
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comprehensive investigation into thermoelectric properties of PEDOT:PSS/Bi0.5Sb1.5Te3 composites†
Saeed
Masoumi
 a,
Kuanysh
Zhussupbekov
b,
Nadezda
Prochukhan
c,
Michael A.
Morris
c and
Amir
Pakdel
*a
a,
Kuanysh
Zhussupbekov
b,
Nadezda
Prochukhan
c,
Michael A.
Morris
c and
Amir
Pakdel
*a
aDepartment of Mechanical, Manufacturing, and Biomedical Engineering, Trinity College Dublin, The University of Dublin, D02PN40 Dublin, Ireland. E-mail: pakdela@tcd.ie
bSchool of Physics, CRANN and AMBER Research Centres, Trinity College Dublin, The University of Dublin, D02PN40 Dublin, Ireland
cSchool of Chemistry, CRANN and AMBER Research Centres, Trinity College Dublin, The University of Dublin, D02PN40 Dublin, Ireland
First published on 30th July 2024
Abstract
The demand for sustainable power sources for wearable electronic devices has spurred exploration into novel energy harvesting technologies. Among these, flexible thermoelectric (TE) materials and generators (TEGs) show promise for transforming body heat into electrical power. For this purpose, organic and polymeric semiconducting materials, such as poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS), have been at the forefront of research recently due to their flexibility, nontoxicity, easy processability, high electrical conductivity, and low thermal conductivity. However, hybridization with inorganic materials, optimization of doping levels, and structural modifications to enhance their TE conversion efficiency are still ongoing areas of investigation, and a comprehensive study encompassing all these approaches is lacking. This research presents the first comprehensive and encyclopedic study of the TE properties of PEDOT:PSS thin films hybridized with Bi0.5Sb1.5Te3 particles to explore the relationship between particle size/concentration, chemical post-treatment strategies, and the resulting microstructural, morphological, and TE properties. Initially, PEDOT:PSS was hybridized with Bi0.5Sb1.5Te3 particles with a wide range of size (0.3–1.4 μm) and concentration (5–60 wt%), and the microstructure and TE properties of the resulting composite films were systematically studied. The composite with 40 wt% Bi0.5Sb1.5Te3 particles exhibited the highest power factor at room temperature, was chosen as the optimal sample, and underwent various single and sequential chemical post-treatments using secondary dopants (DMSO, EG, H2SO4) and a dedoping agent (NaOH). While single post-treatment with 0.5 M H2SO4 could increase the power factor up to 240 times compared to the pristine composite, a sequential pos-treatment with 0.5 M H2SO4 and 0.1 M NaOH resulted in an 864-fold increase in the power factor at room temperature. Furthermore, the temperature-dependent electrical conductivity and Seebeck coefficient of the composite thin films were explored in each case (between 300 K and 375 K) to determine the dominant charge transport mechanism. It was revealed that the charge carrier transport mechanism was three-dimensional variable range hopping in pristine composite samples; however, metallic conduction was observed in the samples after sequential chemical post-treatment. The findings of this paper make it a thorough reference for understanding the TE properties of inorganic/polymeric composites, and offer a detailed perspective on strategies to optimize their power factor for applications in flexible TEGs.
1 Introduction
Over the last few decades, there has been great progress in the evolution of wearable electronic devices that are tailored for integration with the human body.1 These portable devices commonly rely on lightweight power sources, typically batteries. However, the batteries require regular recharging from power outlets and also increase the weight and size of the product considerably.2,3 Consequently, the pursuit of lightweight, adaptable, portable, and eco-friendly power alternatives to these wearables has become imperative. Energy harvesting technologies, with the ability to capture and convert surrounding energies (e.g. solar, thermal, and mechanical) into electrical power, represent promising solutions for powering wearable devices.4 One such technology relies on the Seebeck effect to convert a temperature difference into electricity using a thermoelectric generator (TEG).5 A TEG is composed of p-type and n-type semiconducting materials, whose TE conversion efficiency at any temperature is closely related to a dimensionless figure of merit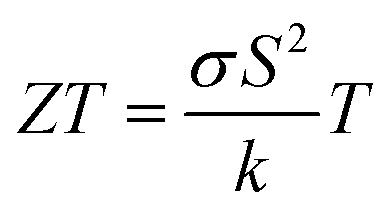 , where σ is the electrical conductivity, S is the Seebeck coefficient, k is the thermal conductivity, and T is the absolute temperature. The ZT equation dictates that simultaneous high values of S and σ and low values of k are required to achieve a high conversion efficiency. The numerator in the ZT equation is called the TE power factor, and is a measure of the material's capacity to efficiently convert thermal gradients into electrical power. In order to capture thermal energy from the human body, TEGs need to be made of flexible materials.6 They should also be durable, lightweight, cost-effective, and safe.7
, where σ is the electrical conductivity, S is the Seebeck coefficient, k is the thermal conductivity, and T is the absolute temperature. The ZT equation dictates that simultaneous high values of S and σ and low values of k are required to achieve a high conversion efficiency. The numerator in the ZT equation is called the TE power factor, and is a measure of the material's capacity to efficiently convert thermal gradients into electrical power. In order to capture thermal energy from the human body, TEGs need to be made of flexible materials.6 They should also be durable, lightweight, cost-effective, and safe.7
Over the past two decades, significant advancements have been made in the performance of inorganic TE materials, including chalcogenides, skutterudite, half-Heusler, Zintl phases, and oxides. Recently, state-of-the-art TE materials such as GeTe,8 SnSe,9 and Cu2Se10 have demonstrated promising results, with ZT values exceeding 2.4. However, ensuring consistent reproducibility of these outcomes remains a challenge, and integrating these new materials effectively into practical devices is still difficult.11 For near-room-temperature applications, Bi2Te3 alloys have been the most promising materials, offering high ZT values for both n- and p-type TE materials. By alloying Bi2Te3 with Sb2Te3 and Bi2Se3, optimal electrical conductivity and Seebeck coefficient levels can be achieved, effectively reducing lattice thermal conductivity.12 The most frequently used p- and n-type compositions are Bi0.5Sb1.5Te313 and Bi2Te2.7Se0.3,14 respectively. However, the scarcity of tellurium (Te) element has emerged as a challenge; thus, alternative alloys and compounds such as Mg3Sb2/MgAgSb15 and Mg3Bi216 have gained much attention as new Te-free high-ZT materials. Nevertheless, the application of such inorganic TE materials in flexible TEGs is hindered by high costs, complex fabrication processes, and mechanical rigidity.17–19
An alternate strategy is to use organic semiconducting materials in TEGs, which offer flexibility, intrinsically low thermal conductivity, but a moderate electrical conductivity and Seebeck coefficient.20 These organic TE materials include conducting polymers, coordination polymers, carbon-based nanomaterials, small molecules, and single molecules.21 Among them, conducting polymers such as poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) have demonstrated superior TE performance at room temperature, especially when hybridized with inorganic TE materials.22,23 Hybridization enhances the electrical conductivity and Seebeck coefficient of PEDOT:PSS by increasing the charge carrier concentration and filtering low-energy charge carriers at the interfaces, respectively. Examples include incorporation of various concentrations of Bi2Te324–26 and Bi0.4Te3Sb1.627 microparticles, Bi0.5Sb1.5Te3 nanosheets,28,29 and Bi2Te3 nanowires30 into the PEDOT:PSS matrix. However, there is a lack of a comprehensive study on the size effect of these inorganic particles on the TE properties of the composites.
Another strategy to enhance the electrical conductivity of PEDOT:PSS is by secondary doping through a post-treatment with chemical agents. These include (a) organic polar solvents, e.g. dimethyl sulfoxide (DMSO), dimethylacetamide (DMAC), and ethylene glycol (EG), (b) acids, e.g. sulfuric acid (H2SO4),31 and (c) small molecules, e.g. 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO–OH).32 Such post-treatment processes increase the electrical conductivity due to segregation and removal of excess PSS particles, as well as conformational changes in PEDOT.31 Alternatively, enhancement of the Seebeck coefficient in PEDOT:PSS films can be achieved by chemical dedoping and lowering the charge carrier concentration through the transition of PEDOT chains from bipolaron to polaron and polaron to neutral states.21 The common reducing agents for this purpose are NaOH, hydrazine, sodium borohydride (NaBH4), L-ascorbic acid (LAA), and ammonium formate (AF).33
Wu et al.34 reported an increase in the electrical conductivity of PEDOT:PSS fibres to 4464 S cm−1via post-treating with concentrated H2SO4, which partially removed excessive PSS, leading to a PF of 80.8 μW mK−2. Zhang et al.35 prepared PEDOT:PSS/single-walled carbon nanotube (SWCNT) composites and optimized the TE properties through pre-treatment with DMSO and post-treatment with NaBH4. As a result, a high σ of 1718 S cm−1 and a large S of 49 μV K−1 were obtained. Lim et al.36 developed a PEDOT:PSS/Bi0.5Sb1.5Te3 nanorod composite through a two-step reduction process, followed by a DMSO post-treatment, and achieved a PF of 308 μW mK−2 at room temperature. Kim et al.37 prepared a PEDOT:PSS/Bi2Te3 nanowire composite and after modifying the interfacial energy barrier through polar solvent vapor annealing (PSVA) achieved a PF of 223 μW mK−2 at room temperature. Wang et al.38 optimized interfacial carrier transports in a PEDOT:PSS/Bi0.5Sb1.5Te3 composite by coating the Bi0.5Sb1.5Te3 fillers with a highly conductive CuTe layer, followed by DMSO and H2SO4 post-treatments, resulting in a PF of 312 μW mK−2 at room temperature. However, in each of these studies a composite with a single concentration of inorganic particles was prepared, and the effects of filler concentration and particle size were not investigated.
The most effective approach to improve both the electrical conductivity and the Seebeck coefficient in PEDOT:PSS is sequential post-treatment, which involves applying both secondary doping and chemical dedoping treatments sequentially to achieve a combined effect.39 Luo et al.40 reported a two-step post-treatment, in which a PEDOT:PSS film was first treated by a polar solvent such as DMSO, and then reduced by a reducing agent such as 1-ethyl-3-methylimidazolium tetrafluoroborate (EMIMBF4). Similarly, Fan et al.39 improved the TE properties of PEDOT:PSS thin films through sequential post-treatments with H2SO4 and NaOH. In some cases, sequential post-treatment involving both secondary doping and chemical dedoping agents can be conducted in a one-step process by mixing the chemical solutions. For instance, Liu et al.41 proposed a single-step post-treatment method to enhance the PF of PEDOT:PSS films. They utilized a water-based solution comprising DMAC, deionized water, and LAA. Employing an 80 vol% DMAC solution notably increased the room-temperature electrical conductivity of the films from 5 to 964 S cm−1. Additionally, treatment with 0.5 mol L−1 LAA further elevated the Seebeck coefficient from 18.7 to 25 μV K−1, resulting in a substantially improved PF of 55.3 μW mK−2. However, these studies used PEDOT:PSS thin films without any inorganic fillers. Consequently, there is a lack of comprehensive research on investigating the effect of inorganic filler concentration and particle size followed by sequential post-treatment on the TE properties of composite thin films.
Generally, organic TE materials have inherently low thermal conductivity, typically falling between 0.1 W m−1 K−1 and 1 W m−1 K−1.42 Similarly, the thermal conductivity of conducting polymers ranges between 0.03 W m−1 K−1 and 0.6 W m−1 K−1.43,44 In particular, PEDOT:PSS thin films exhibit thermal conductivity in the range of 0.2–0.4 W m−1 K−1 due to the presence of defects such as polymer chain ends, entanglement, different orientations, voids, and impurities.40,45–49 The thermal conductivity of conducting polymers is predominantly governed by phonon transport, and these defects act as phonon scattering sites for heat transfer.40,45–49 Consequently, the thermal conductivity of PEDOT:PSS is inherently low and much less affected by doping/dedoping processes in comparison with electrical conductivity. Therefore, the power factor has become the dominant measure for TE performance of such materials in the literature.50 In this regard, the main research focus in the TE field has been the enhancing of both σ and S to maximize the power factor.
In this work, a thorough study on the microstructural and TE properties of PEDOT:PSS/Bi0.5Sb1.5Te3 thin film composites was performed, where the thin films had a wide range of filler size and concentration, and underwent various single and sequential post-treatment processes. Bi0.5Sb1.5Te3 (hereon referred to as BST) particles of nano to micro size were prepared by ball milling and sequential centrifugation. Then, the effect of BST particle concentration and size on the surface morphology and TE properties of the pristine thin film composites were investigated. The maximum PF for pristine PEDOT:PSS/BST thin films was obtained in samples with an average BST particle size of 1.389 μm and a concentration of 40 wt%. Given that the Seebeck coefficient and electrical conductivity in a matrix/filler system can be explained using either series or parallel connected models, both models were applied to the pristine thin film composites in this study. The results demonstrated that the electrical properties of the samples closely align with the series connected model. The temperature dependency of the electrical conductivity and Seebeck coefficient in the pristine thin film composites suggested that their dominant charge charrier transport mechanism was 3D variable range hopping (VRH). Later, the composite films with the optimum particle size and concentration were subjected to single and sequential post-treatments with secondary dopants such as dimethyl sulfoxide (DMSO), ethylene glycol (EG), and sulfuric acid (H2SO4) and chemical dedoping agents such as sodium hydroxide (NaOH). Single post-treatment with 0.5 M H2SO4 boosted the power factor 240 times and the sequential post-treatment with 0.5 M H2SO4 and 0.1 M NaOH improved the PF 864 times as compared to the pristine thin film. The temperature dependency of electrical conductivity and Seebeck coefficient in post-treated samples showed a metallic transport in the charge carriers, in contrast with the 3D VRH mechanism in pristine samples.
2 Experimental
2.1 Materials
A highly conductive PEDOT:PSS suspension (1.0 wt% in H2O, high-conductivity grade), dimethyl sulfoxide (DMSO; 99.9%), ethylene glycol (EG; 99.8%), sulfuric acid (H2SO4; 95–97%), sodium hydroxide (NaOH; ≥98%), hydrochloric acid (HCl; ≥37%), ethanol (99.99%), and silver paste (1.59 μΩ cm, 20 °C) were purchased from Sigma-Aldrich. Bi0.5Sb1.5Te3 (BST) ingot was purchased from Thermonamic Electronics (Jiangxi) Corp., Ltd. Glass and quartz microscope slides were purchased from VWR International LLC. and Präzisions Glas & Optik GmbH, respectively. Nichrome wire was purchased from RS PRO.2.2 Preparation of BST alloy powders
The micro to nano range particle size of BST powders was fabricated in three steps: crushing and grinding the BST ingot, ball milling (Retch, Planetary Ball Mill, PM 100), and centrifugation (Eppendorf, Centrifuge 5804). Ball milling was performed using 1200 zirconium oxide balls with a diameter of 5 mm, at 600 rpm, for 20 h with 15-minute intervals to avoid excessive heating of the powders. The jar capacity was 250 mL, and 190 g of BST powder was used as the feed material for ball milling. Then, a suspension of the ball-milled BST powder was prepared with a concentration of 0.05 g mL−1 in ethanol, followed by sonication (Elma Schmidbauer GmbH, TI-H-10) for 4 hours. Then, four different sets of sequential centrifugations were performed with different speeds: Set1 (500 rpm, 1000 rpm, and 5000 rpm), Set2 (500 rpm, 750 rpm, 1000 rpm, and 2000 rpm), Set3 (250 rpm, 500 rpm, and 8000 rpm), and Set4 (2000 rpm, 5000 rpm, and 9000 rpm), each for 2 minutes. This process aimed to separate the particle sizes. The Set2 centrifugation process was proved most efficient for separating particles with sizes ranging from nm to μm. Subsequently, the powders were rinsed with an HCl solution (5% HCl and 95% DI water) followed by drying at a low temperature to avoid oxidation.2.3 Preparation of PEDOT:PSS/BST thin films
PEDOT:PSS/BST composite thin films, containing 0–60 wt% BST, were fabricated following a procedure outlined in the ESI,† Fig. S1. The BST powder used in these composite thin films was prepared by ball milling and centrifugation at 500 rpm, resulting in an average particle size of 1.389 μm, followed by HCl rinsing. Various amounts of the BST powder were added to 1 mL of the PEDOT:PSS suspension in DI water, followed by sonication for 1 hour. This ensured that the BST particles were evenly dispersed throughout the suspension. To validate the long-term stability of the PEDOT:PSS/BST suspension, aging tests were conducted, as shown in the ESI,† Fig. S1b and c. The thin films were fabricated on 1.5 cm × 2.5 cm glass substrates by spin coating 200 μL of the suspension at 3000 rpm for 30 seconds. It should be noted that the suspension volume was optimized to ensure full surface coverage on the glass substrate, as shown in the ESI,† Fig. S2a and b. The fabricated films were baked on a hot plate at 120 °C for 20 minutes. The thin film samples were then put in contact with silver paste and Ni–Cr wire for electrical characterization followed by baking on a hot plate at 120 °C for 1 hour. Similarly, another set of PEDOT:PSS/BST thin films was fabricated using the optimum concentration of BST powders produced by sequential centrifugation at 500 rpm, 750 rpm, 1000 rpm, and 2000 rpm (Set2).2.4 Post-treatment of PEDOT:PSS/BST thin films
Different sets of PEDOT:PSS/BST thin films underwent single post-treatment with either DMSO, EG, H2SO4 or NaOH, as described in the ESI,† Fig. S3. First, thin films containing 40 wt% BST were fabricated as described in Section 2.3. For the DMSO post-treatment, 200 μL solutions with different concentrations of DMSO in DI water (0–100 vol%) were drop-casted onto the films. The films were left at room temperature for 30 minutes, followed by baking at 120 °C for 30 minutes, rinsing with DI water three times, and baking at 120 °C for 10 minutes. The final step involved creating electrical contacts with the samples for electrical characterization, using Ni–Cr wires and silver paste. The post-treatment with EG (0–100 vol%) followed a similar procedure to the DMSO post-treatment. However, for post-treatment with H2SO4, PEDOT:PSS/BST thin films were placed on a hot plate at 160 °C. Then, 200 μL of H2SO4 solutions in DI water with various concentrations (0–1.5 M) were drop-casted onto the films, followed by drying at 160 °C for 1 hour. In the case of NaOH, solutions with concentrations of 0–5 M in DI water were prepared in beakers. Then, the thin films were immersed in the solutions for 5 seconds. The rinsing, baking, and contacting procedures of H2SO4 and NaOH post-treated samples were similar to those for DMSO and EG treatments. The process of rinsing composite thin films with DI water is schematically illustrated in the ESI,† Fig. S4a. The rinsing process involved sequential immersion of thin films into three beakers containing DI water, using tweezers. This process was performed gently and slowly, keeping the substrate and film in a horizontal position at all times to minimize potential BST particle loss during washing.Besides the single post-treatment procedures, sequential post-treatments were performed on the composite thin films by repeating the post-treatment with DMSO several times, as shown in the ESI,† Fig. S5a. This was repeated 1 to 4 times, using the optimum concentration of DMSO. The subsequent steps in the sequential post-treatment followed a procedure similar to the single post-treatment.
Then, another type of sequential post-treatment was performed on the thin film composites through post-treating with secondary dopants (DMSO, EG, and H2SO4) at an optimum concentration and number of post-treatments followed by post-treating with chemical dedoping agent (NaOH) at different concentrations from 0 M to 2 M, as described in the ESI,† Fig. S5b. The optimum concentration of each chemical agent achieved during single post-treatment was used for maximizing the power factor in sequential post-treatments as well. Also, the number of post-treatments with secondary dopants matched those with DMSO.
2.5 Materials characterization
The surface morphologies of the thin films were analysed using atomic force microscopy (AFM) (Park Systems, XE7 and Asylum MFP-3D microscope) and field emission scanning electron microscopy (FE-SEM) (Carl Ziess ultra). The AFM utilized a silicon microcantilever probe with an ambient force constant of 42 N m−1, operating in noncontact mode. A high-resolution FE-SEM with a secondary electron detector, operating at an accelerating voltage of 5 kV, was used to acquire images of the BST nanoparticles and PEDOT:PSS/BST composites. X-Ray diffraction (XRD) spectra were obtained using a PanAlytical X'Pert Pro diffractometer with Cu Kα radiation (1.5406 Å). Small-angle X-ray scattering (SAXS) and wide-angle X-ray scattering (WAXS) were performed using a BRUKER N8 Horizon with a 2-dimensional VÅNTEC-500 detector for SAXS and a Vantec-1: 1D-linear detector for WAXS. The film thicknesses were measured using AFM, as schematically illustrated in Fig. S6 in the ESI,† using typical step-height profiles within a defined area of 20 μm × 20 μm.The absorption spectra of the films on a quartz substrate were obtained using a PerkinElmer Lambda 1050 spectrometer, ranging from 190 to 1600 nm, with a step size of 3 nm. A purpose-built automated system was used for measuring the Seebeck coefficient and electrical conductivity of the samples.51–53 Two independently controlled microheaters were used for providing the desired temperature difference along the samples. Two K-type thermocouples were placed on top of the sample at the cold and hot ends for measuring the temperatures. The electrical resistance and Seebeck voltage were measured by obtaining the current–voltage characteristic of the samples. An interface circuit and graphical user interface were developed to obtain the current–voltage characteristic, measure temperature differences along the sample, measure the Seebeck voltage and electrical resistance, and control the entire measurement process. Morphological and thermoelectric characterization was performed on all samples to enhance the validity of the experimental data in this study. The error bars in all graphs represent the range between minimum and maximum values of the data relative to the average value.
3 Results and discussion
3.1 Characterization of BST alloy powders
Fig. 1a shows an SEM micrograph and the particle size distribution of BST powder after ball milling. The average particle size of the powder after ball milling was 0.99 μm. As the produced powder by ball milling contained a mixture of nano and micro-size particles, centrifugation was applied to separate them. The SEM micrograph, particle distributions, and average particle size of the powders are illustrated in Fig. 1b–f. An average particle size of 1.389 μm, 0.792 μm, 429 nm, and 316 nm was obtained for the centrifuging speeds of 500 rpm, 750 rpm, 1000 rpm, and 2000 rpm, respectively. This sequential centrifugation experiment resulted in particle size separation from the micro to nano range, paving the way for studying the effect of particle size on the TE properties of the composites in the next stage of research. As mentioned in Section 2.2, additional sets of sequential centrifugations were performed with different speeds, and their particle size analysis is presented in the ESI,† Fig. S7–S9.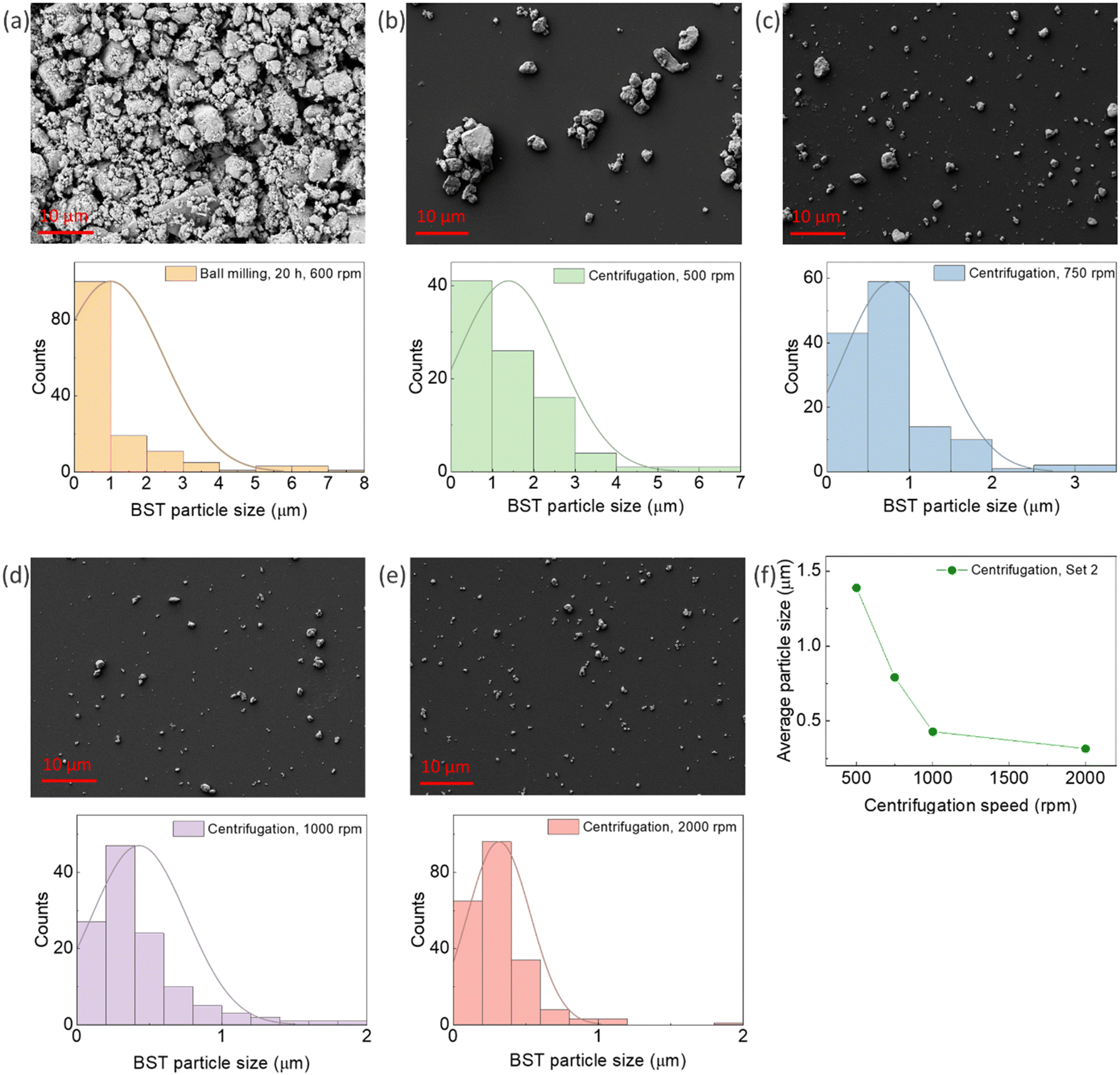 | ||
| Fig. 1 SEM micrograph and particle size distribution of BST powder produced by (a) ball milling for 20 h, 600 rpm, and Set2 centrifugation at four speeds sequentially: (b) 500 rpm, (c) 750 rpm, (d) 1000 rpm, and (e) 2000 rpm. (f) The variation in average particle size with centrifugation speed. | ||
3.2 Morphological and thermoelectric properties of pristine PEDOT:PSS/BST composite thin films
It is known that PEDOT:PSS films exhibit a low Seebeck coefficient, typically in the range of 15–30 μV K−1.33 The combination of a low Seebeck coefficient and high electrical resistance in a sample contributes to reduced accuracy in electrical measurements.51–53 Hence, determining the appropriate amount of DI water in a PEDOT:PSS suspension for preparing a thin film is crucial. Various pristine PEDOT:PSS films were fabricated by diluting the PEDOT:PSS stock solution and DI water. The fabricated samples and their resistance at room temperature are illustrated in the ESI,† Fig. S2c and d. The resistance of samples increased by further addition of DI water into the PEDOT:PSS suspension; therefore, the PEDOT:PSS stock solution was used as the baseline in this study.It should also be noted that many powdered materials, including the present BST powder, can form oxide layers during ball milling. Rinsing BST powders with an HCl solution can effectively remove such oxide layers;54 thus enhancing the power factor of PEDOT:PSS/BST composites. Zhang et al.54 performed X-ray photon spectroscopy (XPS) on BST surfaces before and after HCl rinsing. They concluded that the characteristic peaks of Bi–O and Te–O chemical states are completely removed after HCl rinsing and the peaks associated with BST remain at the same position. Consequently, the HCl rinsing process is very important before mixing BST powders with PEDOT:PSS. To investigate the effect of HCl rinsing in this work, two sets of BST powders (with an average particle size of 1.389 μm) were prepared – one rinsed with HCl and the other without any rinsing. Then, PEDOT:PSS/40 wt% BST thin films were fabricated from both sets of powders and their TE properties were investigated (Fig. S10k, l and S11a–c, ESI†).
It was confirmed that using HCl-rinsed BST in the composite decreased the electrical conductivity while increasing the Seebeck coefficient and power factor. This is important not only for the present research but also for other TE nanocomposite studies, as many other semiconducting or metallic nanomaterials often face surface oxidation during processing. In this study, the BST powders were rinsed with HCl before being added to the PEDOT:PSS matrix.
Fig. 2a–c display typical SEM micrographs of PEDOT:PSS thin films containing 0, 20, and 40 wt% BST particles, respectively. It is evident that particle distribution is homogenous in the composite films. Fig. 2d–f show the AFM surface topology of the films. More SEM and AFM images of PEDOT:PSS films containing 0–60 wt% BST are provided in the ESI,† Fig. S12 and S13. The variation of root mean square (RMS) roughness of the films with increasing BST particle concentration and size is shown in Fig. 2g and h. The PEDOT:PSS thin film exhibited a relatively smooth and uniform surface with no obvious phase separation in the structure and an RMS of 1.69 nm, consistent with values reported in the literature.55 The RMS roughness of the films increased gradually by adding 5, 10, and 20 wt% BST, but a sharp increase was observed thereafter. The increase in RMS roughness with BST concentration across the scanning area can be extrapolated to the entire thin film. Similarly, when the BST concentration was held constant, an increase in BST particle size resulted in higher RMS roughness. AFM step-height profiles were used to calculate the thickness of all the composite films, as shown in the ESI,† Fig. S14 and S15. There was not a considerable change in the average thickness of the composite films containing 5–60 wt% BST (i.e. 105–125 nm).
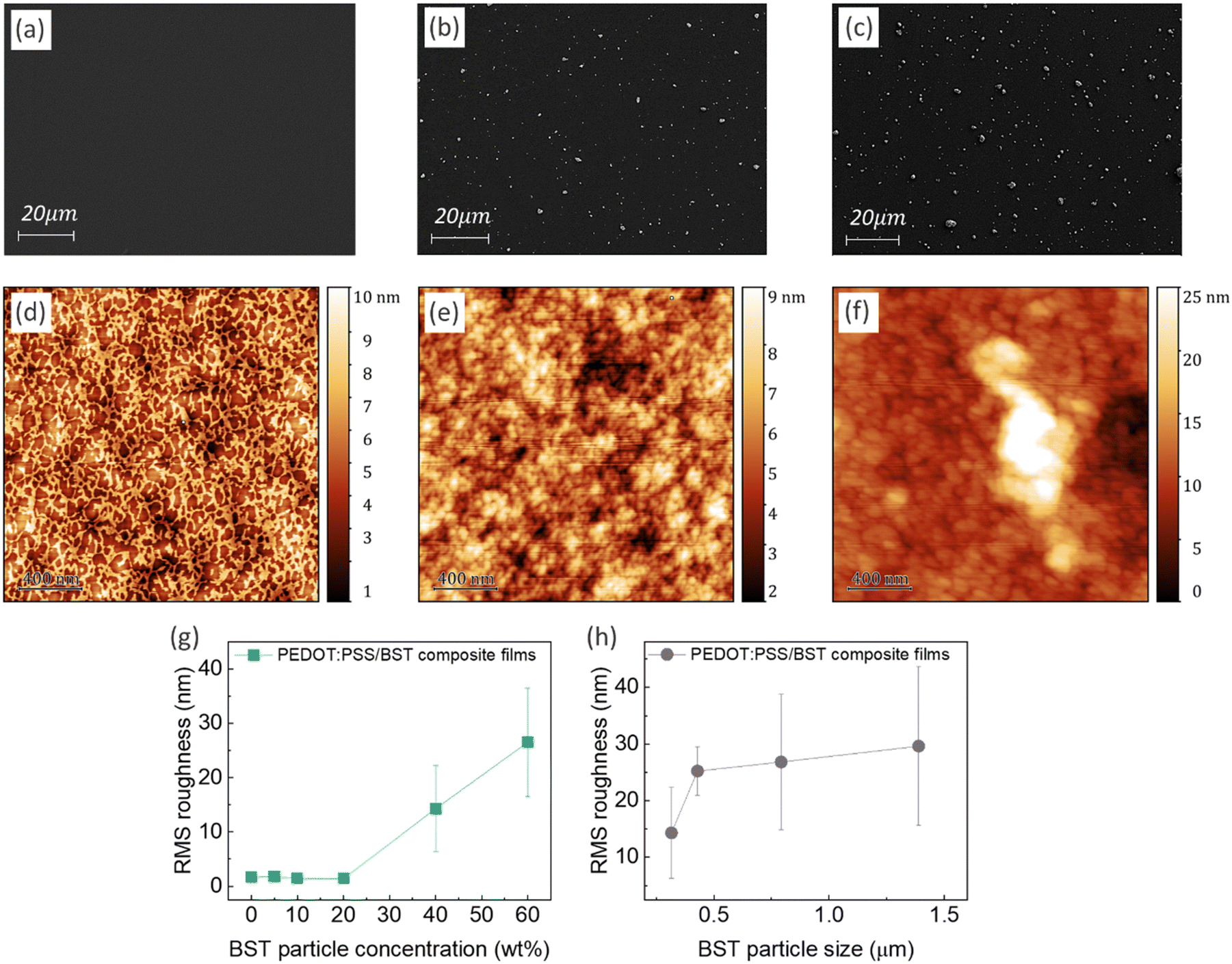 | ||
| Fig. 2 (a)–(c) SEM micrographs of PEDOT:PSS thin films with 0, 20, and 40 wt% BST, respectively. (d)–(f) Corresponding AFM images of the films. The RMS roughness of the films vs. (g) BST concentration, and (h) average size of BST particles. All the scan areas are: 2 × 2 μm2. The error bars in (g) and (h) were calculated using RMS values obtained from two different AFM images, with scan areas of 2 × 2 μm2 and 5 × 5 μm2. | ||
To investigate the effect of filler concentration on TE properties, PEDOT:PSS/BST composite thin films containing 0–60 wt% BST with an average particle size of 1.389 μm were fabricated via the procedure outlined in Section 2.3 (Fig. S10a–f, ESI†). The variation in electrical conductivity, Seebeck coefficient, and power factor of the films with BST concentration is illustrated in Fig. 3a. The electrical conductivity of the films increased with BST concentration from 0 wt% to 40 wt%, but it decreased afterwards. The Seebeck coefficient however decreased with BST concentration from 0 wt% to 10 wt%, and gradually increased afterwards. The maximum power factor of 0.05 μW m K−2 at 300 K was achieved for the PEDOT:PSS/40 wt% BST composite film.
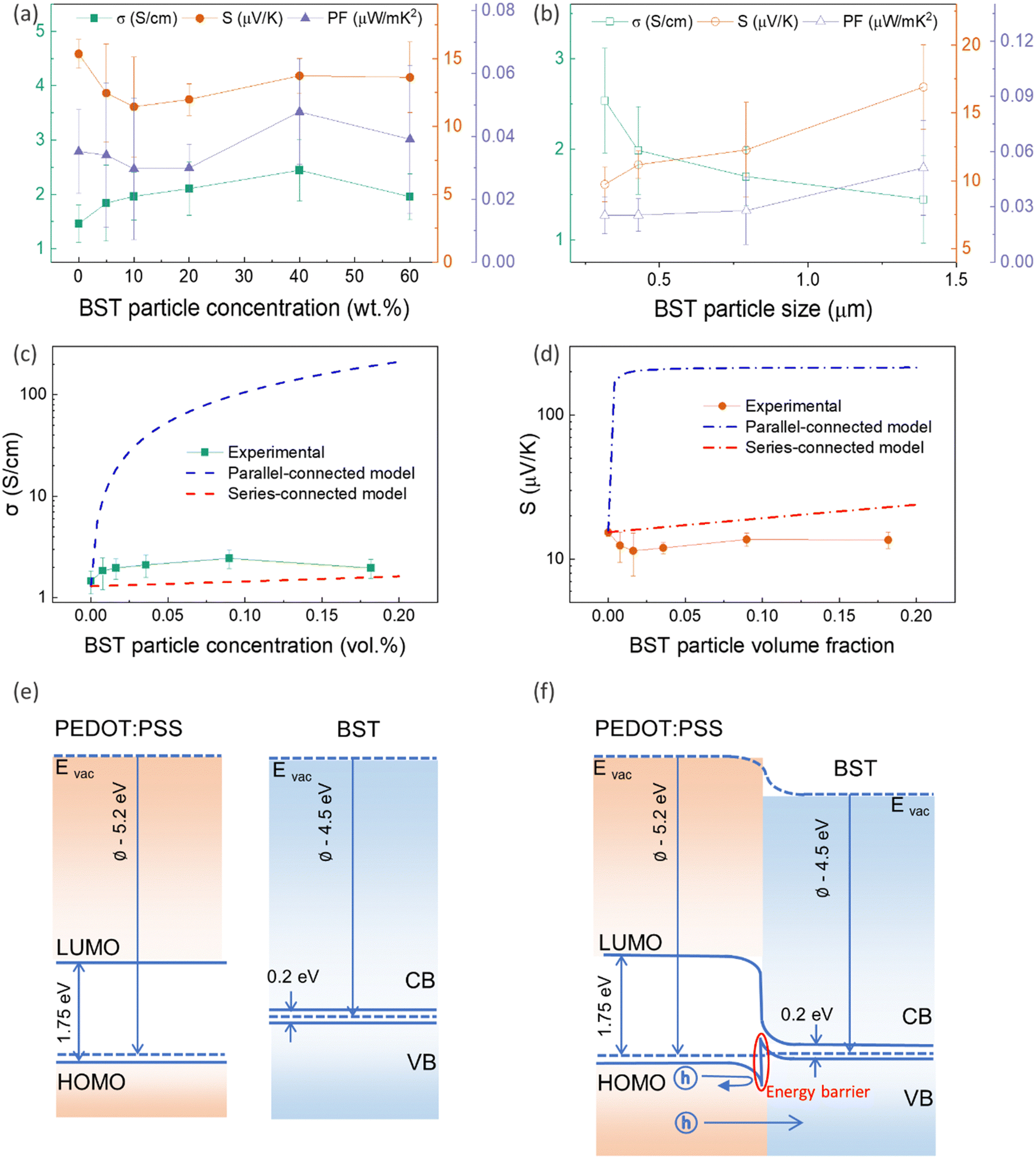 | ||
| Fig. 3 The variation in electrical conductivity, Seebeck coefficient, and power factor of PEDOT:PSS/BST thin films with (a) BST particle concentration, and (b) BST particle size. (c) and (d) Comparing the Seebeck coefficient of the films with two models (series- and parallel-connected models) as a function of the BST particle volume fraction. The error bars of experimental data were calculated using three repetitions of the measurements on two sets of samples. (e) Equilibrium energy band diagram of PEDOT:PSS, BST, and (f) PEDOT:PSS/BST interface, respectively. | ||
To explore the effect of filler particle size on TE properties, PEDOT:PSS/40 wt% BST composite films containing BST powders with different average particle sizes were fabricated (Fig. S10g–j, ESI†). The variation in electrical conductivity, Seebeck coefficient, and power factor in the samples with BST particle size is illustrated in Fig. 3b. The electrical conductivity of the composite thin films decreased with BST particle size, while the Seebeck coefficient increased. The maximum power factor of 0.05 μW mK−2 at 300 K was achieved for the composite with an average BST particle size of 1.389 μm.
Using a simple modelling approach to evaluate the electrical properties of polymer nanocomposite, the Seebeck coefficient and electrical conductivity can be described as either series- or parallel-connected models as a basic guideline, supposing that interfacial interactions are negligible.38,54,56–58 The equations for series- and parallel-connected models for PEDOT:PSS (matrix: M) and BST (filler: F) can be written as:
| σparallel = σM(1 − x) + σFx | (1) |
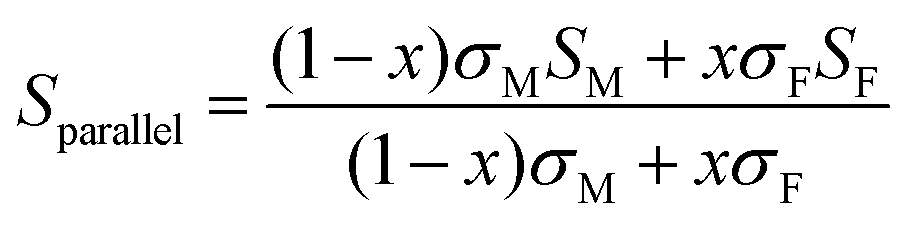 | (2) |
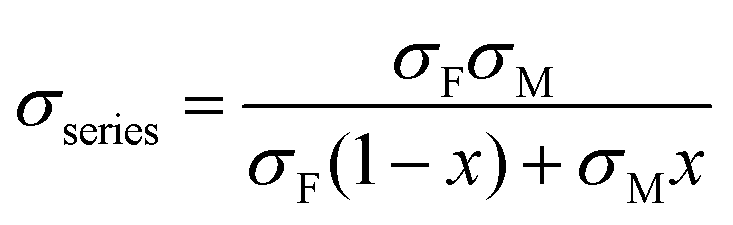 | (3) |
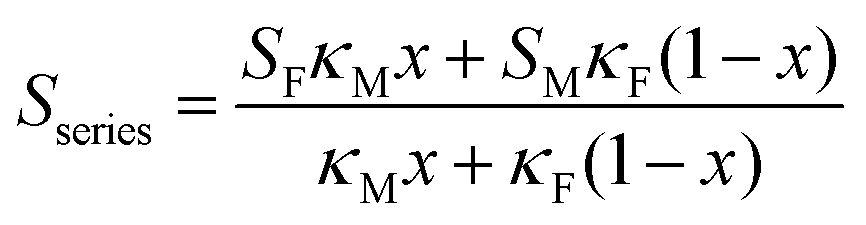 | (4) |
The lower values of the Seebeck coefficient in experiment compared to the predicted line in Fig. 3d could be due to the numerous nanointerfaces that exist between the different materials.38,56 In fact, a potential barrier can exist between these materials, attributed to energetic mismatches at the ceramic/polymer interfaces, and leading to preferential filtering of low-energy charge carriers. Consequently, only high-energy carriers can effectively pass through the interface, resulting in an increase in the Seebeck coefficient. The band diagrams of BST and PEDOT:PSS before and after connection are illustrated in Fig. 3e and f. One of the most common ways to study the charge carrier transport characteristics of organic conjugated systems is to analyse the temperature-dependency of their electrical conductivity and Seebeck coefficient.59 In organic and polymeric materials, such as PEDOT:PSS, these properties can be typically described in the framework of Mott's variable range hopping (VRH) theory (instead of band transport used for ordered solids) as:60
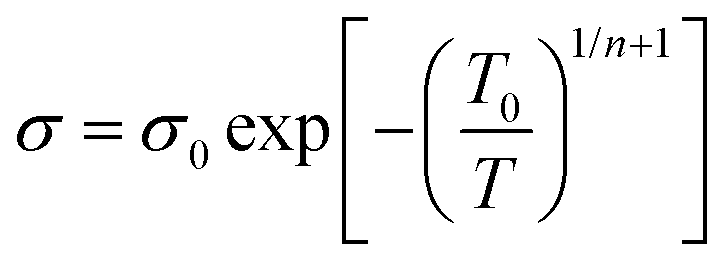 | (5) |
| S ∝ Tn−1/n+1 | (6) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) σ and S of all pristine PEDOT:PSS/BST thin films could be well-fitted linearly, assuming n = 3 in the VRH process. Therefore, the carrier transport path of the composite thin films remains in the 3D mode, indicating that the particle-to-particle variable range plays an important role in the films.41
σ and S of all pristine PEDOT:PSS/BST thin films could be well-fitted linearly, assuming n = 3 in the VRH process. Therefore, the carrier transport path of the composite thin films remains in the 3D mode, indicating that the particle-to-particle variable range plays an important role in the films.41
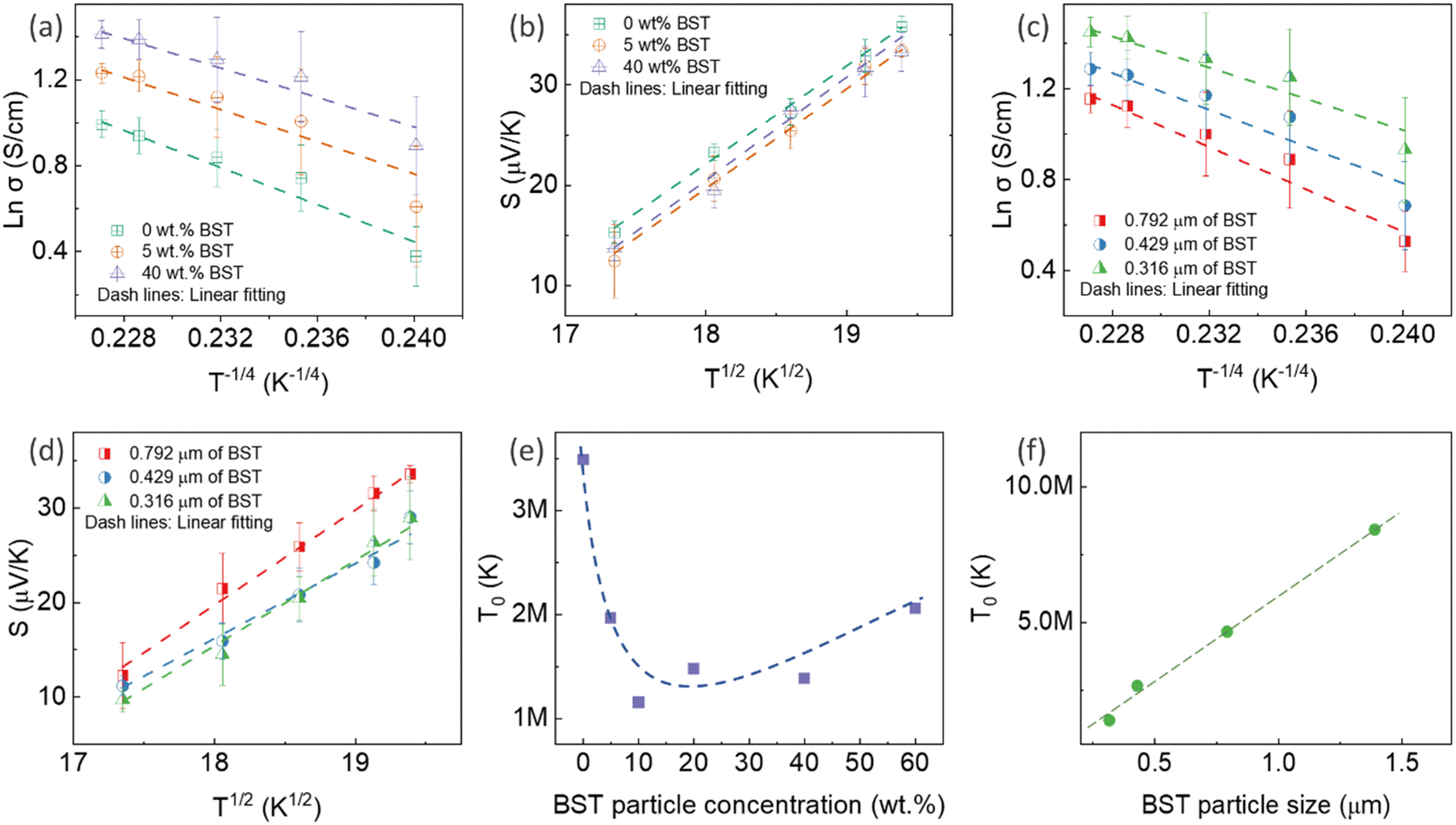 | ||
| Fig. 4 lnσ as a function of T−1/4, and S as a function of T1/2 for PEDOT:PSS/BST thin films with various (a) and (b) BST concentrations, and (c) and (d) BST particle sizes (the dashed lines in (a)–(d) represent linear fits to the Mott VRH model of the experimental data). The derived T0 for PEDOT:PSS/BST thin films with various (e) BST concentrations, and (f) BST particle sizes (the dashed lines in (e) and (f) indicate the eye-tracking). The error bars on the experimental data were calculated based on three measurement repetitions on two sets of samples in each case. | ||
The mechanism is further elucidated by T0, which characterizes the energy barrier for effective carrier hopping. The T0 values were derived from the linear fitting of lnσ versus T−1/4 for pristine PEDOT:PSS/BST thin film composites with various BST particle concentrations and sizes (Fig. 4e and f). The T0 value consistently increased with BST particle size but decreased with BST particle concentration until 10 wt%, after which it began to increase. The decrease in T0 correlates with enhanced electrical conductivity and reduced Seebeck coefficient values, indicating a lower energy barrier for charge carrier transport and an enlarged localization length in the films. On the other hand, the increase in T0 is associated with reduced electrical conductivity and enhanced Seebeck coefficient values, suggesting an elevated energy barrier for charge carrier transport. The presented results in Fig. 4e and f substantiate the electrical conductivity and Seebeck coefficient dependency with BST particle concentration and size shown in Fig. 3a and b.
3.3 Thermoelectric, morphological, and optical properties of post-treated PEDOT:PSS/BST composite thin films
Thin films of PEDOT:PSS/40 wt% BST were post-treated with multiple concentrations of DMSO, EG, H2SO4, and NaOH, following the procedure outlined in Section 2.4. The variations in electrical conductivity, Seebeck coefficient, and power factor of the films with the concentration of these secondary doping/dedoping agents at room temperature are shown in Fig. 5. The pristine sample, PEDOT:PSS/40 wt% BST, was the control sample. Fig. 5a shows that the electrical conductivity of the post-treated films massively increased with DMSO concentration up to 80 vol%, but decreased afterwards. A 134-fold increase was observed in the electrical conductivity of the sample treated with DMSO at 80 vol%, as compared to the pristine untreated sample. Meanwhile, the Seebeck coefficient gradually decreased with DMSO concentration up to 80 vol%, but increased after that. A maximum power factor of 11.6 μW mK−2 at 300 K was achieved at 40 vol% of DMSO. Similarly, Fig. 5b indicates that the electrical conductivity of the post-treated films largely increased with EG concentration, with a 72-fold maximum at 80 vol%. On the other hand, the Seebeck coefficient decreased continuously by increasing the EG concentration. A maximum power factor of 5.2 μW mK−2 at 300 K was achieved at 40 vol% of EG. In the case of post-treatment with H2SO4, Fig. 5c shows that the electrical conductivity increased with H2SO4 concentration up to 0.5 M, where a 309-fold enhancement was observed compared to the untreated film. The Seebeck coefficient however decreased gradually with H2SO4 concentration. A maximum power factor of 12.1 μW mK−2 at 300 K was achieved after treatment with 0.5 M H2SO4. The electrical conductivity enhancement in all these cases is due to secondary doping of PEDOT:PSS.62 The secondary dopants play a crucial role in modifying the PEDOT:PSS morphology through: (a) segregation of large amounts of excessive PSS, and (b) conformational changes in PEDOT from a benzoid to a quinoid structure.33 These can improve the charge transport within the structure significantly, as schematically shown in Fig. 6a and b. In general, secondary doping has minimal impact on the doping level of PEDOT:PSS. The utilized organic polar solvents and acid can weaken the Coulombic interaction between PEDOT and PSS through a screening effect, thus facilitating the phase segregation of PSS from PEDOT on a microscopic scale.61 Accompanied with this phase segregation, the PEDOT chains also change from a coil to a linear or expanded coil conformation.63,64 The BST particles within the PEDOT:PSS matrix can also assist in both the removal of detached PSS and the alignment of PEDOT chains.26,29,38 For example, increasing the BST concentration has shown a rise in the PEDOT/PSS mass ratio in composite thin films,29 suggesting that BST particles contribute to removing excess PSS from the PEDOT:PSS matrix (alongside the removal by secondary dopants). This reduction can lead to enhanced ordering (due to π–π interactions) and connectivity of conducting PEDOT chains.35,38,39,45,49,65–68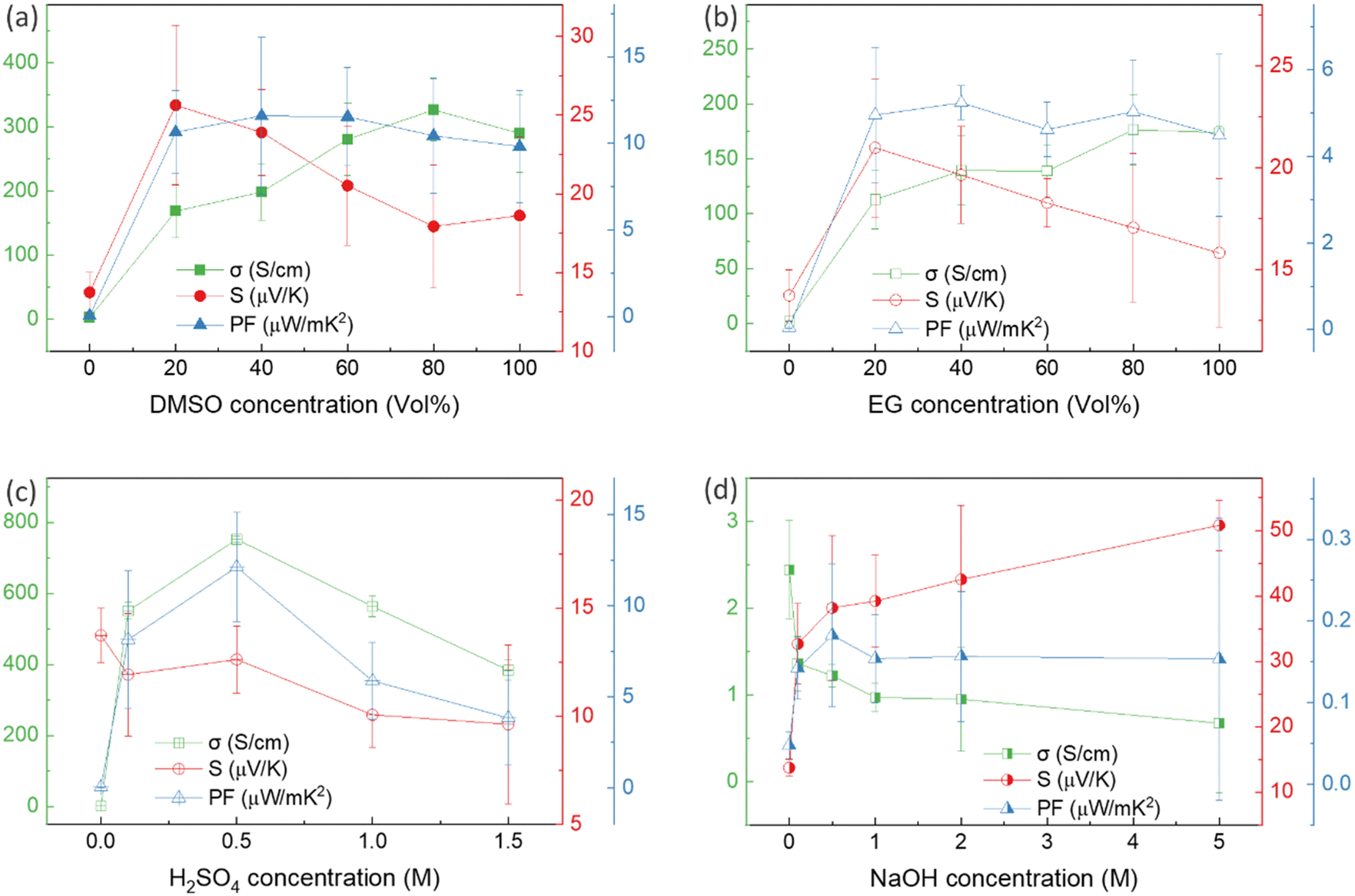 | ||
| Fig. 5 The variation in electrical conductivity, Seebeck coefficient, and power factor of single post-treated PEDOT:PSS thin films with: (a) DMSO, (b) EG, (c) H2SO4, and (d) NaOH concentrations at room temperature. The error bars on the experimental data were calculated based on three repetitions of the measurements on the samples. | ||
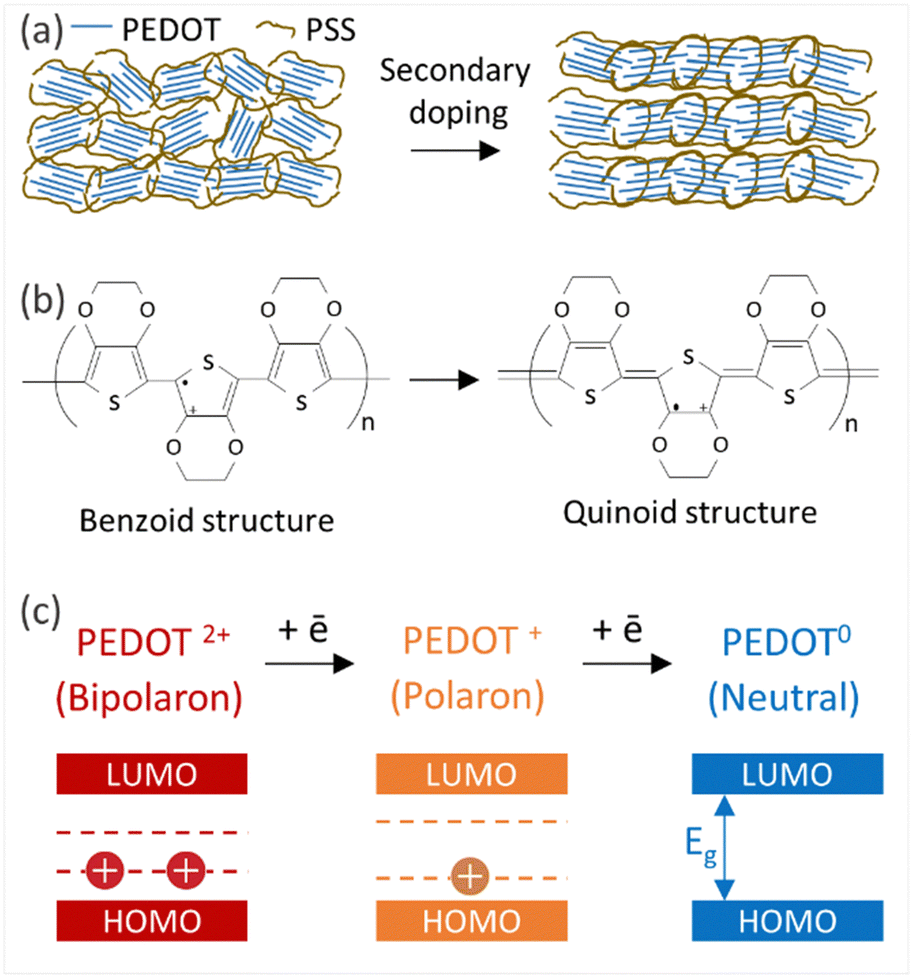 | ||
| Fig. 6 (a) Schematic diagram illustrating morphological changes in PEDOT:PSS films by secondary doping. (b) Conformational changes in PEDOT from a benzoid to quinoid structure. (c) Transition of PEDOT chains from bipolaron to polaron and neutral states during the dedoping process. | ||
In contrast to the above, post-treatment with NaOH (as a dedoping agent) led to a decrease in the electrical conductivity and an increase in the Seebeck coefficient in PEDOT:PSS films, as shown in Fig. 5d. For instance, post-treatment with a 5 M NaOH solution led to a four-fold decrease in the electrical conductivity, a nearly four-fold increase in the Seebeck coefficient, and achieving a power factor of 0.2 μW mK−2 at 300 K. Chemical dedoping has been widely proven to be an effective approach for Seebeck coefficient enhancement in PEDOT:PSS films by modifying the oxidation level of PEDOT and tuning the carrier concentration.69 During the dedoping process, PEDOT chains undergo transition through three states, as depicted in Fig. 6c. Highly oxidized PEDOT chains in the bipolaron state (PEDOT2+) are gradually dedoped to a less oxidized polaron state (PEDOT+), and eventually to a neutral state (PEDOT0) where PEDOT chains have no net charge.40 The overall impact of the dedoping process on PEDOT chains involved a trade-off between electrical conductivity and the Seebeck coefficient. This dynamic behavior is crucial for tailoring the thermoelectric properties of PEDOT:PSS films.
To analyse the effect of BST particles on the overall structure of PEDOT:PSS thin films, XRD characterization was performed on the BST powder, pristine PEDOT:PSS and PEDOT:PSS/40 wt% BST films, and 0.5 M H2SO4–0.1 M NaOH post-treated PEDOT:PSS and PEDOT:PSS/40 wt% BST films. As shown in Fig. S22a–d (ESI†), the diffraction peaks of the powder can be indexed as rhombohedral structured BST with a space group of R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m and the main diffraction peak at 2θ = 28.3° corresponding to the (015) lattice plane (JCPDS no. 49-1713). The pristine films however showed a broad peak at 2θ of ∼20°–32°, attributed to interchain planar stacking of PEDOT planes.38,68,70 The weak intensity and broad diffraction peak is due to the small thickness of the films and also moderate crystallinity, consistent with the literature.24,29,38 The XRD patterns did not show a considerable change after the addition of BST particles due to their low volume percentage as compared to the matrix material. However, literature suggests that micrometre-thick and free-standing composites of PEDOT:PSS/BST can show sharper diffraction peaks, indicating a higher crystallinity in composite samples due to the presence of BST particles.24,29,36,38 Moreover, to see the effect of sequential post-treatment with H2SO4 and NaOH on improved crystallinity of the films, analysis of weak peaks at low 2θ of ∼3° and 7° is required.38,68 For that purpose, SAXS and WAXS measurements were undertaken, however the results did not indicate a considerable change after the addition of BST particles and post-treatment (Fig. S22, ESI†), which could be due to insufficient resolution of the utilized equipment.
m and the main diffraction peak at 2θ = 28.3° corresponding to the (015) lattice plane (JCPDS no. 49-1713). The pristine films however showed a broad peak at 2θ of ∼20°–32°, attributed to interchain planar stacking of PEDOT planes.38,68,70 The weak intensity and broad diffraction peak is due to the small thickness of the films and also moderate crystallinity, consistent with the literature.24,29,38 The XRD patterns did not show a considerable change after the addition of BST particles due to their low volume percentage as compared to the matrix material. However, literature suggests that micrometre-thick and free-standing composites of PEDOT:PSS/BST can show sharper diffraction peaks, indicating a higher crystallinity in composite samples due to the presence of BST particles.24,29,36,38 Moreover, to see the effect of sequential post-treatment with H2SO4 and NaOH on improved crystallinity of the films, analysis of weak peaks at low 2θ of ∼3° and 7° is required.38,68 For that purpose, SAXS and WAXS measurements were undertaken, however the results did not indicate a considerable change after the addition of BST particles and post-treatment (Fig. S22, ESI†), which could be due to insufficient resolution of the utilized equipment.
Thus far, the optimum concentration of every doping/dedoping chemical agent was determined to maximize the power factor of PEDOT:PSS/40 wt% BST composite thin films after a single post-treatment process. These concentrations were 40 vol%, 40 vol%, 0.5 M, and 0.5 M for DMSO, EG, H2SO4, and NaOH, respectively. Subsequently, the thin films underwent a sequential post-treatment process with 40 vol% DMSO, 1 to 4 rounds, as described in the ESI,† Fig. S5a. The variation in electrical conductivity, Seebeck coefficient, and power factor of these thin films at room temperature with the number of post-treatment process repetitions is presented in Fig. 7a. The electrical conductivity improved with increasing the number of treatment repetitions, while the Seebeck coefficient decreased slightly. The electrical conductivity improved nearly 40% after post-treatment with DMSO four times, whereas the Seebeck coefficient deceased by 18%. The maximum power factor of 18.5 μW mK−2 at 300 K was achieved after PEDOT:PSS/40 wt% BST thin films were sequentially post-treated three times with a 40 vol% solution of DMSO.
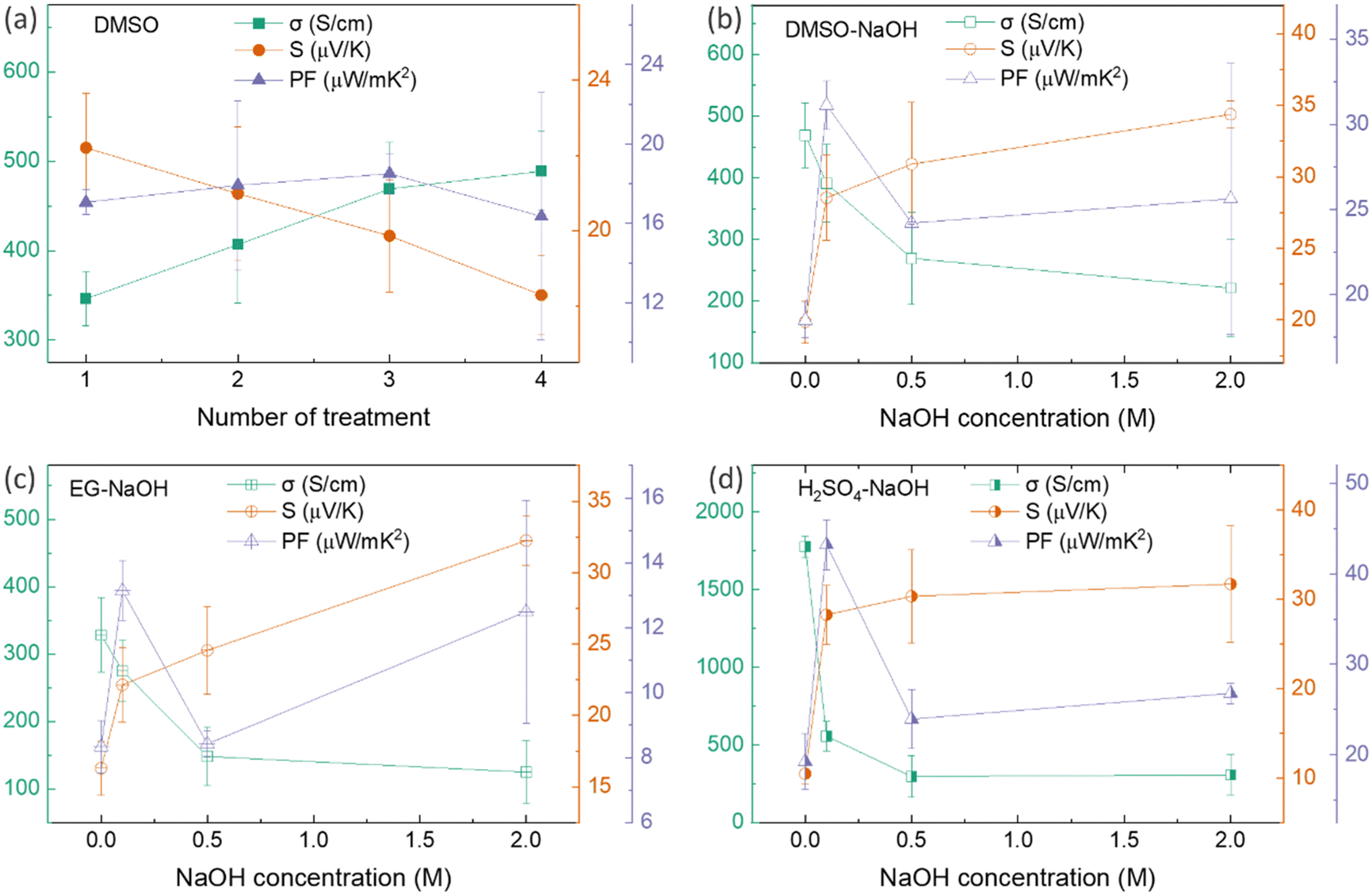 | ||
| Fig. 7 The variation in electrical conductivity, Seebeck coefficient, and power factor of PEDOT:PSS/40 wt% BST thin films post-treated with: (a) 40 vol% DMSO (1 to 4 rounds), (b) 40 vol% DMSO (3 rounds) and various concentrations of NaOH (1 round), (c) 40 vol% EG (3 rounds) and various concentrations of NaOH (1 round), and (d) 0.5 M H2SO4 (3 rounds) and various concentrations of NaOH (1 round) at room temperature. The error bars on experimental data were calculated based on three repetitions of measurements on the samples. | ||
Likewise, three rounds of sequential post-treatment with other secondary dopants (i.e. EG and H2SO4) were concluded to yield the maximum power factor in the composite films, given the similar performance of all those secondary dopants.39 In this regard, a different set of sequential post-treatments was developed, including three rounds of treatment with secondary dopants (DMSO, EG, and H2SO4), followed by a final round of treatment with the chemical dedoping agent NaOH (Fig. S5b, ESI†). The effect of NaOH concentration on electrical conductivity, Seebeck coefficient, and power factor of composite thin films sequentially treated with 40 vol% DMSO–NaOH, 40 vol% EG–NaOH, and 0.5 M H2SO4–NaOH at room temperature is shown in Fig. 7b–d. In the case of DMSO–NaOH treatment, the electrical conductivity decreased with NaOH concentration while the Seebeck coefficient increased, leading to a maximum power factor of 31.1 μW mK−2 at 300 K after sequential treatment with 40 vol% DMSO and 0.1 M NaOH. This indicates a 1.7 times improvement in the power factor after sequential post-treatment with DMSO–NaOH, as compared to single post-treatment with DMSO. Similarly, the electrical conductivity in EG–NaOH and H2SO4–NaOH treated samples reduced with NaOH concentration while the Seebeck coefficient improved. Maximum power factors of 13.1 μW mK−2 and 43.2 μW mK−2 at 300 K were achieved after sequential post-treatment with EG–0.1 M NaOH and H2SO4–0.1 M NaOH. These values are 1.5 times and 2.5 times higher than those achieved after single post-treatment with EG and H2SO4, respectively. The combination of enhanced electrical conductivity arising from secondary dopants (DMSO, EG, and H2SO4) and improved Seebeck coefficient facilitated by NaOH dedoping led to the huge boost in the power factor in sequentially post-treated PEDOT:PSS/40 wt% BST thin films. The best TE performance was observed after three rounds of secondary doping with 0.5 M H2SO4 and one round of dedoping with 0.1 M NaOH, showcasing how the TE behavior in PEDOT:PSS/BST thin films can be the fine-tuned through strategic manipulation of chemical processes and careful choices of doping/dedoping agents.
The temperature dependence of electrical conductivity and Seebeck coefficient was measured for both single and sequentially post-treated PEDOT:PSS/40 wt% BST thin films in the temperature range of 300 K to 375 K to explore charge transport mechanisms in these thin films. The results, presented in Fig. 8 and Fig. S17 (ESI†), show that electrical conductivities of the thin films continuously decreased with temperature, while the Seebeck coefficients steadily increased. This trend was observed in all single and sequentially post-treated thin films, suggesting a metallic transport mechanism, characterized by a decrease in electrical conductivity with increasing temperature. This is contrary to the expected behavior in thermally assisted hopping mechanisms, where a simultaneous increase in both temperature and electrical conductivity is observed in the material. In degenerate semiconductors exhibiting metallic charge transport behavior, the relationship between the Seebeck coefficient and electrical conductivity (based on a parabolic band theory and an energy-independent scattering approximation71) is expressed as:72
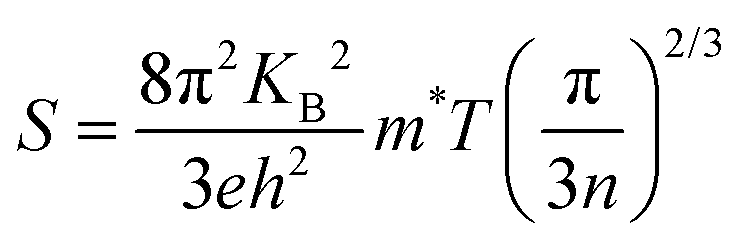 | (7) |
| σ = neμ | (8) |
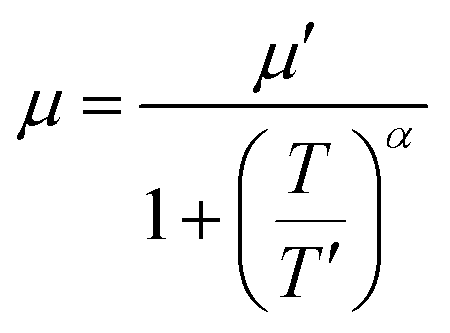 | (9) |
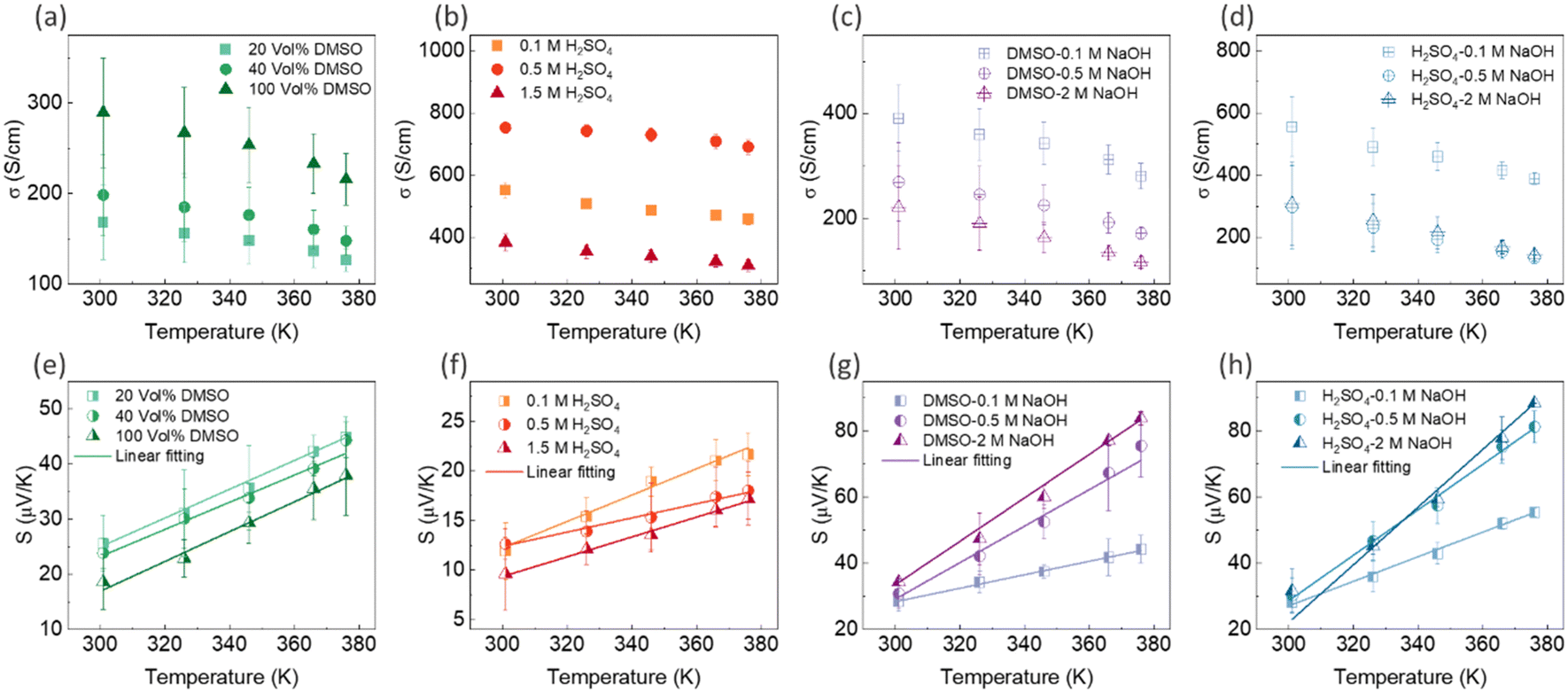 | ||
| Fig. 8 The variation in electrical conductivity and Seebeck coefficient with temperature for PEDOT:PSS/40 wt% BST thin films after: single post-treatment with DMSO (a) and (e), H2SO4 (b) and (f), sequential post-treatment with DMSO–NaOH (c) and (g), and H2SO4–NaOH (d) and (h). The dashed lines in (e)–(h) represent the linear fitting of eqn (7) to the experimental data. The error bars on the experimental data were calculated based on three repetitions of measurements on the samples. | ||
In general, for lattice scattering, α is often taken as 1.5. As the temperature increases, the mobility μ decreases, which leads to a decrease in electrical conductivity. Eqn (7)–(9) characterize the metallic transport behaviour in samples with an increase in the Seebeck coefficient with temperature, accompanied by a decrease in electrical conductivity, which was observed in the single and sequentially post-treated thin films. Also, the linear fittings of Seebeck coefficient versus temperature, illustrated in Fig. 8e–h and Fig. S17d–f (ESI†), further confirm the metallic transport in these films. These results demonstrate that post-treatment of PEDOT:PSS/40 wt% BST changes the charge transport mechanism from 3D VRH to metallic transport.
Post-treatment of PEDOT:PSS thin films with secondary dopants, e.g. DMSO, EG, and H2SO4, can lead to phase separation between PEDOT and PSS. This process can induce partial removal of the insulating PSS shell and extend the PEDOT chains from the coiled benzenoid structure to a linear quinonoid structure, creating more ordered PEDOT-rich chains. This results in higher RMS roughness, demonstrating that excess PSS is effectively removed during the post-treatment process.35,40,46,47,49 The oriented PEDOT domains and PSS removal facilitate the transport of charge carriers due to reduced energy barriers, thereby boosting the electrical conductivity of PEDOT:PSS after secondary doping.35,40,46,47,49 It should be noted that secondary doping generally has a small effect on the doping level of PEDOT:PSS, hence the Seebeck coefficient. The characterization of the surface topology of post-treated thin films showed a direct correlation among secondary dopants, RMS roughness, and electrical conductivity. In this regard, AFM surface topology images of the composite thin films after single and sequential post-treatments with DMSO, EG, H2SO4, and NaOH are illustrated in the ESI,† Fig. S18 and S19, respectively. The corresponding surface roughness graphs are presented in Fig. 9, revealing the following points: (1) RMS roughness in single-treated samples increased by raising DMSO, EG, and H2SO4 concentrations (Fig. 9a–c), but remained nearly unchanged with NaOH concentration alterations (Fig. 9d), which is in good agreement with previous findings.39 (2) Repeating the sequential treatment with 40 vol% DMSO increased the RMS roughness of the films (Fig. 9e), providing evidence of successful secondary doping in the films. The secondary dopants remove the excessive PSS, causing phase separation in the conductive network of PEDOT, and resulting in a roughened surface. (3) The increase in NaOH concentration raised the roughness of 40 vol% DMSO–NaOH, 40 vol% EG–NaOH, and 0.5 M H2SO4–NaOH thin films (Fig. 9f–h). This could be due to a slight collision-induced agglomeration of the PEDOT chains stemming from the decreased repulsion force between their neighbouring neutralized chains.73 Further investigation into the morphological features of the post-treated films is detailed in the ESI,† Fig. S20 and S21. Moreover, a decrease in the thickness of the films was observed after post-treatment with secondary dopants and rinsing with DI water, as shown in the ESI,† Fig. S20 and S21. As an example, the thickness of post-treated films with DMSO decreased from 116 nm to 56 nm after post-treatment with 20 vol% DMSO. This reduction in thickness can be attributed to the depletion and removal of PSS.36–38,74 Additionally, the SEM micrographs of the thin films before and after rinsing in DI water (Fig. S4b and c, ESI†) did not indicate noticeable changes in the particle concentration and distribution after rinsing.
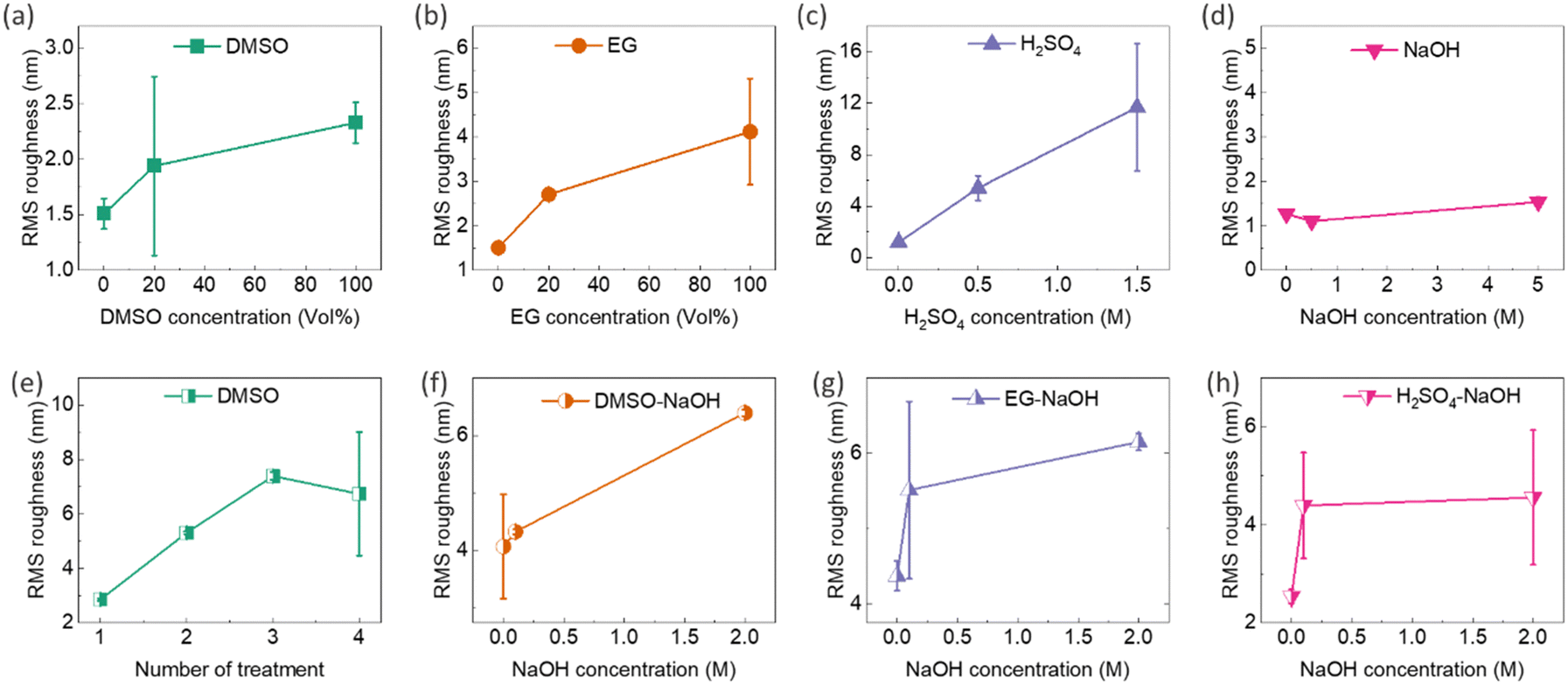 | ||
| Fig. 9 RMS surface roughness profiles of single post-treated PEDOT:PSS/40 wt% BST films with various concentrations of: (a) DMSO, (b) EG, (c) H2SO4, and (d) NaOH. RMS surface roughness profiles of sequentially post-treated PEDOT:PSS/40 wt% BST films with: (e) 40 vol% DMSO (1 to 4 rounds), (f) 40 vol% DMSO (3 rounds) and various concentrations of NaOH (1 round), (g) 40 vol% EG (3 rounds) and various concentrations of NaOH (1 round), and (h) 0.5 M H2SO4 (3 rounds) and various concentrations of NaOH (1 round). The error bars in (g) and (h) were calculated using RMS values obtained from two different AFM images, with scan areas of 2 × 2 μm2 and 5 × 5 μm2. | ||
Ultraviolet-visible-near-infrared (UV-Vis-NIR) absorption spectra of PEDOT:PSS thin films can be used to characterize chemical changes induced by secondary doping and chemical dedoping.33 There are typically two absorption bands between wavelengths of 190 nm and 250 nm, originating from the aromatic rings of PSS.75,76 The reduction in PSS content through the secondary doping process can be indicated by a decreased intensity in these two bands.75,76 Glass substrates (microscope slides) have a cut-off transmission wavelength of 350 nm; thus, to analyse the UV spectra of the PEDOT:PSS-based composites, new samples were fabricated on quartz substrates that have high transmission even at 190 nm. The UV-Vis-NIR absorption spectra of the PEDOT:PSS/40 wt% BST thin films after single post-treatment with different concentrations of DMSO, EG, and H2SO4, and after sequential post-treatment with 40 vol% DMSO are presented in Fig. 10a–d, respectively. It is evident that the two absorption bands between wavelengths of 190 nm and 250 nm decreased after post-treatment with all the secondary dopants. Polar solvents such as H2SO4, DMSO, and EG (with dielectric constants of 84, 46.6, and 37.7 at 20 °C, respectively) induce a strong screening effect in the composite, diminishing Coulomb interactions between positively charged PEDOT and negatively charged PSS dopants. Among them, H2SO4 has a higher dielectric constant that can lead to a stronger screening effect, facilitating easier depletion of PSS after post-treatment compared to DMSO and EG. That is why Fig. 10c exhibits lower absorption intensities in thin films treated with H2SO4, as compared to those treated with DMSO and EG (Fig. 10a and b). No significant change was observed in the visible region of the absorption spectra of PEDOT:PSS thin films after secondary doping.
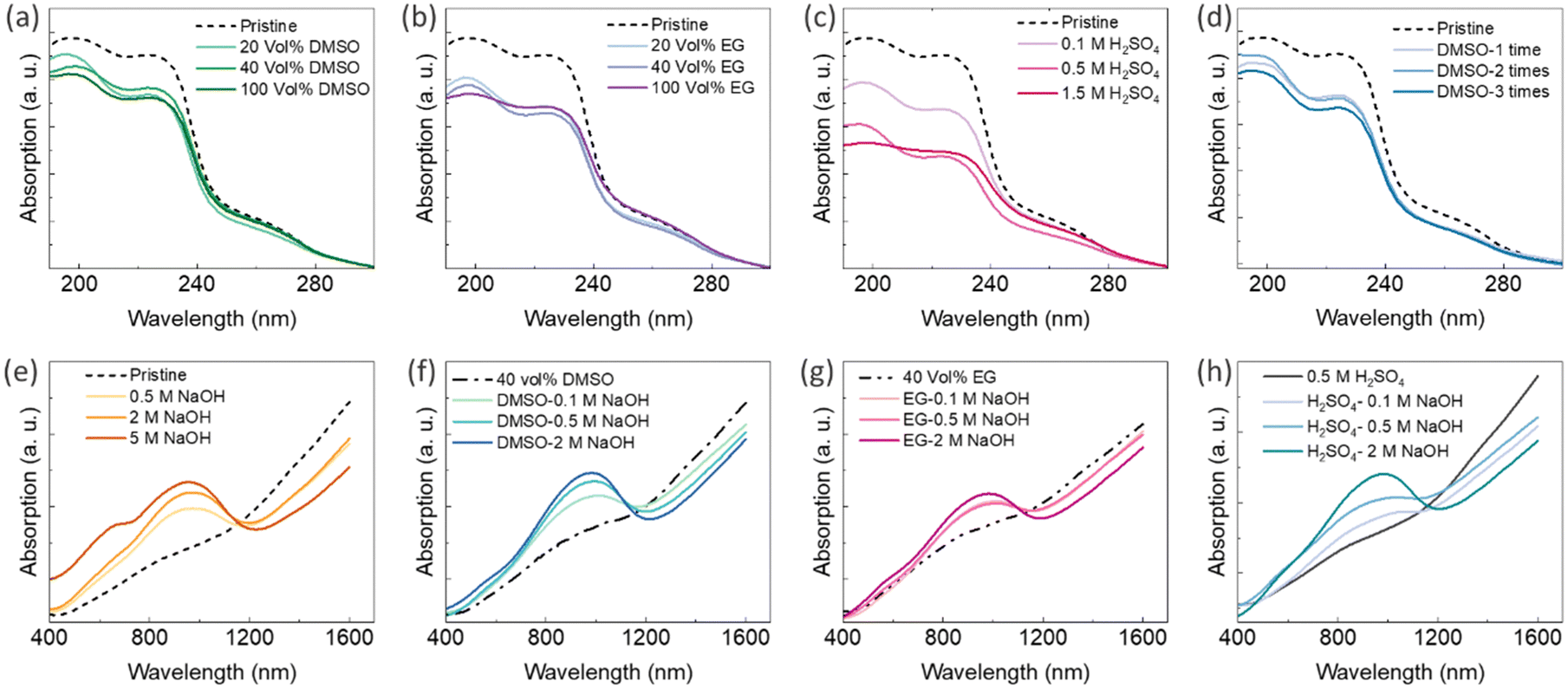 | ||
| Fig. 10 UV-Vis-NIR absorption spectra of PEDOT:PSS/40 wt% BST films after single post-treatment with (a) DMSO, (b) EG, and (c) H2SO4; after sequential post-treatment with (d) 40 vol% DMSO (1–4 rounds), after single post-treatment with (e) various concentrations of NaOH, and after sequential post-treatment with (f) 40 vol% DMSO (3 rounds) and various concentrations of NaOH (1 round), (g) 40 vol% EG (3 rounds) and various concentrations of NaOH (1 round), and (h) 0.5 M H2SO4 (3 rounds) and various concentrations of NaOH (1 round). | ||
In the case of dedoping, the three states of PEDOT chains (i.e. bipolaron, polaron, and neutral states) exhibit absorption at different wavelengths. The neutral polymer chains show absorption around 600 nm, chains in the polaron state show absorption around 900 nm, and chains in the bipolaron state display broad absorption in the near infrared region.40,46,49 The UV-Vis-NIR absorption spectra of the PEDOT:PSS/40 wt% BST thin films after single post-treatment with NaOH and sequential post-treatment with 40 vol% DMSO–NaOH, 40 vol% EG–NaOH, and 0.5 M H2SO4–NaOH are presented in Fig. 10e–h, respectively. As the NaOH concentration increased, a broad polaronic transition peak was intensified at 900 nm in all samples, indicating the dedoping of PEDOT chains. Simultaneously, a new transition evolved at around 600 nm, originating from the absorption of neutral PEDOT segments. This peak became more pronounced at higher NaOH concentrations. The dedoping process by tuning the charge carrier concentration resulted in enlargement of the Seebeck coefficient, a decrease in the electrical conductivity, and maximizing the power factor (Fig. 5d and 7b–d).
4 Outlook
This work and ongoing research into enhancing the efficiency of polymer-based TE materials will pave the way for their widespread adoption in diverse applications. For example, flexible TEGs made of these materials can be integrated into wearable electronics, where they can serve as a power source for portable devices. However further investigation into the mechanical and thermal stability and durability of polymer-based TE materials and devices is essential before they can be practically used in clothing, accessories, and medical devices as a sustainable energy solution for on-the-go users. In addition, subsequent research into innovative post-treatment strategies by exploring new dopants, dedoping agents, or alternative processing techniques is required to enhance the TE performance of conductive polymer-based composites. Moreover, investigation into scalable manufacturing techniques such as roll-to-roll processing or printing methods is critical to facilitate the mass production of flexible TE devices. Finally, beyond material and device-level optimizations, future research should also delve into system-level optimizations such as integrating smart energy management systems to maximize energy harvesting efficiency.5 Conclusions
The present comprehensive investigation into PEDOT:PSS/Bi0.5Sb1.5Te3 composite thin films reveals important insights into their microstructural, morphological, and thermoelectric properties. Bi0.5Sb1.5Te3 particles within micro to nano ranges were prepared by ball milling and centrifugation processes, and a systematic study was conducted on the impact of particle concentration and size on the surface morphology and thermoelectric characteristics of the composite thin films. An optimum composition of 40 wt% BST particles with an average size of 1.389 μm resulted in a maximum power factor of 0.05 μW mK−2 at room temperature. Series and parallel connected models as a guideline in a matrix/filler system were employed for pristine thin films to explain the variation in Seebeck coefficient and electrical conductivity with BST concentration. The results demonstrated that the electrical conductivity and Seebeck coefficient of the samples closely align with the series connected model. Moreover, the temperature-dependent behaviour of electrical conductivity and Seebeck coefficient in the thin films indicated the prevalence of a three-dimensional variable range hopping (VRH) charge carrier transport mechanism. Expanding on these findings, the TE properties were enhanced through various single and sequential post-treatments utilizing secondary dopants (DMSO, EG, H2SO4) and a chemical dedoping agent (NaOH). Among the single post-treatments, 0.5 M H2SO4 demonstrated a 240-fold increase in the PF compared to the pristine thin film. Furthermore, the sequential post-treatment by 0.5 M H2SO4 and 0.1 M NaOH exhibited an 864-fold enhancement in the power factor at room temperature. The measurements of temperature-dependent electrical conductivity and Seebeck coefficient in post-treated samples revealed a metallic charge carrier transport mechanism. These results collectively highlight the potential for optimizing the TE properties of PEDOT:PSS/Bi0.5Sb1.5Te3 composites through BST particle engineering and precise tailoring of the post-treatment methods. This comprehensive study paves the way for developing high-performance, flexible, and efficient TE materials for wearable energy harvesting applications.Author contributions
Conceptualization, A. P. and S. M.; methodology, S. M.; material characterization, S. M., K. Z., N. P. and M. A. M., formal analysis, S. M.; data curation, S. M.; writing, S. M. and A. P.; supervision, A. P.; funding acquisition, A. P. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Further information and data are available upon request from the authors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
A. P. would like to acknowledge the financial support of Science Foundation Ireland (SFI) under grant number 18/SIRG/5621. A. P. and S. M. are grateful to Prof. Jonathan Coleman, Dr Kevin Synnatschke, and Mr Gerard Byrne for their technical support.Notes and references
- D. Won, J. Bang, S. H. Choi, K. R. Pyun, S. Jeong, Y. Lee and S. H. Ko, Chem. Rev., 2023, 123, 9982–10078 CrossRef CAS.
- Y. Sun, Y.-Z. Li and M. Yuan, Nano Energy, 2023, 115, 108715 CrossRef CAS.
- T. Shimura, S. Sato, P. Zalar and N. Matsuhisa, Adv. Electron. Mater., 2023, 9, 2200512 CrossRef CAS.
- Z. Liang, J. He, C. Hu, X. Pu, H. Khani, L. Dai, D. Fan, A. Manthiram and Z.-L. Wang, Adv. Intell. Syst., 2023, 5, 2200045 CrossRef.
- X.-L. Shi, J. Zou and Z.-G. Chen, Chem. Rev., 2020, 120, 7399–7515 CrossRef CAS PubMed.
- T. Cao, X.-L. Shi and Z.-G. Chen, Prog. Mater. Sci., 2023, 131, 101003 CrossRef CAS.
- S. Yang, Y. Li, L. Deng, S. Tian, Y. Yao, F. Yang, C. Feng, J. Dai, P. Wang and M. Gao, Microsyst. Nanoeng., 2023, 9, 106 CrossRef PubMed.
- M. Hong, M. Li, Y. Wang, X. L. Shi and Z. G. Chen, Adv. Mater., 2023, 35, 2208272 CrossRef CAS PubMed.
- D. Liu, D. Wang, T. Hong, Z. Wang, Y. Wang, Y. Qin, L. Su, T. Yang, X. Gao and Z. Ge, Science, 2023, 380, 841–846 CrossRef CAS PubMed.
- Z. Long, Y. Wang, X. Sun, Y. Li, Z. Zeng, L. Zhang and H. Chen, Adv. Mater., 2023, 35, 2210345 CrossRef CAS PubMed.
- X.-L. Shi, S. Sun, T. Wu, J. Tu, Z. Zhou, Q. Liu and Z.-G. Chen, Mater. Futures, 2024, 3, 012103 CrossRef.
- Y. Saberi and S. A. Sajjadi, J. Alloys Compd., 2022, 904, 163918 CrossRef CAS.
- S. I. Kim, K. H. Lee, H. A. Mun, H. S. Kim, S. W. Hwang, J. W. Roh, D. J. Yang, W. H. Shin, X. S. Li and Y. H. Lee, Science, 2015, 348, 109–114 CrossRef CAS.
- K. Kato, Y. Hatasako, M. Uchino, Y. Nakata, Y. Suzuki, T. Hayakawa, C. Adachi and K. Miyazaki, Adv. Mater. Interfaces, 2014, 1, 1300015 CrossRef.
- Z. Liu, N. Sato, W. Gao, K. Yubuta, N. Kawamoto, M. Mitome, K. Kurashima, Y. Owada, K. Nagase and C.-H. Lee, Joule, 2021, 5, 1196–1208 CrossRef CAS.
- Z. Liu, W. Gao, H. Oshima, K. Nagase, C.-H. Lee and T. Mori, Nat. Commun., 2022, 13, 1120 CrossRef CAS PubMed.
- S. Masoumi and A. Pakdel, Adv. Eng. Mater., 2022, 24, 2100955 CrossRef CAS.
- A. Pakdel, Q. Guo, V. Nicolosi and T. Mori, J. Mater. Chem. A, 2018, 6, 21341–21349 RSC.
- A. Pakdel, A. U. Khan, F. Pawula, S. Hébert and T. Mori, Adv. Mater. Interfaces, 2022, 9, 2200785 CrossRef CAS.
- A. Sharma, S. Masoumi, D. Gedefaw, S. O'Shaughnessy, D. Baran and A. Pakdel, Appl. Mater. Today, 2022, 29, 101614 CrossRef.
- S. Masoumi, S. O'Shaughnessy and A. Pakdel, Nano Energy, 2022, 92, 106774 CrossRef CAS.
- L. Deng, Y. Liu, Y. Zhang, S. Wang and P. Gao, Adv. Funct. Mater., 2023, 33, 2210770 CrossRef CAS.
- H. Li, C. Zhang, P. Li, S. Liu, H. Zhang and C. He, Mater. Today Chem., 2023, 34, 101774 CrossRef CAS.
- H. Song, C. Liu, H. Zhu, F. Kong, B. Lu, J. Xu, J. Wang and F. Zhao, J. Electron. Mater., 2013, 42, 1268–1274 CrossRef CAS.
- A. A. Abd Rahman, A. A. Umar and M. H. U. Othman, Physica E, 2015, 66, 293–298 CrossRef.
- G. Goo, G. Anoop, S. Unithrattil, W. S. Kim, H. J. Lee, H. B. Kim, M. H. Jung, J. Park, H. C. Ko and J. Y. Jo, Adv. Electron. Mater., 2019, 5, 1800786 CrossRef.
- K. Kato, H. Hagino and K. Miyazaki, J. Electron. Mater., 2013, 42, 1313–1318 CrossRef CAS.
- Y. Du, K. Cai, S. Chen, P. Cizek and T. Lin, ACS Appl. Mater. Interfaces, 2014, 6, 5735–5743 CrossRef CAS PubMed.
- M. Bharti, A. Singh, G. Saini, S. Saha, A. Bohra, Y. Kaneko, A. Debnath, K. Muthe, K. Marumoto and D. Aswal, J. Power Sources, 2019, 435, 226758 CrossRef CAS.
- J. Xiong, L. Wang, J. Xu, C. Liu, W. Zhou, H. Shi, Q. Jiang and F. Jiang, J. Mater. Sci.: Mater. Electron., 2016, 27, 1769–1776 CrossRef CAS.
- J. Alam, X. Xu, P. C. O. Adu, Q. Meng, K. Zuber, S. Afshar, H.-C. Kuan and J. Ma, Adv. Nanocompos., 2024, 1, 16–38 CrossRef.
- E. P. Tomlinson, M. J. Willmore, X. Zhu, S. W. Hilsmier and B. W. Boudouris, ACS Appl. Mater. Interfaces, 2015, 7, 18195–18200 CrossRef CAS PubMed.
- Z. Fan and J. Ouyang, Adv. Electron. Mater., 2019, 5, 1800769 CrossRef CAS.
- T. Wu, X.-L. Shi, W.-D. Liu, M. Li, F. Yue, P. Huang, Q. Liu and Z.-G. Chen, Adv. Fiber Mater., 2024, 1–12 Search PubMed.
- L. Zhang, B. Xia, X.-L. Shi, W.-D. Liu, Y. Yang, X. Hou, X. Ye, G. Suo and Z.-G. Chen, Carbon, 2022, 196, 718–726 CrossRef CAS.
- J. Y. Lim, S. Cho, H. Kim and Y. Seo, ACS Appl. Energy Mater., 2019, 2, 8219–8228 CrossRef CAS.
- W. S. Kim, G. Anoop, I.-S. Jeong, H. J. Lee, H. B. Kim, S. H. Kim, G. W. Goo, H. Lee, H. J. Lee and C. Kim, Nano Energy, 2020, 67, 104207 CrossRef CAS.
- Y. Wang, M. Hong, W.-D. Liu, X.-L. Shi, S.-D. Xu, Q. Sun, H. Gao, S. Lu, J. Zou and Z.-G. Chen, Chem. Eng. J., 2020, 397, 125360 CrossRef CAS.
- Z. Fan, P. Li, D. Du and J. Ouyang, Adv. Energy Mater., 2017, 7, 1602116 CrossRef.
- J. Luo, D. Billep, T. Waechtler, T. Otto, M. Toader, O. Gordan, E. Sheremet, J. Martin, M. Hietschold and D. R. Zahn, J. Mater. Chem. A, 2013, 1, 7576–7583 RSC.
- X. Liu, X.-L. Shi, L. Zhang, W.-D. Liu, Y. Yang and Z.-G. Chen, J. Mater. Sci. Technol., 2023, 132, 81–89 CrossRef CAS.
- H. Wang and C. Yu, Joule, 2019, 3, 53–80 CrossRef CAS.
- X. Gao, K. Uehara, D. D. Klug, S. Patchkovskii, S. T. John and T. M. Tritt, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 125202 CrossRef.
- P. B. Kaul, K. A. Day and A. R. Abramson, J. Appl. Phys., 2007, 101, 083507 CrossRef.
- J. Wang, K. Cai and S. Shen, Org. Electron., 2014, 15, 3087–3095 CrossRef CAS.
- H. Park, S. H. Lee, F. S. Kim, H. H. Choi, I. W. Cheong and J. H. Kim, J. Mater. Chem. A, 2014, 2, 6532–6539 RSC.
- Z. Zhu, C. Liu, Q. Jiang, H. Shi, F. Jiang, J. Xu, J. Xiong and E. Liu, J. Mater. Sci.: Mater. Electron., 2015, 26, 8515–8521 CrossRef CAS.
- Y. Xu, Z. Liu, X. Wei, J. Wu, J. Guo, B. Zhao, H. Wang, S. Chen and Y. Dou, Synth. Met., 2021, 271, 116628 CrossRef CAS.
- S. Liu, H. Deng, Y. Zhao, S. Ren and Q. Fu, RSC Adv., 2015, 5, 1910–1917 RSC.
- J. Li, A. B. Huckleby and M. Zhang, J. Materiomics, 2022, 8, 204–220 CrossRef.
- S. Masoumi, A. Noori and A. Pakdel, Measurement, 2024, 236, 115162 CrossRef.
- F. Hossein-Babaei, S. Masoumi and A. Noori, Meas. Sci. Technol., 2017, 28, 115002 CrossRef.
- R. Xiong, S. Masoumi and A. Pakdel, Energies, 2023, 16, 6319 CrossRef CAS.
- B. Zhang, J. Sun, H. Katz, F. Fang and R. Opila, ACS Appl. Mater. Interfaces, 2010, 2, 3170–3178 CrossRef CAS PubMed.
- D. A. Mengistie, C.-H. Chen, K. M. Boopathi, F. W. Pranoto, L.-J. Li and C.-W. Chu, ACS Appl. Mater. Interfaces, 2015, 7, 94–100 CrossRef CAS PubMed.
- H. Ju and J. Kim, ACS Nano, 2016, 10, 5730–5739 CrossRef CAS PubMed.
- Y. Gelbstein, J. Appl. Phys., 2009, 105, 023713 CrossRef.
- Z. Liang, M. J. Boland, K. Butrouna, D. R. Strachan and K. R. Graham, J. Mater. Chem. A, 2017, 5, 15891–15900 RSC.
- C. Chen, I. E. Jacobs, K. Kang, Y. Lin, C. Jellett, B. Kang, S. B. Lee, Y. Huang, M. BaloochQarai and R. Ghosh, Adv. Energy Mater., 2023, 13, 2202797 CrossRef CAS.
- X. Huang, L. Deng, F. Liu, Z. Liu and G. Chen, Chem. Eng. J., 2021, 417, 129230 CrossRef CAS.
- A. M. Nardes, R. A. Janssen and M. Kemerink, Adv. Funct. Mater., 2008, 18, 865–871 CrossRef CAS.
- H. Yao, Z. Fan, H. Cheng, X. Guan, C. Wang, K. Sun and J. Ouyang, Macromol. Rapid Commun., 2018, 39, 1700727 CrossRef PubMed.
- J. Ouyang, Q. Xu, C.-W. Chu, Y. Yang, G. Li and J. Shinar, Polymer, 2004, 45, 8443–8450 CrossRef CAS.
- K. Sun, S. Zhang, P. Li, Y. Xia, X. Zhang, D. Du, F. H. Isikgor and J. Ouyang, J. Mater. Sci.: Mater. Electron., 2015, 26, 4438–4462 CrossRef CAS.
- Y. Xu, Y. Jia, P. Liu, Q. Jiang, D. Hu and Y. Ma, Chem. Eng. J., 2021, 404, 126552 CrossRef CAS.
- S. Mahato, J. Puigdollers, C. Voz, M. Mukhopadhyay, M. Mukherjee and S. Hazra, Appl. Surf. Sci., 2020, 499, 143967 CrossRef CAS.
- M. Bharti, A. Singh, S. Samanta, A. Debnath, K. Marumoto, D. Aswal, K. Muthe and S. Gadkari, Vacuum, 2018, 153, 238–247 CrossRef CAS.
- K. Aasmundtveit, E. Samuelsen, L. Pettersson, O. Inganäs, T. Johansson and R. Feidenhans, Synth. Met., 1999, 101, 561–564 CrossRef CAS.
- O. Bubnova, Z. U. Khan, A. Malti, S. Braun, M. Fahlman, M. Berggren and X. Crispin, Nat. Mater., 2011, 10, 429–433 CrossRef CAS PubMed.
- R. Kroon, D. A. Mengistie, D. Kiefer, J. Hynynen, J. D. Ryan, L. Yu and C. Müller, Chem. Soc. Rev., 2016, 45, 6147–6164 RSC.
- M. Cutler, J. Leavy and R. Fitzpatrick, Phys. Rev., 1964, 133, A1143 CrossRef.
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 CrossRef CAS PubMed.
- S. H. Lee, H. Park, W. Son, H. H. Choi and J. H. Kim, J. Mater. Chem. A, 2014, 2, 13380–13387 RSC.
- G.-H. Kim, L. Shao, K. Zhang and K. P. Pipe, Nat. Mater., 2013, 12, 719–723 CrossRef CAS PubMed.
- Y. Xia, K. Sun and J. Ouyang, Energy Environ. Sci., 2012, 5, 5325–5332 RSC.
- Y. Xia, K. Sun and J. Ouyang, Adv. Mater., 2012, 24, 2436–2440 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc00881b |
| This journal is © The Royal Society of Chemistry 2024 |