
A reflection on ‘Protein coronas suppress the hemolytic activity of hydrophilic and hydrophobic nanoparticles’
Cristina-Maria
Hirschbiegel
 ,
Mingdi
Jiang
,
Jungmi
Park
and
Vincent M.
Rotello
*
,
Mingdi
Jiang
,
Jungmi
Park
and
Vincent M.
Rotello
*
Department of Chemistry, University of Massachusetts Amherst, Amherst, 01002, MA, USA. E-mail: rotello@umass.edu
First published on 13th February 2024
Abstract
Nanomaterials are at the forefront of modern therapeutics. The systemic administration of nanomaterials, however, can disrupt red blood cells. This hemolysis impacts the applicability of nanomaterials for biomedical applications. Our 2014 Materials Horizons communication (K. Saha, D. F. Moyano and V. M. Rotello, Mater. Horiz., 2014, 1, 102-105, https://doi.org/10.1039/C3MH00075C) highlighted the importance of nanoparticle hydrophobicity in determining hemolytic activity and how the formation of a protein corona can blunt the hemolytic response. This reflection looks at how the findings of this paper are intertwined with ongoing research in nanotherapeutics.
1. Introduction
The systemic administration of nanoparticles may cause the rupture of red blood cells (hemolysis), leading to hemolytic anemia, renal toxicity, and hypertension.1,2 Quantifying and minimizing the hemolytic properties of nanoparticles is crucial for advancing nanomedicine.3 Significantly, the formation of protein coronas on nanoparticle surfaces during blood circulation will alter the physiochemical properties of these nanoparticles and define their physiological responses.4,5 We probed the effect of nanoparticle hydrophobicity on hemolysis, looking at hemolysis in the absence and presence of serum proteins. Our 2014 communication in Materials Horizons (https://doi.org/10.1039/C3MH00075C)6 demonstrated a strong correlation between increased nanoparticle surface hydrophobicity and hemolysis (Fig. 1). What was surprising, however, was that this correlation was substantially attenuated by serum proteins, dramatically increasing the range of particle hydrophobicity that could be used without hemolysis.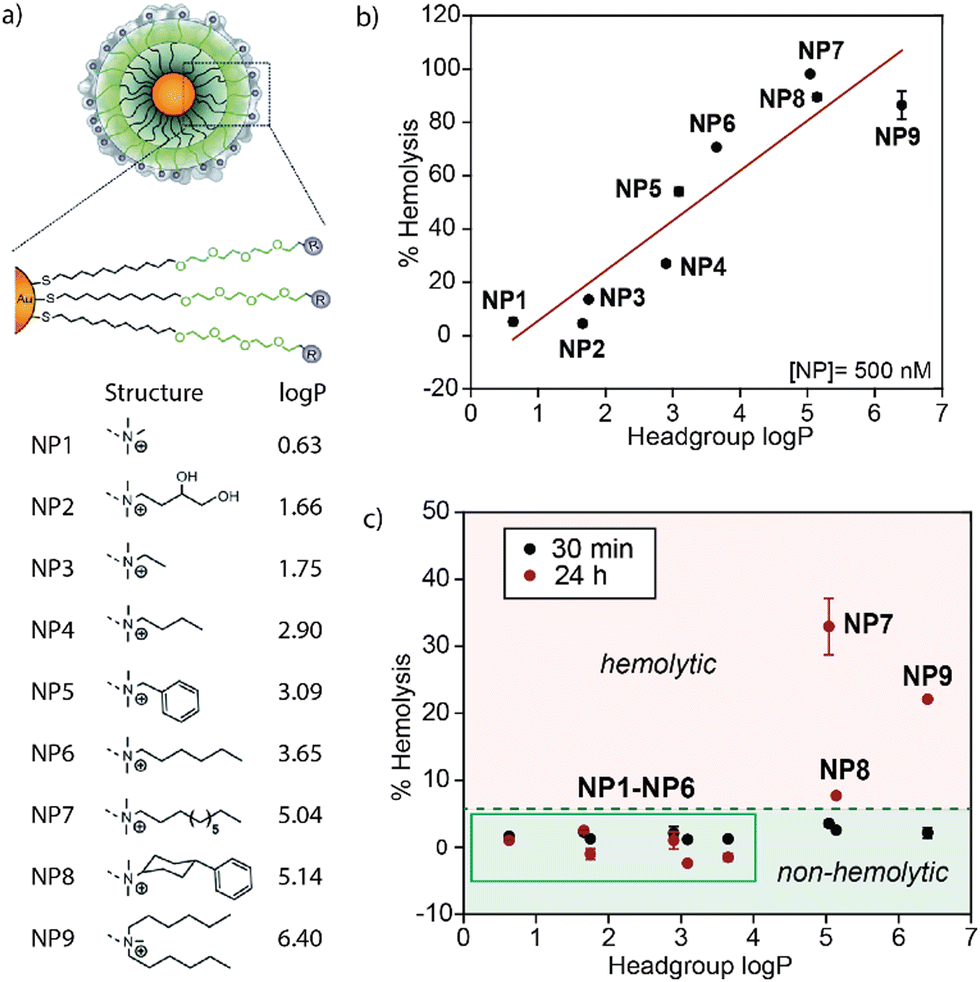 | ||
| Fig. 1 (a) Structure of Au-NPs with the respective headgroups and log P. With increasing log P of the headgroup, the overall hydrophobicity of the Au-NP increases. (b) The hemolytic activity of Au-NPs in serum-free buffer increases with the log P of the headgroup. (c) The hemolytic properties of the nanoparticles are strongly blunted in the presence of serum. Reproduced from ref. 6 with permission from The Royal Society of Chemistry. | ||
The inert surface and ease of functionalization of gold nanoparticles (Au-NPs) make them ideal candidates for biomedical applications such as biosensing and drug delivery.7 For the 2014 study, we synthesized ultrasmall 2 nm Au-NPs decorated with an organic ligand consisting of a hydrophobic segment, a hydrophilic segment, and a cationic headgroup (Fig. 1a).6 This design strategy proved useful for synthesizing stable nanoparticles with high water solubility.7 The cationic headgroup also enabled nanoparticle–protein interactions for protein delivery and sensing. This versatile ligand structure was used to assess the impact of surface hydrophobicity on the hemolysis of erythrocytes and protein corona formation by changing the headgroup structure. An increase in the hydrophobicity of organic ligands attached to the gold nanoparticle surface drastically increased the hemolytic effect of the particles in the absence of plasma proteins (Fig. 1b). In the presence of serum proteins, however, hemolysis was observed with only the most hydrophobic particles, demonstrating the protective role of protein coronas for improved blood compatibility of therapeutic nanomaterials (Fig. 1c).
The particle design strategies highlighted the critical role of hydrophobicity on nanoparticle physiochemical behaviors, encouraging researchers to explore the modulation of the biological functions of nanoparticles by tuning their hydrophobicity.8–10 The research described in this work has impacted the surface design of nanoparticles and facilitated the development of design strategies that consider blood biocompatibility. Also, these studies provide an example of how protein corona formation can positively affect nanoparticle behavior by inhibiting hemolysis.
The research published in Materials Horizons has facilitated the development of new nanomedicine platforms, and facilitated the transition between in vitro and in vivo studies.11,12 In particular, by establishing that the protein corona formation and hemolysis depend on the nanoparticle surface design, this early research has paved the way for future studies assessing (1) the biocompatibility of different nanomaterials, (2) the use of protein coronas for creating responsive systems, and (3) the impact of protein identity on biocompatibility and intracellular uptake. As an example, inorganic and polymeric nanoparticles that showed a minimal hemolytic response in the presence of erythrocytes were developed.1,13,14 Other researchers have likewise demonstrated a significant decrease in the toxicity of polystyrene nanoparticles by forming a protein corona.15 Additionally, studies found that forming a protein corona around nanoparticles increases the colloidal stability of metal nanoparticles.16 This reflection will discuss studies applying nanoparticles and protein corona interplay for biological and medical applications.1,13,14 Future applications and the importance of the previous findings for the biomedical future of nanoparticle-based systems will be discussed.
2. Increasing the biocompatibility of nanomaterials
Our research provided a detailed understanding of the hemolytic activity of gold nanoparticles. These studies provided a model system that inspired later studies that assessed how forming a protein corona around different types of nanoparticles dictated their therapeutic efficacy.6,17–19 In particular, inorganic nanomaterials often possess antimicrobial or anti-tumor activity, making them promising therapeutic agents.13,20–22 As an example, silver nanoparticles (Ag-NPs) act as non-specific antimicrobial agents and are useful for treating multi-drug-resistant bacteria.23,24 However, Ag-NPs have demonstrated toxicity toward mammalian cells, significantly hampering their applicability in biomedicine.25 Giacomelli and co-workers fabricated polymer-coated Ag-NPs that form protein coronas when exposed to fetal bovine serum (FBS).13 The coatings consisted of polyethylimine (PEI), polyvinylpyrrolidine (PVP), or poly(ethylene oxide)-b-poly(2-vinyl pyridine) (PEO-b-P2VP), respectively. Protein coronas were isolated after incubation in FBS and analyzed using liquid chromatography-mass spectrometry (LC-MS). The results indicated that the choice of polymer coating determines the identity of the protein corona and that forming a protein corona significantly decreased the cytotoxicity of these particles to fibroblast cells (Fig. 2a). The impact of the polymer-coated Ag-NPs on antimicrobial performance against E. coli was determined (Fig. 2b). At a higher polymer-to-Ag-NP ratio, the antimicrobial activity decreased due to the slower escape of silver ions.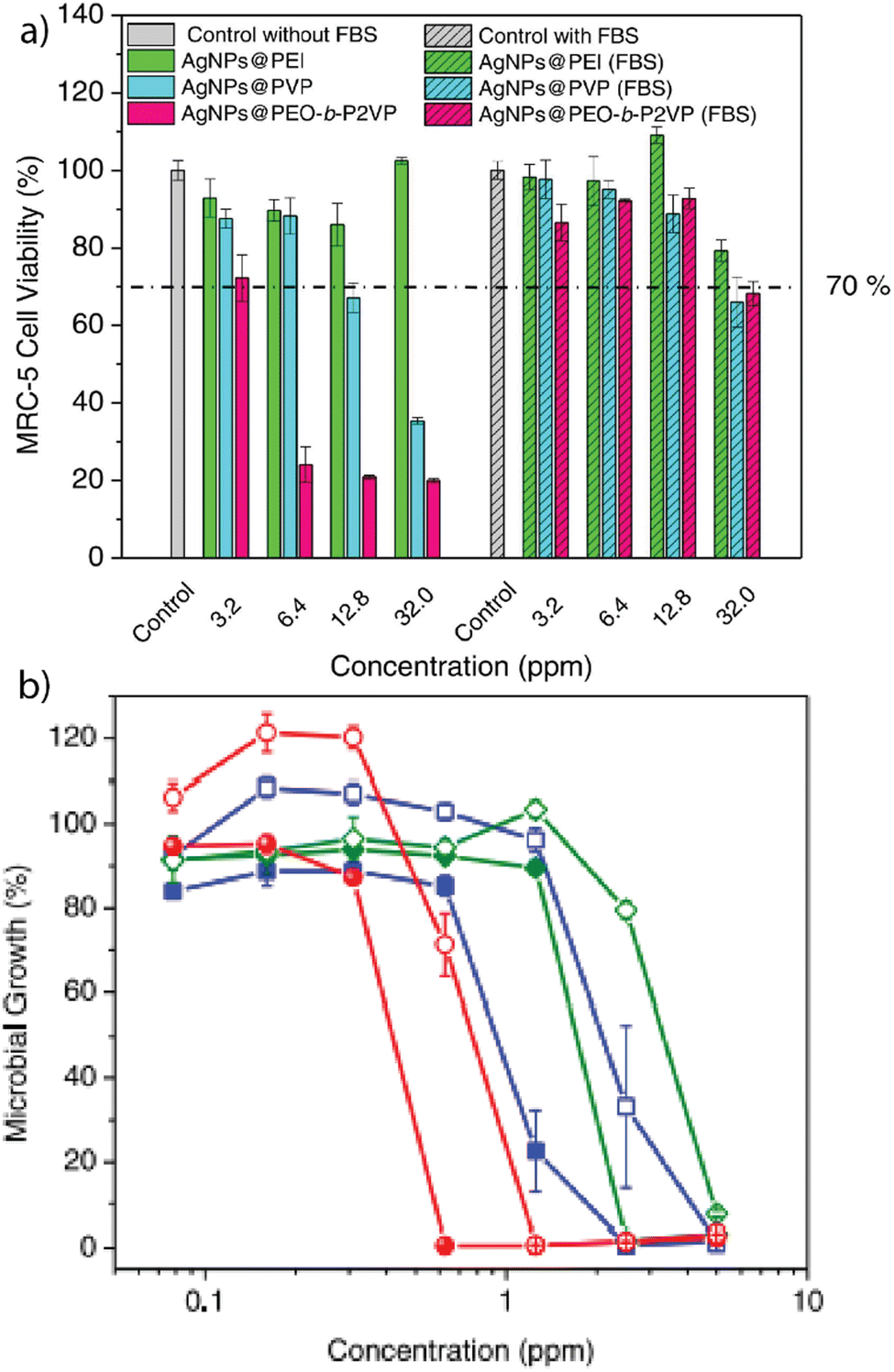 | ||
| Fig. 2 (a) Viability of MRC-5 fibroblast cells after incubation with Ag-NPs or polymer-stabilized Ag-NPs, based on the mass concentration of silver. (b) Effect of the respective Ag-NPs and polymer-coated Ag-NPs on the microbial growth of E. coli after 24 h incubation. Adapted from ref. 13 with permission from Elsevier. | ||
Cedervall and co-workers explored the changes in protein corona identity depending on the nanoparticle surface design using hydrophobic polystyrene (PS) nanoparticles in vivo.15 Spherical PS nanoparticles (53 or 200 nm) were fabricated and functionalized with either amine or carboxylate moieties. Cell viability studies indicated higher cytotoxicity of the 53 nm PS nanoparticles than the 200 nm particles. The planktonic crustacean Daphnia magna ingests nanoparticles of less than 5 μm. After ingestion, protein coronas were formed around the particles. The particles were isolated via centrifugation, and the corona was desorbed from the particle surface and analyzed using LC-MS. Larger proteins (58–171 kDa) preferably bound to the larger cationic nanoparticles, while smaller proteins (11–72 kDa) bound to smaller nanoparticles. The changes in cytotoxicity between the differently sized nanoparticles were explained by the fact that more proteins essential to the survival of the organism formed a protein corona around the 53 nm PS nanoparticles. While the nanoparticle size played an essential role in the protein corona identity, the charge impacted the number of proteins adsorbed to the surface. Negatively charged particles bound to significantly fewer proteins due to charge–charge repulsion of the particle with the negatively charged protein surfaces. However, carboxylate functionalized PS nanoparticles bound triglycerides, forming a lipid corona around the particle. These studies illustrate the importance of the lipid corona, an aspect that was not considered in our 2014 work.
3. Protein corona formation and drug release
Building upon the insights from our previous research, researchers have delved deeper into understanding the properties influencing the formation of protein coronas of nanoparticles. Understanding which properties of the nanomaterials lead to the formation of a protein corona is crucial for employing the corona to enable and regulate therapeutic activity. Mukherjee and co-workers created monometallic and bimetallic gold nanorods (Au-NRs) composed of either gold and palladium or gold and copper.19 The bimetallic composition enhanced the formation of a protein corona due to stronger adsorption of proteins to the surface. The nanorods were incubated with human serum albumin (HSA) or transferrin, respectively. HSA and gold–palladium nanorods formed a stable protein corona due to the interaction of palladium with aromatic amino acids on the protein surface. The anti-cancer drug doxorubicin (Dox) was loaded into the protein corona of the Au-NRs. Drug release was monitored by measuring the absorption of Dox in the supernatant at 485 nm (Fig. 3a). The cytotoxicity of the nanoparticles with and without loaded Dox was tested on MCF-7 breast cancer cells (Fig. 3b). Protein corona-coated Au-NRs showed decreased cytotoxicity. However, upon loading of the drug, the cytotoxicity (i.e., chemotherapeutic efficacy) of all Au-NRs increased significantly, using the protein corona to create a sustained drug release system.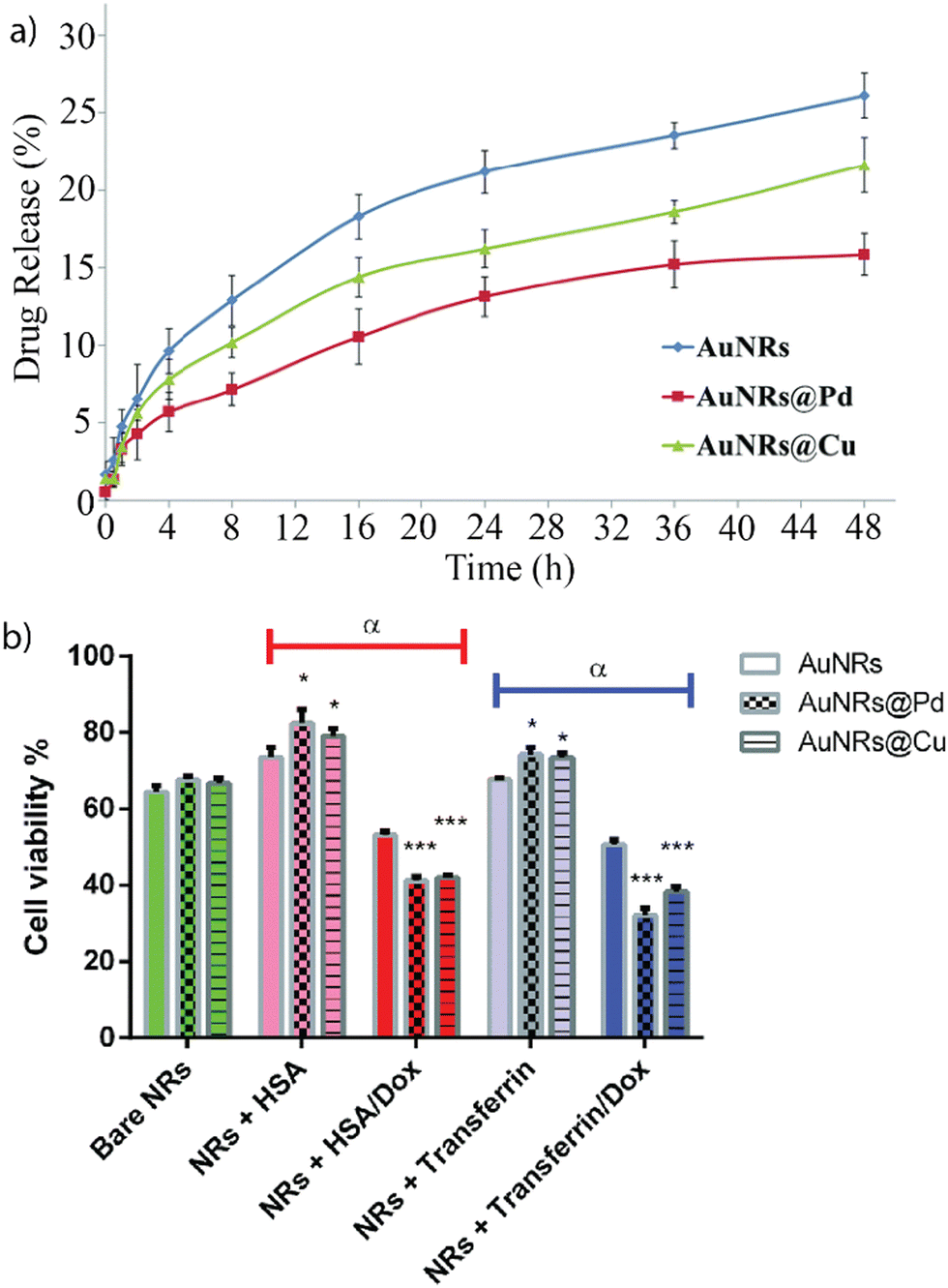 | ||
| Fig. 3 (a) Dox release for bare Au-NRs and bimetallic Au-NRs over 48 h. (b) Impact of nanomaterial with and without encapsulated Dox on cell viability of MCF-7 cells after 48 h incubation. Dox encapsulation significantly increases the cytotoxicity of the nanomaterial. Adapted from ref. 19 with permission from the Royal Society of Chemistry. | ||
Depending on the surface design of the nanomaterials, protein coronas forming around the nanoparticle can be “hard” or “soft”.26,27 Hard protein coronas are irreversible due to permanent adsorption to the nanoparticle surface.28 Soft corona formation is reversible, and proteins can dissociate from the particle surface.27 We engineered 2 nm gold nanoparticles to form a hard or a soft corona in a serum-containing medium (Fig. 4).29 A bioorthogonal ruthenium catalyst that chemically transforms non-toxic pro-drugs into active drugs was encapsulated into the ligand layer. The catalytic activity depends on the access of the pro-drug to the catalyst. A non-fluorescent allyl carbamate-protected rhodamine derivative (pro-Rho) was used to determine the catalytic potential of the respective Au-NP species. Au-NPs were decorated with a hydrophobic C11 chain and a cationic trimethylamine headgroup (NZ1). This surface design promoted the formation of a hard corona, inhibiting the access of the substrate to the catalyst and quenching catalysis in the presence of serum proteins. However, proteases degrade the protein corona upon endosomal uptake of the nanoparticle, restoring catalytic activity. Including a hydrophilic tetra(ethylene glycol) (TEG) linker into the ligand design led to the formation of a soft corona (NZ2), partially inhibiting catalysis. Upon endosomal uptake and corona degradation, catalysis was enhanced. Additionally, the exchange of the cationic trimethylamine headgroup with a zwitterionic moiety prevented the formation of a protein corona and endosomal uptake (NZ3). The results highlight the potential of engineering catalytically active nanoparticles that selectively activate upon endosomal uptake in cells.
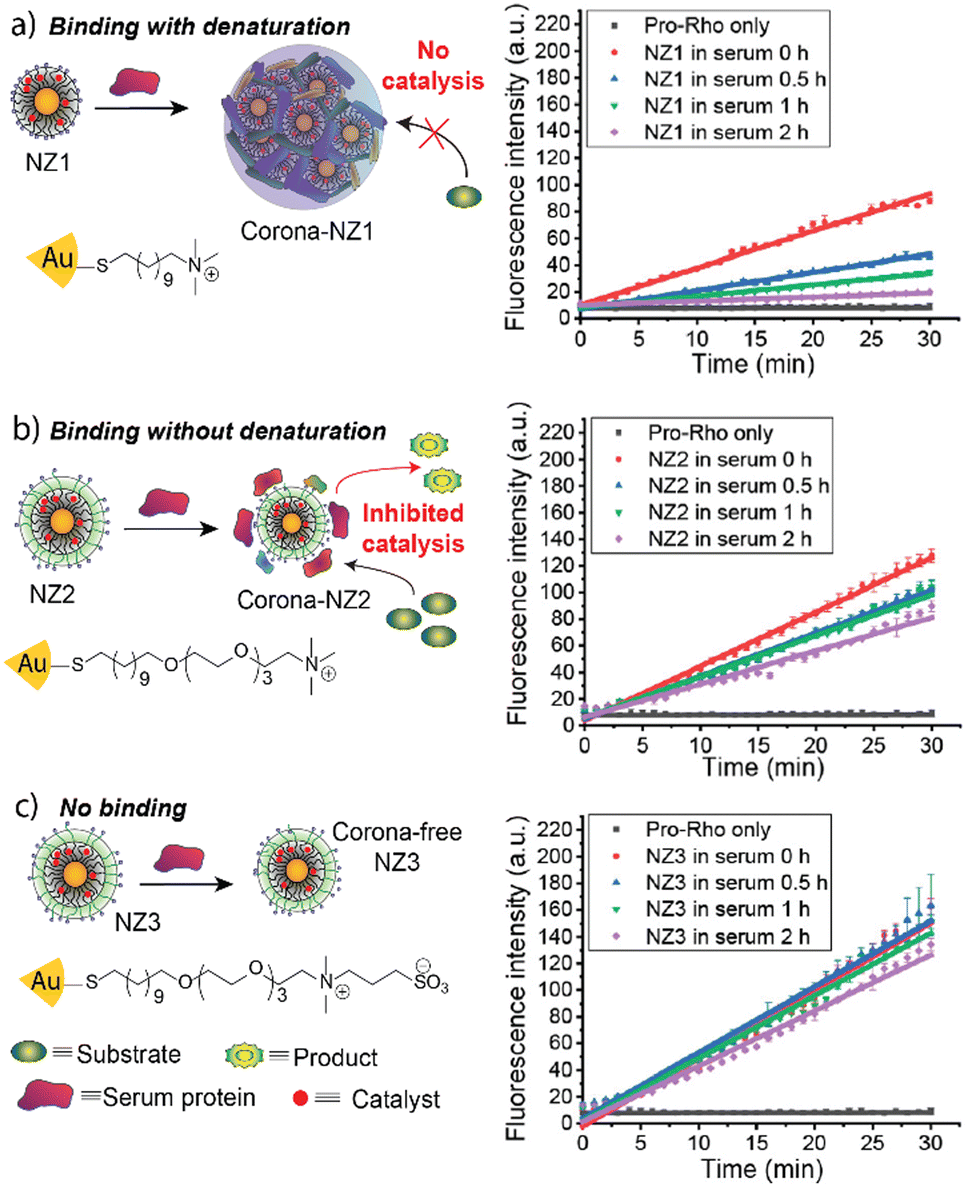 | ||
| Fig. 4 Schematic representation of corona-formation around different nanoparticles and catalytic activity of respective nanoparticles after 0, 0.5, 1, and 2 h serum incubation. NZ1 experiences complete deactivation in the presence of serum proteins (a), while NZ2 only loses partial activity (b). NZ3 retains almost all its activity (c). Adapted from ref. 29 with permission from the American Chemical Society. | ||
4. In vivo translatability of nanoparticles
Despite rapid advancement in the development of therapeutic nanomaterials, many nanomaterials that show promising results in vitro fail to pass through the FDA pipeline, causing immense financial loss for drug developers.30 Challenges for the translatability of nanomaterials between in vitro and in vivo applications include issues with stability, activity, and biocompatibility of nanomaterials.31 The interplay of hemolysis and protein corona formation can contribute to this discrepancy between in vitro and in vivo studies.32 Mailänder and co-workers compared the protein corona profile between coronas formed in mouse blood and in vivo. Magnetite nanoparticles were coated with hydroxyethyl starch and incubated with citrate, EDTA, heparin, or serum plasma from C57BL/6 mice. Nanoparticles were administered into the tail vein of C57BL/6 mice and recovered from the bloodstream using magnetic separation. The protein identity was determined using LC-MS (Fig. 5a). The results showed that the protein corona formed in vivo was significantly more complicated, consisting of approximately a three-fold greater variety of proteins than the protein corona formed in vitro in mouse blood. Furthermore, the overlap in protein identity between the in vitro- and in vivo-formed protein corona was less than 20% of the in vivo protein identity, indicating the complexity of protein coronas in vivo (Fig. 5b). The study presents a complicated issue for the future of clinical applications of nanomaterials and the need to better understand the in vivo corona.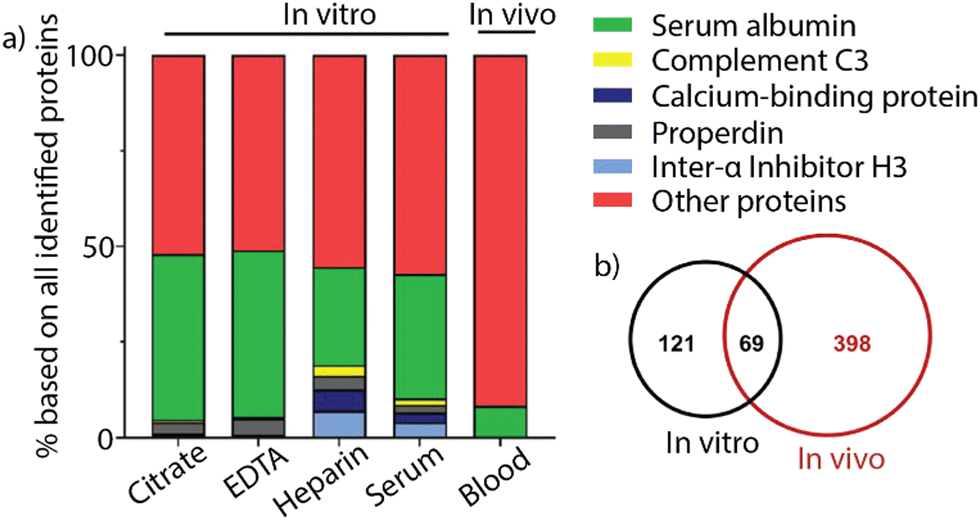 | ||
| Fig. 5 (a) Comparison between the most abundant proteins in the protein corona of nanoparticles after incubation with citrate, EDTA, heparin, or serum plasma and nanoparticles recovered from in vivo experiments. (b) Venn diagram showing the protein identity and identity overlap between in vitro and in vivo protein coronas. Adapted from ref. 32 with permission from MDPI. | ||
Mailänder and co-workers correlated the identity of the protein corona with enhanced or decreased uptake in HeLa cells and hMS cells (human mesenchymal stem cells) by functionalizing the surface of polystyrene nanoparticles.33 Briefly, nanoparticles were functionalized with either carboxy, amine, sulfonate, or phosphate moieties, as well as a fluorophore. Furthermore, sodium dodecyl sulfate (SDS) and lutensol A50 (Lut) were non-covalently attached to phosphate-functionalized nanoparticles. The respective particles were exposed to human blood serum to form protein coronas, which were identified using LC-MS (Fig. 6a). Carboxy- and phosphate-nanoparticles showed an enriched surface coverage of apolipoprotein H (ApoH), while the protein corona of amine- and sulfonate-nanoparticles was depleted in ApoH. HeLa cells or hMS cells were incubated with the protein-coated nanoparticles, and their intracellular uptake was quantified using flow cytometry. Carboxy- and phosphate-functionalized nanoparticles demonstrated increased uptake in both cell lines compared to amine- and sulfonate-functionalized nanoparticles (Fig. 6b). A single protein coating experiment using only carboxy-functionalized nanoparticles was performed as a control. The nanoparticle was incubated with different protein species, respectively, and the uptake in hMS cells was quantified. Nanoparticles coated exclusively with ApoH showed the highest intracellular uptake compared to nanoparticles coated with other proteins. This research demonstrates the importance of the protein corona identity and how the design of nanoparticle surfaces can contribute to differences in intracellular uptake, which may reflect on treatment efficiency.
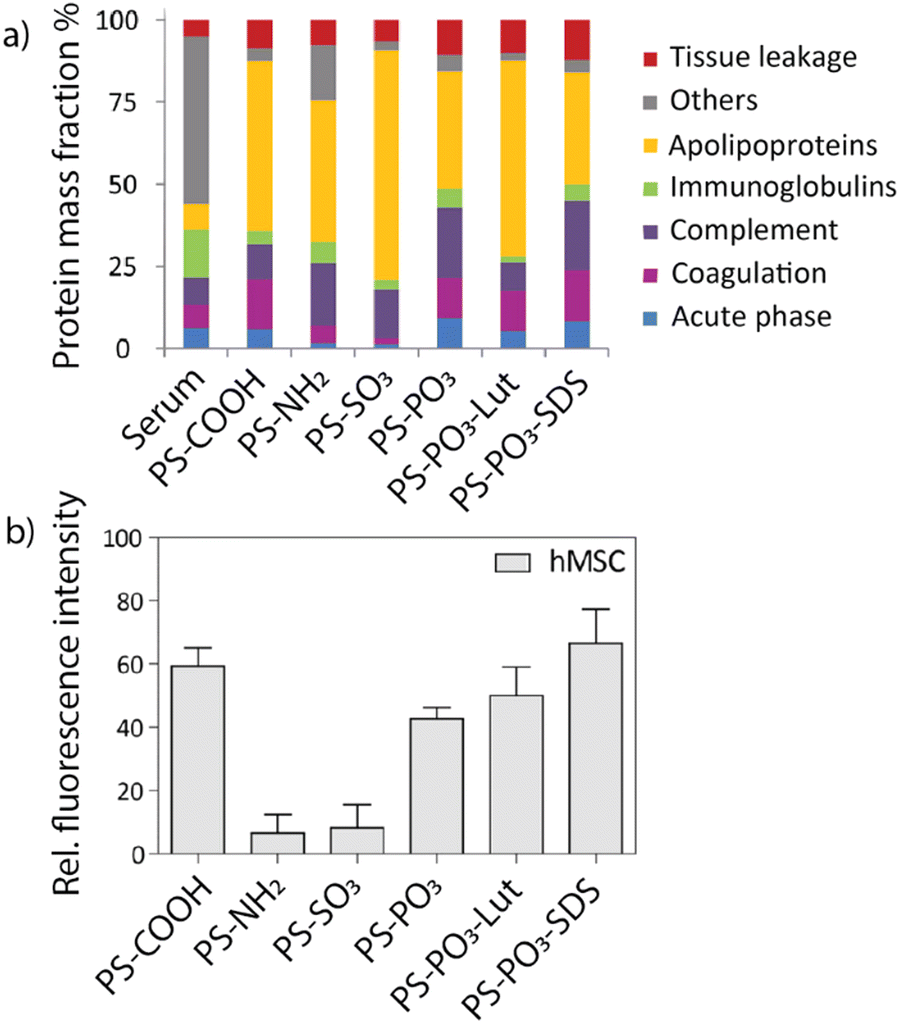 | ||
| Fig. 6 (a) Identity of protein coronas for respective nanoparticles analyzed by LC-MS. (b) Uptake of respective nanomaterials after incubation in human blood serum in hMS cells determined via flow cytometry. Carboxy- and phosphate-functionalized nanoparticles showed the highest uptake compared to other nanoparticles. Adapted from ref. 33 with permission from the American Chemical Society. | ||
5. Conclusion and future perspective
Our initial research on nanoparticle surface hydrophobicity helped to identify the design aspects that impact hemolysis and the role of protein corona formation on hemolytic activity. Later studies analyzed other nanoparticle characteristics, such as size, surface charge, and shape, on hemolysis and biocompatibility.34 This stream of discovery is an essential process for leveraging protein corona formation to optimize the therapeutic activity of nanoparticles.Our initial study, and the subsequent studies described in the Reflection, provide a foundation for understanding and harnessing protein corona formation for nanomedicine. We anticipate future studies will further explore the interplay between nanomaterial corona formation and biological activity. These empirical studies can be leveraged using tools such as artificial intelligence to identify the impact of changes in nanoparticle surface design on hemolysis and other biological behaviors.35 Together, this research will provide insight that allows us to better harness protein corona formation, enabling the creation of effective new nanomedicines.
Furthermore, the ability to direct protein corona identity expands the possibility of creating smart nanotherapeutics where corona formation dictates activity.36 Taken together, the interplay between nanoparticle structure and in vivo behavior (including hemolysis) remains a crucial topic in nanomedicine.
Author contributions
C.-M. H., M. J., J. P., and V. M. R. developed the concept. C.-M. H., M. J., J. P., and V. M. R. wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the National Institutes of Health R01 EB022641, DK121351, and R01 AI134770 (VMR). C.-M. H. was partially supported by a fellowship from the University of Massachusetts as part of the Chemistry-Biology Interface Training Program (National Research Service Award T32 GM139789).References
- F. Xiao, B. Cao, L. Wen, Y. Su, M. Zhan, L. Lu and X. Hu, Chin. Chem. Lett., 2020, 31, 2516–2519 CrossRef CAS.
- J. Choi, V. Reipa, V. M. Hitchins, P. L. Goering and R. A. Malinauskas, Toxicol. Sci., 2011, 123, 133–143 CrossRef CAS PubMed.
- S. Gavas, S. Quazi and T. M. Karpinski, Nanoscale Res. Lett., 2021, 16, 173–194 CrossRef CAS PubMed; N. Elahi, M. Kamali and M. H. Baghersad, Talanta, 2018, 184, 537–556 CrossRef PubMed.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- K. Saha, D. F. Moyano and V. M. Rotello, Mater. Horiz., 2014, 1, 102–105 RSC.
- A. Gupta, W. Ndugire, C.-M. Hirschbiegel, L. Grigerly and V. M. Rotello, Acc. Chem. Res., 2023, 56, 2151–2169 CrossRef CAS PubMed.
- C. Janko, J. Zaloga, M. Pottler, S. Durr, D. Eberbeck, R. Tietze, S. Lyer and C. Alexiou, J. Magn. Magn. Mater., 2017, 431, 281–284 CrossRef CAS.
- A. Valsesia, C. Desmet, I. Ojea-Jimenez, A. Oddo, R. Capomaccio, F. Rossi and P. Colpo, Commun. Chem., 2018, 1, 53 CrossRef.
- D. F. Moyano, K. Saha, G. Prakash, B. Yan, H. Kong, M. Yazdani and V. M. Rotello, ACS Nano, 2014, 8, 6748–6755 CrossRef CAS PubMed.
- P. Jain, R. S. Pawar, R. S. Pandey, J. Madan, S. Pawar, P. K. Lashmi and M. S. Sudheesh, Biotechnol. Adv., 2017, 35, 889–904 CrossRef CAS PubMed.
- Q. Xiao, M. Zoulikha, M. Qiu, C. Teng, C. Lin, Z. Li, M. A. Sallam, Q. Xu and W. He, Adv. Drug Delivery Rev., 2022, 186, 114356 CrossRef CAS PubMed.
- C. C. S. Batista, K. Panico, J. Trousil, O. Janoušková, C. E. de Castro, P. Štěpánek and F. C. Giacomelli, Colloids Surf., B, 2022, 218, 112778 CrossRef CAS PubMed.
- V. Dahanayake, T. Lyons, B. Kerwin, O. Rodriguez, C. Albanese, E. Parasido, Y. Lee, E. V. Keuren, L. Li, E. Maxey, T. Paunesku, G. Woloschak and S. L. Stoll, ACS Appl. Mater. Interfaces, 2021, 13, 39042–39054 CrossRef CAS PubMed.
- E. Kelpsiene, I. Brandts, K. Bernfur, M. T. Ekvall, M. Lundqvist, M. Teles and T. Cedervall, Environ. Sci.: Nano, 2022, 9, 2500–2509 RSC.
- A. Iqbal, S. M. S. Abidi, S. Randhawa, R. Joshi, R. Kumar and A. Acharya, ACS Appl. Mater. Interfaces, 2022, 14, 337–349 CrossRef PubMed.
- M. M. Yallapu, N. Chauhan, S. F. Othman, V. Khalilzad-Sharghi, M. C. Ebeling, S. Khan and S. C. Chauhan, Biomaterials, 2015, 46, 1–12 CrossRef CAS PubMed.
- C. Corbo, R. Molinaro, M. Tabatabaei, O. C. Farokhzad and M. Mahmoudi, Biomater. Sci., 2017, 5, 378–397 RSC.
- D. Chakraborty, L. Mohan, S. A. Alex, N. Chandrasekaran and A. Mukherjee, Biomater. Sci., 2019, 7, 63–75 RSC.
- M. P. Vinardell and M. Mitjans, Nanomaterials, 2015, 5, 1004–1021 CrossRef CAS PubMed.
- A. Gupta, R. F. Landis and V. M. Rotello, F1000Research, 2016, 5, 364 Search PubMed.
- S. M. Dizaj, F. Lotfipour, M. Barzegar-Jalali, M. H. Zarrintan and K. Adibkia, Mater. Sci. Eng., C, 2014, 44, 278–284 CrossRef CAS PubMed.
- S. Tang and J. Zheng, Adv. Healthcare Mater., 2018, 7, 1701503 CrossRef PubMed.
- A. Gupta, S. Mumtaz, C.-H. Li, I. Hussain and V. M. Rotello, Chem. Soc. Rev., 2019, 48, 415–427 RSC.
- L. Q. Chen, L. Fang, J. Ling, C. Z. Ding, B. Kang and C. Z. Huang, Chem. Res. Toxicol., 2015, 28, 501–509 Search PubMed.
- W. Liu, J. Rose, S. Plantevin, M. Auffan, J.-Y. Bottero and C. Vidaud, Nanoscale, 2013, 5, 1658–1668 RSC.
- M. Lundqvist and T. Cedervall, Small, 2020, 16, 2000892 CrossRef CAS PubMed.
- H. Wang, L. Shang, P. Maffre, S. Hohmann, F. Kirschhofer, G. Brenner-Weiss and G. U. Nienhaus, Small, 2016, 12, 5836–5844 CrossRef CAS PubMed.
- X. Zhang, Y. Liu, S. Gopalakrishnan, L. Castellanos-Garcia, G. Li, M. Malassine, I. Uddin, R. Huang, D. C. Luther, R. W. Vachet and V. M. Rotello, ACS Nano, 2020, 14, 4767–4773 CrossRef CAS PubMed.
- D. Sun, W. Gao, H. Hu and S. Zhou, Acta Pharm. Sin. B, 2022, 12, 3049–3062 CrossRef CAS PubMed.
- K. Von Petersdorff-Campen and M. Schmid Daners, ASAIO J., 2022, 68, 3–13 CrossRef PubMed.
- J. Simon, G. Kuhn, M. Fichter, S. Gehring, K. Landfester and V. Mailänder, Cells, 2021, 10, 132 CrossRef CAS PubMed.
- S. Ritz, S. Schottler, N. Kotman, G. Baier, A. Musyanovych, J. Kuharev, K. Landfester, H. Schild, O. Jahn, S. Tenzer and V. Mailänder, Biomacromolecules, 2015, 16, 1311–1321 CrossRef CAS PubMed.
- R. Garcia-Alvarez, M. Hadjidemetriou, A. Sanchez-Iglesias, L. M. Liz-Marzan and K. Kostarelos, Nanoscale, 2018, 10, 1256–1264 RSC.
- A. K. Chew, J. A. Pedersen and R. C. Van Lehn, ACS Nano, 2022, 16, 6282–6292 CrossRef CAS PubMed.
- K. Saha, M. Rahimi, M. Yazdani, S. T. Kim, D. F. Moyano, S. Hou, R. Das, R. Mout, F. Rezaee, M. Mahmoudi and V. M. Rotello, ACS Nano, 2016, 10, 4421–4430 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |