
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron oxide nanozymes enhanced by ascorbic acid for macrophage-based cancer therapy†
Zhongchao
Yi
a,
Xiaoyue
Yang
a,
Ying
Liang
b and
Sheng
Tong
 *a
*a
aDepartment of Biomedical Engineering, University of Kentucky, Lexington, Kentucky 40536, USA. E-mail: sheng.tong@uky.edu
bNew York Blood Center, New York, New York 10065, USA
First published on 15th July 2024
Abstract
In recent years, using pharmacological ascorbic acid has emerged as a promising therapeutic approach in cancer treatment, owing to its capacity to induce extracellular hydrogen peroxide (H2O2) production in solid tumors. The H2O2 is then converted into cytotoxic hydroxyl free radicals (HO˙) by redox-active Fe2+ inside cells. However, the high dosage of ascorbic acid required for efficacy is hampered by adverse effects such as kidney stone formation. In a recent study, we demonstrated the efficient catalytic conversion of H2O2 to HO˙ by wüstite (Fe1−xO) nanoparticles (WNPs) through a heterogenous Fenton reaction. Here, we explore whether WNPs can enhance the therapeutic potential of ascorbic acid, thus mitigating its dose-related limitations. Our findings reveal distinct pH dependencies for WNPs and ascorbic acid in the Fenton reaction and H2O2 generation, respectively. Importantly, WNPs exhibit the capability to either impede or enhance the cytotoxic effect of ascorbic acid, depending on the spatial segregation of the two reagents by cellular compartments. Furthermore, our study demonstrates that treatment with ascorbic acid promotes the polarization of WNP-loaded macrophages toward a pro-inflammatory M1 phenotype, significantly suppressing the growth of 4T1 breast cancer cells. This study highlights the importance of orchestrating the interplay between ascorbic acid and nanozymes in cancer therapy and presents a novel macrophage-based cell therapy approach.
Introduction
Ascorbic acid, also known as vitamin C, is widely recognized for its antioxidant properties.1,2 Interestingly, recent clinical trials have unveiled that pharmacological ascorbic acid, i.e., ascorbic acid administrated intravenously at high doses, can effectively suppress tumour growth across various types of cancer by inducing oxidative stress.3–11 However, its therapeutic efficacy is hindered by poor bioavailability, necessitating repeated administration of high doses (ca. 4 g per kg body weight). Moreover, a high intake of ascorbic acid poses a risk of kidney stone formation in susceptible individuals, as one of its metabolites, oxalate, can interact with calcium in the body to form calcium oxalate stones.12 Hence, there is an urgent need for strategies to enhance the effectiveness of ascorbic acid, thereby alleviating its dose-related limitation.Previous research has demonstrated that pharmacological ascorbic acid, when in the presence of proteins, undergoes decomposition into ascorbic radicals, which in turn generate extracellular hydrogen peroxide (H2O2) in the tumour.3,13,14 H2O2 then infiltrates cells, where it is converted into highly reactive hydroxyl free radicals (HO˙) by redox-active Fe2+via the Fenton reaction. As cancer cells typically exhibit elevated levels of intrinsic oxidative stress, their growth is selectively inhibited by ascorbic acid.10,15,16 This fundamental mechanism suggests that the therapeutic efficacy of ascorbic acid could be enhanced by promoting the hydrolysis of hydrogen peroxide into hydroxyl free radicals. Along these lines, Petronek et al. observed that the tumour-suppressing effect of ascorbic acid was enhanced upon intraperitoneal injection of ferumoxytol, an FDA-approved magnetite nanoparticle formulation of iron supplement, in a mouse glioblastoma model.17 The study revealed that ferumoxytol increased the labile iron pool in the brain tumour, thereby facilitating the Fenton reaction.
Alternatively, research has established the intrinsic peroxidase-like activity of iron oxide nanoparticles (IONPs), which catalyses the hydrolysis of H2O2 to hydroxyl free radicals on the nanoparticle surface via a heterogeneous Fenton reaction.18–20 Compared to labile iron ions, IONPs exhibit a prolonged half-life in the body and offer opportunities for image-guided and targeted delivery to specific tissues or cells.21 Numerous investigations have delved into harnessing the enzymatic activity of IONPs for innovative cancer therapies.22–26 For instance, Zanganeh et al. demonstrated that intratumoral injection of ferumoxytol could induce the polarization of tumour-associated macrophages toward a pro-inflammatory M1 phenotype, thereby suppressing breast cancer growth and reducing lung metastasis.23 Additionally, Shen et al. showcased how magnetite nanoparticles (MNPs) could impede tumour progression through ferroptosis in a mouse orthotopic brain tumour model.24 However, the anticancer efficacy of IONPs relies on their catalytic efficiency as well as the availability of endogenous hydrogen peroxide within the tumour tissue. There is a burgeoning interest in combining IONPs with H2O2-inducing reagents to enhance the effectiveness of cancer treatment strategies.
In a recent study, we have discovered that wüstite nanoparticles (WNPs) exhibit markedly superior catalytic activity compared to magnetite (Fe3O4) and maghemite (Fe2O3) nanoparticles, including ferumoxytol.27 The enhanced catalytic activity of WNPs can be attributed to the presence of internal low-valence iron (Fe0 and Fe2+), stemming from the disproportionation of wüstite at ambient temperature. Here, we explore the potential of WNP nanozymes to augment the efficacy of ascorbic acid in cancer treatment. Surprisingly, our study unveils distinct pH dependencies in the Fenton reaction and hydrogen peroxide generation between WNPs and ascorbic acid, respectively. Further study reveals synergistic effects in cytotoxicity and macrophage polarization when the two reagents are compartmentalized by cellular organelles. Leveraging this unique mechanism, we engineer magnetically targeted M1-polarized macrophages, potentially presenting a promising immune adjuvant for ascorbic acid-based cancer therapy.
Materials and methods
Chemicals
Iron acetylacetonate (99%), 1,2-hexadecanediol, benzyl ether (99%), oleic acid (technical grade, 90%), oleylamine (technical grade, 70%), hydrochloric acid, hydroxylamine hydrochloride, ammonium acetate, ferrozine, iron standard solution, horseradish peroxidase (HRP), hydrogen peroxide, sodium hydroxide, 3,3′,5,5′-tetramethylbenzidine (TMB), and ascorbic acid were purchased from Sigma-Aldrich. 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[malemide(polyethylene glycol)-2000] (DSPE-PEG-mal) were purchased from Avanti Polar Lipids. Fetal bovine serum (FBS), CM-H2DCFDA, Amplex Red, DiO, DiI, DiD, Hoechst33342, and ActinGreen 488 ReadyProbes were purchased from ThermoFisher Scientific. Anti-CD86 antibody (ab119857), and secondary goat anti-rat IgG H&L (AlexaFluor 555, ab150158) were purchased from Abcam. TAT peptide (YGRKKRRQRRRGC) was synthesized by GenScript. Ferumoxytol was a gift from AMAG Pharmaceuticals. All reagents were used without further modifications.Synthesis of IONPs
Wüstite nanocrystals were synthesized by thermo-decomposition of iron acetylacetonate using a published method.27,28 In brief, a mixture of iron acetylacetonate (12 mmol), oleic acid (15 mmol), oleylamine (105 mmol), and benzyl ether (10 mL) was heated at 120 °C under vacuum for 2 hours to remove moisture and air. Next, the solution was heated to 220 °C at a ramping rate of 5 °C min−1 under nitrogen flow and held at 220 °C for 30 min. Then the solution was heated to 250 °C at a ramping rate of 2 °C min−1 and held at this temperature for 30 minutes. After that, the solution was cooled down to room temperature. Wüstite nanocrystals were collected by precipitation with acetone. The supernatant was discarded, and the pellets were dispersed with toluene. The nanocrystals were washed three times with ethanol. After the last wash, the nanocrystals were dispersed in toluene and stored in a glove box under nitrogen. Magnetite nanocrystals were synthesized using a similar thermodecomposition method published previously.29,30Water-dispersible IONPs were generated by coating nanocrystals with DSPE-PEG and DSPE-PEG-mal (molar ratio, 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) via a dual solvent exchange method.31 To enhance the cellular uptake, IONPs were conjugated with TAT peptides by mixing IONPs and peptides in PBS overnight. The efficiency of peptide conjugation was evaluated using gel electrophoresis. IONPs were labelled with fluorophores (DiO, DiI, or DiD) for fluorescence microscopy.32 Unbound DSPE-PEG, fluorophores, and peptides were removed by ultracentrifugation.
:5) via a dual solvent exchange method.31 To enhance the cellular uptake, IONPs were conjugated with TAT peptides by mixing IONPs and peptides in PBS overnight. The efficiency of peptide conjugation was evaluated using gel electrophoresis. IONPs were labelled with fluorophores (DiO, DiI, or DiD) for fluorescence microscopy.32 Unbound DSPE-PEG, fluorophores, and peptides were removed by ultracentrifugation.
Characterization of IONPs
Wüstite and magnetite nanocrystals were imaged with a transmission electron microscope (Talos F200X) connected to a charge-coupled device (CCD) camera. The size distribution of the nanocrystals was quantified using Image J software. The crystal structure of iron oxide nanocrystals was analysed with an X-ray diffractometer (Bruker D8 Advance). The hydrodynamic size of water-dispersible IONPs was measured by dynamic light scattering (Wyatt Technology DynaPro Nanostar). The iron content of IONPs was measured using a Ferrozine assay.31Quantification of hydroxyl free radical and hydrogen peroxide
The hydroxyl free radicals (HO˙) generated by IONPs were measured by TMB assay.27,33 In brief, IONPs solution and H2O2 were diluted to designated concentrations in 0.2 M acetate buffer (pH 4 unless otherwise specified). TMB powder was dissolved in dimethylformamide (DMF). 100 μL of TMB solution was mixed with 50 μL of H2O2 solution in 96-well plate. Then 100 μL of IONPs was added to the mixture. The final concentrations of IONPs and TMB were 20 μg Fe mL−1 and 1 mM, respectively. Light absorbance at 652 nm was recorded every minute using a microplate reader (Tecan Spark). The reaction kinetics was calculated using the Michaelis–Menten equation,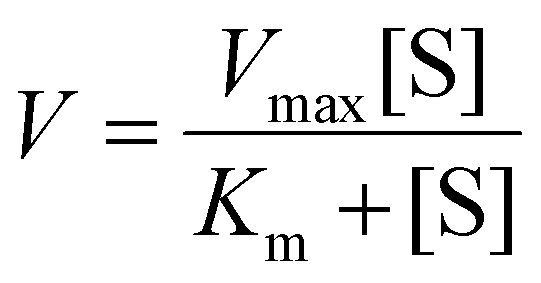 | (1) |
The H2O2 generated by ascorbic acid was measured using an Amplex Red assay. In brief, Amplex Red (50 μM), HRP (0.5 mU), ascorbic acid (25 mM), and FBS (5%) were mixed in reaction buffers in a 96-well plate. The reaction buffer was either 0.2 M acetate buffer at designated pH or PBS (pH 7.2). In the control group, Amplex Red (50 μM), HRP (0.5 mU) and H2O2 were mixed in corresponding reaction buffers. The fluorescence signal (excitation/emission = 530/590 nm) generated during the reaction was measured with the microplate reader. The fluorescence intensity was corrected according to the pH dependency of Amplex Red (ESI Fig. S1†).
Cytotoxicity of WNPs and ascorbic acid
Mouse 4T1 breast adenocarcinoma cells and mouse RAW 264.7 cells were purchased from ATCC. All cells were cultured according to the standard protocols provided by the distributors. RAW 264.7 cells were used as a model macrophage in this study.The synergistic effect of WNPs and ascorbic acid was examined by incubating 4T1 cells and RAW 264.7 cells with various combinations of WNPs and ascorbic acid and subsequently, evaluating the cell viability. The cells were seeded in 96-well plates and incubated for 4 hours. The cells were divided into three groups: (i) the cells were incubated with ascorbic acid alone at designated concentrations for 2 hours; (ii) the cells were incubated with WNPs (50 μg Fe mL−1) for 24 hours, washed with PBS to remove the WNPs in the medium, and incubated with ascorbic acid for 2 hours; (iii) the cells were incubated with the medium containing both WNPs and ascorbic acid for 2 hours. After that, all groups of cells were washed with PBS and incubated in fresh medium for another 24 hours. At the end of incubation, cell viability was evaluated with the MTT assay.
Cellular uptake of WNPs and MNPs
1.25 × 106 RAW 264.7 cells were seeded per well in 6-well plate overnight before they were incubated with the fresh medium containing wüstite nanoparticles (50 μg Fe mL−1) and magnetite nanoparticles (50 μg Fe mL−1) for 4 hours, respectively. After incubation, the cells were washed twice with PBS to remove free nanoparticles. Next, the cells were collected and the IONPs inside the cells was measured using a modified ferrozine method.34To visualize the intracellular distribution of internalized IONPs, RAW 264.7 cells were seeded in a 4-well chambered coverglass (2.0 × 105 cells per well) (Nunc™ Lab-Tek™ II, ThermoFisher) and incubated with IONPs labelled with DiI or DiD (50 μg Fe mL−1) for 24 hours. After incubation, the cells were washed with PBS twice to remove free IONPs and counterstained with Hoechst33342, and ActinGreen 488 ReadyProbes. Fluorescence images were acquired with an inverted fluorescence microscope (Nikon Eclipse Ti2) connected to a CCD camera.
Detection of intracellular hydroxyl free radical
RAW 264.7 cells were seeded in a 4-well chambered coverglass and incubated with nanoparticles as mentioned above. After that, cells were washed with PBS twice and incubated with fresh medium containing ascorbic acid (4 mM) for 2 hours. The intracellular hydroxyl free radicals were detected by incubation with fresh medium containing CM-H2DCFDA (10 μM) for 1 hour. The cells were imaged with the fluorescence microscope. At the end, the cells were detached, and the fluorescence was quantified using a BD FACSymphony cell analyser.Analysis of RAW 264.7 cell polarization
Polarization of RAW 264.7 cells toward pro- or anti-inflammatory phenotypes was analyzed using RT-qPCR. In brief, macrophages were treated with ascorbic acid and IONPs. 24 hours later, the cells were harvested, and the total RNA was extracted using a Quick-RNA MiniPrep Kit (Zymo Research) and was reverse-transcribed to cDNA using the PrimeScript™ RT Master Mix (Takara Bio). RT-qPCR was performed using TB Green Premix Ex Taq II (Tli RNase H Plus) (Takara Bio) with a Biorad CFX96 Real-Time System. PCR primers were ordered from Eurofins (ESI Table S1†). The 2−ΔΔCt method was used to calculate relative gene expression.35Polarization of RAW 264.7 cells was further evaluated based on the expression of CD86 on the cell surface. Briefly, the cells were seeded in chambered coverglass and treated with ascorbic acid and IONPs. The cells were fixed with 4% paraformaldehyde. After blocked with 1% BSA for 30 minutes, the cells were incubated with CD86 antibody (2 μg mL−1) at 4 °C overnight, followed by incubation with secondary AlexaFluor 555 goat anti-rat IgG antibody (10 μg mL−1) at ambient temperature for 1 hour. Then the cells were counterstained with Hoechst33342 and imaged with a fluorescence microscope.
Transwell study of RAW 264.7 cell released cytokines
The transwell assay was utilized to assess the impact of the cytokines released by RAW 264.7 cells on 4T1 cells. Briefly, 4T1 cells were seeded in the upper chamber and RAW 264.7 cells containing IONPs were seeded in the lower chamber of the transwell. The cells were cultured in DMEM medium supplemented with 10% FBS overnight. After that, the cells were incubated with the medium containing 4 mM ascorbic acid for 24 hours. The viability of 4T1 cells in the upper chamber were measured with a CCK8 assay (Dojindo Lab).36Magnetically targeted RAW 264.7 cell therapy
Magnetic targeting and macrophage-mediated damage to cancer cells were investigated under a physiologically relevant flow condition using a flow channel (μ-Slide VI, ibidi). First, 4T1 cells labelled with DiI were seeded in the flow channel at a density of 2 × 104 cells per channel and incubated for 24 hours to allow cell attachment. After that, the channels were connected to a syringe pump (NE-1000, New Era Pump Systems). RAW 264.7 cells loaded with IONPs were infused at a flow rate of 273 μL min−1 for 10 minutes, equivalent to a mean velocity of 3 mm s−1 in the channel. For magnetic targeting, the medial line of the channel was aligned with the edge of a NdFeB block magnet (1/4 × 1/4 × 1′′, N52, K&J Magnetics) in a 3D printed platform. After the flow, the channel was infused with the medium containing ascorbic acid (4 mM) every 24 hours. After 48 hours of incubation, the viability of 4T1 cells were evaluated based on the fluorescence intensity of DiI.Statistics
All measurements were performed at least in triplicate. GraphPad Prism was used for statistical analysis. Data were analysed using one-tailed Student's t-tests or one-way ANOVA with post-hoc Tukey tests. p < 0.05 was considered statistically significant. In all figures, ns denotes not significant, *, p < 0.05, **, p < 0.01, ***, p < 0.001, and ****, p < 0.0001.Results and discussion
IONP-catalysed Fenton reaction
Iron oxide nanocrystals of various sizes and compositions were synthesized by thermodecomposition of iron acetylacetonate. TEM and XRD analyses showed the two types of nanocrystals were 8 nm wüstite and 15 nm magnetite nanocrystals, respectively (Fig. 1a and b and ESI Fig. S2†). It is noteworthy that the wüstite nanocrystals also contained magnetite phase due to surface oxidation and disproportionation at the room temperature. Water-dispersible WNPs and MNPs were synthesized by encapsulating the respective nanocrystals with copolymers of phospholipid and poly (ethylene glycol), as outlined in the method section. Gel electrophoresis confirmed the near-complete conjugation of TAT peptide onto IONPs (ESI Fig. S3†). The key properties of WNPS and MNPs are listed in ESI Table S2.†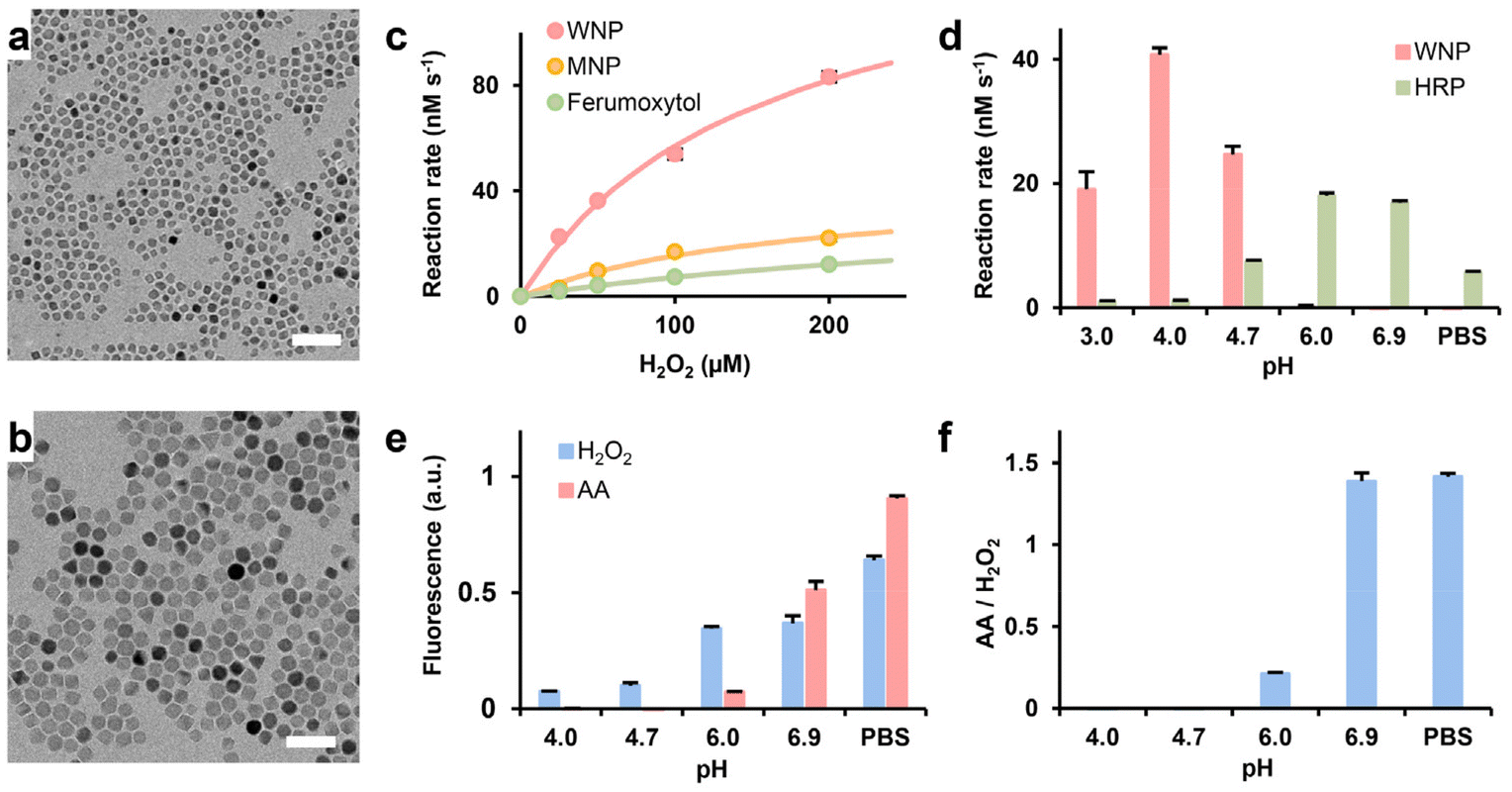 | ||
| Fig. 1 Characterization of IONP-mediated Fenton reaction and ascorbic acid-induced hydrogen peroxide generation. (a) and (b) Representative TEM images of wüstite and magnetite nanocrystals. Scale bars: 50 nm. (c) The catalytic activity of WNPs, MNPs, and ferumoxytol. The data were fitted with the Michaelis–Menten equation. (d) pH dependencies of WNP and HRP catalysis. (e) pH-dependency of ascorbic acid-mediated H2O2 generation. (f) The ratio of the fluorescence intensity shown in (e) data represent mean ± standard deviation (n = 3). AA: ascorbic acid. | ||
The catalytic efficiency of WNPs was evaluated under varying concentrations of H2O2 within a pharmacologically relevant range (0–200 μM) utilizing a TMB assay. The measurements shown in Fig. 1c and ESI Table S3† demonstrated that the catalytic efficiency of WNPs significantly surpassed that of MNPs and ferumoxytol, aligning with prior research.27 An inherent characteristic of the classical Fenton reaction is its pH sensitivity.37 Thus, we examined the pH dependency of WNP-catalysed heterogenous Fenton reaction using the TMB assay. Since most colorimetric assays are sensitive to alterations in salt compositions and pH values, we employed HRP as a reference to validate the TMB assay in buffers of different compositions and pH values. Our study found that WNPs exhibited optimal catalytic activity at pH 4, while no detectable activities at pH levels exceeding 6 (Fig. 1d). Conversely, we observed that WNPs consumed H2O2 in neutral pH buffers (ESI Fig. S4†). This finding concurs with prior report that both magnetite and maghemite nanoparticles mimic peroxidase behaviour in acidic buffers, whereas they catalyse the decomposition of H2O2 into H2O and O2 at neutral pH, akin to a catalase.38
pH dependency of ascorbic acid-mediated H2O2 generation
Chen et al. previously demonstrated that ascorbic acid could generate H2O2 in a protein-dependent manner in various biological fluid.15 In this study, fetal bovine serum (FBS) served as the protein source. Intriguingly, directly mixing ascorbic acid, WNPs, and FBS in acetate buffers spanning pH 3 to 7 failed to generate H2O2 detectable by the TMB assay. Subsequent investigation unveiled that ascorbic acid could bleach oxTMB, the chromogen generated in the TMB assay. Using a pH-calibrated fluorogenic Amplex Red assay, we observed that ascorbic acid-mediated H2O2 generation exhibited optimal efficiency at neutral pH (Fig. 1e and f). After 10 minutes of reaction, the fluorescence signal generated by 25 mM ascorbic acid was equivalent to that of 1.5 μM of H2O2. However, H2O2 generation decreased significantly at pH 6 and became undetectable at lower pH levels.Interaction between WNPs and ascorbic acid in cell culture
The disparity in pH-dependent behaviours between WNPs and ascorbic acid prompted our investigation into whether these agents might synergistically induce cytotoxic effects on cancer cells. The cells were incubated with ascorbic acid and WNPs in different sequences (see Methods). In the first scenario, ascorbic acid alone demonstrated dose-dependent toxicity to both 4T1 and RAW 264.7 cells, with 4T1 cells showing slightly higher sensitivity compared to RAW 264.7 cells (Fig. 2). In the second scenario, when ascorbic acid was introduced to cultured cells, only intracellular WNPs were present. Conversely, in the last scenario, most WNPs were present extracellularly in the medium. Our study revealed that intracellular WNPs significantly amplified the cytotoxicity of ascorbic acid. However, when WNPs and ascorbic acid coexisted in the medium, WNPs completely nullified the cytotoxic effects of ascorbic acid across all tested concentrations. MNPs and ascorbic acid exhibit similar interactions in cultured cells (ESI Fig. S5†).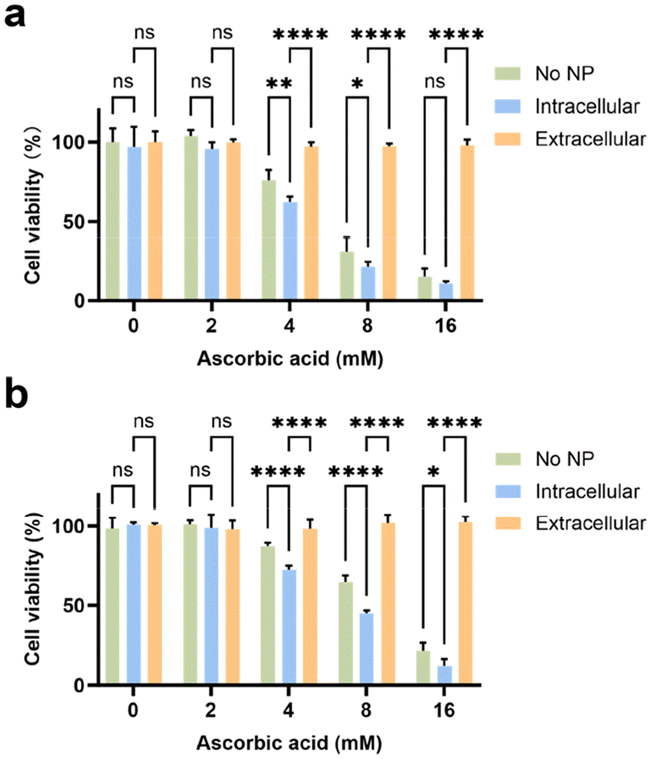 | ||
| Fig. 2 Cytotoxicity of ascorbic acid and WNPs. 4T1 (a) and RAW 264.7 cells (b) were treated with ascorbic acid alone or in the presence of intracellular or extracellular WNPs. Data represent mean ± standard deviation (n = 4). | ||
Previous studies have established that nanoparticles encapsulated in phospholipid–PEG coating are internalized by cells through endocytic pathways, accumulating in endosomes and lysosomes with pH levels ranging from 4 to 5.39 Taken together, when only intracellular WNPs are present, ascorbic acid can generate H2O2 in the medium, which then permeates cells and encounters WNPs accumulated in the acidic cell organelles. Under optimal pH conditions, WNPs efficiently convert H2O2 into toxic hydroxyl free radicals. Conversely, when WNPs are mixed with ascorbic acid in a neutral pH medium, they act as catalase, converting H2O2 into H2O and O2. This discovery underscores the significance of coordinating IONPs and ascorbic acid in cancer treatment. It's plausible that patients undergoing IONP-based tumour detection may not respond to ascorbic acid treatment unless extracellular IONPs in the tumour are cleared via cellular uptake or degradation.
IONP and ascorbic acid-induced RAW 264.7 cell polarization
Given the tendency of systemically administered IONPs to distribute to the extracellular space, potentially diminishing the therapeutic efficacy of ascorbic acid, we investigated the possibility of utilizing IONPs to engineer cell therapy alongside ascorbic acid treatment. In conventional cell therapy, effector cells are modified ex vivo before being administered into the body. An emerging approach involves the ex vivo engineering of macrophages.40,41 Macrophages, being highly plastic cells, can be polarized to a proinflammatory (M1) phenotype that responds to the chemoattractants released from cells undergoing viral infection or various types of irreversible damage, including cancer cells.42,43 On the other hand, macrophages are prominent constituents in most solid tumours, often exhibiting an anti-inflammatory (M2) phenotype within the tumour microenvironment due to intricate crosstalk with cancer and stromal cells. Macrophage-based cell therapy, therefore, relies on engineered macrophages’ ability to target tumour tissue and sustain the M1 phenotype despite the immune-suppressive tumour microenvironment. Previous studies have demonstrated that IONPs could induce the polarization of macrophages towards an M1 phenotype, likely through the production of free radicals.44,45 Hence, we hypothesize that when IONP-loaded macrophages encounter ascorbic acid within the tumour, intracellular IONPs may facilitate the generation of hydroxyl free radicals, subsequently promoting M1 polarization in host macrophages.As a proof of concept, RAW 264.7 cells, a type of immortalized mouse macrophages, were utilized as the model system for macrophages due to their retention of key features resembling native macrophages, including polarization, cytokine release, and cytotoxicity in response to classical stimulants. Fig. 1c illustrates that WMPs exhibit higher catalytic activity compared to MNPs, while magnetite demonstrates significantly higher magnetization than wüstite.28 We explored the potential of RAW 264.7 cells to be simultaneously loaded with WMPs and MNPs, thus enabling both free radical generation and magnetic targeting. We discovered that the uptake of WNPs and MNPs could be augmented through conjugation with a cell-penetrating peptide and facilitated by magnetic field-assisted cellular uptake. With both measures combined, each cell could uptake approximately 4 pg of Fe from IONPs (Fig. 3a). Notably, flow cytometry analysis revealed that nearly all cells contained both WNPs and MNPs (Fig. 3b). Intracellular WNPs and MNPs co-localized within the vesicles scattered around the cell nuclei (Fig. 3c and ESI Fig. S6†). Our previous study showed that DSPE-PEG-coated nanoparticles entered the cells via the endocytic pathway and accumulated in the lysosomes.27,39 The lysosomes provide an acidic environment that favours the IONP-mediated Fenton reaction.
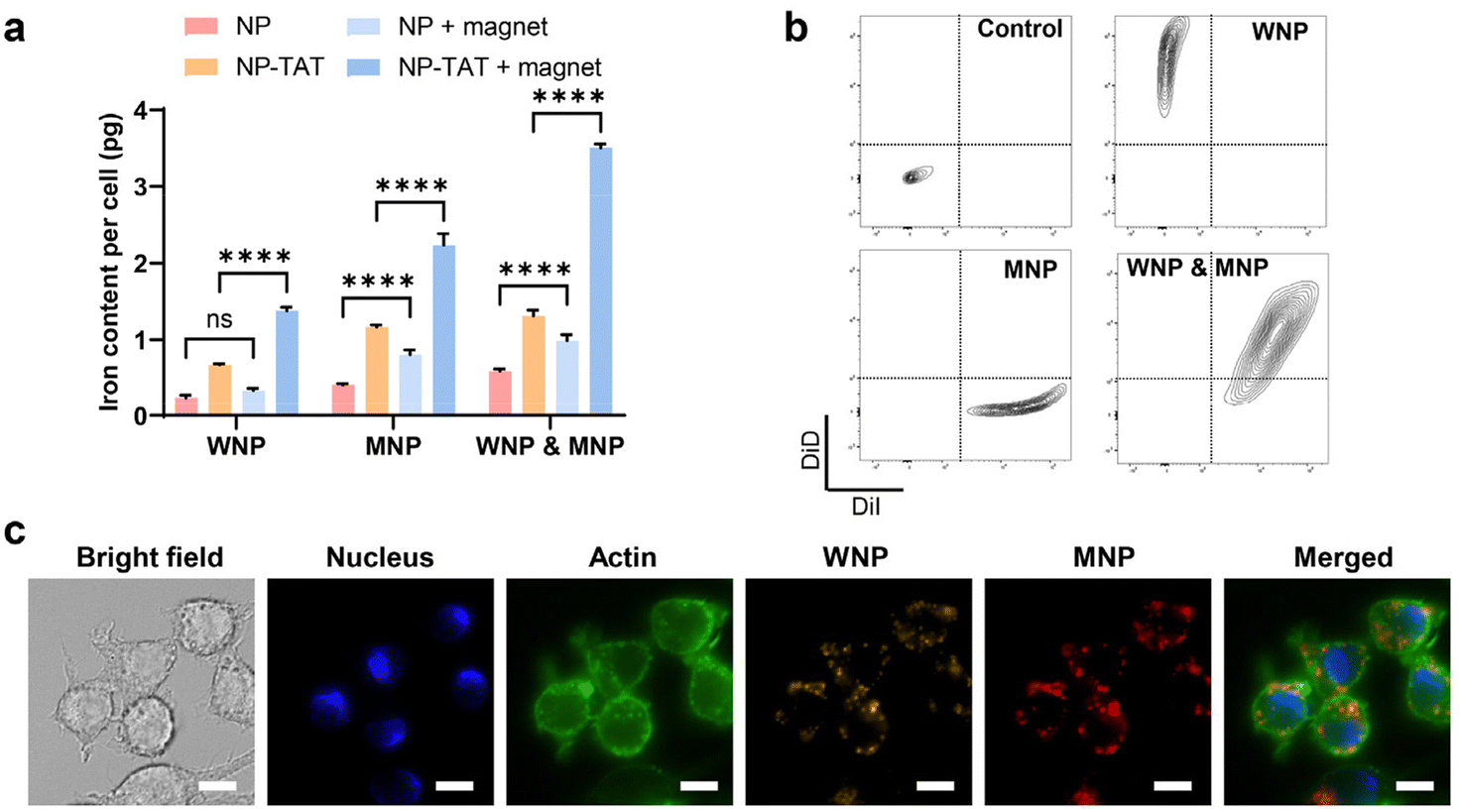 | ||
| Fig. 3 Enhanced cellular uptake of IONPs. RAW 264.7 cells were incubated with WNPs and MNPs for 4 hours under designated conditions. (a) Intracellular IONPs quantified by the Ferrozine assay. (b) Evaluation of IONPs uptake with flow cytometry. (c) Intracellular distribution of IONPs. Scale bar: 10 μm. Data represent mean ± standard deviation (n = 3). | ||
The RAW 264.7 cells underwent further treatment with ascorbic acid at a concentration achievable in vivo within the tumour microenvironment.3 While ascorbic acid alone was capable of generating detectable hydroxyl free radicals, their quantity notably increased in the presence of intracellular IONPs (Fig. 4a and b). Cells containing WNPs exhibited significantly higher levels of free radicals compared to those with ferumoxytol or MNPs, aligning with the catalytic activity of IONPs depicted in Fig. 1c. Moreover, cells harbouring both WNPs and MNPs displayed the highest amount of free radicals, as both contributed to their generation.
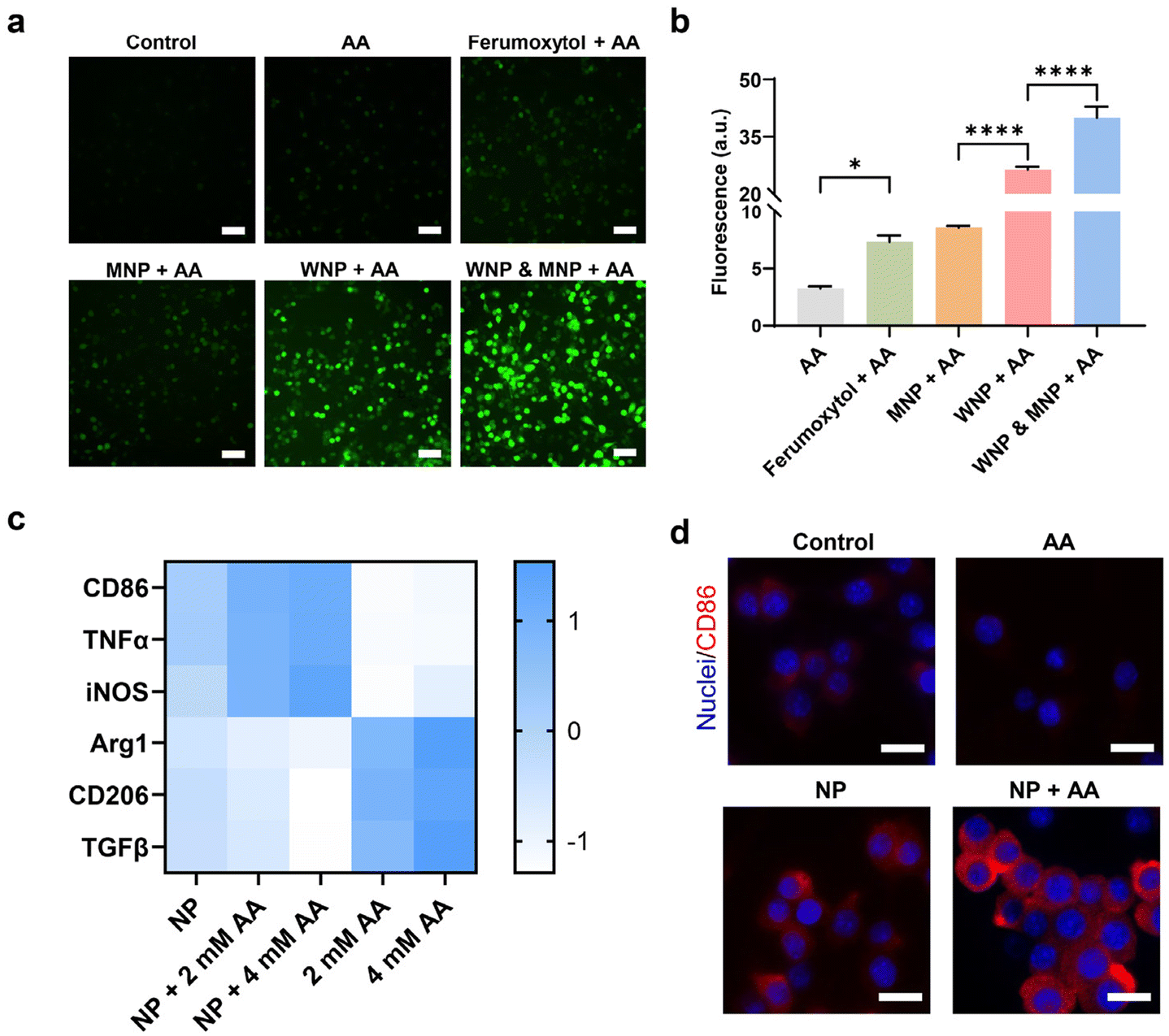 | ||
| Fig. 4 IONPs and ascorbic acid-induced macrophage polarization. RAW 264.7 cells with intracellular IONPs were treated with ascorbic acid (4 mM). (a) and (b) Intracellular hydroxyl free radicals were detected with fluorescence microscopy and flow cytometry. Scale bar: 20 μm. (c) Gene expression in RAW 264.7 cells was evaluated with RT-qPCR. (d) Expression of CD86, an M1 macrophage marker, on the surface of RAW 264.7 cells. Scale bar: 10 μm. Data represent mean ± standard deviation (n = 4). | ||
We investigated the effects of intracellular IONPs and ascorbic acid on the polarization of RAW 264.7 cells, utilizing two sets of gene markers (Fig. 4c). Upregulation of CD86, TNF-α, and iNOS indicates polarization toward the M1 phenotype, while upregulation of Arg1, CD206, and TGF-β is associated with the M2 phenotype.42 Compared to untreated RAW 264.7 cells (M0), those treated with IONPs alone displayed a moderate shift toward the M1 phenotype. The M1 polarization became more following ascorbic acid treatment. Conversely, ascorbic acid alone prompted RAW 264.7 cells to polarize toward the M2 phenotype. The polarization of RAW 264.7 toward the M1 phenotype was confirmed by the upregulation of CD86 on the cell surface (Fig. 4d, ESI Fig. S7 and S8†).
RAW 264.7 cell-based cell therapy
We investigated the effects of RAW 264.7 cells containing IONPs on 4T1 cells, particularly in response to ascorbic acid exposure, which polarized these cells towards the M1 phenotype. The cytotoxicity effect was evaluated utilizing both a transwell and a flow channel system. In the transwell configuration, the porous membrane (0.4 μm pore size) facilitated cytokine exchange while keeping the cell types separate. 4T1 cells and RAW 264.7 cells, with or without IONPs, were respectively seeded into the upper and lower chambers (Fig. 5a). Our findings indicated that untreated RAW 264.7 cells had minimal impact on 4T1 cell viability, whereas those with IONPs decreased 4T1 cell viability by approximately 20% (Fig. 5b). RAW 264.7 cells preloaded with IONPs significantly enhanced the cytotoxicity of ascorbic acid. Moreover, RAW 264.7 cells preloaded with IONPs displayed enhanced polarization towards the M1 phenotype after coculture with 4T1 cells for 4 days (Fig. 5c).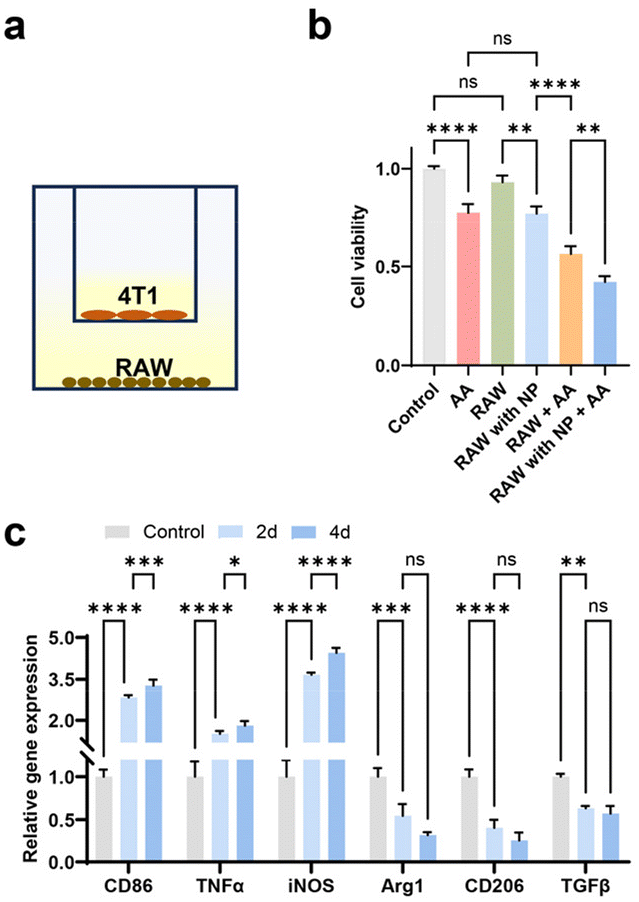 | ||
| Fig. 5 Transwell study of the interaction between RAW 264.7 and 4T1 cells. (a) Schematic diagram of cell seeding in the transwells. (b) Viability of 4T1 cells. (c) Gene expression of RAW 264.7 cells after 2 and 4 days of coculture with 4T1. Data represent mean ± standard deviation (n = 4). | ||
In assessing the potential of RAW 264.7 cells loaded with WNPs and MNPs for tumour targeting via magnetic guidance, we investigated their synergy with ascorbic acid treatment using a flow channel system. Here, 4T1 cells were seeded within the channel, while RAW 264.7 cells were infused at a rate mimicking postcapillary venues (Fig. 6a). After 10 minutes of infusion, minimal RAW 264.7 cell attachment to 4T1 cells was observed, but magnetic targeting with an NdFeB magnet notably increased cell retention within the channel (Fig. 6b). Subsequent analysis revealed that ascorbic acid alone exhibited moderate cytotoxicity towards 4T1 cells. However, the combination of RAW 264.7 cells and ascorbic acid resulted in the death of over 80% of 4T1 cells, demonstrating a promising synergistic effect (Fig. 6c and d). Previous study has shown that systemic administration of ascorbic acid can preferentially elevate H2O2 levels within the tumour microenvironment.16 Combining magnetically targeted macrophages with ascorbic acid offers a dual targeting approach, mitigating potential adverse effects on normal tissue caused by engineered macrophages. Significantly, this strategy of loading macrophages with IONPs, as opposed to genetically modifying them, represents an effortless and swift cell engineering method, facilitating its practical application in engineering patient-derived macrophages.
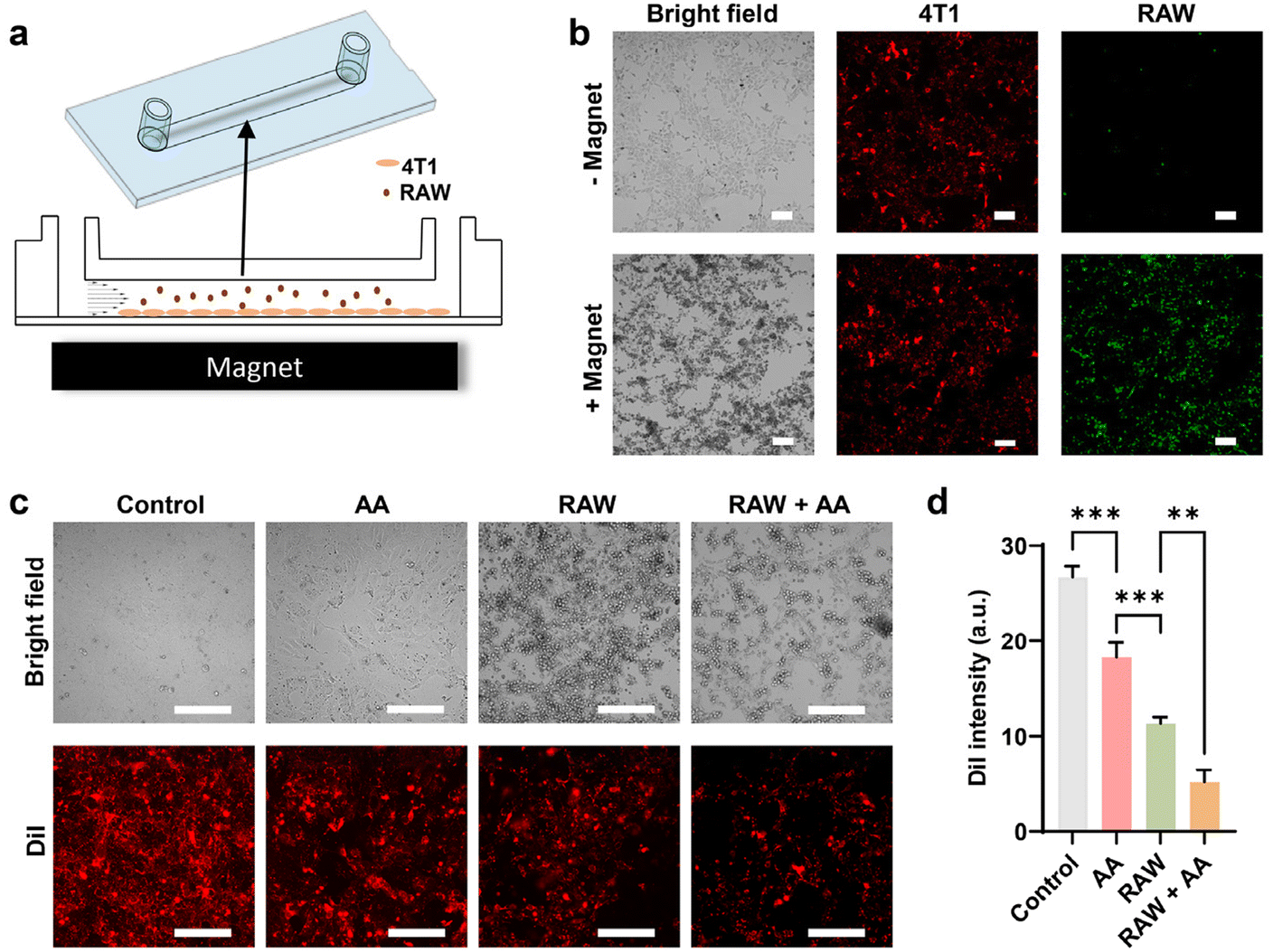 | ||
| Fig. 6 Macrophage-based cell therapy. The potency of macrophage-based cell therapy was evaluated in vitro in a flow channel. (a) Schematic diagram of the flow channel study. RAW 264.7 cells containing IONPs were infused into a flow channel seeded with 4T1 cells. The channel was placed on top of a magnet for magnetic targeting. 4T1 and RAW 264.7 cells were labelled with DiI and DiO respectively for visualization. (b) Representative cell images with and without magnetic targeting. Scale bars: 60 μm. (c) Representative cell images after the treatment with ascorbic acid. Scale bars: 60 μm. (d) Viability of 4T1 cells measured by fluorescence of cell labelling. Data represent mean ± standard deviation (n = 4). | ||
Conclusions
In summary, our investigation illuminates the distinct pH dependency of two promising reagents in cancer therapy. While IONPs exhibit efficacy as nanozymes, effectively curtailing tumour growth through the generation of free radicals, their synergistic utilization with other chemotherapeutic agents necessitates meticulous coordination in accordance with their respective mechanisms of action. Moreover, our study underscores the potential of macrophages engineered with IONPs as potent cellular adjuncts to ascorbic acid in cancer treatment. Here, WNPs and MNPs play pivotal roles in catalysis and magnetic targeting, respectively. This discovery prompts further exploration utilizing native macrophages and in vivo models, charting a path towards an innovative combination therapy.Author contributions
Conceptualization: ZY and ST; methodology: ZY, XY, and ST; investigation: ZY and XY; resources: ST; writing—original draft preparation: ZY; writing—review and editing: XY, YL and ST. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly supported by the NIH/NIBIB funding (R01EB026893 to ST) and by the NIAMS funding (R21AR078447 to ST). The authors would like to thank the U.K. Electron Microscopy Center supported by the National Science Foundation (NNCI-2025075) and the U.K. Flow Cytometry & Immune Monitoring core facility supported by an NCI Center Core Support Grant (P30 CA177558).References
- M. Levine and P.-C. Violet, Cancer Cell, 2017, 31, 467–469 CrossRef CAS PubMed.
- Y. C. Boo, Antioxidants, 2022, 11, 1663 CrossRef CAS.
- Q. Chen, M. G. Espey, A. Y. Sun, J. H. Lee, M. C. Krishna, E. Shacter, P. L. Choyke, C. Pooput, K. L. Kirk, G. R. Buettner and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8749–8754 CrossRef CAS.
- E. Cameron and L. Pauling, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4538–4542 CrossRef CAS.
- E. Cameron and A. Campbell, Chem.-Biol. Interact., 1974, 9, 285–315 CrossRef CAS.
- Y. Ma, J. Chapman, M. Levine, K. Polireddy, J. Drisko and Q. Chen, Sci. Transl. Med., 2014, 6, 222ra218 Search PubMed.
- E. Ma, P. Chen, H. M. Wilkins, T. Wang, R. H. Swerdlow and Q. Chen, Free Radical Biol. Med., 2017, 113, 36–47 CrossRef CAS PubMed.
- C. M. Stephenson, R. D. Levin, T. Spector and C. G. Lis, Cancer Chemother. Pharmacol., 2013, 72, 139–146 CrossRef CAS.
- M. G. Espey, P. Chen, B. Chalmers, J. Drisko, A. Y. Sun, M. Levine and Q. Chen, Free Radical Biol. Med., 2011, 50, 1610–1619 CrossRef CAS PubMed.
- J. D. Schoenfeld, Z. A. Sibenaller, K. A. Mapuskar, B. A. Wagner, K. L. Cramer-Morales, M. Furqan, S. Sandhu, T. L. Carlisle, M. C. Smith, T. Abu Hejleh, D. J. Berg, J. Zhang, J. Keech, K. R. Parekh, S. Bhatia, V. Monga, K. L. Bodeker, L. Ahmann, S. Vollstedt, H. Brown, E. P. Shanahan Kauffman, M. E. Schall, R. J. Hohl, G. H. Clamon, J. D. Greenlee, M. A. Howard, M. K. Schultz, B. J. Smith, D. P. Riley, F. E. Domann, J. J. Cullen, G. R. Buettner, J. M. Buatti, D. R. Spitz and B. G. Allen, Cancer Cell, 2017, 31, 487–500 CrossRef CAS.
- Y. An, J. Zhu, F. Liu, J. Deng, X. Meng, G. Liu, H. Wu, A. Fan, Z. Wang and Y. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 29655–29666 CrossRef CAS.
- L. N. Cossey, F. Rahim and C. P. Larsen, Am. J. Kidney Dis., 2013, 61, 1032–1035 CrossRef PubMed.
- S. J. Padayatty and M. Levine, Oral Dis., 2016, 22, 463–493 CrossRef CAS PubMed.
- S. J. Padayatty, H. Sun, Y. Wang, H. D. Riordan, S. M. Hewitt, A. Katz, R. A. Wesley and M. Levine, Ann. Intern. Med., 2004, 140, 533–537 CrossRef CAS.
- Q. Chen, M. G. Espey, M. C. Krishna, J. B. Mitchell, C. P. Corpe, G. R. Buettner, E. Shacter and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13604–13609 CrossRef CAS.
- Q. Chen, M. G. Espey, A. Y. Sun, C. Pooput, K. L. Kirk, M. C. Krishna, D. B. Khosh, J. Drisko and M. Levine, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11105–11109 CrossRef CAS.
- M. S. Petronek, N. Teferi, J. M. Caster, J. M. Stolwijk, A. Zaher, J. M. Buatti, D. Hasan, E. I. Wafa, A. K. Salem, E. G. Gillan, J. J. St-Aubin, G. R. Buettner, D. R. Spitz, V. A. Magnotta and B. G. Allen, Redox Biol., 2023, 62, 102651 CrossRef CAS.
- J. He, X. Yang, B. Men and D. Wang, J. Environ. Sci., 2016, 39, 97–109 CrossRef CAS.
- L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett and X. Yan, Nat. Nanotechnol., 2007, 2, 577–583 CrossRef CAS.
- Y. Chen, C. J. Miller and T. D. Waite, Environ. Sci. Technol., 2021, 55, 14414–14425 CrossRef CAS PubMed.
- S. Tong, H. Zhu and G. Bao, Mater. Today, 2019, 31, 86–99 CrossRef CAS.
- F. Zhang, F. Li, G.-H. Lu, W. Nie, L. Zhang, Y. Lv, W. Bao, X. Gao, W. Wei, K. Pu and H.-Y. Xie, ACS Nano, 2019, 13, 5662–5673 CrossRef CAS PubMed.
- S. Zanganeh, G. Hutter, R. Spitler, O. Lenkov, M. Mahmoudi, A. Shaw, J. S. Pajarinen, H. Nejadnik, S. Goodman, M. Moseley, L. M. Coussens and H. E. Daldrup-Link, Nat. Nanotechnol., 2016, 11, 986–994 CrossRef CAS.
- Z. Shen, T. Liu, Y. Li, J. Lau, Z. Yang, W. Fan, Z. Zhou, C. Shi, C. Ke, V. I. Bregadze, S. K. Mandal, Y. Liu, Z. Li, T. Xue, G. Zhu, J. Munasinghe, G. Niu, A. Wu and X. Chen, ACS Nano, 2018, 12, 11355–11365 CrossRef CAS PubMed.
- B. Yu, B. Choi, W. Li and D. H. Kim, Nat. Commun., 2020, 11, 3637 CrossRef CAS.
- H. Hou, X. Huang, G. Wei, F. Xu, Y. Wang and S. Zhou, ACS Appl. Mater. Interfaces, 2019, 11, 29579–29592 CrossRef CAS.
- Z. Yi, X. Yang, Y. Liang, F. Chapelin and S. Tong, Small, 2024, 20, e2305974 CrossRef.
- Y. Hou, Z. Xu and S. Sun, Angew. Chem., Int. Ed., 2007, 46, 6329–6332 CrossRef CAS PubMed.
- S. Tong, C. A. Quinto, L. Zhang, P. Mohindra and G. Bao, ACS Nano, 2017, 11, 6808–6816 CrossRef CAS.
- S. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. Li, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS.
- S. Tong, S. Hou, B. Ren, Z. Zheng and G. Bao, Nano Lett., 2011, 11, 3720–3726 CrossRef CAS PubMed.
- L. Zhang, S. Tong, Q. Zhang and G. Bao, ACS Appl. Nano Mater., 2020, 3, 6785–6797 CrossRef CAS.
- B. Jiang, D. Duan, L. Gao, M. Zhou, K. Fan, Y. Tang, J. Xi, Y. Bi, Z. Tong, G. F. Gao, N. Xie, A. Tang, G. Nie, M. Liang and X. Yan, Nat. Protoc., 2018, 13, 1506–1520 CrossRef CAS.
- N. Landazuri, S. Tong, J. Suo, G. Joseph, D. Weiss, D. J. Sutcliffe, D. P. Giddens, G. Bao and W. R. Taylor, Small, 2013, 9, 4017–4026 CrossRef CAS.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- D. J. Son, M. H. Park, S. J. Chae, S. O. Moon, J. W. Lee, H. S. Song, D. C. Moon, S. S. Kang, Y. E. Kwon and J. T. Hong, Mol. Cancer Ther., 2007, 6, 675–683 CrossRef CAS.
- M. L. Kremer, J. Phys. Chem. A, 2003, 107, 1734–1741 CrossRef CAS.
- Z. Chen, J. J. Yin, Y. T. Zhou, Y. Zhang, L. Song, M. Song, S. Hu and N. Gu, ACS Nano, 2012, 6, 4001–4012 CrossRef CAS PubMed.
- L. Zhang, S. Hajebrahimi, S. Tong, X. Gao, H. Cheng, Q. Zhang, D. T. Hinojosa, K. Jiang, L. Hong, J. Huard and G. Bao, ACS Appl. Mater. Interfaces, 2023, 15, 50574–50585 CrossRef CAS.
- C. X. Li, Y. Zhang, X. Dong, L. Zhang, M. D. Liu, B. Li, M. K. Zhang, J. Feng and X. Z. Zhang, Adv. Mater., 2019, 31, 1807211 CrossRef PubMed.
- Y. Xia, L. Rao, H. Yao, Z. Wang, P. Ning and X. Chen, Adv. Mater., 2020, 32, 2002054 CrossRef CAS.
- P. J. Murray, J. E. Allen, S. K. Biswas, E. A. Fisher, D. W. Gilroy, S. Goerdt, S. Gordon, J. A. Hamilton, L. B. Ivashkiv, T. Lawrence, M. Locati, A. Mantovani, F. O. Martinez, J. L. Mege, D. M. Mosser, G. Natoli, J. P. Saeij, J. L. Schultze, K. A. Shirey, A. Sica, J. Suttles, I. Udalova, J. A. van Ginderachter, S. N. Vogel and T. A. Wynn, Immunity, 2014, 41, 14–20 CrossRef CAS PubMed.
- T. A. Wynn, A. Chawla and J. W. Pollard, Nature, 2013, 496, 445–455 CrossRef CAS.
- Z. Gu, T. Liu, J. Tang, Y. Yang, H. Song, Z. K. Tuong, J. Fu and C. Yu, J. Am. Chem. Soc., 2019, 141, 6122–6126 CrossRef CAS PubMed.
- V. Mulens-Arias, J. M. Rojas and D. F. Barber, Front. Immunol., 2021, 12, 2229 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr01208a |
| This journal is © The Royal Society of Chemistry 2024 |