
Combination of dimensional reduction and active site addition strategies for preparing unique {RE9}-cluster-based MOFs: efficient CO2 fixation and Knoevenagel condensation†
Ying
Zhao‡
a,
Dan
Wu‡
bc,
Yidan
Qiao
a,
Guo-Ping
Yang
 *b,
Lu-Fang
Ma
*ab and
Yao-Yu
Wang
b
*b,
Lu-Fang
Ma
*ab and
Yao-Yu
Wang
b
aCollege of Chemistry and Chemical Engineering, Henan Key Laboratory of Function-Oriented Porous Materials, Luoyang Normal University, Luoyang 471934, P.R. China. E-mail: mazhuxp@126.com
bCollege of Chemistry and Materials Science, Northwest University, Xi'an 710127, P.R. China. E-mail: ygp@nwu.edu.cn
cShaanxi Applied Physics and Chemistry Research Institute, Xi'an 710061, P.R. China
First published on 7th March 2024
Abstract
The current application of porous catalytic materials for organic synthesis is always confined to comparatively simple small substrates because of the diffusion barrier. Therefore, in this study, dimensional reduction and active site addition strategies were employed for preparing unique porous {RE9}-cluster-based rare-earth metal–organic frameworks (MOFs) {[Me2NH2]4[RE9(pddb)6(μ3-O)2(μ3-OH)12(H2O)1.5(HCO2)3]·6.5DMF·11H2O}n (MOF-RE, RE = Tb, Y, and Dy) with high-density multiple active sites. It was found that MOF-RE are rare {RE9}-based two-dimensional (2D) networks including triangular-nanoporous (1.3 nm) and triangular-microporous (0.8 nm) channels decorated by abundant Lewis acid–base sites (open RE(III) sites and Npyridine atoms) on the inner surface. As anticipated, due to the coexistence of Lewis acid–base sites, activated samples exhibited better catalytic activity (a yield of 96%, and a TON value of 768 for styrene oxide) than most previously reported 3D MOF materials for the cycloaddition of CO2 and multifarious epoxides under moderate conditions. Moreover, as a heterogeneous catalyst, MOF-Tb, has excellent catalytic performance (with a TON value of 396 for benzaldehyde) for the Knoevenagel condensation reaction of malononitrile and aldehydes with high catalytic stability and recoverability. In addition, both reactions possessed high turnover numbers and frequencies. These dimensional reduction and active site addition tactics may permit the exploitation of new nanoporous MOF catalysts based on rare-earth clusters for useful and intricate organic conversions.
Introduction
One of the most important concerns facing all nations on Earth is global climate change, as the main greenhouse gas, carbon dioxide (CO2) has contributed various severe ecological issues, such as sea level rise and climate change, raising concerns for CO2 capture and utilization technology.1–4 The current efficient methods of using CO2 to create valuable complexes may not only significantly reduce the amount of CO2 in the environment but also help humanity economically, making them one of the most advantageous options.5–7 Since cyclic carbonates are a type of extensively utilized chemical and chemical raw material, it has been discovered that producing cyclic carbonates synthetically from epoxides and CO2 is among the most effective ways to solve environmental issues and realize resource utilization.8–10 Nevertheless, CO2 conversion typically involves synergistic catalysis with numerous active sites. Numerous efforts have been made to date to study promising heterogeneous catalysts. Although some are regarded as advantageous, including zeolites,11 activated carbon,12 metal oxides13 and organic polymers,14 they typically call for high catalytic loadings and harsh conditions, resulting in relatively low yields and conversions with poor recyclability due to the scarcity of catalytic sites.Metal–organic frameworks (MOFs) containing different polynuclear metal-oxo clusters are unique porous crystalline materials with widespread promise for sensing, heterogeneous catalysis and gas storage/separation.15–21 Recent studies have exhibited that polynuclear cluster-based rare-earth (RE) MOFs possess exceedingly high stability and abundant catalytic active sites,22–24 in which the catalytic efficiency for CO2 conversion and Knoevenagel condensation could be greatly accelerated by the synergistic effect of activated metal ions (Lewis acid sites, LASs) and nucleophilic groups (Lewis base sites, LBSs).25,26 However, the accessibility of these RE-MOF materials to sterically demanding substrates is limited by expanding and stabilizing the active sites. Since the substrates cannot easily reach internal LASs, RE-MOFs perform poorly with larger substrates or complex reactions.27,28 It is essential to design functional ligands and precisely build RE-microporous MOFs with a high specific surface area and access to multiple active sites via an in situ function-oriented synthesis strategy to extract the catalytic ability of polynuclear cluster-based RE-MOFs to the greatest extent.
In light of the previous discussion on the standard of functional RE-MOFs as heterogeneous catalysts, this work thoroughly studied the effect of the topological structures of RE-MOFs on the catalytic activity and revealed dimensional reduction and active site addition methods to enhance catalytic reactivity through permitting unrestricted access to Lewis acid–base sites in two-dimensional (2D) MOFs (Scheme 1).29–31 These strategies can accurately predict the structures of MOFs, so as to achieve the regulation of the catalytic performance. Hence, a series of unique porous 2D RE-MOFs {[Me2NH2]4[RE9(pddb)6(μ3-O)2(μ3-OH)12(H2O)1.5(HCO2)3]·6.5DMF·11H2O}n (MOF-RE; RE = Tb, Y and Dy) with {RE9} clusters as secondary building units (SBUs) are successfully prepared from C2-symmetry V-shaped 2,6-bis(4′-carboxyl-phenyl)pyridine (H2pddb) under solvothermal conditions. Notably, MOF-RE have high densities of quantified open metal sites (OMSs) acting as LASs and uncoordinated pyridines as LBSs, all of which are exposed in open channels. Benefiting from these active sites, MOF-RE exhibits excellent catalytic efficiencies for CO2 transformation with high yields, turnover numbers (TONs), and turnover frequencies (TOFs) compared to those of many reported 3D MOF materials. Moreover, they have excellent catalytic properties for Knoevenagel condensations via the synergistic effects of the LASs and LBSs.
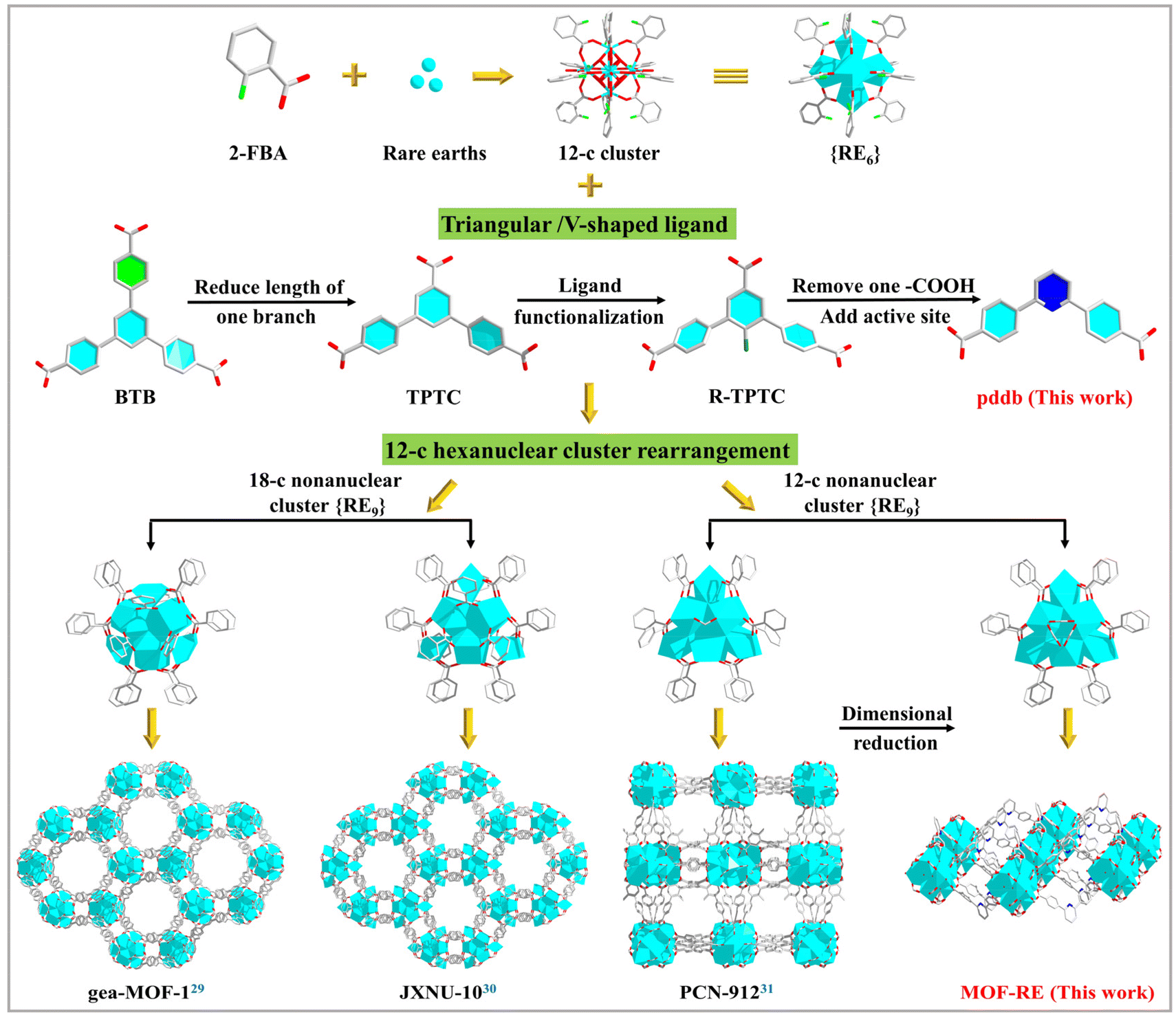 | ||
| Scheme 1 Different polynuclear cluster-based rare-earth MOFs formed by the {RE9} clusters and functional connectors. | ||
Results and discussion
Description of the crystal structure
The reaction of rare-earth salts (RE = Tb, Dy, and Y) with H2pddb in a mixed solution (DMF/H2O) in the presence of HNO3 and 2-fluorobenzoic acid (2-FBA) gave hexagonal crystals of MOF-RE. The presence of 2-FBA as a structure guiding agent is an essential condition for the assembly of the polynuclear cluster-based MOFs. The MOF-RE crystallize in a primitive hexagonal P63/mmc space group (Table 1) and are 2D porous networks. Remarkably, they possess unique polynuclear rare-earth-carboxylate clusters, i.e., a 12-c nonanuclear [RE9(μ3-O)2(μ3-OH)12(O2C-)12(H2O)1.5(HCO2)3] core, in which formates are produced by the breakdown of DMF molecules.32| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2. | |||
|---|---|---|---|
| Complex | MOF-Tb | MOF-Y | MOF-Dy |
| Empirical formula | C117H69N6O45.5Tb9 | C117H69N6O45.5Y9 | C117H69N6O45.5Dy9 |
| Formula mass | 3717.06 | 3086.97 | 3749.28 |
| Crystal system | Hexagonal | Hexagonal | Hexagonal |
| Space group | P63/mmc | P63/mmc | P63/mmc |
| a [Å] | 22.5962(4) | 22.5233(11) | 22.6331(9) |
| b [Å] | 22.5962(4) | 22.5233(11) | 22.6331(9) |
| c [Å] | 22.7452(5) | 22.7551(15) | 22.6392(11) |
| α [°] | 90 | 90 | 90 |
| β [°] | 90 | 90 | 90 |
| γ [°] | 120 | 120 | 120 |
| V [Å3] | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 057.5(4) 057.5(4) |
9997.1(12) | 10043.4(9) |
| Z | 2 | 2 | 2 |
| D calcd. [g cm−3] | 1.227 | 1.026 | 1.240 |
| μ [mm−1] | 3.173 | 2.634 | 3.357 |
| F [000] | 3524 | 3056 | 3542 |
| θ [°] | 2.071–25.349 | 2.072–25.385 | 2.078–25.380 |
| Reflections collected | 34562/3409 |
64794/3391 |
66446/3411 |
| GOOF | 1.050 | 1.061 | 1.041 |
| R , indices [I > 2σ(I)] | R 1 = 0.0261 | R 1 = 0.0419 | R 1 = 0.0270 |
| wR2 = 0.0648 | wR2 = 0.1209 | wR2 = 0.0730 | |
| R indices (all data) | R 1 = 0.0331 | R 1 = 0.0590 | R 1 = 0.0336 |
| wR2 = 0.0680 | wR2 = 0.1303 | wR2 = 0.0766 | |
The [RE9(μ3-O)2(μ3-OH)12(O2C-)12(H2O)1.5(HCO2)3] cluster has a threefold symmetry and is composed of nine RE ions arrayed in a {RE9} tricapped trigonal prism, according to detailed research of the nonanuclear cluster (Fig. S1a†). In contrast to the triangular planes of the tetragonal pyramids of the RE9 tricapped trigonal prism, which are each capped by a μ3-OH, the two triangular planes of the central RE6 trigonal prism are each capped by a μ3-O (Fig. S1b†). The six RE1 ions are each coordinated with eight O atoms: two from carboxylate moieties of two separate pddb2− connecters and four from four μ3-OH, and another two coordination sites are composed of O atoms from one HCO2− ligand and a μ3-O (O1) (Fig. S2a†). The remaining three RE2 cations are each coordinated with nine O atoms: a terminal H2O molecule, four μ3-OH and four carboxylate O from four independent pddb2− ligands (Fig. S2a†). The nonanuclear [RE9(μ3-O)2(μ3-OH)12] cluster is created by twelve μ3-OH and two μ3-O connecting nine RE elements (Fig. 1a) and is terminated through twelve carboxylates from twelve independent pddb2− linkers to generate a 12-connected [RE9(μ3-O)2(μ3-OH)12(O2C-)12] SBU. As the extension point of the [RE9(μ3-O)2(μ3-OH)12(O2C-)12] core, C atoms from the carboxylate groups of twelve independent pddb2− linkers are arranged into a hexagonal prism, which conforms to the d6R vertex diagram of a 12-connected node. There are three H2O molecules and three extra HCO2− bridging linkers coordinated with the RE ions to form a nonanuclear [RE9(μ3-O)2(μ3-OH)12(O2C-)12(H2O)1.5(HCO2)3] cluster (Fig. 1a).
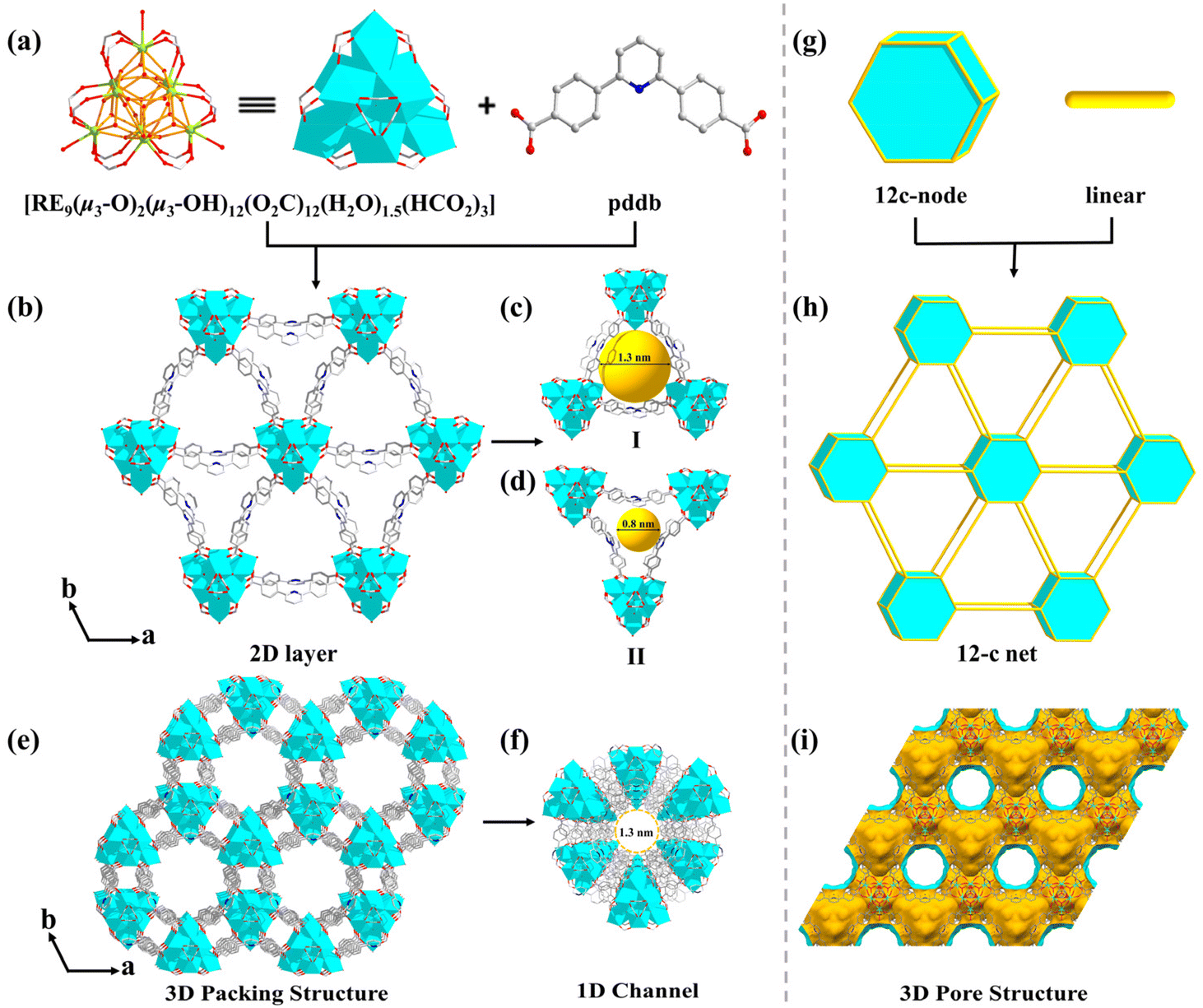 | ||
| Fig. 1 (a and g) Types of {RE9} cluster and ligand and their simplifications; (b and h) 2D structure and corresponding 12-connected net of MOF-RE; (c and d) different channels along the c axis; (e and f) 3D supramolecular arrangement with 1D hexagonal channels; (i) view of the porous structure of MOF-RE. | ||
In the structure of MOF-RE, the pddb2− ligands adopt a bidentate bridging mode to connect two adjacent nonanuclear clusters (Fig. S2b†) to form 2D anionic layers with triangular-microporous (∼0.8 nm) and triangular-nanoporous (∼1.3 nm) channels (Fig. 1b–d). The 2D infinite layers are stacked along the c axis with an average interlayer spacing of 11.37 Å (Fig. 1e and i). Notably, there are honeycomb-like hexagonal channels with a diameter of ∼1.3 nm along the c axis, which are full of protonated [Me2NH2]+ cations (Fig. 1f). The porosity computed by PLATON is ∼56.1% of the overall crystal volume after excluding free solvents. Topologically, the [RE9(μ3-O)2(μ3-OH)12(O2C)12(H2O)1.5(HCO2)3] clusters and ligands may act as 12-connected nodes and linear rods, respectively, and the whole structure can be represented as a 12-connected skeleton (Fig. 1g and h).
Gas adsorption studies
MOF-Tb was chosen as a representative for thorough studies due to its isomorphism. The freshly synthesized sample was vacuum-dried at 200 °C for 4 h prior to the sorption test to obtain the activated sample, MOF-Tba. The thermogravimetric analysis (TGA) curve was used to explore the activation temperature (Fig. S5†). Simultaneously, the framework integrality of activated MOF-Tba was verified via the powder X-ray diffraction (PXRD) pattern (Fig. S3†). A 77 K N2 adsorption experiment was performed to confirm the pore properties of MOF-Tba (Fig. 2a), indicating that it possesses a traditional type-I adsorption isotherm as well as high Langmuir (825.33 m2 g−1) and Brunauer–Emmett–Teller (BET) surface areas (560.35 m2 g−1). The pore size distribution acquired through fitting the 77 K N2-sorption isotherm using nonlocal density functional theory is consistent with the pore diameter provided by X-ray crystal data (Fig. 2a).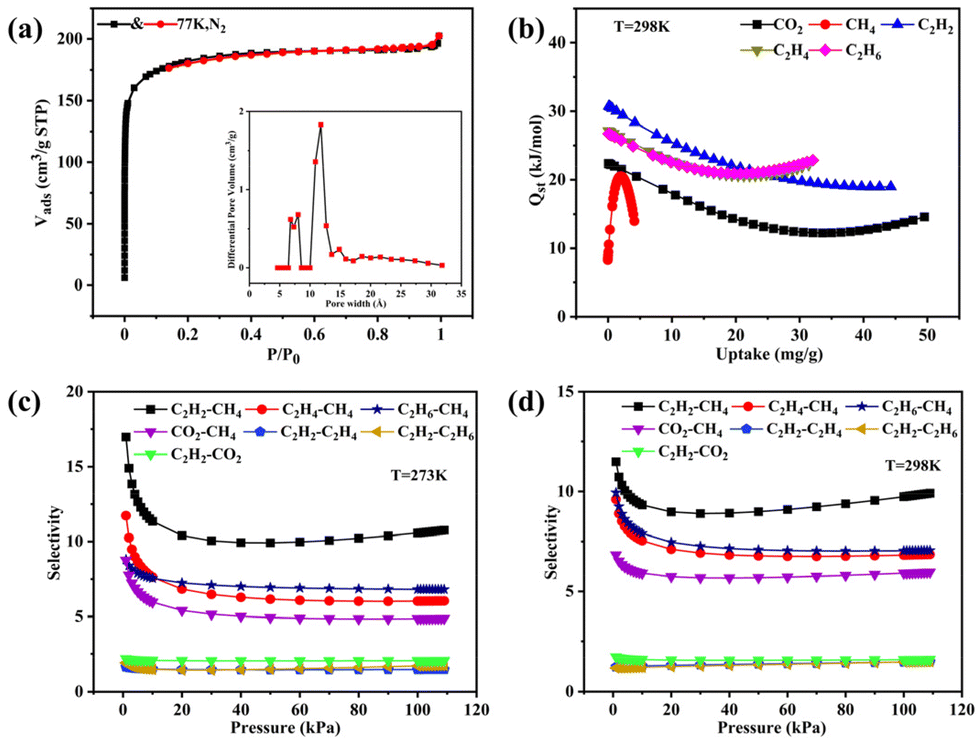 | ||
| Fig. 2 (a) N2 sorption isotherm at 77 K (inset displays the pore size distribution); (b) gas (C2Hn, CO2 and CH4) adsorption heat Qst for MOF-Tba. Adsorption selectivities of MOF-Tba at 273 K (c) and 298 K (d) calculated using IAST for equimolar mixtures of C2Hn/CH4, CO2/CH4, C2H2/C2Hn and C2H2/CO2. | ||
The latent application of MOF-Tba for CO2 and light hydrocarbon (CH4 and C2Hn) sorption (Fig. S6 and 7†)/separation (Fig. 2c and d) has been carefully examined because of its inherent perpetual porosity and channel circumstances. The results displayed that the loading capacity of C2H2 in MOF-Tba is higher than that of other gases, demonstrating the maximum interaction between C2H2 and the framework. The sorption enthalpies (Qst) were determined using the virial approach to establish the adsorption affinity between the five gases and skeleton more accurately (Fig. 2b and Fig. S8†). The Qst of MOF-Tba to C2H2 (37.6 kJ mol−1) is higher than that of CO2 (22.4 kJ mol−1), C2H4 (27.2 kJ mol−1), C2H6 (26.7 kJ mol−1), and CH4 (8.6 kJ mol−1) under zero coverage, which agrees with the measured adsorption amount. Moreover, the possibility of separating CH4 from light hydrocarbons was studied by ideal solution adsorbed theory (IAST) for binary equimolar mixtures (Fig. S9 and 10†).33 The selectivities for CO2, C2H2, C2H4 and C2H6 over CH4 at 1 bar and 298 K are 5.9, 9.8, 6.8 and 7.0, respectively (Fig. 2c), making MOF-Tba an exceptional sorbent for effectively removing CO2/C2 light hydrocarbons from natural gas.
Catalytic performance for CO2 conversion
Considering that MOF-Tba has the advantages of solvent-accessible nanoscale channels, high specific surface area and abundant coexisting Lewis acid–base sites ([Tb9(μ3-O)2(μ3-OH)12(O2C)12(H2O)1.5(HCO2)3] clusters and Npyridine atoms), it was employed as an effective heterogeneous catalyst for the cycloaddition of CO2 and epoxy complexes (Scheme S1†). Our previous study34 has shown that the catalysts were not recycled and exhibited poor activity due to the lack of high-density active sites when selecting the pddb ligand itself and physical mixture of the rare-earth metal-pddb ligand to catalyze this reaction. Therefore, this study conducted a range of control experiments based on styrene oxide, determined ideal reaction conditions, such as time, temperature, and catalyst dosage and identified products through 1H NMR spectroscopy (Fig. S14–25†).Entry 1 (Table 2) displays that when MOF-Tba (0.05 mol%) was added as a catalyst, only a small amount of product with a yield of 5% was detected within 12 h. In addition, only a slight conversion (10%) could be observed under the cocatalyst of n-Bu4NBr (1 mol%) alone, as shown in entry 2. Nevertheless, the yield was tremendously improved to 29% (entry 3) when MOF-Tba (0.05 mol%) and n-Bu4NBr (1 mol%) were concurrently introduced to the reaction, suggesting that MOF-Tba and n-Bu4NBr synergistically activated the second-order reaction of CO2 and epoxides. Increasing the temperature was used to demonstrate that one of the key factors was temperature for influencing the reaction outcome, as shown in entries 4 and 5. Furthermore, entries 6and 7 examined and listed the effect of the cocatalyst n-Bu4NBr dosage, which demonstrated that the quantity of the cocatalyst had a clear impact on the reaction rate. The yield increased to 95% when 5 mol% n-Bu4NBr cocatalyst was added. The amount of MOF-Tba was increased because the practical application of the catalyst will be severely hampered by the 12 h reaction time. Styrene oxide could be converted into 4-phenyl-1,3-dioxolan-2-one more rapidly, as described in entries 8–10. In conclusion, it was found that the ideal reaction conditions are 0.125 mol% MOF-Tba catalyst, 5 mol% n-Bu4NBr cocatalyst, 60 °C and 6 h. In addition, we performed a detailed analysis of the 1H NMR spectrum for entry 10, confirming that no by-products were generated during the reaction (Fig. S19†).
| Entry | MOF (mol%) | n-Bu4NBr (mol%) | T (°C) | t (h) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: solvent-free, styrene oxide (20 mmol), and CO2 (1 atm). b The product yield was determined by 1H NMR. | |||||
| 1 | 0.05 | 0 | 25 | 12 | 5 |
| 2 | 0 | 1 | 25 | 12 | 10 |
| 3 | 0.05 | 1 | 25 | 12 | 32 |
| 4 | 0.05 | 1 | 40 | 12 | 54 |
| 5 | 0.05 | 1 | 60 | 12 | 78 |
| 6 | 0.05 | 3 | 60 | 12 | 86 |
| 7 | 0.05 | 5 | 60 | 12 | 95 |
| 8 | 0.075 | 5 | 60 | 10 | 94 |
| 9 | 0.1 | 5 | 60 | 8 | 95 |
| 10 | 0.125 | 5 | 60 | 6 | 96 |
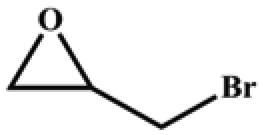
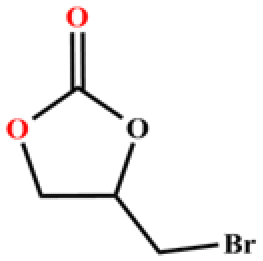
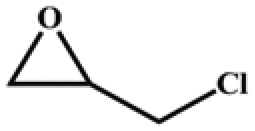
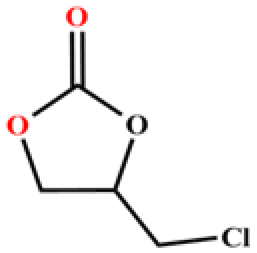
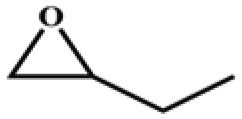
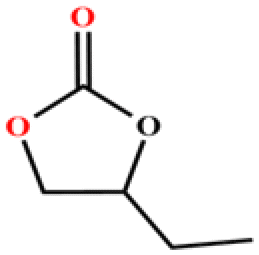
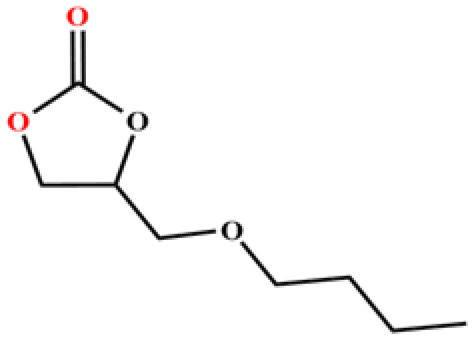
The catalytic universality of MOF-Tba was further evaluated utilizing a range of propylene oxide derivatives with distinct substituents and steric hindrance under determined ideal reaction circumstances (Table 3). The outcomes indicated that there was some regularity in how different substituents affected the yield. It can be seen from the comparison of entries 1 and 2 that the epoxy complexes with electron-withdrawing groups (–Br and –Cl) could improve the efficiency of the cycloaddition reaction, and the yield could reach more than 99%, the reason for which is that the electron-withdrawing group may decrease the electron density of ethylene oxide.35,36 In contrast, electron-donating groups had a disadvantageous impact on this process, as in entry 3.37,38 Furthermore, entries 4–6 exhibited a significant decrease in the conversion of epoxide with bulky substituents, confirming the idea that big substituents restrict the mobility of substrate molecules.39–42 Table S1† lists information about the molecular sizes of all epoxide derivatives. Additionally, the TON of MOF-Tba for styrene oxide was notable compared to most reported TON values for MOF catalysts (Table S2†), which was likely attributable to the profitable contribution of {Tb9} clusters and abundant Npyridine groups in the channels.
| Entry | Epoxide | Product | Yieldb (%) | TONc | TOFd (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: solvent-free, epoxides (20 mmol), n-Bu4NBr (5 mol%), Tb-MOF catalyst (0.125 mol%), CO2 (1 atm), 60 °C, and 6 h. b Yield was determined by 1H NMR. c TON = [product (mmol)]/[catalyst (mmol)]. d TOF = TON/time. | |||||
| 1 |
|
|
>99 | 792 | 132 |
| 2 |
|
|
>99 | 792 | 132 |
| 3 |
|
|
96 | 768 | 128 |
| 4 |
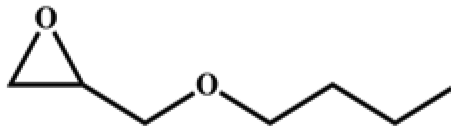
|
|
96 | 768 | 128 |
| 5 |
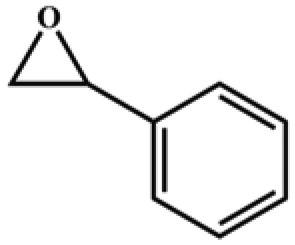
|
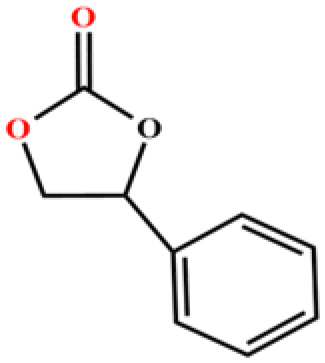
|
96 | 768 | 128 |
| 6 |
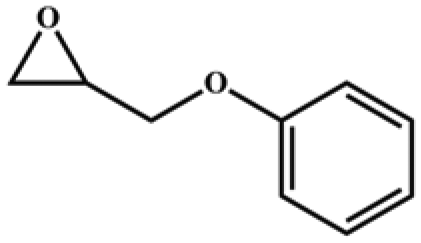
|
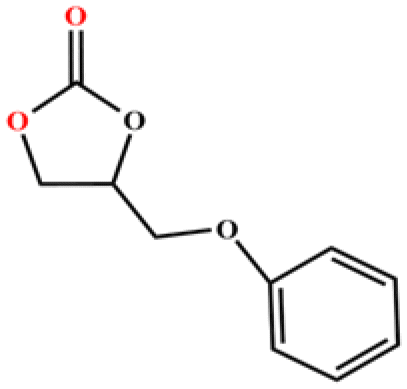
|
95 | 760 | 127 |
The actual organic synthesis industry depends heavily on the stability and recyclability of catalysts;43 hence, additional tests were conducted about hot leaching, recovery, and recycling of MOF-Tba. First, the recycling stability of MOF-Tba for the cyclization reaction of CO2 with styrene oxide was studied under the determined ideal reaction conditions. The conversion of styrene oxide was nearly unchanged for five cycles by the recovered catalyst MOF-Tba (Fig. S11†). Meanwhile, the PXRD pattern of the recovered MOF-Tba sample after five experiments was essentially matched with the newly formed one, showing that the MOF-Tba catalyst maintained the stability of the framework (Fig. S12†). Afterward, inductively coupled plasma (ICP) analysis was then used in leaching experiments. As a result, the recovered filtrate included just a little quantity of Tb(III) ions (0.015%), further demonstrating the stability of MOF-Tba in the organic reaction. Furthermore, a thermal filtration experiment was carried out, and the results showed that the reaction hardly happens when the catalyst is filtered out (Fig. S13†), meaning that MOF-Tba possessed a heterogeneous nature.
The probable catalytic mechanism can be deduced from prior MOF-related literature,44–47 and the distinctive structural characteristics of MOF-Tba, including high specific surface area, functional channel, and plentiful {Tb9} clusters (Fig. 3). First, the epoxide rapidly diffuses into the MOF-Tba catalyst and makes weak contact with its exposed metal sites in a confined environment. Then, the nucleophilic attack of less-obstructed carbon atoms in the epoxide by the Br− anion released by n-Bu4NBr promotes the formation of the alkylcarbonate anion. Subsequently, polarized CO2 molecules tend to undergo nucleophilic addition reactions with alkylcarbonate anions to generate alkylcarbonate salt. Finally, the ring closure behavior results in the production of cyclic carbonate and liberation of catalysts.
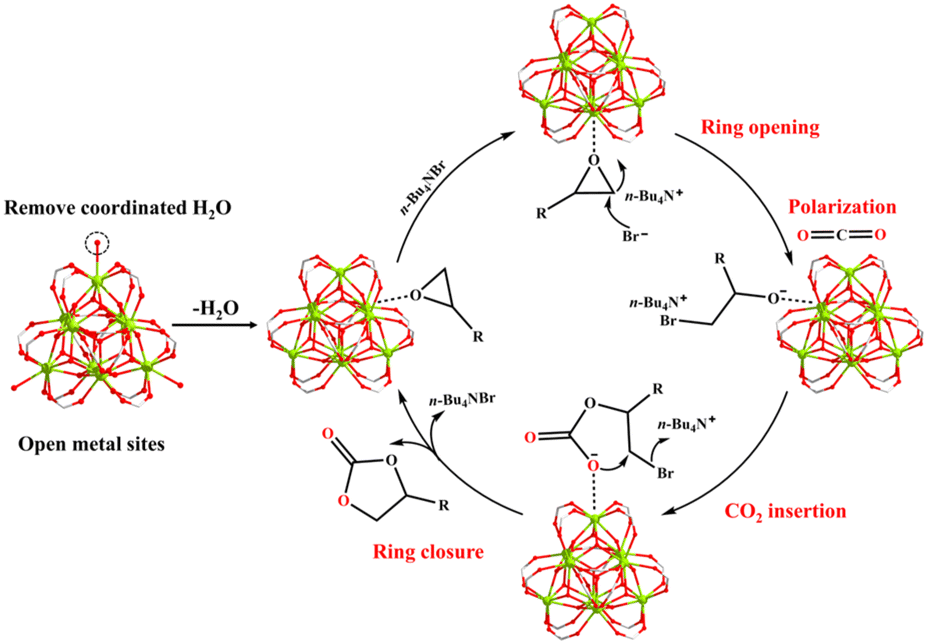 | ||
| Fig. 3 Proposed mechanism of CO2 conversion catalyzed by MOF-Tba. | ||
Catalytic performances for Knoevenagel condensation
Knoevenagel condensation is a classical C–C bond coupling reaction, and its reaction mechanism involves the covalent C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond formed by the carbonyl group combining with the methylene group activated via both electron-withdrawing moieties.40–42,48 Pharmaceuticals and fine compounds are frequently synthesized using this process. Recent research has proven that active metal centers (LASs) and nucleophilic moieties (LBSs) in porous MOFs could work together to significantly speed up the Knoevenagel reaction.49–52 Therefore, more research was done on active porous MOF-Tba to catalyze the Knoevenagel condensation (Scheme S2†).
C bond formed by the carbonyl group combining with the methylene group activated via both electron-withdrawing moieties.40–42,48 Pharmaceuticals and fine compounds are frequently synthesized using this process. Recent research has proven that active metal centers (LASs) and nucleophilic moieties (LBSs) in porous MOFs could work together to significantly speed up the Knoevenagel reaction.49–52 Therefore, more research was done on active porous MOF-Tba to catalyze the Knoevenagel condensation (Scheme S2†).
Initially, perfect reaction conditions were studied with benzaldehyde and malononitrile as substrates in the presence of desolvated MOF-Tba as a heterogeneous catalyst, as seen in Table 4 and Fig. S29–31.† Entry 1 shows that only trace 2-benzylidenemalono-nitrile (3%) was produced at 25 °C without the catalyst MOF-Tba, whereas when MOF-Tba (0.1 mol%) was introduced, a yield of 38% was generated within 2 h (entry 2), indicating that the reaction could scarcely be carried out without the catalyst. The conversion of the substrate increased progressively when all other factors governing the reaction were held constant, and only the catalyst dosage was increased (entries 3 and 4). At 25 °C, the yield was 85% when the MOF-Tba dosage was raised to 0.25 mol% (entry 5). The reaction substrate was virtually entirely converted as the temperature rose from 25 °C to 60 °C in parallel investigations using 0.25 mol% MOF-Tba (entries 6 and 7), demonstrating that temperature was one of the crucial elements in the condensation reaction. Additionally, the connection between the conversion with reaction time was examined under the reaction environments of 0.25 mol% MOF-Tba at 60 °C (entries 8–10). It can be seen from the above results that the ideal reaction conditions were 0.25 mol% MOF-Tba, 60 °C, and 2 h when the ratio of malononitrile to aldehyde was 2:1.
| Entry | MOF-Tba (mol%) | Time (h) | T (°C) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: malononitrile (20 mmol), benzaldehyde (10 mmol). b The product yield was determined by 1H NMR. | ||||
| 1 | 0 | 2 | 25 | 3 |
| 2 | 0.1 | 2 | 25 | 38 |
| 3 | 0.15 | 2 | 25 | 58 |
| 4 | 0.2 | 2 | 25 | 76 |
| 5 | 0.25 | 2 | 25 | 85 |
| 6 | 0.25 | 2 | 40 | 93 |
| 7 | 0.25 | 2 | 60 | 99 |
| 8 | 0.25 | 1.5 | 60 | 95 |
| 9 | 0.25 | 1 | 60 | 88 |
| 10 | 0.25 | 0.5 | 60 | 65 |
Based on aforementioned discoveries, we chose several aldehyde derivatives with various substituents and steric hindrance (Table S3†) to confirm the MOF-Tba catalyst's suitability for the Knoevenagel condensation reaction, and the outcomes are displayed in Table 5 and Fig. S32–38.†
| Entry | Substrate | Product | Yieldb (%) | TONc | TOFd (h−1) |
|---|---|---|---|---|---|
| a Reaction conditions: aldehyde derivatives (10 mmol), malononitrile (20 mmol), catalyst MOF-Tba (0.25 mol%), 2 h, 60 °C. b The yield was calculated by 1H NMR. c TON = [product (mmol)]/[catalyst (mmol)]. d TOF = TON/time. | |||||
| 1 |
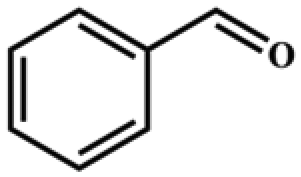
|
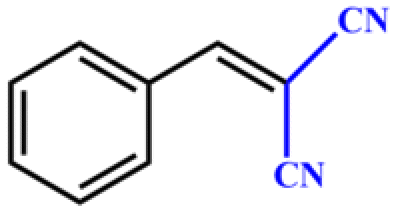
|
>99 | 396 | 198 |
| 2 |
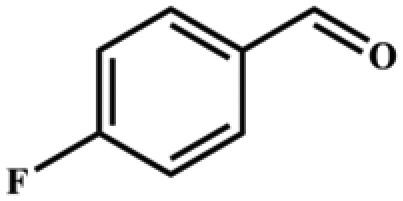
|
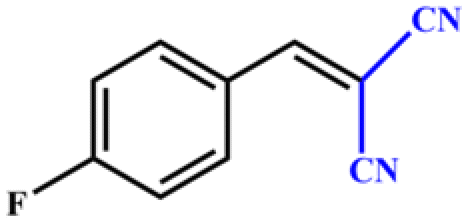
|
>99 | 396 | 198 |
| 3 |
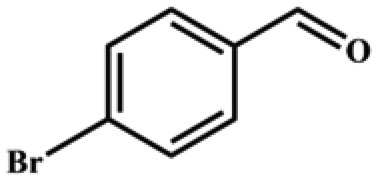
|
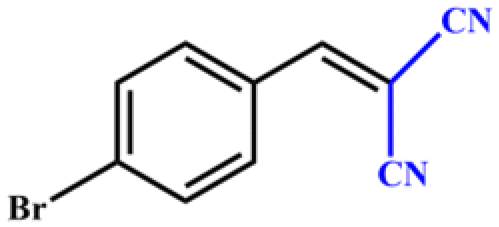
|
>99 | 396 | 198 |
| 4 |
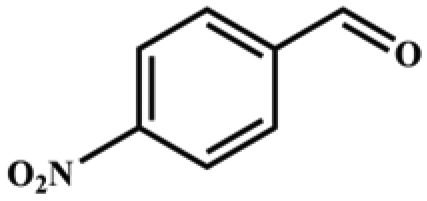
|
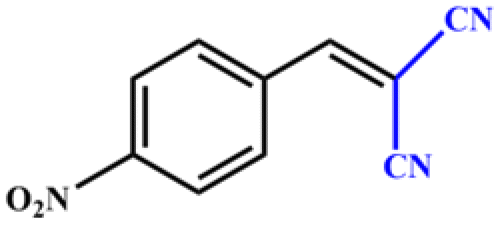
|
>99 | 396 | 198 |
| 5 |
|
|
98 | 384 | 192 |
| 6 |
|
|
94 | 372 | 186 |
| 7 |
|
|
87 | 348 | 174 |
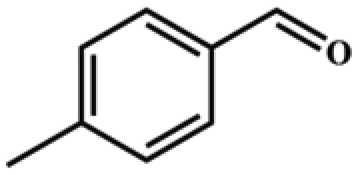
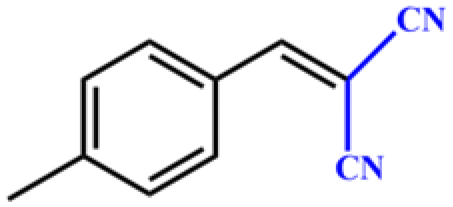
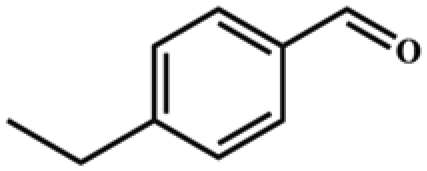
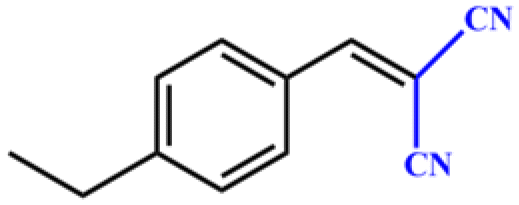
Entries 2–4 show that the conversion efficiency of benzaldehyde with electron-drawing groups (–F, –Br, and –NO2) surpassed 99%, whereas the catalytic yield was slightly decreased due to the presence of the electron-donating groups (–CH3 and –CH2CH3) (entries 5 and 6), indicating that the electron-donating moieties greatly inhibit the Knoevenagel condensation reaction. Additionally, the conversion efficiency clearly reduced as the molecular size and steric hindrance of the substrate increased (entry 7). Remarkably, benzaldehyde had a TON value of 396, which was much higher than the majority of previously documented MOF catalysts (Table S4†).
The best experimental conditions were used to study the stability and recyclability of MOF-Tba. The utilized MOF-Tba catalyst was recovered and repeatedly cleaned with DMF after each reaction. Over 97% of 2-benzylidenemalono-nitrile was produced after five repetitions of the process using MOF-Tba, which retained excellent catalytic activity (Fig. S26†). Furthermore, the PXRD peaks of gathered MOF-Tba demonstrated that the host framework remained unchanged, suggesting the great stability of the microporous heterogeneous catalyst (Fig. S27†). Following the catalytic recycling experiment, leached homogenous Tb(III) was monitored by ICP analysis as well. The probability of leaching metal ions from the MOF-Tba network during Knoevenagel condensation was ruled out when trace Tb(III) of 0.018% was found in the filtrate. Under optimal reaction circumstances, a heat filtration test was conducted to confirm the heterogeneous nature of MOF-Tba. The solid catalyst was filtered out after the reaction had been going on for 0.5 hours. Since the conversion rate barely altered (Fig. S28†), the heterogeneous nature of MOF-Tba was further supported.
Fig. 4 implies a likely catalytic reaction mechanism based on relevant published studies53–59 and structural characteristics of MOF-Tba. First, the carbonyl oxygen of the aldehyde group made a weak interaction with the exposed Tb(III) sites of MOF-Tba, converting its carbon atom into a positive carbon center and initiating the reaction. Simultaneously, Npyridine atoms as the LBSs caused the carbonyl carbon of malononitrile to polarize into a negative center. Second, a covalent bond was formed between two carbon atoms with opposing electric charges, resulting in an imine intermediate. Finally, the recombination of intramolecular electrons combined with the release of H2O molecules and the used catalyst resulted in the formation of the product benzylidenemalononitrile.
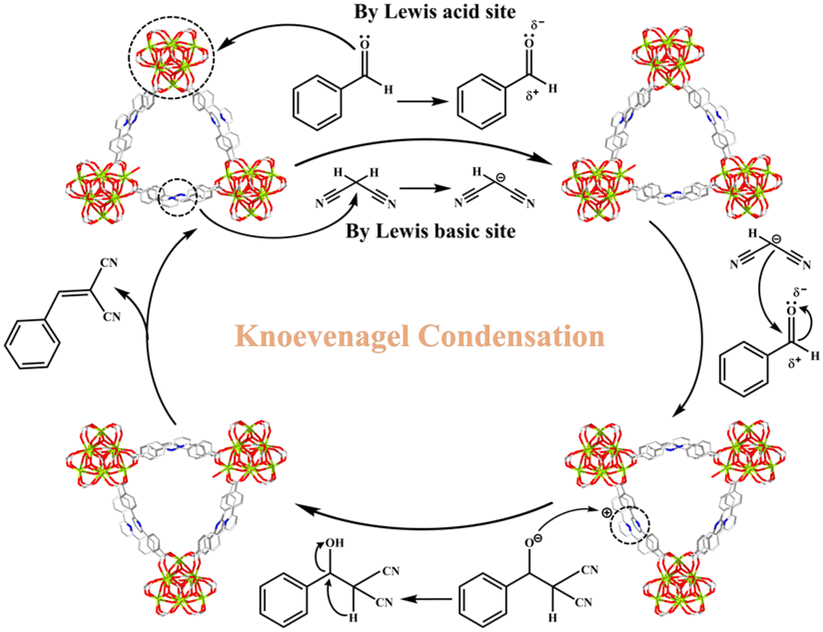 | ||
| Fig. 4 Proposed mechanism for Knoevenagel condensation by MOF-Tba. | ||
Conclusions
A series of 2D {RE9}-cluster-based rare-earth MOF-RE with Lewis acid–base dual functional sites were designed and synthesized by dimensional reduction and active site addition strategies. As expected, they benefited from high-density active sites and had exceptional catalytic properties for the chemical fixation of CO2 with epoxides under moderate conditions, together with satisfactory catalytic efficiencies for Knoevenagel condensation. These strategies proposed in this work not only provide a new method for the preparation of nanoporous cluster-based RE-MOFs with various catalytic activities but also lay a foundation for the research of the catalytic mechanism.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for the financial support from the NSFC (22071194) and Natural Science Foundation of Henan Province (232300421232).References
- C. Wang, B. An and W. Lin, Metal–Organic Frameworks in Solid–Gas Phase Catalysis, ACS Catal., 2018, 9, 130–146 CrossRef.
- J. F. Kurisingal, Y. Rachuri, A. S. Palakkal, R. S. Pillai, Y. Gu, Y. Choe and D. W. Park, Water-Tolerant DUT-Series Metal–Organic Frameworks: A Theoretical–Experimental Study for the Chemical Fixation of CO2 and Catalytic Transfer Hydrogenation of Ethyl Levulinate to γ-Valerolactone, ACS Appl. Mater. Interfaces, 2019, 11, 41458–41471 CrossRef CAS PubMed.
- J. Liu, G. P. Yang, J. Jin, D. Wu, L. F. Ma and Y. Y. Wang, A first new porous d–p HMOF material with multiple active sites for excellent CO2 capture and catalysis, Chem. Commun., 2020, 56, 2395–2398 RSC.
- J. Liu, Y. Z. Fan, X. Li, Y. W. Xu, L. Zhang and C. Y. Su, Catalytic Space Engineering of Porphyrin Metal–Organic Frameworks for Combined CO2 Capture and Conversion at a Low Concentration, ChemSusChem, 2018, 11, 2340–2347 CrossRef CAS PubMed.
- P. Das and S. K. Mandal, Unprecedented High Temperature CO2 Selectivity and Effective Chemical Fixation by a Copper-Based Undulated Metal–Organic Framework, ACS Appl. Mater. Interfaces, 2020, 12, 37137–37146 CrossRef CAS PubMed.
- J. Liu, Y. Wei and Y. Zhao, Trace Carbon Dioxide Capture by Metal–Organic Frameworks, ACS Sustainable Chem. Eng., 2018, 7, 82–93 CrossRef CAS.
- S. L. Hou, J. Dong, X. L. Jiang, Z. H. Jiao and B. Zhao, A Noble-Metal-Free Metal–Organic Framework (MOF) Catalyst for the Highly Efficient Conversion of CO2 with Propargylic Alcohols, Angew. Chem., Int. Ed., 2019, 58, 577–581 CrossRef CAS PubMed.
- E. Liu, J. Zhu, W. Yang, F. Liu, C. Huang and S. Yin, PCN-222(Co) Metal–Organic Framework Nanorods Coated with 2D Metal–Organic Layers for the Catalytic Fixation of CO2 to Cyclic Carbonates, ACS Appl. Nano Mater., 2020, 3, 3578–3584 CrossRef CAS.
- Z. Zhang, J.-H. Ye, T. Ju, L.-L. Liao, H. Huang, Y.-Y. Gui, W.-J. Zhou and D.-G. Yu, Visible-Light-Driven Catalytic Reductive Carboxylation with CO2, ACS Catal., 2020, 10, 10871–10885 CrossRef CAS.
- X. Yang, Q. Zou, T. Zhao, P. Chen, Z. Liu, F. Liu and Q. Lin, Deep Eutectic Solvents as Efficient Catalysts for Fixation of CO2 to Cyclic Carbonates at Ambient Temperature and Pressure through Synergetic Catalysis, ACS Sustainable Chem. Eng., 2021, 9, 10437–10443 CrossRef CAS.
- M. R. Hudson, W. L. Queen, J. A. Mason, D. W. Fickel, R. F. Lobo and C. M. Brown, Unconventional, Highly Selective CO2 Adsorption in Zeolite SSZ-13, J. Am. Chem. Soc., 2012, 134, 1970–1973 CrossRef CAS PubMed.
- Y. Zhang, B. Li, K. Williams, W. Y. Gao and S. Ma, A new microporous carbon material synthesized via thermolysis of a porous aromatic framework embedded with an extra carbon source for low-pressure CO2 uptake, Chem. Commun., 2013, 49, 10269–10271 RSC.
- P. R. Tambe and G. D. Yadav, Heterogeneous cycloaddition of styrene oxide with carbon dioxide for synthesis of styrene carbonate using reusable lanthanum–zirconium mixed oxide as catalyst, Clean Technol. Environ. Policy, 2018, 20, 345–356 CrossRef CAS.
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Capture and conversion of CO2 at ambient conditions by a conjugated microporous polymer, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- H. Wang, X. Liu, W. Yang, G. Mao, Z. Meng, Z. Wu and H. L. Jiang, Surface-Clean Au25 Nanoclusters in Modulated Microenvironment Enabled by Metal–Organic Frameworks for Enhanced Catalysis, J. Am. Chem. Soc., 2022, 144, 22008–22017 CrossRef CAS PubMed.
- P. F. Gao, Y. Y. Jiang, H. Liu, M. S. Zhou, T. Li, H. R. Fu, L. F. Ma and D. S. Li, Pillar-Layer Chiral MOFs as a Crystalline Platform for Circularly Polarized Luminescence and Single-Phase White-Light Emission, ACS Appl. Mater. Interfaces, 2022, 14, 16435–16444 CrossRef CAS PubMed.
- R.-Y. Chen, Y.-P. He, G.-H. Chen and J. Zhang, Designing Cage-Supported Cluster-Organic Framework for Highly Efficient Optical Limiting, ACS Mater. Lett., 2022, 4, 1397–1401 CrossRef CAS.
- Z. H. Zhu, Z. L. Liang, Z. H. Jiao, X. L. Jiang, Y. Xie, H. Xu and B. Zhao, A Facile Strategy to Obtain Low-Cost and High-Performance Gold-Based Catalysts from Artificial Electronic Waste by [Zr48Ni6] Nano-Cages in MOFs for CO2 Electroreduction to CO, Angew. Chem., Int. Ed., 2022, 61, e202214243 CrossRef CAS PubMed.
- Q. Y. Wang, Z. B. Sun, M. Zhang, S. N. Zhao, P. Luo, C. H. Gong, W. X. Liu and S. Q. Zang, Cooperative Catalysis between Dual Copper Centers in a Metal–Organic Framework for Efficient Detoxification of Chemical Warfare Agent Simulants, J. Am. Chem. Soc., 2022, 144, 21046–21055 CrossRef CAS PubMed.
- J. L. Li, X. Xiong, D. Luo, Y. B. Wei, W. Lu and D. Li, Formaldehyde recognition through aminal formation in a luminescent metal–organic framework, Chem. Commun., 2022, 58, 6490–6493 RSC.
- H. Yang, Y. Chen, C. Dang, A. N. Hong, P. Feng and X. Bu, Optimization of Pore-Space-Partitioned Metal–Organic Frameworks Using the Bioisosteric Concept, J. Am. Chem. Soc., 2022, 144, 20221–20226 CrossRef CAS PubMed.
- S. A. Younis, N. Bhardwaj, S. K. Bhardwaj, K.-H. Kim and A. Deep, Rare earth metal–organic frameworks (RE-MOFs): Synthesis, properties, and biomedical applications, Coord. Chem. Rev., 2021, 429, 213620 CrossRef CAS.
- H. A. Bicalho, P. R. Donnarumma, V. Quezada-Novoa, H. M. Titi and A. J. Howarth, Remodelling a shp: Transmetalation in a Rare-Earth Cluster-Based Metal–Organic Framework, Inorg. Chem., 2021, 60, 11795–11802 CrossRef CAS PubMed.
- H. A. Bicalho, F. Saraci, J. J. Velazquez-Garcia, H. M. Titi and A. J. Howarth, Unravelling the synthesis of a rare-earth cluster-based metal–organic framework with spn topology, Chem. Commun., 2022, 58, 10925–10928 RSC.
- T. Zhang, H. Chen, S. Liu, H. Lv, X. Zhang and Q. Li, Highly Robust {Ln4}-Organic Frameworks (Ln = Ho, Yb) for Excellent Catalytic Performance on Cycloaddition Reaction of Epoxides with CO2 and Knoevenagel Condensation, ACS Catal., 2021, 11, 14916–14925 CrossRef CAS.
- J. Qiao, B. Zhang, L. Zhang and Y. Liu, Practice of function-oriented synthesis: high-efficiency CO2 conversion and Knoevenagel condensation by two novel In3-based MOFs with high-density active sites under mild conditions, J. Mater. Chem. A, 2022, 10, 17773–17781 RSC.
- D. Yang and B. C. Gates, Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research, ACS Catal., 2019, 9, 1779–1798 CrossRef CAS.
- H. Chen, S. Liu, H. Lv, Q. P. Qin and X. Zhang, Nanoporous {Y2}-Organic Frameworks for Excellent Catalytic Performance on the Cycloaddition Reaction of Epoxides with CO2 and Deacetalization–Knoevenagel Condensation, ACS Appl. Mater. Interfaces, 2022, 14, 18589–18599 CrossRef CAS PubMed.
- V. Guillerm, L. Weselinski, Y. Belmabkhout, A. J. Cairns, V. D'Elia, L. Wojtas, K. Adil and M. Eddaoudi, Discovery and introduction of a (3,18)-connected net as an ideal blueprint for the design of metal–organic frameworks, Nat. Chem., 2014, 6, 673–680 CrossRef CAS PubMed.
- L. Chen, H. J. Hu, Y. L. Wang, X. F. Zhang, L. P. Xu and Q. Y. Liu, Metal–Organic Frameworks Featuring 18-Connected Nonanuclear Rare-Earth Oxygen Clusters and Cavities for Efficient C2H2/CO2 Separation, Inorg. Chem., 2021, 60, 13471–13478 CrossRef CAS PubMed.
- Y. Wang, L. Feng, W. Fan, K. Y. Wang, X. Wang, X. Wang, K. Zhang, X. Zhang, F. Dai, D. Sun and H. C. Zhou, Topology Exploration in Highly Connected Rare-Earth Metal–Organic Frameworks via Continuous Hindrance Control, J. Am. Chem. Soc., 2019, 141, 6967–6975 CrossRef CAS PubMed.
- A. Rossin, G. Giambastiani, M. Peruzzini and R. Sessoli, Amine-Templated Polymeric Lanthanide Formates: Synthesis, Characterization, and Applications in Luminescence and Magnetism, Inorg. Chem., 2012, 51, 6962–6968 CrossRef CAS PubMed.
- D. Alezi, A. M. Peedikakkal, L. J. Weselinski, V. Guillerm, Y. Belmabkhout, A. J. Cairns, Z. Chen, L. Wojtas and M. Eddaoudi, Quest for Highly Connected Metal–Organic Framework Platforms: Rare-Earth Polynuclear Clusters Versatility Meets Net Topology Needs, J. Am. Chem. Soc., 2015, 137, 5421–5430 CrossRef CAS PubMed.
- D. Wu, X. Lu, Y. Tang, F. Gao, G. Yang and Y.-Y. Wang, Light-Assisted CO2 Cycloaddition over a Nanochannel Cadmium–Organic Framework Loaded with Silver Nanoparticles, ACS Appl. Nano Mater., 2023, 6, 6197–6207 CrossRef CAS.
- H. Chen, L. Fan and X. Zhang, Highly Robust 3s–3d {CaZn}–Organic Framework for Excellent Catalytic Performance on Chemical Fixation of CO2 and Knoevenagel Condensation Reaction, ACS Appl. Mater. Interfaces, 2020, 12, 54884–54892 CrossRef CAS PubMed.
- Y. B. N. Tran, P. T. K. Nguyen, Q. T. Luong and K. D. Nguyen, Series of M-MOF-184 (M = Mg, Co, Ni, Zn, Cu, Fe) Metal–Organic Frameworks for Catalysis Cycloaddition of CO2, Inorg. Chem., 2020, 59, 16747–16759 CrossRef CAS PubMed.
- S. Liu, H. Chen and X. Zhang, Bifunctional {Pb10K2}–Organic Framework for High Catalytic Activity in Cycloaddition of CO2 with Epoxides and Knoevenagel Condensation, ACS Catal., 2022, 12, 10373–10383 CrossRef CAS.
- Q. R. Ding, Y. Yu, C. Cao, J. Zhang and L. Zhang, Stepwise assembly and reversible structural transformation of ligated titanium coated bismuth-oxo cores: shell morphology engineering for enhanced chemical fixation of CO2, Chem. Sci., 2022, 13, 3395–3401 RSC.
- N. Seal and S. Neogi, Intrinsic-Unsaturation-Enriched Biporous and Chemorobust Cu(II) Framework for Efficient Catalytic CO2 Fixation and Pore-Fitting Actuated Size-Exclusive Hantzsch Condensation with Mechanistic Validation, ACS Appl. Mater. Interfaces, 2021, 13, 55123–55135 CrossRef CAS PubMed.
- G. Jin, D. Sensharma, N. Zhu, S. Vaesen and W. Schmitt, A highly augmented, (12,3)-connected Zr-MOF containing hydrated coordination sites for the catalytic transformation of gaseous CO2 to cyclic carbonates, Dalton Trans., 2019, 48, 15487–15492 RSC.
- Y. Li, X. Zhang, J. Lan, D. Li, Z. Wang, P. Xu and J. Sun, A High-Performance Zinc-Organic Framework with Accessible Open Metal Sites Catalyzes CO2 and Styrene Oxide into Styrene Carbonate under Mild Conditions, ACS Sustainable Chem. Eng., 2021, 9, 2795–2803 CrossRef CAS.
- B. Ugale, S. S. Dhankhar and C. M. Nagaraja, Construction of 3-Fold-Interpenetrated Three-Dimensional Metal–Organic Frameworks of Nickel(II) for Highly Efficient Capture and Conversion of Carbon Dioxide, Inorg. Chem., 2016, 55, 9757–9766 CrossRef CAS PubMed.
- K. Maity, C. K. Karan and K. Biradha, Porous Metal-Organic Polyhedral Framework containing Cuboctahedron Cages as SBUs with High Affinity for H2 and CO2 Sorption: A Heterogeneous Catalyst for Chemical Fixation of CO2, Chem. – Eur. J., 2018, 24, 10988–10993 CrossRef CAS PubMed.
- Z. Xue, J. Jiang, M.-G. Ma, M.-F. Li and T. Mu, Gadolinium-Based Metal–Organic Framework as an Efficient and Heterogeneous Catalyst To Activate Epoxides for Cycloaddition of CO2 and Alcoholysis, ACS Sustainable Chem. Eng., 2017, 5, 2623–2631 CrossRef CAS.
- B. Parmar, P. Patel, R. S. Pillai, R. I. Kureshy, N.-U. H. Khan and E. Suresh, Efficient catalytic conversion of terminal/internal epoxides to cyclic carbonates by porous Co(II) MOF under ambient conditions: structure–property correlation and computational studies, J. Mater. Chem. A, 2019, 7, 2884–2894 RSC.
- A. Shaabani, R. Mohammadian, H. Farhid, M. K. Alavijeh and M. M. Amini, Multitask Guanidinium Bromide Functionalized Metal–Organic Framework in Chemical Fixation of CO2 at Low Pressure and Temperature, Ind. Eng. Chem. Res., 2019, 58, 8553 CrossRef CAS.
- F. Norouzi and H. R. Khavasi, Diversity-Oriented Metal Decoration on UiO-Type Metal–Organic Frameworks: an Efficient Approach to Increase CO2 Uptake and Catalytic Conversion to Cyclic Carbonates, ACS Omega, 2019, 4, 19037–19045 CrossRef CAS PubMed.
- T. Stolar, A. Prasnikar, V. Martinez, B. Karadeniz, A. Bjelic, G. Mali, T. Friscic, B. Likozar and K. Uzarevic, Scalable Mechanochemical Amorphization of Bimetallic Cu–Zn MOF-74 Catalyst for Selective CO2 Reduction Reaction to Methanol, ACS Appl. Mater. Interfaces, 2021, 13, 3070–3077 CrossRef CAS PubMed.
- C. I. Ezugwu, B. Mousavi, M. A. Asraf, Z. Luo and F. Verpoort, Post-synthetic modified MOF for Sonogashira cross-coupling and Knoevenagel condensation reactions, J. Catal., 2016, 344, 445–454 CrossRef CAS.
- M. Opanasenko, A. Dhakshinamoorthy, M. Shamzhy, P. Nachtigall, M. Horáček, H. Garcia and J. Čejka, Comparison of the catalytic activity of MOFs and zeolites in Knoevenagel condensation, Catal. Sci. Technol., 2013, 3, 500–507 RSC.
- R. Sharma, A. Bansal, C. N. Ramachandran and P. Mohanty, A multifunctional triazine-based nanoporous polymer as a versatile organocatalyst for CO2 utilization and C–C bond formation, Chem. Commun., 2019, 55, 11607–11610 RSC.
- S. Yuan, L. Huang, Z. Huang, D. Sun, J. S. Qin, L. Feng, J. Li, X. Zou, T. Cagin and H. C. Zhou, Continuous Variation of Lattice Dimensions and Pore Sizes in Metal–Organic Frameworks, J. Am. Chem. Soc., 2020, 142, 4732–4738 CrossRef CAS PubMed.
- Y. Luan, Y. Qi, H. Gao, R. S. Andriamitantsoa, N. Zheng and G. Wang, A general post-synthetic modification approach of amino-tagged metal–organic frameworks to access efficient catalysts for the Knoevenagel condensation reaction, J. Mater. Chem. A, 2015, 3, 17320–17331 RSC.
- F. Ghobakhloo, D. Azarifar, M. Mohammadi, H. Keypour and H. Zeynali, Copper(II) Schiff-Base Complex Modified UiO-66-NH2(Zr) Metal–Organic Framework Catalysts for Knoevenagel Condensation–Michael Addition–Cyclization Reactions, Inorg. Chem., 2022, 61, 4825–4841 CrossRef CAS PubMed.
- Q. Xu, B. Xu, H. Kong, P. He, J. Wang, T. Kannan, P. Ma, J. Wang and J. Niu, Synthesis and Characterization of a Crown-Shaped 36-Molybdate Cluster and Application in Catalyzing Knoevenagel Condensation, Inorg. Chem., 2020, 59, 10665–10672 CrossRef CAS PubMed.
- G. Q. Huang, J. Chen, Y. L. Huang, K. Wu, D. Luo, J. K. Jin, J. Zheng, S. H. Xu and W. Lu, Mixed-Linker Isoreticular Zn(II) Metal–Organic Frameworks as Brønsted Acid–Base Bifunctional Catalysts for Knoevenagel Condensation Reactions, Inorg. Chem., 2022, 61, 8339–8348 CrossRef CAS PubMed.
- Z.-S. Zhao, Y. Zhang, T. Fang, Z.-B. Han and F.-S. Liang, Chitosan-Coated Metal–Organic-Framework Nanoparticles as Catalysts for Tandem Deacetalization–Knoevenagel Condensation Reactions, ACS Appl. Nano Mater., 2020, 3, 6316–6320 CrossRef CAS.
- F. Kalantari, S. Rezayati, A. Ramazani, H. Aghahosseini, K. Ślepokura and T. Lis, Proline-Cu Complex Based 1,3,5-Triazine Coated on Fe3O4 Magnetic Nanoparticles: A Nanocatalyst for the Knoevenagel Condensation of Aldehyde with Malononitrile, ACS Appl. Nano Mater., 2022, 5, 1783–1797 CrossRef CAS.
- J. Qiao, B. Zhang, X. Yu, X. Zou, X. Liu, L. Zhang and Y. Liu, A Stable Y(III)-Based Amide-Functionalized Metal–Organic Framework for Propane/Methane Separation and Knoevenagel Condensation, Inorg. Chem., 2022, 61, 3708–3715 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and supporting figures. CCDC 2216930–2216932. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3qi02527f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2024 |