
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZirconium-based metal–organic frameworks: the relation between linker connectivity, structure stability, and catalytic activity towards organophosphates†
Daniel
Bůžek
 *ab,
Jan
Hynek
a,
Matouš
Kloda
a,
Veronika
Zlámalová
b,
Petr
Bezdička
a,
Slavomír
Adamec
b,
Kamil
Lang
a and
Jan
Demel
a
*ab,
Jan
Hynek
a,
Matouš
Kloda
a,
Veronika
Zlámalová
b,
Petr
Bezdička
a,
Slavomír
Adamec
b,
Kamil
Lang
a and
Jan
Demel
a
aInstitute of Inorganic Chemistry of the Czech Academy of Sciences, 250 68 Husinec-Řež č.p. 1001, Czech Republic. E-mail: buzek@iic.cas.cz
bFaculty of Environment, Jan Evangelista Purkyně University in Ústí nad Labem, Pasteurova 3632/15, 400 96 Ústí nad Labem, Czech Republic
First published on 9th July 2024
Abstract
Metal–organic frameworks (MOFs) are studied for many applications, however, there are only a few examples of commercialization. One of the reasons behind this is that the stability of MOFs is still unknown. Much attention has been devoted to the rational synthesis of novel MOFs, yet the predictability of MOF stability is so far limited. The present study compares the stability in a water environment with pH ranging from 3.0 to 11.0 of four zirconium-based MOFs constructed from ditopic, tritopic, and tetratopic linkers, namely UiO-66 (benzene-1,4-dicarboxylic acid), MOF-808 (benzene-1,3,5-tricarboxylic acid), MIP-200 (5,5′-methylenediisophthalic acid), and PCN-222 (5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin). Finally, to delineate the nature of the defects induced by the linker release, we tested the MOFs treated at a certain pH for the degradation of model organic pollutant methyl paraoxon. It is clear that both MOFs based on tetratopic linkers are much more stable than UiO-66 and MOF-808 composed of di- and tritopic linkers, respectively. It should be noted that the kinetics of the linker release were also significantly slower for tetratopic linkers. At the same time, the connectivity of the Zr6 cluster did not play such an important role. MIP-200 proved to be the most stable MOF from the series in an aqueous environment; however, the loss of a small amount of monocarboxylic acid from the structure allowed thermal recrystallization of MIP-200 to an unknown phase so far.
Introduction
Metal–organic frameworks (MOFs) are porous coordination polymers containing potential voids.1 They are composed of inorganic nodes, referred to as secondary building units (SBUs), linked together by multitopic ligands serving as linkers. Due to the numerous possible combinations of metals or SBUs,2 including homometallic or heterometallic clusters,3 with organic linkers of variable geometries and coordinating groups4,5 it is not surprising that over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 MOF structures have been published so far.6 MOFs provide a broad spectrum of structures and topologies,7 leading to tuneable and sometimes predictable properties and functionalities. These advantages, together with their porosity, high specific surface area,8,9 and tuneability of their properties,10 predetermine MOFs for a broad spectrum of potential applications, e.g. gas storage and separation,11–13 separation of volatile organic compounds,14 adsorption of pollutants from liquids,15,16 drug delivery and cancer therapy,17,18 energy applications,19 water harvesting and storage,20,21 sensing,22,23 and catalysis.24,25
000 MOF structures have been published so far.6 MOFs provide a broad spectrum of structures and topologies,7 leading to tuneable and sometimes predictable properties and functionalities. These advantages, together with their porosity, high specific surface area,8,9 and tuneability of their properties,10 predetermine MOFs for a broad spectrum of potential applications, e.g. gas storage and separation,11–13 separation of volatile organic compounds,14 adsorption of pollutants from liquids,15,16 drug delivery and cancer therapy,17,18 energy applications,19 water harvesting and storage,20,21 sensing,22,23 and catalysis.24,25
In spite of their great application potential, the number of industrial processes utilizing MOFs is so far limited. One of the reasons is the majority of MOFs’ insufficient stability, i.e., low resistance to degradation during operating conditions.26 There are several aspects of stability involving thermal, mechanical, or chemical stability, or their combinations. The chemical stability or more specifically, stability in a water environment, is crucial for many applications including adsorption and catalytic degradation of pollutants, sensing, antibacterial and antiviral use,22,27–31 drug delivery,32 or special applications such as using MOFs as pesticide carriers.33 All those applications require aqueous media; therefore, MOFs’ hydrolytic stability is of crucial importance. Laboratory testing is often limited to neat water; however, in real-life applications (the use of real contaminated water, body fluids, etc.) the aqueous medium contains many other dissolved compounds such as buffers or salts, and has different pH values, making the system much more complex and challenging for MOFs.
While the predictability of MOF synthesis has made significant progress in recent years, the predictability of MOF stabilities is so far unclear. It is understood that the MOF stability in aqueous media is strongly influenced by the strength of the coordination bonds between metals and linkers. In accordance with the hard and soft acid and base (HSAB) theory, the utilization of carboxylate linkers in combination with hard metals (Al(III), Zr(IV), Hf(IV), Ti(IV), etc.) leads to the construction of MOFs with increased stability in comparison with, e.g., the original MOF-5.34–39 Alternatively, the utilization of phosphonate linkers40,41 or oxalate42 can result in MOF structures of exceptional stability. The strength of coordination bonds, however, is not the only factor playing a critical role in the stability of MOFs. It is also affected by the length of connecting agents, the pKa of a linker and its solubility in a given environment, or the SBUs’ hydrophobicity and connectivity (the number of linker molecules bound to an SBU). It is assumed that, in general, higher connectivity, shorter connecting agents, and lower solubility of the linker lead to MOFs with a high stability in the water environment.36,37,43–47
Many of the stability studies in aqueous media that were presented in the literature are based on a post-exposure analysis of the corresponding solids using powder X-ray diffraction (XRPD), gas adsorption, scanning electron microscopy (SEM), infrared spectroscopy (FTIR), or their combination with computational methods.26,32,45,46,48–53 For example, when MOFs’ stability is inferred from XRPD patterns before and after the treatment, while ignoring the mass balance, presence of an amorphous phase, and changes in the chemical composition, erroneous conclusions can be drawn. In our recent studies, we have demonstrated that the structure of UiO-66, a Zr-based MOF considered one of the most stable MOFs, is compromised already at neutral pH or in the presence of commonly used buffers, such as TRIS or phosphate-buffered saline.54,55
Here, we investigate the effect of the SBU connectivity and linker topicity on MOF stabilities in aqueous environments of different pH and correlate the changes made to MOFs in those conditions with catalytic activity in organophosphate degradation. We selected four Zr-MOFs constructed using di-, tri- and tetratopic carboxylate linkers – UiO-66, MOF-808, MIP-200, and PCN-222 (see Fig. 1). UiO-66 is based on terephthalate (benzene-1,4-dicarboxylate, BDC2−) linkers and the 12-connected Zr6O8 SBU,56 forming an fcu-topology (in the ideal defect-free crystal).27,57,58 Next, MOF-808 is based on trimesate (benzene-1,3,5-tricarboxylate, BTC3−) linkers and 6-connected SBUs.62 MIP-200, which is a relatively young member of the Zr-MOF family and rated as exceptionally stable, is composed of tetratopic 5,5′-methylenediisophthalate (MDIP4−) linkers and 8-connected SBUs.59 The last of the series, PCN-222/MOF-545, prepared simultaneously by Yaghi et al.60 and Zhou et al.63 (hereafter abbreviated as PCN-222), features 8-connected SBUs and tetratopic linkers (5,10,15,20-tetrakis(4-carboxyphenyl)porphyrin, abbreviated here as TCPP4−), forming a hexagonal structure.
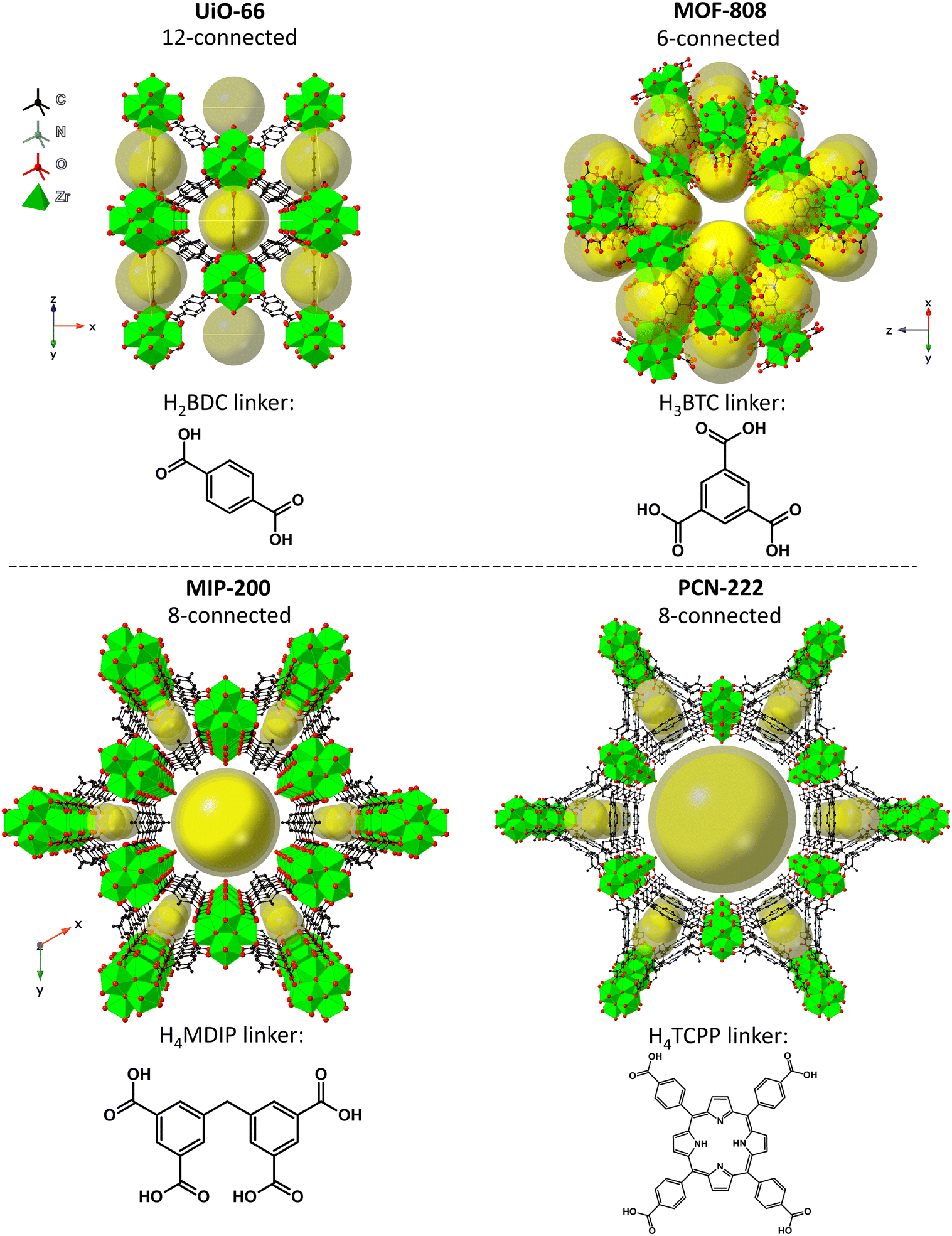 | ||
| Fig. 1 Idealized structures of the tested MOFs: UiO-6661 (top left), MOF-80862 (top right), MIP-20059 (bottom left), and PCN-22263 (bottom right). The references refer to the corresponding CIF sources. | ||
The stability was studied by the combination of HPLC, analysing released linkers from the MOF, and the post-exposure analysis of the remaining solids (XRPD, N2 adsorption, FTIR, differential thermal analysis (TGA/DTA), and chemical composition). Additionally, to put Zr-MOF stability into the perspective of an application, the parent and post-exposure MOFs were used for the catalytic degradation of methyl paraoxon (DMNP), an organophosphate (phosphate ester) commonly used as a simulant of organophosphate-based pesticides64 and nerve chemical warfare agents.65 It is well described that missing-linker or missing-cluster defects in Zr-MOFs play a crucial role in catalytic reactions66,67 because open-metal sites act as catalytic centres for the degradation of organophosphates.68–71 In our previous work,54 we have shown that the removal of BDC2− from UiO-66 increases its catalytic activity considerably due to the increased number of open-metal sites. Here, we present a comparison of the catalytic activities of UiO-66, MOF-808, MIP-200, and PCN-222 after treatment at various pHs on the degradation of DNMP. The catalytic activity in the degradation of DMNP serves as an indication of whether the removal of linker in tested MOFs is connected with the formation of catalytically active defects or rather a structural collapse.
Experimental section
Instruments and materials
The list of chemicals is summarized in Table S1, ESI.† Similarly, instrumentation and measurement conditions (i.e., XRPD, amorphous content, HPLC analyses of the linkers, monocarboxylates, DMNP and 4-NP, ICP-MS analyses of Zr, FTIR, gas adsorption and TGA/DTA, catalytic experiments) are described in ESI.†Syntheses of MOFs
Synthesis of UiO-66,55,56 MOF-808,72 MIP-200,59 and PCN-22260,73 followed the modified procedures published in referenced papers. The procedures are described in detail in ESI.†
Acid–base titration with released linker monitoring
The pH dependence of the amount of released linkers was determined using a procedure published by Klet et al.74 and further elaborated by our group.54 A beaker was charged with 50 mg of activated MOF (heated at 120 °C, 4 h, dynamic vacuum) and 50 mL of water, followed by 3 min of sonication to obtain a suspension that did not sediment within several minutes. The suspension was then stirred, and the pH was adjusted by the addition of 0.1 M NaOH using an automatic titrator Metrohm 906 Titrando. In each step, 0.005 mL of the NaOH solution was added with the rate of addition set to 0.04 mL min−1. During the titration, the pH value gradually increased, and a 0.2 mL sample was taken immediately after reaching a preset pH value. The samples were immediately filtered through a PTFE microfilter (Whatman, 0.2 μm) and analysed using HPLC. These experiments provided data on the immediate release of the linkers at a certain pH; however, the kinetics of the linker release are not reflected in the results.The same procedure was utilized for the determination of the immediate linker solubility at different pHs. 50 mg of the linker was mixed with 50 mL of water, followed by 3 min of sonication, and after that, the suspension of the linker in water was stirred for 2 h to achieve the equilibrium concentration at 25 ± 1 °C. The immediate solubility of the linker during titration experiments indicates how the linker release from the MOF structures is limited by the linker solubility.
Kinetics of the linker release at different pHs
The kinetic stability of selected MOFs was studied at the natural pH of the MOF suspensions and at pH 6.0, 7.0, 8.0, 9.0, 10.0, and 11.0. In the case of MIP-200, a pH value of 12.0 was also used. In each kinetic experiment, 250 mg of MOF was suspended in water and sonicated for 3 min. The exact amount of added water was calculated for each pH to provide the total volume of 250 mL after the addition NaOH. After that, 0.1 M NaOH solution was added to reach the target pH value. The pH was kept constant during the kinetic experiments using an automatic titrator (Metrohm 906 Titrando) which continuously adjusted pH with 0.1 M NaOH. The samples (0.2 mL) were taken out within 0 to 240 min (the periods were shorter during the first 60 min to achieve better rendering of the kinetic curves), filtered through PTFE microfilters (Whatman, 0.2 μm), and analysed by HPLC. Each kinetic experiment was repeated at least three times. After the kinetic experiments, the residual solids were collected by centrifugation (4000 rpm, Hermle ZK 496), washed three times with water and five times with acetone, air-dried, and reactivated by vacuum-drying at 120 °C. Only in the case of MIP-200, the samples were air-dried and reactivated under a vacuum only at laboratory temperature because at higher temperatures, MIP-200 recrystallizes, see below. All these samples were analysed by XRPD, N2 adsorption, FTIR, and TGA/DTA, and the composition was determined by dissolving 10 mg of solid in 10 mL of 0.1 M NaOH and subsequent analysis of the solution. All released carboxylic acids were determined by HPLC-DAD. The metal content was determined after the acidic dissolution of 10 mg of the sample by ICP-MS analysis. Analogous analyses were also performed with the parent MOFs.Results and discussion
Synthesis and characterization of MOFs
Zirconium-based UiO-66 was obtained by the solvothermal reaction of ZrCl4, terephthalic acid (H2BDC), and acetic acid as a modulator in the 1:1:4.8 molar ratio resulting in a microcrystalline powder. The XRPD patterns of the parent UiO-66 confirmed the reported structure with a broad diffraction peak between 2 and 6° 2θ, indicating numerous structural defects caused by the formation of reo-phase (Fig. S1, ESI†).58,75 The N2 adsorption isotherm has the typical shape for UiO-66, with a calculated BET specific surface area (SBET) of 1263 m2 g−1 (Fig. 2D, Table 1). This value is higher than the theoretical surface area due to the presence of structural defects.76 The TGA/DTA curves show a weight loss of approximately 10 wt% below 420 °C that includes release of moisture, residual solvents, and a part of monocarboxylic acids,58 whereas the sharp weight loss of 45 wt% between 420 and 550 °C is, according to the MS analysis of evolved gases, related to the complete combustion of the BDC2− linkers (Fig. S2, ESI†). The result is in good agreement with the content of 48.5% BDC2− determined by HPLC analysis of the MOF dissolved in a basic solution. The mass content of all components is summarized in Tables S3 and S4 in ESI.†
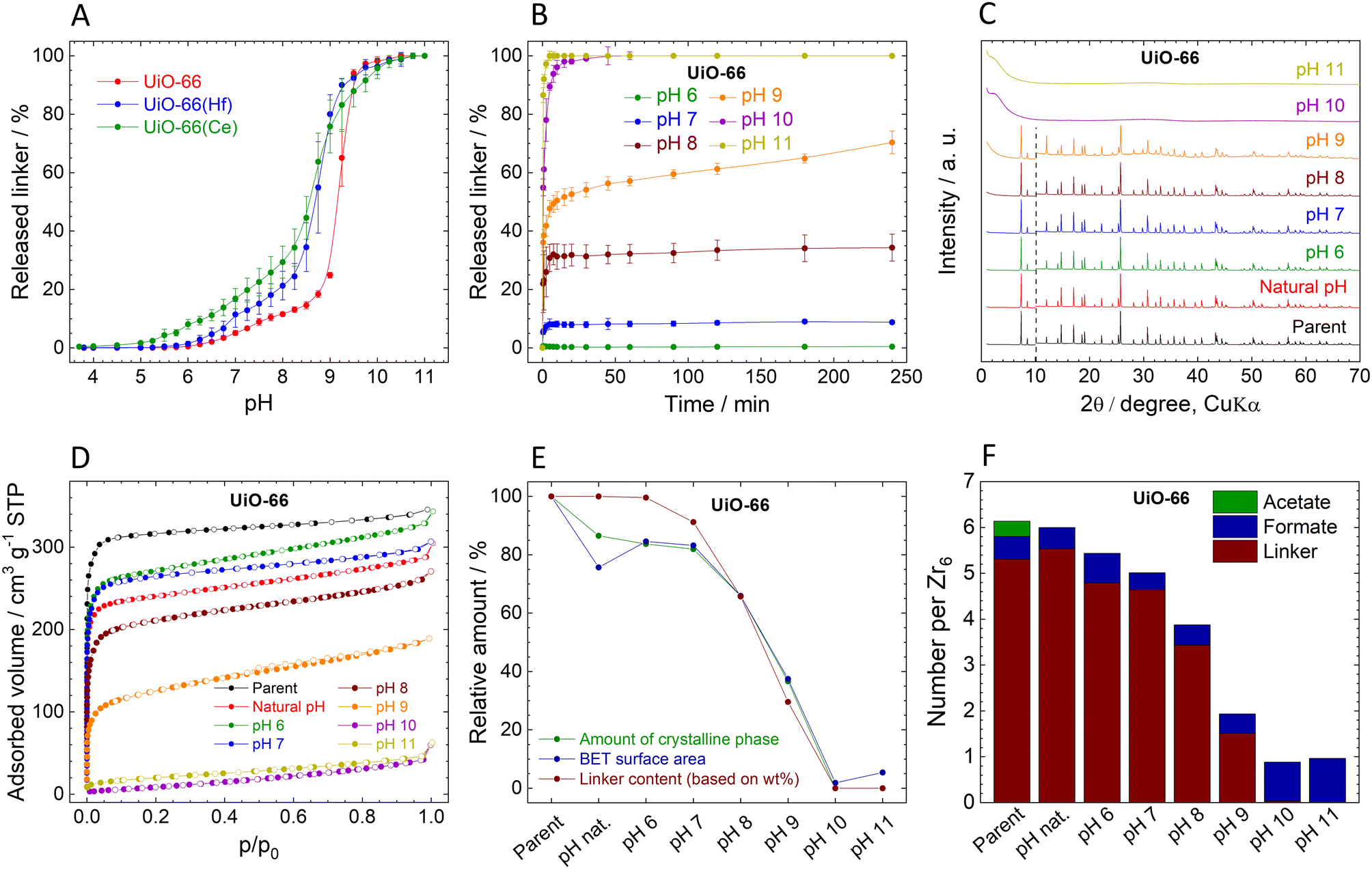 | ||
| Fig. 2 Stability of UiO-66. (A). Immediate release of BDC2− from Zr-based UiO-66 compared with UiO-66(Hf) and UiO-66(Ce). (B). Time-dependent BDC2− release from UiO-66 at different pH values. (C). Comparison of XRPD pattern of the parent and post-exposure UiO-66; the diffractograms are normalized and shifted vertically to avoid overlaps. The dashed line at 10° 2θ separates the zoomed area to visualize the diffractions at higher angles. (D). Adsorption (filled dots) and desorption (empty dots) isotherms of N2 measured at 77 K. (E). Decrease of the crystalline phase percentage, SBET, and linker content (all data are normalized, taking the parent UiO-66 as a default state with 100% values). (F). Molar ratios of the ligands in the parent and post-exposure UiO-66 to the Zr6 metal clusters. | ||
| Sample | Crystalline phase/% | Linker removal after 4 h/% | S BET/m2 g−1 | Conversion DMNP at 30 min/% | Conversion DMNP at 120 min/% |
|---|---|---|---|---|---|
| a Natural pH is 3.8. | |||||
| UiO-66 parent | 100 | 0.0 | 1238 | 66 | 96 |
| UiO-66 nat. pHa | 87 | <LOD | 937 | 73 | 94 |
| UiO-66 pH 6 | 84 | 0.4 | 1047 | 79 | 97 |
| UiO-66 pH 7 | 82 | 8.8 | 1030 | 90 | 98 |
| UiO-66 pH 8 | 66 | 34.3 | 816 | 96 | 100 |
| UiO-66 pH 9 | 37 | 70.4 | 464 | 70 | 92 |
| UiO-66 pH 10 | 0 | 100 | 23 | 4 | 6 |
| UiO-66 pH 11 | 0 | 100 | 67 | 5 | 6 |
MOF-808 was prepared according to the optimized procedure published previously.72 ZrCl4 and H3BTC were mixed in a 3:1 molar ratio in a 1:1 DMF/acetic acid mixture, followed by crystallization at 120 °C for 72 h. The XRPD pattern corresponds to that of a well-crystalline MOF-808 (Fig. S3, ESI†).72 The N2 adsorption isotherm exhibits the shape typical for MOF-808 containing multiple types of pores (Fig. 3D).77 The thermogravimetric curve did not show any sharp features as in the case of UiO-66, which is typical for MOF-808 (Fig. S4, ESI†).72,77 The mass plateaued at 580 °C with a final weight loss of approximately 49 wt%. The MS analysis of evolved gases indicated that residual DMF, acetic acid, acetone, and moisture are released below 400 °C, representing approximately 20% of the total weight. The weight loss assigned to the linker combustion at higher temperatures is approximately 29%, which is in good agreement with 28.2 wt% of the BTC3− determined by HPLC after the solid dissolution. The mass content of all components is summarized in Tables S5 and S6 in ESI.†
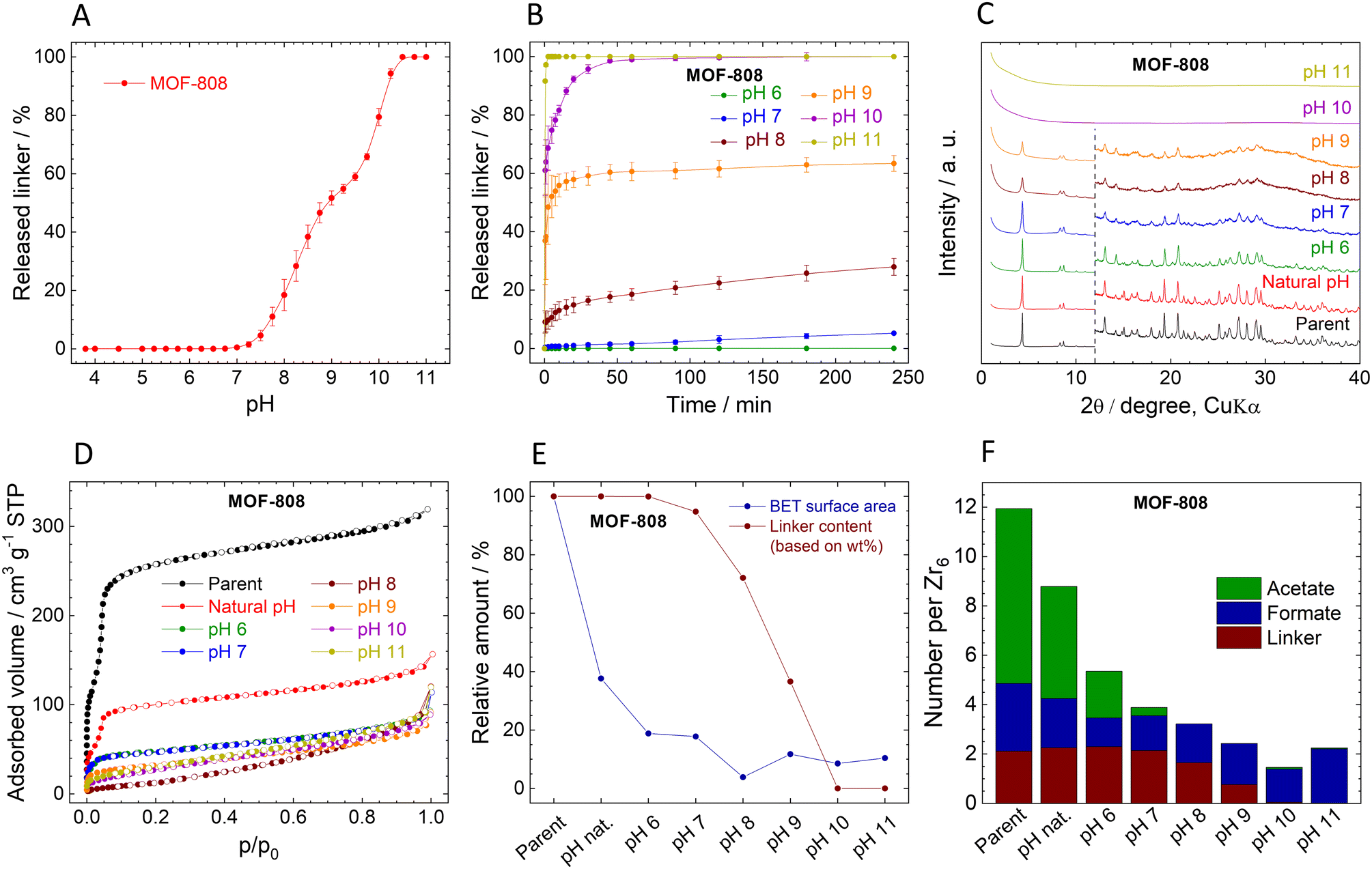 | ||
| Fig. 3 Stability of MOF-808. (A). Immediate release of BTC3− from MOF-808. (B). Time-dependent BTC3− release from MOF-808 at different pH values. (C). Comparison of XRPD patterns of the parent and post-exposure MOF-808; the diffractograms are normalized and shifted vertically to avoid overlaps. The dashed line at 12° 2θ separates the zoomed area to visualize the diffractions at higher angles. (D). Adsorption (filled dots) and desorption (empty dots) isotherms of N2 measured at 77 K. (E). Decrease of SBET, and linker content (all data are normalized, taking the parent MOF-808 as a default state with 100% values). (F). Molar ratios of the ligands in the parent and post-exposure MOF-808 to the Zr6 metal clusters. | ||
MIP-200 was prepared by a slight modification of the published procedure.59 When following the original synthetic protocol,59 we obtained a mixture of two crystalline phases. Several additional diffraction lines (for example 7.15, 8.73, and 12.14° 2θ) that appeared in the XRPD pattern correspond to MIP-201,78 another MOF with 6-connected Zr6 clusters (Fig. S5, ESI†). To obtain pure MIP-200 phase, the amounts of reactants and solvents were reduced to half with respect to the original procedure while the ZrCl4 to H4MDIP ratio was kept at 3:1. The crystallization was performed in DURAN glass bottles instead of a Teflon-lined autoclave. Possibly the surface of the Teflon insert promoted preferential crystallization of the MIP-201 phase. The SBET area of 733 m2 g−1 calculated from N2 adsorption isotherm (Fig. 4D) is below the reported value of ∼1000 m2 g−1,59 which is probably caused by the modified washing procedure. In the original report, MIP-200 was treated with boiling water before the adsorption measurements. We decided to forego this treatment to avoid dissolution of the linker and/or monocarboxylic acids. This hypothesis was confirmed by treating MIP-200 in neat water which increased SBET to 938 m2 g−1 (see stability study below). The thermogravimetric analysis (Fig. S6, ESI†) displayed a total weight loss of 51% at 570 °C. The initial 12% being released between 50 and 210 °C can be attributed to the residual solvents, mainly acetone and moisture. The further 39 wt% released above 210 °C can be attributed to the linker combustion, which is in line with 42.1 wt% of the MDIP4− determined by HPLC after the solid dissolution. For the total mass content of all components, see Tables S7 and S8 in ESI.†
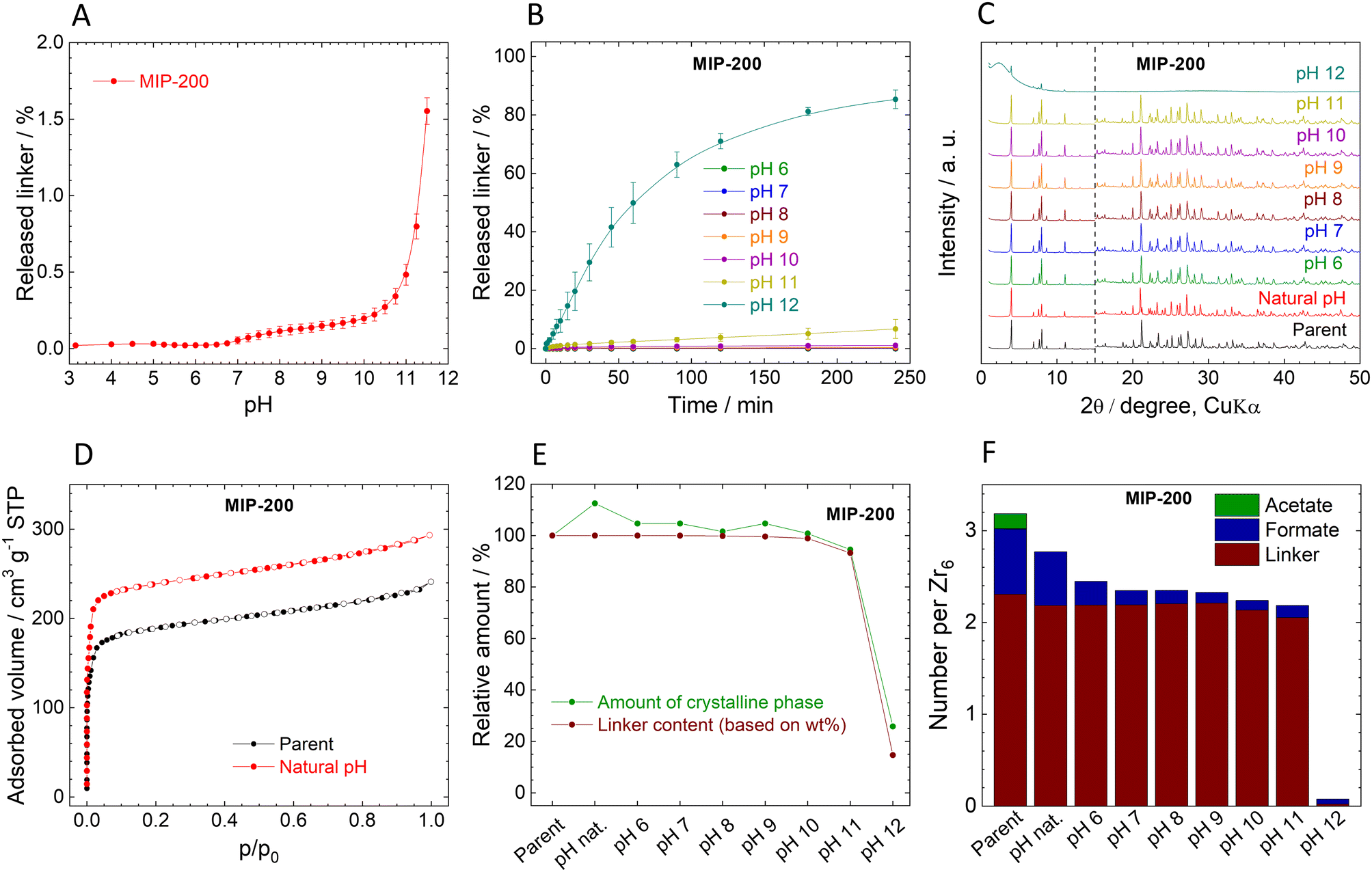 | ||
| Fig. 4 Stability of MIP-200. (A). Immediate release of MDIP4− from MIP-200. (B). Time-dependent MDIP4− release from MIP-200 at different pH values. (C). Comparison of XRPD pattern of the parent and post-exposure MIP-200, the diffractograms are normalized and shifted vertically to avoid overlaps. The dashed line at 15° 2θ separates the zoomed area to visualize the diffractions at higher angles. (D). Adsorption (filled dots) and desorption (empty dots) isotherms of N2 measured at 77 K. (E). Changes of the crystalline phase percentage and linker content (all data are normalized, taking the parent MIP-200 as a default state with 100% values). (F). Molar ratios of the ligands in the parent and post-exposure MIP-200 to the Zr6 metal clusters. | ||
PCN-222 was synthesized by a modified and scaled-up procedure60,73 from zirconyl chloride octahydrate and H4TCPP in a molar ratio of 3:1 under modulated solvothermal conditions. The XRPD pattern (Fig. S7, ESI†) of the parent PCN-222 confirms its phase purity. The adsorption isotherm of N2 showed a typical shape for PCN-222, which contains two types of pores in the structure with a calculated SBET of 1869 m2 g−1 (Fig. 6D). Thermogravimetric analysis is rather inconclusive (Fig. S8, ESI†); there is not a sharp drop attributable to the combustion of the linker; moreover, mass loss is not finished at 800 °C, in which is consistent with earlier reports.79,80 The content of TCPP4− determined by HPLC after the solid dissolution was 57.2 wt%. For total mass content of all components, see Tables S9 and S10 in ESI.†
Chemical stability of the MOFs in water
The chemical stability of UiO-66, MOF-808, MIP-200, and PCN-222 predetermines their applicability in many catalytic reactions, drug delivery systems, adsorption and water treatments, i.e., in all applications that require an aquatic environment. The stability, evaluated by the release of the linkers from the MOF structure, was studied in pHs ranging from the natural value (pH of unadjusted water–MOF suspensions) to 11.0. In our stability studies, we used two approaches:(i) Immediate release of the linkers – the titration of the MOF suspensions from the natural pH by the consecutive addition of 0.1 M NaOH solution. After reaching a selected pH, the immediate concentration of the released linker in the water phase was determined by HPLC. Clearly, the obtained titration curves describe the immediate linker release and bear no information about the long-term behaviour, which is relevant to reaction conditions in many applications.
(ii) Kinetic stability assessed by the time-dependent release of the linkers under constant pH. The target pH was adjusted by the addition of 0.1 M NaOH solution and then the pH value was kept constant during the experiment by an automatic titrator. The samples were taken out at predefined times and filtered, and concentration of the released linker was determined using HPLC. After 4 h, the remaining solids were collected and analysed and catalytic tests were done using these samples. The pH values for the kinetic experiments were selected based on the results from the titration curves, i.e., at the MOFs’ natural pH, at pH 6.0, which appears to be the pH at which some of the MOFs start to decompose; and at pH values from 7.0 to 11.0.
Stability of UiO-66
The observed immediate linker release from UiO-66 is in good agreement with our previous work.54 The titration curve can be divided into four characteristic regions (Fig. 2A): (i) at pH 3.8 (natural pH) to 5.0, the amount of the released BDC2− linker is below the detection limit (<0.01 mg L−1), indicating the stability of UiO-66 in this pH region. (ii) At pH 5.25–8.75, moderate linker release rising from 0.05% to 18.4% of the total amount indicates relatively low pH sensitivity of UiO-66 in this region. (iii) pH 9.00–9.75 is characterized by high sensitivity towards pH with a sharp increase of the released linker amount – up to 97.3%; (iv) above pH 9.75, the linker release is complete. In general, UiO-66 can be considered stable only at pHs below 5, since higher pH values lead to gradual linker release and finally collapse of the structure at 10.0. Having the complete data on the Zr-based UiO-66 (hereafter labelled as UiO-66), we performed the same experiments with the Hf- and Ce-analogues of UiO-66, hereafter labelled as UiO-66(Hf) and UiO-66(Ce) (syntheses in ESI,† XRPD patterns in Fig. S1, ESI†). The curves of immediate linker release (Fig. 2A) from UiO-66(Hf) and UiO-66(Ce) have a similar shape to that of UiO-66; however, they indicate that the linker release is more pronounced. In the case of UiO-66(Ce), a small amount of BDC2− linker (0.4%) was already released at the natural pH of 3.7. The lower pH stability of UiO-66(Hf) in comparison with UiO-66 is surprising because it is contrary to the generally accepted predictions based on the higher dissociation enthalpy of the Hf–O bond, according to which carboxylate-based Hf-MOFs should be more stable than their Zr analogues.81,82 It should be noted that the crystallite sizes, determined by the integral breadth of the first two diffraction lines, follow the trend UiO-66 ∼ UiO-66(Hf) > UiO-66(Ce), see Fig. S1, ESI† and detailed discussion therein. Therefore, the smaller crystallite size of UiO-66(Ce) when compared to UiO-66 and UiO-66(Hf) can be partly responsible for the lower chemical stability. Further studies were performed only with Zr-based UiO-66.The measured kinetics of the linker release from UiO-66 suggest that it is stable at its natural pH (3.8) for 4 h, as the H2BDC concentration was below the detection limit during the entire experiment. Therefore, the corresponding curve is omitted in Fig. 2B. It is noteworthy, that at pH 6.0, the BDC2− release is still below 0.5%. However, increasing pH led to much higher amounts of BDC2− released. The kinetic curves of BDC2− release are very fast, with BDC2− release reaching a plateau within 30 min. The only exception is pH 9.0, where the BDC2− is released throughout the 4 h of the experiment, probably due to the high sensitivity of UiO-66 to pH change in this region (see above).
The UiO-66 samples collected after the 4 h treatment at different pHs display XRPD patterns with the preserved original structure up to pH 8.0, where 34% of the BDC2− linker was already released (Fig. 2C). This observation is in good agreement with our previous works.54,55 The diffraction line at 12° 2θ, which is related to the degree of hydroxylation of the Zr6 clusters83 increases with the increasing pH. We highlight that the diffraction pattern of the sample treated at pH 9.0 still corresponds to the structure of UiO-66 even though there is 70% linker release and a significant reduction of the sample crystallinity (see below). When UiO-66 was treated at pH 10.0 and 11.0, the diffraction lines disappeared completely accompanied by the detected 100% linker release, indicating total destruction of the UiO-66 framework. Since no crystalline phase was detected in these samples, the products of the hydrolysis are probably amorphous zirconium oxide-hydroxide species with partially coordinated formate anions (Fig. 2F and Table S3, ESI†).
The content of the amorphous phase estimated using XRPD (parameters and results in Table S11, ESI†) follows the same trend as the linker release (Fig. 2E; the decrease in the crystalline phase is presented relative to the parent UiO-66-100%). Surprisingly, the formation of an amorphous phase occurred also in UiO-66 treated only with the natural pH, where no linker release was detected. Because this change cannot be caused by the BDC2− release, we attribute the process to the removal of the acetate ligands, which are commonly bound to the defects on the zirconium coordination sites. As can be seen from HPLC analyses, acetates were immediately and completely washed out already at the natural pH, whereas the formate ligands are strongly bound even above pH 10.0 (Fig. 2F, Tables S3 and S4, ESI†). The content of formate ligands is even increased at pH 10.0 and 11.0, probably due to the alkaline hydrolysis of residual DMF in UiO-66.84 The increase of the amorphous phase content at higher pH correlates well with the extent of the linker release (Fig. 2E). We assume that linker and ligands are partially removed from the framework and the crystal structure is still preserved the vacated coordination sites are then occupied by water and OH− ligands, to satisfy the charge and coordination number of the Zr6 SBU as it is generally suggested for all Zr-MOFs with lower cluster connectivity than 12.85 However, as soon as the structure starts to collapse this assumption is not valid.
The adsorption isotherms of N2 are shown in Fig. 2D. Calculated SBET after pH treatment is in good agreement with the structural changes described above (Fig. 2D and E, Table 1). At pH 6.0 and 7.0, SBET slightly increased in comparison with the natural pH sample, which can be attributed to the increasing number of defects caused by the slight BDC2− release.58 At higher pH, SBET gradually decreased and finally at pH 10.0 and 11.0 displayed no measurable porosity. After the normalization, the decrease in the SBET corresponds to the decrease in the crystalline phase content, as shown in Fig. 2E. Also, FTIR spectra of samples treated at pH 10 and 11 (Fig. S9, ESI†) show only broad peaks in the region of 1400–1600 cm−1, corresponding to adsorbed water molecules and coordinated OH− ligands, and weak unresolved peaks in the region below 1000 cm−1 attributed to the Zr–O stretching vibrations. The complete absence of vibrations of the linker molecules confirms the observed 100% BDC2− release (Tables S3 and S4, ESI†).
Stability of MOF-808
The immediate linker release from MOF-808 is similar to that of UiO-66 (Fig. 3A). The titration curve can be divided into three regions: (i) at pH 3.8 (natural pH) to pH 6.5, the MOF is stable, and the concentration of the released linker is below the detection limit (i.e., 0.05 mg L−1). (ii) At pH 6.5–10.25, there is a steep increase in released BTC3−, from approximately 0.03% for pH 6.5 to 94% at pH 10.25. The curve has stair-like characteristics in this range, with a region of lower sensitivity from pH 8.75 to 9.5. (iii) Above pH 10.25, the structure collapses, as documented by the complete release of the linker.The kinetic curves of BTC3− release (Fig. 3B) confirm the stability of MOF-808 at natural pH 3.8, i.e., no released linker was detected, and therefore, the curve is not included in Fig. 3B. Only traces of BTC3− were detected at pH 6.0 after 4 h (0.03%, close to the LOQ), indicating that MOF-808 is more stable in acidic environment than UiO-66. At pH 7.0 and 8.0, 5% and 33% of BTC3− were released, respectively, which is comparable to UiO-66. At pH 10.0, MOF-808 was totally destroyed as evidenced by 100% BTC3 release. Importantly, in contrast to UiO-66, the kinetics of the release are much slower. The fast release at the beginning is followed by a slow continuous release over the following 240 min period.
The MOF-808 samples collected after the 4 h treatment at different pHs display XRPD patterns with the preserved original structure up to pH 9.0 even though the material already lost 63% of BTC3− (Fig. 3C). However, the XRPD patterns of the samples treated at pH 7.0 and higher indicate a decrease in crystallinity. Probably due to the lower Zr6 cluster connectivity of MOF-808 even small releases of BTC3− have detrimental effects on the XRPD patterns. At pH 10.0 and 11.0, when all the BTC3− was released, the material became amorphous. The FTIR spectra also confirm the loss of organic matter and formation of zirconium oxide-hydroxide (Fig. S10, ESI†). Unfortunately, MOF-808 cannot withstand homogenization with internal standard (ZnO), see Table S12, ESI† for additional data, and therefore, the determination of the crystalline phase content was omitted in Fig. 3E.
The composition of MOF-808 changes after the treatment at different pHs (Fig. 3F, Tables S5 and S6, ESI†). Parent MOF-808 contains approximately 7 molecules of acetates and 2.7 of formates per Zr6 oxometallic cluster, which is more than the 6-connected Zr6 cluster can theoretically accommodate together with BTC3−. Thus, we assume that the excess of monocarboxylic acids is adsorbed in the pores even though the MOF was thoroughly washed with acetone. Interestingly, simple washing with water removed 33% of acetates and 29% of formates; the rest of the acetates were gradually washed at higher pHs together with a part of formates that are bound strongly even at highly basic pHs. The presence of adsorbed acetates and formates was confirmed by TGA/DTA with MS detection of evolved gases. At temperatures over 120 °C evolution of acetate was observed, (see Fig. S4, ESI†).
The adsorption isotherms of N2 are shown in Fig. 3D. The adsorbed amount of N2 and the calculated SBET significantly decreased even after treatment at natural pH, with further decreases at increasing pH leading to complete loss of porosity at pH 8.0. The decrease in SBET is in stark contrast with the loss of BTC3− (see Fig. 3E and Table 2). However, it is in line with the evident decrease of crystallinity seen by XRPD. Moreover, the low amount of released BTC3− might be due to re-adsorption to the remaining MOF-808 where it is blocking pores. In other words, based on linker release, the stability of MOF-808 seems similar to that of UiO-66; however, the ongoing structural changes showed that it is much lower.
| Sample | Linker removal after 4 h/% | S BET/m2 g−1 | Conversion DMNP at 30 min/% | Conversion DMNP at 120 min/% |
|---|---|---|---|---|
| a Natural pH is 3.8. | ||||
| MOF-808 parent | 0.0 | 962 | 89 | 98 |
| MOF-808 nat. pHa | <LOD | 362 | 74 | 97 |
| MOF-808 pH 6 | 0.04 | 181 | 32 | 63 |
| MOF-808 pH 7 | 5.2 | 171 | 14 | 29 |
| MOF-808 pH 8 | 27.9 | 37 | 14 | 27 |
| MOF-808 pH 9 | 63.4 | 113 | 15 | 29 |
| MOF-808 pH 10 | 100 | 82 | 6 | 11 |
| MOF-808 pH 11 | 100 | 100 | 6 | 11 |
Stability of MIP-200
In contrast to UiO-66 and MOF-808, MIP-200 based on 8-connected Zr6 clusters was significantly more stable during the titration experiment. Up to pH 11.0, the amount of the released MDIP4− was close to the detection limit, and at pH 11.5 only 1.6% of the linker was released into the solution (Fig. 4A).The exceptional hydrolytic stability of MIP-200 is further confirmed by the kinetic curves (Fig. 4B). At pH 6.0, MDIP4− release was 0.013%, which was only slightly above the limit of detection (0.05 mg L−1). A more evident MDIP4− release occurred at pH 9.0 and 11.0; however, it did not exceed 0.5% and 7%, respectively. Since MIP-200 can withstand harsh conditions up to pH 11.0, we extended the testing pH range up to pH 12.0 where the amount of released MDIP4− rapidly increased to 85%. In contrast to UiO-66 and MOF-808, the kinetic curves for MIP-200 showed continuous progress. For this reason, in the case of pH 11.0 and 12.0, we prolonged the time range to 72 h and 120 h, respectively. Interestingly, at pH 11.0, MDIP4− was released during the whole timeframe of the experiment, while at pH 12.0 the release was completed within 8 h (Fig. 5A). The XRPD patterns of the resulting solids confirmed that the structure of MIP-200 is partially preserved with a high degree of amorphization at pH 11.0, while at pH 12.0, the structure is completely destroyed (Fig. 5B).
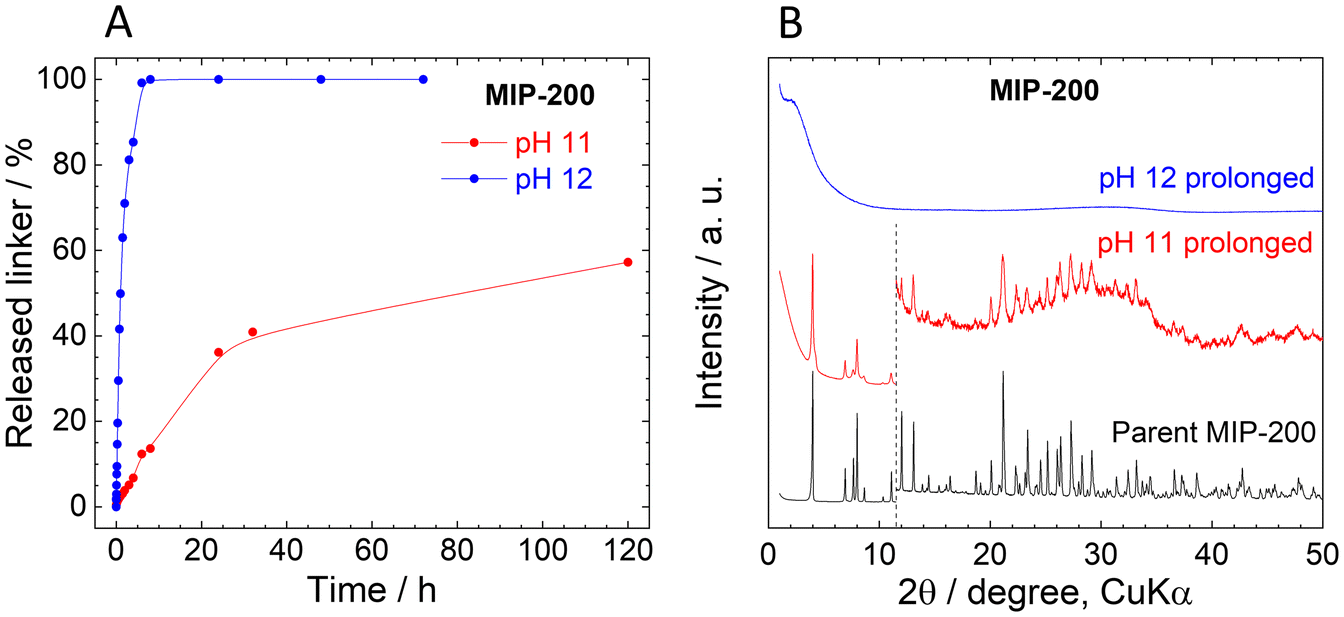 | ||
| Fig. 5 (A). Prolonged time-dependent MDIP4− release from MIP-200 at pH 11.0 and 12.0. (B). XRPD patterns of the parent and post-exposure MIP-200 measured after kinetic experiments presented in panel A; the diffractograms are normalized and shifted vertically to avoid overlaps. The dashed line at 11.5° 2θ separates the zoomed area to visualize the diffractions at higher angles. | ||
The XRPD patterns of the parent and post-exposure MIP-200 (Fig. 4C) indicate good preservation of the crystallinity up to pH 11.0. However, when samples treated at pH 6.0 and higher were reactivated at 100 °C overnight in a vacuum, the XRPD patterns revealed rather broad peaks of a new crystalline phase (compare Fig. 4C with Fig. S12A†). Meanwhile, the sample treated in neat water (natural pH) preserved the original structure even after the activation. As can be seen from Fig. 4F (see also Tables S7 and S8, ESI†), treatment with pH 6.0 and higher is related to the release of monocarboxylate ligands. To confirm our hypothesis that the loss of monocarboxylates causes thermal instability, we have taken MIP-200 treated at pH 7.0 for 4 h and treated it overnight in 1 M acetic acid, 1 M formic acid or the same medium that is used for MIP-200 synthesis (mixture of acetic anhydride with formic acid). In all three cases, when the sample was then activated at 100 °C, the XRPD pattern did not change from the parent MIP-200 (see Fig. S12C†). Moreover, we found that the temperature is the responsible parameter for these changes; a vacuum alone did not induce a phase change (see Fig. S12B†). This was also confirmed by temperature-resolved XRPD (Fig. S13, ESI†). For this reason, the reactivation of MIP-200 was done only in a vacuum at laboratory temperature, and we omitted the measurement of adsorption isotherms.
The behaviour of MIP-200, which can be rearranged by simple heating when monocarboxylic acids are removed, while their reintroduction leads to restoring temperature stability, is quite unusual. A similar phenomenon was recently observed by Zhou et al.86 where the rearrangement to a new phase was observed upon treatment with strongly coordinating solvents followed by activation under mild conditions. The process led to higher surface area, and moreover, in the published study the process was reversible.
The changes in the amorphous phase content are in line with the release of MDIP4− (see Fig. 4E). It is evident that the amorphous content did not increase too much when MIP-200 was treated with pH from natural to 11.0. Treatment with pH 12.0 is connected with high linker release, which led to partial destruction of the structure, as confirmed by an increase in the amorphous phase content. Adsorption isotherms of N2 were measured only for the parent MIP-200 and the sample treated at natural pH (Fig. 4D). As discussed above, the increase in the adsorption amount is probably related to washing off the monocarboxylic acids. The FTIR spectra of MIP-200 (Fig. S11, ESI†) showed no significant changes even when the new crystalline phase was formed, indicating that the formation is not connected with visible changes in the building block constitution. After treating MIP-200 at pH 12.0, the FTIR spectrum showed a partial disappearance of vibrations belonging to organic matter which is in good agreement with the intensive loss of linker.
Stability of PCN-222
The immediate release of TCPP4− from PCN-222 shows a similar trend to MIP-200 (Fig. 6A). High linker release was observed only at high pHs when the amount of released TCPP4− reached 1%, 4%, and 43% at pH 10.5, 11.25, and 11.5, respectively.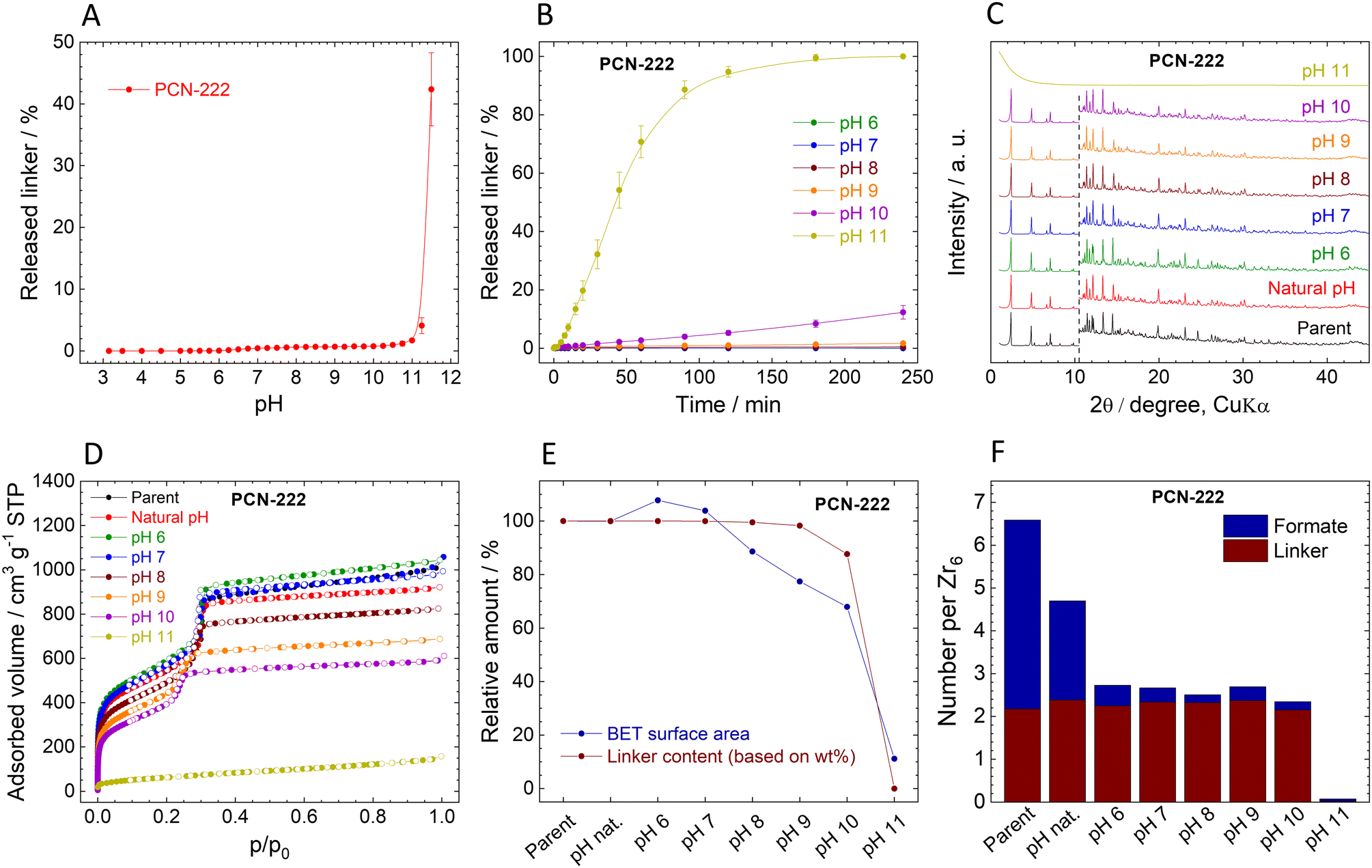 | ||
| Fig. 6 Stability of PCN-222. (A). Immediate release of TCPP4− from PCN-222. (B). Time-dependent TCPP4− release from PCN-222 at different pH values. (C). Comparison of XRPD pattern of the parent and post-exposure PCN-222, the diffractograms are normalized and shifted vertically to avoid overlaps. The dashed line at 10.5° 2θ separates the zoomed area to visualize the diffractions at higher angles. (D). Adsorption (filled dots) and desorption (empty dots) isotherms of N2 measured at 77 K. (E). Decrease in SBET and linker content (all data are normalized, taking the parent PCN-222 as a default state with 100% values). (F). Molar ratios of the ligands in the parent and post-exposure PCN-222 to the Zr6 metal clusters. | ||
The kinetic curves of TCPP4− release confirm the low amounts of released linker (Fig. 6B). At the natural pH of 3.2, the release was below the detection limit of 0.005 mg L−1, while at pH 6.0 and 7.0, the initially released TCPP4− was later reincorporated back into the MOF structure. More significant linker release was observed at pH 8.0 and 9.0, but still, it was only 0.5% and 1.7% of the total amount, respectively. PCN-222 released 12.3% of the linker at pH 10.0 while partially keeping the crystal structure. The complete destruction of the network at pH 11.0 was accompanied by the 100% release of the linker. Similarly to MIP-200, the kinetic curves of PCN-222 showed continuous progress.
The comparison of post-exposure XRPD patterns with that of parent PCN-222 showed that the PCN-222 structure was preserved (although slightly modified) even at pH 10.0, when 12.3% of TCPP4− was released (Fig. 6C). pH 11.0 led to amorphization, which is in line with complete TCPP4− release. Unfortunately, because of low mechanical stability of PCN-222, the amorphous content was not determined; for details, see Table S12, ESI.†
The composition of PCN-222 changed after the treatment at different pH (Fig. 6F, Tables S9 and S10, ESI†). Similarly to MOF-808, the number of formate ligands exceeds the number of coordination sites of the Zr6 clusters, indicating that formates are partially adsorbed in the pores. This is supported by the fact that approximately 50% of formates were quickly removed at natural pH. In this case, no acetate ligands were present, since the synthesis was performed without the addition of acetic acid.
N2 adsorption isotherms confirm the stability of PCN-222 (Fig. 6D, Table 4). Very small changes up to pH 7.0 can be associated with the release of formates. Up to pH 10.0, the characteristic shape of the isotherms is preserved, indicating that two types of pores with different sizes are still present in the structure; however, the adsorbed gas volume drops significantly. The collapse of the structure at pH 11.0 is associated with loss of porosity. The correlation of the linker contents and SBET show that both parameters are applicable for analysing the stability of PCN-222 and pH-induced changes to its structure (Fig. 6E). However, the N2 adsorption seems to be more sensitive because it recorded decrease in SBET before the significant amount of linker was released (Fig. 6E). The FTIR spectra also confirm the loss of organic matter and formation of zirconium oxide–hydroxide at pH 11.0 (Fig. S14, ESI†).
Summary of the MOFs stability
To summarize the above results, it is clear that the linker topicity and cluster connectivity have significant effects on the hydrolytic stability of Zr-MOFs. All MOFs are stable in neat water; however, around neutral pH, UiO-66 and MOF-808 (based on ditopic and tritopic linkers, respectively) start to degrade (compare Fig. S15 and S16, ESI†). Meanwhile, the MOFs based on tetratopic linkers, MIP-200 and PCN-222, survived up to pH 10.0 and 9.0, respectively. At the same time, it should be noted that the kinetics of the linker release get slower with increasing topicity. In the presented study, we investigated stability only in the time range of up to 4 h, which might be too short for a true assessment of MOFs based on tetratopic linkers.Based on the literature,36,37,45,46 it is expected that, in general, higher connectivity leads to more stable MOFs. Here, we report that the relationship between stability and Zr6 cluster connectivity is not as straightforward. The structure of UiO-66, due to the 12-connected clusters, can survive a higher degree of linker removal accompanied by the formation of structural defects, whereas in the case of MOF-808 with 6-connected clusters, even trace amounts of the released linker lead to the collapse of the structure instead of the formation of defects. PCN-222 and MIP-200 with 8-connected clusters can also resist partial removal of linkers. It seems that a cluster connectivity of 6 is the critical point.
To prove that the linker release from MOFs is not governed by the limited solubility of the linkers, we determined the solubility of all four objective linkers as a function of pH (Fig. S17, ESI†). In all cases, the solubility was much higher than the concentrations detected in the tested solutions.
Finally, it is evident that a complex analysis based on linker release together with detailed post-exposure analysis is necessary to accurately describe the stability of MOFs and an approach limited to only some of those aspects may not be sufficient.
Catalysis using MOFs pre-treated at different pH values
For the catalytic tests, the parent and post-exposure MOFs with a different number of removed linkers and/or monocarboxylate ligands were used. Wide range of Zr-MOFs including UiO-66,68,71 MOF-808,70,87 and PCN-22268,86 has been tested for catalytic degradation of organophosphates so far. MIP-200 was tested as a catalyst for other reactions,88 although it has not been tested for organophosphate degradation to date. In our previous work,54 we described that the removal of BDC2− from UiO-66 increased the activity because the creation of missing linker defects is directly related to new open-metal sites acting as catalytic centres.69 Here, we took the recovered MOFs treated for 4 h at a given pH to test if the formed defects are connected with the formation of catalytically active centres or rather a structural collapse. It should be noted that we assume that the formed missing linker defects are compensated by coordinated water and OH− ligands.
As a substrate for the model degradation reaction, we chose methyl paraoxon (DMNP), an organophosphate used as a simulant of pesticides or nerve warfare agents. DMNP is a suitable model compound because of its good solubility in water and ease of differentiating between substrate adsorption and catalytic degradation leading to 4-nitrophenol (4-NP) as the main product (see Fig. 7). Commonly, the catalytic degradation of DMNP is done in a buffer environment. However, buffers very often induce new defects or even MOF collapse.54 For this reason, we tested all MOFs in neat water without pH adjustment (at natural pH values ranging from 3.9 to 4.6). After all catalytic experiments, we analysed the linker content in the solution. In all cases, the concentration of the released linker during the catalytic experiments was below the detection limit. Fig. 8 depicts the conversion of DMNP to 4-NP using different parent and post-exposure MOFs. The curves represent the formation of 4-NP. For the comparison of DMNP decrease with 4-NP formation, see Fig. S18, ESI.†
 | ||
| Fig. 7 Hydrolysis of DMNP to 4-NP and dimethylphosphate (DMP) catalysed by a Zr-MOF. | ||
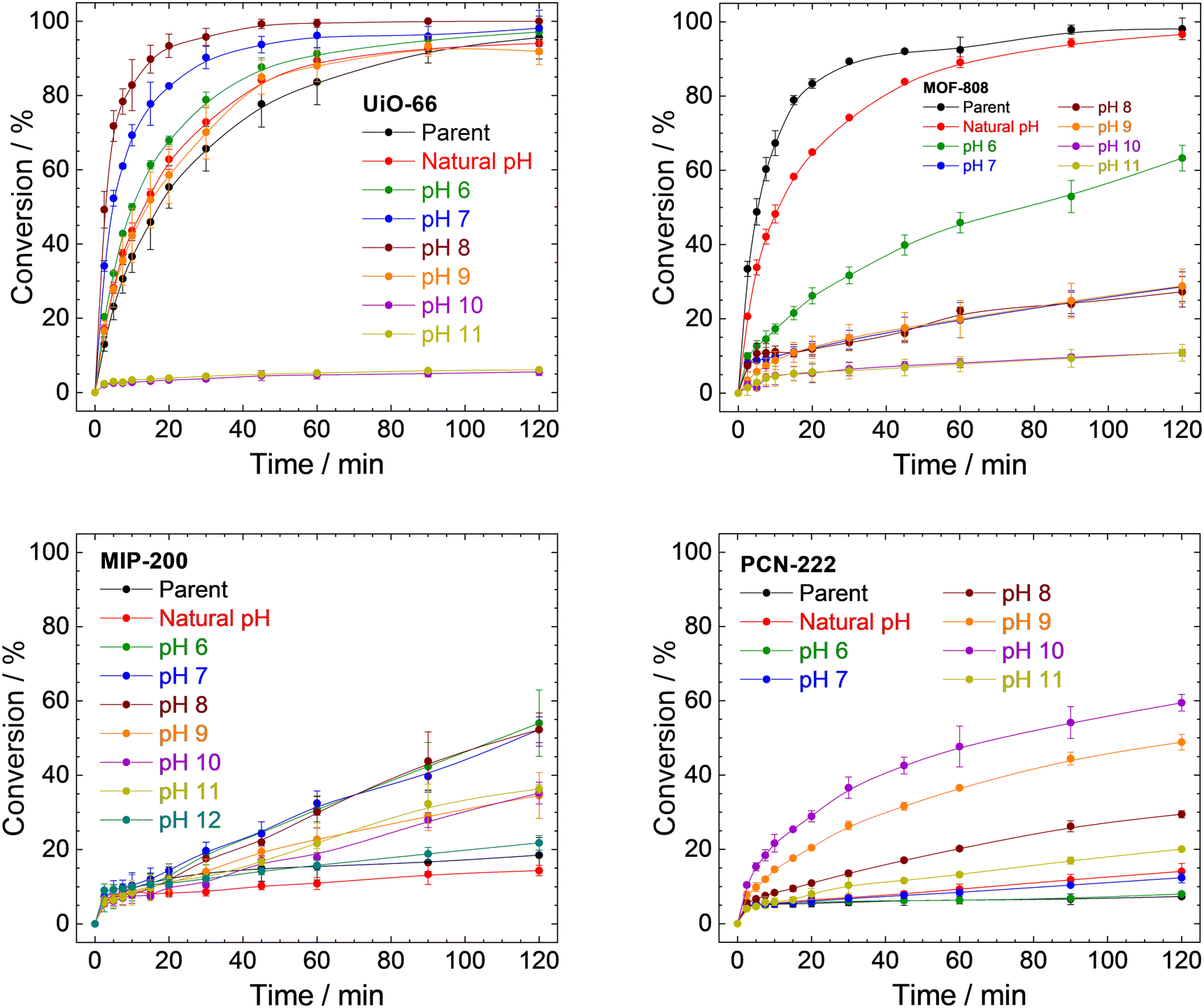 | ||
| Fig. 8 Catalytic conversions of DMNP to 4-NP using parent UiO-66, MOF-808, MIP-200, PCN-222, and samples prepared by treating MOFs with a different pH for 4 h. The conversions are represented as an increase of 4-NP concentration related to the total amount of DMNP. The corresponding decrease in DMNP concentrations is presented in Fig. S18 in ESI.† | ||
Conversion of DMNP to 4-NP using the UiO-66 catalyst followed the trends described earlier (Fig. 8 top left and Table 1). The parent UiO-66 is active; however, upon treatment with neat water (natural pH) or at pH 6.0, the reaction rate accelerated slightly. At this pH, the changes in the structure involve mainly the removal of acetate ligands from metal sites (see Table S3, ESI†). Treatment at pH 7.0 and 8.0 led to a significant increase in the catalytic activity, which is in good agreement with the higher amount of released BDC2−, while the MOF structure was still preserved. Obviously, the removal of the linker leads to the formation of catalytically active centres. However, at pH 9.0, the high linker release (70%) is already connected with a considerable degradation of the structure which results in a slower rate of reaction. Complete linker release at pH 10.0 and 11.0 is associated with a significant loss of catalytic activity. The remaining catalytic activity can be attributed to the resulting zirconium oxide-hydroxides.89–91
MOF-808 does not follow the same trend as UiO-66 (Fig. 8 top right). The highest rate of degradation was recorded for the parent MOF-808, and any treatment led to a decrease in activity. MOF-808 treated with neat water at natural pH is still comparable to the parent MOF-808; however, already treatment with pH 6.0, where only 0.04% of BTC3− was released along with acetate and formate anions, displays a significant decrease in activity. A plausible explanation is that it is related to the damage to the structure (as evidenced by adsorption isotherms of N2 and XRPD patterns; see Table 2 and Fig. 3C) instead of the formation of catalytically active defects even when a small portion of linker is removed.
MIP-200 does catalyse DMNP degradation; however, the decrease in DMNP concentration is much faster than the creation of 4-NP (compare Fig. S18, ESI†) in this case. This is probably due to the fast adsorption of DMNP coupled with a very slow degradation. As shown in Fig. 8 (bottom left) and Table 3, the parent MIP-200 and MIP-200 treated at natural pH provided only negligible activity. Acceleration of DMNP conversion was observed in the case of MIP-200 treated at pH between 6.0 and 11.0. In these cases, only a negligible amount of MDIP4− was released; therefore, the removal of acetate and formate anions is mainly responsible for the activity enhancement. The catalytic activity of MIP-200 treated at pH 12.0 decreased again to levels of the parent MIP-200, probably due to the large number of removed MDIP4− accompanied by a loss of crystallinity.
| Sample | Crystalline phase/% | Linker removal after 4 h/% | S BET/m2 g−1 | Conversion DMNP at 30 min/% | Conversion DMNP at 120 min/% |
|---|---|---|---|---|---|
| a Natural pH is 3.2. | |||||
| MIP-200 parent | 100 | 0 | 733 | 14 | 19 |
| MIP-200 nat. pHa | 112 | <LOD | 938 | 9 | 14 |
| MIP-200 pH 6 | 104 | 0.01 | n.a. | 19 | 54 |
| MIP-200 pH 7 | 104 | 0.04 | n.a. | 20 | 52 |
| MIP-200 pH 8 | 102 | 0.17 | n.a. | 18 | 52 |
| MIP-200 pH 9 | 104 | 0.40 | n.a. | 14 | 35 |
| MIP-200 pH 10 | 101 | 1.11 | n.a. | 11 | 35 |
| MIP-200 pH11 | 95 | 6.77 | n.a. | 12 | 36 |
| MIP-200 pH12 | 26 | 85.34 | n.a. | 12 | 22 |
Similarly to MIP-200, simultaneous adsorption and catalytic degradation were observed for PCN-222 (see Fig. 8 and S18, ESI†). The parent PCN-222 and PCN-222 treated at natural pH and pH 6.0 and 7.0 are only slightly active due to the negligible removal of the linker. The removal of formate ligands did not play any significant role in this case. The increase in the reaction rate was observed mainly when PCN-222 treated at pH 8.0, 9.0, or 10.0 was used. This matches well with the higher linker removal (compare conversion with linker removal in Table 4) accompanied by the formation of new catalytically active defects, as was observed for UiO-66. When PCN-222 was treated at pH 11.0, we observed total release of the linker and structural collapse, as well as a steep decline in the catalytic activity.
| Sample | Linker removal after 4 h/% | S BET/m2 g−1 | Conversion DMNP at 30 min/% | Conversion DMNP at 120 min/% |
|---|---|---|---|---|
| a Natural pH is 3.2. | ||||
| PCN-222 parent | 0 | 1870 | 6 | 7 |
| PCN-222 nat. pHa | <LOD | 1869 | 7 | 14 |
| PCN-222 pH 6 | 0.008 | 2015 | 6 | 8 |
| PCN-222 pH 7 | 0.04 | 1942 | 7 | 12 |
| PCN-222 pH 8 | 0.48 | 1657 | 14 | 29 |
| PCN-222 pH 9 | 1.73 | 1448 | 26 | 49 |
| PCN-222 pH 10 | 12.34 | 1270 | 37 | 59 |
| PCN-222 pH 11 | 100 | 209 | 10 | 20 |
In general, the removal of linker molecules leads to an increase in catalytic activity, with the exception of MOF-808. This can be explained by the lower Zr6 cluster connectivity in MOF-808 when compared to the other tested MOFs. While the structure of UiO-66 with 12-connected clusters can be preserved even when a relatively high percentage of BDC2− is removed and open metal sites are formed, the structure of MOF-808 collapses, and no catalytic centres are formed. For this reason, we assume that defect engineering based on the removal of linkers is not suitable for MOF-808. MOFs based on tetratopic linkers have in their parent form very low activity, which can be somewhat enhanced, however, even fine tuning of defects in the structure does not lead to catalytic activity comparable to that of UiO-66 or parent MOF-808. To summarize the catalytic part, defect engineering through linker removal can be used as a tool to increase the catalytic activity only for MOFs with higher connectivity.
Conclusions
This work examined the stability of UiO-66, MOF-808, MIP-200, and PCN-222 in pHs between 3.0 and 11.0 to delineate the relationship between linker topicity, Zr6 cluster connectivity, and stability in aqueous media of various pHs. It should be noted that each MOF has its specifics and therefore we were changing more parameters at once.We found that MOFs constructed from tetratopic linkers have significantly enhanced stability. Even though PCN-222 is a mesoporous MOF with SBET close to 2000 m2 g−1, it could tolerate a neutral or slightly basic environment much better than UiO-66 formed from BDC2−. At the same time, tetratopic linkers have much slower kinetics of release and we cannot rule out damage to their frameworks if MIP-200 or PCN-222 would be treated at neutral pH for days or weeks. Nevertheless, MIP-200 was the most stable MOF in aqueous media from the tested series.
The relationship between Zr6 connectivity and stability is much more complicated. 12-connected UiO-66 can tolerate the loss of more than 30% of linker without detectable collapse of the structure. At the same time, when MOF-808 with 6-connected clusters loses even a small amount of the linker or monocarboxylic acid the structure starts to collapse. 8-connected MIP-200 and PCN-222 can tolerate a small linker release without structural damage. Studies with more MOFs would be needed to have more definite conclusion; however, from these results, it seems that as long as the actual connectivity in the Zr6 cluster is higher than 6, defects can be created in the structure of a MOF without a structural collapse. It is, however, necessary to evaluate the effect of monocarboxylic acids as demonstrated on MIP-200 where their absence resulted in loss of thermal stability.
Additionally, we demonstrated that the determination of linker release is a sensitive tool for stability assessment only in the case of UiO-66. This is probably because, in all other studied cases, the MOFs were able to adsorb the released linker. In those cases, measurement of the adsorption isotherm or determination of the catalytic activity in organophosphate degradation gave a better picture of the structure. Analysis of the amorphous content is a powerful tool; however, it cannot be widely applied because of low mechanical stability of many MOFs.
Finally, the obtained results show that MOFs based on tetratopic linkers are not very active in organophosphate degradation. This might be related to the slow linker release, which makes the Zr6 cluster less accessible to the organophosphate substrate. As there is a strong relationship between the preservation of the structure and catalytic properties, testing catalytic activity can also disclose whether defect engineering leads to the formation of defects or directly to the collapse of the structure.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (No. 23-06562S). The students collaborating on this work were supported by the Student Grant Agency of J. E. Purkyně University in Ústí nad Labem (No. UJEP-SGS-44201 15 2095 01). The authors also acknowledge the assistance provided by the Research Infrastructure NanoEnviCz, supported by the Ministry of Education, Youth and Sports of the Czech Republic under Project No. LM2023066. We are also grateful to collaborating staff, namely: Jitka Bezdičková and Petr Ryšánek (XRPD), Pavla Kurhajcová (TGA and FTIR), Michal Valter and Petra Veronesi Dáňová (catalytic tests), and Anna Litovčenková (help with stability tests).References
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. P. Suh and J. Reedijk, Terminology of metal–organic frameworks and coordination polymers (IUPAC Recommendations 2013), Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- A. Schoedel, Chapter 2 – Secondary building units of MOFs, in Metal-Organic Frameworks for Biomedical Applications, 2020, pp. 11–44 Search PubMed.
- J. Ha, J. H. Lee and H. R. Moon, Alterations to secondary building units of metal–organic frameworks for the development of new functions, Inorg. Chem. Front., 2020, 7, 12–27 RSC.
- H. Ghasempour, K.-Y. Wang, J. A. Powell, F. ZareKarizi, X.-L. Lv, A. Morsali and H.-C. Zhou, Metal–organic frameworks based on multicarboxylate linkers, Coord. Chem. Rev., 2021, 426, 213542 CrossRef CAS.
- S. A. A. Razavi and A. Morsali, Linker functionalized metal-organic frameworks, Coord. Chem. Rev., 2019, 399, 213023 CrossRef.
- S. M. Moosavi, A. Nandy, K. M. Jablonka, D. Ongari, J. P. Janet, P. G. Boyd, Y. Lee, B. Smit and H. J. Kulik, Understanding the diversity of the metal-organic framework ecosystem, Nat. Commun., 2020, 11, 4068 CrossRef CAS.
- M. J. Kalmutzki, N. Hanikel and O. M. Yaghi, Secondary building units as the turning point in the development of the reticular chemistry of MOFs, Sci. Adv., 2018, 4, eaat9180 CrossRef PubMed.
- X. Zhang, Z. Chen, X. Liu, S. L. Hanna, X. Wang, R. Taheri-Ledari, A. Maleki, P. Li and O. K. Farha, A historical overview of the activation and porosity of metal–organic frameworks, Chem. Soc. Rev., 2020, 49, 7406–7427 RSC.
- W. Zhang, R. Taheri-Ledari, M. Saeidirad, F. S. Qazi, A. Kashtiaray, F. Ganjali, Y. Tian and A. Maleki, Regulation of porosity in MOFs: A review on tunable scaffolds and related effects and advances in different applications, J. Environ. Chem. Eng., 2022, 10, 108836 CrossRef CAS.
- A. D. Burrows, L. K. Cadman, W. J. Gee, H. A. Hamzah, J. V. Knichal and S. Rochat, Chapter 2: Tuning the properties of metal–organic frameworks by post-synthetic modification, in Metal–Organic Frameworks: Applications in separations and Catalysis, 2018, pp. 29–56 Search PubMed.
- H. Li, K. Wang, Y. Sun, C. T. Lollar, J. Li and H.-C. Zhou, Recent advances in gas storage and separation using metal–organic frameworks, Mater. Today, 2018, 21, 108–121 CrossRef CAS.
- T. Jia, Y. Gu and F. Li, Progress and potential of metal-organic frameworks (MOFs) for gas storage and separation: A review, J. Environ. Chem. Eng., 2022, 10, 108300 CrossRef CAS.
- C. Jiang, X. Wang, Y. Ouyang, K. Lu, W. Jiang, H. Xu, X. Wei, Z. Wang, F. Dai and D. Sun, Recent advances in metal–organic frameworks for gas adsorption/separation, Nanoscale Adv., 2022, 4, 2077–2089 RSC.
- B. Siu, A. R. Chowdhury, Z. Yan, S. M. Humphrey and T. Hutter, Selective adsorption of volatile organic compounds in metal-organic frameworks (MOFs), Coord. Chem. Rev., 2023, 485, 215119 CrossRef CAS.
- M. J. Uddin, R. E. Ampiaw and W. Lee, Adsorptive removal of dyes from wastewater using a metal-organic framework: A review, Chemosphere, 2021, 284, 131314 CrossRef CAS PubMed.
- M. Feng, P. Zhang, H.-C. Zhou and V. K. Sharma, Water-stable metal-organic frameworks for aqueous removal of heavy metals and radionuclides: A review, Chemosphere, 2018, 209, 783–800 CrossRef CAS.
- H. D. Lawson, S. P. Walton and C. Chan, Metal−organic frameworks for drug delivery: A design perspective, ACS Appl. Mater. Interfaces, 2021, 13, 7004–7020 CrossRef CAS PubMed.
- S. Mallakpour, E. Nikkhoo and C. M. Hussain, Application of MOF materials as drug delivery systems for cancer therapy and dermal treatment, Coord. Chem. Rev., 2022, 451, 214262 CrossRef CAS.
- T. Qiu, Z. Liang, W. Guo, H. Tabassum, S. Gao and R. Zou, Metal–organic framework-based materials for energy conversion and storage, ACS Energy Lett., 2020, 5, 520–532 CrossRef CAS.
- W. Xu and O. M. Yaghi, Metal–organic frameworks for water harvesting from air, anywhere, anytime, ACS Cent. Sci., 2020, 6, 1348–1354 CrossRef CAS PubMed.
- L. Cheng, Y. Dang, Y. Wang and K.-J. Chen, Recent advances in metal–organic frameworks for water absorption and their applications, Mater. Chem. Front., 2024, 8, 1171–1194 RSC.
- J. F. Olorunyomi, S. T. Geh, R. A. Caruso and C. M. Doherty, Metal–organic frameworks for chemical sensing devices, Mater. Horiz., 2021, 8, 2387–2419 RSC.
- A. Zuliani, N. Khiar and C. Carrillo-Carrión, Recent progress of metal–organic frameworks as sensors in (bio)analytical fields: towards real-world applications, Anal. Bioanal. Chem., 2023, 415, 2005–2023 CrossRef CAS PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Metal–organic frameworks in heterogeneous catalysis: Recent progress, new trends, and future perspectives, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- T. A. Goetjen, J. Liu, Y. Wu, J. Sui, X. Zhang, J. T. Hupp and O. K. Farha, Metal–organic framework (MOF) materials as polymerization catalysts: a review and recent advances, Chem. Commun., 2020, 56, 10409–10418 RSC.
- G. Mouchaham, S. Wang and C. Serre, Chapter 1: The stability of metal–organic frameworks, in Metal–Organic Frameworks: Applications in Separations and Catalysis, 2018, pp. 1–28 Search PubMed.
- S. Daliran, A. R. Oveisi, C.-W. Kung, U. Sen, A. Dhakshinamoorthy, C.-H. Chuang, M. Khajeh, M. Erkartal and J. T. Hupp, Defect-enabling zirconium-based metal–organic frameworks for energy and environmental remediation applications, Chem. Soc. Rev., 2024, 53, 6244–6294 RSC.
- F. Ahmadijokani, H. Molavi, M. Rezakazemi, S. Tajahmadi, A. Bahi, F. Ko, T. M. Aminabhavi, J.-R. Li and M. Arjmand, UiO-66 metal–organic frameworks in water treatment: A critical review, Prog. Mater. Sci., 2022, 125, 100904 CrossRef CAS.
- E. M. Dias and C. Petit, Towards the use of metal–organic frameworks for water reuse: a review of the recent advances in the field of organic pollutants removal and degradation and the next steps in the field, J. Mater. Chem. A, 2015, 3, 22484–22506 RSC.
- Z. Mo, H. Zhang, A. Shahab, F. A. Khan, J. Chen and C. Huang, Functionalized metal-organic framework UiO-66 nanocomposites with ultra-high stability for efficient adsorption of heavy metals: Kinetics, thermodynamics, and isothermal adsorption, J. Taiwan Inst. Chem. Eng., 2023, 146, 104778 CrossRef CAS.
- K. Kirakci, D. Bůžek, P. Peer, V. Liška, J. Mosinger, I. Křížová, M. Kloda, S. Ondrušová, K. Lang and J. Demel, Polymeric membranes containing iodine-loaded UiO-66 nanoparticles as water-responsive antibacterial and antiviral surfaces, ACS Appl. Nano Mater., 2022, 5, 1244–1251 CrossRef CAS.
- H. Bunzen, Chemical stability of metal-organic frameworks for applications in drug delivery, ChemNanoMat, 2021, 7, 998–1007 CrossRef CAS.
- N. H. Ly, N. B. Nguyen, H. N. Tran, T. T. H. Hoang, S.-W. Joo, Y. Vasseghian, H. Kamyab, S. Chelliapan and J. J. Klemeš, Metal-organic framework nanopesticide carrier for accurate pesticide delivery and decrement of groundwater pollution, J. Cleaner Prod., 2023, 402, 136809 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Design and synthesis of an exceptionally stable and highly porous metal-organic framework, Nature, 1999, 402, 276–279 CrossRef CAS.
- S. Yuan, J.-S. Qin, C. T. Lollar and H.-C. Zhou, Stable metal–organic frameworks with group 4 metals: Current status and trends, ACS Cent. Sci., 2018, 4, 440–450 CrossRef CAS.
- T. He, X.-J. Kong and J.-R. Li, Chemically stable metal–organic frameworks: Rational construction and application expansion, Acc. Chem. Res., 2021, 54, 3083–3094 CrossRef CAS.
- Y. An, X. Lv, W. Jiang, L. Wang, Y. Shi, X. Hang and H. Pang, The stability of MOFs in aqueous solutions—research progress and prospects, Green Chem. Eng., 2024, 5, 187–204 CrossRef.
- L. Wang, J. Li, L. Cheng, Y. Song, P. Zeng and X. Wen, Application of hard and soft acid base theory to uncover the destructiveness of Lewis bases to UiO-66 type metal organic frameworks in aqueous solutions, J. Mater. Chem. A, 2021, 9, 14868–14876 RSC.
- A. M. Hamisu, A. Ariffin and A. C. Wibowo, Cation exchange in metal-organic frameworks (MOFs): The hard-soft acid-base (HSAB) principle appraisal, Inorg. Chim. Acta, 2020, 511, 119801 CrossRef CAS.
- M. Taddei, F. Costantino, F. Marmottini, A. Comotti, P. Sozzani and R. Vivani, The first route to highly stable crystalline microporous zirconium phosphonate metal–organic frameworks, Chem. Commun., 2014, 50, 14831–14834 RSC.
- S. J. I. Shearan, N. Stock, F. Emmerling, J. Demel, P. A. Wright, K. D. Demadis, M. Vassaki, F. Costantino, R. Vivani, S. Sallard, I. R. Salcedo, A. Cabeza and M. Taddei, New Directions in Metal Phosphonate and Phosphinate Chemistry, Crystals, 2019, 9, 270 CrossRef CAS.
- J.-B. Lin, T. T. T. Nguyen, R. Vaidhyanathan, J. Burner, J. M. Taylor, H. Durekova, F. Akhtar, R. K. Mah, O. Ghaffari-Nik, S. Marx, N. Fylstra, S. S. Iremonger, K. W. Dawson, P. Sarkar, P. Hovington, A. Rajendran, T. K. Woo and G. K. H. Shimizu, A scalable metal-organic framework as a durable physisorbent for carbon dioxide capture, Science, 2021, 374, 1464–1469 CrossRef CAS.
- M. Ding, X. Cai and H.-L. Jiang, Improving MOF stability: approaches and applications, Chem. Sci., 2019, 10, 10209–10230 RSC.
- L. Feng, K.-Y. Wang, G. S. Day, M. R. Ryder and H.-C. Zhou, Destruction of metal–organic frameworks: Positive and negative aspects of stability and lability, Chem. Rev., 2020, 120, 13087–13133 CrossRef CAS.
- E. Moumen, A. H. Assen, K. Adil and Y. Belmabkhout, Versatility vs stability. Are the assets of metal–organic frameworks deployable in aqueous acidic and basic media?, Coord. Chem. Rev., 2021, 443, 214020 CrossRef CAS.
- L. Wang, X. Li, B. Yang, K. Xiao, H. Duan and H. Zhao, The chemical stability of metal-organic frameworks in water treatments: Fundamentals, effect of water matrix and judging methods, J. Chem. Eng., 2022, 450, 138215 CrossRef CAS.
- P. Lu, Y. Wu, H. Kang, H. Wei, H. Liu and M. Fang, What can pKa and NBO charges of the ligands tell us about the water and thermal stability of metal organic frameworks?, J. Mater. Chem. A, 2014, 2, 16250–16267 RSC.
- D. Cartagenova, F. A. P. Esteves, N. T. Fischer, J. A. van Bokhoven and M. Ranocchiari, Solvent-dependent textural properties of defective UiO-66 after acidic and basic treatment, Inorg. Chem. Front., 2022, 9, 70–77 RSC.
- C. D. Fast, J. Woods, J. Lentchner and T. A. Makal, Stabilizing defects in metal–organic frameworks: pendant Lewis basic sites as capping agents in UiO-66-type MOFs toward highly stable and defective porous materials, Dalton Trans., 2019, 48, 14696–14704 RSC.
- J. Y. Kim, J. Kang, S. Cha, H. Kim, D. Kim, H. Kang, I. Choi and M. Kim, Stability of Zr-Based UiO-66 metal–organic frameworks in basic solutions, Naomaterials, 2024, 14, 110 CrossRef CAS.
- Z. Wang, A. Bilegsaikhan, R. T. Jerozal, T. A. Pitt and P. J. Milner, Evaluating the robustness of metal–organic frameworks for synthetic chemistry, ACS Appl. Mater. Interfaces, 2021, 13, 17517–17531 CrossRef CAS.
- K. Leus, T. Bogaerts, J. De Decker, H. Depauw, K. Hendrickx, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Systematic study of the chemical and hydrothermal stability of selected “stable” Metal Organic Frameworks, Microporous Mesoporous Mater., 2016, 226, 110–116 CrossRef CAS.
- M. E. A. Safy, M. Amin, R. R. Haikal, B. Elshazly, J. Wang, Y. Wang, C. Wöll and M. H. Alkordi, Probing the water stability limits and degradation pathways of metal–organic frameworks, Chem. – Eur. J., 2020, 26, 7109–7117 CrossRef CAS PubMed.
- D. Bůžek, J. Demel and K. Lang, Zirconium metal–organic framework UiO-66: Stability in an aqueous environment and its relevance for organophosphate degradation, Inorg. Chem., 2018, 57, 14290–14297 CrossRef.
- D. Bůžek, S. Adamec, K. Lang and J. Demel, Metal–organic frameworks vs. buffers: case study of UiO-66 stability, Inorg. Chem. Front., 2021, 8, 720–734 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, A new zirconium inorganic building brick forming metal organic frameworks with exceptional stability, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef.
- R. M. Rego, M. D. Kurkuri and M. Kigga, A comprehensive review on water remediation using UiO-66 MOFs and their derivatives, Chemospehere, 2022, 302, 134845 CrossRef CAS PubMed.
- G. C. Shearer, S. Chavan, S. Bordiga, S. Svelle, U. Olsbye and K. P. Lillerud, Defect engineering: Tuning the porosity and composition of the metal–organic framework UiO-66 via modulated synthesis, Chem. Mater., 2016, 28, 3749–3761 CrossRef CAS.
- S. Wang, J. S. Lee, M. Wahiduzzaman, J. Park, M. Muschi, C. Martineau-Corcos, A. Tissot, K. H. Cho, J. Marrot, W. Shepard, G. Maurin, J.-S. Chang and C. Serre, A robust large-pore zirconium carboxylate metal–organic framework for energy-efficient water-sorption-driven refrigeration, Nat. Energy, 2018, 3, 985–993 CrossRef CAS.
- W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Synthesis, structure, and metalation of two new highly porous zirconium metal–organic frameworks, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Detailed structure analysis of atomic positions and defects in zirconium metal–organic frameworks, Cryst. Growth Des., 2014, 14, 5370–5372 CrossRef.
- H. Furukawa, F. Gándara, Y.-B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, Water adsorption in porous metal–organic frameworks and related materials, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Zirconium-metalloporphyrin PCN-222: Mesoporous metal–organic frameworks with ultrahigh stability as biomimetic catalysts, Angew. Chem., Int. Ed., 2012, 51, 10307–10310 CrossRef CAS.
- H. Mali, C. Shah, B. H. Raghunandan, A. S. Prajapati, D. H. Patel, U. Trivedi and R. B. Subramanian, Organophosphate pesticides an emerging environmental contaminant: Pollution, toxicity, bioremediation progress, and remaining challenges, J. Environ. Sci., 2023, 127, 234–250 CrossRef CAS PubMed.
- S. Mukherjee and R. D. Gupta, Organophosphorus nerve agents: Types, toxicity, and treatments, J. Toxicol., 2020, 3007984 CAS.
- J. Wang, L. Liu, C. Chen, X. Dong, Q. Wang, L. Alfilfil, M. R. AlAlouni, K. Yao, J. Huang, D. Zhang and Y. Han, Engineering effective structural defects of metal–organic frameworks to enhance their catalytic performances, J. Mater. Chem. A, 2020, 8, 4464–4472 RSC.
- X. Hou, J. Wang, B. Mousavi, N. Klomkliang and S. Chaemchuen, Strategies for induced defects in metal–organic frameworks for enhancing adsorption and catalytic performance, Dalton Trans., 2022, 51, 8133–8159 RSC.
- A. M. Ploskonka and J. B. DeCoste, Insight into organophosphate chemical warfare agent simulant hydrolysis in metal-organic frameworks, J. Hazard. Mater., 2019, 375, 191–197 CrossRef CAS.
- Y. Liao, T. R. Sheridan, J. Liu, Z. Lu, K. Ma, H. Yang, O. K. Farha and J. T. Hupp, Probing the mechanism of hydrolytic degradation of nerve agent simulant with zirconium-based metal–organic frameworks, ACS Catal., 2024, 14, 437–448 CrossRef CAS.
- S.-Y. Moon, Y. Liu, J. T. Hupp and O. K. Farha, Instantaneous hydrolysis of nerve-agent simulants with a six-connected zirconium-based metal–organic framework, Angew. Chem., Int. Ed., 2015, 54, 6795–6799 CrossRef CAS PubMed.
- M. J. Katz, S.-Y. Moon, J. E. Mondloch, M. H. Beyzavi, C. J. Stephenson, J. T. Hupp and O. K. Farha, Exploiting parameter space in MOFs: a 20-fold enhancement of phosphate-ester hydrolysis with UiO-66-NH2, Chem. Sci., 2015, 6, 2286–2291 RSC.
- J. Xu, J. Liu, Z. Li, X. Wang, Y. Xu, S. Chen and Z. Wang, Optimized synthesis of Zr(IV) metal organic frameworks (MOFs-808) for efficient hydrogen storage, New J. Chem., 2019, 43, 4092–4099 RSC.
- D. Bůžek, J. Zelenka, P. Ulbrich, T. Ruml, I. Křížová, J. Lang, P. Kubát, J. Demel, K. Kirakci and K. Lang, Nanoscaled porphyrinic metal–organic frameworks: photosensitizer delivery systems for photodynamic therapy, J. Mater. Chem. B, 2017, 5, 1815–1821 RSC.
- R. C. Klet, Y. Liu, T. C. Wang, J. T. Hupp and O. K. Farha, Evaluation of Brønsted acidity and proton topology in Zr- and Hf-based metal–organic frameworks using potentiometric acid–base titration, J. Mater. Chem. A, 2016, 4, 1479–1485 RSC.
- B. T. Yost, B. Gibbons, A. Wilson, A. J. Morris and L. E. McNeil, Vibrational spectroscopy investigation of defects in Zr- and Hf-UiO-66, RSC Adv., 2022, 12, 22440–22447 RSC.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Tuned to Perfection: Ironing Out the Defects in Metal–Organic Framework UiO-66, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- O. Basu, S. Mukhopadhyay, S. Laha and S. K. Das, Defect Engineering in a metal–organic framework system to achieve super-protonic conductivity, Chem. Mater., 2022, 34, 6734–6743 CrossRef CAS.
- S. Wang, H. G. T. Ly, M. Wahiduzzaman, C. Simms, I. Dovgaliuk, A. Tissot, G. Maurin, T. N. Parac-Vogt and C. Serre, A zirconium metal-organic framework with SOC topological net for catalytic peptide bond hydrolysis, Nat. Commun., 2022, 13, 1284 CrossRef CAS.
- J. Zhang, Y. Zeng, L. Chen, X. Lei, Y. Yang, Z. Chen, L. Guo and L. Li, A novel core-shell composite of PCN-222@MIPIL for ultrasensitive electrochemical sensing 4-nonylphenol, Environ. Res., 2023, 225, 115499 CrossRef CAS PubMed.
- A. Zuliani, M. C. Castillejos and N. Khiar, Continuous flow synthesis of PCN-222 (MOF-545) with controlled size and morphology: a sustainable approach for efficient production, Green Chem., 2023, 25, 10596–10610 RSC.
- H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, A hafnium-based metal–organic framework as an efficient and multifunctional catalyst for facile CO2 fixation and regioselective and enantioretentive epoxide activation, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed.
- Z. Hu, Y. Wang and D. Zhao, The chemistry and applications of hafnium and cerium(IV) metal–organic frameworks, Chem. Soc. Rev., 2021, 50, 4629–4683 RSC.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Disclosing the complex structure of UiO-66 metal organic framework: A synergic combination of experiment and theory, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- E. Buncel and E. A. Symons, The inherent instability of dimethylformamide–water systems containing hydroxide ion, J. Chem. Soc. D, 1970, 164–165 RSC.
- Y. Bai, Y. Dou, L.-H. Xie, W. Rutledge, J.-R. Li and H.-C. Zhou, Zr-based metal-organic framework: design, synthesis, structure and applications, Chem. Soc. Rev., 2016, 45, 2327–2367 RSC.
- S.-H. Lo, L. Feng, K. Tan, Z. Huang, S. Yuan, K.-Y. Wang, B.-H. Li, W.-L. Liu, G. S. Day, S. Tao, C.-C. Yang, T.-T. Luo, C.-H. Lin, S.-L. Wang, S. J. L. Billinge, K.-L. Lu, Y. J. Chabal, X. Zou and H.-C. Zhou, Rapid desolvation-triggered domino lattice rearrangement in a metal–organic framework, Nat. Chem., 2020, 12, 90–97 CrossRef.
- S. Wu, L. Wang, H. Zhu, J. Liang, L. Ge, C. Li, T. Miao, J. Li and Z. Cheng, Catalytic degradation of CWAs with MOF-808 and PCN-222: Toward practical application, J. Chem. Res., 2022, 46, 6 Search PubMed.
- N. V. Maksimchuk, I. D. Ivanchikova, K. H. Cho, O. V. Zalomaeva, V. Y. Evtushok, K. P. Larionov, T. S. Glazneva, J.-S. Chang and O. A. Kholdeeva, Catalytic performance of Zr-based metal–organic frameworks Zr-abtc and MIP-200 in selective oxidations with H2O2, Chem. – Eur. J., 2021, 27, 6985–6992 CrossRef CAS PubMed.
- H. Wang, J. Zhong, C. Zhao, X. Guo and Y. Zhao, A tightly-bonded and wettable self-detoxifying Zr(OH)4/fiber composite for decontamination of chemical warfare agent simulants, J. Environ. Chem. Eng., 2021, 9, 105938 CrossRef CAS.
- E. Denet, M. B. Espina-Benitez, I. Pitault, T. Pollet, D. Blaha, M.-A. Bolzinger, V. Rodriguez-Nava and S. Briançon, Metal oxide nanoparticles for the decontamination of toxic chemical and biological compounds, Int. J. Pharm., 2020, 583, 119373 CrossRef CAS.
- T. J. Bandosz, M. Laskoski, J. Mahle, G. Mogilevsky, G. W. Peterson, J. A. Rossin and G. W. Wagner, Reactions of VX, GD, and HD with Zr(OH)4: Near instantaneous decontamination of VX, J. Phys. Chem. C, 2012, 116, 11606–11614 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi01366b |
| This journal is © the Partner Organisations 2024 |