DOI:
10.1039/D3MD00737E
(Research Article)
RSC Med. Chem., 2024,
15, 839-847
N-Arylsulfonamide-based adenosine analogues to target RNA cap N7-methyltransferase nsp14 of SARS-CoV-2†
Received
22nd December 2023
, Accepted 25th January 2024
First published on 26th January 2024
Abstract
RNA cap methylations have been shown to be crucial for the life cycle, replication, and infection of ssRNA viruses, as well as for evading the host's innate immune system. Viral methyltransferases (MTases) therefore represent an attractive target for the development of compounds as tools and inhibitors. In coronaviruses, N7-methyltransferase function is localized in nsp14, which has become an increasingly important therapeutic target with the COVID-19 pandemic. In recent years, we have been developing SAH-derived bisubstrates with adenosine and an N-arylsulfonamide moiety targeting both SAM and RNA binding sites in nsp14. We report here the synthesis of 31 SAH analogues with the N-arylsulfonamide attached to the 5′-position of adenosine via different linkers such as N-ethylthioether, N-ethylsulfone, N-ethylamino or N-methyltriazole. The compounds were obtained efficiently by amine sulfonylation or click chemistry. Their ability to inhibit SARS-CoV-2 N7-MTase was evaluated and the best inhibitors showed a submicromolar inhibitory activity against N7-MTase nsp14.
Introduction
Targeting viral methyltransferases (MTases) has become an increasingly attractive approach to antiviral therapy, as RNA cap methylations have been demonstrated to be critical for the life cycle, replication and infection of certain ssRNA viruses, as well as for evading the host's innate immune system.1–7 In coronaviruses, two MTases catalyze the transfer of a methyl group from S-adenosylmethionine to viral RNA cap substrates, at the N7-position and on the 2′-OH of the first nucleotide, respectively. Indeed, nsp14 catalyzes the N7-methylation of the cap guanosine8 and nsp16, in complex with nsp10, methylates the 2′-OH ribose of the first nucleotide of nascent RNA.9 By interacting with these MTases and selectively modulating their activities, we might expect to block viral infection, as previously demonstrated with SAH analogues and derivatives on various viruses.5,10–14 Spurred by the recent SARS-CoV-2 pandemic, the N7-MTase nsp14 is an emerging target of great interest for inhibitor development, and over the past three years numerous groups worldwide have proposed original active compounds against this enzyme.15–28 Most nsp14 inhibitors have been designed as SAM-derived compounds,17,19,21,23–26,28 but high-throughput screening of large libraries18,20 or a structure-based docking approach on large libraries27 have also lead to non-SAM-like inhibitors of nsp14. In the latest docking study,27 the most potent inhibitors are low micromolar, much weaker than the most potent SAM-derived inhibitor, which is active in the subnanomolar range against SARS-CoV-2 nsp14.26
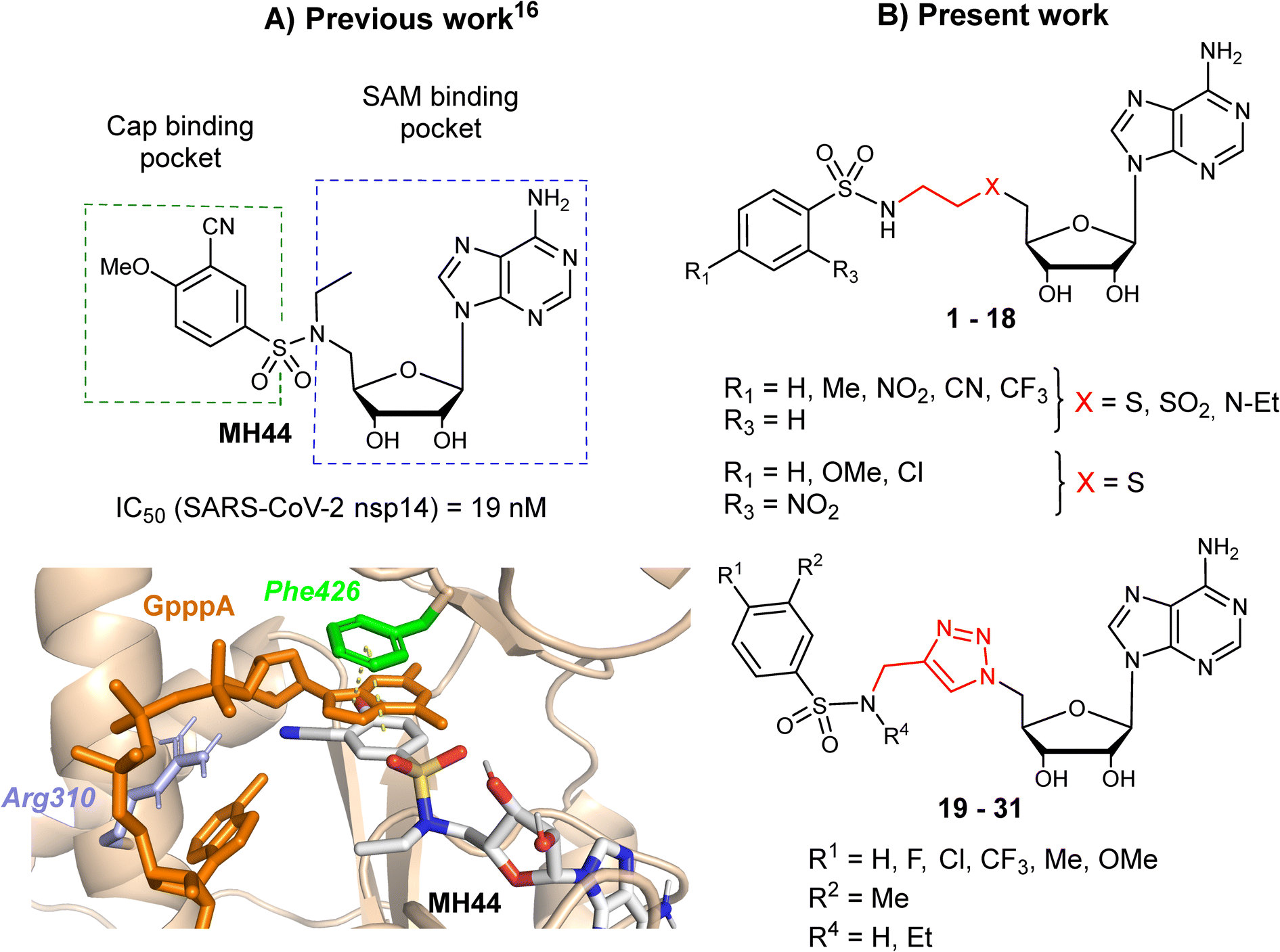
In two earlier studies, we developed N7-MTase competitive bisubstrates composed of an adenosine or its analogues occupying the SAM methyl donor binding pocket, linked to an N-arylsulfonamide scaffold that mimics the guanine moiety of the cap structure in the cap binding site of nsp14.23,26N-Arylsulfonamides, known to be ubiquitous functional scaffolds among medicinally interesting molecules, proved to be here a key component in explaining the inhibitors' high affinity for SARS-CoV-2 nsp14 (tens of nanomolar inhibition) in our work23,26,29 and that of others.24,28 In these potent inhibitors, the N-arylsulfonamide moieties were directly attached to the C5′ ribose atom of adenosine or its derivatives (Fig. 1A).
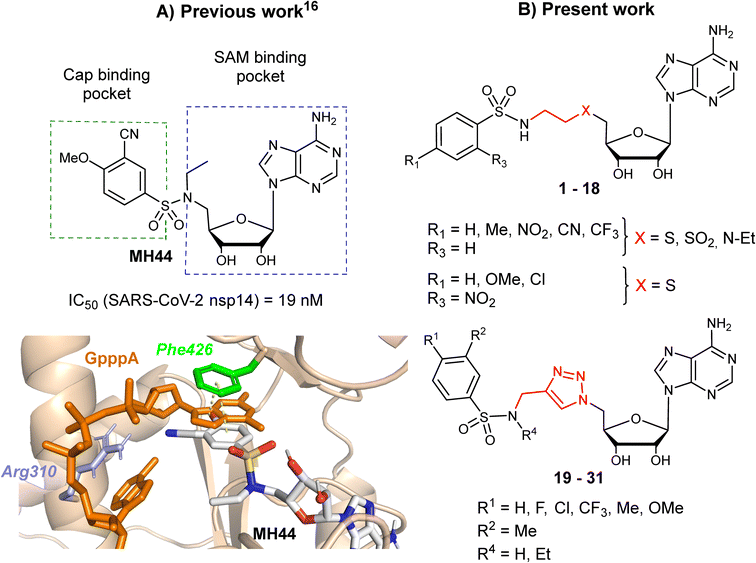 |
| | Fig. 1 A) The structure of SARS CoV-2 N7-MTase nsp14 in which a potent inhibitor (MH44) was docking into the cap-binding site of SARS-CoV nsp14 (PDB ID: 5C8S, resolution 3.3 Å) in a previous work.23 B) Chemical structures of novel SARS-CoV-2 N7-MTase nsp14 bisubstrates 1–31 with different linkers between the N-arylsulfonamide moiety and adenosine. | |
In the present work, we aimed to separate the N-arylsulfonamide part from adenosine by introducing an N-ethylthioether, an N-ethylsulfone, an N-ethylamino linker or an N-methyltriazole ring attached to the C5′ (Fig. 1B) and to investigate how modification of the initial scaffold by these new linkers would impact on N7-MTase inhibitory activity in vitro. Previously, the N-methyltriazole linker had been used in the same way to synthesize bisubstrates interfering with the protein N-terminal MTase (NTMT1) by Huang and co-workers,30 m6A and m1A RNA MTases by the Ethève-Quelquejeu's group,31,32 or to prepare 5′ cap mimics with a triazole ring inside the oligophosphate chain by Jemielity's group.33 Using click chemistry to combine two parts of a complex molecule represents an attractive approach over conventional multi-step approaches, which are often more time-consuming and tedious. Moreover, replacing an alkyl linker by a 1,2,3-triazole ring provides rigidity between the substrate mimic (N-arylsulfonamide moiety) and adenosine. Here, we synthesized three series of compounds representing over 30 final compounds using either amine N-sulfonylation or copper-catalyzed azide-alkyne cycloaddition (CuAAC) to build the corresponding linkers (Fig. 1B). To decipher the interaction of our compounds with SARS-CoV-2 N7-MTase nsp14, they were screened for their ability to inhibit the enzyme, which is involved in viral RNA cap methylation.
Results and discussion
Design
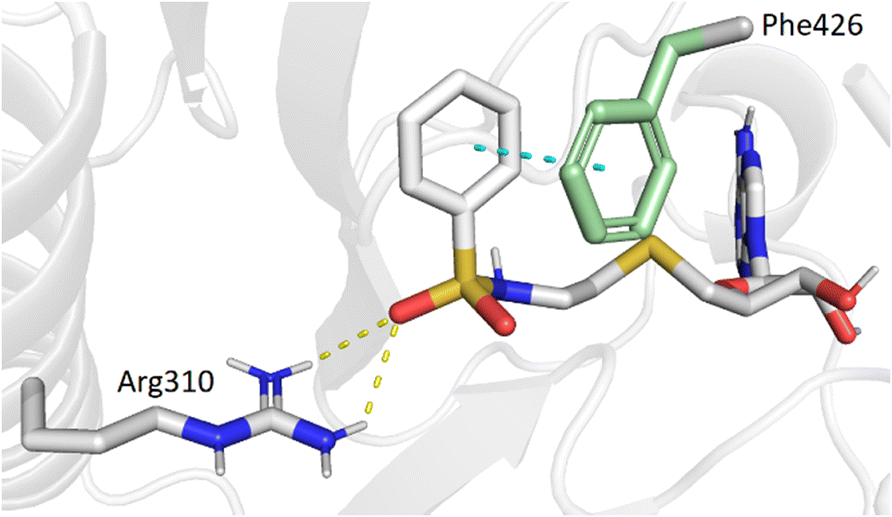
Our previous work, supported by molecular docking studies, has shown that adenosine analogues bearing an arylsulfonamide scaffold at C5′ fit well into the cap-binding pocket of nsp14.23 While the adenosine inserts into the SAM binding pocket, a π–π stacking interaction takes place between the arylsulfonamide moiety and Phe426 residue (Fig. 1A). This predominant interaction supports the bisubstrate approach of our inhibitors described since 2020 and is crucial for nsp14 inhibition.23,26 Phe426 naturally stacks the guanosine of the viral mRNA cap structure to allow its N7-methylation using SAM as methyl donor. By retaining the arylsulfonamide motif in the design of new nsp14 inhibitors, we have also shown that an electron-withdrawing group (EWG) (nitro or cyano) positioned in the meta position of the arylsulfonamide motif leads to the best inhibition of the MTase activity of SARS-CoV-2 nsp14. This is partly due to the good position of the EWG with Arg310 residue in SARS-CoV-2 nsp14, generating a double hydrogen bond.
In the present study, molecular docking of compound 1 with a sulfur atom in the C5′ position of adenosine (to mimic the structure of the natural SAM cofactor of methyltransferase) and arylsulfonamide without a substituent in the phenyl ring showed that, due to linker extension, it is no longer this group that interacts with Arg310 but rather the two S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of the sulfonamide (Fig. 2). Similar results were observed with derivatives 2–5 bearing various para substituents. Interestingly, the space around the ortho position is large enough to accommodate a substituent, therefore we inserted the NO2 group at this position in 6–8, hoping that it would interact, in a dynamic system, with the Arg310 residue. In our static docking model, the distance between NO2 and Arg310 is estimated at 5.9 Å, a too large distance to admit the existence of a true hydrogen bond. With a nitro group in ortho (compound 6), 7–8 were designed with an additional chlorine atom or a methoxy group in para, the latter having proved of major interest for nsp14 inhibition in our previous studies.23,26 We then replaced the sulfur atom by a sulfone or an ethylamino motif at C5′ position of adenosine, leading to compounds 9–13 and 14–18. Indeed, the N-ethyl motif proved crucial in the most potent previous nsp14 inhibitors. Finally, the linker separating the arylsulfonamide motif from adenosine was enlarged and rigidified by inserting a triazole ring, leading to compounds 19–31.
O of the sulfonamide (Fig. 2). Similar results were observed with derivatives 2–5 bearing various para substituents. Interestingly, the space around the ortho position is large enough to accommodate a substituent, therefore we inserted the NO2 group at this position in 6–8, hoping that it would interact, in a dynamic system, with the Arg310 residue. In our static docking model, the distance between NO2 and Arg310 is estimated at 5.9 Å, a too large distance to admit the existence of a true hydrogen bond. With a nitro group in ortho (compound 6), 7–8 were designed with an additional chlorine atom or a methoxy group in para, the latter having proved of major interest for nsp14 inhibition in our previous studies.23,26 We then replaced the sulfur atom by a sulfone or an ethylamino motif at C5′ position of adenosine, leading to compounds 9–13 and 14–18. Indeed, the N-ethyl motif proved crucial in the most potent previous nsp14 inhibitors. Finally, the linker separating the arylsulfonamide motif from adenosine was enlarged and rigidified by inserting a triazole ring, leading to compounds 19–31.
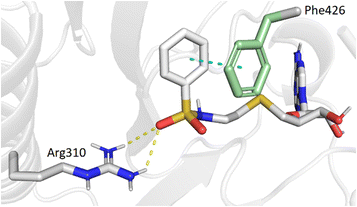 |
| | Fig. 2 Modeling results of docking initial compound 1 with the SAM and cap-binding pocket of SARS-CoV-2 nsp14 (PDB ID: 7R2V, resolution 2.53 Å). Contribution of the non-substituted arylsulfonamide core of 1. Phe426 (green) and Arg310, hydrogen bonds (yellow, 2.9, 2.6 Å) and the π–π stacking interaction (cyan, 4.0 Å) are shown. | |
Synthesis of N-arylsulfonamide adenosine analogues with an N-ethylthioether or N-ethylsulfone linker
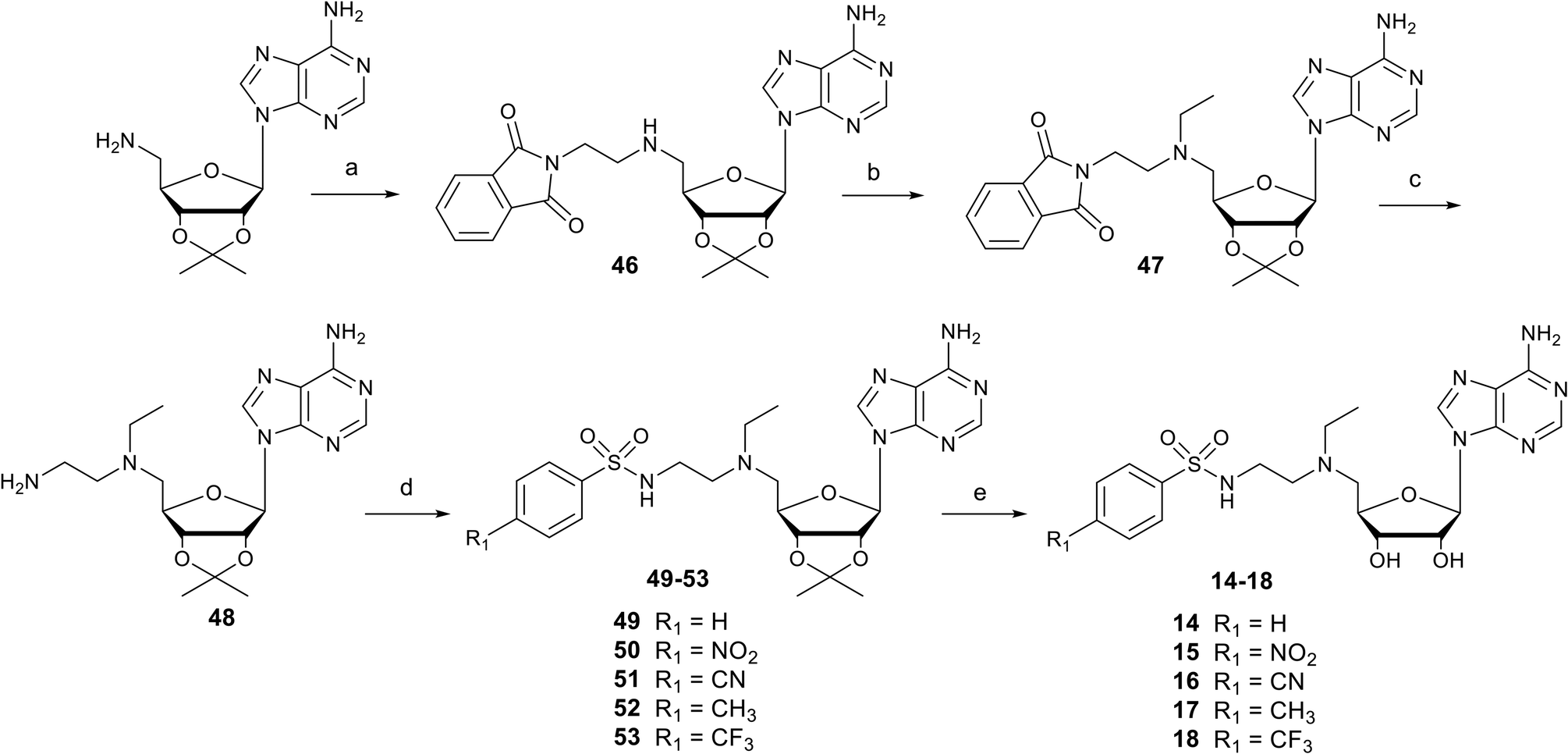
Nucleosides 1–8 and 9–13 bearing N-ethylthioether or N-ethylsulfone linkers respectively were prepared in two and three steps respectively from 2′,3′-O-isopropylidene-5′-(aminoethyl)thio adenosine 32 (Scheme 1).34 Similar to our previous work, the reaction between 32 and appropriate arylsulfonyl chlorides under basic conditions leads to N-arylsulfonamide-containing intermediates 33–40 in 38–80% yield. Then, among various reagents for oxidation of the sulfur atom to sulfone, such as Oxone® or hydrogen peroxide, the m-chloroperbenzoic acid reagent (mCPBA) was preferred mainly for solubility reasons and led to intermediates 41–45 in moderate yields (51–83%).35 Mild acidic deprotection finally liberates the 2′ and 3′ hydroxyls, giving nucleosides 1–13.25
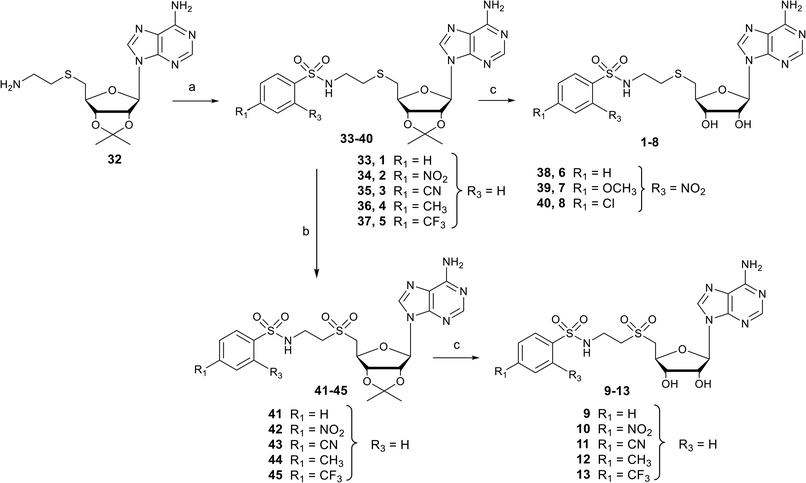 |
| | Scheme 1 Synthesis of N-arylsulfonamide adenosine analogues containing an N-ethylthioether (1–8) or N-ethylsulfone (9–13) linkers. Reagents and conditions: (a) arylsulfonyl chloride, Et3N, DMF, 25 °C, 3 h, 38–80%; (b) mCPBA, DCM, 0 °C, 2 h, 51–83%; (c) HCO2H/H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 25 °C, 24–48 h, 64–96%. :1 v/v, 25 °C, 24–48 h, 64–96%. | |
Synthesis of N-arylsulfonamide adenosine analogues with an N-ethylamino linker
Nucleosides 14–18 bearing the N-ethylamino linker were prepared in 5 steps from 5′-amino-2′,3′-O-isopropylideneadenosine (Scheme 2).15 The first two steps are reductive amination reactions, successively introducing ethylphthalimide and ethyl units on the 5′ nitrogen atom using N-(2-oxoethyl)phthalimide and acetaldehyde respectively.36,37 It should be noted that “one-pot” reductive double amination experiments have not been successful, requiring a step by step process. Furthermore, the order of reactions is important, as reductive amination with acetaldehyde must be carried out last to avoid the issue of double addition to the primary amine.38 The ethyl group was added onto 46 for two reasons: to increase the lipophilicity of nucleosides 14–18 and to mask the reactivity of the amine in the 5′ position, so as to functionalize the amine at the end of the ethyl chain exclusively with arylsulfonamide units. In addition, our previous results showed that an ethyl group functionalizing the 5′ amine improved SARS-CoV-2 N7-MTase nsp14 inhibition by a factor of 10.23 The phthalimide group was removed from 47 in the presence of methylamine and the resulting primary amine of 48 was reacted with various arylsulfonyl chlorides in basic medium to yield the N-arylsulfonamide-containing nucleosides 49–53.34 Like for compounds 1–13, the 2′ and 3′ hydroxyls were deprotected under acidic conditions to afford N-arylsulfonamide N-ethylaminoethyl adenosine analogues 14–18 in 64–96%.
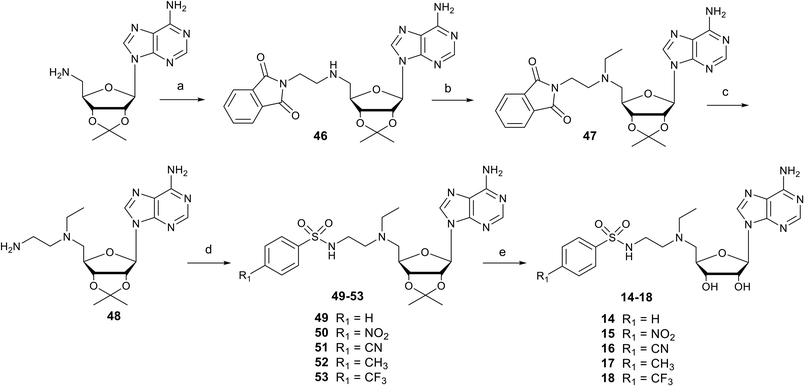 |
| | Scheme 2 Synthesis of N-arylsulfonamide adenosine analogues 14–18 containing an N-ethylamino linker. Reagents and conditions: (a) N-(2-oxoethyl)phthalimide, NaBH(OAc)3, THF, 25 °C, 18 h, 52%; (b) acetaldehyde, CH3CO2H, NaBH(OAc)3, DCE, 25 °C, 18 h, 60%; (c) MeNH2 in EtOH, 25 °C, 18 h, then AcOH, 91%. (d) Arylsulfonyl chloride, Et3N, DMF, 25 °C, 3 h, 38–80%; (e) HCO2H/H2O 1:1 v/v, 25 °C, 24–48 h, 64–96%. | |
Synthesis of N-arylsulfonamide adenosine analogues with an N-methyltriazole linker 19–31
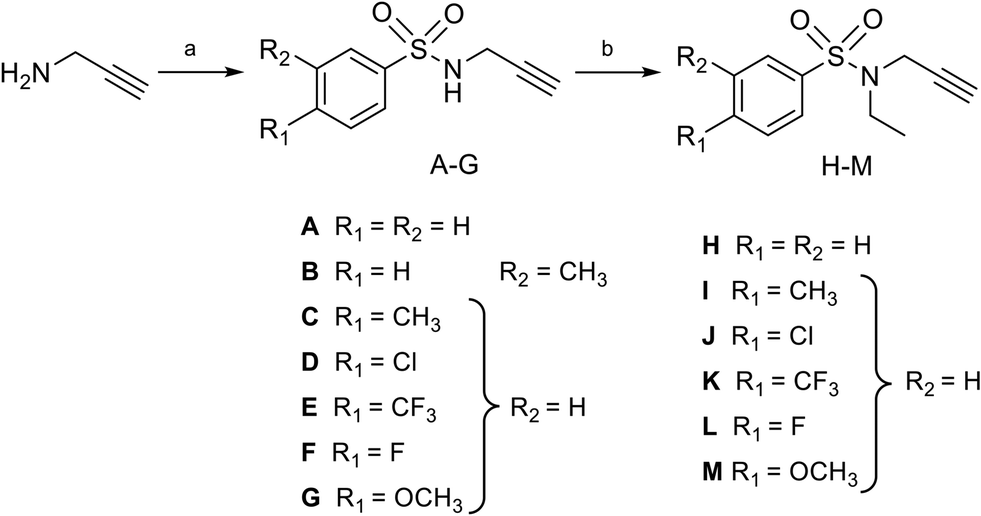
Nucleosides 19–31 bearing the N-methyltriazole linker were prepared in a single step from 5′-azidoadenosine39 and N-propargylsulfonamide derivatives. The latter were obtained in two steps from commercial propargylamine without a purification step (Scheme 3). In fact, the first step, in which the arylsulfonamide group is attached, uses an excess of propargylamine.40,41 After completion of the reaction, the propargylamine is protonated by treatment in an acid medium and removed by aqueous extraction. In the second step, the ethyl group is incorporated into the sulfonamide of A–G using ethyl-p-toluenesulfonate.23 Here, sulfonamide was used in excess and, after completion of the reaction, treatment in a basic medium removed the excess starting material and isolated the H–M derivatives with good purity. A “click” cycloaddition step in the presence of CuSO4 and sodium ascorbate yielded the final derivatives 19–31 after purification (13–61%) (Scheme 4).42
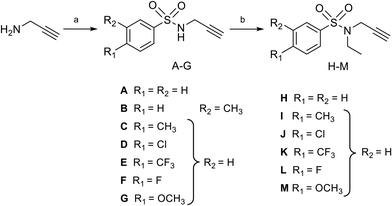 |
| | Scheme 3 Synthesis of N-arylsulfonamide-N-propargyl reagents A–M from commercially available propargylamine. Reagents and conditions: (a) arylsulfonyl chloride, Et3N, DCM, 0 °C then 25 °C, 16 h, quant.; (b) ethyl-p-toluenesulfonate, KI, K2CO3, DMF, 50 °C, 16 h, quant. | |
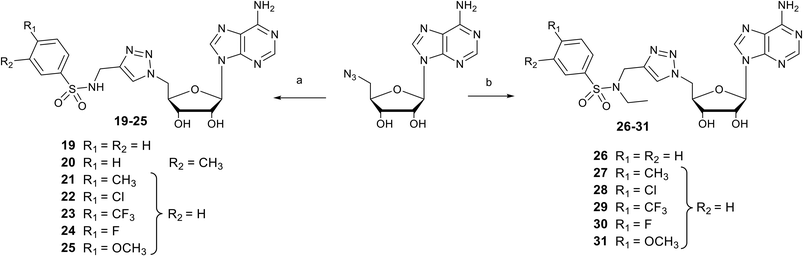 |
| | Scheme 4 Synthesis of N-arylsulfonamide adenosine analogues containing an N-methyltriazole linker 19–25 or an N-ethyl-N-methyltriazole linker 26–31. Reagents and conditions: (a) N-arylsulfonamide-N-propargyl reagents A–G, CuSO4, sodium ascorbate, H2O/Dioxane, 0 °C, 3 h, 13–61%; (b) N-arylsulfonamide-N-propargyl reagents H–M, CuSO4, sodium ascorbate, H2O/Dioxane, 0 °C, 3 h, 23–54%. | |
SARS-CoV-2 RNA cap N7-methyltransferase activity assays
Compounds 1–31 were screened for their ability to inhibit SARS-CoV-2 N7-MTase nsp14 using a radioactive MTase assay. The nsp14 protein was incubated with compounds 1–31 at 50 μM and the transfer of [3H]-radiolabeled methyl from the SAM methyl donor onto the cap structure of a synthetic RNA (GpppAC4) was measured by filter binding assay (FBA).43 The introduction of a N-methyltriazole linker between the N-arylsulfonamide moiety and adenosine is clearly detrimental to the inhibitory activity of compounds 19–31 at 50 μM against N7-MTase nsp14, as enzyme activity remains above 50% at this concentration (Table 1). The N-ethylamino linker in compounds 14–19 also impairs the inhibition of nsp14 activity. In contrast, adenosine analogues 1–13 containing N-ethylthioether or N-ethylsulfone linkers show a noticeable inhibitory effect at 50 μM on N7-MTase, which is more pronounced for compounds 1–8 with the N-ethylthioether linker (MTase activity < 20%).
Table 1 Screening for inhibitory activity of compounds 1–31 at 50 μM and IC50 values of 13 most active compounds 1–13 on SARS-CoV-2 N7-MTase nsp14
| Compounds |
SARS-CoV-2 N7-MTase nsp14 |
| % of MTase activity at 50 μM |
IC50a (μM) |
|
Values are the mean of three independent experiments. The MTase activity was measured using a filter binding assay. Assays were carried out in reaction mixture [40 mM Tris-HCl (pH 8.0), 1 mM DTT, 1 mM MgCl2, 2 μM SAM and 0.1 μM 3H-SAM] in the presence of 0.7 μM synthetic RNA GpppAC4, SARS-CoV-2 nsp14 N7-MTase (10 nM) and incubated at 30 °C. The concentration of MTase was adjusted in order to stay in the linear phase of the enzymatic reaction. Compounds were previously dissolved in 50% DMSO.
n.d. = not determined.
IC50 value from previous work.23
|
|
1
|
13 |
4.6 ± 2.4 |
|
2
|
9 |
4.7 ± 1.4 |
|
3
|
21 |
15.6 ± 4.0 |
|
4
|
10 |
2.2 ± 0.7 |
|
5
|
7 |
7.3 ± 1.1 |
|
6
|
0 |
2.04 ± 0.8 |
|
7
|
0 |
0.9 ± 0.3 |
|
8
|
2 |
0.9 ± 0.2 |
|
9
|
48 |
23.3 ± 18.7 |
|
10
|
27 |
27.4 ± 6.3 |
|
11
|
42 |
52.4 ± 14.7 |
|
12
|
38 |
49.0 ± 9.2 |
|
13
|
37 |
38.4 ± 3.0 |
|
14
|
87 |
n.d.b |
|
15
|
57 |
n.d. |
|
16
|
67 |
n.d. |
|
17
|
66 |
n.d. |
|
18
|
83 |
n.d. |
|
19
|
59 |
n.d. |
|
20
|
82 |
n.d. |
|
21
|
50 |
n.d. |
|
22
|
72 |
n.d. |
|
23
|
81 |
n.d. |
|
24
|
91 |
n.d. |
|
25
|
63 |
n.d. |
|
26
|
84 |
n.d. |
|
27
|
59 |
n.d. |
|
28
|
69 |
n.d. |
|
29
|
47 |
n.d. |
|
30
|
93 |
n.d. |
|
31
|
60 |
n.d. |
| Sinefungin |
0 |
0.278 ± 0.008c |
The most active compounds 1–13 were then tested in a dose–response assay with increasing compound concentration and MTase activity was measured using a FBA to determine the corresponding IC50 values (Table 1). N-Arylsulfonamide adenosine analogues 9–13 bearing a sulfone-containing linker showed IC50 values in the double-digit micromolar range, irrespective of the substituent (Ø, NO2, CN, Me, CF3) in the para position of the phenyl ring. In contrast, most of compounds, except for 3, with the N-ethylthioether linker and only one substituent at the para-position of the phenyl ring displayed micromolar IC50 values in the same range, irrespective of the substituent. Remarkably, two nucleosides 7–8 with the same S-containing linker but with an additional nitro group in the ortho-position of the phenyl exhibited higher inhibitory activity in the submicromolar range, close to that of the broad-spectrum inhibitor, sinefungin (IC50 = 278 nM). All these results endorse the N-ethylthioether linkage, a substituent in the para-position (Cl or OMe) and a NO2 group at the ortho-position of the phenyl ring, in the optimized scaffold of these series of SARS-CoV-2 N7-MTase inhibitors. The active site of nsp14 is highly conserved among coronaviruses, and the compounds described here are highly likely to inhibit also nsp14 from SARS-CoV, MERS-CoV, or OC43 as recently shown with previous inhibitors by us and other groups.26,44
Molecular docking studies of SARS-CoV-2 N7-MTase nsp14 in complex with one of the best inhibitor 7
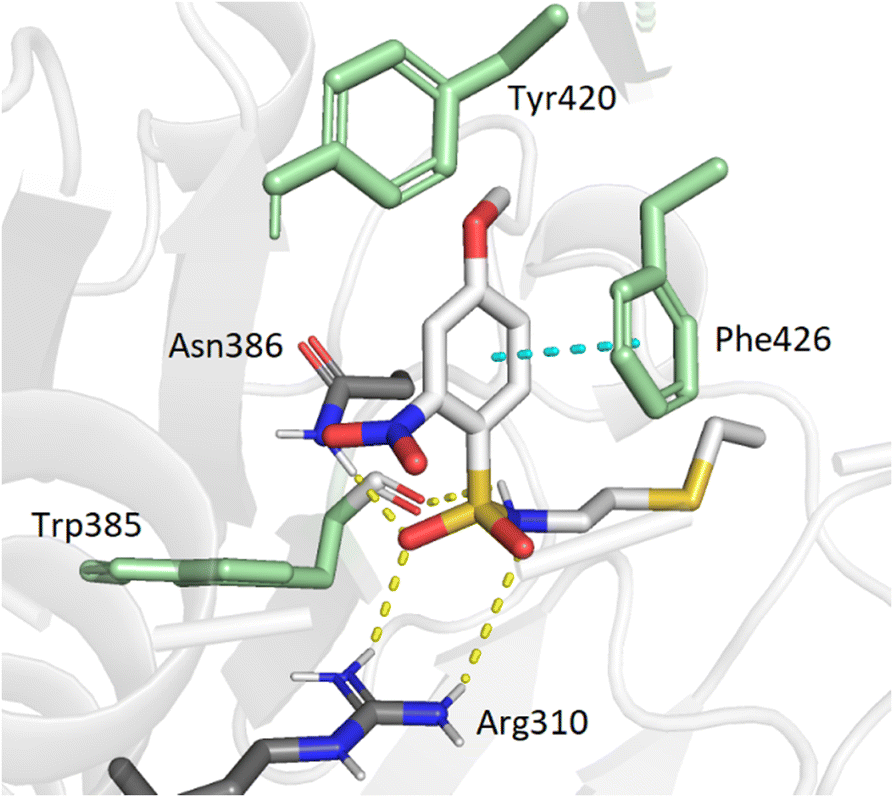
To support our results in enzymatic assays, we carried out computational docking studies with one of the most potent inhibitor 7, using Autodock Vina.45 Nucleoside 7 was modeled in the SAM- and cap-binding pockets of the SARS-CoV-2 N7-MTase nsp14 structure (PDB ID: 7R2V). At first sight, the overlay of the 5′ S-adenosine of 7 with the adenosine of SAM-bound structure is suitable and all interactions between SAM and nsp14 are equivalent to those observed between the adenosine core and the viral protein. As with previous bisubstrate inhibitors,23,26 the N-arylsulfonamide moiety interacts with Phe426 through π–π staking interactions (4.0 Å) (Fig. 3). Several hydrogen bond interactions were observed between the sulfonamide bond SO2-NH and the residues Arg310 (2.7 Å and 3.3 Å), Asn386 (2.9 Å) and Trp385 (2.8 Å), the latter never having been observed in our previous studies, which brings a positive view concerning the design of 7 with its enlarged linker.
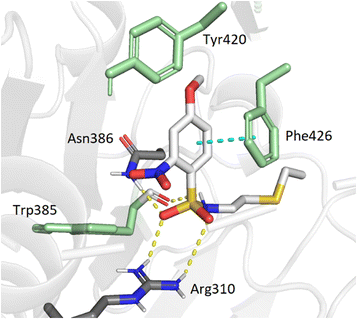 |
| | Fig. 3 Modeling docking of the best inhibitor 7 with the cap-binding pocket of SARS-CoV-2 nsp14 (PDB ID: 7R2V, resolution 2.53 Å). Contribution of the o,p-substituted arylsulfonamide core of 7. Trp385, Tyr420, Phe426, (green), Arg310, hydrogen bonds (SO2 – Asn386, 2.9 Å; SO2 – Arg310, 2.7 Å, 3.3 Å; NH – Trp385, 2.8 Å) and the π–π stacking interaction (cyan, 4.0 Å) are shown. 5′-N-adenosine moiety is not shown for clarity purposes. | |
Conclusions
We performed a structure-guided design approach to develop inhibitors of the SARS-CoV-2 nsp14 N7-MTase. In this study, we have designed a new series of bisubstrates as adenosine analogues carrying in the 5′position the N-arylsulfonamide scaffold previously shown to be necessary for inhibitory activity against nsp14. Here, the linker between N-arylsulfonamide and adenosine has been lengthened by an ethyl group or a methyltriazole moiety compared with the original inhibitors whose sulfonamide function was directly connected to the C4′ of the ribose. We have synthesized 31 compounds either by amine sulfonylation to obtain adenosine analogues with N-ethylthioether, N-ethylsulfone, N-ethylamino linkers, or by click chemistry to obtain the N-methyltriazole-containing derivatives. They were evaluated as potential inhibitors of SARS-CoV-2 N7-MTase nsp14. N-ethylamino or N-methyltriazole moieties in adenosine analogues drastically impair the inhibition of nsp14 activity, while compounds with N-ethylthioether or N-ethylsulfone showed inhibitory activity over a wide micromolar range. Two compounds with two substituents in the ortho- and para-position of the phenyl ring and an N-ethylthioether linking the arylsulfonamide moiety to adenosine exhibit submicromolar inhibitory activity close to that of broad-spectrum sinefungin, but lower than that of our previous inhibitors in the double-digit nanomolar range.23 Although interesting, these results seem to support that the best design of SAH analogues as nsp14 inhibitors remain one in which the distance between arylsulfonamide and adenosine is minimal.
Experimental
Detailed synthetic procedures and spectral characterization data for compounds 1–53 are included in the ESI.† Only general synthetic procedures are reported in this experimental section.
General method A for the synthesis of compounds 33–40
To a solution at 0 °C under argon of 32 (1.00 eq) in anhydrous DMF (0.1 M) were successively added Et3N (2.00 eq) and the corresponding arylsulfonyl chloride reactant (1.25 eq) in three portions. After stirring at r.t for 3 hours, the reaction mixture was diluted with AcOEt and H2O. The aqueous layer was extracted with AcOEt and the combined organic extracts were washed with brine, dried over Na2SO4 and concentrated under vacuum. The residue was purified by flash column chromatography (silica gel, linear gradient 0–4% MeOH in DCM) to give the desired compound as a white solid.
General method B for the synthesis of compounds 41–45
To a solution at 0 °C under argon of corresponding 33–37 (1.00 eq) in anhydrous DCM (0.03 M) was added mCPBA over 25 min. After stirring at 0 °C for 1.5 h, the reaction mixture was diluted with DCM and saturated Na2SO3. The aqueous layer was extracted with DCM and the combined organic extracts were washed with brine, dried over Na2SO4 and concentrated under vacuum. The residue was purified by flash column chromatography (silica gel, linear gradient 0–4% MeOH in DCM) to give the desired compound as a white solid.
General method C for the synthesis of compounds 1–18
A solution of 33–45 or 49–53 in a 1:1 mix of HCOOH/H2O (0.05 M) was stirred for 24–48 h at 25 °C. The reaction mixture was concentrated under vacuum and co-evaporated 3 times with absolute EtOH. The residue was purified by flash column chromatography (silica gel, linear gradient 0–14% MeOH in DCM) to give the desired compound as a white solid.
General method D for the synthesis of N-ethyl-N-propargyl reagents J–M
A suspension of NH-propargyl reagents D–G (1.00 eq), ethyl p-toluenesulfonate (1.00 eq), KI (0.10 eq) and K2CO3 (3.00 eq) in anhydrous DMF was stirred under argon at 50 °C for 16 hours. After cooling to room temperature, the reaction mixture was diluted with AcOEt. The organic phase was washed with 1 N NaOH, brine, dried over Na2SO4 and concentrated under vacuum.
General method E for the synthesis of compounds 19–31
To a solution at room temperature under argon of 5′-deoxy-5′-azido-adenosine (1.0 eq) in 1,4-dioxane (C = 0.02 M) were successively added the suitable propargyl reagent (1.4 eq) and a freshly pre-mixed solution of CuSO4 (0.4 eq, C = 0.06 M) and sodium ascorbate (1.0 eq, C = 0.2 M) in water. After stirring at room temperature for 2 hours, solvents were removed under reduced pressure and the residue was co-evaporated twice with acetonitrile. Then, the residue was re-suspended in a mixture of dichloromethane and methanol (1/1, v:v) and filtered to remove the salts. Silica was added and solvents were removed under vacuum. The residue was purified by flash column chromatography (dry sample, silica gel, linear gradient 0–18% MeOH in CH2Cl2) to give the desired compound, after lyophilization, as a white powder.
Expression and purification of recombinant protein
SARS-CoV-2 nsp14 (N7-MTase) coding sequence was cloned in fusion with a N-terminus hexa-histidine tag in pET28 plasmids.23 The protein was expressed in E. coli C2566 and purified in a two-step IMAC using cobalt beads. Briefly, cells were lysed by sonication in a buffer containing 50 mM Tris pH 6.8, 300 mM NaCl, 10 mM imidazole, 5 mM MgCl2, and 1 mM BME, supplemented with 0.25 mg mL−1 lysozyme, 10 μg mL−1 DNase, and 1 mM PMSF. The protein was next purified through affinity chromatography with HisPur Cobalt resin 480 (Thermo Scientific), washing with an increased concentration of salt (1 M NaCl) and imidazole (20 mM), prior to elution in buffer supplemented with 250 mM imidazole. The second step of purification was performed by size exclusion chromatography (GE Superdex S200) in a final buffer of 50 mM Tris pH 6.8, 300 mM NaCl, 5 mM MgCl2, and 1 mM BME and the protein was subsequently concentrated up to 12.5 μM and conserved at −20 °C in a buffer containing 50% of glycerol.
Determination of the MTase activity by filter binding assay (FBA)
The SARS-CoV-2 nsp14 MTase assay was carried out in reaction mixture [40 mM Tris-HCl (pH 8.0), 1 mM DTT, 1 mM MgCl2, 1.9 μM SAM, and 0.1 μM 3H-SAM (Perkin Elmer)] in the presence of 0.7 μM GpppAC4 synthetic RNA23 and the MTase (10 nM). Briefly, the enzyme was first mixed with the compound suspended in 50% DMSO (2.5% final DMSO) before the addition of RNA substrate and SAM and then incubated at 30 °C. Reactions mixtures were stopped after 30 min by their 10-fold dilution in ice-cold water. Samples were transferred to diethylaminoethyl (DEAE) filtermat (Perkin Elmer) using a Filtermat Harvester (Packard Instruments). The RNA-retaining mats were washed twice with 10 mM ammonium formate pH 8.0, twice with water and once with ethanol. They were soaked with scintillation fluid (Perkin Elmer), and 3H-methyl transfer to the RNA substrates was determined using a Wallac MicroBeta TriLux liquid scintillation counter (Perkin Elmer). For IC50 measurements, values were normalized and fitted with Prism (GraphPad software) using the following equation: Y = 100/[1 + ((X/IC50)^Hillslope)]. IC50 is defined as the inhibitory compound concentration that causes 50% reduction in enzyme activity.
Author contributions
R. A.-B.: conceptualization, investigation, methodology, validation, writing – original draft. J. T.: investigation, methodology, validation. A. D.: investigation, methodology, validation. B. C.: resources, writing – review & editing. J.-J. V.: resources, supervision, writing – review & editing. E. D.: data curation, supervision, validation, funding acquisition, project administration, writing – review & editing. F. D.: funding acquisition, project administration, supervision, validation, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors (R. A.-B., J. T., J.-J. V., F. D.) thank the Agence Nationale de la Recherche (ANR) (Project MetInCoV: ANR-21-CO14-0004-01) for financial support and Amina Tahir for HPLC analyses of the final compounds. The team viral Replicase (A.D, B.C, E. D.) has received funding from the Fondation pour la Recherche Médicale (Aide aux Équipes), SCORE project H2020 SC1-PHE-Coronavirus-2020, grant#101003627 and the Innovative Medicines Initiative 2 Joint Undertaking (JU) under grant agreement No. 101005077 (IMI-CARE).
Notes and references
- T. R. Fischer, L. Meidner, M. Schwickert, M. Weber, R. A. Zimmermann, C. Kersten, T. Schirmeister and M. Helm, Nucleic Acids Res., 2022, 50, 4216–4245 CrossRef CAS PubMed.
- P. Ramdhan and C. Li, Viruses, 2022, 14, 379 CrossRef CAS PubMed.
- R. Nencka, J. Silhan, M. Klima, T. Otava, H. Kocek, P. Krafcikova and E. Boura, Nucleic Acids Res., 2022, 50, 635–650 CrossRef CAS PubMed.
- L. R. Silva, P. F. da Silva Santos-Junior, J. de Andrade Brandao, L. Anderson, E. J. Bassi, J. Xavier de Araujo-Junior, S. H. Cardoso and E. F. da Silva-Junior, Bioorg. Med. Chem., 2020, 28, 115745 CrossRef CAS PubMed.
- E. Decroly and B. Canard, Curr. Opin. Virol., 2017, 24, 87–96 CrossRef CAS PubMed.
- A. Decombe, P. El Kazzi and E. Decroly, Curr. Opin. Virol., 2023, 59, 101302 CrossRef CAS PubMed.
- J. E. Thames, C. D. Waters, C. Valle, M. Bassetto, W. Aouadi, B. Martin, B. Selisko, A. Falat, B. Coutard, A. Brancale, B. Canard, E. Decroly and K. L. Seley-Radtke, Bioorg. Med. Chem., 2020, 28, 115713 CrossRef CAS PubMed.
- Y. Chen, H. Cai, J. Pan, N. Xiang, P. Tien, T. Ahola and D. Guo, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3484–3489 CrossRef CAS PubMed.
- M. Bouvet, C. Debarnot, I. Imbert, B. Selisko, E. J. Snijder, B. Canard and E. Decroly, PLoS Pathog., 2010, 6, e1000863 CrossRef PubMed.
- C. S. Pugh, R. T. Borchardt and H. O. Stone, J. Biol. Chem., 1978, 253, 4075–4077 CrossRef CAS PubMed.
- E. Benghiat, P. A. Crooks, R. Goodwin and F. Rottman, J. Pharm. Sci., 1986, 75, 142–145 CrossRef CAS PubMed.
- J. Balzarini, E. De Clercq, P. Serafinowski, E. Dorland and K. R. Harrap, J. Med. Chem., 1992, 35, 4576–4583 CrossRef CAS PubMed.
- M. Zgarbová, T. Otava, J. Silhan, R. Nencka, J. Weber and E. Boura, Antiviral Res., 2023, 218, 105714 CrossRef PubMed.
- J. Silhan, M. Klima, T. Otava, P. Skvara, D. Chalupska, K. Chalupsky, J. Kozic, R. Nencka and E. Boura, Nat. Commun., 2023, 14, 2259 CrossRef CAS PubMed.
- R. Ahmed-Belkacem, P. Sutto-Ortiz, M. Guiraud, B. Canard, J. J. Vasseur, E. Decroly and F. Debart, Eur. J. Med. Chem., 2020, 201, 112557 CrossRef CAS PubMed.
- S. Basu, T. Mak, R. Ulferts, M. Wu, T. Deegan, R. Fujisawa, K. W. Tan, C. T. Lim, C. Basier, B. Canal, J. F. Curran, L. S. Drury, A. W. McClure, E. L. Roberts, F. Weissmann, T. U. Zeisner, R. Beale, V. H. Cowling, M. Howell, K. Labib and J. F. X. Diffley, Biochem. J., 2021, 478, 2481–2497 CrossRef CAS PubMed.
- O. Bobileva, R. Bobrovs, I. Kanepe, L. Patetko, G. Kalnins, M. Sisovs, A. L. Bula, S. Gri Nberga, M. R. Boroduskis, A. Ramata-Stunda, N. Rostoks, A. Jirgensons, K. Ta Rs and K. Jaudzems, ACS Med. Chem. Lett., 2021, 12, 1102–1107 CrossRef CAS PubMed.
- R. Bobrovs, I. Kanepe, N. Narvaiss, L. Patetko, G. Kalnins, M. Sisovs, A. L. Bula, S. Grinberga, M. Boroduskis, A. Ramata-Stunda, N. Rostoks, A. Jirgensons, K. Tars and K. Jaudzems, Pharmaceuticals, 2021, 14, 1243 CrossRef CAS PubMed.
- K. Devkota, M. Schapira, S. Perveen, A. Khalili Yazdi, F. Li, I. Chau, P. Ghiabi, T. Hajian, P. Loppnau, A. Bolotokova, K. J. F. Satchell, K. Wang, D. Li, J. Liu, D. Smil, M. Luo, J. Jin, P. V. Fish, P. J. Brown and M. Vedadi, SLAS Discovery, 2021, 26, 1200–1211 CrossRef CAS PubMed.
- R. Kasprzyk, T. J. Spiewla, M. Smietanski, S. Golojuch, L. Vangeel, S. De Jonghe, D. Jochmans, J. Neyts, J. Kowalska and J. Jemielity, Antiviral Res., 2021, 193, 105142 CrossRef CAS PubMed.
- T. Otava, M. Šála, F. Li, J. Fanfrlík, K. Devkota, S. Perveen, I. Chau, P. Pakarian, P. Hobza, M. Vedadi, E. Boura and R. Nencka, ACS Infect. Dis., 2021, 7, 2214–2220 CrossRef CAS PubMed.
- L. A. Pearson, C. J. Green, D. Lin, A. P. Petit, D. W. Gray, V. H. Cowling and E. A. F. Fordyce, SLAS Discovery, 2021, 26, 749–756 CrossRef CAS PubMed.
- R. Ahmed-Belkacem, M. Hausdorff, A. Delpal, P. Sutto-Ortiz, A. M. G. Colmant, F. Touret, N. S. Ogando, E. J. Snijder, B. Canard, B. Coutard, J. J. Vasseur, E. Decroly and F. Debart, J. Med. Chem., 2022, 65, 6231–6249 CrossRef CAS PubMed.
- E. Jung, R. Soto-Acosta, J. Xie, D. J. Wilson, C. D. Dreis, R. Majima, T. C. Edwards, R. J. Geraghty and L. Chen, ACS Med. Chem. Lett., 2022, 13, 1477–1484 CrossRef CAS PubMed.
- O. Bobileva, R. Bobrovs, E. E. Sirma, I. Kanepe, A. L. Bula, L. Patetko, A. Ramata-Stunda, S. Grinberga, A. Jirgensons and K. Jaudzems, Molecules, 2023, 28, 768 CrossRef CAS PubMed.
- M. Hausdorff, A. Delpal, S. Barelier, L. Nicollet, B. Canard, F. Touret, A. Colmant, B. Coutard, J. J. Vasseur, E. Decroly and F. Debart, Eur. J. Med. Chem., 2023, 256, 115474 CrossRef CAS PubMed.
- I. Singh, F. Li, E. A. Fink, I. Chau, A. Li, A. Rodriguez-Hernández, I. Glenn, F. J. Zapatero-Belinchón, M. L. Rodriguez, K. Devkota, Z. Deng, K. White, X. Wan, N. A. Tolmachova, Y. S. Moroz, H. Ü. Kaniskan, M. Ott, A. García-Sastre, J. Jin, D. G. Fujimori, J. J. Irwin, M. Vedadi and B. K. Shoichet, J. Med. Chem., 2023, 66, 7785–7803 CrossRef CAS PubMed.
- M. Štefek, D. Chalupská, K. Chalupský, M. Zgarbová, A. Dvořáková, P. Krafčíková, A. S. M. Li, M. Šála, M. Dejmek, T. Otava, E. Chaloupecká, J. Kozák, J. Kozic, M. Vedadi, J. Weber, H. Mertlíková-Kaiserová and R. Nencka, ACS Omega, 2023, 8, 27410–27418 CrossRef PubMed.
- R. Amador, A. Delpal, B. Canard, J. J. Vasseur, E. Decroly, F. Debart, G. Clave and M. Smietana, Org. Biomol. Chem., 2022, 20, 7582–7586 RSC.
- G. Zhang, S. L. Richardson, Y. Mao and R. Huang, Org. Biomol. Chem., 2015, 13, 4149–4154 RSC.
- C. Atdjian, D. Coelho, L. Iannazzo, M. Ethève-Quelquejeu and E. Braud, Molecules, 2020, 25, 3241 CrossRef CAS PubMed.
- D. Coelho, L. Le Corre, K. Bartosik, L. Iannazzo, E. Braud and M. Ethève-Quelquejeu, Chem. – Eur. J., 2023, 29, e202301134 CrossRef CAS PubMed.
- S. Walczak, A. Nowicka, D. Kubacka, K. Fac, P. Wanat, S. Mroczek, J. Kowalska and J. Jemielity, Chem. Sci., 2017, 8, 260–267 RSC.
- M. van Haren, L. Q. van Ufford, E. E. Moret and N. I. Martin, Org. Biomol. Chem., 2015, 13, 549–560 RSC.
- G. Liu, W. Chen, Z. Xu, F. Ye, Y. Pan, X. Chen, S. H. Liu, L. Zeng and J. Yin, Org. Biomol. Chem., 2018, 16, 5517–5523 RSC.
- A. Safavy, D. C. Smith, A. Bazooband and D. J. Buchsbaum, Bioconjugate Chem., 2002, 13, 317–326 CrossRef CAS PubMed.
- J. Xiang, Z.-K. Wan, H.-Q. Li, M. Ipek, E. Binnun, J. Nunez, L. Chen, J. C. McKew, T. S. Mansour, X. Xu, V. Suri, M. Tam, Y. Xing, X. Li, S. Hahm, J. Tobin and E. Saiah, J. Med. Chem., 2008, 51, 4068–4071 CrossRef CAS PubMed.
- S.-J. Qu, G.-F. Wang, W.-H. Duan, S.-Y. Yao, J.-P. Zuo, C.-H. Tan and D.-Y. Zhu, Bioorg. Med. Chem., 2011, 19, 3120–3127 CrossRef CAS PubMed.
- R. Ahmed-Belkacem, P. Sutto-Ortiz, A. Delpal, J. Troussier, B. Canard, J. J. Vasseur, E. Decroly and F. Debart, Bioorg. Chem., 2024, 143, 107035 CrossRef CAS PubMed.
- J. Teske and B. Plietker, ACS Catal., 2016, 6, 7148–7151 CrossRef CAS.
- H. M. Peng, J. Zhao and X. Li, Adv. Synth. Catal., 2009, 351, 1371–1377 CrossRef CAS.
- K. Liu, H. Shi, H. Xiao, A. G. L. Chong, X. Bi, Y.-T. Chang, K. S. W. Tan, R. Y. Yada and S. Q. Yao, Angew. Chem., Int. Ed., 2009, 48, 8293–8297 CrossRef CAS PubMed.
- F. Peyrane, B. Selisko, E. Decroly, J. J. Vasseur, D. Benarroch, B. Canard and K. Alvarez, Nucleic Acids Res., 2007, 35, e26 CrossRef CAS PubMed.
- F. Li, P. Ghiabi, T. Hajian, M. Klima, A. S. M. Li, A. Khalili Yazdi, I. Chau, P. Loppnau, M. Kutera, A. Seitova, A. Bolotokova, A. Hutchinson, S. Perveen, E. Boura and M. Vedadi, Biochim. Biophys. Acta, Gen. Subj., 2023, 1867, 130319 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Françoise
Debart
*b and
Françoise
Debart