
Photophysics and its application in photon upconversion
Yutong
Zhang
ab,
Wenna
Du
 *ab and
Xinfeng
Liu
*ab
*ab and
Xinfeng
Liu
*ab
aCAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, P. R. China. E-mail: zhangyt2021@nanoctr.cn; duwn@nanoctr.cn; liuxf@nanoctr.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 4th January 2024
Abstract
Photoluminescence (PL) upconversion is a phenomenon involving light–matter interaction, where the energy of the emitted photons is higher than that of the incident photons. PL upconversion has promising applications in optoelectronic devices, displays, photovoltaics, imaging, diagnosis and treatment. In this review, we summarize the mechanism of PL upconversion and ultrafast PL physical processes. In particular, we highlight the advances in laser cooling, biological imaging, volumetric displays and photonics.
 Yutong Zhang | Yutong Zhang received his BS degree from Nankai University in 2021. Now he is a MS student at the National Center for Nanoscience and Technology (NCNST) under the supervision of Prof. Xinfeng Liu. His research focuses on the physical mechanisms of two-dimensional perovskite electron–phonon coupling. |
 Wenna Du | Wenna Du is an associate professor at the National Center for Nanoscience and Technology (NCNST), China. She received her Ph.D. degree in materials physics and chemistry from the Institute of Semiconductors, Chinese Academy of Sciences in 2016. After that, she joined the NCNST as a research assistant professor and became an associate professor in 2019. Her research focuses on light–matter interactions of perovskites by optical spectroscopy approaches. |
 Xinfeng Liu | Prof. Xinfeng Liu graduated from Northeast Normal University in 2007, and obtained his doctoral degree from the National Nanoscience Center in 2011, and engaged in postdoctoral research at Nanyang Technological University in Singapore from 2011 to 2105. In 2015, he joined the NCNST through the Overseas Talent Program of the Chinese Academy of Sciences. In 2023, he was supported by the Outstanding Youth Program of the National Science Foundation of China. His research focuses on the study of high spatial and temporal resolution ultrafast dynamics and related physical and chemical properties. |
Introduction
Photoluminescence (PL) upconversion is a phenomenon involving light–matter interactions. In contrast to conventional Stokes emission, upconversion luminescence converts low-frequency photons into high-frequency photons. The origins of upconversion can be attributed to a number of mechanisms including triplet–triplet annihilation, higher-order harmonic generation, multiphoton absorption, Auger recombination, and phonon-assisted anti-Stokes emission. This phenomenon has been widely observed in organic dyes, nanotubes, quantum wells, quantum dots (QDs), II–VI semiconductors, rare-earth-doped materials and two-dimensional materials. Biological imaging, optical cooling in semiconductors and potential applications of upconversion photovoltaic devices are motivations to study PL upconversion.In this review, we highlight recent work on PL upconversion in semiconductor nanomaterials. A brief overview of the main mechanisms of PL upconversion is presented first. Subsequently, we highlight the ultrafast photophysical processes involved in PL upconversion and discuss how we can utilize PL upconversion for advanced applications. Finally, we discuss the main challenges and opportunities for future applications in this area.
Mechanisms of PL upconversion
During the PL upconversion process, additional energy is required to propel the electrons excited by low energy photons to the conduction or defect bands of the material.1–8 In general, this extra energy can be gained from phonon absorption,9–11 multiphoton absorption12–15 and Auger recombination.16,17 For multicomponent systems, such as lanthanide-doped nanoparticles, the upconversion mechanism is more complex and may involve excited state absorption (ESA), energy transfer upconversion (ETU), collaborative sensitization upconversion (CSU), and photon avalanche (PA).8,18–20 Considering the organic nature of quantum dot-sensitized triple–triple annihilation upconversion, this will not be discussed in this review article. Here, we discuss the three main upconversion mechanisms including phonon-assisted upconversion, multiphoton absorption, and energy transfer.Phonon-assisted upconversion
In the process of phonon-assisted PL upconversion, an incident photon excites an electron from the ground state to a virtual or real intermediate state, and absorbs one or more phonons from the lattice to the final state (Fig. 1(a)). The real intermediate state can be defect states, surface states, or electron sub-bands of the semiconductor materials.21–24Fig. 2(a) shows the observation of PL upconversion from defects in h-BN.11 The intensities of the normal downconversion and the upconversion PL at different excitation wavelengths are presented in the PLE spectrum, revealing an energy gain of up to 162 meV. In addition, chemical treatments (annealing, doping) can create or passivate defect states or surface states.25 However, the intensity of the upconversion PL will change after chemical treatments.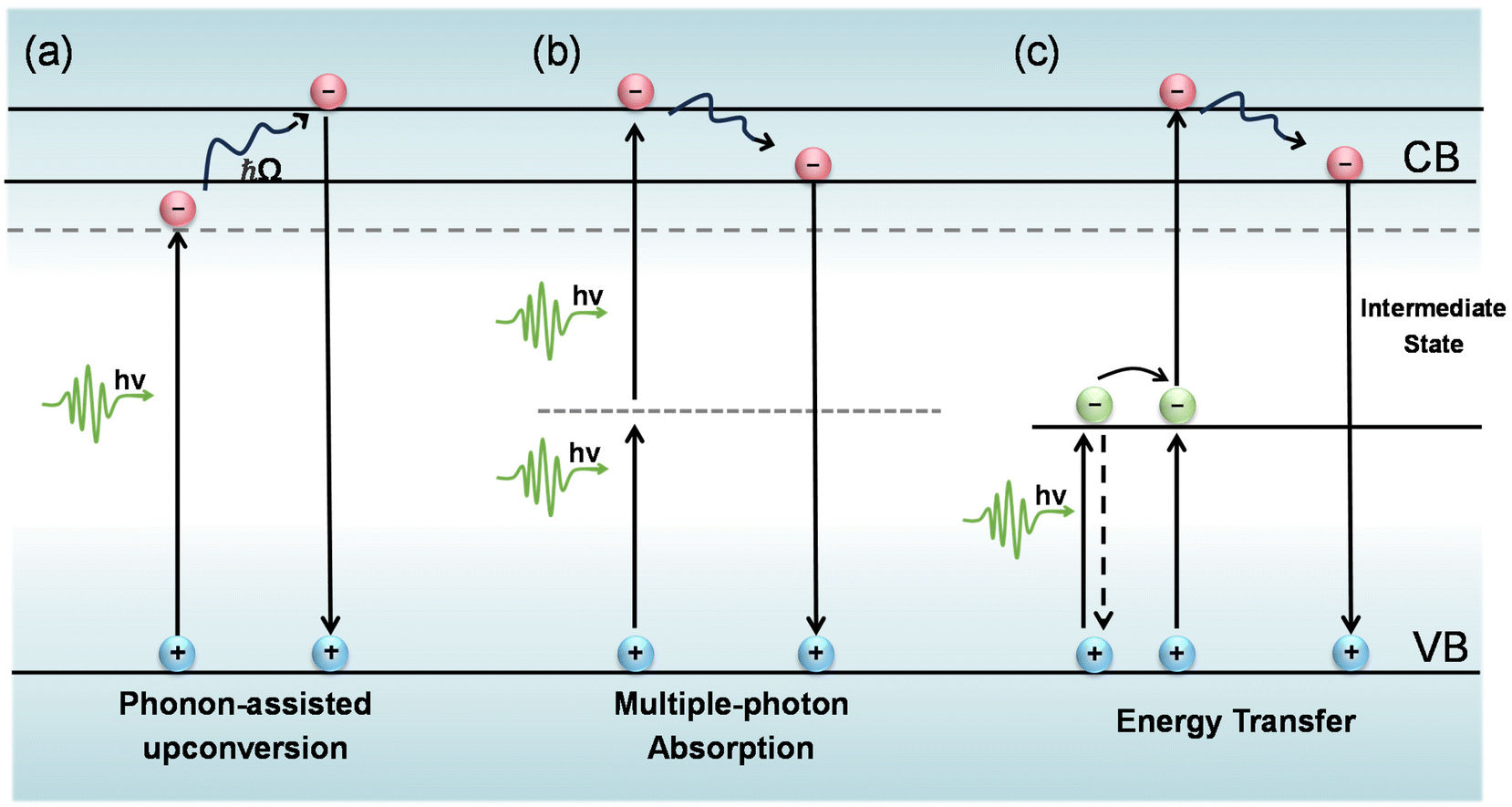 | ||
| Fig. 1 Mechanisms of PL upconversion. (a) Phonon-assisted upconversion. (b) Multiphoton absorption. (c) Energy transfer. | ||
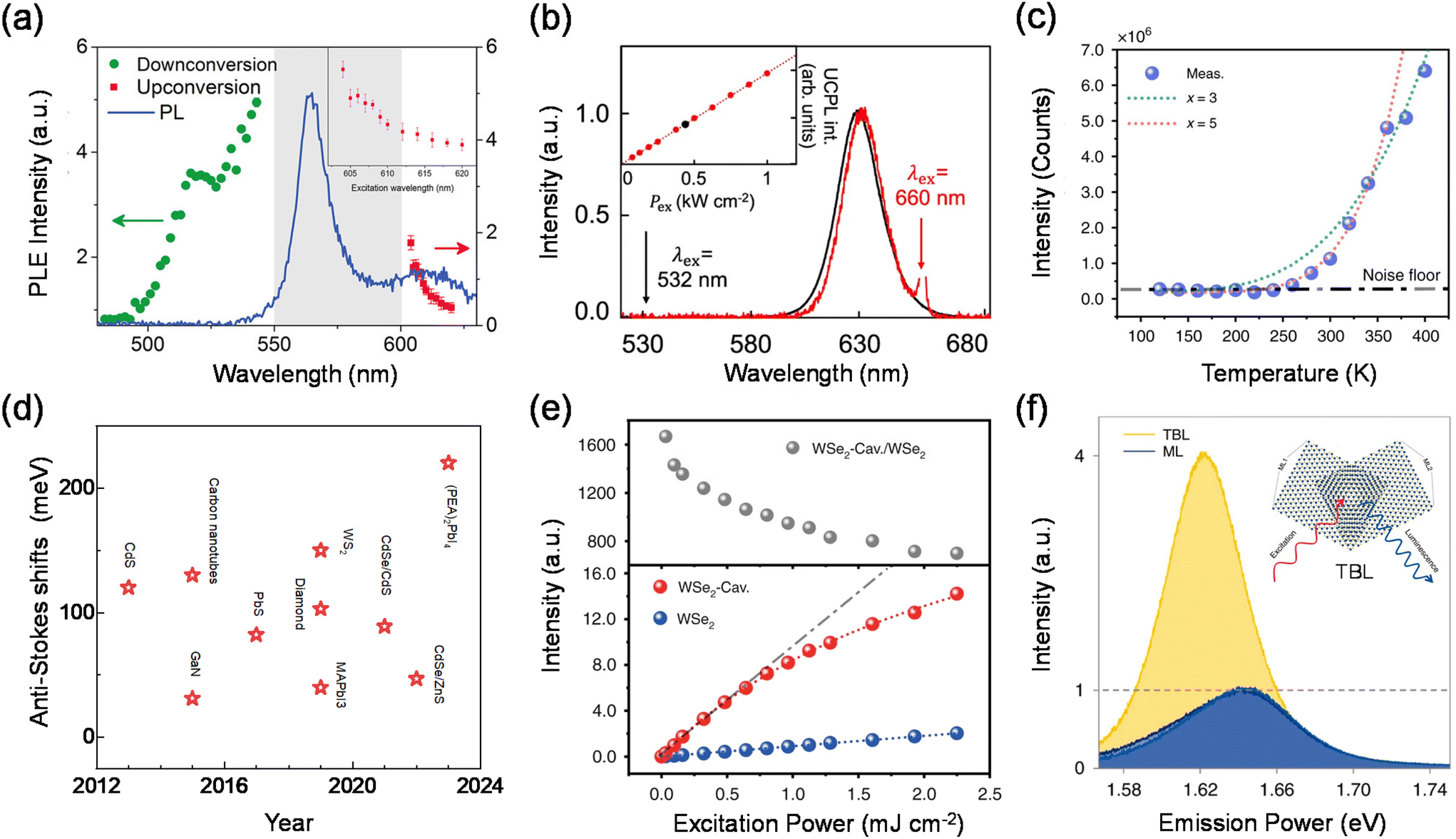 | ||
| Fig. 2 Phonon-assisted upconversion. (a) Photoluminescence excitation spectrum of h-BN. (b) Normalized UCPL and PL spectra of monodisperse CdSe/CdS core/shell quantum dots. Inset: excitation power density-(Pex) dependent upconversion PL intensity. (c) Temperature-dependent integrated upconverted emission intensity of monolayer WSe2. (d) Summary of anti-Stokes shifts for recently emerging low-dimensional upconversion materials. (e) Excitation power-dependent integrated upconverted emission intensity (down) of WSe2 and enhancement (top) in the plasmonic WSe2 cavity. (f) PL spectra of the lower WSe2 monolayer (dark blue lines), the upper WSe2 monolayer (blue lines) and the twisted WSe2 bilayer (yellow lines). Inset: schematic of the device structure. | ||
A typical characteristic of phonon-assisted PL upconversion is the linear dependence of the upconversion PL intensity on the excitation power.1,3,11,26 The relationship can be expressed by the equation I = β × Pα, where I is the integrated PL intensity, P is the laser power density, α is an exponent, and β is a constant. At low excitation power, the upconversion PL intensity increases almost linearly with increasing laser power. Fig. 2(b) demonstrates that the upconversion and downconversion PL spectra of monodisperse CdSe/CdS core/shell quantum dot samples are very similar in the FWHM and peak position. The upconversion PL intensity is linearly related to the excitation power density, suggesting that this is a single-photon upconversion process with the energy discrepancy compensated by phonons.24 However, for the phonon-assisted upconversion of defects as intermediate states, the PL intensity is sublinearly related to the pump power at low excitation power densities, while the PL saturation phenomenon is observed with the further elevation of the pump fluence due to the trap filling effect and/or multibody Auger recombination.
In addition, phonon-assisted upconversion is extremely sensitive to temperature. The intensity of the upconverted PL increases as the temperature increases, due to the fact that there are more phonons at higher temperatures. The phonon modes involved in upconversion can be extracted by using temperature-dependent PL intensity and Raman spectroscopy.1,3,11,27–29 In contrast, the intensity of the downconversion PL increases with decreasing temperature due to the decrease in non-radiative transitions.2,11Fig. 2(c) shows the temperature-dependent upconversion emission intensity of the 2D semiconductor monolayer WSe2.30 The total upconversion emission intensity declines sharply with decreasing temperature, and the detectable upconverted emission (above the noise floor) cannot be found for temperatures lower than 250 K, thereby providing undoubtable evidence for phonon-assisted excitonic upconverted emission.
There are two primary ways to enhance the efficiency of phonon-assisted upconversion. One valid way is that the selected material itself has a strong electron–phonon coupling interaction, which makes it possible to efficiently utilize phonons in upconversion. Fig. 2(d) summarizes experimental nanomaterial anti-Stokes shifts in the literature. Upon inspection, it is evident that many nanomaterial systems exhibit efficient absorption of phonons and then generate upconversion PL. Newer systems such as CdS, WS2, and hybrid/all inorganic lead halide perovskites show much more promising anti-Stokes shifts. These systems should therefore be focused on future laser cooling studies. Xiong et al. presented a systematic study on the upconversion properties of a set of high-quality all-inorganic CsPbA3 perovskite semiconductor colloidal nanocrystals. High-quality nanomaterials and structures with strong electron–phonon interactions and low thermal conductivity were found to strongly contribute to the upconversion phenomenon. The step-like behaviour of β clearly delimits different mechanisms at room temperature, where the phonon-assisted regime appears in an energy window from 0 to 250 meV and the multiphoton absorption process takes place beyond 250 meV.2 The surprisingly large threshold of ∼200 meV exceeds those of other semiconductor nanocrystals.
The efficiency of phonon-assisted upconversion can also be enhanced by emitters with a high optical transition strength or by realizing resonant conditions, i.e., the incident and/or emitted photon energy matches the resonance level of the material system. Fang et al. reported the enhanced upconverted emission of two-dimensional excitons in doubly resonant plasmonic nanocavities.30 The phonon-mediated optical absorption is enhanced by integrating monolayer WSe2 into the designed plasmonic nanocavities that doubly resonate with the incident and/or emitted photons. Eventually, up to 1100-fold enhancement of the upconversion PL in monolayer WSe2 is achieved (Fig. 2(e)), which decreases energy consumption. Recently, they have also studied phonon-assisted upconversion in WSe2 twisted bilayers.28 A 4-fold upconversion enhancement was achieved in a 5.5° twisted bilayer while the PL weakened by half (Fig. 2(f)). Through Raman spectroscopy and time-resolved fluorescence spectroscopy, they attributed the phenomenon to a mutual influence of both the upconversion pump efficiency increase caused by a spectral redshift and the interlayer exciton conversion efficiency decrease resulting from lattice relaxation in WSe2 twisted bilayers. As a new phenomenon in twisted-angle TMDs, the results provide a novel insight into a unique way by which the twisted angle affects upconversion and light–matter interactions.
Multiphoton absorption
In semiconductor nanomaterials, multiphoton absorption is a process of exciting electrons from the valence band to the conduction band by absorbing multiple photons simultaneously without populating any real intermediate states. The single photon energy can be lower than the band gap of the material due to the fact that transition states conserve energy. The initial excitation has significant energy above the band edge and will rapidly relax to the lowest excited state at which the charge carriers can radiatively recombine and emit photons with energy higher than that of the excitation photons (Fig. 1(b)).4,5,14,31–33 The low energy photons demonstrate typically longer penetration depths and lower biological damage compared to the high energy photons in the one-photon absorption process.15,34,35 In this section, we focus on the PL upconversion of two-photon absorption (TPA), which is the most common mechanism.The PL intensity of TPA typically shows a quadratic dependence on the excitation power (α ≈ 2).36,37 This fingerprint can be used to distinguish between the two-photon upconversion and phonon-assisted upconversion processes. Fig. 3(a) shows the two-photon excitation of hybrid organic–inorganic lead halide perovskites.5 At the excitation photon energy ħω = 2.80 eV, which is above the bandgap, one-photon absorption occurs. They further obtained α = 2.0 at ħω = 1.24 eV and only a slightly lower α of 1.9 at ħω = 1.50 eV.
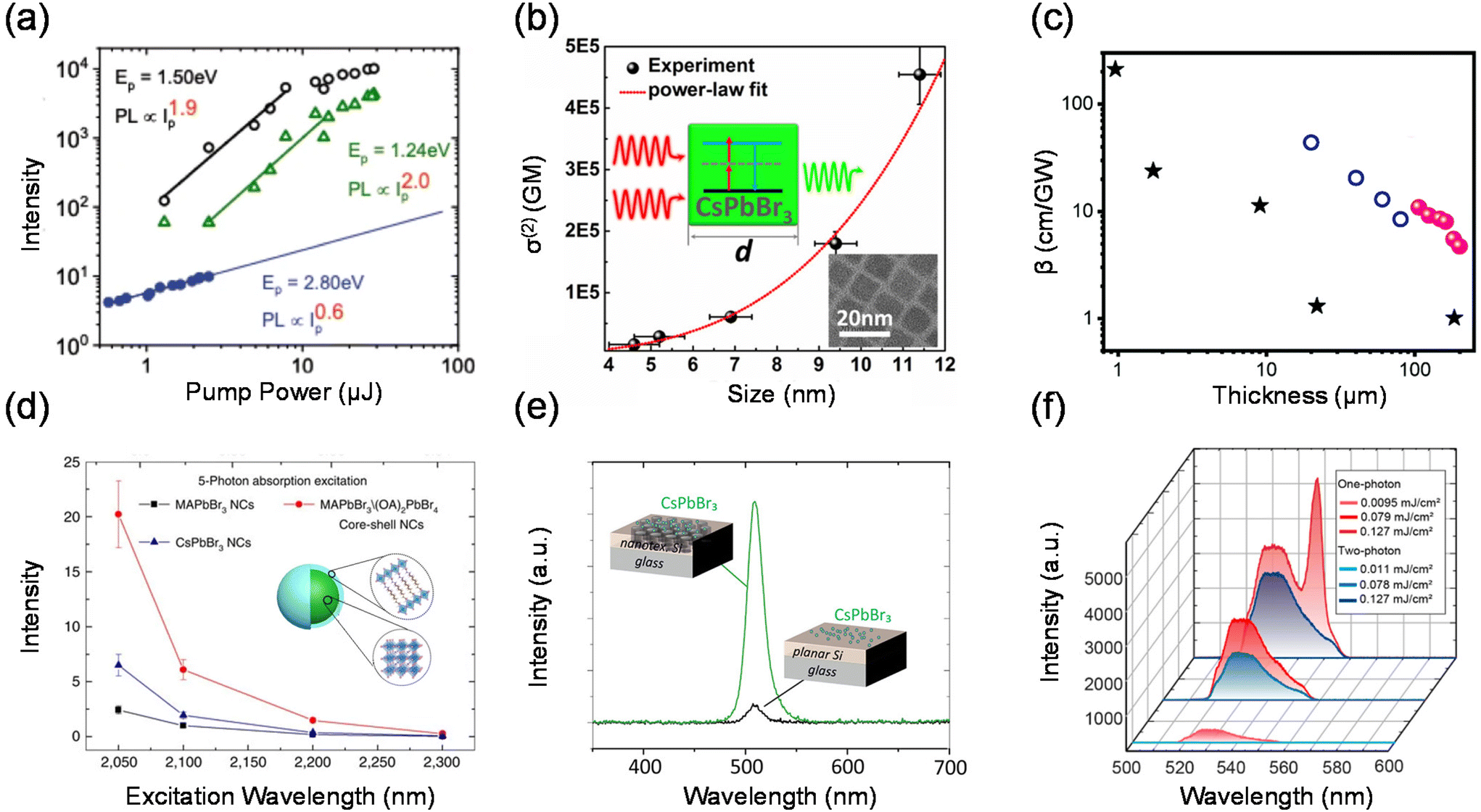 | ||
| Fig. 3 Multiphoton absorption. (a) Log–log plot of the pump intensity (Ip) dependence of the PL emission from MAPbBr3 crystals. (b) TPA cross-section σ(2) dependence of CsPbBr3 QD size d in a log–log scale. (c) Thickness-dependent TPA coefficients of halide perovskites. Data 1–3 are from (PEA)2PbI4, (n-C4H9NH3)2(CH3NH3)Pb2I7 and CsPbBr3. (d) Five-photon action cross-section (ησ5) spectra of core–shell MAPbBr3/(OA)2PbBr4 nanocrystals, MAPbBr3 nanocrystals and CsPbBr3 nanocrystals. (e) Examples of photoluminescence spectra with the excitation beam hitting CsPbBr3 quantum dots located on either a nanotextured area or a planar area of the silicon layer. (f) Emission spectra of MAPbBr3 metasurfaces under one-photon and two-photon excitation at three pump fluences. | ||
The TPA process can be impacted by different factors such as the chemical composition, structure, size and external physical environment.31,38–42 These factors provide plenty of possibilities to adjust and optimize the TPA properties of materials in a wide range. Tõnu Pullerits et al. used transient absorption spectroscopy to measure the TPA cross-sections of CsPbBr3 QDs of different sizes (4.6–11.4 nm) as shown in Fig. 3(b).41 The TPA cross-section of CsPbBr3 QDs shows a power-law size dependence of σ(2) ∝ d3.3. This result demonstrates the strong connection between the TPA process and the sample size. Chen et al. summarized that 2D perovskite nanosheets have stronger saturable absorption properties with a large modulation depth and a very low saturation intensity compared with those of bulk perovskite films.43 The TPA coefficient decreases as the sample thickness increases from 0.95 μm to 200 μm (Fig. 3(c)).
In order to enhance the TPA, a straightforward approach is to enhance the TPA coefficient of the material.34,44,45 Tze Chien Sum et al. developed a core–shell structure to enhance the TPA cross-section of lead halide perovskite nanocrystals (core–shell MAPbBr3/(OA)2PbBr4).46 The multidimensional core–shell perovskite nanocrystals offer amazing five-photon action cross-sections that are at least 9 orders larger than the state-of-the-art specially designed organic molecules (Fig. 3(d)). In addition, due to the nonlinearity of TPA, two-photon excited PL can be promoted externally by enhancing light–matter interactions, such as concentrating the excitation electromagnetic field,47,48 near-field enhancement,49 and surface plasmon exciton coupling.50 Tõnu Pullerits et al. investigated the two-photon excited PL of CsPbBr3 perovskite quantum dots on a silicon photonic crystal slab.49 Due to the near-field enhancement effect on nanostructured silicon surfaces, they observed an enhancement of two-photon-pumped PL by more than 1 order of magnitude when compared to using a bulk silicon film (Fig. 3(e)). Lu et al. reported a perovskite–microsphere hybrid dielectric structure. The two-photon absorption-induced PL emission from the 2D perovskite is increased by two orders of magnitude in the hybrid dielectric structure as a result of the cooperative effects of the dielectric microspheres, which increase the excitation intensity and internal quantum efficiency.47 Xiao et al. have studied experimentally two-photon PL in perovskite metasurfaces.48 They observed a giant enhancement of two-photon emission from MAPbBr3 perovskite metasurfaces comparable with the one-photon processes (Fig. 3(f)). This effect has been attributed to the local-field enhancement in metasurfaces and a significant increase of an overlap between stimulated emission and excitation modes.
Energy transfer mechanism
Lanthanides’ 4fn electronic configurations are split by appreciable electronic repulsion and spin–orbit coupling, leading to a rich energy-level pattern. This attribute makes lanthanides ideal candidates for achieving photon upconversion.18,51–53 Early studies of lanthanide-doped bulk materials allowed a deep understanding of the underlying physics.54 Unlike TPA, lanthanide nanocrystal upconversion is characterized by sequential absorption of incident photons.55 The 4f orbital electrons of lanthanides are partially filled and shielded by outer 5s and 5p electrons, giving rise to a weak electron–phonon coupling and hence sharp emission lines resulting from electronic transitions (Fig. 4(b)).53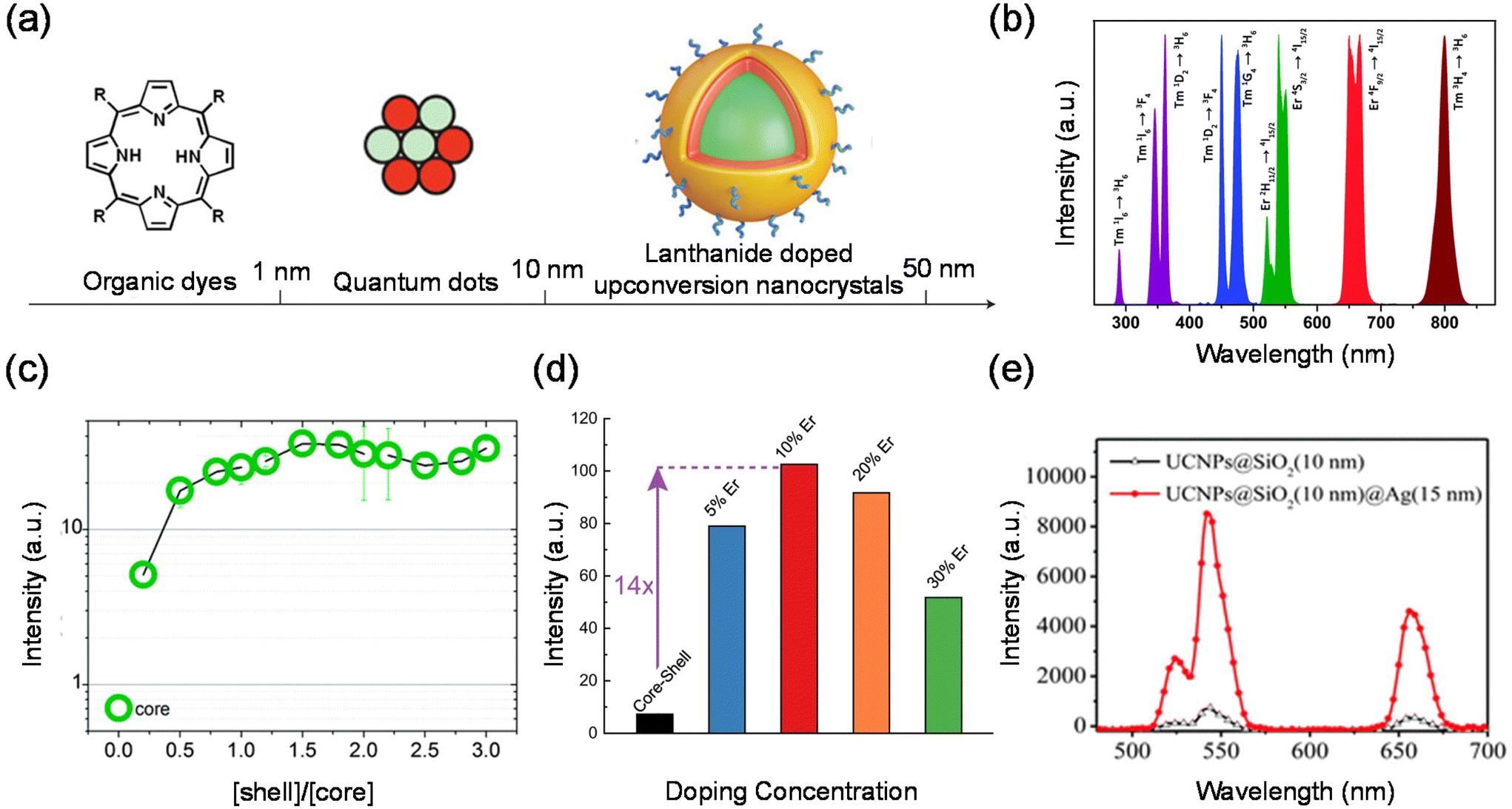 | ||
| Fig. 4 Cooperative energy transfer. (a) A comparison of three distinct classes of luminescent nanomaterials: organic dyes (left), semiconductor quantum dots (middle) and lanthanide-doped nanoparticles (right). (b) Typical UC emissions, ranging from the UV to NIR regions from co-doped UCNPs under 980 nm excitation. (c) Dependence of emission intensity on the shell–core molar ratio in colloidal α-NaYF4:Yb3+,Tb3+-doped nanocrystals. (d) Single-particle brightness of the conventional core–shell design and the outside-in architectures of β-NaLu0.78Yb0.2Er0.02F4@NaLuF4 UCNPs with different doping concentrations in the core. (e) Enhanced upconversion luminescence spectra of core–shell NaYF4:Yb,Er@SiO2@Ag nanocomposites. | ||
Organic dyes were among the first classical fluorescent materials. They are widely popular due to their low cost, availability and ease of use. However, recent studies have reported organic dyes with better chemical and optical properties. The inherent drawbacks of organic dyes remain intractable, such as short Stokes shifts, poor photochemical stability, susceptibility to photobleaching, and decomposition under repeated excitation. Quantum dots (QDs) are semiconductor nanocrystals. They are usually defined as particles with physical dimensions smaller than the exciton Bohr radius (typically 1–5 nm). The small size of these QDs leads to quantum confinement effects that confer unique optical and electronic properties to the nanocrystals. Compared to conventional organic dyes such as rhodamine, QDs are much brighter and more stable against photobleaching, with a narrower spectral linewidth. Second, the emission wavelength of QDs changes as the size or chemical composition of the QDs changes. Thus, the emission wavelength can be tuned by changing their size and chemical composition, resulting in multiple emission colors. However, QDs themselves are not biocompatible and have to be surface modified before they are used in live cell or animal experiments.
Compared with traditional fluorescent QDs and organic fluorescent materials, lanthanide upconversion nanoparticles are composed of an inorganic crystalline host matrix and trivalent lanthanide ions (Ln3+) embedded in the host lattice (Fig. 4(a)). They possess many advantages, such as high chemical stability, excellent photostability, narrow-band peak emission, and continuous emission tunability.56–58 An NIR laser as their excitation light source brings many advantages, such as deeper light penetration depth, almost no damage to biological tissues, no luminescence of biological tissues (no background fluorescence), etc.59,60 Lanthanide upconversion nanoparticles also have luminescence decay times on the order of milliseconds. Therefore, using a time-selective intensity detector, the short-lived autofluorescence emitted by a biological specimen upon excitation can be completely separated from the long-lived luminescence of lanthanide upconversion nanoparticles.61–63 In addition, lanthanide upconversion luminescence requires only low power density near-infrared continuous lasers, which are much more versatile than TPA required expensive high power density pulsed laser excitation. The TPA efficiency is theoretically proportional to the square of the excitation light intensity.
In principle, the upconversion mechanisms involving lanthanides can be mainly categorized into five classes: excited-state absorption, energy transfer upconversion, cooperative upconversion and photon avalanche.19,64–68 Here, we focus on the mechanisms of energy transfer upconversion. Fluorescence upconversion of lanthanides (e.g., Er3+, Tm3+, and Ho3+) is usually realized via ETU,19,20 where long-lived intermediate states exist to store energy (Fig. 1(c)). The ETU process typically occurs in a sensitizer–activator (S–A) system. In such a system, the energy transfer from the sensitizer to the activator populates not only the emitting levels but also the intermediate levels, as evidenced in the case of Yb3+–X3+ (X = Er, Tm or Ho) couples. According to Dexter's theory,8,69 the energy transfer probability WS–A under a dipole–dipole interaction can be expressed by:
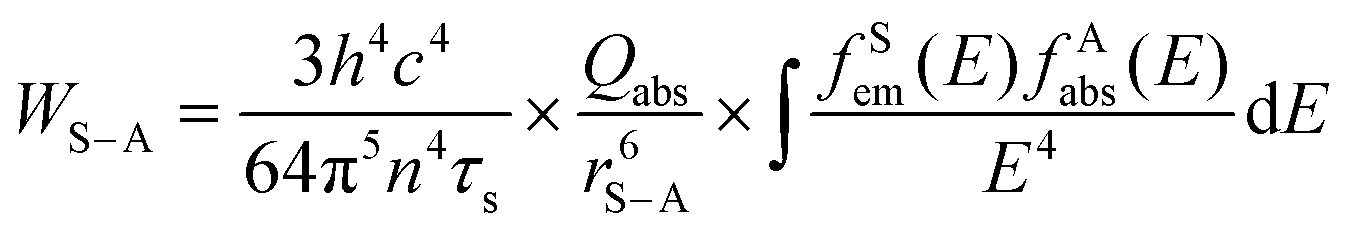
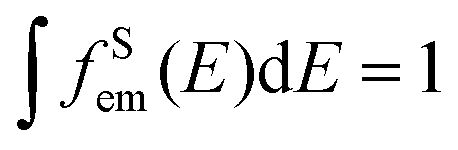 and the absorption band of the activator at intermediate levels
and the absorption band of the activator at intermediate levels 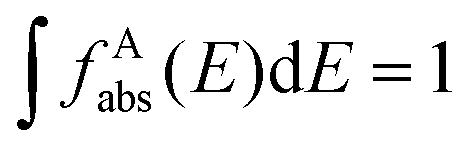 , respectively. Therefore, the S–A distance (rS–A) and the spectral overlap (the integration term in the above equation) are the two critical parameters governing the energy transfer. Using a well-established rate equation model for lanthanum-doped bulk materials and devices, it is possible to quantitatively predict photon upconversion and its dynamic processes in nanocrystals.18 The microscopic parameters in the rate equation models can be determined using the Judd–Ofelt theoretical approximation.70 Importantly, recent developments in computational methods allow for the robust design of complex material systems and the accurate prediction of new properties by incorporating subtle electronic perturbations in the physical properties of these materials.71,72
, respectively. Therefore, the S–A distance (rS–A) and the spectral overlap (the integration term in the above equation) are the two critical parameters governing the energy transfer. Using a well-established rate equation model for lanthanum-doped bulk materials and devices, it is possible to quantitatively predict photon upconversion and its dynamic processes in nanocrystals.18 The microscopic parameters in the rate equation models can be determined using the Judd–Ofelt theoretical approximation.70 Importantly, recent developments in computational methods allow for the robust design of complex material systems and the accurate prediction of new properties by incorporating subtle electronic perturbations in the physical properties of these materials.71,72
Lanthanide upconversion emissions can be modulated by incorporating energy extractors or passivators, decorating the surface of nanoparticles. Energy extractors would capture partial energy from activators doped in the system, resulting in a decreased emission intensity. Energy passivators can minimize the excitation energy loss induced by surface effects and solvents, leading to an increased emission intensity as a result.19,73 Luminescence quenching from the surface can be solved by forming a uniform shell over the core. After the epitaxial growth of the shell layer, energy transfer from activators in the core to surface defects, capping ligands, and solvent molecules is blocked.74–76 The quenching effects can thus be significantly suppressed, resulting in improved emissions. For the perfect growth of a shell, it should be similar in composition, structure or lattice constant to the core.77–80 In recent studies, luminescent centres have been introduced into the shell layer to realize efficient sensitization and novel energy transfer occurring through the core and shell. Wieslaw Strek et al. have shown an enhancement of the upconversion owing to the passivation of their surface.81 They have studied quantitatively the influence of the shell type, its thickness, and the shell deposition method on the luminescence properties of the nanoparticles. Fig. 4(c) shows that up to 40-fold upconversion intensity enhancement may be obtained for the core–shell nanoparticles in comparison with the bare core nanoparticles. Steven Chu et al. systematically characterized single-particle luminescence for core–shell–shell structure UCNPs with various formulations. They showed a representative ensemble upconversion luminescence spectrum with 980 nm laser excitation of a solution containing 22 nm NaY0.78F4:Yb0.2Er0.02 UCNPs.82 Emission peaks centred at 520 nm, 541 nm and 654 nm are attributed to the transitions from the 2H11/2, 4S3/2 and 4F9/2 excited states to the 4I15/2 ground state, respectively. In contrast, when an inert shell was added to NaYF4@NaYb0.98F4:Er0.02 to form the core–shell–shell structure, they observed an extraordinary 740-fold green emission enhancement. The core–shell–shell particles exhibit a giant enhancement, given their high sensitizer Yb3+ content and the presence of an inert shell to prevent energy migration to defects. These brighter probes open the possibility of cellular and single-molecule tracking at low irradiance.
The distance between the energy donor and the acceptor is a key factor that significantly affects the energy transfer efficiency. Different doping concentrations of sensitizers or activators lead to various spatial distances.83–85 It has been found that the radiative transitions from high-lying excited states depend greatly on the distance between the luminescent centres.86,87 As a result, distinctive colour outputs with different emission intensities may be observed. Qian Liu et al. reported an inorganic optical nano-system composed of NaErF4 and NaYbF4, in which topological arrangement enhanced the upconversion luminescence. They precisely demonstrated that the outside-in architecture is the brightest in term of single-particle measurement.88 Cross-relaxation between Er3+ ions may hamper upconversion luminescence emission by keeping the emitter ions in an intermediate energy level. In order to find the optimal Er3+ doping concentration for upconversion luminescence, the same three nanoparticle architectures studied above were made with 5, 10, 20 or 30% Er3+ doping. They compared the luminescence with different Er3+ doping concentrations. UCNPs with 10% Er3+ doping were the brightest in both ensemble and single-particle measurements (Fig. 4(d)). At all irradiances examined, Lu0.9Er0.1@Yb@Lu UCNPs showed the brightest single-particle emission with a 14-fold enhancement compared to a conventional core–shell architecture of β-NaLu0.78Yb0.2Er0.02F4@NaLuF4 UCNPs with the same size.
Another interesting approach to enhancing photon upconversion is harnessing the effect of surface plasmon resonance (SPR). For one thing, the amplification of the localized incident electromagnetic field leads to an increase in the excitation rate, which originates from the resonant coupling with the noble metal SPR. On the other hand, the coupled surface plasma emission increases the emissivity and thus the emission efficiency, which will effectively contribute to the radiative and nonradiative decay rates.54,89–91 Therefore, a number of schemes have been explored to tune and optimize the interaction between metals and upconversion nanoparticles, including the coating of a metal shell on the surface of upconversion nanoparticles, the coupling of Au nanoparticles to upconversion nanoparticles and the construction of 3D plasmonic nanoantennas.92–95 Xu et al. prepared NaYF4:Yb,Er@SiO2@Ag core–shell nanocomposites to investigate metal-enhanced upconversion luminescence.91 The emission intensity of the upconversion nanocrystals was found to be strongly modulated by34 the presence of Ag NPs on the outer shell layer of the nanocomposites. Fig. 4(e) shows that a maximum upconversion luminescence enhancement of 14.4-fold was observed when 15 nm Ag nanoparticles were used. Chen et al. constructed a gold-nanorod dimer nanocavity with a single UCNP in the gap and achieved upconversion luminescence coupling with a single nanocavity mode. The UCNP has a core/shell/shell structure consisting of a core of undoped NaYF4, a shell of Yb/Tm co-doped NaYF4 and an outermost shell of undoped NaYF4. By controlling the polarization of excitation and the doping concentrations in the nanocrystals,96 they showed upconversion luminescence enhancement factors spanning seven orders of magnitude, from 5 × 10−3 (∼200-fold suppression) to 2.3 × 105. These works pave the way to establish a link between optical physics and materials science associated with lanthanide-doped nanocrystals, which is appealing for a range of applications, including ultrasensitive biochemical sensing and ultralow-threshold lasing emission from single UCNPs.
Here, we count the UCQY, upconversion thresholds, anti-Stokes shifts, and upconversion fluorescence lifetimes in different systems (Table 1). The abundant energy levels of lanthanide element ions serve as intermediate excited states in the upconversion process. The upconversion center wavelength can be easily adjusted by designing the structure of the nanoparticles and adjusting the concentration of doped rare earth elements. The major problem of current UCNPs lies in their low UC QYs, which inevitably limits their commercial bio-applications. Despite a high QY already achieved in some UCNPs, one should bear in mind that it was obtained upon high power density excitation that is not acceptable for some in vivo applications. TPA is upconversion emission through a virtual intermediate state. However, the nonlinear nature of TPA requires the excitation intensity of several orders of magnitude higher than the OPA process. This limits the application of TPA-based sub-bandgap photodetection only for high power laser and, perhaps, short-pulsed laser light detection. To overcome such limitations, we should seek for materials with a large TPA coefficient. In contrast, phonon-assisted upconversion places no restrictions on the choice of light source, but requires the sample itself to have a high electroacoustic coupling coefficient, which itself limits the phonon-assisted upconversion luminescence to lower anti-Stokes shifts.
| Mechanism | System | UC threshold | UCQY | Anti-Stokes shift (meV) | Fluorescence lifetime |
|---|---|---|---|---|---|
| Phonon absorption | Germanium vacancy101 | 0.44 mJ cm−2 | 103 | ||
| Carbon nanotubes27 | 200 mJ cm−2 | 130 | |||
| CdS127 | 0.048 | 120 | |||
| CdSe/CdS24 | 89 | 10 ns | |||
(PEA)2PbI4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29 29 |
0.031 | 386 | |||
| (PEA)2PbI4164 |
1 μJ cm−2 | 220 | 4 ns | ||
| WS23 |
150 | ||||
| WSe228 |
101 | ||||
| h-BN11 | 0.0056 | 162 | |||
| Multiphoton absorption | (PEA)2PbI447 |
0.875 | 780 | 3.74 ns | |
| MAPbBr348 |
0.13 mJ cm−2 | 432 | 1.5 ns | ||
| CsPbBr3116 |
0.8 | 890 | 7.3 ns | ||
| MAPbBr3/(OA)2PbBr4 core–shell46 | 2.31 mJ cm−2 | 0.92 | 1794 | 12.7 ns | |
| CsPbBr3 NCs107 | 0.2 μJ cm−2 | 0.6 | 871 | 18 ns | |
| MAPbI35 |
940 | ||||
| CdSe/CdS heterostructures165 | 3.4 mJ cm−2 | 0.75 | 516 | 10 ns | |
| CdSe/CdS/ZnS core–shell quantum dots166 | 3 mJ cm−2 | 998 | 16.5 ns | ||
| CdSe/CdS heterostructures167 | 0.61 | 1203 | |||
| Energy transfer | NaYF4:Yb/Tm96 | 0.5 J cm−2 | 0.16 | 284 | 110 us |
| NaLuErF@NaYbF4@NaLuF4 core–shell88 | 0.019 | 1114 | 1.2 ms | ||
| NaYF4@NaYbF4:Er@NaYF4 single-particle core–shell82 | 8 J cm−2 | 0.054 | 1119 | 1.5 ms | |
| NaYF4:Yb3+,Nd3+,Tm3+,Ho3+ and Ce3+ doped core–shell142 | 1103 | 0.5 ms | |||
| NaGdF4:Yb/Tm core–shell75 | 1490 | 0.26 ms | |||
| YbEr@Yb@NdYb core–shell139 | 0.5 J cm−2 | 0.0011 | 830 | ||
| YbEr@Yb@NdYb@Y@Gd core–shell168 | 0.0075 | 830 | 0.9 ms | ||
| YbEr@Yb@NdYb@Yb core–shell169 | 0.0018 | 1489 |
Ultrafast photophysics in upconversion fluorescence
Phonon-assisted upconversion with intrinsic electron–phonon coupling
Phonon-assisted PL upconversion forms the basis for solid-state laser cooling, as well as numerous other applications, such as photovoltaics, fluorescence imaging, anti-counterfeiting, and lasers.97–99 A few recent studies have shown that some low-dimensional materials such as nanotubes, nanobelts, QDs, transition metal dichalcogenide monolayers, and diamond colour centres, can achieve large energy gains of around 100 meV.1,3,24,27,100,101 However, the role of phonons in enabling such a large anti-Stokes shift in these materials is not yet fully understood. The upconversion is a multi-phonon process occurring through intermediate levels such as sub-gap traps, surface states, and the Urbach tail,25,102,103 and can be enhanced by doubly resonant Raman scattering and excitonic effects.28,30,104 Further investigation into the microscopic ultrafast photophysics of photon–electron–phonon interactions behind the upconversion processes is crucial to enhance the efficiency and energy gain of thermal-assisted upconversion.Peng and Qin et al. reported efficient upconversion luminescence based on solution QDs, revealing their upconversion luminescence mechanism based on intrinsic electron–phonon coupling energy levels.24 For almost defect-free core–shell structured QDs (CdSe/CdS), upconversion luminescence occurs efficiently and the decay kinetics of PL and upconversion PL follow the same monoexponential function (Fig. 5(a)). With ultrafast fluorescence spectroscopy, they measured the time constant of the transition process on the QDs which was found to be less than 200 fs (Fig. 5(b)), much faster than the usual recombination composition and the rate of defect trapping. Femtosecond-resolved absorption and resonant Raman spectra were recorded to identify the features of the strong coherent phonon coupling accompanying upconversion excitation. The frequency of the coherent vibration matches that of the longitudinal optical phonon of CdSe QDs measured by Raman spectroscopy, indicating that phonon-assisted photon absorption occurs within the CdSe core as shown in Fig. 5(c). To this end, the authors proposed an upconversion luminescence mechanism based on intrinsic electron–phonon coupling energy levels to explain the observed upconversion luminescence phenomenon and realize the optical cooling of QDs.
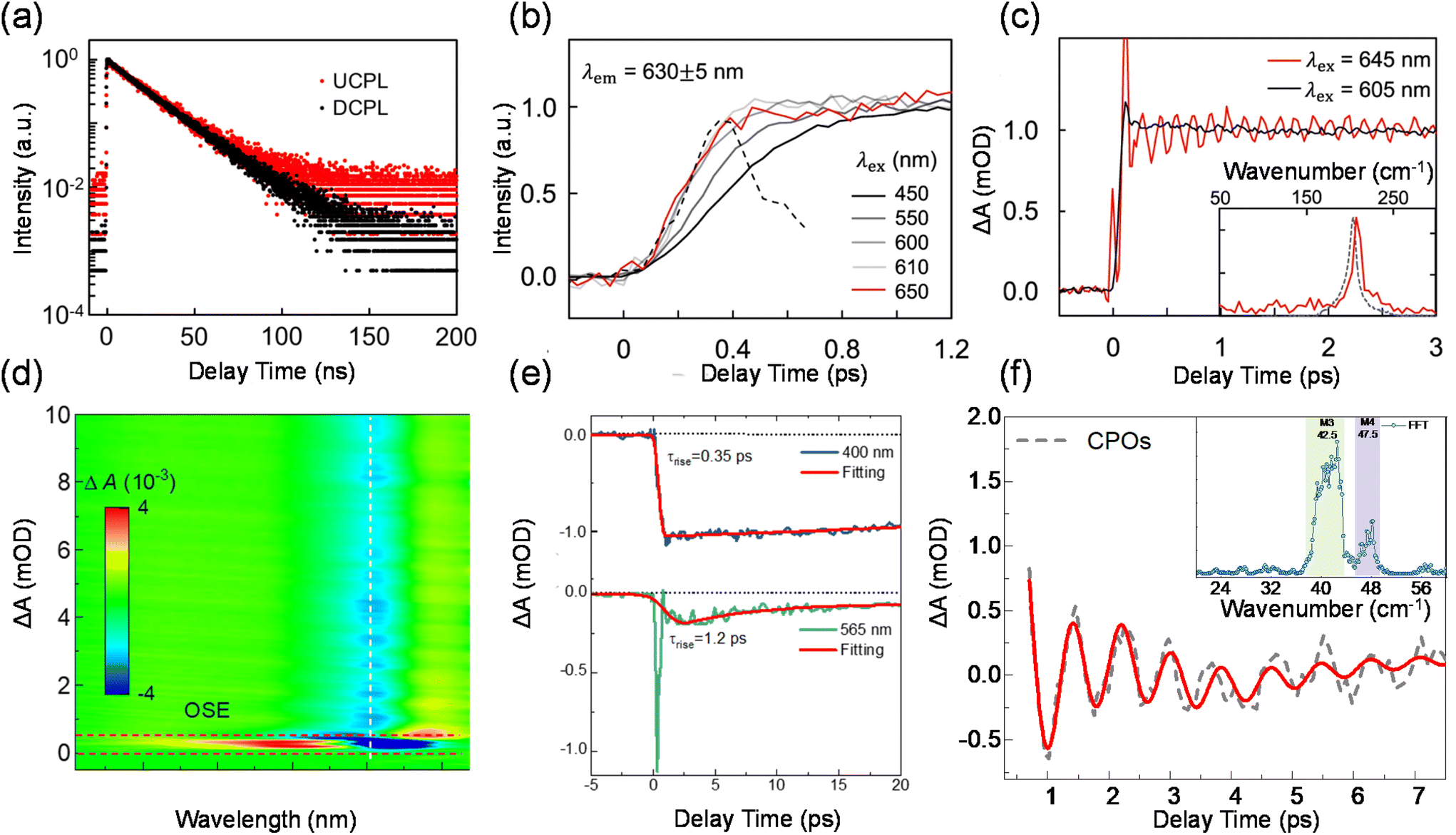 | ||
| Fig. 5 Phonon-assisted upconversion with intrinsic electron–phonon coupling. (a) Upconversion and downconversion decay dynamics of ensemble CdSe QDs. (b) Normalized femtosecond-resolved photoluminescence spectra of ensemble CdSe QDs at different excitation wavelengths. The black dashed line is the instrument response function. (c) Normalized femtosecond-resolved absorption spectra monitored at the first absorption peak under upconversion (red) and downconversion (black) excitation. Inset: the Fourier transform of the red curve in the main figure along with the resonant Raman spectrum of CdSe QDs. (d) TA spectra of a PEPI micro platelet upon upconversion excitation. (e) Exciton band bleach dynamics monitored for downconversion and upconversion excitation. (f) Damping of coherent phonon oscillations monitored at 524 nm and its fitting using a multi-oscillator model. Inset: the Fourier transform of the coherent phonon oscillations showing prominent contribution. | ||
Recently, Liu and Wu et al. showed that a quasi-two-dimensional perovskite, namely phenethylammonium lead iodide perovskite ((PEA)2PbI4), can facilitate efficient upconversion with an enormous anti-Stokes shift of up to 220 meV. By using microscopic transient absorption spectroscopy, the rise time for the exciton band bleach following downconversion excitation was found to be around 0.35 ps. For upconversion excitation, after removing the strong nonlinear response by the optical Stark effect, it is found that the rise of exciton band bleach is relatively slower with a time constant of 1.2 ps (Fig. 5(d) and (e)). The slow rise implies that the photon upconversion is not a one-step process via the absorption of a high-energy organic molecule vibration mode (>100 meV), which would occur at a timescale on the order of a phonon period (∼10 fs). The slow rise of the exciton band bleach is accompanied by the damping of the oscillatory component. As shown in Fig. 5(f), through fast Fourier transformation (FFT), we identified two dominant oscillation frequencies in the inset: 42.5 cm−1 and 47.5 cm−1. These two modes correspond well with M3 and M4 phonon modes reported previously. M3 can be attributed to the Pb–I–Pb bending and Pb–I stretching modes. M4 is assigned to the scissoring of the Pb–I–Pb angle. Given that the energy of excitons generated upon upconversion excitation is strongly modulated by Pb–I phonon modes, they inferred that the excitons relax to the thermal equilibrium state by coupling to the lattice vibrations during its anharmonic motion in a few ps. By using density functional theory calculations, they propose that the strong lattice fluctuation produces abundant phonon-dressed electronic density of states below the band gap, enabling considerable sub-gap photon absorption. The electron–hole pairs generated below the band gap then couple to the picosecond fast lattice deformation and move to the equilibrium states where they radiatively recombine.
Carrier dynamics of two-photon excitation
The initial carrier dynamics of two-photon excitation may differ from single-photon excitation.41,105–108 As mentioned before, two-photon excitation has a longer penetration depth compared to single-photon excitation. Due to the limited penetration depth of the excitation light, single-photon excitation charge carriers are mostly located at the surface of the material, where the resulting charge carriers are ready to recombine or diffuse into the material in a non-radiative/radiative regime.109–112 Two-photon excitation charge carriers are located inside the material and exhibit different dynamics, requiring consideration of large differences between the surface and volume of the material, such as trap density, structural dimensions and mobility.113–115 Tõnu Pullerits et al. reported that perovskite nanocrystals show a red-shifted PL spectrum and different kinetics under TPA and one-photon absorption excitation by utilizing time-resolved PL and femtosecond transient absorption spectroscopy techniques (Fig. 6(a)).116 They carried out TA experiments for NCs with different mean sizes (d = 4.6, 9.4, and 11.4 nm) as shown in Fig. 6(b). The ground-state bleaching (GSB) decay signal of NCs with small sizes can be fitted with double exponential functions. GSB signals with 800 nm excitation typically show longer decay compared to those with 400 nm excitation. In NCs with smaller sizes (4.6 and 9.4 nm), such differences are pronouncedly reflected as prolonged lifetimes. In larger NCs (11.4 nm), however, the difference is subtle. They attributed it to the different size selections between one-photon absorption and TPA excitation. One-photon absorption and TPA cross-sections of CsPbBr3 nanocrystals show different power-law dependences on the size of the nanocrystals. One-photon absorption cross-section increases with nanocrystal size in the third power-law dependence, whereas the TPA cross-section increases in the 3.6th power-law dependence. For nanocrystals with the same size distribution, TPA always leads to preferential excitation of larger particles within the collection compared with the one-photon absorption. Wu et al. similarly observed that two-photon excited PL shows slower decay compared to single-photon excitation. They identified the surface and bulk photophysics of inorganic organohalide chalcogenide single crystals by selective excitation of single- and two-photon absorptions.113 The surface regions of large single crystals have relatively high trap densities and short diffusion lengths. On the other hand, the bulk regions with lower lattice strain and lower trap density yield longer diffusion lengths.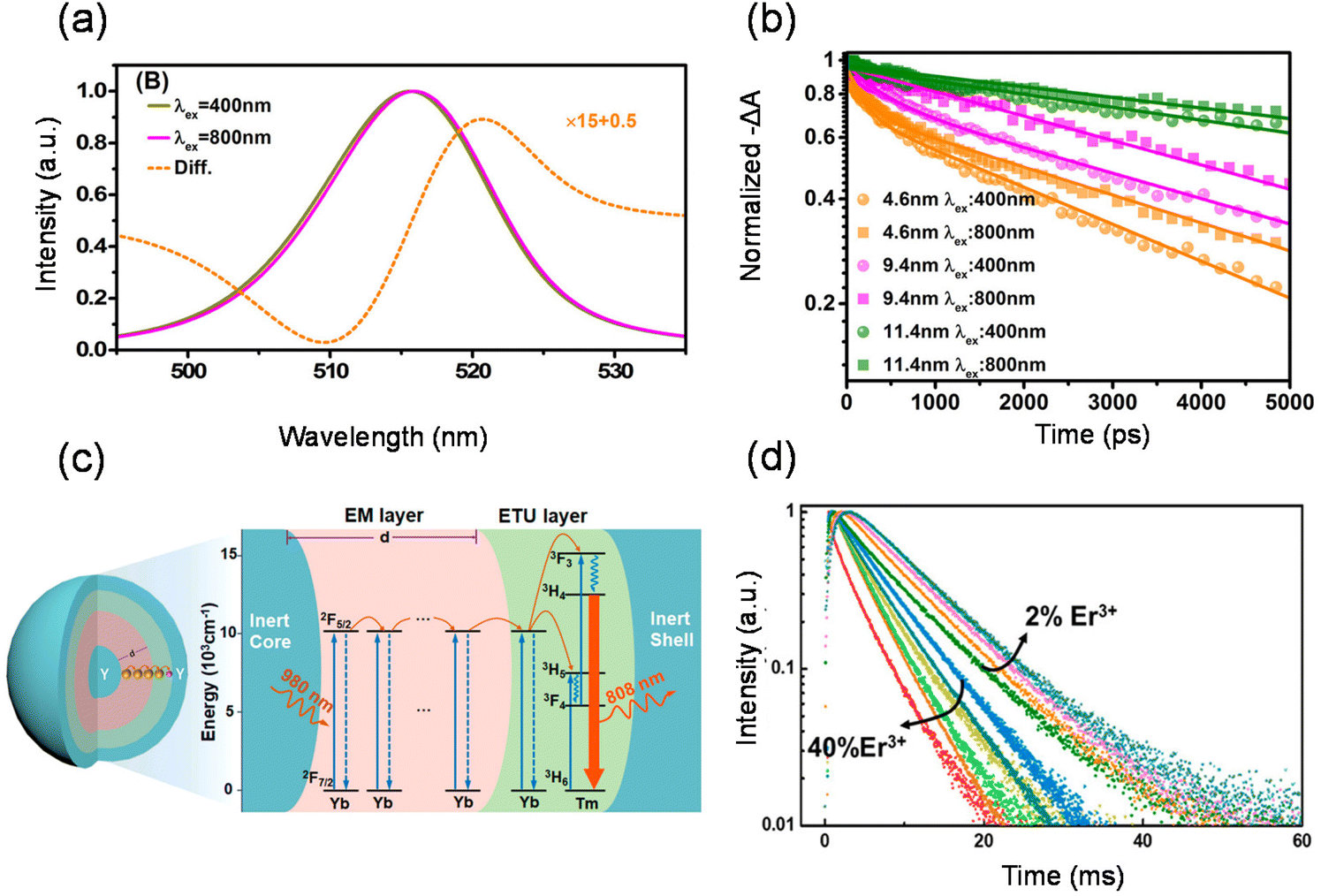 | ||
| Fig. 6 Carrier dynamics of two-photon excitation and the transport of energy migration in lanthanide upconversion. (a) Simulated PL emission with 400 nm and 800 nm excitation and the simulated difference between the two- and one-photon excited PL spectra of CsPbBr3 NCs. (b) Transient absorption kinetics for CsPbBr3 NCs with three different sizes. (c) Schematic diagram illustrating designated energy transfer processes. (d) Controlled luminescence lifetime of NaYF4:Yb3+/Er3+@NaYbF4@NaYF4@NaYF4:Nd3+ core/multi-shell nanoparticles through energy regulation. | ||
The transport of energy migration in lanthanide upconversion
Ionic interactions are widely present in lanthanide-doped systems. Energy migration is another interesting and important process in the various interactions between lanthanide ions. In the same lanthanide ion, there is an inherent spectral overlap between the absorption and emission associated with the energy-migrating states, making energy migration easy to occur under the right conditions (Fig. 6(c)). A particular advantage of energy migration is transporting energy over long distances, which can be observed in the spectra in real time.117,118 The design of energy migration has contributed greatly to the study of new upconversion mechanisms as well as to the precise control of upconversion kinetics.119–121 Chen et al. devised a core/shell/shell/shell structure.119 The inert shell acts as an energy-retarding layer to regulate the energy flow from the outmost light-harvesting layer to the inner core domain to produce long-lived luminescence. As shown in Fig. 6(d), a precise control of the NaYF4 layer thickness enables yielding a precisely defined lifetime tunable between ∼3 and 10 ms, yet without compromising luminescence intensities. Zhang et al. designed a smart nanosystem termed a “dopant ion’ spatial separation” (DISS) nanostructure to separate the three basic processes of lanthanide upconversion.77 They prepared a nanostructure, YbEr@Yb@Nd, to explore the temporal effect of energy migration. The upconversion emission of this structure relies fully on the Yb3+ → Yb3+ energy transfer to transport the absorbed energy from the outer layer to the core area. Increasing the thickness of the middle layer from 0 to about 4.5 nm results in a prolongation of the rise of around 540 nm upconversion emission from 195 to 390 μs. They demonstrated a close connection between excitation energy migration and upconversion emission dynamics. Similarly, Fan Zhang et al. designed a systematic approach based on controlled energy relay in a core–multi-shell nanostructure.122 The sensitizers confined in the outer layer harvest the excitation photons and transfer the energy to the Yb3+ ions in the same layer via inter-ionic cross-relaxation. The energy is then relayed via diffusive energy migration among the Yb3+ sublattices, until reaching the inner layer, where it is entrapped by the Er3+ emitters to generate upconversion luminescence. With the increasing thickness of the energy relay layer, the luminescence lifetime at 1525 nm was effectively extended from 1.25 ms to 7.21 ms. By adjusting the thickness of the energy relay layer and the concentration of doping elements, a tuneable lifetime range spanning three orders of magnitude can be realized in a single emission band.Broad implications and applications
The rapid development of upconversion nanomaterials has driven a wide range of applications from the biological field to nanophotonics. In this section, we summarize and highlight the wide range of uses of upconversion nanomaterials in laser cooling, full-colour displays, upconversion lasers, 3D printing, and other potential applications.Laser cooling
The basic principle of laser cooling (also called optical refrigeration) is that during the excitation of anti-Stokes fluorescence of a solid with a monochromatic light, the emission of anti-Stokes fluorescence takes away some of the heat energy from the solid, thus reducing the temperature of the material because the energy of the anti-Stokes photons is higher than the energy of the excitation photons. This principle was first proposed by a German physicist Peter Pringsheim in 1929.123 Phonon-assisted upconversion absorbs one or more phonons from lattice thermal vibration to achieve an energy gain of light emission, which has been regarded as a promising way to realize optical micro-zone refrigeration. The advantages of compactness, no vibrations, no moving parts or fluids, high reliability, and no need for cryogenic fluids have motivated laser cooling research.9At a fixed temperature, when a sample is irradiated with a laser, the photo-excited electron–hole pair satisfies the following rate equation:
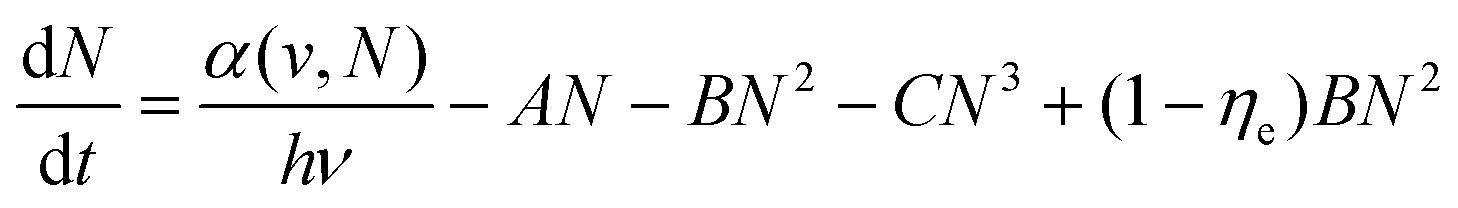
Here α(ν,N) is the interband absorption coefficient, and the combination processes include nonradiative combination (AN), radiative combination (BN2), and the Auger (CN3) processes, all of which are temperature-dependent functions.124,125 The last term on the right-hand side of the equation indicates that fluorescence photons cannot escape out of the sample and give rise to reabsorption and thermal effects. According to the above equation, the upconversion cooling efficiency ηc(hν,T) can be written as follows:
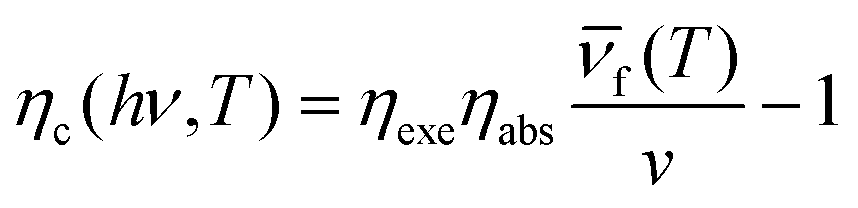
Here, ν is the pump laser frequency, T is the sample temperature, and 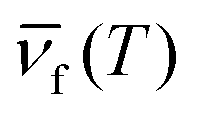 is the average emission frequency of the escaping fluorescent photons. ηexe is the fluorescence external quantum efficiency of the sample. ηabs is the absorption coefficient, which represents the percentage of the contribution of all absorbed pump photons to the cooling.126 According to the equation, the net laser cooling requires the laser cooling efficiency ηc(hν,T) > 0, and thus the following three conditions need to be satisfied: (1) a large upconversion energy difference
is the average emission frequency of the escaping fluorescent photons. ηexe is the fluorescence external quantum efficiency of the sample. ηabs is the absorption coefficient, which represents the percentage of the contribution of all absorbed pump photons to the cooling.126 According to the equation, the net laser cooling requires the laser cooling efficiency ηc(hν,T) > 0, and thus the following three conditions need to be satisfied: (1) a large upconversion energy difference 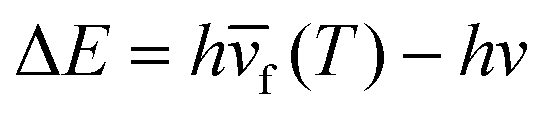 , (2) an external quantum efficiency ηexe close to 100%, and (3) an absorption rate ηabs close to 100%. The first condition requires the material to have a strong electron–phonon coupling coefficient, so that in the photon conversion process as efficient as possible to take away the thermal energy (phonon). The second condition requires the material to emit light not only has a high internal quantum efficiency, but also requires that the emitted photons can have a high escape efficiency, which requires the semiconductor's refractive index is relatively small, or the use of nanostructures to enhance photon escape. The third condition requires the material to be very small non-intrinsic absorption, which requires very high purity, very few defects, and very demanding sample processing requirements.10,24,29,127–129
, (2) an external quantum efficiency ηexe close to 100%, and (3) an absorption rate ηabs close to 100%. The first condition requires the material to have a strong electron–phonon coupling coefficient, so that in the photon conversion process as efficient as possible to take away the thermal energy (phonon). The second condition requires the material to emit light not only has a high internal quantum efficiency, but also requires that the emitted photons can have a high escape efficiency, which requires the semiconductor's refractive index is relatively small, or the use of nanostructures to enhance photon escape. The third condition requires the material to be very small non-intrinsic absorption, which requires very high purity, very few defects, and very demanding sample processing requirements.10,24,29,127–129
Zhang et al. in 2013 proposed that in semiconductors with strong electron–phonon coupling, the upconversion energy can be greatly increased by the resonant annihilation of multiple longitudinal optical phonons.127 Using this principle, they reported a net cooling by about 40 kelvin in a semiconductor using group II–VI cadmium sulphide nanoribbons, or nanobelts, starting from 290 kelvin. Perovskite single crystals possess a low trap-state density and a high external quantum efficiency, both of which are advantageous for laser cooling. In 2016, Ha et al. found that lead halide perovskite crystals in both 3D (MAPbI3) and 2D (PhEPbI4) forms show strong PL upconversion, suggesting the possibility of laser cooling.29 They demonstrated experimentally that MAPbI3 platelets grown by vapour phase synthesis and PhEPbI4 samples exfoliated from a bulk crystal by solution synthesis can be laser cooled by ∼23.0 and 58.7 K, respectively, from room temperature. Qin et al. measured the optical cooling effects of CdSe/CdS core/shell QDs.24 The QDs with an emission peak at 632 nm excited with 450 and 532 nm lasers show strong and excitation-wavelength dependent heating effects. When the excitation wavelength is further shifted to the tail of the absorption spectrum (671 nm), corresponding to an average upconversion energy gain of 110 meV, a maximum temperature decrease of 0.18 K under 300 mW irradiation relative to the control specimen is observed as shown in Fig. 7(a).
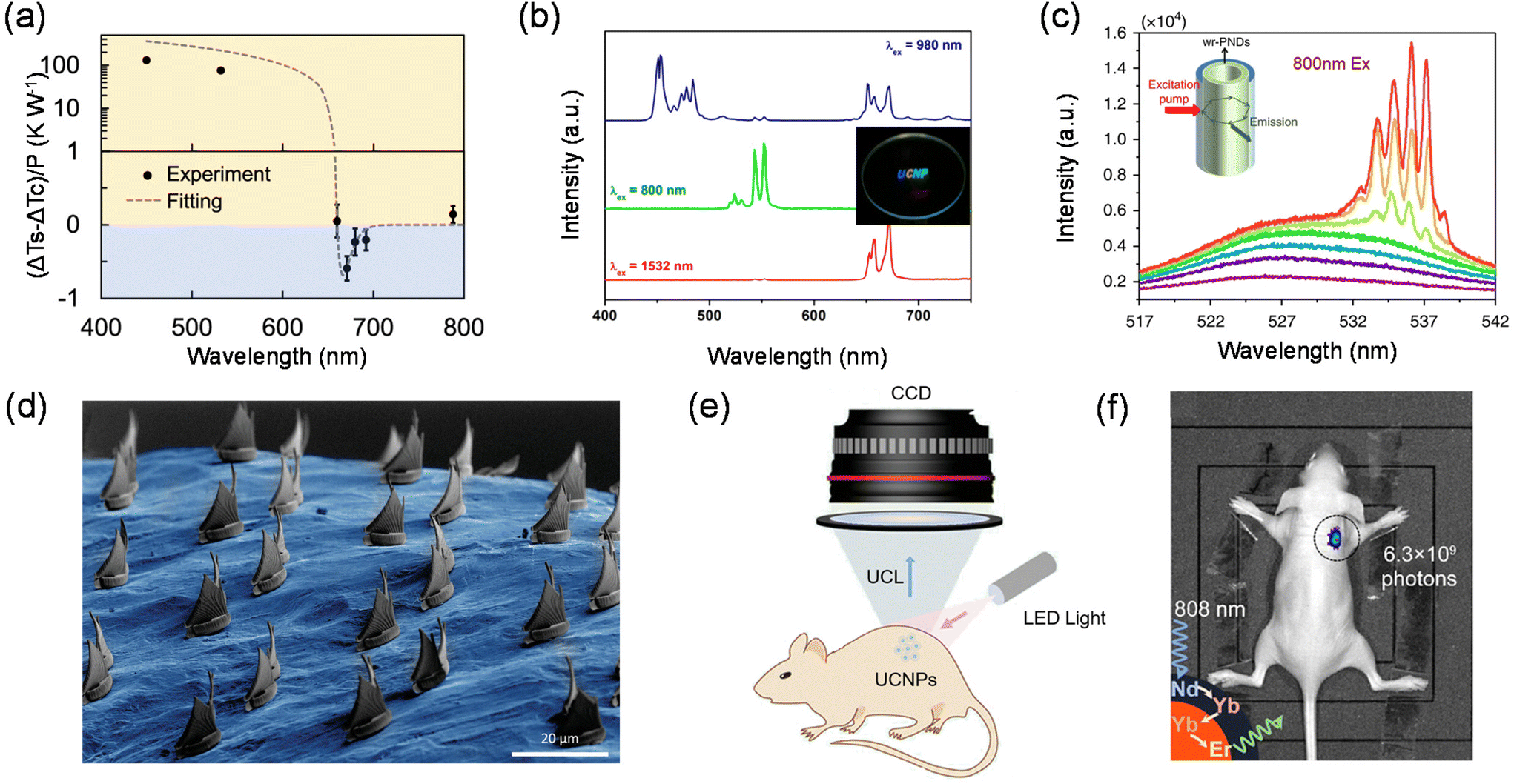 | ||
| Fig. 7 Applications of upconversion nanomaterials. (a) Relative temperature changes of the CdSe/CdS QD sample compared with the control specimen at different excitation wavelengths with a unit excitation power. (b) PL spectra under 980, 800, and 1532 nm excitation and the upconversion image obtained from the LiREF4-based core/sextuple-shell composite. (c) Emission spectra, integrated intensity and emission FWHM of CsPbBr3@SiO2 nanodots immersed in water for 13 h as a function of pumping intensity under 800 nm laser excitation. (d) SEM image of the transferred elements on the complex surface of a specific target. (e) Schematic diagram of LED light excited bioimaging in vivo. (f) In vivo UC imaging of a nude mouse subcutaneously injected with Er@Nd NPs. | ||
Full-colour displays
The full-colour display technology requires multi-colour emitting materials that can potentially be translated into displays with higher resolution. Therefore, the development of light-harvesting materials with tuneable emission colours has been at the forefront of colour display technology.130–132 The variation in materials composition, phase and structure can provide a useful tool for producing a wide range of emission colours, but controlling the colour gamut in a material with a fixed composition remains a daunting challenge.133–136 Fortunately, multi-colour tuneable emissions have been widely available in lanthanide-doped nanomaterials, showing great promise for displays with much higher spatial resolutions.134–138Over the past few years, a single lanthanide activator featuring ladder-like arranged energy levels can produce a set of emission bands that span the wavelength range from ultraviolet to near-infrared (NIR). Under single wavelength excitation, the emission colour can be controlled by adjusting the intensity ratio of different emission peaks.19,132,139 However, this colour tuning technique requires repeated and rigorous experiments to obtain the optimal doping concentration.140,141 Liu et al. demonstrated a pulse width-modulated approach that offers exquisite control over the emission colour of lanthanide-doped upconversion nanocrystals.142 Hexagonal phase NaYF4 was chosen as the host material for the synthesis of colour-tuneable core–shell nanocrystals and they incorporated five sets of lanthanide ions into specific layers at precisely defined concentrations. Under 808 nm excitation with a continuous-wave (c.w.) diode laser, the nanocrystals gave rise to a dominant blue emission band at 470 nm. When irradiated at 980 nm with a diode laser, these nanocrystals showed a marked suppression in the blue emission, and exhibited a weak green emission band at 541 nm and a strong red emission band at 646 nm. The green/red emission intensity ratio can be tuned by using a pulsed diode laser with different pulse widths. The two lasers were aligned using a 900 nm short-pass dichroic mirror to be combined into one beam, which was focused to draw 3D images in the transparent monolith matrix under the control of laser scanning software. Different emission colours can be realized by simply modulating the on/off and pulse width of the two excitation lasers. Jang et al. synthesized LiREF4-based core/sextuple-shell (C/6S) upconversion nanoparticles. By combining incident near-infrared light with various wavelengths, full-color upconversion, including blue, cyan, green, yellow, orange, red, purple, and white upconversion, was achieved from the single composition (Fig. 7(b)).135 This method is capable of generating multi-view colour maps with high spatial resolution and accuracy and will provide attractive opportunities for applications in optical memory, multiplexed optical-chemical sensing, secure printing, etc.
Upconversion lasers
Upconversion nanomaterials enable large anti-Stokes shift emission generally through multiphoton absorption of low-energy photons (i.e., near-infrared light) to produce high-energy photons (i.e., ultraviolet or visible light), which can effectively invert the population of multiphoton-excited states. These properties make them an attractive gain medium for the development of anti-Stokes shift microlasers.143,144 The offer deeper penetration depth, higher signal-to-noise ratios and less damage to devices than conventional Stokes shift microlasers.108,145,146 Therefore, many attempts have been made in the last decade to realize anti-Stokes shift microlasers, mainly by coating upconversion nanomaterials on the surface of microresonators based on spherical or cylindrical dielectric cavities.147–149 However, they have lagged behind other microlasers due to their higher lasing thresholds and propagation losses in metallic cavities.Dong-Hwan Kim et al. integrated upconversion nanomaterials into monolithic microspheres with diameters as small as 2.44 μm by the liquid-quenching method.150 The microspheres have a highly smooth surface texture, resulting in a reduced intracavity loss and high pump-to-gain interactions for efficient light coupling to whispering gallery mode resonators. The superior properties of the liquid-quenched upconversion microspheres they developed lead to an ultra-low upconversion lasing threshold of 4.7 W cm−2 with a linewidth as narrow as 0.27 nm, which makes the anti-Stokes shift-based microlasers competitive with the state-of-the-art Stokes shift microlasers. Alex K.-Y. Jen et al. developed a Pb–S bonding approach to synthesize water-resistant perovskite@silica nanodots to keep their emission in water for over six weeks.151 The device exhibits a low lasing threshold and a high Q factor under two-photon pumping through coupling the amplified spontaneous emission of perovskite QDs to the whispering gallery modes of a tubular microcavity (Fig. 7(c)). Furthermore, a two-photon-pumped random laser device based on the perovskite@silica nanodot powder could still operate after the nanodots were dispersed in water for up to 15 days. The water-proof capability and outstanding optical properties make the perovskite@silica nanodots an ideal active material system for a variety of photonics and optoelectronics applications, which will significantly extend the application scenario of upconversion nanomaterials in solid-state laser devices.
3D printing
Stereolithographic 3D printing has become an indispensable part of human society and contributes greatly to aerospace materials, coatings, microfluidics, drug delivery, and so on.152–155 Typically, in conventional stereolithography (SLA) systems, for one-photon absorption dye-triggered polymerization, high spatial resolution is difficult to achieve due to the lack of a clear threshold for the excitation power, and in most cases all molecules in the entire beam path are excited.156 In addition, the print volume is limited by the poor penetration of UV or blue light. For photonic upconversion technology, the nonlinear dependence of the upconversion intensity on the excitation power opens up the possibility of spatially confined optical excitation.157 Polymerization based on photonic upconversion can only occur in the focused region where the light intensity exceeds the threshold, and selective photoreaction at the focusing point can be achieved by correctly designing the photoresist formulation.158–160Shih-Chi Chen et al. used ultrafast laser pulses to print 3D polymer materials on the micron/nano scale via TPA lithography.158 They developed a femtosecond projection two-photon lithography technique that ensures simultaneous spatial and temporal focusing of ultrafast light to achieve parallel printing of arbitrarily complex 3D structures with submicrometer resolution. Two-photon lithography-enabled 3D printing has good penetration ability and spatial resolution when compared to conventional polymerization using UV light. While conventional nanofabrication is usually limited to planar surfaces, the fabrication of arbitrary structures directly on morphologically complex surfaces is highly demanded for the development of flexible electronics and photonics devices. In recent years, the preparation of unconventional targets such as cylindrical surfaces, optical fibers, micro-optics, etc. has been achieved. Mattoli et al. have realized the printing of 3D microstructures on arbitrarily complex surfaces. A nice trick for this technology is the development of ultrathin polymer films as conformal substrates, allowing direct laser writing on the film surface via a TPP process. In addition, microstructures printed on polymer films can be spontaneously released in water and then efficiently transferred to specific targets (Fig. 7(d)).
Bioimaging
Photonic upconversion, as one of the most important non-invasive bioimaging techniques, has a positive role in the early diagnosis of various diseases and bioimaging-guided therapy. UCNPs with good chemical and optical stability, low toxicity and good biocompatibility can usually be irradiated with near-infrared light with higher tissue penetration depth than short-wavelength UV or visible light, avoiding the interference of background fluorescence from the organism (Fig. 7(e)).161 The characteristic emission of UCNPs is determined by the stepped atomic energy levels of each lanthanide-doped ion. Therefore, UCNPs with different sharp emission peaks can be prepared with only lanthanide-doped ions. By utilizing a rational UCNP design, multifunctional optical properties can be integrated into a single UCNPs with flexible external excitation, which in turn will bring upconversion-based bioimaging to a new stage of development.162Yan et al. reported the synthesis of Nd3+-doped UCNPs, which can be activated using an 808 nm laser.163 The 808 nm laser excites Nd3+ to its 4F5/2 state, followed by nonradiative relaxation to the 4F3/2 state. The energy could transfer to nearby Yb3+ and populate its 2F5/2 state and further transfer to nearby Yb3+ ions crossing the shell, and then upconversion between Yb3+ and Er3+ occurs. Because the absorption cross-section of Nd3+ in the NIR region is larger than that of Yb3+, energy transfer from Nd3+ to Yb3+ is highly efficient, and the resulting UCNPs exhibit a high upconversion efficiency under 808 nm excitation. This efficient Nd3+-doped emission of UCNPs was also demonstrated in mouse imaging experiments (Fig. 7(f)). The NIR image of mouse injected with Er@Nd nanoparticles shows a high signal-to-noise ratio under 808 nm laser excitation with a greatly minimized tissue overheating effect. The reduction of localized overheating facilitates the minimization of tissue damage during serial in vivo imaging. Zhang et al. presented a luminescence lifetime multiplexed detection/imaging approach based on lanthanide-doped nanocrystals with selectable luminescence lifetimes.122 They devised a core/multi-shell structure, which absorbs 808 nm photons to generate emission centered at 1525 nm through an efficient Nd3+ → Yb3+ → Er3+ energy transfer. This structure allows the luminescence lifetime to be tuned systematically by increasing the thickness of the energy relay layer and increasing the Er3+ concentration. The method creates a tunable lifetime range spanning three orders of magnitude in a single emission band. The lifetime is in good agreement with it compared to conventional spectral channels used for deep tissue imaging (6–8 mm). These studies have greatly advanced the bioimaging applications of UCNPs in the NIR region.
Conclusions and perspectives
In this review, we presented a brief overview of the three main mechanisms in upconversion luminescence. Furthermore, we introduced the ultrafast process in upconversion luminescence. We have summarized the ultrafast photophysics of phonon-assisted upconversion with intrinsic electron–phonon coupling as an intermediate energy level, the different dynamics between two-photon excitation and single-photon excitation because of the penetration depth and strategies for regulating the dynamics of energy migration in lanthanide upconversion. To date, various applications, such as photovoltaics, fluorescence imaging, anti-counterfeiting and lasers, have been demonstrated and have highlighted the elegance of upconversion nanomaterials. Despite these achievements, there are still many issues that need to be tackled in this area. Compared to downconversion, there is the significant drawback of low efficiency for upconversion. Since phonon-assisted upconversion usually involves intermediate states, new methods can be developed to generate new intermediate states or to extend the lifetime and reduce the complexation of intermediate state excitons through heterostructure and photonic or phononic crystal engineering. For TPA, the nonlinear nature requires an excitation strength several orders of magnitude higher than that of the OPA process, which limits its optoelectronic applications. To overcome this limitation, we should look for materials with large TPA cross-sections, high PL quantum yields and stability. UCNP nanostructure designs are often complex, which can lead to some difficulties in integrating multifunctional components into a single particle. Therefore, it is necessary to simplify the complex design of UCNP nanostructures to further reduce the difficulty of materials synthesis while meeting practical requirements. Nanostructures trade-off between the tunable optical properties, the specific surface area and the light-emitting quantum efficiency. In the past few years, emerging strategies such as inert shell modification, surface plasmon resonance, photonic crystal engineering, etc. have been used to enhance upconversion. One urgent task is to generate upconversion nanostructures with high fluorescence quantum yields to reduce energy loss during migration. We believe that investigations of UC will continue to be a top spot in interdisciplinary fields for the foreseeable future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. F. L. and W. N. D. acknowledge the support from the National Natural Science Foundation of China (12074086 and 22073022), the Beijing Municipal Natural Science Foundation (1222030), the Strategic Priority Research Program of Chinese Academy of Sciences (XDB36000000) and the National Key Research and Development Program (2022YFA1603701).References
- A. M. Jones, H. Yu, J. R. Schaibley, J. Yan, D. G. Mandrus, T. Taniguchi, K. Watanabe, H. Dery, W. Yao and X. Xu, Nat. Phys., 2016, 12, 323–327 Search PubMed.
- A. Granados del Águila, T. T. H. Do, J. Xing, W. J. Jee, J. B. Khurgin and Q. Xiong, Nano Res., 2020, 13, 1962–1969 CrossRef.
- J. Jadczak, L. Bryja, J. Kutrowska-Girzycka, P. Kapuściński, M. Bieniek, Y. S. Huang and P. Hawrylak, Nat. Commun., 2019, 10, 107 CrossRef CAS PubMed.
- Q. Wang and A. T. S. Wee, ACS Nano, 2021, 15, 10437–10443 CrossRef CAS PubMed.
- E. Lafalce, E. Amerling, Z.-G. Yu, P. C. Sercel, L. Whittaker-Brooks and Z. V. Vardeny, Nat. Commun., 2022, 13, 483 CrossRef CAS PubMed.
- Q. Zhang, X. Liu, M. I. B. Utama, G. Xing, T. C. Sum and Q. Xiong, Adv. Mater., 2016, 28, 276–283 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976–989 RSC.
- B. Zhou, B. Shi, D. Jin and X. Liu, Nat. Nanotechnol., 2015, 10, 924–936 CrossRef CAS PubMed.
- S. Zhang, M. Zhukovskyi, B. Jankó and M. Kuno, NPG Asia Mater., 2019, 11, 54 CrossRef CAS.
- J. B. Khurgin, Appl. Phys. Lett., 2014, 104, 221115 CrossRef.
- Q. Wang, Q. Zhang, X. Zhao, X. Luo, C. P. Y. Wong, J. Wang, D. Wan, T. Venkatesan, S. J. Pennycook, K. P. Loh, G. Eda and A. T. S. Wee, Nano Lett., 2018, 18, 6898–6905 CrossRef CAS PubMed.
- W. Chen, A. G. Joly and D. E. McCready, J. Chem. Phys., 2005, 122, 224708 CrossRef PubMed.
- Y. Zhou, Y. Huang, X. Xu, Z. Fan, J. B. Khurgin and Q. Xiong, Appl. Phys. Rev., 2020, 7, 041313 CAS.
- J. Olesiak-Banska, M. Waszkielewicz, P. Obstarczyk and M. Samoc, Chem. Soc. Rev., 2019, 48, 4087–4117 RSC.
- M. Drobizhev, N. S. Makarov, S. E. Tillo, T. E. Hughes and A. Rebane, Nat. Methods, 2011, 8, 393–399 CrossRef CAS PubMed.
- D. Kovalev, J. Diener, H. Heckler, G. Polisski, N. Künzner, F. Koch, A. L. Efros and M. Rosen, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 15841–15847 CrossRef CAS.
- W. Seidel, A. Titkov, J. P. André, P. Voisin and M. Voos, Phys. Rev. Lett., 1994, 73, 2356–2359 CrossRef CAS PubMed.
- F. Auzel, Chem. Rev., 2004, 104, 139–174 CrossRef CAS PubMed.
- S. Liu, L. Yan, J. Huang, Q. Zhang and B. Zhou, Chem. Soc. Rev., 2022, 51, 1729–1765 RSC.
- H. Dong, L.-D. Sun and C.-H. Yan, Chem. Soc. Rev., 2015, 44, 1608–1634 RSC.
- B. J. Roman and M. Sheldon, Chem. Commun., 2018, 54, 6851–6854 RSC.
- Y. V. Morozov, S. Zhang, M. C. Brennan, B. Janko and M. Kuno, ACS Energy Lett., 2017, 2, 2514–2515 CrossRef CAS.
- S. Ye, M. Zhao, M. Yu, M. Zhu, W. Yan, J. Song and J. Qu, J. Phys. Chem. C, 2018, 122, 3152–3156 CrossRef CAS.
- Z. Ye, X. Lin, N. Wang, J. Zhou, M. Zhu, H. Qin and X. Peng, Nat. Commun., 2021, 12, 4283 CrossRef CAS.
- D. Zhu, Y. Sun, S. Yuan, R. Gao, Y. Wang, X.-C. Ai and J.-P. Zhang, J. Phys. Chem. C, 2022, 126, 2447–2453 CrossRef CAS.
- Q. Wang, Q. Zhang, X. Zhao, Y. J. Zheng, J. Wang, X. Luo, J. Dan, R. Zhu, Q. Liang, L. Zhang, P. K. J. Wong, X. He, Y. L. Huang, X. Wang, S. J. Pennycook, G. Eda and A. T. S. Wee, Nano Lett., 2019, 19, 5595–5603 CrossRef CAS PubMed.
- N. Akizuki, S. Aota, S. Mouri, K. Matsuda and Y. Miyauchi, Nat. Commun., 2015, 6, 8920 CrossRef CAS.
- Y. Dai, P. Qi, G. Tao, G. Yao, B. Shi, Z. Liu, Z. Liu, X. He, P. Peng, Z. Dang, L. Zheng, T. Zhang, Y. Gong, Y. Guan, K. Liu and Z. Fang, Light: Sci. Appl., 2023, 12, 6 CrossRef CAS PubMed.
- S.-T. Ha, C. Shen, J. Zhang and Q. Xiong, Nat. Photonics, 2016, 10, 115–121 CrossRef CAS.
- P. Qi, Y. Dai, Y. Luo, G. Tao, L. Zheng, D. Liu, T. Zhang, J. Zhou, B. Shen, F. Lin, Z. Liu and Z. Fang, Light: Sci. Appl., 2022, 11, 176 CrossRef CAS PubMed.
- L. Li, X. Shang, S. Wang, N. Dong, C. Ji, X. Chen, S. Zhao, J. Wang, Z. Sun, M. Hong and J. Luo, J. Am. Chem. Soc., 2018, 140, 6806–6809 CrossRef CAS PubMed.
- Z. Wei, D. Guo, J. Thieme, C. Katan, V. M. Caselli, J. Even and T. J. Savenije, Nat. Commun., 2019, 10, 5342 CrossRef PubMed.
- A. C. Tam and C. K. N. Patel, Nature, 1979, 280, 304–306 CrossRef CAS.
- Y. Liu, J. Zhao, X. Xu, Y. Xu, W. Cui, Y. Yang and J. Li, Angew. Chem., Int. Ed., 2023, 62, e202308019 CrossRef CAS PubMed.
- Z. Zheng, T. Zhang, H. Liu, Y. Chen, R. T. K. Kwok, C. Ma, P. Zhang, H. H. Y. Sung, I. D. Williams, J. W. Y. Lam, K. S. Wong and B. Z. Tang, ACS Nano, 2018, 12, 8145–8159 CrossRef CAS PubMed.
- V. M. Caselli, Z. Wei, M. M. Ackermans, E. M. Hutter, B. Ehrler and T. J. Savenije, ACS Energy Lett., 2020, 5, 3821–3827 CrossRef CAS.
- S. Zhang, N. Dong, N. McEvoy, M. O'Brien, S. Winters, N. C. Berner, C. Yim, Y. Li, X. Zhang, Z. Chen, L. Zhang, G. S. Duesberg and J. Wang, ACS Nano, 2015, 9, 7142–7150 CrossRef CAS.
- W. Liu, J. Xing, J. Zhao, X. Wen, K. Wang, P. Lu and Q. Xiong, Adv. Opt. Mater., 2017, 5, 1601045 CrossRef.
- J. Wang, Y. Mi, X. Gao, J. Li, J. Li, S. Lan, C. Fang, H. Shen, X. Wen, R. Chen, X. Liu, T. He and D. Li, Adv. Opt. Mater., 2019, 7, 1900398 CrossRef.
- G. Walters, B. R. Sutherland, S. Hoogland, D. Shi, R. Comin, D. P. Sellan, O. M. Bakr and E. H. Sargent, ACS Nano, 2015, 9, 9340–9346 CrossRef CAS.
- J. Chen, K. Žídek, P. Chábera, D. Liu, P. Cheng, L. Nuuttila, M. J. Al-Marri, H. Lehtivuori, M. E. Messing, K. Han, K. Zheng and T. Pullerits, J. Phys. Chem. Lett., 2017, 8, 2316–2321 CrossRef CAS PubMed.
- G. Nagamine, J. O. Rocha, L. G. Bonato, A. F. Nogueira, Z. Zaharieva, A. A. R. Watt, C. H. de Brito Cruz and L. A. Padilha, J. Phys. Chem. Lett., 2018, 9, 3478–3484 CrossRef CAS PubMed.
- J. Chen, W. Zhang and T. Pullerits, Mater. Horiz., 2022, 9, 2255–2287 RSC.
- S. Castro-Fernández, C. M. Cruz, I. F. A. Mariz, I. R. Márquez, V. G. Jiménez, L. Palomino-Ruiz, J. M. Cuerva, E. Maçôas and A. G. Campaña, Angew. Chem., Int. Ed., 2020, 59, 7139–7145 CrossRef.
- R. Li, Z. Wei, H. Zhao, H. Yu, X. Fang, D. Fang, J. Li, T. He, R. Chen and X. Wang, Nanoscale, 2018, 10, 22766–22774 RSC.
- W. Chen, S. Bhaumik, S. A. Veldhuis, G. Xing, Q. Xu, M. Grätzel, S. Mhaisalkar, N. Mathews and T. C. Sum, Nat. Commun., 2017, 8, 15198 CrossRef CAS PubMed.
- W. Liu, X. Li, Y. Song, C. Zhang, X. Han, H. Long, B. Wang, K. Wang and P. Lu, Adv. Funct. Mater., 2018, 28, 1707550 CrossRef.
- Y. Fan, P. Tonkaev, Y. Wang, Q. Song, J. Han, S. V. Makarov, Y. Kivshar and S. Xiao, Nano Lett., 2021, 21, 7191–7197 CrossRef CAS PubMed.
- C. Becker, S. Burger, C. Barth, P. Manley, K. Jäger, D. Eisenhauer, G. Köppel, P. Chabera, J. Chen, K. Zheng and T. Pullerits, ACS Photonics, 2018, 5, 4668–4676 CrossRef CAS.
- N. Abeyasinghe, S. Kumar, K. Sun, J. F. Mansfield, R. Jin and T. Goodson III, J. Am. Chem. Soc., 2016, 138, 16299–16307 CrossRef CAS PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- X. Liu, C.-H. Yan and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1299–1301 RSC.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- H. Dong, L.-D. Sun and C.-H. Yan, Nanoscale, 2013, 5, 5703–5714 RSC.
- J. F. Suyver, A. Aebischer, D. Biner, P. Gerner, J. Grimm, S. Heer, K. W. Krämer, C. Reinhard and H. U. Güdel, Opt. Mater., 2005, 27, 1111–1130 CrossRef CAS.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- M.-K. Tsang, G. Bai and J. Hao, Chem. Soc. Rev., 2015, 44, 1585–1607 RSC.
- D. Hudry, I. A. Howard, R. Popescu, D. Gerthsen and B. S. Richards, Adv. Mater., 2019, 31, 1900623 CrossRef PubMed.
- F. Wang, S. Wen, H. He, B. Wang, Z. Zhou, O. Shimoni and D. Jin, Light: Sci. Appl., 2018, 7, 18007–18007 CrossRef CAS PubMed.
- X. Zhu, Q. Su, W. Feng and F. Li, Chem. Soc. Rev., 2017, 46, 1025–1039 RSC.
- S. Chen, A. Z. Weitemier, X. Zeng, L. He, X. Wang, Y. Tao, A. J. Y. Huang, Y. Hashimotodani, M. Kano, H. Iwasaki, L. K. Parajuli, S. Okabe, D. B. L. Teh, A. H. All, I. Tsutsui-Kimura, K. F. Tanaka, X. Liu and T. J. McHugh, Science, 2018, 359, 679–684 CrossRef CAS PubMed.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015, 44, 1526–1560 RSC.
- G. Sun, Y. Xie, L. Sun and H. Zhang, Nanoscale Horiz., 2021, 6, 766–780 RSC.
- W. Zheng, P. Huang, D. Tu, E. Ma, H. Zhu and X. Chen, Chem. Soc. Rev., 2015, 44, 1379–1415 RSC.
- W. Zheng, P. Huang, Z. Gong, D. Tu, J. Xu, Q. Zou, R. Li, W. You, J.-C. G. Bünzli and X. Chen, Nat. Commun., 2018, 9, 3462 CrossRef PubMed.
- X. Guo, R. Pu, Z. Zhu, S. Qiao, Y. Liang, B. Huang, H. Liu, L. Labrador-Páez, U. Kostiv, P. Zhao, Q. Wu, J. Widengren and Q. Zhan, Nat. Commun., 2022, 13, 2843 CrossRef CAS PubMed.
- M. Zhang, P. Huang, W. Zheng, X. Song, X. Shang, W. Zhang, D. Yang, X. Yi and X. Chen, Nano Lett., 2023, 23, 8576–8584 CrossRef CAS PubMed.
- D. Yang, W. Zheng, P. Huang, M. Zhang, W. Zhang, Z. Shao, W. Zhang, X. Yi and X. Chen, Aggregate, 2023, 4, e387 CrossRef CAS.
- D. L. Dexter, J. Chem. Phys., 2004, 21, 836–850 CrossRef.
- B. R. Judd, Phys. Rev., 1962, 127, 750–761 CrossRef CAS.
- C. M. Dodson and R. Zia, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 125102 CrossRef.
- E. M. Chan, D. J. Gargas, P. J. Schuck and D. J. Milliron, J. Phys. Chem. B, 2012, 116, 10561–10570 CrossRef CAS PubMed.
- X. Chen, D. Peng, Q. Ju and F. Wang, Chem. Soc. Rev., 2015, 44, 1318–1330 RSC.
- T. Sun, Y. Li, W. L. Ho, Q. Zhu, X. Chen, L. Jin, H. Zhu, B. Huang, J. Lin, B. E. Little, S. T. Chu and F. Wang, Nat. Commun., 2019, 10, 1811 CrossRef PubMed.
- F. Wang, R. Deng, J. Wang, Q. Wang, Y. Han, H. Zhu, X. Chen and X. Liu, Nat. Mater., 2011, 10, 968–973 CrossRef CAS PubMed.
- X. Xie, N. Gao, R. Deng, Q. Sun, Q.-H. Xu and X. Liu, J. Am. Chem. Soc., 2013, 135, 12608–12611 CrossRef CAS PubMed.
- J. Zuo, D. Sun, L. Tu, Y. Wu, Y. Cao, B. Xue, Y. Zhang, Y. Chang, X. Liu, X. Kong, W. J. Buma, E. J. Meijer and H. Zhang, Angew. Chem., Int. Ed., 2018, 57, 3054–3058 CrossRef CAS PubMed.
- L. Tu, K. Wu, Y. Luo, E. Wang, J. Yuan, J. Zuo, D. Zhou, B. Li, J. Zhou, D. Jin and H. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202217100 CrossRef CAS PubMed.
- B. Zhou, L. Yan, J. Huang, X. Liu, L. Tao and Q. Zhang, Nat. Photonics, 2020, 14, 760–766 CrossRef CAS.
- C. Dong, A. Korinek, B. Blasiak, B. Tomanek and F. C. J. M. van Veggel, Chem. Mater., 2012, 24, 1297–1305 CrossRef CAS.
- K. Prorok, A. Bednarkiewicz, B. Cichy, A. Gnach, M. Misiak, M. Sobczyk and W. Strek, Nanoscale, 2014, 6, 1855–1864 RSC.
- Q. Liu, Y. Zhang, C. S. Peng, T. Yang, L.-M. Joubert and S. Chu, Nat. Photonics, 2018, 12, 548–553 CrossRef CAS PubMed.
- I. Taarit, F. Alves, A. Benchohra, L. Guénée, B. Golesorkhi, A. Rosspeintner, A. Fürstenberg and C. Piguet, J. Am. Chem. Soc., 2023, 145, 8621–8633 CAS.
- X. Cheng, H. Ge, Y. Wei, K. Zhang, W. Su, J. Zhou, L. Yin, Q. Zhan, S. Jing and L. Huang, ACS Nano, 2018, 12, 10992–10999 CrossRef CAS PubMed.
- F. Wang, Y. Han, C. S. Lim, Y. Lu, J. Wang, J. Xu, H. Chen, C. Zhang, M. Hong and X. Liu, Nature, 2010, 463, 1061–1065 CrossRef CAS PubMed.
- A. Yin, Y. Zhang, L. Sun and C. Yan, Nanoscale, 2010, 2, 953–959 RSC.
- H. Qiu, G. Chen, R. Fan, L. Yang, C. Liu, S. Hao, M. J. Sailor, H. Ågren, C. Yang and P. N. Prasad, Nanoscale, 2014, 6, 753–757 RSC.
- Y. Zhang, R. Wen, J. Hu, D. Guan, X. Qiu, Y. Zhang, D. S. Kohane and Q. Liu, Nat. Commun., 2022, 13, 5927 CrossRef CAS PubMed.
- H. Chen, T. Ming, L. Zhao, F. Wang, L.-D. Sun, J. Wang and C.-H. Yan, Nano Today, 2010, 5, 494–505 CrossRef CAS.
- W. Feng, L.-D. Sun and C.-H. Yan, Chem. Commun., 2009, 4393–4395, 10.1039/B909164E.
- P. Yuan, Y. H. Lee, M. K. Gnanasammandhan, Z. Guan, Y. Zhang and Q.-H. Xu, Nanoscale, 2012, 4, 5132–5137 RSC.
- S. Schietinger, T. Aichele, H.-Q. Wang, T. Nann and O. Benson, Nano Lett., 2010, 10, 134–138 CrossRef CAS PubMed.
- Y. Ji, W. Xu, D. Li, D. Zhou, X. Chen, N. Ding, J. Li, N. Wang, X. Bai and H. Song, Nano Energy, 2019, 61, 211–220 CrossRef CAS.
- K. Deng, L. Xu, X. Guo, X. Wu, Y. Liu, Z. Zhu, Q. Li, Q. Zhan, C. Li and Z. Quan, Small, 2020, 16, 2002066 CrossRef CAS PubMed.
- F. Kang, J. He, T. Sun, Z. Y. Bao, F. Wang and D. Y. Lei, Adv. Funct. Mater., 2017, 27, 1701842 CrossRef.
- Y. Meng, D. Huang, H. Li, X. Feng, F. Li, Q. Liang, T. Ma, J. Han, J. Tang, G. Chen and X.-W. Chen, Nat. Photonics, 2023, 17, 73–81 CrossRef CAS.
- B. S. Richards, D. Hudry, D. Busko, A. Turshatov and I. A. Howard, Chem. Rev., 2021, 121, 9165–9195 CrossRef CAS PubMed.
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC.
- A. Bhardwaj, J. Kaur, M. Wuest and F. Wuest, Nat. Commun., 2017, 8, 1 CrossRef PubMed.
- X. Wang, W. W. Yu, J. Zhang, J. Aldana, X. Peng and M. Xiao, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 125318 CrossRef.
- T. T. Tran, B. Regan, E. A. Ekimov, Z. Mu, Y. Zhou, W.-B. Gao, P. Narang, A. S. Solntsev, M. Toth, I. Aharonovich and C. Bradac, Sci. Adv., 2019, 5, eaav9180 CrossRef CAS PubMed.
- E. Poles, D. C. Selmarten, O. I. Mićić and A. J. Nozik, Appl. Phys. Lett., 1999, 75, 971–973 CrossRef CAS.
- J. B. Khurgin, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 235206 CrossRef.
- G. Rupper, N. H. Kwong and R. Binder, Phys. Rev. Lett., 2006, 97, 117401 CrossRef CAS PubMed.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- Q. Han, W. Wu, W. Liu and Y. Yang, RSC Adv., 2017, 7, 35757–35764 RSC.
- Y. Xu, Q. Chen, C. Zhang, R. Wang, H. Wu, X. Zhang, G. Xing, W. W. Yu, X. Wang, Y. Zhang and M. Xiao, J. Am. Chem. Soc., 2016, 138, 3761–3768 CrossRef CAS PubMed.
- Y. Wang, X. Li, X. Zhao, L. Xiao, H. Zeng and H. Sun, Nano Lett., 2016, 16, 448–453 CrossRef CAS PubMed.
- B. Yang, F. Zhang, J. Chen, S. Yang, X. Xia, T. Pullerits, W. Deng and K. Han, Adv. Mater., 2017, 29, 1703758 CrossRef PubMed.
- M. Sebastian, J. A. Peters, C. C. Stoumpos, J. Im, S. S. Kostina, Z. Liu, M. G. Kanatzidis, A. J. Freeman and B. W. Wessels, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 92, 235210 CrossRef.
- Y. Yamada, T. Yamada, L. Q. Phuong, N. Maruyama, H. Nishimura, A. Wakamiya, Y. Murata and Y. Kanemitsu, J. Am. Chem. Soc., 2015, 137, 10456–10459 CrossRef CAS PubMed.
- T. Yamada, T. Aharen and Y. Kanemitsu, Phys. Rev. Lett., 2018, 120, 057404 CrossRef CAS PubMed.
- B. Wu, H. T. Nguyen, Z. Ku, G. Han, D. Giovanni, N. Mathews, H. J. Fan and T. C. Sum, Adv. Energy Mater., 2016, 6, 1600551 CrossRef.
- Y. Fang, H. Wei, Q. Dong and J. Huang, Nat. Commun., 2017, 8, 14417 CrossRef CAS PubMed.
- Q. Wei, B. Du, B. Wu, J. Guo, M. J. Li, J. Fu, Z. Zhang, J. Yu, T. Hou, G. Xing, T. C. Sum and W. Huang, Adv. Opt. Mater., 2017, 5, 1700809 CrossRef.
- J. Chen, P. Chábera, T. Pascher, M. E. Messing, R. Schaller, S. Canton, K. Zheng and T. Pullerits, J. Phys. Chem. Lett., 2017, 8, 5119–5124 CrossRef CAS PubMed.
- K. L. Reddy, R. Balaji, A. Kumar and V. Krishnan, Small, 2018, 14, 1801304 CrossRef PubMed.
- J. Zhao, Z. Lu, Y. Yin, C. McRae, J. A. Piper, J. M. Dawes, D. Jin and E. M. Goldys, Nanoscale, 2013, 5, 944–952 RSC.
- M. Tan, F. Li, X. Wang, R. Fan and G. Chen, ACS Nano, 2020, 14, 6532–6538 CrossRef CAS PubMed.
- H. Li, M. Tan, X. Wang, F. Li, Y. Zhang, L. Zhao, C. Yang and G. Chen, J. Am. Chem. Soc., 2020, 142, 2023–2030 CrossRef CAS PubMed.
- X. Liu, Z.-H. Chen, H. Zhang, Y. Fan and F. Zhang, Angew. Chem., Int. Ed., 2021, 60, 7041–7045 CrossRef CAS PubMed.
- Y. Fan, P. Wang, Y. Lu, R. Wang, L. Zhou, X. Zheng, X. Li, J. A. Piper and F. Zhang, Nat. Nanotechnol., 2018, 13, 941–946 CrossRef CAS PubMed.
- P. Pringsheim, Z. Phys., 1929, 57, 739–746 CrossRef CAS.
- M. Sheik-Bahae and R. I. Epstein, Phys. Rev. Lett., 2004, 92, 247403 CrossRef PubMed.
- D. Li, J. Zhang, X. Wang, B. Huang and Q. Xiong, Nano Lett., 2014, 14, 4724–4728 CrossRef CAS PubMed.
- M. Sheik-Bahae and R. I. Epstein, Nat. Photonics, 2007, 1, 693–699 CrossRef CAS.
- J. Zhang, D. Li, R. Chen and Q. Xiong, Nature, 2013, 493, 504–508 CrossRef CAS PubMed.
- J.-M. Lai, Y.-J. Sun, Q.-H. Tan, P.-H. Tan and J. Zhang, Nano Lett., 2022, 22, 7129–7135 CrossRef CAS PubMed.
- J. Zhang, D. Li and Q. Xiong, Nature, 2019, 570, E62–E64 CrossRef CAS PubMed.
- T.-H. Kim, K.-S. Cho, E. K. Lee, S. J. Lee, J. Chae, J. W. Kim, D. H. Kim, J.-Y. Kwon, G. Amaratunga, S. Y. Lee, B. L. Choi, Y. Kuk, J. M. Kim and K. Kim, Nat. Photonics, 2011, 5, 176–182 CrossRef CAS.
- B. Zhu, B. Qian, Y. Liu, C. Xu, C. Liu, Q. Chen, J. Zhou, X. Liu and J. Qiu, NPG Asia Mater., 2017, 9, e394–e394 CrossRef CAS.
- C. Zhang, L. Yang, J. Zhao, B. Liu, M.-Y. Han and Z. Zhang, Angew. Chem., Int. Ed., 2015, 54, 11531–11535 CrossRef CAS PubMed.
- X. Li, Z. Guo, T. Zhao, Y. Lu, L. Zhou, D. Zhao and F. Zhang, Angew. Chem., Int. Ed., 2016, 55, 2464–2469 CrossRef CAS PubMed.
- M. Wu, L. Yan, T. Wang, B. Zhou and Q. Zhang, Adv. Funct. Mater., 2019, 29, 1804160 CrossRef.
- A. R. Hong, J.-H. Kyhm, G. Kang and H. S. Jang, Nano Lett., 2021, 21, 4838–4844 CrossRef CAS PubMed.
- X. Liu, H. Chen, Y. Wang, Y. Si, H. Zhang, X. Li, Z. Zhang, B. Yan, S. Jiang, F. Wang, S. Weng, W. Xu, D. Zhao, J. Zhang and F. Zhang, Nat. Commun., 2021, 12, 5662 CrossRef CAS PubMed.
- E. Downing, L. Hesselink, J. Ralston and R. Macfarlane, Science, 1996, 273, 1185–1189 CrossRef CAS.
- X. Yin, H. Wang, Y. Tian, M. Xing, Y. Fu and X. Luo, Nanoscale, 2018, 10, 9673–9678 RSC.
- Y. Zhong, G. Tian, Z. Gu, Y. Yang, L. Gu, Y. Zhao, Y. Ma and J. Yao, Adv. Mater., 2014, 26, 2831–2837 CrossRef CAS PubMed.
- B. Zhou, J. Huang, L. Yan, X. Liu, N. Song, L. Tao and Q. Zhang, Adv. Mater., 2019, 31, 1806308 CrossRef PubMed.
- B. Zhou, L. Yan, L. Tao, N. Song, M. Wu, T. Wang and Q. Zhang, Adv. Sci., 2018, 5, 1700667 CrossRef PubMed.
- R. Deng, F. Qin, R. Chen, W. Huang, M. Hong and X. Liu, Nat. Nanotechnol., 2015, 10, 237–242 CrossRef CAS PubMed.
- X. Qin, J. Xu, Y. Wu and X. Liu, ACS Cent. Sci., 2019, 5, 29–42 CrossRef CAS PubMed.
- R. Scheps, Prog. Quantum Electron., 1996, 20, 271–358 CrossRef CAS.
- H. Dong, C. Zhang, X. Liu, J. Yao and Y. S. Zhao, Chem. Soc. Rev., 2020, 49, 951–982 RSC.
- Z. Gu, K. Wang, W. Sun, J. Li, S. Liu, Q. Song and S. Xiao, Adv. Opt. Mater., 2016, 4, 472–479 CrossRef CAS.
- A. Fernandez-Bravo, K. Yao, E. S. Barnard, N. J. Borys, E. S. Levy, B. Tian, C. A. Tajon, L. Moretti, M. V. Altoe, S. Aloni, K. Beketayev, F. Scotognella, B. E. Cohen, E. M. Chan and P. J. Schuck, Nat. Nanotechnol., 2018, 13, 572–577 CrossRef CAS PubMed.
- L. Jin, Y. Wu, Y. Wang, S. Liu, Y. Zhang, Z. Li, X. Chen, W. Zhang, S. Xiao and Q. Song, Adv. Mater., 2019, 31, 1807079 CrossRef PubMed.
- H. Zhu, X. Chen, L. M. Jin, Q. J. Wang, F. Wang and S. F. Yu, ACS Nano, 2013, 7, 11420–11426 CrossRef CAS PubMed.
- B.-S. Moon, T. K. Lee, W. C. Jeon, S. K. Kwak, Y.-J. Kim and D.-H. Kim, Nat. Commun., 2021, 12, 4437 CrossRef CAS PubMed.
- S. Li, D. Lei, W. Ren, X. Guo, S. Wu, Y. Zhu, A. L. Rogach, M. Chhowalla and A. K. Y. Jen, Nat. Commun., 2020, 11, 1192 CrossRef CAS PubMed.
- H. Wang, W. Zhang, D. Ladika, H. Yu, D. Gailevičius, H. Wang, C.-F. Pan, P. N. S. Nair, Y. Ke, T. Mori, J. Y. E. Chan, Q. Ruan, M. Farsari, M. Malinauskas, S. Juodkazis, M. Gu and J. K. W. Yang, Adv. Funct. Mater., 2023, 33, 2214211 CrossRef CAS.
- Y. Yuan, L. Chen, Z. Shi and J. Chen, Nanomaterials, 2022, 12, 391 CrossRef CAS PubMed.
- J.-M. Park, H. Lee, H.-S. Choe, S.-K. Ahn, K.-Y. Seong, S. Y. Yang and J.-H. Kim, J. Mater. Chem. C, 2022, 10, 4584–4589 RSC.
- S. Kawata, H.-B. Sun, T. Tanaka and K. Takada, Nature, 2001, 412, 697–698 CrossRef CAS PubMed.
- Y. Rong, R. Liu, P. Jin, C. Liu, X. Wang, L. Fang, L. Chen, W. Wu and C. Yang, J. Mater. Chem. A, 2023, 11, 5895–5901 RSC.
- V. Hahn, T. Messer, N. M. Bojanowski, E. R. Curticean, I. Wacker, R. R. Schröder, E. Blasco and M. Wegener, Nat. Photonics, 2021, 15, 932–938 CrossRef CAS.
- S. K. Saha, D. Wang, V. H. Nguyen, Y. Chang, J. S. Oakdale and S.-C. Chen, Science, 2019, 366, 105–109 CrossRef CAS PubMed.
- J. Zhou, Q. Liu, W. Feng, Y. Sun and F. Li, Chem. Rev., 2015, 115, 395–465 CrossRef CAS PubMed.
- J. Wong, S. Wei, R. Meir, N. Sadaba, N. A. Ballinger, E. K. Harmon, X. Gao, G. Altin-Yavuzarslan, L. D. Pozzo, L. M. Campos and A. Nelson, Adv. Mater., 2023, 35, 2207673 CrossRef CAS PubMed.
- F. Wang, W. B. Tan, Y. Zhang, X. Fan and M. Wang, Nanotechnology, 2006, 17, R1 CrossRef CAS.
- H. Dong, S.-R. Du, X.-Y. Zheng, G.-M. Lyu, L.-D. Sun, L.-D. Li, P.-Z. Zhang, C. Zhang and C.-H. Yan, Chem. Rev., 2015, 115, 10725–10815 CrossRef CAS PubMed.
- Y.-F. Wang, G.-Y. Liu, L.-D. Sun, J.-W. Xiao, J.-C. Zhou and C.-H. Yan, ACS Nano, 2013, 7, 7200–7206 CrossRef CAS PubMed.
- B. Wu, A. Wang, J. Fu, Y. Zhang, C. Yang, Y. Gong, C. Jiang, M. Long, G. Zhou, S. Yue, W. Ma and X. Liu, Sci. Adv., 2023, 9, eadi9347 CrossRef CAS PubMed.
- G. Xing, Y. Liao, X. Wu, S. Chakrabortty, X. Liu, E. K. L. Yeow, Y. Chan and T. C. Sum, ACS Nano, 2012, 6, 10835–10844 CrossRef CAS PubMed.
- Y. Wang, V. D. Ta, Y. Gao, T. C. He, R. Chen, E. Mutlugun, H. V. Demir and H. D. Sun, Adv. Mater., 2014, 26, 2954–2961 CrossRef CAS PubMed.
- G. Xing, S. Chakrabortty, S. W. Ngiam, Y. Chan and T. C. Sum, J. Phys. Chem. C, 2011, 115, 17711–17716 CrossRef CAS.
- Y. Liu, N. Kang, J. Lv, Z. Zhou, Q. Zhao, L. Ma, Z. Chen, L. Ren and L. Nie, Adv. Mater., 2016, 28, 6411–6419 CrossRef CAS PubMed.
- B. Liu, Y. Chen, C. Li, F. He, Z. Hou, S. Huang, H. Zhu, X. Chen and J. Lin, Adv. Funct. Mater., 2015, 25, 4717–4729 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |