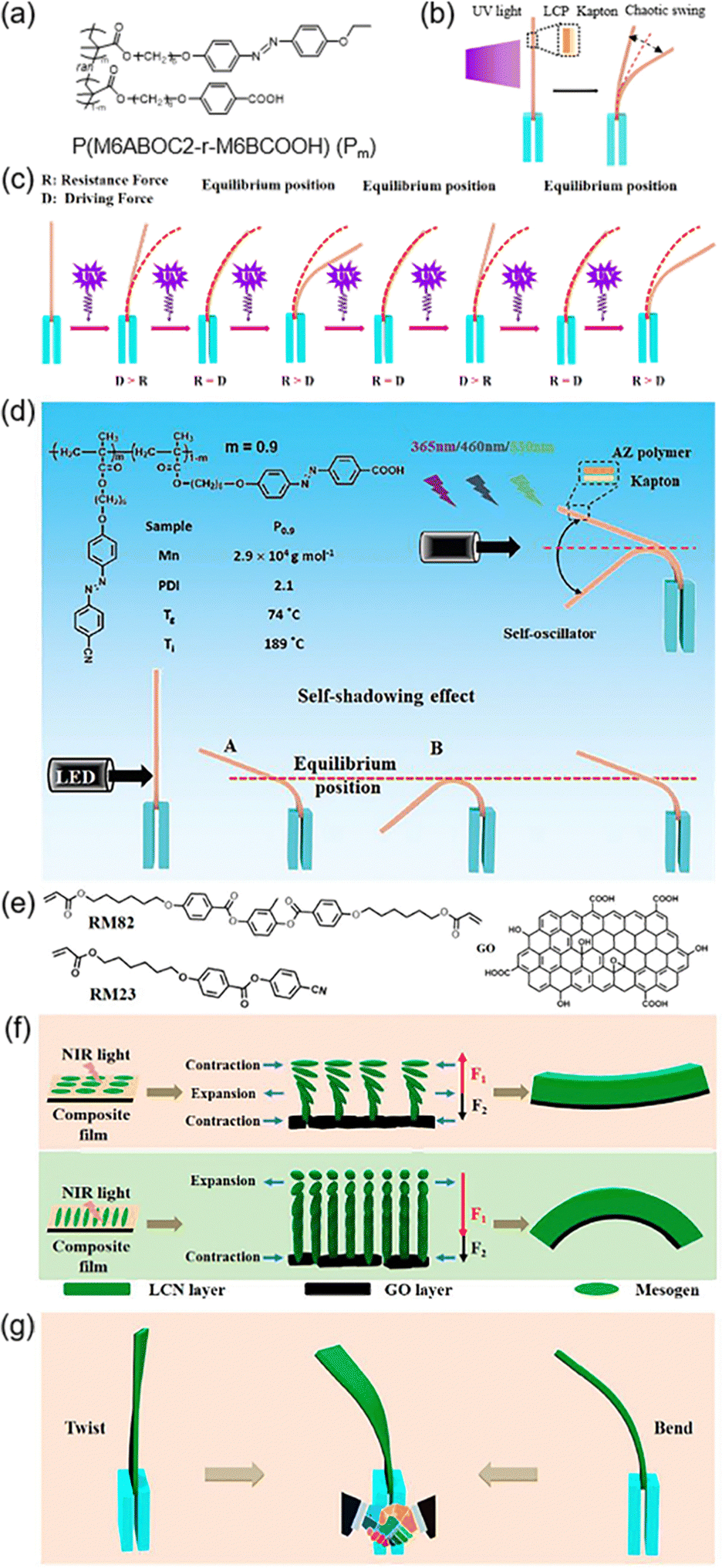 | ||
| Fig. 15 (a) Chemical structure of the random copolymer. (b) Schematic illustration of the chaotic self-oscillation when exposed to UV light. (c) Schematic illustration of the self-oscillation of the bilayer film under continuous UV illumination. Reproduced from ref. 129; copyright 2021, American Chemical Society. (d) Chemical structure of the random copolymer and a schematic illustration of the light-fueled autonomous self-oscillation. Reproduced from ref. 130; copyright 2021, The Royal Society of Chemistry.(e) Chemical structures of the cross-linker RM82, LC monomer RM23, and photothermal agent GO. (f) Schematic diagram of the deformation mechanism of the film cut along or perpendicular to the rubbing direction, respectively. (g) Schematic illustration of the deformation process under NIR light irradiation. Reproduced from ref. 131; copyright 2022, American Chemical Society. | ||
Another system with physical crosslinking sites has been designed, which is responsive to UV illumination and visible-light irradiation (Fig. 15d).130 The maximum deflection angle became smaller as the wavelength of the light increased from 365 nm to 460 nm and 530 nm,130 which is due to the combined influence of the cis-AZ content and the trans–cis dynamic process. Based on these studies, light-powered self-oscillators have been fabricated. When the actuator bent across the equilibrium position (state B in Fig. 15d), the self-shadowing effect dominated and it would recover back to receive light energy again. This repeated behaviour could occur spontaneously under consistent light irradiation and it can be manipulated by tuning the size of the oscillator, the light intensity/wavelength, the light-irradiated position and the loaded mass on the bilayer film.130
Furthermore, GO/liquid-crystalline network (LCN) composite films have been used to fabricate light-activated self-oscillators (Fig. 15e).131 Due to the mismatch of the coefficient of thermal expansion (CTE) between GO and the LCN layer and asymmetric contraction/expansion in the LCN layer, the bending direction and the deflection angle were different when changing the cutting direction upon NIR light irradiation (Fig. 15f). Moreover, an unconventional hybrid oscillation mode consisting of bending and twisting occurred during the self-oscillation process upon exposure to NIR light (Fig. 15g). These results offer the possibility to design a light-powered actuator with various oscillation modes.
5.3. Photo-driven biomimetic motions
Photo-driven biomimetic motions in water or sky have attracted much attention such as water striders and swimmers.132–137 Recently, we fabricated a transportation device based on a bilayer composite film composed of one AZ-containing LCN layer and one Kapton layer (Fig. 16a–c).44 The bilayer composite film would bend towards the Kapton layer upon UV irradiation.45 When it was placed on a liquid surface and the Kapton layer faced the liquid interface, UV irradiation on the left half would allow the film to bend downward, then a reactive force was produced to propel the swimmer moving forward (Fig. 16b). When the swimmer is away from the light source, it returns to its initial state. The flat state remained until the subsequent UV irradiation arrived by moving the light source. As a result, the swimmer would move forward like a dolphin swimming in water. In addition, the transportation device composed of a square propeller and a rectangular direction controller was designed (Fig. 16c). Since it could move forward and rotate at any angle, it was able to move in any direction on the liquid surface and then transportation of cargos was realized efficiently. This system paves the way for applications in transportation of micro-objects such as micro-chemical reactants, catalysts and micro-capsules.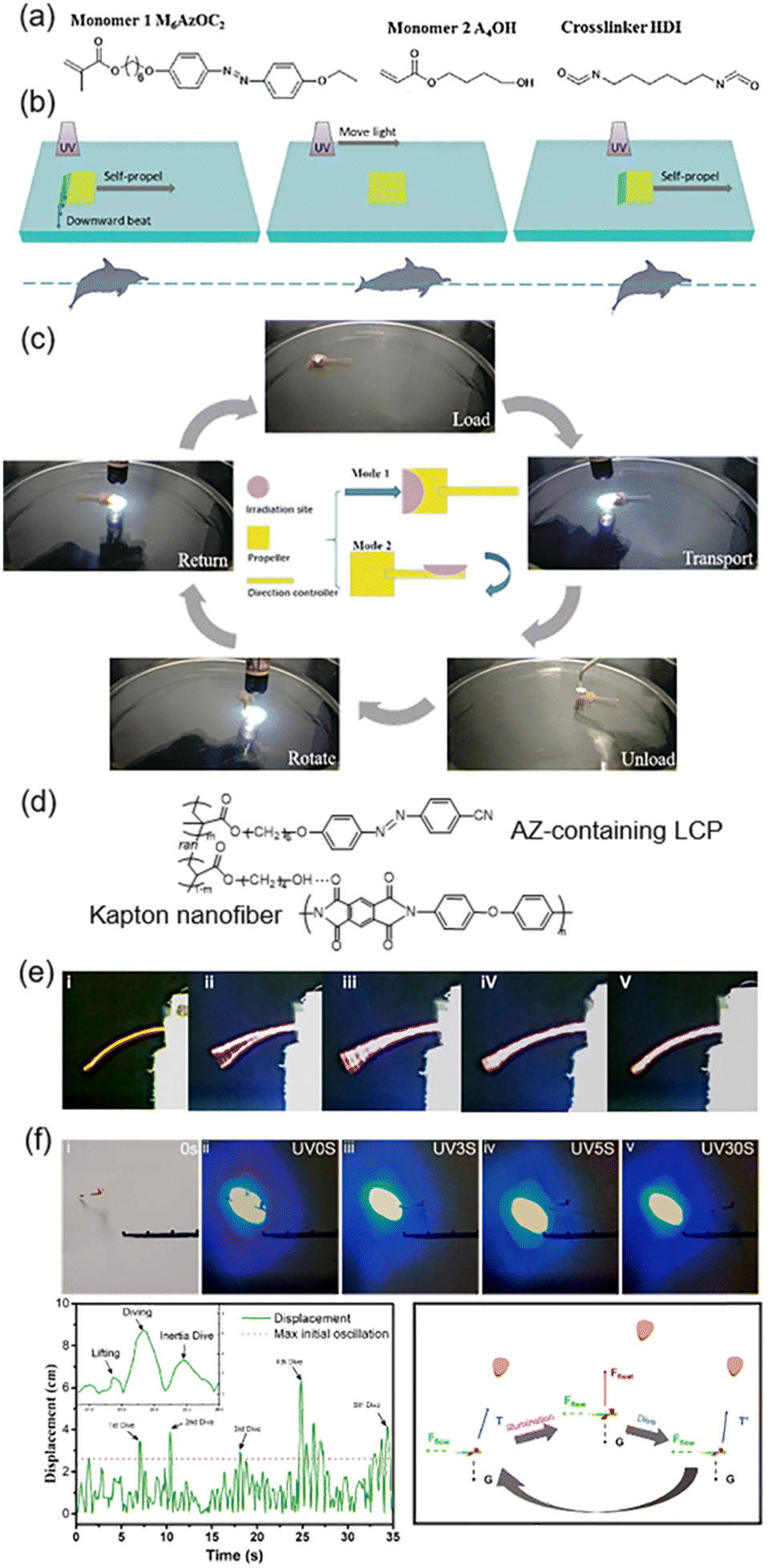 | ||
| Fig. 16 (a) Chemical structures of the materials. (b) Schematic illustration of the photo-controlled motions, which is analogous to a dolphin kick. (c) Photographs of the transportation process of a well-designed device. Reproduced from ref. 44; copyright 2019, Wiley-VCH. (d) Chemical structures of the composite system. (e) Photographs of the composite films exhibiting their flapping properties under different conditions. (f) Digital photographs, trajectory tracking curves, and schematic illustration of the light-driven flight of the artificial dragonfly device. Reproduced from ref. 138; copyright 2021, Elsevier. | ||
Furthermore, photo-actuated flight has become popular in recent years.139–141 As shown in Fig. 16d, a light-powered soft actuator was prepared based on the composite system consisting of AZ-containing LCPs and Kapton nanofibers.138 The mechanical properties were reinforced via the supramolecular HB; therefore, the oscillation induced by photoisomerization was capable of lifting a load by 40 times the weight of the film itself.138 Then, the relationship between the bending angle, vibration frequency and light intensity was explored. As shown in Fig. 16e, (i) is the original composite film without UV illumination; (ii) and (v) are in the gliding region with a low flapping frequency and a large bending angle; and (iii) and (iv) are in the flapping region with a high flapping frequency and a small bending angle. Then, the composite films were assembled into an artificial dragonfly device to demonstrate the realization of light-driven flight (Fig. 16f). The forewings and hindwings were designed to bend upwards and downwards, respectively, simulating the phase difference during the flight of natural dragonfly.138 Upon blue light (460 nm) irradiation, the artificial dragonfly flew away from the light source and five dives could be observed over 30 s (Fig. 16f). This is almost an early demonstration of a light-driven flying device.
6. Potential applications
6.1. Energy storage
The energy issue is one of the imperative problems that needs to be solved in human society. Therefore, manipulation of the storage and release of solar energy is of great importance and has been investigated extensively.142–144 Recently, advances in AZ-containing polymers for solar energy storage and release have been reviewed.145–148 Here, we focus on several typical examples, as shown in Fig. 17. It has been reported that 4-methoxyazobenzene (MO-Azo) is a photo-liquefiable molecule which can achieve solid-to-liquid transition at room temperature (Fig. 17a). Taking advantage of this feature, solid-state solar thermal fuel (STF) devices have been fabricated by thermally melting MO-Azo at 60 °C on one kind of cloth fabric (Fig. 17a).149 The homogeneous dispersion of MO-Azo in the gaps of the fabric texture enhanced the energy-storage capacity.150,151 After UV irradiation, charging of the STF device at room temperature occurred and it would release the stored energy when exposed to blue light or heating (Fig. 17a). It is worth noting that the energy density of this device is not proportional to the cis content since the energy storage (ΔHstorage) includes two parts: the isomerization energy (ΔHcis–trans) and the phase transition energy (ΔHphase transition).149 Then, blue light irradiation or heating was provided to accelerate the energy release, as the external energy input would assist cis-isomers to overcome the energy barrier (Ea), accelerating the cis–trans back-isomerization (Fig. 17b).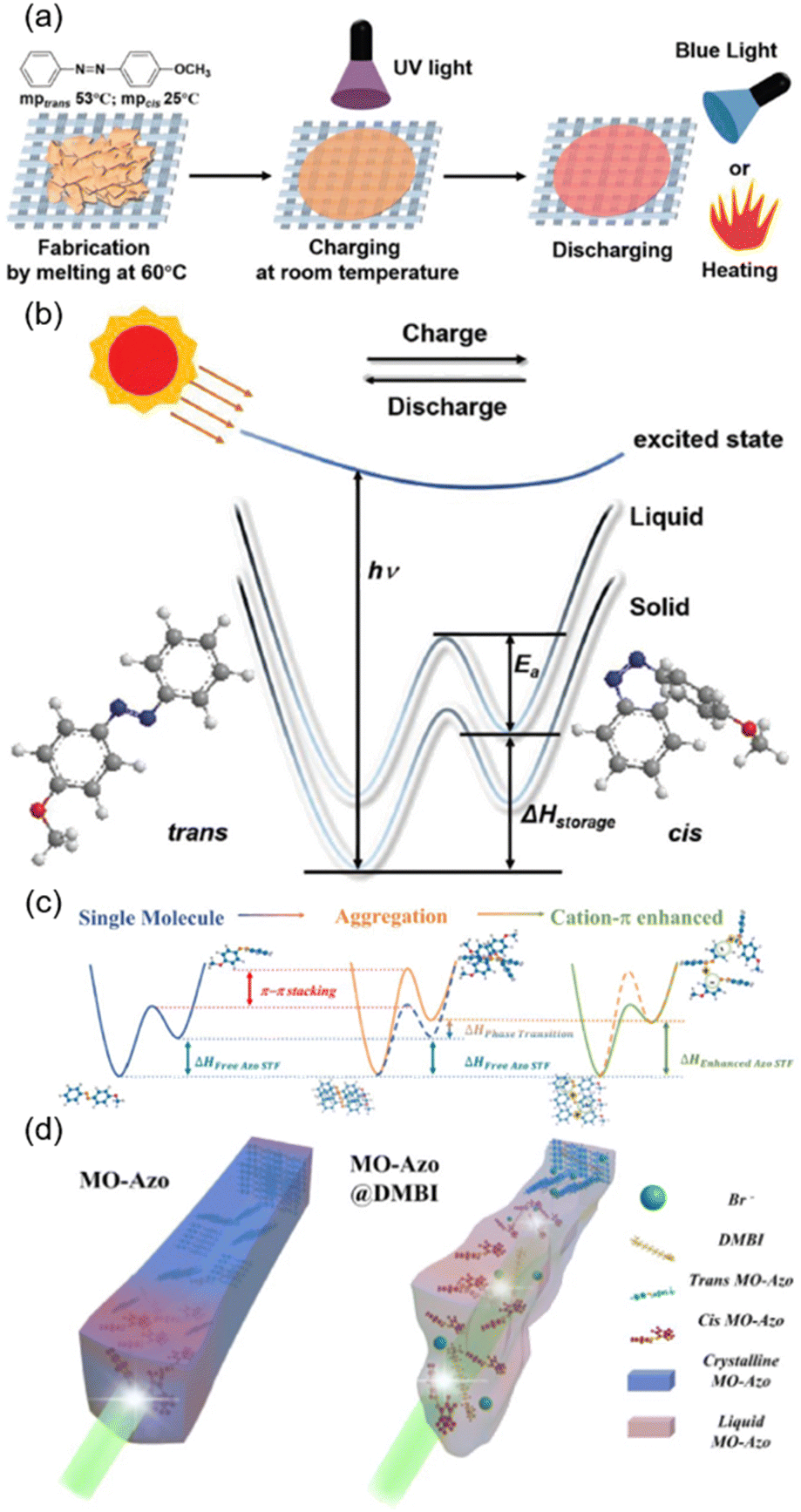 | ||
| Fig. 17 (a) Schematic illustration of the fabrication, charging and discharging processes of the STF device. (b) Mechanism of energy storage in the STF device. Reproduced from ref. 149; copyright 2019, Wiley-VCH. (c) Schematic illustration of the cation–π interaction enhanced STF at the molecular scale. (d) Schematic illustration of the cation–π interaction enhanced STF at the aggregation scale. Reproduced from ref. 60; copyright 2022, American Chemical Society. | ||
Furthermore, a supramolecular system with a cation–π interaction was designed by introducing decanoquaternary amine bromide (DMBI) into the photo-liquefiable MO-Azo, as shown in Fig. 3c.60 Improvement of the STF energy density has been proved in two aspects: the molecular scale and the aggregation scale (Fig. 17c and d). At the molecular scale, there is a competition between cation–π interaction and π–π stacking during trans–cis photoisomerization and photo-caused recovery, whether it is in the solution or just the liquid state (cis-rich state). Therefore, more MO-Azo molecules will undergo trans–cis isomerization under UV illumination even though the molar extinction coefficient of MO-Azo is decreased and the quantum efficiency of photoisomerization varies slightly (Fig. 17c).60 As for the aggregation scale, the cation–π interaction has a great impact on the crystalline structure. It is well-known that photoisomerization occurs much more easily in liquid than in a solid.60 For the pristine MO-Azo, there is a mismatch between trans–cis photoisomerization and crystal structure evolution, hindering the further photoisomerization (Fig. 17d, left). The introduction of the cation–π interaction hinders the π–π stacking of MO-Azo molecules and reduces the isomerization rate, enabling the evolution of the crystal structure to keep pace with the photoisomerization.60 As a result, more MO-Azo molecules can undergo the photoisomerization process efficiently (Fig. 17d, right). In a word, the construction of this supramolecular system opens a door for increasing the energy density of STF and achieving the balance between the molecular design and aggregation state.
Phase-change materials (PCMs) are important for energy storage due to the increase of the energy density resulting from the simultaneous absorption of ambient heat and light energy.152–156 Therefore, we proposed a method to combine one light-inactive PCM with an AZ-containing LCBC to improve the energy storage efficiently.112 First, the LCBC shown in Fig. 5a was used, where the PEO (a repeated unit of 114) used as the separated phase is a kind of PCM and the continuous phase is the AZ-containing LCP where two kinds of energies can be stored: one is arising from photoisomerization of AZ groups and the other is the latent heat of phase transition from the LC to the isotropic phase (Fig. 18a). Then, one small-molecule PEO (sPEO) was introduced into the LCBC system as the PCM, so the energy can be stored in the AZ-containing LCP block, the PEO block and the additional sPEO (Fig. 18a). The size and periodicity of MPS nanostructures would change with different addition ratios of sPEO (Fig. 18b). As the doping ratio of sPEO increased, the size of PEO nanocylinders became larger and then irregular until non-uniform structures at the macroscale appeared. Correspondingly, the crystallization point (Tc) of the composites was completely different when the doping amount of sPEO varied, as shown in Fig. 18c. A significant supercooling was observed since the Tc of the sPEO was 24 °C but the Tc of the PEO block in the cis-rich LCBC was −35 °C.112 Also, the Tc remains almost unchanged when sPEO is at a low doping amount. In this case, the nanoconfinement effect of MPS in the LCBC and the steric hindrance of cis-AZ would limit the crystallization of sPEO, resulting in a decreased Tc of sPEO even at −38 °C (Fig. 18c). Therefore, this composite can store the energy at a suitable temperature, and release at higher and lower temperatures, respectively. In addition, STF devices fabricated using this composite can be charged upon UV illumination and discharged using 460 nm blue light.157 As shown in Fig. 18d, pure cloth without this composite (top) and a charged wearable device (bottom) were wrapped around the wrist for comparison, respectively. There was almost no temperature change in the pure cloth, while for the charged device, the stored energy could be released under blue light irradiation, allowing the temperature to increase from 32.2 °C to 43.6 °C as the irradiation time increased. These results strongly demonstrate that this STF device can be used to keep warm in winter and facilitated applications in the field of wearable smart materials.112
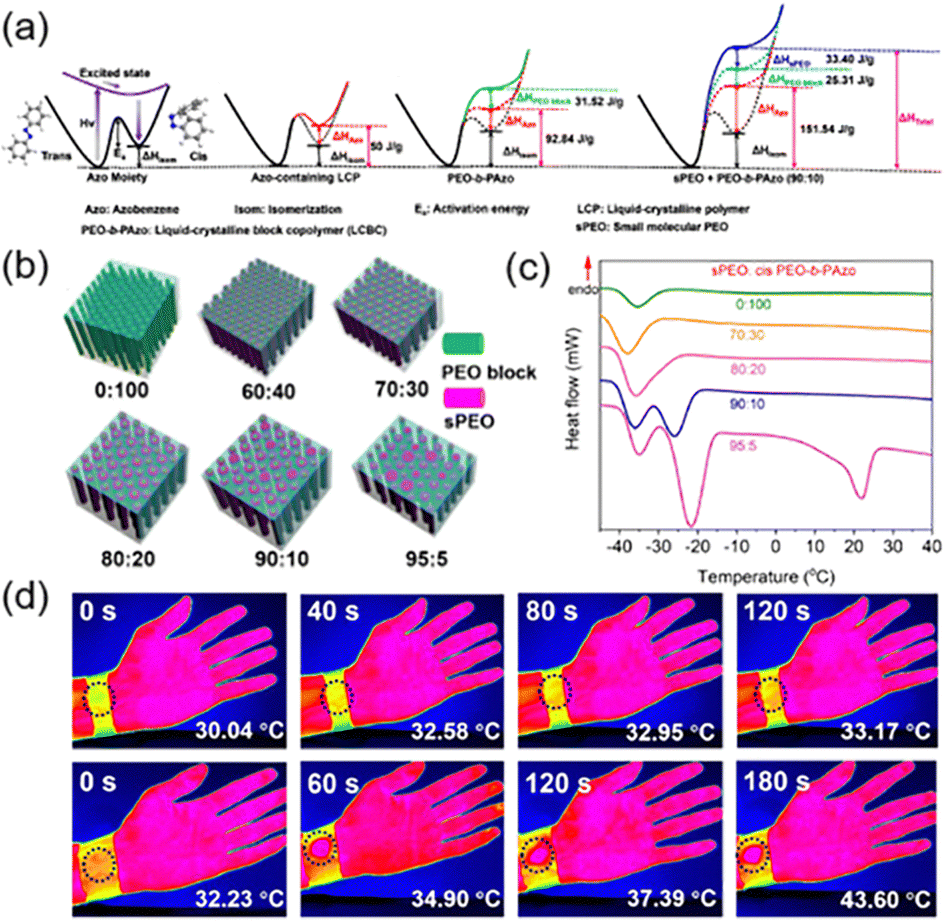 | ||
| Fig. 18 (a) Schematic illustration of the energy storage in the composite system composed of the LCBC and sPEO. (b) Schematic illustration of the composite films with different addition ratios of sPEO. (c) DSC curves of the charged composites with different doping amounts of sPEO. (d) IR thermography images of the pure cloth (top) and charged wearable device (bottom) wrapped around the wrist upon 460 nm blue light irradiation for different times. Reproduced from ref. 112; Copyright 2021, American Chemical Society. | ||
6.2. Advanced anticounterfeiting and information encryption
Nowadays, information security has become an issue of widespread concern in academics and industrial applications. To avoid the risk of information leakage and forgery, advanced anticounterfeiting materials including photonic crystals,158,159 luminescent materials,160,161 and self-erasing materials162,163 have been developed. Typically, taking advantage of the AT-NIL method and photo-switchable Tg of PM6AZC4, our group successfully fabricated clover patterns exhibiting an angle-dependent structural color (Fig. 19a),43 which has covered the whole visible-light range with good monochromaticity. In addition, both positive and negative patterns have been obtained using the same method, so it can be applied in anticounterfeiting.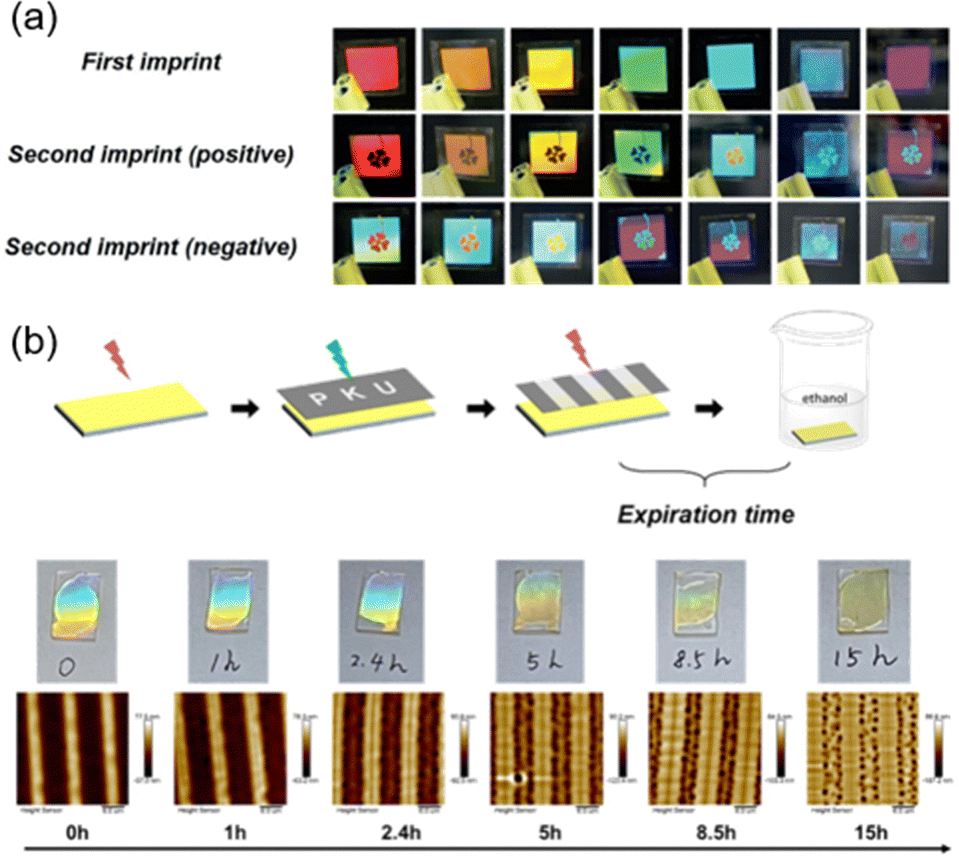 | ||
| Fig. 19 (a) Photographs of clover patterns with an angle-dependent structural color. Reproduced from ref. 43; copyright 2020, Wiley-VCH. (b) Schematic illustration, photographs, and AFM images of the microstructures on the film surface, which can serve as anticounterfeiting labels. Reproduced from ref. 109; copyright 2023, American Chemical Society. | ||
Moreover, combining the two-step method in Fig. 13a with the half-life of the cis-azopolymer, anticounterfeiting labels were prepared.109 It is obvious that the stored information is unable to be read without immersion in a solvent. Therefore, there is a maximum time interval between photo-patterning and immersion in a solvent since the cis-isomer will thermally recover to its trans state if the time interval is long enough, resulting in a little surface-energy gradient and non-occurrence of mass transport. As shown in Fig. 19b, the maximum time interval was about 8 h. If it is more than 8 h, surface morphing will not be generated, let alone the structural color, so this time interval can be called the expiration time. By designing the molecular structure of the azopolymer, the half-life can be modulated in a controlled manner, then anticounterfeiting labels with different expiration times on-demand can be fabricated.
Moreover, a multi-mode anticounterfeiting platform was designed by tailoring the bidirectional flow in the host–guest system.110 Specifically, the first mode is to determine the authenticity of the information by using two photomasks with inversed patterns, where one has alternating 5 μm transparent width and 20 μm opaque width, and the other is just the opposite. If the information is true, the bidirectional polymer flow occurs upon exposure to ethanol vapor, leading to a similar surface topography in both cases (Fig. 20a and b). However, if there is only unidirectional mass transport, the resulting two surface topographies should be reversed, suggesting that the information is false. When there is only one photomask consisting of a three-letter pattern, another anticounterfeiting mode can be applied to distinguish between two patterns that look similar under the observation of an optical microscope (Fig. 20c and d). When the unidirectional flow occurs, the obtained pattern is false information (Fig. 20c). If the host–guest ratio is suitable, bidirectional flow occurs and all the flowing mass moves to the boundary, then the height difference is only reflected in the one-dimensional (1D) boundary, not a 2D area (Fig. 20d), which is different from the traditional patterns, demonstrating the true information. The third mode is that the authenticity of the information can be identified by laser scanning confocal microscopy (LSCM) measurements (Fig. 20e). A doped red fluorescent dye could be considered as a visualization of the polymer flow in fluorescent images and readout signal intensity mapping images. If the information is true, there is little difference in fluorescence intensity between the trans-rich and cis-rich regions due to the bidirectional mass transport (Fig. 20e). Additionally, there is a sharp decrease in red fluorescence intensity at the boundary in the original film plane, since the fluorescent molecules have been delivered to the boundary along with the polymer flow (away from the original film plane). Therefore, true information is allowed to be identified using LSCM, providing opportunities for the design of numerous anticounterfeiting platforms.
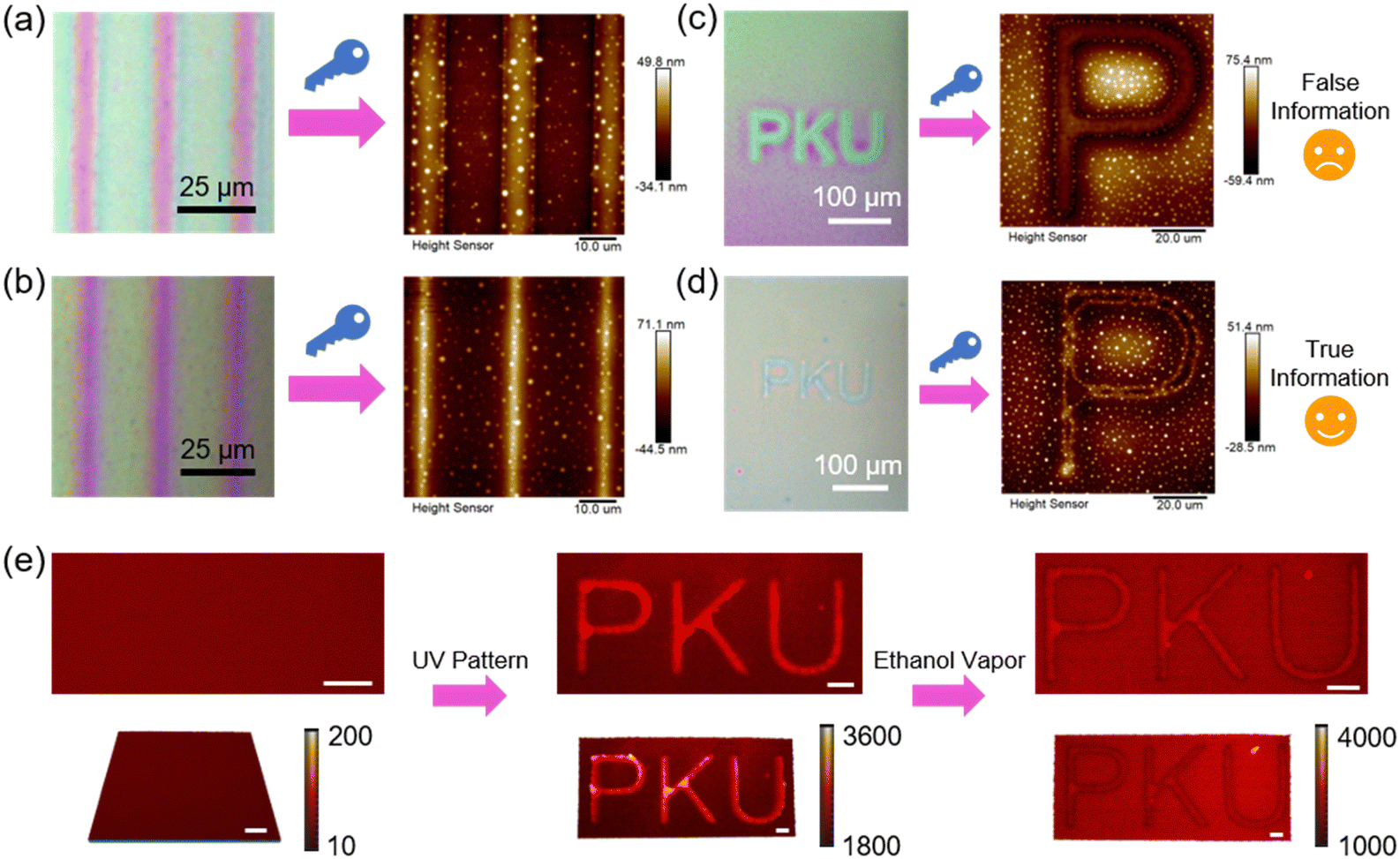 | ||
| Fig. 20 (a) Information decryption when using a photomask with a 5 μm transparent width and a 20 μm opaque width in the UV-patterning process. (b) Information decryption when using a photomask with a 5 μm opaque width and a 20 μm transparent width in the UV-patterning process. (c) False information obtained by using the photomask with a three-letter pattern. (d) True information obtained by using the photomask with a three-letter pattern. (e) Laser scanning confocal microscopy (LSCM) images and readout signal intensity mapping images of the identification process of the true information. Scale bars are all 20 μm. Reproduced from ref. 110; copyright 2024, Cell Press. | ||
Besides multi-mode static anticounterfeiting, multi-level dynamic anticounterfeiting has become popular recently.164 Our group has proposed a facile strategy to realize time-dependent anticounterfeiting and double-lock information encryption.111 Based on the dynamic process upon heating the host–guest film with periodic microstructures at 100 °C, anticounterfeiting labels with a tunable structural color were fabricated. Two pieces of information that look identical could be distinguished by observing the heating process: if the “Love” pattern disappeared and emerged again, it suggests that the information was correct (Fig. 21a), while if vanished gradually and no longer appeared, it indicates that it was false information (Fig. 21b). Also, the time-dependent multi-information display can be achieved using three clover patterns with different host–guest ratios and the dynamic processes of different three-clover-pattern combinations are distinct.111 Taking advantage of this unique feature, information at each time period can be encoded, where the displayed state is 1 and the erased state is 0. As a result, passwords can be encrypted and stored as binary numbers in this dynamic process. To decrypt the password, the whole process of the variation in three-clover-pattern combination and the decoding method should be acquired simultaneously. If different decoding methods are used, different passwords would be obtained even if the dynamic process of the three-clover-pattern combination is the same (Fig. 21c). Similarly, passwords are distinct when the dynamic processes are different but the decoding method is the same (Fig. 21d). Therefore, this host–guest system with time-dependent features can be regarded as a material with a double-lock,111 and information could only be decrypted with the correct dynamic process and correct decoding method, efficiently improving the capability of information encryption with a higher level of security.
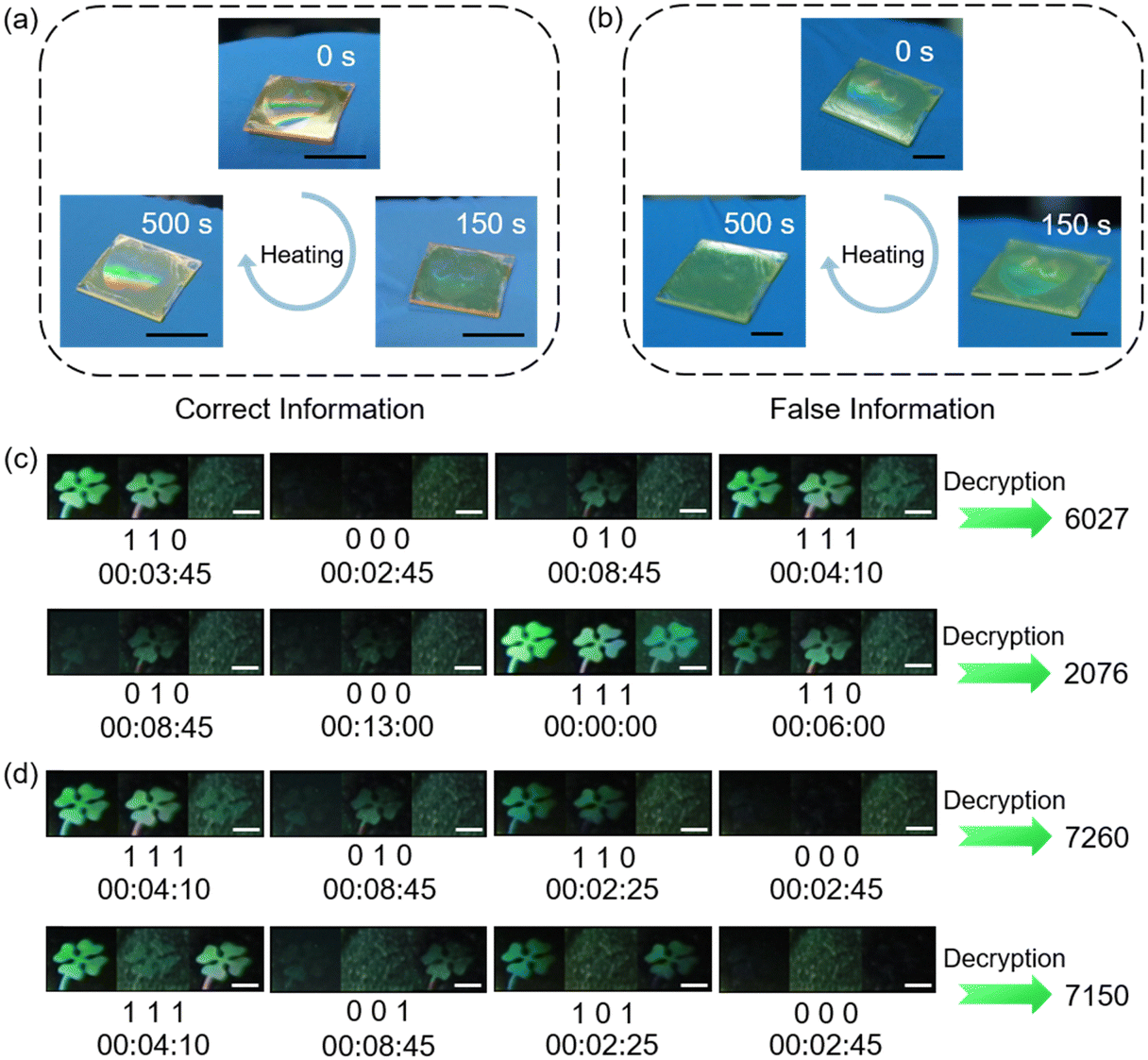 | ||
| Fig. 21 (a) Photographs of the identification process of the correct “Love” pattern in the film. Scale bars are all 1 cm. (b) Photographs of the identification process of the false “Love” pattern in the film. Scale bars are all 5 mm. (c) Decryption of two different passwords using one dynamic process and different decoding methods. Scale bars are all 2 mm. (d) Decryption of two different passwords using different dynamic processes and the same decoding method. Scale bars are all 2 mm. Reproduced from ref. 111; copyright 2024, Wiley-VCH. | ||
6.3. Switchable adhesion
Adhesives are widely used in our daily life, however, strong and ductile adhesive materials are difficult to develop. Recently, an AZ-containing siloxane-based tough adhesive was successfully prepared.165 As shown in Fig. 22a, siloxane chains were introduced into the azopolymer, leading to a lower molar extinction coefficient of the polymer and a higher flexibility of the polymer chains, enabling the adhesive to maintain a high mechanical strength and excellent ductility. Therefore, the polycarbonate (PC) ring can be reversibly opened and closed upon exposure to UV light and green light with pressure on the adhesive position, respectively (Fig. 22b).165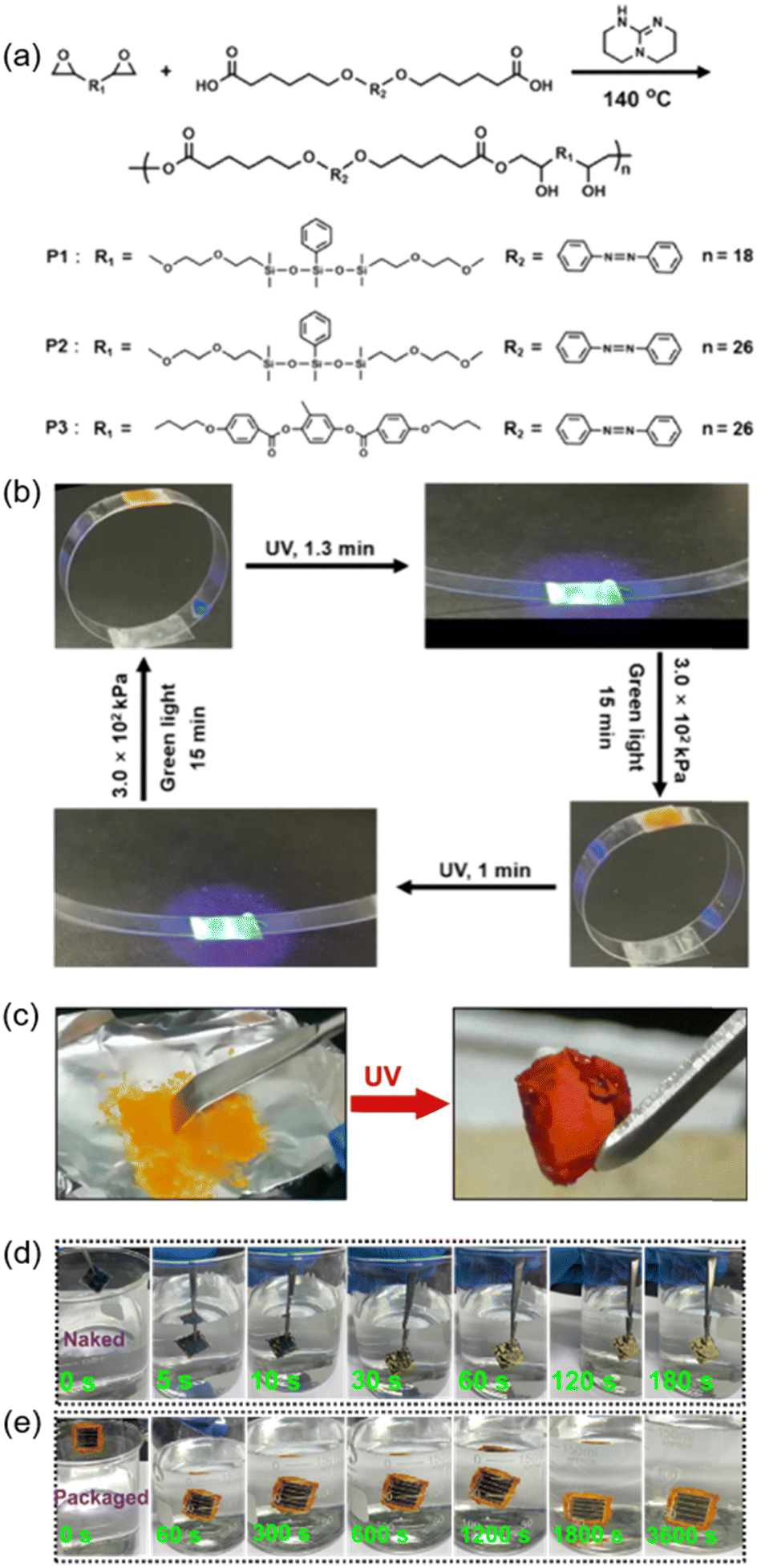 | ||
| Fig. 22 (a) Chemical structures of the siloxane-based materials. (b) Photographs exhibiting photoinduced reversible opening and closing of a ring. Reproduced from ref. 165; copyright 2021, American Chemical Society. (c) Photographs of the TBC film in trans and cis states. (d) Photographs of the naked device immersed in water for different times. (e) Photographs of the packaged device immersed in water for different times. Reproduced from ref. 76; copyright 2022, American Association for the Advancement of Science. | ||
Furthermore, the AZ-containing TBC in Fig. 6a can also be used for photo-switchable adhesion since it has two reversible states with different mechanical properties: stiffness and elasticity (Fig. 22c).76 Due to its stronger capability to achieve the adhesion of two objects through elastic deformation after UV irradiation, the cis-rich TBC film can be used for packaging electronic devices. Compared with other traditional encapsulating materials, this TBC has three advantages: first, the photoinduced transition from stiff to elastic occurs at room temperature; second, this free-standing elastomer can be applied in more situations, whether hard or soft substrates; last but not least, it can be easily removed by using ethanol. As shown in Fig. 22d, the naked device began to decompose when immersed in water for 10 s and completely decomposed at 180 s.76 In contrast, no significant discoloration was observed in the device packaged with the TBC film after immersion in water for 3600 s (Fig. 22e). These results strongly demonstrate that the photo-switchable TBC is an excellent candidate for isolating the device from water efficiently, which is suitable for the fabrication of various devices that need to work underwater.
7. Summary and outlook
In this review, we discuss the recent progress of various photoresponsive LCPs from molecular design and manipulation at multiple scales to potential applications. The intriguing properties of AZ-containing LCPs include photoisomerization, photoalignment, photoinduced phase transition, photo-caused Tg variation, molecular and supramolecular cooperative motion, and the corresponding photo-controlled mass transport, photo-tunable surface energy and light-powered actuation, providing opportunities for applications in energy storage, advanced anticounterfeiting, information encryption and switchable adhesion. Specifically, at the nanoscale, MPS nanostructures in LCBCs resulted from supramolecular self-assembly under certain conditions can be manipulated by mechanical rubbing, UV illumination, LPL alignment and doping chiral additives. This manipulation method enables the smallest nanostructures to be obtained, but it requires relatively complicated molecular design and synthesis. At the micrometre scale, hierarchical stripes, diffraction gratings, and other microstructures could be fabricated by FESA, irradiation with an interference pattern, AT-NIL, Marangoni flow, and capillary flow. The method to fabricate various microstructures is simple, but the accuracy is lower than that of MPS nanostructures in LCBCs, limiting their application fields. At the macroscale, photomechanical deformation arisen from the photoinduced phase transition or volume change enables the preparation of different kinds of biomimetic actuators. This approach provides an opportunity for generating variations visible to the naked eye, but details at the micrometre scale or the nanoscale are neglected. Therefore, photoresponsive LCPs are promising candidates for on-demand manufacturing of advanced optical devices at multiple scales.Although significant processes in this field have been made, challenges still exist and further studies are required. The current research studies on AZ-containing LCPs are limited in scalability and most of the fabricating techniques are still in the laboratory stage. In addition, the preparation of sub-10 nm nanostructures is urgently needed to be well explored, whether from molecular design or external manipulation. Moreover, taking advantage of photo-controlled mass transport directly to fabricate dynamic microstructures at room temperature is expected to be achieved. Furthermore, the stability of the cis isomer should be improved, if it can exist for a very long time, many more situations will be applicable. More importantly, the mechanism of photomechanical deformation should be investigated in depth, then actuators with multiple sophisticated motions can be designed on-demand. Expanding the range of photoinduced changes of mechanical properties is also a key point. In a word, it is imperative for researchers to focus on these issues and contribute to the development of AZ-containing LCPs by improving their performance in industrial applications.
Author contributions
Conceptualization: H. Y. funding acquisition: H. Y. investigation: Y. J. supervision: H. Y. visualization: Y. J. and H. Y. writing – original draft: Y. J. writing – review and editing: Y. J. and H. Y.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No.s 52173066, 92156011 and 51921002).Notes and references
- L. Zhang, X. Chen and Y. Hu, Small, 2024, 20, 2305185 CrossRef CAS.
- S. Wang, R. Liu, J. Li, C. Meng, J. Liu, J. Chen, P. Cheng and K. Wu, Angew. Chem., Int. Ed., 2024, 63, e202403927 CrossRef CAS PubMed.
- X. Yin, H. Li, L. Han, J. Meng, J. Lu and Q. Song, Small, 2021, 17, 2008056 CrossRef CAS PubMed.
- G. Finkelstein-Zuta, Z. A. Arnon, T. Vijayakanth, O. Messer, O. S. Lusky, A. Wagner, G. Zilberman, R. Aizen, L. Michaeli, S. Rencus-Lazar, S. Gilead, S. Shankar, M. J. Pavan, D. A. Goldstein, S. Kutchinsky, T. Ellenbogen, B. A. Palmer, A. Goldbourt, M. Sokol and E. Gazit, Nature, 2024, 630, 368–374 CrossRef CAS PubMed.
- H. Wang, B. Yuan, X. Zhu, X. Shan, S. Chen, W. Ding, Y. Cao, K. Dong, X. Zhang, R. Guo, Y. Yao, B. Wang, J. Tang and J. Liu, Sci. Adv., 2024, 10, eadp5215 CrossRef CAS PubMed.
- H. Zhang, B. Zhang, C. Cai, K. Zhang, Y. Wang, Y. Wang, Y. Yang, Y. Wu, X. Ba and R. Hoogenboom, Nat. Commun., 2024, 15, 2055 CrossRef CAS.
- Y. Ding, X. Wang, M. Tang and H. Qiu, Adv. Sci., 2022, 9, 2103833 CrossRef CAS.
- L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef CAS PubMed.
- W. Kang, Y. Tang, X. Meng, S. Lin, X. Zhang, J. Guo and Q. Li, Angew. Chem., Int. Ed., 2023, 62, e202311486 CrossRef CAS.
- L. Fang, Z. W. Lin, Y. Zhang, B. Ye, J. Li, Q. S. Ran, X. T. Wang, M. J. Yang, Z. K. Yuan, X. F. Lin, D. S. Yu, X. D. Chen and Q. Li, Angew. Chem., Int. Ed., 2024, 63, e202402349 CrossRef CAS.
- J. Bai, L. Zhang, Z. Shi and X. Jiang, Adv. Funct. Mater., 2023, 33, 2212556 CrossRef CAS.
- Y. Wang, C. Yuan, W. Huang, P.-Z. Sun, B. Liu, H. Hu, Z. Zheng, Y. Lu and Q. Li, Adv. Mater., 2023, 35, 2211521 CrossRef CAS PubMed.
- H. Lu, B. Wu, X. Le, W. Lu, Q. Yang, Q. Liu, J. Zhang and T. Chen, Adv. Funct. Mater., 2022, 32, 2206912 CrossRef CAS.
- Y. Yang, Y. Wang, M. Lin, M. Liu and C. Huang, Mater. Horiz., 2024, 11, 2180–2190 RSC.
- Y. Ji, B. Yang, F. Cai and H. Yu, Macromol. Chem. Phys., 2022, 223, 2100418 CrossRef CAS.
- X. Lv, M. Yu, W. Wang and H. Yu, Compos. Commun., 2021, 24, 100651 CrossRef.
- X. Lv, W. Wang, A. J. Clancy and H. Yu, ACS Appl. Mater. Interfaces, 2021, 13, 23030–23037 CrossRef CAS.
- X. Lv, W. Wang and H. Yu, Adv. Eng. Mater., 2019, 21, 1900977 CrossRef CAS.
- H. Shang, X. Le, M. Si, S. Wu, Y. Peng, F. Shan, S. Wu and T. Chen, Chem. Eng. J., 2022, 429, 132290 CrossRef CAS.
- J. Bai, Z. Shi and X. Jiang, Adv. Funct. Mater., 2023, 33, 2301797 CrossRef CAS.
- Z. Li, P. Liu, X. Ji, J. Gong, Y. Hu, W. Wu, X. Wang, H. Peng, R. T. K. Kwok, J. W. Y. Lam, J. Lu and B. Z. Tang, Adv. Mater., 2020, 32, 1906493 CrossRef CAS.
- J. Gao, Y. Tang, D. Martella, J. Guo, D. S. Wiersma and Q. Li, Responsive Mater., 2023, 1, e20230008 CrossRef.
- R. Zheng, Y. Wei, Z. Zhang, Z. Wang, L. Ma, Y. Wang, L. Huang and Y. Lu, Responsive Mater., 2023, 1, e20230017 CrossRef.
- J. Liu, Z. Song, L. Sun, B. Li, Y. Lu and Q. Li, Responsive Mater., 2023, 1, e20230005 CrossRef.
- S. Lin, K. G. Gutierrez-Cuevas, X. Zhang, J. Guo and Q. Li, Adv. Funct. Mater., 2021, 31, 2007957 CrossRef CAS.
- K. Luu and S. Y. Park, Macromolecules, 2023, 56, 1324–1338 CrossRef CAS.
- Z. Ouyang, Y. Yang, C. Zhang, S. Zhu, L. Qin, W. Wang, D. He, Y. Zhou, H. Luo and F. Qin, J. Mater. Chem. A, 2021, 9, 13402–13441 RSC.
- G. S. Hartley, Nature, 1937, 140, 281 CrossRef CAS.
- T. Hugel, N. B. Holland, A. Cattani, L. Moroder, M. Seitz and H. E. Gaub, Science, 2002, 296, 1103–1106 CrossRef PubMed.
- N. B. Holland, T. Hugel, G. Neuert, A. Cattani-Scholz, C. Renner, D. Oesterhelt, L. Moroder, M. Seitz and H. E. Gaub, Macromolecules, 2003, 36, 2015–2023 CrossRef CAS.
- O. M. Tanchak and C. J. Barrett, Macromolecules, 2005, 38, 10566–10570 CrossRef CAS.
- D. Q. Liu and D. J. Broer, Nat. Commun., 2015, 6, 8334 CrossRef CAS PubMed.
- G. N. Mol, K. D. Harris, C. W. M. Bastiaansen and D. J. Broer, Adv. Funct. Mater., 2005, 15, 1155–1159 CrossRef CAS.
- W. Feng, D. J. Broer and D. Q. Liu, Adv. Mater., 2018, 30, 1704970 CrossRef.
- K. D. Harris, R. Cuypers, P. Scheibe, C. L. van Oosten, C. W. M. Bastiaansen, J. Lub and D. J. Broer, J. Mater. Chem., 2005, 15, 5043–5048 RSC.
- T. Ikeda, J. Mater. Chem., 2003, 13, 2037–2057 RSC.
- C. L. van Oosten, C. W. M. Bastiaansen and D. J. Broer, Nat. Mater., 2009, 8, 677–682 CrossRef CAS PubMed.
- A. H. Gelebart, D. J. Mulder, M. Varga, A. Konya, G. Vantomme, E. W. Meijer, R. L. B. Selinger and D. J. Broer, Nature, 2017, 546, 632–636 CrossRef CAS PubMed.
- H. Yu, J. Li, T. Ikeda and T. Iyoda, Adv. Mater., 2006, 18, 2213–2215 CrossRef CAS.
- M. Komura and T. Iyoda, Macromolecules, 2007, 40, 4106–4108 CrossRef CAS.
- T. Wang, X. Li, Z. Dong, S. Huang and H. Yu, ACS Appl. Mater. Interfaces, 2017, 9, 24864–24872 CrossRef CAS.
- X. Li, B. Li, M. He, W. Wang, T. J. Wang, A. Wang, J. Yu, Z. L. Wang, S. W. Hong, M. Byun, S. L. Lin, H. F. Yu and Z. Q. Lin, ACS Appl. Mater. Interfaces, 2018, 10, 4961–4970 CrossRef CAS PubMed.
- B. Yang, F. Cai, S. Huang and H. Yu, Angew. Chem., Int. Ed., 2020, 59, 4035–4042 CrossRef CAS PubMed.
- S. Ma, X. Li, S. Huang, J. Hu and H. Yu, Angew. Chem., Int. Ed., 2019, 58, 2655–2659 CrossRef CAS PubMed.
- X. Li, S. Ma, J. Hu, Y. Ni, Z. Lin and H. Yu, J. Mater. Chem. C, 2019, 7, 622–629 RSC.
- K. Mehta, A. R. Peeketi, L. Liu, D. Broer, P. Onck and R. K. Annabattula, Appl. Phys. Rev., 2020, 7, 041306 CAS.
- W. Feng, D. Liu and D. J. Broer, Small Struct., 2021, 2, 2000107 CrossRef CAS.
- H. Yu, J. Mater. Chem. C, 2014, 2, 3047–3054 RSC.
- W. Wang, Y. Wang and H. Yang, Chem. Soc. Rev., 2016, 45, 2656–2693 RSC.
- H. Ren, D. Chen, Y. Shi, H. Yu and Z. Fu, Polym. Chem., 2015, 6, 270–277 RSC.
- D. Chen, H. Liu, T. Kobayashi and H. Yu, J. Mater. Chem., 2010, 20, 3610–3614 RSC.
- Y. Ni, X. Li, J. Hu, S. Huang and H. Yu, Chem. Mater., 2019, 31, 3388–3394 CrossRef CAS.
- H. Liu, T. Kobayashi and H. Yu, Macromol. Rapid Commun., 2011, 32, 378–383 CrossRef CAS.
- H. Yu, H. Liu and T. Kobayashi, ACS Appl. Mater. Interfaces, 2011, 3, 1333–1340 CrossRef CAS PubMed.
- P. Metrangolo, H. Neukirch, T. Pilati and G. Resnati, Acc. Chem. Res., 2005, 38, 386–395 CrossRef CAS PubMed.
- Y. Chen, H. Yu, L. Zhang, H. Yang and Y. Lu, Chem. Commun., 2014, 50, 9647–9649 RSC.
- O. S. Bushuyev, A. Tomberg, T. Friščić and C. J. Barrett, J. Am. Chem. Soc., 2013, 135, 12556–12559 CrossRef CAS PubMed.
- T. Seki, S. Nagano and M. Hara, Polymer, 2013, 54, 6053–6072 CrossRef CAS.
- T. Seki, Macromol. Rapid Commun., 2014, 35, 271–290 CrossRef CAS PubMed.
- T. Song, H. Lei, F. Cai, Y. Kang, H. Yu and L. Zhang, ACS Appl. Mater. Interfaces, 2022, 14, 1940–1949 CrossRef CAS PubMed.
- D. A. Dougherty, Science, 1996, 271, 163–168 CrossRef CAS PubMed.
- Q. Lu, D. X. Oh, Y. Lee, Y. Jho, D. S. Hwang and H. Zeng, Angew. Chem., Int. Ed., 2013, 52, 3944–3948 CrossRef CAS PubMed.
- R. MacKinnon and G. Yellen, Science, 1990, 250, 276–279 CrossRef CAS PubMed.
- E. V. Pletneva, A. T. Laederach, D. B. Fulton and N. M. Kostić, J. Am. Chem. Soc., 2001, 123, 6232–6245 CrossRef CAS PubMed.
- Q. Lu, D. S. Hwang, Y. Liu and H. Zeng, Biomaterials, 2012, 33, 1903–1911 CrossRef CAS PubMed.
- W. Zhou, T. Kobayashi, H. Zhu and H. Yu, Chem. Commun., 2011, 47, 12768–12770 RSC.
- W. Zhou and H. Yu, ACS Appl. Mater. Interfaces, 2012, 4, 2154–2159 CrossRef CAS PubMed.
- W. Zhou and H. Yu, RSC Adv., 2013, 3, 22155–22159 RSC.
- H. Zhou, C. Xue, P. Weis, Y. Suzuki, S. Huang, K. Koynov, G. K. Auernhammer, R. Berger, H. J. Butt and S. Wu, Nat. Chem., 2017, 9, 145–151 CrossRef CAS PubMed.
- Y. Liu, S. Liang, C. Yuan, A. Best, M. Kappl, K. Koynov, H. J. Butt and S. Wu, Adv. Funct. Mater., 2021, 31, 2103908 CrossRef CAS.
- W. Xu, C. Liu, S. Liang, D. Zhang, Y. Liu and S. Wu, Adv. Mater., 2022, 34, 2202150 CrossRef CAS PubMed.
- H. Yu, T. Iyoda and T. Ikeda, J. Am. Chem. Soc., 2006, 128, 11010–11011 CrossRef CAS PubMed.
- C. C. Honeker and E. L. Thomas, Chem. Mater., 1996, 8, 1702–1714 CrossRef CAS.
- Y. Takahashi, Y. Song, N. Nemoto, A. Takano, Y. Akazawa and Y. Matsushita, Macromolecules, 2005, 38, 9724–9729 CrossRef CAS.
- M. Hayashi, S. Matsushima, A. Noro and Y. Matsushita, Macromolecules, 2015, 48, 421–431 CrossRef CAS.
- F. Cai, B. Yang, X. Lv, W. Feng and H. Yu, Sci. Adv., 2022, 8, eabo1626 CrossRef CAS PubMed.
- Z. Cheng, T. Wang, X. Li, Y. Zhang and H. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 27494–27501 CrossRef CAS PubMed.
- P. Zhang, F. Cai, G. Wang and H. Yu, Chin. J. Polym. Sci., 2022, 40, 166–174 CrossRef CAS.
- P. Zhang, B. Wu, S. Huang, F. Cai, G. Wang and H. Yu, Polymer, 2019, 178, 121644 CrossRef CAS.
- J. Hu, X. Li, Y. Ni, S. Ma and H. Yu, J. Mater. Chem. C, 2018, 6, 10815–10821 RSC.
- Z. Liu, R. Tang, D. Xu, J. Liu and H. Yu, Macromol. Rapid Commun., 2015, 36, 1171–1176 CrossRef CAS PubMed.
- Y. Chen, S. Huang, T. Wang, Z. Dong and H. Yu, Macromolecules, 2019, 52, 1892–1898 CrossRef CAS.
- Y. Chen, S. Huang, T. Wang and H. Yu, Macromolecules, 2020, 53, 1486–1493 CrossRef CAS.
- H. S. Suh, D. H. Kim, P. Moni, S. Xiong, L. E. Ocola, N. J. Zaluzec, K. K. Gleason and P. F. Nealey, Nat. Nanotechnol., 2017, 12, 575–581 CrossRef CAS PubMed.
- R. Milani, N. Houbenov, F. Fernandez-Palacio, G. Cavallo, A. Luzio, J. Haataja, G. Giancane, M. Saccone, A. Priimagi, P. Metrangolo and O. Ikkala, Chem, 2017, 2, 417–426 CAS.
- S. Huang, L. Pang, Y. Chen, L. Zhou, S. Fang and H. Yu, Macromol. Rapid Commun., 2018, 39, 1700783 CrossRef PubMed.
- S. Huang, Y. Chen, S. Ma and H. Yu, Angew. Chem., Int. Ed., 2018, 57, 12524–12528 CrossRef CAS PubMed.
- S. Huang, H. Yu and Q. Li, Adv. Sci., 2021, 8, 2002132 CrossRef PubMed.
- W. Zhang, M. Fujiki and X. Zhu, Chem. – Eur. J., 2011, 17, 10628–10635 CrossRef CAS PubMed.
- D. Yang, C. Liu, L. Zhang and M. Liu, Chem. Commun., 2014, 50, 12688–12690 RSC.
- S. R. Nam, H. Y. Lee and J. I. Hong, Chem. Eur. J., 2008, 14, 6040–6043 CrossRef CAS PubMed.
- L. Yao and J. J. Watkins, ACS Nano, 2013, 7, 1513–1523 CrossRef CAS PubMed.
- S. Guo, X. Liang, C. Zhang, M. Chen, C. Shen, L. Zhang, X. Yuan, B. He and H. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 2942–2947 CrossRef CAS PubMed.
- W. Han, M. He, M. Byun, B. Li and Z. Lin, Angew. Chem., Int. Ed., 2013, 52, 2564–2568 CrossRef CAS PubMed.
- W. Han and Z. Lin, Angew. Chem., Int. Ed., 2012, 51, 1534–1546 CrossRef CAS PubMed.
- J. Xu, J. Xia, S. W. Hong, Z. Lin, F. Qiu and Y. Yang, Phys. Rev. Lett., 2006, 96, 066104 CrossRef PubMed.
- S. I. Na, S. S. Kim, J. Jo, S. H. Oh, J. Kim and D. Y. Kim, Adv. Funct. Mater., 2008, 18, 3956–3963 CrossRef CAS.
- J. H. Park, K. J. Park, T. Jiang, Q. Sun, J. H. Huh, Z. L. Wang, S. Lee and J. H. Cho, Nano Energy, 2017, 38, 412–418 CrossRef CAS.
- C. Rianna, A. Calabuig, M. Ventre, S. Cavalli, V. Pagliarulo, S. Grilli, P. Ferraro and P. A. Netti, ACS Appl. Mater. Interfaces, 2015, 7, 16984–16991 CrossRef CAS PubMed.
- C. Rianna, L. Rossano, R. H. Kollarigowda, F. Formiggini, S. Cavalli, M. Ventre and P. A. Netti, Adv. Funct. Mater., 2016, 26, 7572–7580 CrossRef CAS.
- H. F. Yu, K. Okano, A. Shishido, T. Ikeda, K. Kamata, M. Komura and T. Iyoda, Adv. Mater., 2005, 17, 2184–2188 CrossRef CAS.
- C. B. Kim, J. C. Wistrom, H. Ha, S. X. Zhou, R. Katsumata, A. R. Jones, D. W. Janes, K. M. Miller and C. J. Ellison, Macromolecules, 2016, 49, 7069–7076 CrossRef CAS.
- A. R. Jones, C. B. Kim, S. X. Zhou, H. Ha, R. Katsumata, G. Blachut, R. T. Bonnecaze and C. J. Ellison, Macromolecules, 2017, 50, 4588–4596 CrossRef CAS.
- I. Kitamura, K. Oishi, M. Hara, S. Nagano and T. Seki, Sci. Rep., 2019, 9, 2556 CrossRef PubMed.
- I. Kitamura, K. Kato, R. B. Berk, T. Nakai, M. Hara, S. Nagano and T. Seki, Sci. Rep., 2020, 10, 12664 CrossRef CAS PubMed.
- I. Kitamura, M. Hara, S. Nagano and T. Seki, Mol. Cryst. Liq. Cryst., 2021, 727, 52–64 CrossRef CAS.
- L. E. Scriven and C. V. Sternling, Nature, 1960, 187, 186–188 CrossRef.
- C. V. Sternling and L. E. Scriven, AIChE J., 1959, 5, 514–523 CrossRef CAS.
- B. Yang, Y. Ji, F. Cai and H. Yu, ACS Appl. Mater. Interfaces, 2023, 15, 23804–23812 CrossRef CAS PubMed.
- Y. Ji, B. Yang, F. Cai, T. Song and H. Yu, iScience, 2024, 27, 108790 CrossRef PubMed.
- Y. Ji, T. Song and H. Yu, Angew. Chem., Int. Ed., 2024, 63, e202401208 CrossRef CAS PubMed.
- F. Cai, T. Song, B. Yang, X. Lv, L. Zhang and H. Yu, Chem. Mater., 2021, 33, 9750–9759 CrossRef CAS.
- J. Hu, M. Yu, M. Wang, K. L. Choy and H. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 12951–12963 CrossRef CAS PubMed.
- T. Ikeda, M. Nakano, Y. Yu, O. Tsutsumi and A. Kanazawa, Adv. Mater., 2003, 15, 201–205 CrossRef CAS.
- H. Yu and T. Ikeda, Adv. Mater., 2011, 23, 2149–2180 CrossRef CAS PubMed.
- T. Ube, K. Kawasaki and T. Ikeda, Adv. Mater., 2016, 28, 8212–8217 CrossRef CAS PubMed.
- T. Yoshino, M. Kondo, J. Mamiya, M. Kinoshita, Y. Yu and T. Ikeda, Adv. Mater., 2010, 22, 1361–1363 CrossRef CAS.
- Y. Zhang, J. Xu, F. Cheng, R. Yin, C. C. Yen and Y. Yu, J. Mater. Chem., 2010, 20, 7123–7130 RSC.
- Z. Cheng, S. Ma, Y. Zhang, S. Huang, Y. Chen and H. Yu, Macromolecules, 2017, 50, 8317–8324 CrossRef CAS.
- D. Q. Liu, C. W. M. Bastiaansen, J. M. J. den Toonder and D. J. Broer, Angew. Chem., Int. Ed., 2012, 51, 892–896 CrossRef CAS PubMed.
- A. Priimagi, A. Shimamura, M. Kondo, T. Hiraoka, S. Kubo, J. Mamiya, M. Kinoshita, T. Ikeda and A. Shishido, ACS Macro Lett., 2012, 1, 96–99 CrossRef CAS PubMed.
- K. G. Yager, O. M. Tanchak, C. Godbout, H. Fritzsche and C. J. Barrett, Macromolecules, 2006, 39, 9311–9319 CrossRef CAS.
- D. Q. Liu, C. W. M. Bastiaansen, J. M. J. den Toonder and D. J. Broer, Macromolecules, 2012, 45, 8005–8012 CrossRef CAS.
- Y. Hao, S. Huang, Y. Guo, L. Zhou, H. Hao, C. J. Barrett and H. Yu, J. Mater. Chem. C, 2019, 7, 503–508 RSC.
- Y. Hao, L. Gao, X. Zhang, R. Wei, T. Wang, N. Wang, X. Huang, H. Yu and H. Hao, J. Mater. Chem. C, 2021, 9, 8294–8301 RSC.
- K. Kumar, C. Knie, D. Bléger, M. A. Peletier, H. Friedrich, S. Hecht, D. J. Broer, M. G. Debije and A. P. H. J. Schenning, Nat. Commun., 2016, 7, 11975 CrossRef CAS PubMed.
- S. Serak, N. Tabiryan, R. Vergara, T. J. White, R. A. Vaia and T. J. Bunning, Soft Matter, 2010, 6, 779–783 RSC.
- T. J. White, N. V. Tabiryan, S. V. Serak, U. A. Hrozhyk, V. P. Tondiglia, H. Koerner, R. A. Vaia and T. J. Bunning, Soft Matter, 2008, 4, 1796–1798 RSC.
- J. Wang, S. Huang, Y. Zhang, J. Liu, M. Yu and H. Yu, ACS Appl. Mater. Interfaces, 2021, 13, 6585–6596 CrossRef CAS PubMed.
- J. Wang, T. Song, Y. Zhang, J. Liu, M. Yu and H. Yu, J. Mater. Chem. C, 2021, 9, 12573–12580 RSC.
- J. Wang, B. Yang, M. Yu and H. Yu, ACS Appl. Mater. Interfaces, 2022, 14, 15632–15640 CrossRef CAS PubMed.
- D. Okawa, S. J. Pastine, A. Zettl and J. M. J. Fréchet, J. Am. Chem. Soc., 2009, 131, 5396–5398 CrossRef CAS PubMed.
- C. Maggi, F. Saglimbeni, M. Dipalo, F. De Angelis and R. Di Leonardo, Nat. Commun., 2015, 6, 7855 CrossRef CAS PubMed.
- Z. Wu, T. Si, W. Gao, X. Lin, J. Wang and Q. He, Small, 2016, 12, 577–582 CrossRef CAS PubMed.
- W. Wang, Y. Liu, Y. Liu, B. Han, H. Wang, D. Han, J. Wang, Y. Zhang and H. Sun, Adv. Funct. Mater., 2017, 27, 1702946 CrossRef.
- W. Jiang, D. Niu, H. Liu, C. Wang, T. Zhao, L. Yin, Y. Shi, B. Chen, Y. Ding and B. Lu, Adv. Funct. Mater., 2014, 24, 7598–7604 CrossRef CAS.
- H. Tian, Z. Wang, Y. Chen, J. Shao, T. Gao and S. Cai, ACS Appl. Mater. Interfaces, 2018, 10, 8307–8316 CrossRef CAS PubMed.
- T. Song, H. Lei, A. J. Clancy, S. Ma, H. Yu and L. Zhang, Nano Energy, 2021, 87, 106207 CrossRef CAS.
- Y. Chen, C. Valenzuela, X. Zhang, X. Yang, L. Wang and W. Feng, Nat. Commun., 2023, 14, 3036 CrossRef CAS PubMed.
- J. Yang, H. Zhang, A. Berdin, W. Hu and H. Zeng, Adv. Sci., 2023, 10, 2206752 CrossRef CAS PubMed.
- D. Wang, Z. Chen, M. Li, Z. Hou, C. Zhan, Q. Zheng, D. Wang, X. Wang, M. Cheng, W. Hu, B. Dong, F. Shi and M. Sitti, Nat. Commun., 2023, 14, 5070 CrossRef CAS PubMed.
- X. Xu, B. Wu, P. Zhang, H. Yu and G. Wang, Sol. RRL, 2020, 4, 1900422 CrossRef CAS.
- X. Xu, P. Zhang, B. Wu, Y. Xing, K. Shi, W. Fang, H. Yu and G. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 50135–50142 CrossRef CAS PubMed.
- X. Xu, Y. Xing, Y. Yin, W. Fang, B. Wu, P. Bei, J. Feng, H. Yu, G. Wang and W. Li, Chem. Eng. J., 2023, 466, 143175 CrossRef CAS.
- X. Xu, J. Feng, W. Li, G. Wang, W. Feng and H. Yu, Prog. Polym. Sci., 2024, 149, 101782 CrossRef CAS.
- R. J. Salthouse and K. Moth-Poulsen, J. Mater. Chem. A, 2024, 12, 3180–3208 RSC.
- Z. Wang, P. Erhart, T. Li, Z.-Y. Zhang, D. Sampedro, Z. Hu, H. A. Wegner, O. Brummel, J. Libuda, M. B. Nielsen and K. Moth-Poulsen, Joule, 2021, 5, 3116–3136 CrossRef CAS.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- J. Hu, S. Huang, M. Yu and H. Yu, Adv. Energy Mater., 2019, 9, 1901363 CrossRef CAS.
- D. Zhitomirsky, E. Cho and J. C. Grossman, Adv. Energy Mater., 2016, 6, 1502006 CrossRef.
- E. N. Cho, D. Zhitomirsky, G. G. D. Han, Y. Liu and J. C. Grossman, ACS Appl. Mater. Interfaces, 2017, 9, 8679–8687 CrossRef CAS PubMed.
- H. Liu, J. Tang, L. Dong, H. Wang, T. Xu, W. Gao, F. Zhai, Y. Feng and W. Feng, Adv. Funct. Mater., 2021, 31, 2008496 CrossRef CAS.
- G. G. D. Han, H. Li and J. C. Grossman, Nat. Commun., 2017, 8, 1446 CrossRef.
- G. G. D. Han, J. H. Deru, E. N. Cho and J. C. Grossman, Chem. Commun., 2018, 54, 10722–10725 RSC.
- N. Zhang, Y. Yuan, X. Cao, Y. Du, Z. Zhang and Y. Gui, Adv. Eng. Mater., 2018, 20, 1700753 CrossRef.
- K. Pielichowska and K. Pielichowski, Prog. Mater. Sci., 2014, 65, 67–123 CrossRef CAS.
- L. A. Weinstein, J. Loomis, B. Bhatia, D. M. Bierman, E. N. Wang and G. Chen, Chem. Rev., 2015, 115, 12797–12838 CrossRef CAS PubMed.
- K. Zhong, J. Li, L. Liu, S. Van Cleuvenbergen, K. Song and K. Clays, Adv. Mater., 2018, 30, 1707246 CrossRef PubMed.
- X. Lai, Q. Ren, F. Vogelbacher, W. E. I. Sha, X. Hou, X. Yao, Y. Song and M. Li, Adv. Mater., 2022, 34, 2107243 CrossRef CAS PubMed.
- X. Fu, S. Fu, Q. Lu, J. Zhang, P. Wan, J. Liu, Y. Zhang, C.-H. Chen, W. Li, H. Wang and Q. Mei, Nat. Commun., 2022, 13, 4741 CrossRef CAS PubMed.
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang, Y. Zhou, W. Yao, Y. Zhang, H. Zhang, Z. Zhang, X. Hang, Z. An, X. Liu and W. Huang, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed.
- P. K. Kundu, D. Samanta, R. Leizrowice, B. Margulis, H. Zhao, M. Börner, T. Udayabhaskararao, D. Manna and R. Klajn, Nat. Chem., 2015, 7, 646–652 CrossRef CAS PubMed.
- Z. Chen, Y. Chen, Y. Guo, Z. Yang, H. Li and H. Liu, Adv. Funct. Mater., 2022, 32, 2201009 CrossRef CAS.
- Y. Shen, X. Le, Y. Wu and T. Chen, Chem. Soc. Rev., 2024, 53, 606–623 RSC.
- P. Zhang, F. Cai, W. Wang, G. Wang and H. Yu, ACS Appl. Polym. Mater., 2021, 3, 2325–2329 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |