
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced cerium-catalyzed C–H acylation of unactivated alkanes†
Jing
Cao
,
Joshua L.
Zhu
and
Karl A.
Scheidt
 *
*
Department of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, IL 60208, USA. E-mail: scheidt@northwestern.edu
First published on 25th November 2023
Abstract
Ketones are ubiquitous motifs in the realm of pharmaceuticals and natural products. Traditional approaches to accessing these species involve the addition of metal reagents to carboxyl compounds under harsh conditions. Herein, we report a cerium-catalyzed acylation of unactivated C(sp3)–H bonds using bench-stable acyl azolium reagents under mild and operationally-friendly conditions. This reaction exhibits excellent generality, accommodating a wide range of feedstock chemicals such as cycloalkanes and acyclic compounds as well as facilitating the late-stage functionalization of pharmaceuticals. We demonstrate further applications of our strategy with a three-component radical relay reaction and an enantioselective N-heterocyclic carbene (NHC) and cerium dual-catalyzed reaction.
Introduction
Simple alkanes represent one of the most abundant materials on earth,1 but reactions involving these unactivated substrates typically require prefunctionalization to selectively activate desired C(sp3)–H bonds over others with similar reactivities.2 The resurgence of photoredox reactions has inspired a series of innovative strategies, expanding the boundaries of chemical reactivity beyond traditional two-electron processes.3 Intermolecular hydrogen atom transfer (HAT) is one such strategy which has enabled the direct utilization of simple alkanes as substrates in radical transformations. This process efficiently cleaves normally inert C(sp3)–H bonds under mild conditions, yielding highly reactive radical species.4 One of the pivotal determinants of HAT processes is the bond dissociation energy (BDE); increasing the bond strength (Fig. 1A) leads to a higher activation energy barrier, consequently decelerating the HAT rate.5 This heightened barrier makes it challenging to access unactivated hydrocarbons, which have some of the highest C(sp3)–H BDEs.6 Over the past decade, substantial research endeavors have been directed towards the discovery of novel HAT methodologies for the functionalization of unactivated C(sp3)–H bonds.7 The first asymmetric ketone synthesis via direct HAT process was reported by the Ryu and Fagnoni in 2011, allowing the three-component reaction of simple alkane, carbon monoxide and electron-deficient alkenes.8 In 2018, the MacMillan group reported a direct HAT reaction using a decatungstate photocatalyst, successfully coupling aryl bromides and alkanes with unactivated C(sp3)–H bonds (Fig. 1A).9 Indirect HAT reactions, on the other hand, introduce HAT agents which can be oxidized under photoredox conditions to generate an electrophilic radical that serves as the hydrogen atom abstractor.11 In 2018, the Zuo group reported the use of a ligand-to-metal charge transfer (LMCT) process to generate high energy alkoxy radicals capable of cleaving various unactivated C(sp3)–H bonds.12 More recently, the Hong group combined the photoredox and nickel dual catalysis protocols for the synthesis of unsymmetric ketones using alkane substrates.13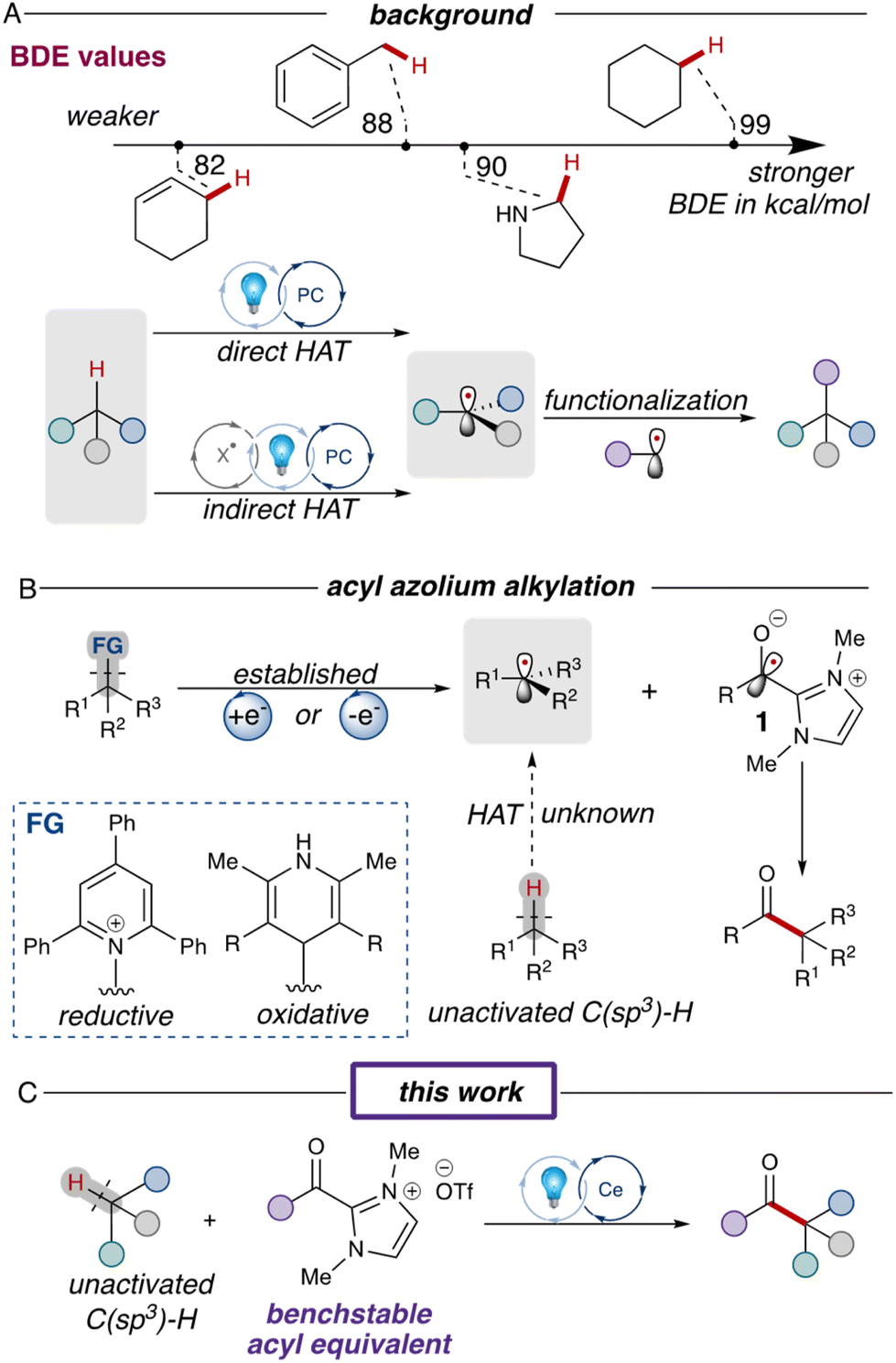 | ||
| Fig. 1 (A) Background for HAT processes and BDE parameters.10 (B) Established photoredox acylations via NHC intermediates and their challenges. (C) This work's strategy utilizing a LMCT process. | ||
Recently, studies merging N-heterocyclic carbene (NHC) catalysis with photoredox catalysis have garnered significant attention from the scientific community.14 One of the key discoveries was the generation of ketyl radical 1 either through single-electron reduction of the acyl azolium or oxidation of the Breslow intermediate, enabling the synthesis of ketones through ketyl radical cross-coupling (Fig. 1B).15 However, this reaction has been limited to prefunctionalized redox-active substrates like Hantzsch esters,16 Katritzky salts,17 and NHPI esters.18 Earlier this year, our group reported an acylation reaction involving unfunctionalized structures, such as proline derivatives.19 However, in this process, the acyl azolium proved unreactive with unactivated C(sp3)–H bonds due to the high energy barrier for HAT. To address this limitation, we sought a more potent HAT agent capable of activating inert C(sp3)–H bonds. In this work,20 we report the cerium-catalyzed acylation reactions of unactivated alkanes, employing in situ LMCT-generated chlorine radicals as the HAT agents (Fig. 1C).
Results and discussion
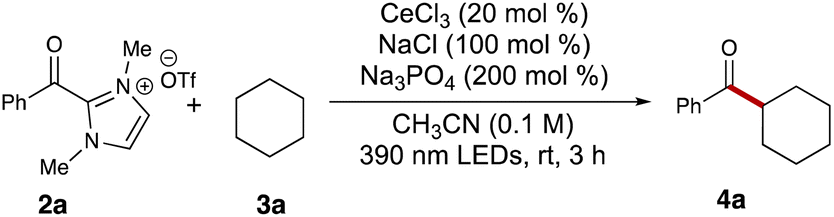
Our initial studies commenced with an examination of the acylation reaction between benzoyl dimethylimidazolium triflate (2a) and cyclohexane (3a, 5 equiv.). Following a series of screenings, we identified optimal conditions using cerium trichloride (0.2 equiv.), sodium chloride (1.0 equiv.), and sodium phosphate (3.0 equiv.). With these conditions, the desired ketone product 4a was isolated with 65% yield after 3 hours of 390 nm LED irradiation in 0.1 M acetonitrile (Table 1, entry 1). Switching cerium trichloride to cerium triflate significantly dropped the yield to 16% (Table 1, entry 2), but changing the cation for the chloride dopant to potassium led to a less dramatic loss in yield (Table 1, entry 3). Using a weaker base like sodium acetate dropped the yield to 35% (Table 1, entry 4), while organic base DBU failed to yield any of the desired product 4a (Table 1, entry 5). This reaction also showed sensitivity to the solvent environment, as seen when using ethyl acetate (48%, Table 1, entry 6) or dimethylformamide (0%, Table 1, entry 7). Tuning the reaction concentration, comparatively, had less of an impact on the yield of ketone 4a (43% and 45%, Table 1, entries 8–9). Control experiments showed no product formation in the absence of light or base (Table 1, entries 14 and 15). Interestingly, the reaction also provided the desired product 4a with 12% yield in the absence of cerium trichloride (Table 1, entry 12). This suggested that photoexcited acyl azolium 2a could serve as a less effective HAT agent in generating the cyclohexyl radical compared to the LMCT process. Finally, switching the light wavelength to 365 nm or 427 nm provided unsatisfying results (Table 1, entries 10 and 11).|
|
||
|---|---|---|
| Entry | Deviation from standard | Yielda (%) |
| a Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as internal standard. b Yield was based on isolation after chromatography. See ESI for reaction details. | ||
| 1 | None | 62 (56b) |
| 2 | Ce(OTf)3 instead of CeCl3 | 16 |
| 3 | KCl insetad of NaCl | 42 |
| 4 | NaOAc instead of Na3PO4 | 35 |
| 5 | DBU instead of Cs2CO3 | 0 |
| 6 | EtOAc instead of CH3CN | 48 |
| 7 | DMF instead of CH3CN | 0 |
| 8 | 0.05 M instead of 0.1 M | 43 |
| 9 | 0.2 M instead of 0.1 M | 45 |
| 10 | 365 nm instead of 390 nm | 11 |
| 11 | 427 nm instead of 390 nm | 0 |
| 12 | No CeCl3 | 12 |
| 13 | No NaCl | 30 |
| 14 | No Na3PO4 | 0 |
| 15 | No irradiation | 0 |
With the optimized conditions established, the reaction scope was explored starting with different ring sizes of the cycloalkanes, which all provided the corresponding ketones in good yields (Table 2, 56% to 64%, 4b to 4e). Cyclohexene 3j selectively reacted at the weak allylic C–H bond, leading to the formation of ketone 4j with 61% yield. Acyclic alkanes like n-pentane 3f coupled with acyl azolium 2f to generate a mixture of regioisomers (60%, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:2 rr), whereas cyclopentanone 3g exclusively provided the 1,4-dicarbonyl 4g in 48% yield. Due to the stabilization effect by adjacent heteroatoms, substr-ates 3h, 3l, 3k and 3g all preferentially furnished the α-acylated products with moderate to good yields. When employing benzylic alkanes as substrates, the conditions exhibited exceptional versatility, accommodating primary, secondary and tertiary radical precursors with consistently good yields. Various functional groups, such as esters (4q and 4r), trimethoxysilane (4s), and pinacolborane (4u), were all found to be well-tolerated under these conditions.
:5:2 rr), whereas cyclopentanone 3g exclusively provided the 1,4-dicarbonyl 4g in 48% yield. Due to the stabilization effect by adjacent heteroatoms, substr-ates 3h, 3l, 3k and 3g all preferentially furnished the α-acylated products with moderate to good yields. When employing benzylic alkanes as substrates, the conditions exhibited exceptional versatility, accommodating primary, secondary and tertiary radical precursors with consistently good yields. Various functional groups, such as esters (4q and 4r), trimethoxysilane (4s), and pinacolborane (4u), were all found to be well-tolerated under these conditions.
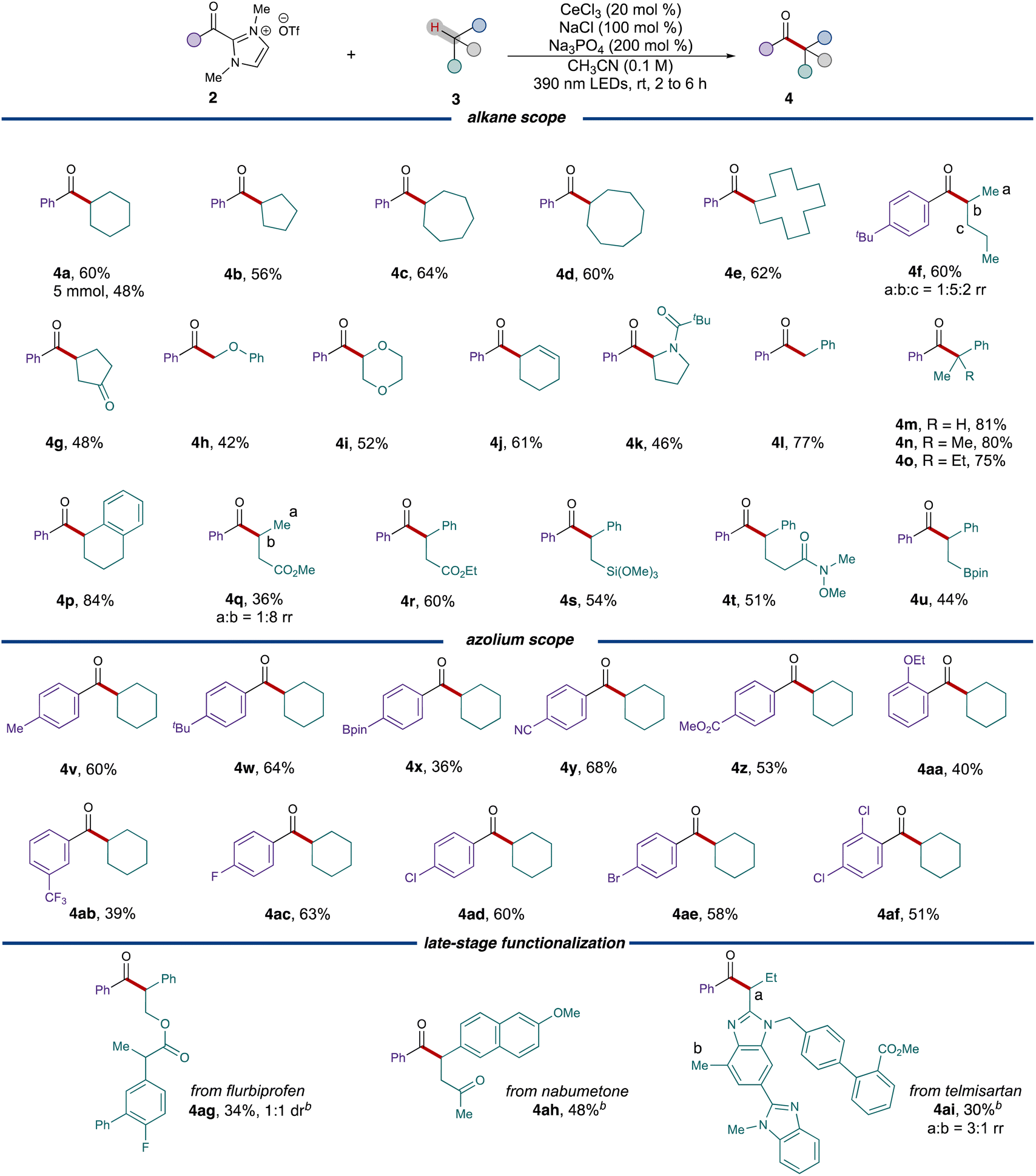
We subsequently proceeded to examine different acyl azolium partners. When modifying the substituents on the para position of the phenyl ring, both electron-poor substituents (3y, 3z) and electron-rich ones (3v, 3w) resulted in the formation of ketones with satisfactory yields. However, for more hindered cases, such as ortho-ethoxy benzoyl azolium 3aa, the yield of the product 4aa decreased to 40%. Different halogenated acyl azoliums (3ac, 3ad, 3ae) were well tolerated under the conditions as well. These conditions could also be extended to the late stage functionalizations of pharmaceuticals. Both nabumetone 3ah and telmisartan derivative 3ai provided the acylated products 4ah and 4ai with moderate yields while only employing 1.0 equivalent of the drug substrate. Flurbiprofen derivative 3ag also provided the corresponding ketone 4ag in 34% yield and 1:1 dr.
To probe the mechanism of this reaction, we conducted two experiments. A kinetic isotope effect study was performed via using equal amounts of cyclohexane 3a and cyclohexane-d125 in a single reaction vial, resulting in products 4a and 6 in a 3.35:1 ratio (Scheme 1A). The high ratio is indicative of a primary kinetic isotope effect, implying the HAT process is the rate-determining step. Next, a TEMPO trapping experiment was performed with the standard reaction. After addition of 3.0 equivalents of TEMPO to the standard conditions, no ketone 4a could be detected. Additionally, TEMPO adducts with the intermediary cyclohexyl radical and the azolium radical intermediate were both observed via ESI-HRMS (TEMPO adducts 7 and 8, Scheme 1B).
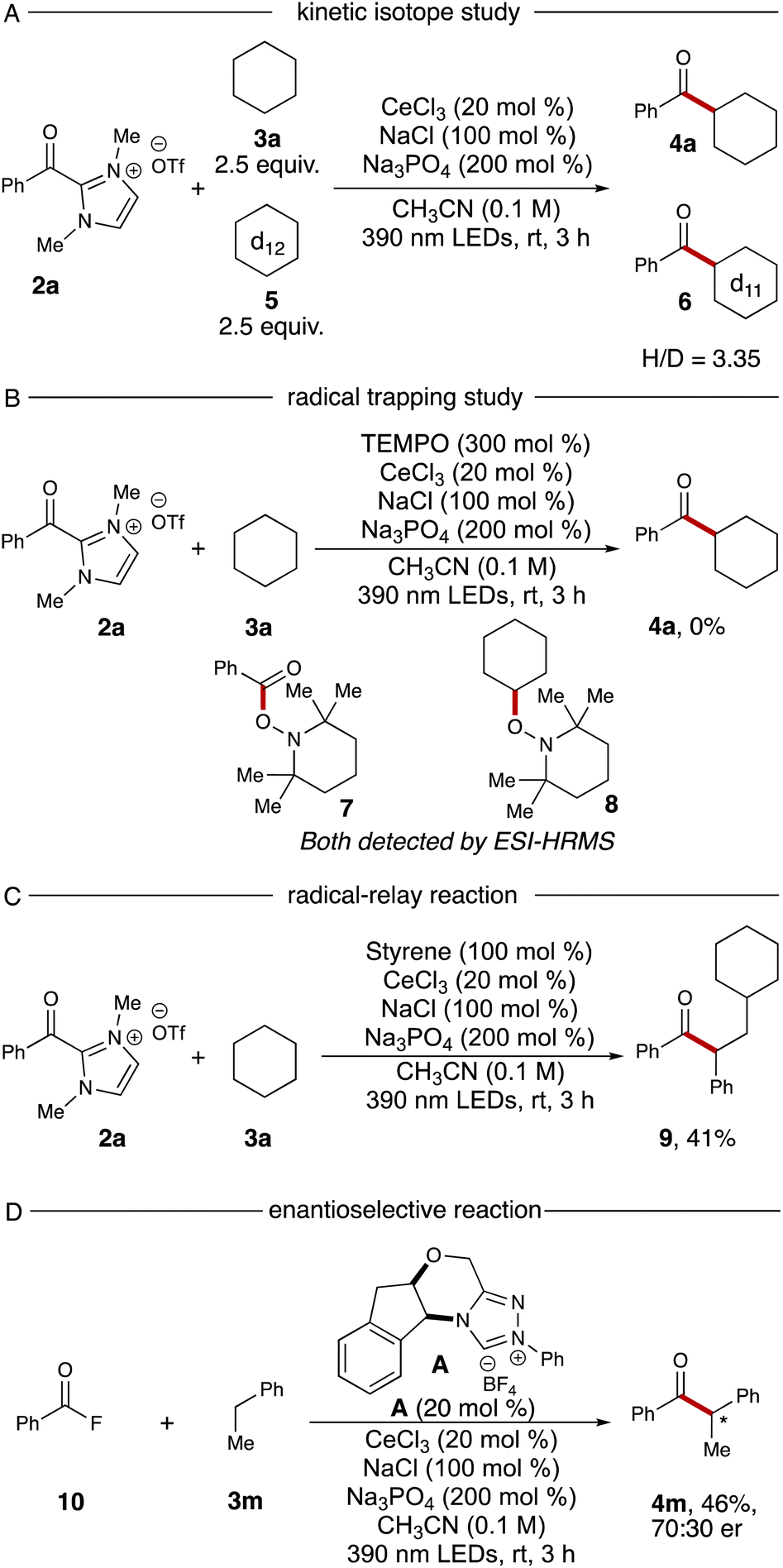 | ||
| Scheme 1 (A) Competition KIE experiment with cyclohexane and cyclohexane-d12. (B) TEMPO trapping experiment. (C) Three-component radical relay reaction. (D) Enantioselective NHC and cerium trichloride dual catalyzed reaction. | ||
The radical–relay reaction of alkenes offers powerful methods for rapidly assembling molecular complexity using readily available materials. Recent work by our group,21 Studer,22 Ohmiya,23 Chi,15e and Wang24 have extended the reactivity pattern of acyl azoliums into three-component radical reactions. When adding 1.0 equivalent of styrene into our standard reaction conditions, the desired relay product 9 was furnished with 41% yield without further optimization (Scheme 1C). Finally, we examined the enantioselective NHC-catalyzed acylation using benzoyl fluoride 10 as the acyl azolium precursor. After screening a series of chiral NHC catalysts, we identified that catalyst A provided the chiral ketone 4m in 46% yield and 70:30 er (Scheme 1D, details for other chiral NHC screening see ESI†).
Given these studies,25 we propose the following reaction mechanism (Scheme 2). Initially, the catalytically-active metal complex [CeIIICl6]3− is generated from cerium chloride. This species, when photoexcited, serves as a single electron reductant 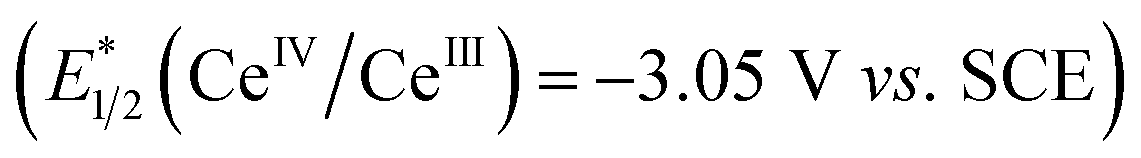 ,26 which can reduce the acyl azolium (benzoyl azolium 2a, E1/2 = −1.29 V vs. SCE).16 to provide the persistent azolium radical intermediate 1 and [CeIVCl6]2−.27 Following this is the LMCT process, wherein [CeIVCl6]2−, after excitation to a short-lived excited state, undergoes homolytic cleavage, yielding the reduced [CeIIICl5]2− complex and a chloride radical.28 The chloride radical then abstracts a hydrogen atom from alkane 3, resulting in alkyl radical 10. Subsequent radical–radical coupling leads to the formation of the tertiary azolium intermediate 11. Under basic conditions, this intermediate ejects the carbene 12, ultimately yielding the ketone 4.
,26 which can reduce the acyl azolium (benzoyl azolium 2a, E1/2 = −1.29 V vs. SCE).16 to provide the persistent azolium radical intermediate 1 and [CeIVCl6]2−.27 Following this is the LMCT process, wherein [CeIVCl6]2−, after excitation to a short-lived excited state, undergoes homolytic cleavage, yielding the reduced [CeIIICl5]2− complex and a chloride radical.28 The chloride radical then abstracts a hydrogen atom from alkane 3, resulting in alkyl radical 10. Subsequent radical–radical coupling leads to the formation of the tertiary azolium intermediate 11. Under basic conditions, this intermediate ejects the carbene 12, ultimately yielding the ketone 4.
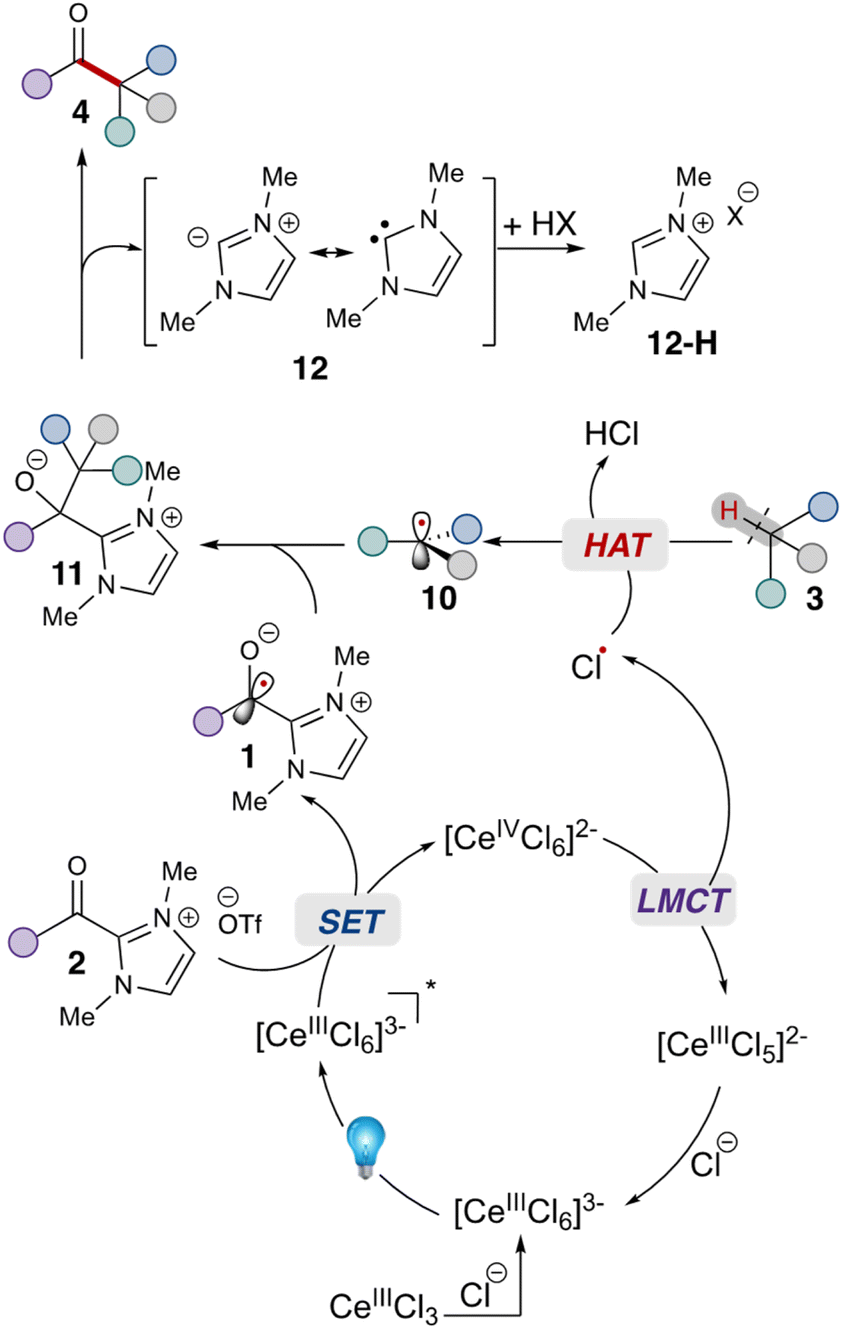 | ||
| Scheme 2 Proposed mechanism for the acylation of alkane 3 with acyl azolium 2. | ||
Conclusions
In summary, we have successfully established a cerium-catalyzed acylation capable of functionalizing unactivated C(sp3)–H bonds using bench-stable acyl azolium reagents. This reaction demonstrates remarkable versatility, accommodating various types of C–H bonds with moderate to good yields. We have further demonstrated the application of these conditions to a three-component reaction without further optimization, thus underscoring their potential applicability in a wide range of acylating transformations. Additionally, the enantioselective NHC and cerium dual-catalyzed reaction serves as a preliminary demonstration of asymmetric ketone synthesis from unactived alkanes. The development of new catalytic applications for cerium, a cheap and earth-abundant lanthanide, has opened up new avenues for sustainable synthesis.29 By integrating cerium catalysis in conjunction with acyl azoliums and NHC catalysis, we are able to achieve the acylation of unactivated C(sp3)–H bonds in simple alkanes. Future studies around developing the highly enantioselective acylation of simple alkanes is ongoing in our group.Data availability
All experimental data, and detailed experimental procedures are available in the published article and ESI.†Author contributions
The work was conceptualized by K. A. S. The experiments were performed by J. C. and J. L. Z. The manuscript was written through contributions of all authors. K. A. S. secured funding and supervised the entire work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Institute of General Medical Sciences (R35 GM136440) and Northwestern University for financial support of this work. We thank Dr Qiupeng Peng (NU) for help with HRMS. We thank Meemie Hwang (NU) for assistance with FT-IR and Cullen R. Schull (NU) for assistance with the manuscript.Notes and references
- A. A. Fokin and P. R. Schreiner, Chem. Rev., 2002, 102, 1551–1594 CrossRef CAS.
- (a) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed; (b) O. K. Rasheed and B. Sun, ChemistrySelect, 2018, 3, 5689–5708 CrossRef CAS; (c) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (d) R. H. Crabtree, Chem. Rev., 2010, 110, 575 CrossRef CAS; (e) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS; (f) H. M. L. Davies, J. Du Bois and J.-Q. Yu, Chem. Soc. Rev., 2011, 40, 1855–1856 RSC.
- (a) J. Twilton, C. Le, P. Zhang, M. H. Shaw, R. W. Evans and D. W. C. MacMillan, Nat. Rev. Chem, 2017, 1, 0052 CrossRef CAS; (b) J. P. Barham and B. König, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS; (c) D. A. Nagib, Chem. Rev., 2022, 122, 15989–15992 CrossRef CAS; (d) K. Zeitler, Angew. Chem., Int. Ed., 2009, 48, 9785–9789 CrossRef CAS PubMed; (e) T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed; (f) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC; (g) D. Ravelli, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2013, 42, 97–113 RSC; (h) J. Xuan and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 6828–6838 CrossRef CAS; (i) M. N. Hopkinson, B. Sahoo, J.-L. Li and F. Glorius, Chem.–Eur. J., 2014, 20, 3874–3886 CrossRef CAS.
- (a) N. Holmberg-Douglas and D. A. Nicewicz, Chem. Rev., 2021, 122, 1925–2016 CrossRef; (b) D. Ravelli, S. Protti and M. Fagnoni, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS PubMed; (c) K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (d) Z. Ye, Y. M. Lin and L. Gong, Eur. J. Org. Chem., 2021, 2021, 5545–5556 CrossRef CAS; (e) L. Capaldo and D. Ravelli, Eur. J. Org. Chem., 2017, 2017, 2056–2071 CrossRef CAS.
- J. M. Tedder, Tetrahedron, 1982, 38, 313–329 CrossRef CAS.
- L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- (a) C. Shu, A. Noble and V. K. Aggarwal, Nature, 2020, 586, 714–719 CrossRef CAS; (b) S. Mukherjee, B. Maji, A. Tlahuext-Aca and F. Glorius, J. Am. Chem. Soc., 2016, 138, 16200–16203 CrossRef CAS PubMed; (c) C. M. Morton, Q. Zhu, H. Ripberger, L. Troian-Gautier, Z. S. D. Toa, R. R. Knowles and E. J. Alexanian, J. Am. Chem. Soc., 2019, 141, 13253–13260 CrossRef CAS; (d) L. K. G. Ackerman, J. I. Martinez Alvarado and A. G. Doyle, J. Am. Chem. Soc., 2018, 140, 14059–14063 CrossRef CAS PubMed; (e) D.-M. Yan, C. Xiao and J.-R. Chen, Chem, 2018, 4, 2496–2498 CrossRef CAS; (f) K. Yahata, S. Yoshioka, S. Hori, S. Sakurai, Y. Kaneko, K. Hasegawa and S. Akai, Chem. Pharm. Bull., 2020, 68, 336–338 CrossRef; (g) D. Mazzarella, A. Pulcinella, L. Bovy, R. Broersma and T. Noel, Angew. Chem., Int. Ed., 2021, 60, 21277–21282 CrossRef CAS.
- I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, M. Fagnoni and A. Albini, Angew. Chem., Int. Ed., 2011, 50, 1869–1872 CrossRef CAS.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Nature, 2018, 560, 70–75 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- (a) F. S. Meger and J. A. Murphy, Mol., 2023, 28 Search PubMed; (b) Y. Abderrazak, A. Bhattacharyya and O. Reiser, Angew. Chem., Int. Ed., 2021, 60, 21100–21115 CrossRef CAS; (c) L. Chang, S. Wang, Q. An, L. Liu, H. Wang, Y. Li, K. Feng and Z. Zuo, Chem. Sci., 2023, 14, 6841–6859 RSC.
- A. Hu, J.-J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS.
- (a) G. S. Lee, J. Won, S. Choi, M.-H. Baik and S. H. Hong, Angew. Chem., Int. Ed., 2020, 59, 16933–16942 CrossRef CAS; (b) G. S. Lee, B. Park and S. H. Hong, Nat. Commun., 2022, 13, 5200 CrossRef CAS.
- (a) A. V. Bay and K. A. Scheidt, Trends Chem., 2022, 4, 277–290 CrossRef CAS PubMed; (b) K. Liu, M. Schwenzer and A. Studer, ACS Catal., 2022, 12, 11984–11999 CrossRef CAS; (c) A. Mavroskoufis, M. Jakob and M. N. Hopkinson, ChemPhotoChem, 2020, 4, 5147–5153 CrossRef CAS.
- (a) A. V. Bay, K. P. Fitzpatrick, G. A. González-Montiel, A. O. Farah, P. H.-Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2021, 60, 17925–17931 CrossRef CAS; (b) M. J. Rourke, C. T. Wang, C. R. Schull and K. A. Scheidt, ACS Catal., 2023, 13, 7987–7994 CrossRef CAS; (c) A. V. Bay, E. J. Farnam and K. A. Scheidt, J. Am. Chem. Soc., 2022, 144, 7030–7037 CrossRef CAS; (d) Q.-Y. Meng, L. Lezius and A. Studer, Nat. Commun., 2021, 12, 2068 CrossRef CAS; (e) S.-C. Ren, X. Yang, B. Mondal, C. Mou, W. Tian, Z. Jin and Y. R. Chi, Nat. Commun., 2022, 13, 2846 CrossRef CAS PubMed; (f) S.-C. Ren, W.-X. Lv, X. Yang, J.-L. Yan, J. Xu, F.-X. Wang, L. Hao, H. Chai, Z. Jin and Y. R. Chi, ACS Catal., 2021, 11, 2925–2934 CrossRef CAS; (g) M.-S. Liu and W. Shu, ACS Catal., 2020, 10, 12960–12966 CrossRef CAS; (h) X. Wang, B. Zhu, Y. Liu and Q. Wang, ACS Catal., 2022, 12, 2522–2531 CrossRef CAS; (i) K. Liu and A. Studer, J. Am. Chem. Soc., 2021, 143, 4903–4909 CrossRef CAS.
- A. V. Bay, K. P. Fitzpatrick, R. C. Betori and K. A. Scheidt, Angew. Chem., Int. Ed., 2020, 59, 9143–9148 CrossRef CAS PubMed.
- I. Kim, H. Im, H. Lee and S. Hong, Chem. Sci., 2020, 11, 3192–3197 RSC.
- T. Ishii, Y. Kakeno, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 3854–3858 CrossRef CAS PubMed.
- J. L. Zhu, C. R. Schull, A. T. Tam, Á. Rentería-Gómez, A. R. Gogoi, O. Gutierrez and K. A. Scheidt, J. Am. Chem. Soc., 2023, 145, 1535–1541 CrossRef CAS PubMed.
- F. Su, F. Lu, K. Tang, X. Lv, Z. Luo, F. Che, H. Long, X. Wu and Y. R. Chi, Angew. Chem., Int. Ed., 2023, e202310072, DOI:10.1002/anie.202310072.
- P. Wang, K. P. Fitzpatrick and K. A. Scheidt, Adv. Synth. Catal., 2022, 364, 518–524 CrossRef CAS.
- Q.-Y. Meng, N. Döben and A. Studer, Angew. Chem., Int. Ed., 2020, 59, 19956–19960 CrossRef CAS PubMed.
- Y. Sato, Y. Goto, K. Nakamura, Y. Miyamoto, Y. Sumida and H. Ohmiya, ACS Catal., 2021, 11, 12886–12892 CrossRef CAS.
- B. Zhang, J.-Q. Qi, Y. Liu, Z. Li and J. Wang, Org. Lett., 2022, 24, 279–283 CrossRef CAS PubMed.
- Due to the similarities in absorption profile between the cerium catalyst and the acyl imidazolium we were unable to obtain a linear Stern–Volmer quenching relationship. This is likely due to an inability to selectively excite the cerium catalyst and potentially due to an inner filter effect.
- H. Yin, Y. Jin, J. E. Hertzog, K. C. Mullane, P. J. Carroll, B. C. Manor, J. M. Anna and E. J. Schelter, J. Am. Chem. Soc., 2016, 138, 16266–16273 CrossRef CAS.
- V. Regnier, E. A. Romero, F. Molton, R. Jazzar, G. Bertrand and D. Martin, J. Am. Chem. Soc., 2019, 141, 1109–1117 CrossRef CAS.
- Q. Yang, Y.-H. Wang, Y. Qiao, M. Gau, P. J. Carroll, P. J. Walsh and E. J. Schelter, Science, 2021, 372, 847–852 CrossRef CAS.
- (a) Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS PubMed; (b) N. Mahieu, J. Piatkowski, T. Simler and G. Nocton, Chem. Sci., 2023, 14, 443–457 RSC; (c) H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2021, 143, 7879–7890 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc05162e |
| This journal is © The Royal Society of Chemistry 2024 |