
Selective oxidation of linear alcohols: the promotional effect of water and inhibiting effect of carboxylates over dilute PdAu catalysts†
Jennifer D.
Lee‡§
 *a,
Amanda
Filie§
b,
Leigh
Wilson
a,
Karin
Nguyen
c,
Tanya
Shirman
ab,
Erjia
Guan
d,
Mathilde
Luneau
a,
Michael
Aizenberg
b,
Joanna
Aizenberg
ab,
Robert J.
Madix¶
b and
Cynthia M.
Friend
*ab
*a,
Amanda
Filie§
b,
Leigh
Wilson
a,
Karin
Nguyen
c,
Tanya
Shirman
ab,
Erjia
Guan
d,
Mathilde
Luneau
a,
Michael
Aizenberg
b,
Joanna
Aizenberg
ab,
Robert J.
Madix¶
b and
Cynthia M.
Friend
*ab
aDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, MA 02138, USA. E-mail: jenniferlee@ucmerced.edu; friend@fas.harvard.edu
bJohn A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA 02138, USA
cDepartment of Chemistry and Pharmacy, University of Freiburg, Freiburg, Germany
dDepartment of Materials Science and Chemical Engineering, Stony Brook University, Stony Brook, New York 11794, USA
First published on 14th May 2024
Abstract
Dilute PdAu alloys are promising catalysts for selective oxidation with high activity and high selectivity. The promotional effect of Pd for selective alcohol oxidation is strongly dependent on the alkyl chain length. Whereas Pd substantially promotes the selective oxidation of methanol, there is essentially no promotion of oxidation of longer-chain alcohols (C2–C4) for Pd3Au97 RCT-SiO2 compared to pure Au RCT-SiO2. The lack of promotion of selective oxidation of higher alcohols by Pd is attributed to their over-oxidation on the Pd3Au97 RCT-SiO2 catalyst, yielding strongly bound carboxylates that poison the Pd sites. The longer-chain carboxylates are stable on the surface under reaction conditions whereas formate, which would form from over-oxidation of methanol, decomposes. The surface carboxylate was identified for 1-propanol oxidation as propionate using in situ Fourier transform infrared (FTIR) spectroscopy. Further evidence for poisoning by carboxylates is the production of CO2 in temperature programmed oxidation following catalyst use. The poisoned catalyst can be regenerated in a flowing methanol/oxygen mixture, restoring the promotional effect of Pd for methanol oxidation. The residual activity of the Au and of the poisoned Pd/Au catalysts may be due to the migration of hydroxyl from the silica support to the nanoparticle. A reservoir of hydroxyl groups on the RCT-SiO2 is identified using FTIR. Hydroxyls on Au are known to spontaneously disproportionate to adsorbed oxygen atoms and water. These results demonstrate the sensitivity of dilute alloy catalyst reactivity to molecular structure.
Introduction
Interest in using Au-based catalysts for selective oxidation reactions has grown since Au was discovered as a highly selective catalyst, enabling more sustainable chemical processes.1–8 Due to the nature of supported Au nanoparticle catalysts, numerous material properties related to the Au nanoparticles and/or the supporting metal oxide could be associated with the activity of Au catalysts.3 Although the probability of molecular O2 dissociation on under-coordinated Au atoms is significantly greater than on extended Au surfaces,9 additional means of activating molecular O2 can be used to deliver oxygen to the Au surfaces of a supported Au nanoparticle catalyst, namely through the metal oxide support3,10 or through a dopant metal active for molecular O2 dissociation, such as Pd.Supports play an important role in determining the activity of supported Au nanoparticle catalysts. Since the initial discovery and studies of its activity for the oxidation of CO,11,12 several more studies on the selective oxidation chemistry of supported Au nanoparticle catalysts have followed.13–18 It has been found that Au/SiO2 has decent activity and high selectivity for the oxidation of aliphatic alcohols (specifically 1-propanol and 1-butanol, among other alcohols) between 523 K and 573 K.19 Typically, the reducible oxide supported Au catalysts are considered more active for oxidation reactions since the oxygen species on the reducible support are considered more mobile than those on the irreducible support, thus enabling the migration of oxygen species from the support to the Au nanoparticle. It has been shown that co-feeding H2O increases the activity of supported Au/SiO2, among other supports, for CO oxidation20,21 and selective propene oxidation.22–25 This raises the question as to what role H2O would play in selective oxidation over Au catalysts, as it is a byproduct in selective oxidation over Au. Only a few studies on the effect of water on the activity of ceria supported Au for oxidative ethanol dehydrogenation26 and the partial oxidation of allyl alcohols on oxygen-covered Au single-crystals have been performed;27,28 much remains to be understood about the role of H2O present on Au catalysts for the numerous selective oxidation reactions Au catalyzes.
The use of bimetallic alloys has proven to be another successful approach to generating selective oxidation Au-based catalysts. Nanoporous AgAu (npAgAu), containing ∼3 at% Ag, is one example of an Au-based bimetallic alloy catalyst that is highly selective to forming methyl ester for the oxidative self-coupling of methanol.29,30 It has been shown that the oxidative methanol self-coupling network, determined from model studies of Au in ultra-high vacuum, can be used as the basis of a microkinetic model to predict the performance of npAgAu over a wide range of pressures and temperatures;31,32 this reinforces the applicability of mechanisms determined over model Au catalysts in ultra-high vacuum to npAgAu catalysts in a flow reactor.33,34 More recently, the reaction steps of the same microkinetic model for the oxidative methanol coupling were modified using density functional theory (DFT) scaling relations to accurately predict the increase in selectivity to aldehyde formation as the alkyl chain length increases for the oxidative self-coupling of C1 to C4 aliphatic alcohols over npAgAu in a plug flow reactor.35 The predicted selectivity towards aldehyde formation in the oxidation of C2 to C4 alcohols on an Au catalyst agrees with gas-phase reactor studies of selective oxidation of 1-propanol and 1-butanol over Au/SiO2,19 suggesting this reaction mechanism can also be applied to supported nanoparticle catalysts. Several studies also show that supported PdAu alloy catalysts are active for the selective oxidation of alcohols,36–40 raising the question as to whether the oxidative coupling mechanism over Au would adequately predict the activity of Pd-doped Au catalysts.
Efforts to further test the applicability of the Au-based oxidative alcohol self-coupling reaction network to supported bimetallic Au-based nanoparticles are needed. As a first step in this endeavor, supported dilute Pd-in-Au (PdxAu1−x, x = 1.7 and 3.4 at%) nanoparticles on raspberry colloid templated silica (RCT-SiO2) were studied for the oxidative self-coupling of methanol.41 The RCT catalysts in general, and the PdxAu1−x RCT-SiO2 catalysts, in particular, are stable under oxidative reaction conditions at elevated temperatures and have remarkable resistance against sintering,41–44 two features that allow for a wide range of temperatures to be tested without compromising the performance of the catalyst material over multiple testing cycles. Introducing as little as 3 at% Pd metal to the Au nanoparticles promotes activity for methanol coupling between 363 K and 448 K, resulting in significant activity and high selectivity to methyl formate.41 The importance of available Pd atoms at the surface to activate molecular oxygen and initiate the catalyst cycle to produce methyl formate was evident from the lack of activity under non-oxidative conditions and the nearly complete suppression of methyl formate formation when CO was co-fed with a methanol/O2 mixture over Pd3Au97 RCT-SiO2. It is demonstrated that the small amount of Pd is responsible for the activation of molecular O2 to initiate the catalyst cycle much like the small amount of Ag in npAgAu is, with nearly 10 times higher activity in methanol conversion comparing Pd3Au97 RCT-SiO2 to npAgAu.41
The capacity of carboxylates, an intermediate in the oxidative alcohol coupling mechanism over Au(111),45 to be strongly bound to the catalytically active metal centers makes them a potential surface poison and motivates the study of their stability in model systems comprising Au, Pd and PdAu in ultra-high vacuum. Co-adsorbed atomic oxygen stabilizes carboxylates on Pd(111) surface,46,47 while it destabilizes the intermediates on Au(111).48–50 The stability of carboxylates on PdAu alloys remains vastly under-investigated, especially in the dilute Pd-in-Au regime. However, it has been shown that acetic acid immediately forms acetate on PdAu alloys where Pd ensembles are present (i.e. 0.4 ML of Au or less on Pd(100) in ultra-high vacuum), while for alloys containing only isolated Pd, the acetic acid molecular adsorbs, only forming some acetate upon heating.51 Furthermore, it was shown that both alloying Au with Pd and the presence of co-adsorbed oxygen increase the stability of the acetate species much like was observed in the studies of acetate decomposition on Pd(110)52 and Pd(111).47 The acetate stability on PdAu alloys reveals a potential challenge in creating PdAu alloy catalysts for selective alcohol oxidation reaction; however, it also brings the opportunity to tune reaction conditions to free Pd ensembles from the active site blocking carboxylate.
Herein, both pure Au and Pd3Au97 RCT-SiO2 are demonstrated to catalyze the oxidative alcohol coupling of longer chain (C2–C4) alcohols. The activity of the Pd-doped Au RCT catalyst was similar to that of the pure Au counterpart. The activity of the pure Au RCT-SiO2 is attributed to the migration of hydroxyl from the SiO2 support to the Au nanoparticles, leading to the activation of the alcohols. A reservoir of hydroxyl groups on the SiO2 support was shown using in situ Fourier transform infrared (FTIR) spectroscopy over a wide temperature range (298 K to 773 K). Furthermore, we show that water promotes the activity of the two catalysts studied in the oxidation of 1-propanol at 423 K. This is the result of hydroxyls on Au reacting with alcohols to produce water and an alkoxy, forming the key intermediate in the overall process. The overall effect of Pd in the Pd3Au97 RCT-SiO2 catalysts on the oxidation of ethanol, 1-propanol and 1-butanol was minimal, whereas previous work showed that Pd substantially promotes methanol oxidation. This is due to the higher stability of carboxylates that formed during the over-oxidation of the longer chain alcohols. The surface carboxylate species was identified as propionate formed under 1-propanol oxidation over Pd3Au97 RCT-SiO2 catalyst based on in situ FTIR studies. The higher stability of the linearly bound propionate results in the poisoning of surface Pd sites on the Pd3Au97 RCT-SiO2 catalyst, leading to a reduction in reaction selectivity. The production of CO2 in temperature programmed oxidation following catalyst use further provides evidence for surface poisoning. The catalyst poisoned during 1-propanol oxidation can be further regenerated in a flowing methanol/oxygen mixture, restoring the promotional effect of Pd for methanol oxidation, as observed from reactor and in situ FTIR studies. These results demonstrate the sensitivity of dilute alloy catalyst reactivity to surface molecular structure.
Experimental methods
Material synthesis and characterization
The SiO2-supported nanoparticle (NP) catalysts were prepared using the raspberry colloid templated (RCT) method as described previously.42 Three catalysts were investigated: pure Au, 3 at% Pd-in-Au, and pure Pd (Au RCT-SiO2, Pd3Au97 RCT-SiO2, and Pd RCT-SiO2, respectively). In brief, the bimetallic NP catalysts were prepared by first synthesizing pure Au NPs53 and then adding a calculated volume of an aqueous solution of Pd nitrate to yield the specified Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au ratio together with ascorbic acid aqueous solution as the reducing agent. The pure Pd NPs were synthesized based on a published method.42 The so-called “raspberry colloid templates” (RCTs) were prepared by adding specific amounts of Au or Pd3Au97 NPs to a solution containing thiol-modified polystyrene colloids. Once the RCTs underwent self-assembly during evaporation of water, the voids between RCTs were filled with pre-hydrolyzed tetraethyl orthosilicate (TEOS) and calcined in air at 773 K. The resulting catalyst materials are referred to as “as-prepared.” The final calcination step helps reduce the defect sites that might be present on bimetallic Pd-in-Au NP catalysts during the synthesis. The raspberry colloid templated SiO2 supported bimetallic Pd-in-Au NP catalysts (Pd3Au97 RCT-SiO2) are comprised of a highly ordered and porous SiO254,55 with substantially embedded nanoparticles (Fig. S1†).56 SiO2 support was selected to allow evaluation of the catalytic activity to the metal nanoparticles and to provide high sinter resistance for the materials. The average particle diameter and the Pd atomic percent (at%), and the total metal loading (wt%) of each catalyst have already been reported41 and are summarized here (Table 1). Transmission electron microscopy was performed with an aberration-corrected JEOL ARM 200F, operating at 200 kV and in scanning transmission electron microscopy (STEM) mode. The elemental composition of Pd and Au, and the total metal loading were determined by inductively coupled plasma−mass spectrometry (ICP-MS, Agilent 7900). The PdAu NPs of Pd3Au97 RCT-SiO2 catalysts are well-alloyed and its support structure highly ordered.41
:Au ratio together with ascorbic acid aqueous solution as the reducing agent. The pure Pd NPs were synthesized based on a published method.42 The so-called “raspberry colloid templates” (RCTs) were prepared by adding specific amounts of Au or Pd3Au97 NPs to a solution containing thiol-modified polystyrene colloids. Once the RCTs underwent self-assembly during evaporation of water, the voids between RCTs were filled with pre-hydrolyzed tetraethyl orthosilicate (TEOS) and calcined in air at 773 K. The resulting catalyst materials are referred to as “as-prepared.” The final calcination step helps reduce the defect sites that might be present on bimetallic Pd-in-Au NP catalysts during the synthesis. The raspberry colloid templated SiO2 supported bimetallic Pd-in-Au NP catalysts (Pd3Au97 RCT-SiO2) are comprised of a highly ordered and porous SiO254,55 with substantially embedded nanoparticles (Fig. S1†).56 SiO2 support was selected to allow evaluation of the catalytic activity to the metal nanoparticles and to provide high sinter resistance for the materials. The average particle diameter and the Pd atomic percent (at%), and the total metal loading (wt%) of each catalyst have already been reported41 and are summarized here (Table 1). Transmission electron microscopy was performed with an aberration-corrected JEOL ARM 200F, operating at 200 kV and in scanning transmission electron microscopy (STEM) mode. The elemental composition of Pd and Au, and the total metal loading were determined by inductively coupled plasma−mass spectrometry (ICP-MS, Agilent 7900). The PdAu NPs of Pd3Au97 RCT-SiO2 catalysts are well-alloyed and its support structure highly ordered.41
Nanoporous dilute Ag-in-Au (npAgAu) catalysts were also prepared for comparison using a previously reported method.30 Briefly, the Ag of 6 karat white Au sheets (15% Au in Ag) was removed by chemical etching with a nitric acid solution, followed by washing with water and drying under He at 323 K. The npAgAu was activated by heating the sample from 298 to 423 K and holding at 423 K for 1 h under an ozone environment. The npAgAu materials have been characterized thoroughly in previous studies, specifically their ligament diameters and pore size.30
Infrared spectroscopy
Fourier transform infrared (FTIR) spectra of Pd3Au97 RCT-SiO2 during heating and under alcohol oxidation reactions were collected by loading catalysts into a Harrick Praying Mantis low temperature reaction chamber with KBr windows mounted inside a vacuum Fourier transform infrared (FTIR) spectrometer (Bruker Vertex 70V) with a mercury cadmium telluride (MCT) detector cooled by liquid nitrogen. All samples were measured in diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) configuration. IR spectra between 4000 cm−1 and 650 cm−1 were collected with 2 cm−1 resolution. For IR characterization during heating, samples were heated at 1 bar with 20 mL min−1 O2 to 773 K using a heating rate of 10 K min−1 while continuously collecting FTIR signals. For characterization under alcohol oxidation reactions, catalysts were pretreated at 1 bar with 20 mL min−1 O2 at 773 K for 15 min using a heating rate of 10 K min−1, followed by cooling in He to 423 K. The alcohol oxidation reaction tests and regeneration experiments were then carried out in the same DRIFTS cell at 1 bar by introducing reactants (4% alcohol and 10% O2 balanced in He; total flow rate = 20 mL min−1) and maintaining the temperature at 423 K until the FTIR signal stabilizes, typically takes ∼1–1.5 h.Catalytic measurements
Catalyst testing was conducted in a fixed bed gas-phase flow reactor under atmospheric pressure, and the amount of alcohol vapor in the reactant stream was controlled as described previously.57 The powders of as-prepared Au and Pd3Au97 RCT-SiO2 catalysts synthesized in the same manner as in prior studies,41 were crushed and sieved to obtain particle sizes between 100–300 μm in diameter. The catalyst beds with ∼10 or 40 mg of the crushed powder were loaded into a tubular reactor (inner diameter: 1 cm) and diluted with quartz sand (particle sizes 210–300 μm) to a total catalyst bed length of 1 cm.Prior to activity measurements, all catalysts were pretreated at 1 bar with 20% O2 balanced in He with a total flow rate of 50 mL min−1 (gas hourly space velocity of 3800 h−1) at 773 K for 15 min using a heating rate of 10 K min−1. Thereafter, the O2-pretreated catalyst was cooled in He to 423 K, at which point reaction conditions (4% alcohol and 10% O2 balanced in He; total flow rate = 50 mL min−1) were initiated to stabilize the catalyst activity; for the oxidation of ethanol, 1-propanol and 1-butanol the catalyst activity stabilized immediately, while in the case of methanol stabilization took ∼1.5 h. The standard procedure described was followed for experiments where 1% H2O was co-fed during 1-propanol oxidation reaction at 423 K, and where regeneration was performed with methanol replacing 1-propanol at 423 K. Only the experiments where 2% CO was co-fed during alcohol oxidation deviated slightly from the standard procedure described; the reaction was initiated at 473 or 573 K as opposed to 423 K.
For testing npAgAu catalysts, the sample (18.0 mg) was stabilized under methanol oxidation at 423 K (10% methanol and 20% O2 balanced in He; total flow rate = 50 mL min−1) for ∼18 h. Then the partial pressure of O2 was decreased (10% methanol and 10% O2 balanced in He; total flow rate = 50 mL min−1) and allowed to stabilize. The change in partial pressure was to allow the co-feeding of H2O at the same total flow rate without having to heat the water saturator and reactor lines, thus reducing the possibility of water condensation that would lead to erroneous vapor concentrations.
The reactor effluent was analyzed using a gas chromatograph (Agilent 7890A series, Column HP-Plot/Q) coupled with a mass spectrometer (Agilent 5975C series, inert MSD with triple axis detector) (equations for conversion of alcohol and selectivity to each product are provided in ESI†). The deposition of carbon containing species onto the surface was determined by an online mass spectrometer (MS, Hiden HAL 301/3F Series) after the alcohol oxidation reaction at 423 K for ∼2 h by measuring the evolution of CO2 (m/z = 44) during the temperature programmed oxidation (TPO) of the catalyst (at 1 bar with 10% O2 balanced with He; total flow rate = 20 mL min−1) from 323 to 773 K using a heating rate of 10 K min−1.
Results
The oxidation of alcohols over Au RCT-SiO2
The silica (RCT-SiO2) supporting nanoparticles was found to be hydroxyl-rich from room temperature up to 773 K (Fig. 1a), which ensured that it can serve as a reservoir of hydroxyls over this temperature range. At room temperature (298 K), the in situ Fourier transform infrared (FTIR) spectra of Au RCT-SiO2 exhibited a peak centered at ν ∼3650 cm−1 and a peak centered at ν ∼3520 cm−1; the former is assigned to isolated, non-interacting silica hydroxyl groups (i.e. silanol, Si–OH) and the latter is assigned to H-bonded Si–OH.58,59 The shoulder at ν ∼3250 cm−1 is assigned to Si–OH groups perturbed by adsorbed molecular H2O.58,59 | ||
| Fig. 1 (a) In situ FTIR spectra of as-prepared Au RCT-SiO2 in the 4000–2700 cm−1 region collected as a function of temperature from 298 to 773 K in O2. (b) The rate of 1-propanol oxidation over Au RCT-SiO2 in the absence and presence of co-fed 1% H2O at 423 K. Dashed lines in (a) denoted the assignments of silanol groups. Reaction conditions in (a): 100% O2; flow rate = 20 mL min−1, (b): 4% 1-propanol, 10% O2, 0 or 1% H2O, balanced in He; total flow rate = 50 mL min−1; mcat = 10.0 mg. | ||
The silica hydroxyls of the as-prepared Au RCT-SiO2 between 298–473 K were found by FTIR to be either non-interacting or hydrogen-bonded with either neighboring silanols or adsorbed water. Between 473–573 K, the isolated, non-interacting Si–OH bonds (ν ∼3650 cm−1) persisted and the H-bonded Si–OH (ν ∼3520 cm−1) decreased. As the temperature was further increased to 673–773 K, the signal corresponding to the non-interacting Si–OH (ν ∼3650 cm−1) decreased slightly and that corresponding to the H-bonded Si–OH (ν ∼3520 cm−1) disappeared entirely. This agrees with the notion that as the silica-hydroxyl coverage decreases there is less chance for hydrogen bonding as silica hydroxyls are depleted. Non-interacting Si–OH (ν ∼3650 cm−1) are still present even after holding the cell temperature at 773 K for 15 min (same conditions as the pretreatment carried out before alcohol oxidation reactions). Altogether, these in situ FTIR data show the presence of hydroxyls on the support of the Au RCT-SiO2 catalysts over a wide temperature range, from 298 to 773 K. Furthermore, the hydroxyl-rich environment of RCT-SiO2 between 298 and 773 K is independent of the nanoparticle composition as shown with a similar trend observed over RCT-SiO2 and Pd3Au97 RCT-SiO2 (Fig. S2†).
Evidence that the hydroxyls on the RCT-SiO2 support play a significant role in the activity of the Au RCT-SiO2 catalyst in the oxidation of alcohols was demonstrated by co-feeding H2O with the reactant stream during the experiment (Fig. 1b). The O2-pretreated Au RCT-SiO2 was initially stabilized under reaction conditions (4% 1-propanol, 10% O2) for ∼1.4 h, exhibiting a propanol oxidation rate of 4 μmol 1-propanol gcat−1 s−1. When 1% H2O was co-fed with the reactant mixture, the activity towards 1-propanol increased by a factor of ∼5 over the course of 2.5 h. The Au RCT-SiO2 is necessary for the oxidation reaction to occur because the control experiment using blank (no Au NPs) RCT-SiO2 support showed only negligible activity under the same reaction conditions (Fig. S3a†). Similarly to the Au RCT-SiO2 case, the reaction rate over Pd3Au97 RCT-SiO2 increased by a factor of ∼3 when 1% H2O was co-fed at 423 K (Fig. S3b†). The hypothesis that the SiO2 plays an essential role in the activity of the Au-based RCT-SiO2 catalysts by Si–OH directly activating alcohols or supplying hydroxyls to the Au nanoparticle surface, is reinforced by the lack of promotional effect co-fed water had on the activity of a support-free dilute Ag-in-Au catalyst (npAgAu) for oxidative methanol coupling (Fig. S4†). Furthermore, the Au nanoparticle-SiO2 interface was essential to the observed activity of the Au RCT-SiO2 catalyst for the oxidation of 1-propanol between 423 K and 573 K, see ESI, Tables S1 and S2† for a more detailed discussion.
In addition to promoting the conversion of 1-propanol over the Au RCT-SiO2 at 423 K, co-feeding water also increased the selectivity to propyl propionate (Table S1†). A similar increase in selectivity towards the coupled product was observed on Pd3Au97 RCT-SiO2 (Table S1†). The increase in conversion of 1-propanol and selectivity towards coupled product when 1% H2O was co-fed can be explained by a disproportionation reaction of hydroxyls over the Au NPs, as has been observed on single crystal studies of Au.60,61 The disproportionation of hydroxyls over Au to form water and adsorbed atomic oxygen results in a greater surface coverage of active sites, in this case, adsorbed atomic oxygen, and, consequently, adsorbed reactant intermediates that would lead to production of propanal and propyl propionate. A more detailed discussion of how hydroxyls could be formed in higher amounts on PdxAu1−x RCT-SiO2 catalysts is presented in the Discussion section.
The oxidation of alcohols over Pd3Au97 RCT-SiO2
Unlike its significant promotional effect in the oxidation of methanol (C1 alcohol) at 423 K, the Pd in Pd3Au97 RCT-SiO2 had little effect in promoting activity towards the higher chain alcohols (C2 to C4 alcohols) under the same reaction conditions (Fig. 2). The activity of Au RCT-SiO2 towards the C1 to C4 alcohols varied between ∼12% and 28% conversion (Fig. 2a). As expected, based on previous studies of the PdxAu1−x RCT-SiO2 systems for oxidative methanol coupling,41 Pd3Au97 RCT-SiO2 (Fig. 2b) showed a much greater activity towards methanol than Au RCT-SiO2. However, the conversion of ethanol, 1-propanol, and 1-butanol was similar over both catalysts. The activity of Pd3Au97 RCT-SiO2 towards methanol was greater than that of Au RCT-SiO2 by a factor of ∼3, but this factor collapsed to ∼1 for the higher chain alcohols. This result strongly suggests the Pd active site, which has been proven essential to activating molecular O2 and consequently promoting oxidative methanol coupling,41 was blocked at 423 K under reaction conditions for the oxidation of higher chain alcohols. Pure Pd RCT-SiO2 was also compared as a control and showed greater activity towards methanol than Au RCT-SiO2 (Table S3†). However, due to its highly exothermic behavior and instability under alcohol oxidation reaction conditions, sudden increases in CO2 production were observed over time and a steady-state conversion was not achievable for pure Pd RCT-SiO2. | ||
| Fig. 2 The conversion of C1 to C4 alcohols and the rate of alcohol oxidation at 423 K over (a and c) Au and (b and d) Pd3Au97 RCT-SiO2, and the corresponding selectivity over (e) Au and (f) Pd3Au97 RCT-SiO2. Reaction products in (e and f) are listed in Table S4.† Complete data sets are listed in Table S5.† Reaction conditions: 4% alcohol, 10% O2, balanced in He; total flow rate = 50 mL min−1; mcat = 40.0 and 40.5 mg for Au RCT-SiO2 and Pd3Au97 RCT-SiO2, respectively. | ||
Oxygen is required for alcohol conversion at the temperatures investigated as demonstrated by the lack of methanol and 1-propanol conversion under non-oxidative conditions over Au and Pd3Au97 RCT-SiO2 (Table S6†). The rates of methanol or 1-propanol conversion over Au and Pd3Au97 RCT-SiO2 catalysts under non-oxidative conditions were negligible. The necessity for O2 to convert alcohols is consistent with previous report on methanol oxidation,41 and Pd itself does not induce O–H bond activation under these conditions.
The small amount of alkene products with one carbon less than the parent alcohol (C3 and C4 alcohol), observed in this study, and near equimolar amount of CO2 formed (Fig. 2f) suggest the Pd in Pd3Au97 RCT-SiO2 might not be entirely blocked and thus creates an alternative reaction pathway involving a carbon–carbon bond scission. The selectivity for the oxidation of the C1 to C4 alcohols over Au RCT-SiO2 (Fig. 2e) agrees with the selectivity predicted for the oxidation of these alcohols over npAgAu based on a combination of microkinetic modelling and plug flow reactor simulation.35 The network predicts a drastic decline in selectivity to coupled product and a concomitant increase in selectivity to aldehyde from oxidizing the C1 alcohol to oxidizing the C2 alcohol and above. A similar trend for the aldehyde and coupled products was observed over Pd3Au97 RCT-SiO2 as the molecular weight of the alcohol increased (Fig. 2f). However, ethylene and propylene were observed during the oxidation of 1-propanol and 1-butanol, respectively. No hydrocarbon was formed when testing ethanol. The formation of the alkene products is not predicted by the Au-based reaction network35 and suggests the reaction pathway exists on Pd3Au97 RCT-SiO2 as a result of its Pd.
To investigate reaction conditions that might render the Pd containing catalyst more active than the pure Au catalyst, the activity of 1-propanol was tested over a temperature range spanning 423 K and 573 K over both catalysts (Fig. S5†). The conversion of methanol over Au RCT-SiO2 at 423 K was ∼11%, whereas over Pd3Au97 RCT-SiO2 this was ∼32% (Fig. S5a and b,† respectively). The conversion of 1-propanol over Au RCT-SiO2 was ∼3%, whereas over Pd3Au97 RCT-SiO2 was ∼6%. Thus, the conversion of methanol and 1-propanol increased by a factor of 3 and 2, respectively, over the Pd-containing catalyst as compared to the pure Au catalyst. When oxidizing either alcohol, the promotional effect of Pd3Au97 RCT-SiO2 diminished as the temperature increased from 423 K to 573 K so that this factor approached 1. It should be noted that the difference in activity between methanol and 1-propanol over Au RCT-SiO2 approaches zero at 573 K while over Pd3Au97 RCT-SiO2 this occurs near 523 K. The Pd3Au97 RCT-SiO2 catalyst had little effect in promoting desirable activity towards oxidation of 1-propanol even at temperatures greater than 423 K (Fig. S5†) calling into question why Pd does not promote alcohol activation even when catalyst surface is free of residual surface carbon. At least two arguments could be made to explain the similar activity of Au RCT-SiO2 and Pd3Au97 RCT-SiO2 at 573 K. First, the activity due to the Au–SiO2 interface may dominate at this temperature. Second, Pd at the surface may have been driven to the bulk of the nanoparticle as a result of the elevated temperature. However, the dependence of selectivity on temperature suggests substantial Pd is at the surface of the catalysts even at 573 K.
The dependence of selectivity on temperature over Pd3Au97 RCT-SiO2 during both the oxidative self-coupling of methanol and the oxidation of 1-propanol reveals Pd promotes oxygen dissociation on the predominantly Au nanoparticle. The selectivity for methyl formate and CO2 was 95% and 5%, respectively, over Au at 423 K (Fig. S5e†). Under the same conditions, Pd3Au97 RCT-SiO2 exhibited a selectivity for methyl formate and CO2 of 82% and 16%, respectively (Fig. S5f†). The selectivity for methyl formate over both catalysts decreased as the temperature increased to 573 K (Fig. S5e and f†), but the selectivity to methyl formate over Pd3Au97 RCT-SiO2 was always lower than that of Au RCT-SiO2. A similar increase in conversion of methanol over the Pd-containing catalyst at 423 K that comes at a cost to selectivity for methyl formate qualitatively agrees with previous studies of oxidative methanol self-coupling under equimolar conditions (10% methanol, 10% O2) over these same PdxAu1−x RCT-SiO2 catalysts.41 Both Au and Pd3Au97 RCT-SiO2 produce significant formaldehyde under the reaction conditions investigated herein (4% methanol, 10% O2, temperature between 423 K and 573 K). Microkinetic models of the Au-based oxidative methanol coupling network predict an increase in selectivity for formaldehyde under the pressure conditions of the gas phase flow reactor as the temperature increases.31,32 This provides further evidence the Au-based oxidative alcohol self-coupling mechanism is generally applicable to dilute alloy systems where Au is the majority element.
The enhanced molecular oxygen dissociation over Pd3Au97 RCT-SiO2 that promoted activity towards methanol (Fig. 2b and S5b†) at a cost to selectivity to methyl formate (Fig. S5f†) seemed to have no impact on conversion of 1-propanol (Fig. S5b†) and introduced undesired byproducts, ethylene and equimolar amounts of CO2, as the temperature increased (Fig. S5h†). Unlike Au RCT-SiO2 which produces only propanal, propyl propionate, and small amounts of propyl acrylate (Fig. S5g†), Pd3Au97 RCT-SiO2 also produced ethylene accompanied by equimolar amounts of CO2 (Fig. S5h†). The selectivity for the olefin and CO2 increased with temperature between 473 K and 573 K, as would be expected if the surface bound intermediate leading these products were also associated with the residual surface carbon. Because studies of oxidative methanol coupling show molecular oxygen dissociation is promoted on Pd3Au97 RCT-SiO2,41 it is possible that sufficient oxygen is present on the surface to over-oxidize propanal to form propanoate; the consequences of forming this carboxylate are discussed later.
The effect of co-fed CO
The nature of the surface poison over Pd3Au97 RCT-SiO2 under high chain alcohol oxidation reaction conditions was further probed by co-feeding CO during the oxidation of methanol and 1-propanol (Fig. 3). After the activity of the Pd3Au97 RCT-SiO2 for oxidation of methanol was stabilized at 473 K, 2% CO was introduced into the reactant mixture and the rate of methanol oxidation decreased significantly (Fig. 3a) as expected based on previous studies of Pd3Au97 RCT-SiO2.41 When the same procedure was conducted at 573 K, the 2% CO minimally reduced the rate of methanol oxidation. Analogous experiments for the oxidation of 1-propanol showed that the poisoning effect of CO on the conversion of 1-propanol was significant at 473 K (Fig. 3b), but less significant than that observed for the oxidation of methanol. At 573 K the co-fed CO had minimal impact on the rate of 1-propanol reacted. It is important to note that dilute Pd-in-Au RCT-SiO2 catalysts show significant activity for CO oxidation at 523 K and above,42,43 thus it is expected that the poisoning effect at 573 K would be in part associated with increased turnover of CO at this temperature. | ||
| Fig. 3 The rate of (a) methanol and (b) 1-propanol oxidation in the absence (w/o CO) and presence (w/CO) of co-fed 2% CO at 473 K and 573 K over Pd3Au97 RCT-SiO2, and the corresponding yield of products during (c) methanol and (d) 1-propanol oxidation. Reaction conditions: 4% 1-propanol, 10% O2, 0 or 2% CO, balanced in He; total flow rate = 50 mL min−1; mcat = 10.0 mg. | ||
The corresponding product yields for the co-fed CO experiments suggest that CO binds significantly to Pd3Au97 RCT-SiO2 at both 473 K and 573 K (Fig. 3c and d). At 473 K, the presence of CO reduced the production of methyl formate and CO2 in methanol oxidation reaction (Fig. 3c), and to some extent the formaldehyde. At 573 K the dominant product is CO2. Although the dilute Pd-in-Au RCT-SiO2 catalysts oxidize CO at this temperature,42 co-feeding CO suppressed the over-oxidation pathway since the yield of CO2 decreased despite adding the pathway of CO oxidation (Fig. 3c). It should be noted the production of methyl formate and formaldehyde increased as well. In the case of 1-propanol (Fig. 3d), a similar effect of CO was observed at 473 K where the presence of CO reduced the production of propanal, the dominant selective oxidation product for this reaction. Interestingly, at 573 K the amount of propanal increased by nearly the same amount that the ethylene decreased; the amount of ethylene was reduced by a factor of 2. These changes in selectivity agree with the presence of CO binding to Pd ensembles, and the Pd arrangements that have been proven essential for oxygen dissociation,41,62,63 that reduces the availability of atomically adsorbed oxygen on the surface to initiate the selective oxidation mechanism and drive the reaction selectivity.
Identification of surface bound carboxylates
The chemical species detected during methanol and 1-propanol oxidation reactions over Pd3Au97 RCT-SiO2 at 423 K using infrared spectroscopy (Fig. 4) correlate well with the product distributions measured in reactor studies (Fig. 2f). The in situ FTIR spectra were monitored as a function of time and collected until the peak intensity no longer changed, typically after 1–1.5 h under reaction conditions (Fig. S6†). For methanol oxidation at 423 K over Pd3Au97 RCT-SiO2 the FTIR spectra in the 2500–1100 cm−1 region revealed main features corresponding to the presence of CO2 [ν (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 2360 and 2342],64 methyl formate [ν (CO) at 1740 and ν (C–O) at 1200–1150 cm−1],37,65 formate [ν (O–CO) at 1570 cm−1],37,66 and methoxy [δ (C–H) at 1480 cm−1]65,67 (Fig. 4a) (Table 2). Products including methyl formate and CO2 assigned by the FTIR spectra are consistent with the selectivity measured in the reactor studies, which showed a selectivity of 82% and 16% for methyl formate and CO2, respectively. In addition, intermediates for the coupling and over-oxidation pathways are also observed, namely methoxy and formate species, respectively. The peak assignments for the products were also confirmed by the difference spectra by subtracting the steady-state spectra of methanol oxidation over Pd3Au97 RCT-SiO2 with support SiO2 to eliminate the contribution from gas phase reactants (Fig. S7a†).
O) at 2360 and 2342],64 methyl formate [ν (CO) at 1740 and ν (C–O) at 1200–1150 cm−1],37,65 formate [ν (O–CO) at 1570 cm−1],37,66 and methoxy [δ (C–H) at 1480 cm−1]65,67 (Fig. 4a) (Table 2). Products including methyl formate and CO2 assigned by the FTIR spectra are consistent with the selectivity measured in the reactor studies, which showed a selectivity of 82% and 16% for methyl formate and CO2, respectively. In addition, intermediates for the coupling and over-oxidation pathways are also observed, namely methoxy and formate species, respectively. The peak assignments for the products were also confirmed by the difference spectra by subtracting the steady-state spectra of methanol oxidation over Pd3Au97 RCT-SiO2 with support SiO2 to eliminate the contribution from gas phase reactants (Fig. S7a†).
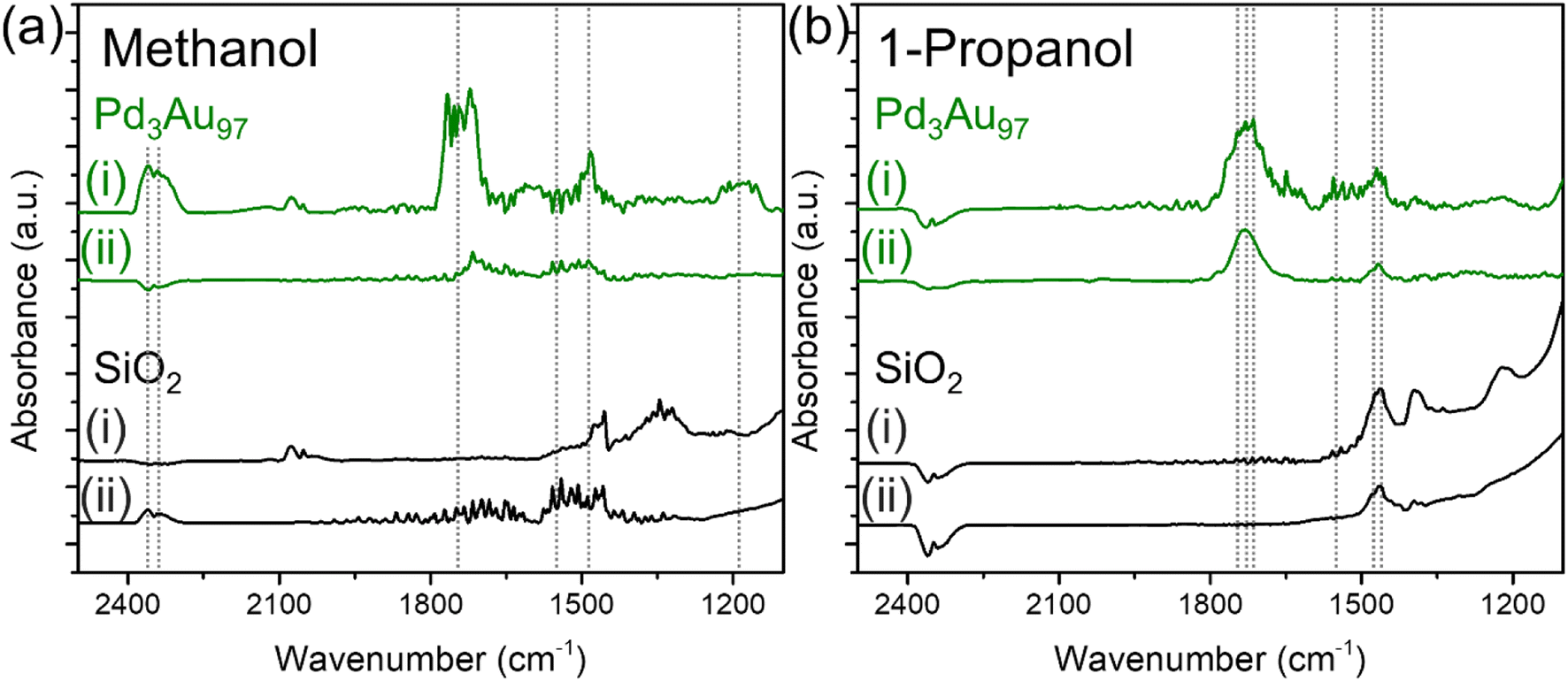 | ||
| Fig. 4 In situ FTIR spectra in the 2500–1100 cm−1 region collected (i) during the oxidation of (a) methanol and (b) 1-propanol over Pd3Au97 RCT-SiO2 (green) and RCT-SiO2 (black) at 423 K for 1.5 h and followed by (ii) purging in He for 2 h. Reaction conditions: 4% alcohol and 10% O2, balanced in He; total flow rate = 20 mL min−1. Dashed lines denoted the peaks of interest with peak assignments summarized in Table 2. | ||
| Reaction | Frequency (cm−1) | Vibrational mode | Assignment |
|---|---|---|---|
| Methanol oxidation | 2360 |
ν (CO) |
CO2 |
| 2342 |
ν (CO) |
CO2 | |
| 1740 |
ν (CO) |
Methyl formate | |
| 1570 |
ν (O–CO) |
Formate | |
| 1480 | δ (C–H) | Methoxy | |
| 1200–1150 | ν (C–O) | Methyl formate | |
| 1-Propanol oxidation | 1760 |
ν (CO) |
Propyl propionate |
| 1730 |
ν (CO) |
Propionate | |
| 1725 |
ν (CO) |
Propanal | |
| 1558–1540 |
ν
as (O–CO) |
Propionate | |
| 1471 |
ν
s (O–CO) |
Propionate | |
| 1460 | ν (C–O) | Propanal |
It should be noted that it is not possible to distinguish between gas phase and surface-bound species from the measurements, so the spectra reported is the result of a sum of both species. The FTIR spectra in the C–H region (3100–2700 cm−1) for oxidation of methanol over Pd3Au97 RCT-SiO2 at 423 K showed a combination of gas and surface species (Fig. S8†). Bands for adsorbed methanol are assigned at 2994, 2955 and 2856 (νas (CH3) 2994, 2955, νs (CH3) 2856 cm−1),68 a band at 2844 is assigned to gas phase methanol (νs (CH3) 2844 cm−1),68 and adsorbed methoxy (νs (CH3) 2923 and 2δ (CH3) 2822 cm−1)37 was also detected under methanol oxidation conditions.
Analogous IR experiments for detecting species present during the oxidation of 1-propanol (Fig. 4b) showed good correlations with the product distributions measured in reactor studies (Fig. 2f). In situ FTIR spectra collected for 1-propanol oxidation over Pd3Au97 RCT-SiO2 at 423 K in the 2500–1100 cm−1 region showed main features corresponding to the presence of propyl propionate [ν (CO) at 1760 cm−1],69 propanal [ν (CO) at 1725 cm−1 and ν (C–O) at 1460 cm−1],70 propionate [ν (CO) at 1730 cm−1, νas (O–CO) at 1558–1540 cm−1 and νs (O–CO) at 1471 cm−1] (Table 2).70,71 Based on the reactor studies of 1-propanol oxidation over Pd3Au97 RCT-SiO2, the major product is propanal (79% selectivity) with a minor product of propyl propionate (16% selectivity); both products are detected in the in situ FTIR spectra for 1-propanol oxidation. Additionally, several features confirmed the presence of propionate under 1-propanol oxidation conditions, the assignments are based on previously reported spectra.70
The surface of Pd3Au97 RCT-SiO2 under the oxidation of 1-propanol at 423 K was populated with strongly bound spectator species (carboxylates), but not in the case of methanol oxidation. The accumulation of surface species under 1-propanol oxidation reactions over Pd3Au97 RCT-SiO2 was identified by comparing the in situ FTIR spectra collected after 1.5 h of reaction (Fig. 4bi) with the spectra collected after purging with an inert gas (He) for 2 h (Fig. 4bii). A major peak centered at ∼1730 cm−1 and a smaller peak at ∼1470 cm−1 persisted after purging in He for 2 h. In contrast, peaks observed during methanol oxidation (Fig. 4ai) decreased drastically after purging in He for 2 h (Fig. 4aii). As noted earlier, it is not possible to distinguish between gas phase and surface-bound species over steady-state FTIR spectra; therefore, by collecting spectra after purging with He, the remaining observable species on the catalyst surface are expected to be strongly bound spectator species formed under reaction conditions. The accumulation of surface species on Pd3Au97 RCT-SiO2 under 1-propanol oxidation reaction was further confirmed using the integrated area in spectral region characteristic for CO and C–O stretches (1900–1100 cm−1) collected as a function of time (Fig. S9, see ESI† for a more detailed discussion).
The surface species accumulated on Pd3Au97 RCT-SiO2 under 1-propanol oxidation reactions were identified as propionates based on the ν (CO) region in the in situ FTIR spectra. The presence of propyl propionate (1760 cm−1), propionate (1730 cm−1), and propanal (1725 cm−1) were detected during 1-propanol oxidation (Fig. 4bi) while two features (1730 and 1470 cm−1) persisted after purging in He for 2 h, which can be attributed to linearly bound propionate species.66,67 This is consistent with the reported frequency of linearly adsorbed aliphatic esters, where the ν (CO) frequency typically appears in the range of 1750–1720 cm−1.72 No peaks were observed in the CO region of in situ FTIR spectra collected over SiO2 under 1-propanol oxidation followed by purging in He (Fig. 4b, black). The band at 1730 cm−1 is unlikely to be gas phase propanal as the ν (CO) frequency will redshift to below 1700 cm−1 for surface-bound propanal. Single crystal study of propanal adsorption on Pd(111) showed a ν (CO) frequency at 1695 cm−1 as compared to the original feature at 1728 cm−1.70,73 In addition, ν (C–H) of aldehyde is expected to appear as doublet in the range of 2850–2750 cm−1,74 which was not observed in the FTIR spectra after purging in He.
Propionate species bound on the catalyst surface after purging mainly exist in the form of linearly bound geometry. Bands characteristic of the bidentate binding of the carboxylate (COO−) group were only observed in the steady-state FTIR spectrum collected under 1-propanol oxidation (Fig. 4bi), which typically appears in the range of 1590–1560 and 1470–1440 cm−1 for the asymmetric and symmetric stretches, respectively.67 No bands were observed in the range of 1590–1560 cm−1 and only a small peak in the 1470–1440 cm−1 region was observed in the FTIR spectrum after purging in He (Fig. 4bii), suggesting propionate adsorbed on the catalyst surface is mainly in the form of linearly coordinated. This is consistent with stronger linear adsorption of aliphatic ester than bidentate binding geometry.66,67
The build-up of carbonaceous species on Pd3Au97 RCT-SiO2 under 1-propanol oxidation reactions is confirmed by the production of CO2 in temperature programmed oxidation (TPO) over spent catalysts (Fig. S10).† The analysis of residual surface carbon using temperature programmed oxidation reactions over Pd3Au97 RCT-SiO2 catalysts after the oxidation of C1 to C4 alcohols showed the amount of CO2 formed was inversely related to the activity towards the alcohol (Fig. S10†). Only small amounts of CO2 formed after methanol oxidation because the conversion of methanol was nearly 100% at 423 K. In contrast, clear CO2 formation peaks were detected during TPO for Pd3Au97 RCT-SiO2 catalysts after the high chain alcohols reactions. The residual surface carbon formed during the oxidation of C2–C4 alcohols starts to decompose ∼473 K and the catalyst surface would be free of the residual carbon ∼573 K. Thus, testing the oxidation of C2 to C4 alcohols at elevated temperatures under oxidative conditions could eliminate the surface poison over Pd3Au97 RCT-SiO2 leading to a greater availability of active sites and a greater conversion of alcohol as compared to Au RCT-SiO2.
Regeneration of the poisoned Pd3Au97 RCT-SiO2 surface
The Pd3Au97 RCT-SiO2 catalysts poisoned by surface-bound propionates formed under 1-propanol oxidation conditions can be regenerated in a flowing methanol/oxygen mixture, restoring the promotional effect of Pd for methanol oxidation. The regeneration is monitored by the reactor studies with the conversion of alcohols collected in the following sequence: (i) methanol oxidation for 2 h, (ii) 1-propanol oxidation after replacing methanol with 1-propanol for 1 h, and (iii) methanol oxidation after replacing 1-propanol with methanol for 5 h at 423 K (Fig. 5a). The conversion was 89% after (i) methanol oxidation and dropped immediately to 27% after establishing (ii) 1-propanol oxidation, consistent with the steady-state results (Fig. 1b). The decrease in conversion is due to surface poisoning of propionate species, which can be removed by re-establishing (iii) methanol oxidation conditions. The conversion of methanol oxidation in step (iii) gradually increased after 5 h under reaction conditions and reached the initial conversion as in step (i), suggesting a regeneration of the catalyst surface free of strongly bound propionate species. The longer time scale for step (iii) (up to 5 h) suggests a slow removal of the surface propionate species formed under 1-propanol oxidation reaction, further confirming the strong binding nature of carboxylate groups. | ||
| Fig. 5 (a) The conversion of (i) methanol oxidation for 2 h, (ii) 1-propanol oxidation for 1 h after replacing methanol with 1-propanol, and (iii) re-establish methanol oxidation for 5 h after replacing 1-propanol with methanol at 423 K over Pd3Au97 RCT-SiO2. (b–f) The corresponding in situ FTIR spectra in the 3100–2700 cm−1 region representing different stages of the experiment, including during (b) (i) methanol oxidation, (c) (ii) 1-propanol oxidation, and (d–f) (iii) re-establishing methanol oxidation after replacing 1-propanol with methanol. Dashed lines in (a) indicated the replacement of reactants, and in (c–f) denoted the IR peak of interest. Reaction conditions: 4% alcohol and 10% O2 balanced in He; total flow rate = (a) 50 and (b) 20 mL min−1; mcat in (a) = 40.5 mg. | ||
Following the same reaction sequence (i–iii) as in reactor studies, the disappearance of the characteristic feature of propionate observed in in situ FTIR spectra confirms the removal of surface-bound species on Pd3Au97 RCT-SiO2 by re-establishing methanol oxidation conditions (Fig. 5b–f). The characteristic feature at 2888 cm−1 (ν (C–H)) was used to monitor the replacement of propionate in the (C–H) region (3100–2700 cm−1).75 Note that it is not possible to use the ν (CO) region as it overlaps with the ν (CO) of methyl formate and is rather weak in intensity compared to the absorbance at the ν (C–H) region. Propionate was not observed during (i) methanol oxidation (Fig. 5b) but appeared after establishing (ii) 1-propanol oxidation (Fig. 5c), as expected from the steady-state results (Fig. 1b). A strong correlation was observed between the slow disappearance of the feature at 2888 cm−1 with time (Fig. 5c and d) after re-establishing (iii) methanol oxidation conditions and the gradually increase in methanol oxidation conversion during time-on-stream in reactor studies (Fig. 5a), confirming the removal of surface bound propionate species regenerates the Pd3Au97 RCT-SiO2 catalyst.
Discussion
Disproportionation of hydroxyls: explaining the activity of Au and Pd3Au97 RCT-SiO2
One explanation for the enhanced activity of Au and Pd3Au97 RCT-SiO2 towards oxidation of 1-propanol by co-feeding H2O could be the creation of additional adsorbed atomic oxygen, the active site initiating the oxidative self-coupling mechanism over Au,76 as a result of hydroxyl (OH) disproportionation over Au. Previous studies of H2O reacting with oxygen-covered Au(111) show that when OH groups form, they disproportionate rapidly numerous times.60 This results in short-lived OH species on Au surfaces that, although could theoretically participate in the oxidative alcohol coupling mechanism mechanism,77 likely form water and adsorbed atomic oxygen quickly.60,61 It is reasonable to conclude that if more OH groups formed on the predominantly Au nanoparticle of PdxAu1−x RCT-SiO2 catalysts, disproportionation of Au–OHs would create atomic oxygen on Au promoting activity for selective oxidation chemistry.Based on the in situ FTIR studies over Pd3Au97 RCT-SiO2 and the activity of the Au and Pd3Au97 RCT-SiO2 catalyst, the Au nanoparticle-SiO2 support interface is proposed to be important to the activity of the PdxAu1−x RCT-SiO2 catalysts for the oxidation of high chain alcohols. The RCT-SiO2 support is rich in hydroxyls between 298 K and 773 K (Fig. 1a). The presence of OH groups on the support at the nanoparticle-support interface can lead to the disproportionation of OH groups and form atomically adsorbed oxygen. Because nanoparticle catalysts prepared using the RCT approach are ∼85% embedded in the support,56 the nanoparticle-support interface area of these materials are significant. Consequently, the PdxAu1−x RCT-SiO2 catalysts have an ample supply of Si–OH and significant interface area that could enable Si–OHs migration from the RCT-SiO2 support to the predominantly Au nanoparticle possible.
The importance of H2O and the perimeter of the nanoparticle for the activity of Au/SiO2 catalysts for the oxidation of CO has been reported.20 In that study it is suggested that reaction between molecular O2 at the interface and water adsorbed on SiO2 support results in atomic oxygen and 2 hydroxyls groups at the interface; the hydroxyls then further disproportionate to form another atomic oxygen at the perimeter and H2O is reformed. Indeed, this constitutes another mechanism by which O2 activation can occur at the Au nanoparticle-SiO2 support interface. Regardless of the mechanism by which hydroxyls are supplied to the Au nanoparticles, there is strong evidence for the formation of adsorbed atomic oxygen as a result of the Au nanoparticle-SiO2 support interface, rendering the Au RCT-SiO2 active for the oxidation of alcohols (Fig. 1b, 2a, c, S5a and c†).
Carboxylates: the case for an infamous surface poison
The similar activity of Pd3Au97 RCT-SiO2 to Au RCT-SiO2 for the oxidation of C2–C4 alcohols at 423 K (Fig. 2) strongly suggests the availability of surface Pd atoms is hindered in a way that is not the case for the oxidation of the C1 alcohol. The inverse dependence of residual surface carbon on activity for oxidation of alcohols (Fig. S10†) and the similarity of the peak temperature of the CO2 to carboxylate equivalents decomposing over Au single-crystals (Table S7†) would indicate the residual surface carbon, and potential surface poison, is a surface carboxylate. Indeed, surface bound propionate species were detected on Pd3Au97 RCT-SiO2 catalysts during 1-propanol oxidation reaction based on the in situ FTIR studies (Fig. 4b). By comparing the FTIR spectra collected under reaction conditions to those after purging in an inert environment (removal of gas phase species), bands characteristic of propionates is the only features that persisted on Pd3Au97 RCT-SiO2. This is unique to the oxidation of 1-propanol over Pd3Au97 RCT-SiO2, no residual peaks were detected in the region of interest for methanol oxidation reaction or for alcohol oxidation over RCT-SiO2.The regeneration experiments support the strongly bound nature of propionate intermediate formed under 1-propanol oxidation over Pd3Au97 RCT-SiO2 but also demonstrate reactivation. The surface species can be gradually removed by re-establishing the methanol/oxygen stream (up to 5 hours). Once the catalyst surface is free of propionates, the methanol oxidation activity of Pd3Au97 RCT-SiO2 can be restored to its initial state, suggesting a promising reactivation process. Similar surface poisoning by reaction intermediates or products has been reported for the deactivation of catalysts in flow reactor studies and electrocatalysis.78,79 Studies of oxidative cross-coupling of methanol and acrolein over npAgAu catalysts have shown that the formation of methyl acrylate blocks the active sites and resulted in a decrease in acrolein conversion.78 In formic acid fuel cells, the Pd catalysts surface is poisoned by methyl formate and acetic acid but can be regenerated by reoxidizing the surface species electrochemically.79
The peak temperatures of the CO2 produced during the post-catalysis TPO of ethanol, 1-propanol and 1-butanol over Pd3Au97 are remarkably similar to those reported for the carboxylate equivalents decomposing over Au single-crystals, specifically under limited oxygen conditions (Table S7).† The lesser extent of CO poisoning over Pd3Au97 RCT-SiO2 for the oxidation of 1-propanol at 473 K (Fig. 3b) further proves surface Pd atoms are not as available to activate molecular oxygen as they are under oxidative methanol conditions; they are likely blocked by the residual surface carbon at this temperature, which would result in an oxygen-limited surface (Table S7†). Because reaction conditions are O2-rich, it is expected that the residual surface carbon would be continually removed at 573 K, thus allowing for CO to bind to Pd at the surface, decreasing the amount of O2 available at the surface, which increases the yield of selective products (Fig. 3c and d).
Because previous studies have shown carboxylates to be an intermediate in the combustion pathway over Au76 and are similar to carbonates that are often associated with the deactivation of Au catalysts,20,80,81 we consider how the decomposition of carboxylates over Au elucidates the formation of the alkene products (i.e. ethylene and propylene) over Pd3Au97 RCT-SiO2. Studies of acetate on Au(111) have shown the mechanism for acetate decomposition involves a C–C bond scission under limited O2 conditions producing mostly CO2 and methyl radicals, the latter species being able to re-adsorb on the surface to react further with other surface-bound intermediates.49 Temperature programmed reaction of isotopically labeled ethanol on an oxygen covered PdAu single crystal showed irrevocable evidence of C–C bond scission and methyl radical re-adsorption enabling further reaction on the surface.82 Applying the same decomposition mechanism (i.e. a C–C bond scission between the α- and β-carbon, resulting in an alkyl radical) to propanoate or butanoate it is clear that the alkyl radical containing one carbon less than the parent alcohol could re-adsorb on the surface and further react. Applying this further to the Pd3Au97 RCT-SiO2 catalyst, the formation and adsorption of an ethyl or propyl radical could precede the formation of ethylene and propylene, respectively. Because the formation of the alkene products was only observed over the Pd containing catalysts (Fig. 2f and S5h†), this suggests that the carboxylates at or near the Pd ensembles inhibit its availability to activate molecular oxygen and promote activity over the catalyst.
The evidence herein suggests carboxylates block the Pd active site, which is supported by the studies of ethanol dehydrogenation on model PdAu alloys, which recognize the relationship between Pd ensemble size and susceptibility to surface poisoning to carbon contamination.83 The activity of model PdAu alloys (formed by deposition of Pd on Au(111) followed by high temperature annealing to alloy) for non-oxidative dehydrogenation of ethanol showed a volcano-like dependence of activity on Pd concentration, where activity was lowest for 1 ML Pd–Au(111), increased to a maximum for 2 ML Pd–Au(111) and decreased slightly for 3 ML and 4 ML Pd–Au(111).83 A combination of experiments and DFT calculations of the barriers for the initial dehydrogenation of ethanol (non-oxidatively) shows as the Pd ensemble size increases the binding strength of all intermediates, except ethanol and acetaldehyde, increases and associates the build-up of surface carbons with Pd ensembles.83 The Pd3Au97 RCT-SiO2 showed no significant conversion of methanol or 1-propanol under non-oxidative conditions at 423 K and only minor conversion (<3%) at 573 K that was unrelated to the Pd in the system since the activity of the Au RCT-SiO2 was nearly identical (Table S6†). Thus, the residual surface carbon is a result of further dehydrogenation of the alcohol.
The CO-poisoning experiment can be considered as a qualitative measure of available Pd ensembles because analogous experiments over the Au RCT-SiO2 showed a small decrease in conversion of alcohols at 473 K and no change at 573 K (Fig. S11†). The poisoning effect of CO during oxidation of methanol over Pd3Au97 RCT-SiO2 at 473 K suggests substantial Pd is at the surface of the catalyst, even at this elevated temperature. In previous work,41 the decrease in conversion of the methanol was due to the strong binding of CO to Pd ensembles that inhibited the molecular O2 activation needed to propagate the selective oxidation cycle; it was implied that Pd ensembles must have been free of surface bound intermediates under reaction conditions prior to CO exposure. Extending this reasoning to the co-fed CO during oxidation of 1-propanol over Pd3Au97 RCT-SiO2, the presence of a strongly bound surface intermediate (propionates) would reduce the availability of Pd ensembles and consequently the CO poisoning effect. Although the poisoning effect at 573 K was insignificant, the changes in the yield of selective products suggest CO adsorbed substantially on the surface, enough to change the product distribution, especially in the case of 1-propanol. Based on the residual carbon analysis (Table S7†), it is expected that at 573 K the surface carbon would not build up on the catalyst surface, thus greater availability of surface Pd. The decrease in ethylene yield at this temperature when CO was co-fed also suggests that Pd sites became freed of a strongly bound intermediate and the CO can effectively bind to Pd.
Conclusion
In this work, the activity of Pd3Au97 RCT-SiO2 for the oxidation of higher chain alcohols (C2–C4 linear alcohols) was similar to the activity of pure Au RCT-SiO2 even though the Pd-containing catalyst promotes the oxidation of methanol at 423 K. The similarity originates from (1) the activity of the Au RCT-SiO2 that is attributed to the activation of alcohols due to hydroxyl migration from the hydroxyl-rich RCT-SiO2 support and (2) the accumulation of a strongly bound species that reduces the availability of Pd ensembles under reaction conditions for the oxidation of high molecular weight aliphatic alcohols. Linearly-bound, propionate species were identified as the surface poison formed via the over-oxidation pathway during higher chain alcohol oxidation, based on the close correlations between in situ FTIR characterization and reactor studies. The selectivity for the oxidation of C1–C4 alcohols over Pd3Au97 RCT-SiO2 changed from the coupling product to aldehydes when increasing alcohol chain length from C1 to C2–C4, consistent with the selectivity predicted for other Au-based catalysts. Furthermore, the surface poisoned by carboxylate species can be regenerated by re-establishing methanol oxidation conditions to replace higher molecular weight alcohols. This study provides molecular-level evidence for the presence of surface-bound carboxylate species on Pd active sites during higher chain alcohol oxidation reactions and underscores the challenge of developing a dilute alloy catalyst capable of catalyzing the oxidation of a wide variety of alcohols.Author contributions
Jennifer D. Lee: investigation, methodology, formal analysis, visualization, writing – original draft, writing – review & editing, project administration. Amanda Filie: investigation, methodology, formal analysis, visualization, writing – original draft, writing – review & editing. Leigh Wilson: formal analysis, investigation. Karin Nguyen: formal analysis, investigation. Tanya Shirman: investigation, resources. Erjia Guan: formal analysis, investigation, resources. Mathilde Luneau: formal analysis, investigation. Michael Aizenberg: supervision, writing – review & editing. Joanna Aizenberg: conceptualization, supervision, writing – review & editing. Robert J. Madix: conceptualization, supervision, project administration, writing – review & editing. Cynthia M. Friend: conceptualization, supervision, project administration, funding acquisition, writing – review & editing. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported as part of the IMASC, an Energy Frontier Research Center funded by the U. S. Department of Energy, Office of Science, Basic Energy Sciences under award #DE-SC0012573. The authors acknowledge the useful discussions on FTIR results with Prof. Anatoly I. Frenkel at Stony Brook University and Brookhaven National Laboratory.References
- M. Haruta, Nature, 2005, 437, 1098–1099 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- B. K. Min and C. M. Friend, Chem. Rev., 2007, 107, 2709–2724 CrossRef CAS PubMed.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- C. G. Freyschlag and R. J. Madix, Mater. Today, 2011, 14, 134–142 CrossRef CAS.
- C. Della Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369 RSC.
- A. S. Sharma, H. Kaur and D. Shah, RSC Adv., 2016, 6, 28688–28727 RSC.
- X. Deng, B. K. Min, A. Guloy and C. M. Friend, J. Am. Chem. Soc., 2005, 127, 9267–9270 CrossRef CAS PubMed.
- M. Haruta, Cattech, 2002, 6, 102–115 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- M. Haruta, S. Tsubota, T. Kobayashi, H. Kageyama, M. J. Genet and B. Delmon, J. Catal., 1993, 144, 175–192 CrossRef CAS.
- A. Abad, P. Concepción, A. Corma and H. García, Angew. Chem., Int. Ed., 2005, 44, 4066–4069 CrossRef CAS PubMed.
- I. S. Nielsen, E. Taarning, K. Egeblad, R. Madsen and C. H. Christensen, Catal. Lett., 2007, 116, 35–40 CrossRef CAS.
- A. Abad, A. Corma and H. García, Chem.–Eur. J., 2008, 14, 212–222 CrossRef CAS PubMed.
- B. N. Zope, D. D. Hibbitts, M. Neurock and R. J. Davis, Science, 2010, 330, 74–78 CrossRef CAS PubMed.
- V. I. Sobolev, K. Yu. Koltunov, O. A. Simakova, A.-R. Leino and D. Yu. Murzin, Appl. Catal., A, 2012, 433–434, 88–95 CrossRef CAS.
- P. Wu, P. Bai, Z. Yan and G. X. S. Zhao, Sci. Rep., 2016, 6, 18817 CrossRef PubMed.
- S. Biella and M. Rossi, Chem. Commun., 2003, 378–379 RSC.
- M. Daté, M. Okumura, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 2129–2132 CrossRef PubMed.
- M. Daté and M. Haruta, J. Catal., 2001, 201, 221–224 CrossRef.
- T. A. Nijhuis and B. M. Weckhuysen, Chem. Commun., 2005, 6002–6004 RSC.
- J. Huang, T. Akita, J. Faye, T. Fujitani, T. Takei and M. Haruta, Angew. Chem., Int. Ed., 2009, 48, 7862–7866 CrossRef CAS PubMed.
- S. Lee, L. M. Molina, M. J. López, J. A. Alonso, B. Hammer, B. Lee, S. Seifert, R. E. Winans, J. W. Elam, M. J. Pellin and S. Vajda, Angew. Chem., Int. Ed., 2009, 48, 1467–1471 CrossRef CAS PubMed.
- M. Ojeda and E. Iglesia, Chem. Commun., 2009, 352–354 RSC.
- G. M. Mullen, E. J. Evans Jr, I. Sabzevari, B. E. Long, K. Alhazmi, B. D. Chandler and C. B. Mullins, ACS Catal., 2017, 7, 1216–1226 CrossRef CAS.
- G. M. Mullen, L. Zhang, E. J. Evans Jr, T. Yan, G. Henkelman and C. B. Mullins, J. Am. Chem. Soc., 2014, 136, 6489–6498 CrossRef CAS PubMed.
- G. M. Mullen, L. Zhang, E. J. Evans, T. Yan, G. Henkelman and C. B. Mullins, Phys. Chem. Chem. Phys., 2015, 17, 4730–4738 RSC.
- A. Wittstock, V. Zielasek, J. Biener, C. M. Friend and M. Bäumer, Science, 2010, 327, 319–322 CrossRef CAS PubMed.
- M. L. Personick, B. Zugic, M. M. Biener, J. Biener, R. J. Madix and C. M. Friend, ACS Catal., 2015, 5, 4237–4241 CrossRef CAS.
- C. Reece, E. A. Redekop, S. Karakalos, C. M. Friend and R. J. Madix, Nat. Catal., 2018, 1, 852–859 CrossRef CAS.
- C. Reece, M. Luneau and R. J. Madix, ACS Catal., 2019, 9, 4477–4487 CrossRef CAS.
- K. J. Stowers, R. J. Madix and C. M. Friend, J. Catal., 2013, 308, 131–141 CrossRef CAS.
- M. L. Personick, R. J. Madix and C. M. Friend, ACS Catal., 2017, 7, 965–985 CrossRef CAS.
- C. Reece, M. Luneau, C. M. Friend and R. J. Madix, Angew. Chem., 2020, 132, 10956–10959 CrossRef.
- R. Wang, Z. Wu, C. Chen, Z. Qin, H. Zhu, G. Wang, H. Wang, C. Wu, W. Dong, W. Fan and J. Wang, Chem. Commun., 2013, 49, 8250–8252 RSC.
- G. T. Whiting, S. A. Kondrat, C. Hammond, N. Dimitratos, Q. He, D. J. Morgan, N. F. Dummer, J. K. Bartley, C. J. Kiely, S. H. Taylor and G. J. Hutchings, ACS Catal., 2015, 5, 637–644 CrossRef CAS.
- N. Al-Rifai, P. J. Miedziak, M. Morad, M. Sankar, C. Waldron, S. Cattaneo, E. Cao, S. Pattisson, D. Morgan, D. Bethell, G. J. Hutchings and A. Gavriilidis, Ind. Eng. Chem. Res., 2017, 56, 12984–12993 CrossRef CAS.
- T. A. G. Silva, C. P. Ferraz, R. V Gonçalves, E. Teixeira-Neto, R. Wojcieszak and L. M. Rossi, ChemCatChem, 2019, 11, 4021–4027 CrossRef CAS.
- C. J. Wrasman, A. Boubnov, A. R. Riscoe, A. S. Hoffman, S. R. Bare and M. Cargnello, J. Am. Chem. Soc., 2018, 140, 12930–12939 CrossRef CAS PubMed.
- A. Filie, T. Shirman, A. C. Foucher, E. A. Stach, M. Aizenberg, J. Aizenberg, C. M. Friend and R. J. Madix, J. Catal., 2021, 404, 943–953 CrossRef CAS.
- M. Luneau, T. Shirman, A. Filie, J. Timoshenko, W. Chen, A. Trimpalis, M. Flytzani-Stephanopoulos, E. Kaxiras, A. I. Frenkel, J. Aizenberg, C. M. Friend and R. J. Madix, Chem. Mater., 2019, 31, 5759–5768 CrossRef CAS.
- A. Filie, T. Shirman, M. Aizenberg, J. Aizenberg, C. M. Friend and R. J. Madix, Catal. Sci. Technol., 2021, 11, 4072–4082 RSC.
- T. Shirman, T. J. Toops, E. Shirman, A. V Shneidman, S. Liu, K. Gurkin, J. Alvarenga, M. P. Lewandowski, M. Aizenberg and J. Aizenberg, Catal. Today, 2021, 360, 241–251 CrossRef CAS.
- X. Liu, B. Xu, J. Haubrich, R. J. Madix and C. M. Friend, J. Am. Chem. Soc., 2009, 131, 5757–5759 CrossRef CAS PubMed.
- J. L. Davis and M. A. Barteau, Surf. Sci., 1992, 268, 11–24 CrossRef CAS.
- J. L. Davis and M. A. Barteau, Surf. Sci., 1991, 256, 50–66 CrossRef CAS.
- T. Cremer, C. G. F. Siler, J. C. F. Rodríguez-Reyes, C. M. Friend and R. J. Madix, J. Phys. Chem. Lett., 2014, 5, 1126–1130 CrossRef CAS PubMed.
- C. G. F. Siler, T. Cremer, J. C. F. Rodriguez-Reyes, C. M. Friend and R. J. Madix, ACS Catal., 2014, 4, 3281–3288 CrossRef CAS.
- C. R. O. O'Connor, Elucidating Determining Factors in Heterogeneous Catalysis Using Surface Science Models, Dr thesis, Harvard University, 2020 Search PubMed.
- Z. Li and W. T. Tysoe, Surf. Sci., 2012, 606, 1934–1941 CrossRef CAS.
- N. Aas and M. Bowker, J. Chem. Soc., Faraday Trans., 1993, 89, 1249–1255 RSC.
- K. C. Grabar, K. J. Allison, B. E. Baker, R. M. Bright, K. R. Brown, R. G. Freeman, A. P. Fox, C. D. Keating, M. D. Musick and M. J. Natan, Langmuir, 1996, 12, 2353–2361 CrossRef CAS.
- E. Shirman, T. Shirman, A. V. Shneidman, A. Grinthal, K. R. Phillips, H. Whelan, E. Bulger, M. Abramovitch, J. Patil, R. Nevarez and J. Aizenberg, Adv. Funct. Mater., 2018, 28, 1–20 CrossRef.
- T. Shirman, J. Lattimer, M. Luneau, E. Shirman, C. Reece, M. Aizenberg, R. J. Madix, J. Aizenberg and C. M. Friend, Chem.–Eur. J., 2018, 24, 1743 CrossRef CAS.
- J. E. S. van der Hoeven, S. Krämer, S. Dussi, T. Shirman, K.-C. K. Park, C. H. Rycroft, D. C. Bell, C. M. Friend and J. Aizenberg, Adv. Funct. Mater., 2021, 31, 2106876 CrossRef CAS.
- L.-C. Wang, K. J. Stowers, B. Zugic, M. M. Biener, J. Biener, C. M. Friend and R. J. Madix, Catal. Sci. Technol., 2015, 5, 1299–1306 RSC.
- S. P. Zhdanov, L. S. Kosheleva and T. I. Titova, Langmuir, 1987, 3, 960–967 CrossRef CAS.
- V. Dugas and Y. Chevalier, J. Colloid Interface Sci., 2003, 264, 354–361 CrossRef CAS PubMed.
- R. G. Quiller, T. A. Baker, X. Deng, M. E. Colling, B. K. Min and C. M. Friend, J. Chem. Phys., 2008, 129, 64702 CrossRef CAS PubMed.
- F. Xu, I. Fampiou, C. R. O'Connor, S. Karakalos, F. Hiebel, E. Kaxiras, R. J. Madix and C. M. Friend, Phys. Chem. Chem. Phys., 2018, 20, 2196–2204 RSC.
- F. Gao, Y. Wang and D. W. Goodman, J. Am. Chem. Soc., 2009, 131, 5734–5735 CrossRef CAS PubMed.
- F. Gao, Y. Wang and D. W. Goodman, J. Phys. Chem. C, 2009, 113, 14993–15000 CrossRef CAS.
- D. Pavia, G. Lampman, G. Kriz and J. Vyvyan, Introduction to Spectroscopy, Brooks/Cole Cengage Learning, 2008 Search PubMed.
- R. Wojcieszak, A. Karelovic, E. M. Gaigneaux and P. Ruiz, Catal. Sci. Technol., 2014, 4, 3298–3305 RSC.
- S. Shan, J. Li, Y. Maswadeh, C. O'Brien, H. Kareem, D. T. Tran, I. C. Lee, Z. P. Wu, S. Wang, S. Yan, H. Cronk, D. Mott, L. Yang, J. Luo, V. Petkov and C. J. Zhong, Nat. Commun., 2020, 11, 1–15 CrossRef CAS PubMed.
- C. P. O'Brien and I. C. Lee, J. Catal., 2017, 347, 1–8 CrossRef.
- M. A. Natal-Santiago, J. J. de Pablo and J. A. Dumesic, Catal. Lett., 1997, 47, 119–128 CrossRef CAS.
- A. S. Malik, S. F. Zaman, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, Appl. Catal., A, 2018, 560, 42–53 CrossRef CAS.
- K. H. Dostert, C. P. O'Brien, F. Mirabella, F. Ivars-Barceló and S. Schauermann, Phys. Chem. Chem. Phys., 2016, 18, 13960–13973 RSC.
- J. D. Kammert, A. Chemburkar, N. Miyake, M. Neurock and R. J. Davis, ACS Catal., 2021, 11, 1435–1455 CrossRef CAS.
- D. Pavia, G. Lampman, G. Kriz and J. Vyvyan, Introduction to Spectroscopy, Brooks/Cole Cengage Learning, 2008 Search PubMed.
- R. Shekhar, M. A. Barteau, R. V. Plank and J. M. Vohs, J. Phys. Chem. B, 1997, 101, 7939–7951 CrossRef CAS.
- D. Pavia, G. Lampman, G. Kriz and J. Vyvyan, Introduction to Spectroscopy, Brooks/Cole Cengage Learning, 2008 Search PubMed.
- S. R. Tong, L. Y. Wu, M. F. Ge, W. G. Wang and Z. F. Pu, Atmos. Chem. Phys., 2010, 10, 7561–7574 CrossRef CAS.
- B. Xu, X. Liu, J. Haubrich, R. J. Madix and C. M. Friend, Angew. Chem., Int. Ed., 2009, 48, 4206–4209 CrossRef CAS PubMed.
- B. Xu, J. Haubrich, T. A. Baker, E. Kaxiras and C. M. Friend, J. Phys. Chem. C, 2011, 115, 3703–3708 CrossRef CAS.
- S. Karakalos, B. Zugic, K. J. Stowers, M. M. Biener, J. Biener, C. M. Friend and R. J. Madix, Surf. Sci., 2016, 652, 58–66 CrossRef CAS.
- W. L. Law, A. M. Platt, P. D. C. Wimalaratne and S. L. Blair, J. Electrochem. Soc., 2009, 156, B553 CrossRef CAS.
- M. M. Schubert, A. Venugopal, M. J. Kahlich, V. Plzak and R. J. Behm, J. Catal., 2004, 222, 32–40 CrossRef CAS.
- Y. Hao, M. Mihaylov, E. Ivanova, K. Hadjiivanov, H. Knözinger and B. C. Gates, J. Catal., 2009, 261, 137–149 CrossRef CAS.
- E. J. Jr. Evans, H. Li, S. Han, G. Henkelman and C. B. Mullins, ACS Catal., 2019, 9, 4516–4525 CrossRef CAS.
- E. J. Evans, H. Li, W.-Y. Yu, G. M. Mullen, G. Henkelman and C. B. Mullins, Phys. Chem. Chem. Phys., 2017, 19, 30578–30589 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S11, Tables S1–S8, and equations for conversion of alcohol and selectivity. See DOI: https://doi.org/10.1039/d4ta01383b |
| ‡ Current address: Department of Chemistry and Biochemistry, University of California, Merced, Merced, CA 95343, USA. |
| § These authors contributed equally. |
| ¶ Deceased. |
| This journal is © The Royal Society of Chemistry 2024 |