
Small molecules modulating RNA splicing: a review of targets and future perspectives
Léa
Bouton
ab,
Agathe
Ecoutin
ab,
Florian
Malard
*ab and
Sébastien
Campagne
 *ab
*ab
aInserm U1212, CNRS UMR5320, ARNA Laboratory, University of Bordeaux, 146 rue Léo Saignat, 33076 Bordeaux Cedex, France. E-mail: florian.malard@inserm.fr; sebastien.campagne@inserm.fr
bInstitut Européen de Chimie et de Biologie, F-33600 Pessac, France
First published on 11th January 2024
Abstract
In eukaryotic cells, RNA splicing is crucial for gene expression. Dysregulation of this process can result in incorrect mRNA processing, leading to aberrant gene expression patterns. Such abnormalities are implicated in many inherited diseases and cancers. Historically, antisense oligonucleotides, which bind to specific RNA targets, have been used to correct these splicing abnormalities. Despite their high specificity of action, these oligonucleotides have drawbacks, such as lack of oral bioavailability and the need for chemical modifications to enhance cellular uptake and stability. As a result, recent efforts focused on the development of small organic molecules that can correct abnormal RNA splicing event under disease conditions. This review discusses known and potential targets of these molecules, including RNA structures, trans-acting splicing factors, and the spliceosome – the macromolecular complex responsible for RNA splicing. We also rely on recent advances to discuss therapeutic applications of RNA-targeting small molecules in splicing correction. Overall, this review presents an update on strategies for RNA splicing modulation, emphasizing the therapeutic promise of small molecules.
Introduction
In eukaryotic organisms, RNA splicing constitutes a critical maturation process.1,2 This mechanism achieves the excision of non-coding intronic sequences and the subsequent ligation of coding exonic sequences within the precursor mRNA (pre-mRNA), yielding a mature, spliced mRNA (Fig. 1A). The splicing process is mediated by small nuclear ribonucleoproteins (snRNPs) complexes and associated proteins (Fig. 1B) that sequentially assemble within a large and highly dynamic macromolecular machinery known as the spliceosome (Fig. 1C).1,3 Within the pre-mRNA, cis-acting regulatory sequences, such as 5′ and 3′ splice sites, branch point (BP), intronic and exonic splicing enhancers (ISE, ESE) and silencers (ISS, ESS), delineate the recruitment sites for spliceosome components and trans-splicing factors.4 RNA splicing is modulated by the interplay between cis-acting regulatory sequences and trans-acting splicing factors, leading to alternative splicing.5,6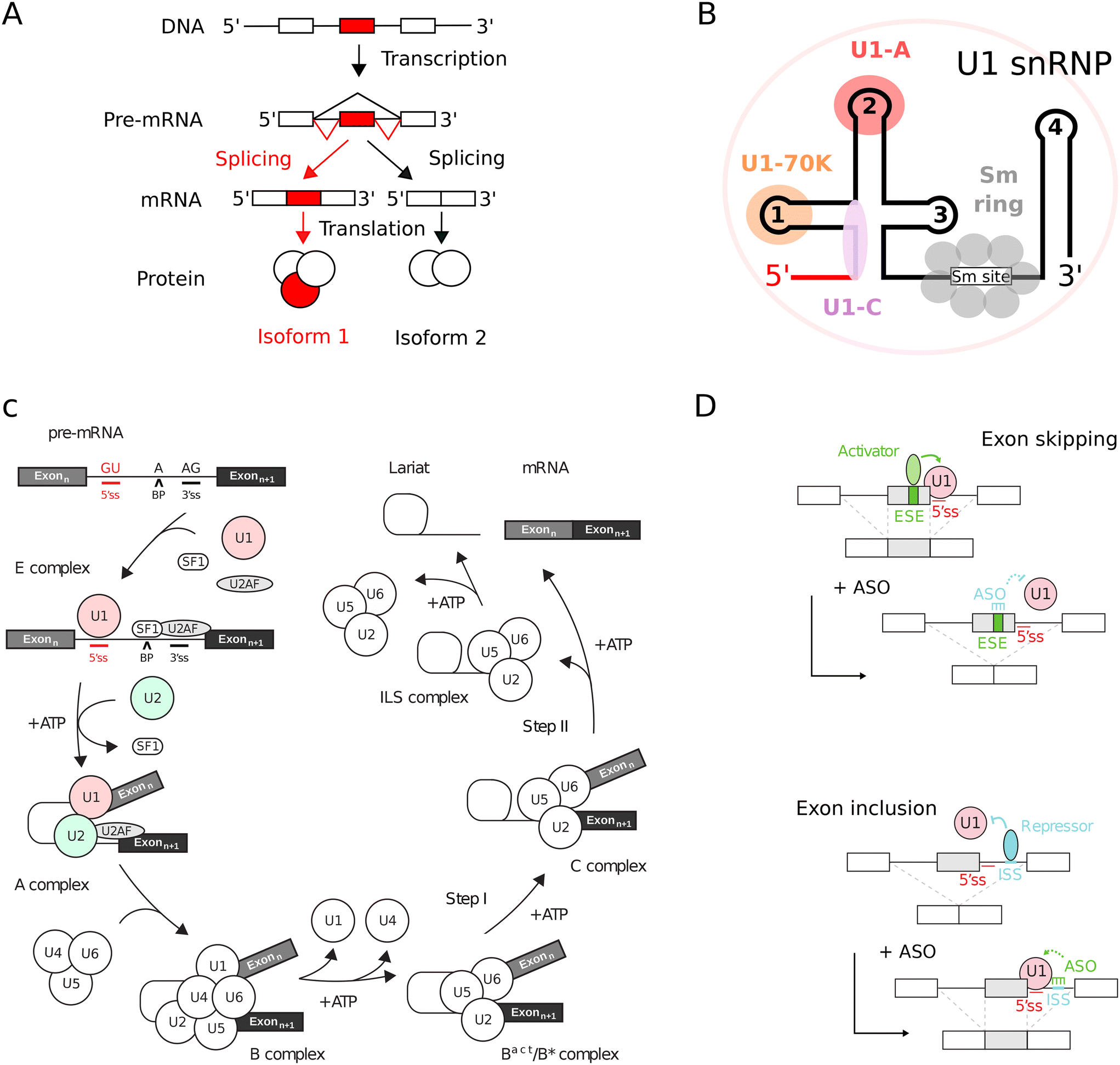 | ||
| Fig. 1 Principles of RNA splicing and correction with ASO splicing switches. (A) RNA splicing in eukaryotes. Non-coding introns (lines) are removed and coding exons (boxes) are connected. Regulation can produce distinct protein isoforms via alternative splicing. (B) Topology of snRNPs. An example is given with U1 snRNP which contains U1 snRNA (lines), three U1-specific proteins (U1-A, U1-C and U1-70K) and seven shared Sm proteins (Sm B/B', Sm D1, Sm D2, Sm D3, Sm E, Sm F and Sm G). In all spliceosomal snRNPs, the Sm proteins are assembled in a ring around the Sm site of snRNAs. In particular for U1 snRNP, the 5′ end of U1 snRNA hybridizes with the 5′ splice site (red line). (C) Splicing reaction. The pre-mRNA needs two exons (boxes) separated by an intron (line) with defined 5′ and 3′ splice sites (5′ss, 3′ss) and a branch point (BP) adenosine. The spliceosome includes five snRNPs (U1/2/4/5/6) and auxiliary factors like SF1 and U2AF.1,2 (D) ASO in splicing correction. ASOs can adjust the usage of 5′ splice sites by masking cis-regulatory sequences. Exon skipping occurs with enhancer-targeting ASOs, while silencer-targeting promotes exon inclusion.18,19 | ||
Correct gene expression is closely tied to physiological splicing patterns. These patterns can vary depending on the tissue type and on the developmental stages, as well as in other situations.5–7 Mutations in certain trans-acting splicing factors, specifically those that act in a tissue-specific manner, can disrupt these physiological patterns.8 In cancer, some of these splicing factors can function as either oncogenic proteins or as tumor suppressors. Mutations in these factors are often linked to the onset and progression of cancers.9–11 Moreover, both germline and somatic mutations can alter cis-regulatory sequences within pre-mRNA. This can lead to incorrect splicing patterns. Such patterns can underlie various diseases. For example, aberrant splicing is implicated in inherited conditions like Huntington's disease, a rare neurdegenative disorder.12 It is also involved in specific types of breast and colorectal cancers, among others.13–16
The therapeutic modulation of RNA splicing emerged as a promising avenue for the treatment of several diseases previously refractory to medical intervention.2,17 Historically, antisense oligonucleotides (ASOs) have been used to target cis-regulatory sequences in the pre-mRNA (Fig. 1D).18–20 In this paradigm, RNA analog sequences are designed to hybridize with complementary cis-acting regulatory elements on the target pre-mRNA. By doing so, ASO drugs can inhibit exon inclusion events by masking a splicing enhancer cis-acting regulatory sequence, hence preventing the recruitment of an activator trans-acting splicing factor (e.g., eteplirsen19). Complementary, ASOs can also anneal with splicing silencer cis-acting regulatory sequences, thereby promoting exon inclusion (e.g., nusinersen18). Although ASOs have manifested efficacy in clinical contexts, their deployment typically necessitates more intensive therapeutic protocols, including recurrent injections in the hospital setting.2,21
Over the past decade, the pharmacological modulation of RNA splicing using small organic molecules has emerged as a viable and innovative complement to traditional methodologies.2 A significant advantage of this approach lies in its therapeutic administration, because these molecules are orally bioavailable and do not necessitate specific clinical environments for administration.22,23 These small molecules can be designed to target an array of actors within the context of splicing, from spliceosome components to diverse trans-acting splicing factors, and even RNA targets themselves, including both regulatory and precursor messenger RNAs.24–28 In particular, the task of designing small molecules that specifically bind to RNA and modulate splicing is a substantial challenge. Indeed, RNA molecules have markedly fewer hydrophobic surfaces relative to proteins and they present more restricted binding pockets.29,30 Yet, they remain amenable to drug targeting by exploiting their distinct three-dimensional conformations and inherent sequence element specificity.31,32
In this comprehensive review, we examine small molecules that can influence RNA splicing. We group and discuss them based on their target types: the spliceosome, trans-acting splicing factors, and RNA targets. For each target type, we describe its specific role and highlight key small molecules that can adjust splicing by interacting with that target. We finally discuss future perspectives with respect to each target type. Overall, our main goal is to picture the landscape of innovative strategies aiming to modulate RNA splicing with small molecules in the context of diseases.
Small molecules targeting the spliceosome
RNA splicing is a complex and dynamic biochemical process wherein the spliceosome, a large and highly dynamic ribonucleoprotein assembly, acts sequentially on the pre-mRNA to achieve the splicing reaction (Fig. 1C).1 The major spliceosome contains small nuclear ribonucleoproteins (snRNPs) specifically denoted as U1, U2, U4, U5, and U6. Each snRNP is composed of a small nuclear RNA (snRNA), an heptameric ring of Sm proteins and several snRNP-specific proteins bound to the snRNA (Fig. 1B). Small molecules targeting spliceosome biogenesis and those binding to snRNP components have been identified as potential anti-cancer therapeutics and as investigative tools both in vitro and in vivo.24,25Inhibitors of U2 snRNP via SF3B complex
The SF3b complex is an essential component of U2 snRNP and is required for pre-mRNA splicing to complete.33,34 While U2 snRNP recognizes the branch point via base-pairing mediated by U2 snRNA, the stability of this interaction is enhanced by the SF3b complex, which also connects with the intron. The SF3b complex includes seven proteins, namely SF3b1-6 and PHF5A (Fig. 2A).35,36 In particular, SF3b1 interacts with the nucleotides surrounding the branch point adenosine and, alongside PHF5A, creates a binding pocket to accommodate the corresponding intron. Several inhibitors targeting SF3b1 were developed, with significant therapeutic possibilities, especially in cancer treatments.37 These inhibitors can be broadly divided into three classes: pladienolides, herboxidienes, and spliceostatins. | ||
| Fig. 2 Small molecules targeting the spliceosome to modulate splicing. (A) Crystal structure of the human SF3b core complex.35 The region for binding of inhibitors is highlighted (red dashed frame) and corresponds to the close-up view shown in (C). (B) Chemical structure of molecules targeting the SF3b complex. (C) Crystal structure of the SF3b complex bound to the splicing inhibitor pladienolide B.37 (D) UHM–ULM interactions in pre-spliceosomal complexes. ULM ligands (W-containing yellow circles) and UHM domains (red-contoured circles) are highlighted. (E) Chemical structure of molecules targeting UHM-containing proteins. (F) Crystal structure of PUF60-UHM in complex with 7,8-dimethoxyperphenazine.67 (G) Inhibition of PRMT5 during snRNP biogenesis. The 20S methylosome (PRMT5-containing complex) catalyzes the arginine-methylation of Sm proteins D1, B/B′ and D3 using S-adenosylmethionine (SAM) as methyl-donor. Upon nuclear export of snRNA, the Sm site is the location around which mature Sm proteins are assembled in a ring. | ||
Pladienolides originate from natural products that contain a large macrocyclic lactone ring, so they are referred to as macrolides (Fig. 2B). Pladienolides include molecules such as pladienolides A–G, H3B-8800,38 and E7107.39,40 Correspondingly, herboxidienes are also of natural origin and they are polyketide compounds containing a tetrahydropyran ring and an epoxy group that is important for splicing inhibition (Fig. 2B).41 Finally, the spliceostatins group comprises spliceostatin A, sudemycins C, D1, D6, E, F, meayamycin B, and other synthetic compounds.42–45 Spliceostatins contain two functionalized tetrahydropyran rings joined by a diene moiety and an acyclic side chain linked with an amide bond (Fig. 2B).46 Pladienolides, herboxidienes and spliceostatins are highly cytotoxic and they are reported to exhibit strong antiproliferative effects against multiple cancer cell lines.47–49 These splicing inhibitors target SF3b1 within the SF3b complex, but they also interact with PHF5A, another component of the SF3b complex.37,49–52 Interestingly, mutations of Y36 in PHF5A and of K1071, R1074, V1078 in SF3b1 can be associated with resistance to these inhibitors, suggesting a shared binding site.51,53 These resistance-related mutations are localized around the branch point adenosine binding pocket, suggesting a competitive mechanism of inhibition that locks the SF3b complex into an open, inactive conformation.37,51 Furthermore, all-atom simulations of SF3b in apo form and in complex with splicing modulators not only predicted these results but also revealed the impact of recurrent drug resistance and sensitising mutations in SF3B1 or PHF5A on the functional dynamics of SF3b1.53
In the crystal structure of the human SF3b core in complex with pladienolide B, the molecule is accommodated within an hourglass-shaped tunnel that matches the binding site of the branch point adenosine (Fig. 2C).37 Accordingly, the cryogenic electron microscopy (cryo-EM) structure of the SF3b complex bound to the pladienolide derivative E7107 showed that the drug also binds to the branch point adenosine binding pocket.52 Notably, E7107 closely interacts with two residues, R1074 in SF3b1 and Y36 in PHF5A. When these residues are altered, resistance occurs. Overall, pladienolide B and E7107 stall SF3b in an open conformation, preventing the formation of a stable complex with the duplex between the branch point and U2 snRNP.37,52 E7107 was the first spliceosome inhibitor to enter a clinical trial but was discontinued due to adverse effects, including vision loss for two participants.54 Since then, the pladienolide derivative H3B-8800 was discovered as an orally administered modulator of the SF3b complex, which had shown specificity and efficacy in targeting spliceosome-mutant cells in murine models of various cancers (Fig. 2B).38 Interestingly, H3B-8800 induced preferential lethality in cancer cells bearing the frequent K700E mutation in SF3b1 by triggering aberrant splicing.55,56 H3B-8800 entered a clinical trial but, despite a more favorable safety profile compared to E7107, failed to recapitulate the murine experiments with no complete or partial responses observed.57 Despite the chemical similarities between H3B-8800 and E7107, and the assumption that they act through a similar mode of action, the mechanisms underlying their distinct efficacy and safety profiles require further investigations.
In summary, the discovery and development of spliceosome inhibitors such as pladienolides, herboxidienes, and spliceostatins, have highlighted the therapeutic potential of targeting the SF3b complex for cancer treatments. The structural insights from both crystallography and cryo-EM studies have advanced our understanding of how these inhibitors bind and inhibit the SF3b complex, providing a structural basis for further developments in this field.37,52 Despite the initial setback in clinical trials with E7107 and H3B-8800,54,57 the development of safer and more effective spliceosomal inhibitors is underway and could hopefully lead to new options in cancer-therapy.
Inhibitors of U2AF homology motif (UHM) domains
During the early phases of spliceosome assembly, essential interactions occur between U2AF homology motif (UHM) domains found in spliceosome components and their corresponding U2AF ligand motif (ULM) ligands.58,59 UHM domains, structurally equivalent to RNA recognition motifs (RRMs), differ functionally as they bind to peptidic sequences instead of ribonucleic acids.60 A defining feature of UHMs is the Arg-X-Phe motif located in the loop connecting helix α2 and strand β4.61 They preferentially recognize ULM peptides, which contain a conserved tryptophan surrounded by basic residues.61 UHM domains are found in several spliceosome components, and their mutations have implications in diseases like myelodysplastic syndromes.62,63 For instance, U2AF, an essential spliceosome heterodimer, comprises U2AF2 and U2AF1, each containing a UHM domain.60 During the pre-spliceosome complex E formation, UHM-containing components arrange on the intron: U2AF2 binds to the polypyrimidine tract, U2AF1 to the 3′ splice site, and SF1 to the branch point, which enables UHM–ULM interactions since U2AF2 and SF1 each contain an ULM (Fig. 2D).58,59 In the pre-spliceosome complex A, the multiple ULMs of SF3b1 can interact with U2AF2-UHM and with UHM-containing splicing factors (Fig. 2D).64 Overall, UHM–ULM interactions are pivotal in the early stages of spliceosome assembly and for splicing to occur,61,62 introducing pharmacological targets for splicing modulation by small molecules.Using virtual screening on a large small molecule database and in vitro competition assays, a novel small molecule, UHMCP1, was identified (Fig. 2E).65 UHMCP1 efficiently blocks the SF3b1/U2AF2 interaction in the pre-spliceosome complex A, as supported by both NMR spectroscopy analyses and molecular dynamics simulations.65 The effect of UHMCP1 on RNA splicing modulation is correlated with antiproliferative properties, which makes the compound a potential novel anticancer drug candidate. UHMCP1 was shown to interact with the hydrophobic pocket of U2AF2-UHM domain.65 The molecule features a polycyclic group that binds to the tryptophan binding pocket of UHM, analogous to the indole group of the ULM tryptophan residue.61,65 In the structural model of the complex, the tail of UHMCP1 extends towards loops connecting β1 to α1 and α2 to β4 in U2AF2-UHM domain, closely resembling to the binding mode of the native ULM peptide.61,65 In addition to spliceosomal SF1, U2AF1 and U2AF2, the splicing factors RBM39, SPF45 and PUF60 also contain an UHM domain that can be targeted with small molecules. In a recent study, the binding activity profiles of UHMCP1 derivatives were determined using binding assays.66 Unmodified UHMCP1 showed IC50 values of 74.85 μM for SPF45-UHM, 159.35 μM for RBM39-UHM, 175.61 μM for PUF60-UHM and 239.9 μM for U2AF35-UHM, highlighting the limited selectivity of UHMCP1 to target UHM domains.66
Phenothiazines have been identified as broad inhibitors of UHM–ULM interactions (Fig. 2E).67 Specifically, derivatives of 7,8-dihydroxyperphenazine can prevent the formation of the pre-spliceosome complex A, leading to a stalled spliceosome. NMR spectroscopy studies confirmed that these compounds bind to the tryptophan binding pocket within the UHM domain of SPF45.67 This is further supported by a co-crystal structure that shows 7,8-dimethoxyperphenazine bound to the UHM domain of PUF60 (Fig. 2F).67 In this structure, the phenothiazine moiety fills the tryptophan binding pocket of PUF60-UHM, which is slightly extended compared to the recognition of ULM peptide in order to fit the three aromatic rings of the phenothiazine. The hexagonal ring at the 7,8-dimethoxy position engages in π-stacking with F534 in the PUF60-UHM domain. Additionally, the N14-amine group of the piperazine part interacts with the E483 side chain. This bond is further stabilized by an intramolecular electrostatic interaction with the side chain of K486. Compared to UHMCP1, 7,8-dimethoxyperphenazine showed lower IC50 values with 10.4 μM for SPF45-UHM and less specificity with 10.4 μM for PUF60-UHM and 9.2 μM for U2AF2-UHM.67 Other derivatives reported in the study showed reduced or increased binding activities but without convincingly improving drug specificity.
In summary, small molecule UHM inhibitors, together with synthetic ULM peptides,68 modulate UHM–ULM interactions. Despite challenges in target specificity, these inhibitors can block early spliceosome assembly by affecting interactions between components like SF1, U2AF1, U2AF2, and specific trans-acting splicing factors. They can be used as investigative tools to study the role of UHM-mediated interactions in splicing, awaiting the development of more selective UHM inhibitors.
Inhibitors of snRNPs biogenesis
The small nuclear ribonucleoprotein (snRNP) particles that make the spliceosome are each composed of a small nuclear RNA (snRNA) that contains a so-called Sm site around which a ring of seven Sm proteins is assembled (Fig. 1B).1 Among the Sm proteins, the heterodimers D1/D2 and B/D3 are methylated by the type II arginine methyl-transferase PRMT5 from the 20S methylosome complex.69,70 Methylation enables the association of the seven Sm proteins with the survival of motor neurons (SMN) complex, prior to ring assembly around the Sm site found in cytosolic snRNAs (Fig. 2G). Importantly, the involvement of PRMT5 in the regulation of pre-mRNA splicing is evolutionary conserved across diverse organisms.71 Since core components of the splicing machinery are arginine-methylated by PRMT5, the development of small molecule inhibitors of PRMT5 represents an additional route to inhibit splicing by targeting snRNP biogenesis rather than spliceosome assembly itself.Selective inhibitors of PRMT5 were developed, showing in vitro and in vivo potency in lymphoma-derived cells (Fig. 2G).72 Antiproliferative effects and cell death were observed with IC50 values in the nanomolar range for the reported GSK3235025 and GSK3203591 compounds.72,73 Importantly, PRMT5 inhibition was monitored through levels of Sm D3 methylation, which means that decreased methylation of Sm proteins was correlated with anti-tumor effect upon anti-PRMT5 treatment.72,73 Structural studies highlighted the importance of the tetrahydroisoquinoline core of the designed ligands for cation–π interactions, as shown in the crystal structure of PRMT5/MEP50 in complex with GSK3235025.72 Even though PRMT family members regulate the activity of many proteins, including splicing factors, it is known that inhibition may affect splicing activities.74,75 Moreover, decreased Sm protein methylation correlates with antiproliferative effects, which encourages to investigate how strong is the causal link between inhibition of snRNP biogenesis itself and antiproliferative effect of PRMT5 inhibitors.
Yet, RNA-binding proteins are the most enriched cellular substrates of PRMTs and inhibition of RNA splicing as a whole underlies the cytotoxic effects of PRMT inhibitors.76 Many splicing factors can be methylated by PRMT family members and mutations can result in non-physiological splicing patterns that contribute to diseases. In acute myeloid leukemia cells, mutations in splicing factors SRSF2, SF3b1 and U2AF35 can sensitize cells to PRMT1 inhibition with MS023,77 PRMT5 inhibition with GSK3203591,73 or both.76 A combined treatment using type I PRMT inhibitors (e.g., GSK3368715) with PRMT5 inhibitors has shown superior tumor growth suppression both in vitro and in vivo than using either inhibitor in isolation.76,78,79 Protein profiling reveals that separate treatments target different protein groups, while combined treatments impact a more extensive protein selection, potential substrates for both PRMT types. Transcriptomic studies consistently showed that combined treatments lead to an increase in aberrant splicing events compared to individual treatments.76,78
In summary, PRMT5 inhibition with small molecules result in splicing defects associated with antiproliferative and cytotoxic properties in cancer cells. Whether the inhibition of snRNP biogenesis is a sufficient route to achieve this effect remain an open question, as it remains highly intricate with changes in the methylation of splicing factors as a whole, and subsequent effect on splicing and cell viability.
Small molecules targeting trans-splicing factors
Small molecules can modulate RNA splicing by targeting the spliceosome. However, due to the ubiquitous nature of the spliceosome, achieving tissue-specific modulation remains a challenge. An alternative strategy is to target trans-acting splicing factors, particularly serine–arginine-rich (SR) splicing factors. These factors are crucial in determining tissue-specific splicing patterns and are implicated in inherited diseases and cancer.5,7,80,81 Therefore, we will describe in this section small molecules designed to target trans-acting splicing factors to correct RNA splicing. We will first address their impact on the phosphorylation of SR proteins, emphasizing molecules that affect SR protein kinases (SRPKs) and CDC2-like kinases (CLKs). Then, we will discuss molecules that directly interact with SR-rich splicing factors to modulate their function. Finally, we will introduce small molecules degraders of splicing factors that can be used to restore physiological splicing patterns.Inhibition of kinase activities required for RNA splicing
Recent studies have identified several small molecules that target kinases responsible for phosphorylating splicing factors. The therapeutic potential of targeting serine/arginine-rich splicing factor protein kinases (SRPKs) and CDC2-like kinases (CLKs) is large, especially for addressing abnormal SR protein expression patterns associated with diseases.82SRPKs form a distinct subfamily of kinases, notable for phosphorylating serine residues within ser–arg and arg–ser dipeptide motifs. This phosphorylation is essential to both constitutive and alternative mRNA splicing.83 SRPK1, in particular, is a key modulator of pro-angiogenic VEGF splicing, achieving this through the phosphorylation of serine-rich splicing factor 1 (SRSF1), which then associates with VEGF pre-mRNA.84VEGF pre-mRNA undergoes alternative splicing to produce two major VEGF isoforms. The pro-angiogenic isoform is generated when exon 8a is included, a process promoted by the binding of phosphorylated SRSF1 to a specific regulatory element in exon 8a of the pre-mRNA (Fig. 3A).85 SRPK1 knockdown via a lentiviral approach has been shown to reduce VEGF-mediated angiogenesis in vivo. Pharmacological inhibition of SRPK1 selectively decreases the expression of pro-angiogenic VEGF isoforms, while sparing the anti-angiogenic variants.86 The kinase catalytic domain in SRPK family members features a unique domain insert within the kinase hinge region. Structural studies of SRPK kinases reveal a helix in this insertion domain, positioned parallel to the kinase hinge, offering a potential anchoring site for inhibitors.84 This structural feature has been used to develop inhibitors of SRPK family members. SRPIN340, an inhibitor characterized by a trifluoromethylphenyl group essential for kinase inhibition, has shown efficacy in inhibiting the kinase activity of SRPK1 (Fig. 3A and D).84In vivo studies in a melanoma mouse model demonstrated the antiproliferative effects of SRPIN340 on tumor cells upon peritumoral administration (IC50 = 0.96 μM).86 Furthermore, overexpression of SRPK1 in leukemia models and the subsequent treatment with SRPIN340 led to both early and late apoptotic events.87 Finally, the SRPIN340 derivative SPHINX, exhibiting more potent SRPK1 inhibition (IC50 = 0.58 μM) and improved selectivity over SRPIN340, underscores the importance of pursuing the development of highly selective SRPK1 inhibitors.84
 | ||
| Fig. 3 Mechanisms of small molecules targeting splicing factors. (A) Mechanism of SRPK1 kinase activity inhibition by SRPIN340. SRPK1 inhibition leads to SRSF1 phosphorylation inhibition, and a decrease of VEGF pro-angiogenic isoform production. (B) Mechanism of pre-mRNA cytoplasmic retention by quercetin. Quercetin competes with the transportin Tnpo1 (purple) for binding to hnRNP A1, inhibiting the nuclear re-import of hnRNP A1, promoting the recruitment of stress granules and leading to apoptosis. (C) Mechanism of RBM39 targeted degradation by indisulam (ArS). Indisulam acts as a molecular glue between RBM39 and DCAF15-U3 Ub ligase, leading to the poly-ubiquitination of RBM39 and its degradation by the proteasome. It triggers AML cell death. (D) Chemical structures of SRPIN340, quercetin and indisulam (ArS). | ||
Similarly, CLKs (Cdc2-like kinases) are essential in regulating mRNA splicing through the phosphorylation of SR-rich domains in splicing factors, which leads to the production of protein isoforms crucial for cell growth and survival.88 CLK targets include SR proteins, SPF45, and U1-70k. CLK-mediated phosphorylation results in increased alternative splicing of certain genes, such as the exclusion of exon 6 in FAS and skipping of exon 6.3 in CCNL2. Typically, this phosphorylation process is guided by the R-x-x-S/T consensus sequence.88 Chlorexidine, a member of the cationic bisbiguanide class with antimicrobial properties, has been identified as a potent modulator of SR protein-mediated alternative splicing. It achieves this by selectively inhibiting CLK family members that phosphorylate SR protein splicing factors.89,90 Exon array experiments revealed that chlorexidine induces significant alternative splicing changes, affecting 1 444 transcripts, including those of RON, caspase 9, JAK3, and HIV Tat2-3.90 In the context of HIV-1, CLK1 has been shown to enhance the expression of HIV-Gag genes, which are crucial for the assembly, release, and maturation of virus particles. The inhibitory effect of chlorexidine on CLK1 leads to altered HIV-1 RNA processing, significantly reducing HIV-1 Gag processing and virus replication.91 Other notable CLK inhibitors, such as TG-003 and TG693, have been effective in suppressing CLK1 kinase activity, resulting in splicing alterations both in vitro and in vivo. These compounds have been shown to facilitate the exclusion of the mutated exon 31 of dystrophin in Duchenne muscular dystrophy (DMD), while preserving the wild-type protein expression. This action promotes the production of functional dystrophin protein, offering potential therapeutic benefits.92,93
In summary, the exploration of small molecule inhibitors targeting kinases essential for RNA splicing, particularly SRPKs and CLKs, has opened new therapeutic avenues. The mechanism of SRPK1-mediated SRSF1 phosphorylation and its regulation of VEGF splicing, alongside the potential of inhibitors like SRPIN340 and SPHINX, offers a promising strategy for treating angiogenesis-related conditions and cancer. Concurrently, CLK inhibitors, including chlorexidine, TG-003, and TG693, are showing promise in modifying alternative splicing patterns, presenting potential treatments for conditions like HIV-1 and DMD.
Inhibitors of trans-splicing factors
Small molecules can regulate the function of trans-acting splicing factors, leading to diverse effects, including either inhibition or activation of these factors.94In the context of splicing repressors, particularly the heterogeneous nuclear ribonucleoproteins (hnRNPs), several inhibitory small molecules have been identified. hnRNP A1, one of these targets, is known for its ability to bind with high affinity to the UAGGGA/U consensus sequence present within pre-mRNA exons.95 When it binds to the pre-mRNA, hnRNP A1 acts as an exportin, interacting with the nuclear pore complex to facilitate the export of the pre-mRNA into the cytoplasm.26 Quercetin, a phytochemical from the flavonoid class, has been found to inhibit hnRNPs.96,97 This compound has shown effectiveness in various cancers, including breast and pancreatic tumors. Under normal conditions, transportin 1 (Tnpo1) binds to the C-terminal region of hnRNP A1, enabling the nuclear re-import of hnRNP A1.98 However, quercetin competes with Tnpo1 for binding to the C-terminal domain of hnRNP A1, thus preventing nuclear re-import and causing cytoplasmic retention of hnRNP A1.26 This interaction triggers cellular changes such as recruitment of stress granules, cell cycle arrest, enhancement of apoptosis, and inhibition of angiogenesis (Fig. 3B).26,96 Additionally, the related hnRNP A18 can recognize a unique RNA signature motif in the 3′ UTR of transcripts.99 This motif is critical for cancer progression and is found in genes like VEGF, TRX, RPA, and the CTLA4 immune checkpoint.99 Chemical probes such as Chembridge 5224046 or 6823240, which bind to the RNA recognition motif (RRM) of hnRNPA18, have been discovered. Their binding disrupts the interaction between hnRNP A18 and the pre-mRNA, leading to reduced levels of Trx and CTLA4 proteins, among others. Such treatments inhibit the growth of various cancer cells while sparing normal epithelial cell viability.99
On the other hand, splicing activators such as the splicing factor SRSF6 are known to be overexpressed in various cancers, correlating with poor prognostic outcomes.100 Studies have demonstrated that SRSF6 promotes colorectal cancer (CRC) proliferation, migration, and invasion in vitro, as well as tumor development and metastasis in NOD/SCID mice.101 SRSF6 contributes to tumorigenesis by modulating the alternative splicing of key tumor-associated genes, including ZO-1 exon 23, a critical cell adhesion molecule.101 The overexpression of SRSF6, leading to cellular transformation, underscores its role as an oncogene and its potential as a therapeutic target. In the context of drug discovery, virtual screening methods have identified indacaterol, a β2 adrenergic receptor-agonist typically used for treating chronic obstructive pulmonary disease (COPD), as a potential pharmacological inhibitor of SRSF6.101,102 Indacaterol inhibits the function of the SRSF6 protein by binding to its RRM2 domain, forming two hydrogen bonds with the side chains of residues E180 and D167, and two more with the backbone of residue L166.101In vitro experiments have shown that this inhibition alters ZO-1 splicing patterns, leading to an increase in the inclusion of exon 23.101 Consequently, this resulted in reduced growth of colorectal cancer cells, as well as decreased tumor development and metastasis in mouse models.101
In summary, the modulation of splicing factors by small molecules, as exemplified by the inhibition of hnRNPs with quercetin and the targeting of SRSF6 with indacaterol, represents a significant advance in oncological therapeutics. These interventions, impacting pre-mRNA splicing and function, demonstrate the potential of precise molecular targeting in cancer treatment, offering pathways for innovative drug development and therapeutic strategies.
Degraders of splicing factors
The targeted degradation of splicing factors is an innovative and highly promising approach for RNA splicing regulation. The typical methodology for targeted protein degradation involves the use of bimodal small molecules that enable a target protein to bind to an E3 Ubiquitin ligase, leading to the ubiquitination of the protein and subsequent proteasomal degradation. A notable breakthrough in this area is the emergence of proteolysis targeting chimeras (PROTACs).103,104 PROTACs are characterized by a bifunctional chemical linker that joins a molecule, which specifically binds to the target protein, with another that recruits an E3 ubiquitin ligase. In the field of RNA splicing, significant advancements have been made with a PROTAC targeting the SF3b complex within the core spliceosome.105 Furthermore, RNA–PROTACs have been introduced, functioning similarly to PROTACs but using an RNA segment to target splicing factors. These RNA–PROTACs have been successfully used to induce the degradation of Lin28A in myelogenous leukemia K562 cells. Lin28A, a key player in development, metabolism, and pluripotency, is overexpressed in various tumors and tumor-derived cell lines. It is known for interacting with let-7 precursors and the 3′-untranslated regions (UTRs) of mRNAs related to cell proliferation.106 A specially engineered 7-nt oligonucleotide analogue (5′-AGGAGAU-3′), modified with a PS-MOE backbone, has shown strong affinity for the zinc finger domain of Lin28A, effectively competing with native RNA for Lin28 binding in cells.106Alternatively, molecular glues that act as degraders of splicing factors have demonstrated significant efficacy, making them attractive candidates for drug development.107,108 In the context of acute myeloid leukemia (AML), RNA binding motif 39 (RBM39) has been identified as a promising target due to its overexpression and vital role in the maintenance and survival of AML cells. RBM39 is essential for the proper splicing of RNA-binding proteins (RBPs) preferentially required in AML, thus making its degradation a feasible strategy in AML treatment.109,110 Arylsulfonamides, such as indisulam, are particularly effective in inducing the proteasomal degradation of RBM39 (Fig. 3C).111 These compounds function as molecular glues, facilitating the interaction between the RRM2 domain of RBM39 and the DCAF15-CRL4 ubiquitin ligase complex.108,112,113 This interaction leads to the ubiquitination of RBM39 in its N-terminal region by the CRL4-U3 ubiquitin ligase, resulting in proteasome-mediated degradation. The depletion of RBM39 disrupts normal splicing patterns of transcriptional regulators crucial for AML cell survival, triggering an apoptotic cascade that leads to the death of these cells.109,113,114 Furthermore, the versatility of arylsulfonamides extends to the degradation of other RNA binding motif (RBM) proteins, including RBM23, a paralog of RBM39.108
Overall, the development of proteolysis targeting chimeras (PROTACs) and RNA–PROTACs marks a significant leap in RNA splicing regulation, offering targeted approaches for degrading specific splicing factors. These novel strategies have shown promise in treating diseases like myelogenous leukemia and acute myeloid leukemia (AML), highlighting a new frontier in therapeutic interventions and drug development.103,105–108
Small molecules targeting RNA
In recent years, efforts have been made to design small molecules that target spliceosomal proteins and splicing factors to regulate alternative splicing. An emerging trend, however, consists in designing molecules that bind RNA to alter RNA metabolism. Structurally, targeting RNA is more challenging than proteins because RNA is highly dynamic and flexible.29 Additionally, the polyanionic RNA backbone hinders the formation of deep hydrophobic pockets, resulting in smaller binding surfaces compared to proteins.30 Furthermore, RNA offers limited structural diversity, which complicates the design of highly specific molecules. Yet, several examples of small molecules interacting with RNA to modulate splicing in disease were reported in the last years. This discussion reviews small molecules acting through bulge-repair at the 5′ splice site,27 highlights representative examples of molecules targeting RNA structure to modulate splicing, and delves into the emerging RNA degrader molecules.115Splicing modifiers acting through bulge-repair at the 5′ splice site
Mutations that disrupt the splicing process can lead to various diseases by altering exon selection and splicing patterns, thereby impacting functional protein production. Such mutations can occur in the 5′ splice site, recognized by U1 snRNP (Fig. 1C), with deviations from the canonical sequence being associated with multiple genetic diseases.13,116,117A prototypical disease associated with RNA splicing is spinal muscular atrophy (SMA), a leading cause of child mortality.118 Over 95% of SMA cases are caused by homozygous inactivation of the SMN1 gene, which encodes the survival motor neuron (SMN) protein.119 Humans also have a paralog gene, SMN2. A higher SMN2 gene copy number often correlates with milder SMA phenotypes.120 However, a silent mutation in SMN2 exon 7 weakens its 5′ splice site, resulting in about 85% of the transcripts excluding exon 7.121 This exclusion produces a truncated, non-functional SMN protein. SMA was historically the first condition for which RNA-targeting small molecule splicing modifiers were developed, showing potency in enhancing motor function and lifespan in animal models and humans with SMA.122–125
The molecular basis for splicing correction by related compounds was established using the selective SMN2 splicing modifier SMN-C5, a substituted pyrimidone, as model (Fig. 4A).27,126 SMN-C5 interacts with the RNA helix formed between the 5′ splice site and the 5′ end of U1 snRNA in the solution structure of the complex (Fig. 4B). The carbonyl group of SMN-C5 provides specificity by forming a direct hydrogen bond with the primary amine of the bulged adenine at position −1. The positively charged amine in the piperazine moiety forms a salt bridge with the negatively charged oxygen in the phosphate group of C9 in the 5′ end of U1 snRNA. By stabilizing an unpaired adenine at the exon-intron junction, SMN-C5 is reported to act through the bulge-repair mechanism.27 This interaction induces an allosteric shift, enhancing the attachment of the U1-C zinc finger, and subsequently U1 snRNP, to the 5′ splice site of SMN2 exon 7, and thus promoting exon inclusion and the production of a functional SMN protein from SMN2 gene (Fig. 4C).
 | ||
| Fig. 4 Small molecules targeting RNA and its metabolism. (A) Chemical structure of splicing modifiers SMN-C5, Risdiplam, and Branaplam for the treatment of spinal muscular atrophy and Huntington's disease. (B) The bulge-repair concept.27 A weak 5′ splice site may be strengthened by small-molecule splicing modifiers acting as a glue between the 5′ end of U1 snRNA and the 5′ splice site. (C) A close-up view of the SMN-C5 binding pocket.27 (D) Chemical structure of tau splicing regulatory element (SRE) stabilizers based on dibenzothiopene (left), quinoline (middle) or pyrimidoindole (right) scaffolds. (E) A close-up view of the quinoline derivative in complex with tau SRE.28 (F) Mechanism of small molecule that stabilize tau SRE to promote exon 10 exclusion. (G) Principle of RNA degraders. | ||
The related splicing modifier, risdiplam, is a substituted pyrimidone already in use for SMA treatment (Fig. 4A).23,125 This early development paved the way for branaplam, a substituted phenol, which targets the huntingtin (HTT) gene to induce the inclusion of a poison exon, leading to the degradation of mRNA instead of the production of a pathological HTT mRNA isoform (Fig. 4A).22,127 Branaplam is in advanced clinical trials for Huntington's disease (HD) treatment, another inherited neurodegenerative disorder. Both SMA and HD share a feature: an unpaired adenine at position −1 (A−1) weakens the 5′ splice site. Splicing modifiers, believed to act via the bulge-repair mechanism, can correct this anomaly, leading to therapeutic effects.22,127
However, there remains an urgent need to discover splicing modifiers that target other bulged bases in the 5′ splice site. Drugs that could correct bulged −1 uracil or cytosine seem especially important, as mutations here are linked to diseases like X-linked lymphoproliferative syndrome and erythropoietic protoporphyria.116 The prevalence of genetic disorders associated with bulged 5′ splice sites emphasizes the need for drugs that can target and rectify these specific mutations.
Small molecules targeting RNA structure and metabolism
Compounds that target RNA structures to alter RNA metabolism and splicing are increasingly developed, expanding beyond small molecules that act via bulge-repair mechanisms.27 These compounds may inspire the development of therapies relying on alternative splicing correction to address related pathologies.In neurodegenerative diseases, the pre-mRNA of tau, an essential microtubule-associated protein, emerges as a promising therapeutic target.128 Mutations in the related gene have been linked to frontotemporal dementia and parkinsonism related to chromosome 17 (FTDP-17), with potential implications for Alzheimer's disease.128 Specifically, a mutation characterized by a C-to-U alteration in intron 10 of tau pre-mRNA, located 14 nucleotides downstream from the 5′ splice site, is noteworthy. This mutation destabilizes the hairpin structure at the junction of exon 10 and intron 10, a sensitive splicing regulatory element (SRE).129 This structural alteration enhances U1 snRNP binding to the 5′ splice site, leading to increased exon 10 inclusion. Small molecules that strengthen this hairpin could be therapeutically advantageous, as they might promote exon 10 skipping and diminish the generation of pathogenic tau variant.28,130–132 Recently, an InfoRNA database search identified compounds that stabilizes the tau pre-mRNA SRE (Fig. 4D).28,133 These molecules interact with a pocket at the A-bulge within the SRE stem, echoing the binding mode of splicing modifiers that target A−1 bulged splice sites (Fig. 4E). Interestingly, while some of these compounds are derived from dibenzothiopene, others contain a quinoline or pyrimidoindole core. Of these, only the quinoline derivatives demonstrated interaction, yet unspecific, with the bulged base in the tau SRE (Fig. 4E). The stabilization provided by dibenzothiopene and pyrimidoindole derivatives arised mainly from π–π and Van der Waals interactions, not direct engagement with the bulged adenine. Yet, all these compounds effectively promoted tau exon 10 exclusion, thus mitigating the effect of C-to-U mutation in the SRE of intron 10 in tau pre-mRNA (Fig. 4F).28
In the context of inherited diseases, myotonic dystrophy type 1 (DM1) originates from an extended RNA repeat expansion, termed r(CUG), located in the 3′ untranslated region of the dystrophia myotonica protein kinase (DMPK) mRNA. This mutation causes the disease through a gain-of-function mechanism.134 The r(CUG) repeat serves as a binding site that captures the muscle-blind like protein 1 (MBNL1). MBNL1 is crucial for alternative splicing of multiple transcripts, especially the insulin receptor (IR) and muscle-specific chloride ion channel (Clnc1).135–137 When MBNL1 is trapped by the r(CUG) repeats, it results in improperly spliced products, central to DM1 pathology. The affinity of MBNL1 to these repeats ranges from 3–200 nM.138 Thus, discovering molecules that can bind to the r(CUG) repeats more strongly than MBNL1 could potentially prevent MBNL1 capture and restore its physiological activity on splicing. A particular study identified a cell-permeable compound with two triaminotriazine units connected by a bisamidinium segment that can compete effectively with MBNL1 for r(CUG) repeat binding.139 Consequently, researchers developed multivalent cell-penetrating ligands targeting r(CUG) repeats. Designed to mirror the repetitive target sequence, they alternate recognition sites with bisamidinium groove binders, yielding an amphiphilic, polycationic structure reminiscent of cell-penetrating peptides.140 These compounds significantly alleviated DM1 symptoms in DM1 cells and a DM1 mouse liver model.140 Additionally, improvements in climbing defects were noted in adult DM1 Drosophila models.
In summary, targeting RNA structures with small molecules opens new therapeutic avenues for diseases driven by RNA mutations and splicing anomalies. This approach represents a significant step forward in developing innovative therapies for RNA-related pathologies, indicating a promising direction for future medical research and drug design.
RNA degraders
Small molecules that specifically bind RNA with high affinity do not always have intrinsic biological activity. Yet, when conjugated with a compound that recruits a ribonuclease, these otherwise inactive molecules can potently modulate RNA metabolism by inducing specific RNA decay. This innovative strategy offers a fresh perspective on RNA modulation using bifunctional small molecules.115,141,142 Although it has not yet been employed for RNA splicing modulation, it presents a promising alternative for managing toxic mRNAs, such as in Huntington's disease (HD). Current treatments for HD rely on splicing modifiers that mediate mRNA decay by promoting the inclusion of a toxic exon.In RNA degradation research, ribonuclease-targeting chimeras (RIBOTACs) represent a significant advancement. These chimeras combine an RNA-binding molecule with a compound that recruits RNase L locally (Fig. 4G).141,142 RNase L preferentially cleaves RNAs with a UNN pattern, especially those with an unpaired uracil. In RIBOTACs, the RNA-binding section specifically recognizes the structured RNA segment, due to the broader binding surfaces offered by folded RNAs. Successful examples include RIBOTACs developed to selectively target the precursor to oncogenic microRNA-21 (pre-miR-21) for cleavage and degradation with high selectivity.142 One part of the compound specifically binds pre-miR-21 and the other contains a heterocycle that recruits and activates a ribonuclease to degrade pre-miR-21, leading to antiproliferative effect in cell models of breast cancer.142 Using high-throughput approaches, recent studies have also investigated the interaction dynamics of natural product-inspired small molecules with RNA structures.115 While many identified interactions were presumed biologically inactive, the RIBOTAC strategy enabled conversion into degraders, with some exhibiting bioactivity. Specific degraders were designed for disease-related pre-miR-155, JUN mRNA, and MYC mRNA. Applying this strategy to a miRNA precursor revealed that RIBOTAC degraders decreased miRNA concentrations in disease-associated cell lines and in vivo.115 Additionally, RIBOTAC degraders effectively targeted JUN and MYC mRNAs, inhibiting pathways regulated by these oncogenes.
In parallel, the proximity-induced nucleic acid degrader (PINAD) approach is an alternative method for RNA targeting and degradation.143 Designed in part to bypass the necessity for localized RNAse L expression, PINADs are composed of an RNA-binding small molecule, an elongated flexible linker, and an imidazole, the RNA-degrading component found in many ribonucleases. Previous studies indicated that attaching imidazole groups to RNA leads to their degradation.144 This suggests the PINAD work through spatial proximity of the imidazole to the cleavage site. Consistently, a study using the aminoglycoside antibiotic neomycin, coupled with an amino acid histidine, demonstrated RNA degradation capability under physiological conditions.145 Using the PINAD methodology, two RNA degrader classes targeting unique SARS-CoV-2 genome structures were formulated against G-quadruplexes and betacoronaviral pseudoknots.143 Their efficacy in degrading targets was validated through in vitro, in cellulo, and in vivo SARS-CoV-2 models.
In summary, small molecules that target RNA have emerged as innovative tools to manipulate RNA metabolism. These molecules can be used to modulate alternative splicing and address several diseases associated with aberrant RNA splicing. The development of these small molecules is based on specific interactions with the target RNA sequence and structure. Recent advancements, such as the discovery of small molecules that act through bulge-repair27 and RNA degraders like RIBOTACs,115,142 not only highlight the potential of RNA as a drug target but also underscore the therapeutic promise of these molecules.
Discussion
RNA splicing modulation with small molecules offers promising clinical potential. In line, a recent review article provides a complementary structural view based on computational and structural biology data, focusing on spliceosome inhibitors.146 Herein, we proposed a comprehensive overview of small molecules that modulate splicing by reviewing distinct targets, including the core spliceosome, specific splicing factors, and particular RNA sequences (Table 1). Additionally, we discussed case examples of small molecules targeting these components to modulate splicing.| Name | Target class | Molecular target | Clinical trials | Chemical formula |
|---|---|---|---|---|
| H3B-8800 (ref. 38) | Spliceosome (U2 snRNP) | SF3b1 | NCT02841540 |

|
| E7107 (ref. 39) | SF3b1 | NCT00499499, NCT00459823 |
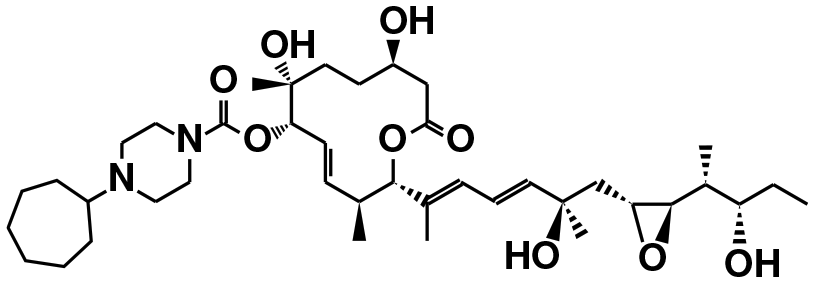
|
|
| Herboxidiene41 | SF3b1 | — |

|
|
| FR901464 (ref. 50) | SF3b1 | — |
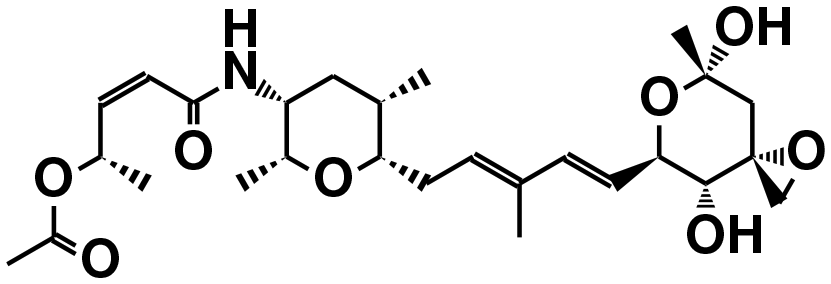
|
|
| UHMCP1 (ref. 65) | Spliceosome (UHM/ULM) | UHM domains (U2AF1, U2AF2, PUF60,…) | — |

|
| 7,8-dimethoxyperphenazine67 | — |

|
||
| GSK3203591 (ref. 72 and 73) | Spliceosome (snRNP biogenesis) | PRMT5 | — |

|
| SRPIN340 (ref. 84) | Splicing factors (via kinases) | SRPK1 | — |

|
| Quercetin96,97 | Splicing factors | hnRNP A1 | No relevant trial |

|
| Indisulam111 | RBM39, RBM23 | NCT00014625, NCT00165594, see https://clinicaltrials.gov/ |

|
|
| SMN-C5 (ref. 27 and 125) | RNA | A−1 bulged | — |

|
| 5′ splice sites | ||||
| Risdiplam23,125 | A−1 bulged | NCT02908685, NCT03779334, see https://clinicaltrials.gov/ |

|
|
| 5′ splice sites | ||||
| Branaplam22,127 | A−1 bulged | NCT05111249, NCT05330286, NCT02268552 |
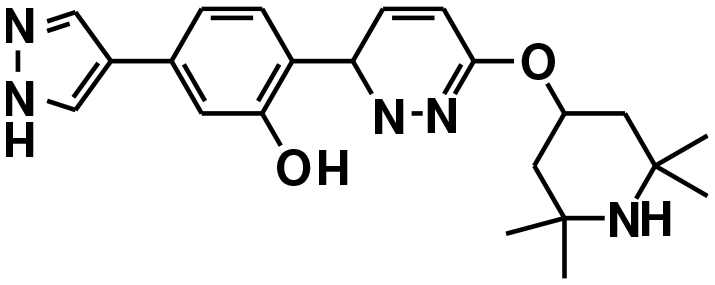
|
|
| 5′ splice sites | ||||
| Cmp5 (ref. 28) | Tau SRE | — |

|
In this review, we highlighted how small molecules that target the spliceosome can function as potential anti-cancer agents and investigative tools.24,25 Several inhibitors have been identified to target SF3b1 within the SF3b complex of U2 snRNP, including pladienolides, herboxidienes, and spliceostatins. Notably, pladienolides have moved from laboratory investigations to clinical trials, though with mixed results regarding safety and efficacy.54,57 Recent research also explained how spliceostatin A can disrupt prespliceosome formation by forming a covalent link with the zinc finger of PHF5A, a component of the SF3b complex, as revealed by crystallographic studies.147 Alternatively, recent efforts demonstrated that proteolysis-targeting chimeras (PROTACs) can be developed to selectively target SF3b1, leading to proteasome-mediated degradation and cell death.105 Moreover, it was recently shown that the cyclin-dependent kinase 11 (CDK11) phosphorylates SF3b1, and that selective inhibition of CDK11 led to widespread intron retention and accumulation of non-functional spliceosomes on pre-mRNAs and chromatin.148 Therefore, the development of inovative approaches to target the SF3b complex is ongoing. In parallel, the U2AF homology motif (UHM) domains play a crucial role in spliceosome assembly. Recent molecules like UHMCP1 have shown potential to inhibit UHM-mediated interactions within pre-spliceosomal complexes.65 Yet, achieving specificity remains challenging, as demonstrated by limited selectivity of UHMCP1 and phenothiazine derivatives for target UHMs.66,67 Future studies aim to optimize these molecules for specificity and safety. Alternatively, it should be noted that UHM-containing proteins can also be targeted on distinct regions. For instance, the small molecule NSC 194308 interacts with the tandem RRMs of U2AF2, thereby promoting an early-stage U2AF2–RNA complex and triggering a stall in pre-mRNA splicing.149,150
Efforts to modulate tissue-specific splicing can involve targeting trans-acting splicing factors with small molecules.26,100,109 Among the most promising developments are PROTACs and molecular glues, both of which promoting proteasome-mediated degradation of the target.103,151 These drug agents can induce almost complete protein degradation even when present at substochiometric concentration with respect to the target splicing factor. In this review, we have explored PROTACs bearing an RNA component that target the splicing factor Lin28A.106 Subsequently, the RNA–PROTAC triggers proteasome-mediated degradation through its E3 ligase recruitment warhead. RNA–PROTACs leverage the RNA binding capabilities of splicing factors, and it is thus plausible to imagine developing PROTACs purely based on small molecules for targeting splicing factors. For instance, attaching an E3 ligase-recruiting component to a small molecule known to target UHM domain in splicing factors could enable directed degradation of UHM-containing splicing factors.65,103 On another front, the scientific community has shown increasing interest in expanding the list of molecular glues that target RNA-binding proteins (RBPs) beyond arylsulfonamides and their representative compound indisulam, with a potential therapeutic benefits for a range of conditions.109,151 Molecular glues have unlocked opportunities to target proteins previously considered undruggable.113,152 Indeed, molecular glues work by inducing a tripartite complex between the target and the E3 ligase adapter (e.g., DCAF15), without the need for targeting a specific functional site in the target splicing factor, and while being effective at substochiometric concentrations.153 This means that, in principle, it is not necessary for the drug to fully saturate its target to achieve the desired effect on splicing modulation, thus encouraging to focus on ligand specificity rather than affinity in the design of future molecular glues.
When it comes to target RNA in order to modulate its metabolism, a recent groundbreaking progress is the development of RNA degraders, either ribonuclease-targeting chimeras (RIBOTACs)141,142 or proximity-induced nucleic acid degraders (PINADs).143 This is primarily due to their potential to selectively diminish the levels of disease-associated, target RNAs.115 While such approaches were not yet applied to splicing modulation, one could easily imagine designing RNA degraders directed toward, for instance, the toxic mRNA responsible for Huntington's disease. More generally, RNA degraders could be used in principle to selectively degrade mRNA containing the toxic exon or motif, based on specific sequence and structure elements. In parallel, there is a growing interest directed toward splicing modifiers acting through the bulge-repair mechanism at the junction between U1 snRNP and the 5′ splice site. Recent research highlights that three distinct chemotypes consistently strengthening the 5′ splice site toward U1 snRNP.152 Consistently, these chemotypes appear to share a common mode of action recapitulated by the bulge-repair concept at the 5′ splice site.27,154 In the future, a notable challenge will be the diversification of these modifiers to target other bulged bases for any position in the splice site, as bulged sites are associated with various inherited diseases and several types of cancer.116 Given the systematic nature of the associated pharmacological targets, which are short RNA duplexes with single point differences, it is anticipated that leveraging high-throughput screening combined with both generative and deterministic artificial intelligence methods will speed-up the identification of new compounds. These compounds will target the spectrum of bulged bases in 5′ splice sites related to diseases.116
In conclusion, recent studies highlight the significant promise of small molecules for modulating RNA splicing in clinical settings. Their adaptability to engage with a spectrum of targets with diverse mode of action hopefully lays the groundwork for upcoming therapeutic breakthroughs.
Conflicts of interest
There are no conflicts of interest to declare.References
- M. E. Wilkinson, C. Charenton and K. Nagai, Annu. Rev. Biochem., 2020, 89, 359–388 CrossRef CAS PubMed.
- F. Malard, C. D. Mackereth and S. Campagne, RNA Biol., 2022, 19, 943–960 CrossRef CAS PubMed.
- K. S. Rodrigues, L. P. Petroski, P. H. Utumi, A. Ferrasa and R. H. Herai, Life Sci. Alliance, 2023, 6, e202201593 CrossRef CAS PubMed.
- Z. Wang and C. B. Burge, RNA, 2008, 14, 802–813 CrossRef CAS PubMed.
- J. M. Rodriguez, F. Pozo, T. di Domenico, J. Vazquez and M. L. Tress, PLoS Comput. Biol., 2020, 16, e1008287 CrossRef CAS PubMed.
- N. H. Gehring and J. Y. Roignant, Trends Genet., 2021, 37, 355–372 CrossRef CAS PubMed.
- J. D. Ellis, M. Barrios-Rodiles, R. Colak, M. Irimia, T. Kim, J. A. Calarco, X. Wang, Q. Pan, D. O'Hanlon, P. M. Kim, J. L. Wrana and B. J. Blencowe, Mol. Cell, 2012, 46, 884–892 CrossRef CAS PubMed.
- H. Dvinge, E. Kim, O. Abdel-Wahab and R. K. Bradley, Nat. Rev. Cancer, 2016, 16, 413–430 CrossRef CAS PubMed.
- E. Papaemmanuil, M. Cazzola, J. Boultwood, L. Malcovati, P. Vyas, D. Bowen, A. Pellagatti, J. S. Wainscoat, E. Hellstrom-Lindberg, C. Gambacorti-Passerini, A. L. Godfrey, I. Rapado, A. Cvejic, R. Rance, C. McGee, P. Ellis, L. J. Mudie, P. J. Stephens, S. McLaren, C. E. Massie, P. S. Tarpey, I. Varela, S. Nik-Zainal, H. R. Davies, A. Shlien, D. Jones, K. Raine, J. Hinton, A. P. Butler, J. W. Teague, E. J. Baxter, J. Score, A. Galli, M. G. Della Porta, E. Travaglino, M. Groves, S. Tauro, N. C. Munshi, K. C. Anderson, A. El-Naggar, A. Fischer, V. Mustonen, A. J. Warren, N. C. Cross, A. R. Green, P. A. Futreal, M. R. Stratton and P. J. Campbell, N. Engl. J. Med., 2011, 365, 1384–1395 CrossRef CAS PubMed.
- T. A. Graubert, D. Shen, L. Ding, T. Okeyo-Owuor, C. L. Lunn, J. Shao, K. Krysiak, C. C. Harris, D. C. Koboldt, D. E. Larson, M. D. McLellan, D. J. Dooling, R. M. Abbott, R. S. Fulton, H. Schmidt, J. Kalicki-Veizer, M. O'Laughlin, M. Grillot, J. Baty, S. Heath, J. L. Frater, T. Nasim, D. C. Link, M. H. Tomasson, P. Westervelt, J. F. DiPersio, E. R. Mardis, T. J. Ley, R. K. Wilson and M. J. Walter, Nat. Genet., 2011, 44, 53–57 CrossRef PubMed.
- F. Damm, O. Kosmider, V. Gelsi-Boyer, A. Renneville, N. Carbuccia, C. Hidalgo-Curtis, V. Della Valle, L. Couronné, L. Scourzic, V. Chesnais, A. Guerci-Bresler, B. Slama, O. Beyne-Rauzy, A. Schmidt-Tanguy, A. Stamatoullas-Bastard, F. Dreyfus, T. Prébet, S. de Botton, N. Vey, M. A. Morgan, N. C. Cross, C. Preudhomme, D. Birnbaum, O. A. Bernard and M. Fontenay, Blood, 2012, 119, 3211–3218 CrossRef CAS PubMed.
- K. Sathasivam, A. Neueder, T. A. Gipson, C. Landles, A. C. Benjamin, M. K. Bondulich, D. L. Smith, R. L. M. Faull, R. A. C. Roos, D. Howland, P. J. Detloff, D. E. Housman and G. P. Bates, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2366–2370 CrossRef CAS PubMed.
- M. M. Scotti and M. S. Swanson, Nat. Rev. Genet., 2016, 17, 19–32 CrossRef CAS PubMed.
- Y. Zhang, J. Qian, C. Gu and Y. Yang, Signal Transduction Targeted Ther., 2021, 6, 78 CrossRef CAS PubMed.
- R. K. Bradley and O. Anczuków, Nat. Rev. Cancer, 2023, 23, 135–155 CrossRef CAS PubMed.
- S. C. Bonnal, I. López-Oreja and J. Valcárcel, Nat. Rev. Clin. Oncol., 2020, 17, 457–474 CrossRef PubMed.
- E. El Marabti and O. Abdel-Wahab, Trends Mol. Med., 2021, 27, 643–659 CrossRef CAS PubMed.
- N. N. Singh, M. D. Howell, E. J. Androphy and R. N. Singh, Gene Ther., 2017, 24, 520–526 CrossRef CAS PubMed.
- Y. Y. Syed, Drugs, 2016, 76, 1699–1704 CrossRef CAS PubMed.
- J. Kim, C. Hu, C. Moufawad El Achkar, L. E. Black, J. Douville, A. Larson, M. K. Pendergast, S. F. Goldkind, E. A. Lee, A. Kuniholm, A. Soucy, J. Vaze, N. R. Belur, K. Fredriksen, I. Stojkovska, A. Tsytsykova, M. Armant, R. L. DiDonato, J. Choi, L. Cornelissen, L. M. Pereira, E. F. Augustine, C. A. Genetti, K. Dies, B. Barton, L. Williams, B. D. Goodlett, B. L. Riley, A. Pasternak, E. R. Berry, K. A. Pflock, S. Chu, C. Reed, K. Tyndall, P. B. Agrawal, A. H. Beggs, P. E. Grant, D. K. Urion, R. O. Snyder, S. E. Waisbren, A. Poduri, P. J. Park, A. Patterson, A. Biffi, J. R. Mazzulli, O. Bodamer, C. B. Berde and T. W. Yu, N. Engl. J. Med, 2019, 381, 1644–1652 CrossRef CAS PubMed.
- M. Gagliardi and A. T. Ashizawa, Biomedicines, 2021, 9, 433 CrossRef CAS PubMed.
- C. G. Keller, Y. Shin, A. M. Monteys, N. Renaud, M. Beibel, N. Teider, T. Peters, T. Faller, S. St-Cyr, J. Knehr, G. Roma, A. Reyes, M. Hild, D. Lukashev, D. Theil, N. Dales, J. H. Cha, B. Borowsky, R. Dolmetsch, B. L. Davidson and R. Sivasankaran, Nat. Commun., 2022, 13, 1150 CrossRef CAS PubMed.
- S. Dhillon, Drugs, 2020, 80, 1853–1858 CrossRef CAS PubMed.
- M. Salton and T. Misteli, Trends Mol. Med., 2016, 22, 28–37 CrossRef CAS PubMed.
- K. A. Effenberger, V. K. Urabe and M. S. Jurica, WIREs RNA, 2017, 8(2), e1381 CrossRef PubMed.
- Y. Lu, X. Wang, Q. Gu, J. Wang, Y. Sui, J. Wu and J. Feng, Cell Death Discovery, 2022, 8, 337 CrossRef CAS PubMed.
- S. Campagne, S. Boigner, S. Rüdisser, A. Moursy, L. Gillioz, A. Knörlein, J. Hall, H. Ratni, A. Cléry and F. H. Allain, Nat. Chem. Biol., 2019, 15, 1191–1198 CrossRef CAS PubMed.
- J. L. Chen, P. Zhang, M. Abe, H. Aikawa, L. Zhang, A. J. Frank, T. Zembryski, C. Hubbs, H. Park, J. Withka, C. Steppan, L. Rogers, S. Cabral, M. Pettersson, T. T. Wager, M. A. Fountain, G. Rumbaugh, J. L. Childs-Disney and M. D. Disney, J. Am. Chem. Soc., 2020, 142, 8706–8727 CrossRef PubMed.
- C. M. Connelly, M. H. Moon and J. S. Schneekloth, Cell Chem. Biol., 2016, 23, 1077–1090 CrossRef CAS PubMed.
- E. Menichelli, B. J. Lam, Y. Wang, V. S. Wang, J. Shaffer, K. F. Tjhung, B. Bursulaya, T. N. Nguyen, T. Vo, P. B. Alper, C. S. McAllister, D. H. Jones, G. Spraggon, P. Y. Michellys, J. Joslin, G. F. Joyce and J. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2213117119 CrossRef CAS PubMed.
- A. M. Yu, Y. H. Choi and M. J. Tu, Pharmacol. Rev., 2020, 72, 862–898 CrossRef CAS PubMed.
- C. L. Haga and D. G. Phinney, Expert Opin. Drug Discovery, 2023, 18, 135–147 CrossRef CAS PubMed.
- D. Maji, A. Grossfield and C. L. Kielkopf, Biochim. Biophys. Acta, Gene Regul. Mech., 2019, 1862, 194440 CrossRef CAS PubMed.
- C. van der Feltz and A. A. Hoskins, Crit. Rev. Biochem. Mol. Biol., 2019, 54, 443–465 CrossRef CAS PubMed.
- C. Cretu, J. Schmitzová, A. Ponce-Salvatierra, O. Dybkov, E. I. De Laurentiis, K. Sharma, C. L. Will, H. Urlaub, R. Lührmann and V. Pena, Mol. Cell, 2016, 64, 307–319 CrossRef CAS PubMed.
- C. Sun, Cell. Mol. Life Sci., 2020, 77, 3583–3595 CrossRef CAS PubMed.
- C. Cretu, A. A. Agrawal, A. Cook, C. L. Will, P. Fekkes, P. G. Smith, R. Lührmann, N. Larsen, S. Buonamici and V. Pena, Mol. Cell, 2018, 70, 265–273.e8 CrossRef CAS PubMed.
- M. Seiler, A. Yoshimi, R. Darman, B. Chan, G. Keaney, M. Thomas, A. A. Agrawal, B. Caleb, A. Csibi, E. Sean, P. Fekkes, C. Karr, V. Klimek, G. Lai, L. Lee, P. Kumar, S. C. Lee, X. Liu, C. Mackenzie, C. Meeske, Y. Mizui, E. Padron, E. Park, E. Pazolli, S. Peng, S. Prajapati, J. Taylor, T. Teng, J. Wang, M. Warmuth, H. Yao, L. Yu, P. Zhu, O. Abdel-Wahab, P. G. Smith and S. Buonamici, Nat. Med., 2018, 24, 497–504 CrossRef CAS PubMed.
- Y. Kotake, K. Sagane, T. Owa, Y. Mimori-Kiyosue, H. Shimizu, M. Uesugi, Y. Ishihama, M. Iwata and Y. Mizui, Nat. Chem. Biol., 2007, 3, 570–575 CrossRef CAS PubMed.
- E. G. Folco, K. E. Coil and R. Reed, Genes Dev., 2011, 25, 440–444 CrossRef CAS PubMed.
- A. Gamboa Lopez, S. R. Allu, P. Mendez, G. Chandrashekar Reddy, H. M. Maul-Newby, A. K. Ghosh and M. S. Jurica, ACS Chem. Biol., 2021, 16, 520–528 CrossRef CAS PubMed.
- J. Bajsa-Hirschel, Z. Pan, P. Pandey, R. N. Asolkar, A. G. Chittiboyina, L. Boddy, M. C. Machingura and S. O. Duke, Front. Plant Sci., 2022, 13, 1019938 CrossRef PubMed.
- Y. Yoshikawa, A. Ishibashi, T. Takehara, T. Suzuki, K. Murai, Y. Kaneda, K. Nimura and M. Arisawa, ACS Med. Chem. Lett., 2020, 11, 1310–1315 CrossRef CAS PubMed.
- K. C. Nicolaou, D. Rhoades and S. M. Kumar, J. Am. Chem. Soc., 2018, 140, 8303–8320 CrossRef CAS PubMed.
- R. Yoshimoto, J. K. Chhipi-Shrestha, T. Schneider-Poetsch, M. Furuno, A. M. Burroughs, S. Noma, H. Suzuki, Y. Hayashizaki, A. Mayeda, S. Nakagawa, D. Kaida, S. Iwasaki and M. Yoshida, Cell Chem. Biol., 2021, 28, 1356–1365.e4 CrossRef CAS PubMed.
- A. K. Ghosh, J. L. Mishevich and M. S. Jurica, J. Nat. Prod., 2021, 84, 1681–1706 CrossRef CAS PubMed.
- B. J. Albert, P. A. McPherson, K. O'Brien, N. L. Czaicki, V. Destefino, S. Osman, M. Li, B. W. Day, P. J. Grabowski, M. J. Moore, A. Vogt and K. Koide, Mol. Cancer Ther., 2009, 8, 2308–2318 CrossRef CAS PubMed.
- C. Lagisetti, A. Pourpak, T. Goronga, Q. Jiang, X. Cui, J. Hyle, J. M. Lahti, S. W. Morris and T. R. Webb, J. Med. Chem., 2009, 52, 6979–6990 CrossRef CAS PubMed.
- H. Kakeya, D. Kaida, H. Sekiya, K. Nagai, M. Yoshida and H. Osada, J. Antibiot., 2016, 69, 121–123 CrossRef CAS PubMed.
- D. Kaida, H. Motoyoshi, E. Tashiro, T. Nojima, M. Hagiwara, K. Ishigami, H. Watanabe, T. Kitahara, T. Yoshida, H. Nakajima, T. Tani, S. Horinouchi and M. Yoshida, Nat. Chem. Biol., 2007, 3, 576–583 CrossRef CAS PubMed.
- T. Teng, J. H. Tsai, X. Puyang, M. Seiler, S. Peng, S. Prajapati, D. Aird, S. Buonamici, B. Caleb, B. Chan, L. Corson, J. Feala, P. Fekkes, B. Gerard, C. Karr, M. Korpal, X. Liu, J. T. Lowe, Y. Mizui, J. Palacino, E. Park, P. G. Smith, V. Subramanian, Z. J. Wu, J. Zou, L. Yu, A. Chicas, M. Warmuth, N. Larsen and P. Zhu, Nat. Commun., 2017, 8, 15522 CrossRef CAS PubMed.
- L. I. Finci, X. Zhang, X. Huang, Q. Zhou, J. Tsai, T. Teng, A. Agrawal, B. Chan, S. Irwin, C. Karr, A. Cook, P. Zhu, D. Reynolds, P. G. Smith, P. Fekkes, S. Buonamici and N. A. Larsen, Genes Dev., 2018, 32, 309–320 CrossRef CAS PubMed.
- J. Borišek, A. Saltalamacchia, A. Spinello and A. Magistrato, J. Chem. Inf. Model., 2020, 60, 2510–2521 CrossRef PubMed.
- D. S. Hong, R. Kurzrock, A. Naing, J. J. Wheler, G. S. Falchook, J. S. Schiffman, N. Faulkner, M. J. Pilat, J. O'Brien and P. LoRusso, Invest. New Drugs, 2014, 32, 436–444 CrossRef CAS PubMed.
- A. Spinello, J. Borišek, L. Malcovati and A. Magistrato, Int. J. Mol. Sci., 2021, 22, 11222 CrossRef CAS PubMed.
- A. Spinello, P. Janos, R. Rozza and A. Magistrato, J. Phys. Chem. Lett., 2023, 14, 6263–6269 CrossRef CAS PubMed.
- D. P. Steensma, M. Wermke, V. M. Klimek, P. L. Greenberg, P. Font, R. S. Komrokji, J. Yang, A. M. Brunner, H. E. Carraway, L. Ades, A. Al-Kali, J. M. Alonso-Dominguez, A. Alfonso-Piérola, C. C. Coombs, H. J. Deeg, I. Flinn, J. M. Foran, G. Garcia-Manero, M. B. Maris, M. McMasters, J. B. Micol, J. P. De Oteyza, F. Thol, E. S. Wang, J. M. Watts, J. Taylor, R. Stone, V. Gourineni, A. J. Marino, H. Yao, B. Destenaves, X. Yuan, K. Yu, S. Dar, L. Ohanjanian, K. Kuida, J. Xiao, C. Scholz, A. Gualberto and U. Platzbecker, Leukemia, 2021, 35, 3542–3550 CrossRef CAS PubMed.
- C. L. Kielkopf, N. A. Rodionova, M. R. Green and S. K. Burley, Cell, 2001, 106, 595–605 CrossRef CAS PubMed.
- P. Selenko, G. Gregorovic, R. Sprangers, G. Stier, Z. Rhani, A. Krämer and M. Sattler, Mol. Cell, 2003, 11, 965–976 CrossRef CAS PubMed.
- C. L. Kielkopf, S. Lücke and M. R. Green, Genes Dev., 2004, 18, 1513–1526 CrossRef CAS PubMed.
- S. Loerch and C. L. Kielkopf, RNA, 2016, 22, 1795–1807 CrossRef CAS PubMed.
- A. Tefferi, C. M. Finke, T. L. Lasho, C. A. Hanson, R. P. Ketterling, N. Gangat and A. Pardanani, Leukemia, 2018, 32, 2274–2278 CrossRef CAS PubMed.
- K. Yoshida, M. Sanada, Y. Shiraishi, D. Nowak, Y. Nagata, R. Yamamoto, Y. Sato, A. Sato-Otsubo, A. Kon, M. Nagasaki, G. Chalkidis, Y. Suzuki, M. Shiosaka, R. Kawahata, T. Yamaguchi, M. Otsu, N. Obara, M. Sakata-Yanagimoto, K. Ishiyama, H. Mori, F. Nolte, W. K. Hofmann, S. Miyawaki, S. Sugano, C. Haferlach, H. P. Koeffler, L. Y. Shih, T. Haferlach, S. Chiba, H. Nakauchi, S. Miyano and S. Ogawa, Nature, 2011, 478, 64–69 CrossRef CAS PubMed.
- J. W. Galardi, V. N. Bela, N. Jeffery, X. He, E. Glasser, S. Loerch, J. L. Jenkins, M. J. Pulvino, P. L. Boutz and C. L. Kielkopf, J. Biol. Chem., 2022, 298, 102224 CrossRef CAS PubMed.
- A. Kobayashi, M. J. Clément, P. Craveur, K. El Hage, J. M. Salone, G. Bollot, D. Pastré and A. Maucuer, FEBS J., 2022, 289, 682–698 CrossRef CAS PubMed.
- X. Yuan, K. L. Howie, M. Kazemi Sabzvar, K. Chinnaswamy, J. A. Stuckey and C. Y. Yang, ACS Med. Chem. Lett., 2023, 14, 450–457 CrossRef CAS PubMed.
- P. K. A. Jagtap, T. Kubelka, K. Soni, C. L. Will, D. Garg, C. Sippel, T. G. Kapp, H. K. Potukuchi, K. Schorpp, K. Hadian, H. Kessler, R. Lührmann, F. Hausch, T. Bach and M. Sattler, Nat. Commun., 2020, 11, 5621 CrossRef CAS PubMed.
- P. K. Jagtap, D. Garg, T. G. Kapp, C. L. Will, O. Demmer, R. Lührmann, H. Kessler and M. Sattler, J. Med. Chem., 2016, 59, 10190–10197 CrossRef CAS PubMed.
- W. J. Friesen, S. Paushkin, A. Wyce, S. Massenet, G. S. Pesiridis, G. Van Duyne, J. Rappsilber, M. Mann and G. Dreyfuss, Mol. Cell. Biol., 2001, 21, 8289–8300 CrossRef CAS PubMed.
- G. Meister, C. Eggert, D. Bühler, H. Brahms, C. Kambach and U. Fischer, Curr. Biol., 2001, 11, 1990–1994 CrossRef CAS PubMed.
- S. E. Sanchez, E. Petrillo, E. J. Beckwith, X. Zhang, M. L. Rugnone, C. Esteban Hernando, J. C. Cuevas, M. A. Godoy Herz, A. Depetris-Chauvin, C. G. Simpson, J. W. Brown, P. D. Cerdán, J. O. Borevitz, P. Mas, M. F. Ceriani, A. R. Kornblihtt and M. J. Yanovsky, Nature, 2010, 468, 112–116 CrossRef CAS PubMed.
- E. Chan-Penebre, K. G. Kuplast, C. R. Majer, P. A. Boriack-Sjodin, T. J. Wigle, L. D. Johnston, N. Rioux, M. J. Munchhof, L. Jin, S. L. Jacques, K. A. West, T. Lingaraj, K. Stickland, S. A. Ribich, A. Raimondi, M. P. Scott, N. J. Waters, R. M. Pollock, J. J. Smith, O. Barbash, M. Pappalardi, T. F. Ho, K. Nurse, K. P. Oza, K. T. Gallagher, R. Kruger, M. P. Moyer, R. A. Copeland, R. Chesworth and K. W. Duncan, Nat. Chem. Biol., 2015, 11, 432–437 CrossRef CAS PubMed.
- K. W. Duncan, N. Rioux, P. A. Boriack-Sjodin, M. J. Munchhof, L. A. Reiter, C. R. Majer, L. Jin, L. D. Johnston, E. Chan-Penebre, K. G. Kuplast, M. Porter Scott, R. M. Pollock, N. J. Waters, J. J. Smith, M. P. Moyer, R. A. Copeland and R. Chesworth, ACS Med. Chem. Lett., 2016, 7, 162–166 CrossRef CAS PubMed.
- C. M. Koh, M. Bezzi, D. H. Low, W. X. Ang, S. X. Teo, F. P. Gay, M. Al-Haddawi, S. Y. Tan, M. Osato, A. Sabò, B. Amati, K. B. Wee and E. Guccione, Nature, 2015, 523, 96–100 CrossRef CAS PubMed.
- L. Zhang, N. T. Tran, H. Su, R. Wang, Y. Lu, H. Tang, S. Aoyagi, A. Guo, A. Khodadadi-Jamayran, D. Zhou, K. Qian, T. Hricik, J. Côté, X. Han, W. Zhou, S. Laha, O. Abdel-Wahab, R. L. Levine, G. Raffel, Y. Liu, D. Chen, H. Li, T. Townes, H. Wang, H. Deng, Y. G. Zheng, C. Leslie, M. Luo and X. Zhao, eLife, 2015, 4, e07938 CrossRef PubMed.
- J. Y. Fong, L. Pignata, P. A. Goy, K. C. Kawabata, S. C. Lee, C. M. Koh, D. Musiani, E. Massignani, A. G. Kotini, A. Penson, C. M. Wun, Y. Shen, M. Schwarz, D. H. Low, A. Rialdi, M. Ki, H. Wollmann, S. Mzoughi, F. Gay, C. Thompson, T. Hart, O. Barbash, G. M. Luciani, M. M. Szewczyk, B. J. Wouters, R. Delwel, E. P. Papapetrou, D. Barsyte-Lovejoy, C. H. Arrowsmith, M. D. Minden, J. Jin, A. Melnick, T. Bonaldi, O. Abdel-Wahab and E. Guccione, Cancer Cell, 2019, 36, 194–209.e9 CrossRef CAS PubMed.
- M. S. Eram, Y. Shen, M. Szewczyk, H. Wu, G. Senisterra, F. Li, K. V. Butler, H. Ü. Kaniskan, B. A. Speed, C. Dela Seña, A. Dong, H. Zeng, M. Schapira, P. J. Brown, C. H. Arrowsmith, D. Barsyte-Lovejoy, J. Liu, M. Vedadi and J. Jin, ACS Chem. Biol., 2016, 11, 772–781 CrossRef CAS PubMed.
- A. Fedoriw, S. R. Rajapurkar, S. O'Brien, S. V. Gerhart, L. H. Mitchell, N. D. Adams, N. Rioux, T. Lingaraj, S. A. Ribich, M. B. Pappalardi, N. Shah, J. Laraio, Y. Liu, M. Butticello, C. L. Carpenter, C. Creasy, S. Korenchuk, M. T. McCabe, C. F. McHugh, R. Nagarajan, C. Wagner, F. Zappacosta, R. Annan, N. O. Concha, R. A. Thomas, T. K. Hart, J. J. Smith, R. A. Copeland, M. P. Moyer, J. Campbell, K. Stickland, J. Mills, S. Jacques-O'Hagan, C. Allain, D. Johnston, A. Raimondi, M. P. Scott, N. Waters, K. Swinger, A. Boriack-Sjodin, T. Riera, G. Shapiro, R. Chesworth, R. K. Prinjha, R. G. Kruger, O. Barbash and H. P. Mohammad, Cancer Cell, 2019, 36, 100–114.e25 CrossRef CAS PubMed.
- H. Kim and Z. A. Ronai, Cell Stress, 2020, 4, 199–215 CrossRef CAS PubMed.
- M. Seiler, S. Peng, A. A. Agrawal, J. Palacino, T. Teng, P. Zhu, P. G. Smith, S. Buonamici and L. Yu, Cell Rep., 2018, 23, 282–296.e4 CrossRef CAS PubMed.
- R. Cheng, L. Xiao, W. Zhou, X. Jin, Z. Xu, C. Xu, P. Wang, M. Luo, M. Wang, K. Ma, H. Cao, Y. Huang, X. Lin, F. Pang, Y. Li and Q. Jiang, Oncogene, 2021, 40, 5441–5450 CrossRef CAS PubMed.
- A. J. Murphy, A. H. Li, P. Li and H. Sun, Front. Oncol., 2022, 12, 868664 CrossRef CAS PubMed.
- T. Giannakouros, E. Nikolakaki, I. Mylonis and E. Georgatsou, FEBS J., 2011, 278, 570–586 CrossRef CAS PubMed.
- M. V. Gammons, O. Fedorov, D. Ivison, C. Du, T. Clark, C. Hopkins, M. Hagiwara, A. D. Dick, R. Cox, S. J. Harper, J. C. Hancox, S. Knapp and D. O. Bates, Invest. Ophthalmol. Visual Sci., 2013, 54, 6052–6062 CrossRef CAS PubMed.
- Q. Li, C. Zeng, H. Liu, K. W. Y. Yung, C. Chen, Q. Xie, Y. Zhang, S. W. C. Wan, B. S. W. Mak, J. Xia, S. Xiong and J. C. K. Ngo, iScience, 2021, 24, 102423 CrossRef CAS PubMed.
- M. V. Gammons, R. Lucas, R. Dean, S. E. Coupland, S. Oltean and D. O. Bates, Br. J. Cancer, 2014, 111, 477–485 CrossRef CAS PubMed.
- R. P. Siqueira, A. Barbosa Éde, M. D. Polêto, G. L. Righetto, T. V. Seraphim, R. L. Salgado, J. G. Ferreira, M. V. Barros, L. L. de Oliveira, A. B. Laranjeira, M. R. Almeida, A. S. Júnior, J. L. Fietto, J. Kobarg, E. B. de Oliveira, R. R. Teixeira, J. C. Borges, J. A. Yunes and G. C. Bressan, PLoS One, 2015, 10, e0134882 CrossRef PubMed.
- M. Song, L. Pang, M. Zhang, Y. Qu, K. V. Laster and Z. Dong, Signal Transduction Targeted Ther., 2023, 8, 148 CrossRef PubMed.
- H. Alqarni, A. Jamleh and M. S. Chamber, Cureus, 2022, 14, e30566 Search PubMed.
- I. Younis, M. Berg, D. Kaida, K. Dittmar, C. Wang and G. Dreyfuss, Mol. Cell. Biol., 2010, 30, 1718–1728 CrossRef CAS PubMed.
- A. A. Waheed and E. O. Freed, AIDS Res. Hum. Retroviruses, 2012, 28, 54–75 CrossRef CAS PubMed.
- A. Nishida, N. Kataoka, Y. Takeshima, M. Yagi, H. Awano, M. Ota, K. Itoh, M. Hagiwara and M. Matsuo, Nat. Commun., 2011, 2, 308 CrossRef PubMed.
- Y. Sako, K. Ninomiya, Y. Okuno, M. Toyomoto, A. Nishida, Y. Koike, K. Ohe, I. Kii, S. Yoshida, N. Hashimoto, T. Hosoya, M. Matsuo and M. Hagiwara, Sci. Rep., 2017, 7, 46126 CrossRef CAS PubMed.
- J. Soret, N. Bakkour, S. Maire, S. Durand, L. Zekri, M. Gabut, W. Fic, G. Divita, C. Rivalle, D. Dauzonne, C. H. Nguyen, P. Jeanteur and J. Tazi, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 8764–8769 CrossRef CAS PubMed.
- C. G. Burd and G. Dreyfuss, EMBO J., 1994, 13, 1197–1204 CrossRef CAS PubMed.
- A. J. Vargas and R. Burd, Nutr. Rev., 2010, 68, 418–428 CrossRef PubMed.
- C. C. Ko, Y. J. Chen, C. T. Chen, Y. C. Liu, F. C. Cheng, K. C. Hsu and L. P. Chow, J. Biol. Chem., 2014, 289, 22078–22089 CrossRef CAS PubMed.
- S. Nakielny, M. C. Siomi, H. Siomi, W. M. Michael, V. Pollard and G. Dreyfuss, Exp. Cell Res., 1996, 229, 261–266 CrossRef CAS PubMed.
- E. Solano-Gonzalez, K. M. Coburn, W. Yu, G. M. Wilson, E. Nurmemmedov, S. Kesari, E. T. Chang, A. D. MacKerell, D. J. Weber and F. Carrier, Nucleic Acids Res., 2021, 49, 1235–1246 CrossRef CAS PubMed.
- D. Li, W. Yu and M. Lai, Acta Pharm. Sin. B, 2023, 13, 3181–3207 CrossRef CAS PubMed.
- L. Wan, W. Yu, E. Shen, W. Sun, Y. Liu, J. Kong, Y. Wu, F. Han, L. Zhang, T. Yu, Y. Zhou, S. Xie, E. Xu, H. Zhang and M. Lai, Gut, 2019, 68, 118–129 CrossRef CAS PubMed.
- H. K. Yum, H. R. Kim, Y. S. Chang, K. C. Shin, S. Kim and Y. M. O. Tuberc, Respir. Dis., 2017, 80, 52–59 Search PubMed.
- M. S. Gadd, A. Testa, X. Lucas, K. H. Chan, W. Chen, D. J. Lamont, M. Zengerle and A. Ciulli, Nat. Chem. Biol., 2017, 13, 514–521 CrossRef CAS PubMed.
- G. Weng, X. Cai, D. Cao, H. Du, C. Shen, Y. Deng, Q. He, B. Yang, D. Li and T. Hou, Nucleic Acids Res., 2023, 51, D1367–D1372 CrossRef PubMed.
- R. A. Gama-Brambila, J. Chen, J. Zhou, G. Tascher, C. Münch and X. Cheng, Cell Chem. Biol., 2021, 28, 1616–1627.e8 CrossRef CAS PubMed.
- A. Ghidini, A. Cléry, F. Halloy, F. H. T. Allain and J. Hall, Angew. Chem., Int. Ed., 2021, 60, 3163–3169 CrossRef CAS PubMed.
- G. Petzold, E. S. Fischer and N. H. Thomä, Nature, 2016, 532, 127–130 CrossRef CAS PubMed.
- T. C. Ting, M. Goralski, K. Klein, B. Wang, J. Kim, Y. Xie and D. Nijhawan, Cell Rep., 2019, 29, 1499–1510.e6 CrossRef CAS PubMed.
- T. Han, M. Goralski, N. Gaskill, E. Capota, J. Kim, T. C. Ting, Y. Xie, N. S. Williams and D. Nijhawan, Science, 2017, 356(6336), eaal3755 CrossRef PubMed.
- S. Campagne, D. Jutzi, F. Malard, M. Matoga, K. Romane, M. Feldmuller, M. Colombo, M. D. Ruepp and F. H. Allain, Nat. Commun., 2023, 14, 5366 CrossRef CAS PubMed.
- T. Uehara, Y. Minoshima, K. Sagane, N. H. Sugi, K. O. Mitsuhashi, N. Yamamoto, H. Kamiyama, K. Takahashi, Y. Kotake, M. Uesugi, A. Yokoi, A. Inoue, T. Yoshida, M. Mabuchi, A. Tanaka and T. Owa, Nat. Chem. Biol., 2017, 13, 675–680 CrossRef CAS PubMed.
- X. Du, O. A. Volkov, R. M. Czerwinski, H. Tan, C. Huerta, E. R. Morton, J. P. Rizzi, P. M. Wehn, R. Xu, D. Nijhawan and E. M. Wallace, Structure, 2019, 27, 1625–1633.e3 CrossRef CAS PubMed.
- D. E. Bussiere, L. Xie, H. Srinivas, W. Shu, A. Burke, C. Be, J. Zhao, A. Godbole, D. King, R. G. Karki, V. Hornak, F. Xu, J. Cobb, N. Carte, A. O. Frank, A. Frommlet, P. Graff, M. Knapp, A. Fazal, B. Okram, S. Jiang, P. Y. Michellys, R. Beckwith, H. Voshol, C. Wiesmann, J. M. Solomon and J. Paulk, Nat. Chem. Biol., 2020, 16, 15–23 CrossRef CAS PubMed.
- C. Xu, X. Chen, X. Zhang, D. Zhao, Z. Dou, X. Xie, H. Li, H. Yang, Q. Li, H. Zhang and C. Di, Cell Death Discov., 2021, 7, 214 CrossRef CAS PubMed.
- Y. Tong, Y. Lee, X. Liu, J. L. Childs-Disney, B. M. Suresh, R. I. Benhamou, C. Yang, W. Li, M. G. Costales, H. S. Haniff, S. Sievers, D. Abegg, T. Wegner, T. O. Paulisch, E. Lekah, M. Grefe, G. Crynen, M. Van Meter, T. Wang, Q. M. R. Gibaut, J. L. Cleveland, A. Adibekian, F. Glorius, H. Waldmann and M. D. Disney, Nature, 2023, 618, 169–179 CrossRef CAS PubMed.
- X. Roca, M. Akerman, H. Gaus, A. Berdeja, C. F. Bennett and A. R. Krainer, Genes Dev., 2012, 26, 1098–1109 CrossRef CAS PubMed.
- E. Buratti, M. Chivers, G. Hwang and I. Vorechovsky, Nucleic Acids Res., 2011, 39, D86–D91 CrossRef CAS PubMed.
- M. R. Lunn and C. H. Wang, Lancet, 2008, 371, 2120–2133 CrossRef PubMed.
- S. Lefebvre, L. Bürglen, S. Reboullet, O. Clermont, P. Burlet, L. Viollet, B. Benichou, C. Cruaud, P. Millasseau and M. Zeviani, Cell, 1995, 80, 155–165 CrossRef CAS PubMed.
- M. D. Mailman, J. W. Heinz, A. C. Papp, P. J. Snyder, M. S. Sedra, B. Wirth, A. H. Burghes and T. W. Prior, Genet. Med., 2002, 4, 20–26 CrossRef CAS PubMed.
- S. Cho and G. Dreyfuss, Genes Dev., 2010, 24, 438–442 CrossRef CAS PubMed.
- N. A. Naryshkin, M. Weetall, A. Dakka, J. Narasimhan, X. Zhao, Z. Feng, K. K. Ling, G. M. Karp, H. Qi, M. G. Woll, G. Chen, N. Zhang, V. Gabbeta, P. Vazirani, A. Bhattacharyya, B. Furia, N. Risher, J. Sheedy, R. Kong, J. Ma, A. Turpoff, C. S. Lee, X. Zhang, Y. C. Moon, P. Trifillis, E. M. Welch, J. M. Colacino, J. Babiak, N. G. Almstead, S. W. Peltz, L. A. Eng, K. S. Chen, J. L. Mull, M. S. Lynes, L. L. Rubin, P. Fontoura, L. Santarelli, D. Haehnke, K. D. McCarthy, R. Schmucki, M. Ebeling, M. Sivaramakrishnan, C. P. Ko, S. V. Paushkin, H. Ratni, I. Gerlach, A. Ghosh and F. Metzger, Science, 2014, 345, 688–693 CrossRef CAS PubMed.
- J. Palacino, S. E. Swalley, C. Song, A. K. Cheung, L. Shu, X. Zhang, M. Van Hoosear, Y. Shin, D. N. Chin, C. G. Keller, M. Beibel, N. A. Renaud, T. M. Smith, M. Salcius, X. Shi, M. Hild, R. Servais, M. Jain, L. Deng, C. Bullock, M. McLellan, S. Schuierer, L. Murphy, M. J. Blommers, C. Blaustein, F. Berenshteyn, A. Lacoste, J. R. Thomas, G. Roma, G. A. Michaud, B. S. Tseng, J. A. Porter, V. E. Myer, J. A. Tallarico, L. G. Hamann, D. Curtis, M. C. Fishman, W. F. Dietrich, N. A. Dales and R. Sivasankaran, Nat. Chem. Biol., 2015, 11, 511–517 CrossRef CAS PubMed.
- M. G. Woll, H. Qi, A. Turpoff, N. Zhang, X. Zhang, G. Chen, C. Li, S. Huang, T. Yang, Y. C. Moon, C. S. Lee, S. Choi, N. G. Almstead, N. A. Naryshkin, A. Dakka, J. Narasimhan, V. Gabbeta, E. Welch, X. Zhao, N. Risher, J. Sheedy, M. Weetall and G. M. Karp, J. Med. Chem., 2016, 59, 6070–6085 CrossRef CAS PubMed.
- H. Ratni, M. Ebeling, J. Baird, S. Bendels, J. Bylund, K. S. Chen, N. Denk, Z. Feng, L. Green, M. Guerard, P. Jablonski, B. Jacobsen, O. Khwaja, H. Kletzl, C. P. Ko, S. Kustermann, A. Marquet, F. Metzger, B. Mueller, N. A. Naryshkin, S. V. Paushkin, E. Pinard, A. Poirier, M. Reutlinger, M. Weetall, A. Zeller, X. Zhao and L. Mueller, J. Med. Chem., 2018, 61, 6501–6517 CrossRef CAS PubMed.
- M. Sivaramakrishnan, K. D. McCarthy, S. Campagne, S. Huber, S. Meier, A. Augustin, T. Heckel, H. Meistermann, M. N. Hug, P. Birrer, A. Moursy, S. Khawaja, R. Schmucki, N. Berntenis, N. Giroud, S. Golling, M. Tzouros, B. Banfai, G. Duran-Pacheco, J. Lamerz, Y. Hsiu Liu, T. Luebbers, H. Ratni, M. Ebeling, A. Cléry, S. Paushkin, A. R. Krainer, F. H. Allain and F. Metzger, Nat. Commun., 2017, 8, 1476 CrossRef PubMed.
- A. Bhattacharyya, C. R. Trotta, J. Narasimhan, K. J. Wiedinger, W. Li, K. A. Effenberger, M. G. Woll, M. B. Jani, N. Risher, S. Yeh, Y. Cheng, N. Sydorenko, Y. C. Moon, G. M. Karp, M. Weetall, A. Dakka, V. Gabbeta, N. A. Naryshkin, J. D. Graci, T. Tripodi, A. Southwell, M. Hayden, J. M. Colacino and S. W. Peltz, Nat. Commun., 2021, 12, 7299 CrossRef CAS PubMed.
- Y. Zhang, K. M. Wu, L. Yang, Q. Dong and J. T. Y. Mol, Neurodegeneration, 2022, 17, 28 CrossRef CAS PubMed.
- C. P. Donahue, C. Muratore, J. Y. Wu, K. S. Kosik and M. S. Wolfe, J. Biol. Chem., 2006, 281, 23302–23306 CrossRef CAS PubMed.
- C. P. Donahue, J. Ni, E. Rozners, M. A. Glicksman and M. S. Wolfe, J. Biomol. Screening, 2007, 12, 789–799 CrossRef CAS PubMed.
- S. Zheng, Y. Chen, C. P. Donahue, M. S. Wolfe and G. Varani, Chem. Biol., 2009, 16, 557–566 CrossRef CAS PubMed.
- Y. Luo and M. D. Disney, ChemBioChem, 2014, 15, 2041–2044 CrossRef CAS PubMed.
- M. D. Disney, A. M. Winkelsas, S. P. Velagapudi, M. Southern, M. Fallahi and J. L. Childs-Disney, ACS Chem. Biol., 2016, 11, 1720–1728 CrossRef CAS PubMed.
- H. G. Harley, S. A. Rundle, J. C. MacMillan, J. Myring, J. D. Brook, S. Crow, W. Reardon, I. Fenton, D. J. Shaw and P. S. Harper, Am. J. Hum. Genet., 1993, 52, 1164–1174 CAS.
- H. Jiang, A. Mankodi, M. S. Swanson, R. T. Moxley and C. A. Thornton, Hum. Mol. Genet., 2004, 13, 3079–3088 CrossRef CAS PubMed.
- Y. Yuan, S. A. Compton, K. Sobczak, M. G. Stenberg, C. A. Thornton, J. D. Griffith and M. S. Swanson, Nucleic Acids Res., 2007, 35, 5474–5486 CrossRef CAS PubMed.
- M. Nakamori, K. Sobczak, A. Puwanant, S. Welle, K. Eichinger, S. Pandya, J. Dekdebrun, C. R. Heatwole, M. P. McDermott, T. Chen, M. Cline, R. Tawil, R. J. Osborne, T. M. Wheeler, M. S. Swanson, R. T. Moxley and C. A. Thornton, Ann. Neurol., 2013, 74, 862–872 CrossRef CAS PubMed.
- S. G. Rzuczek, Y. Gao, Z. Z. Tang, C. A. Thornton, T. Kodadek and M. D. Disney, ACS Chem. Biol., 2013, 8, 2312–2321 CrossRef CAS PubMed.
- C. H. Wong, L. Nguyen, J. Peh, L. M. Luu, J. S. Sanchez, S. L. Richardson, T. Tuccinardi, H. Tsoi, W. Y. Chan, H. Y. Chan, A. M. Baranger, P. J. Hergenrother and S. C. Zimmerman, J. Am. Chem. Soc., 2014, 136, 6355–6361 CrossRef CAS PubMed.
- J. Lee, Y. Bai, U. V. Chembazhi, S. Peng, K. Yum, L. M. Luu, L. D. Hagler, J. F. Serrano, H. Y. E. Chan, A. Kalsotra and S. C. Zimmerman, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 8709–8714 CrossRef CAS PubMed.
- M. G. Costales, Y. Matsumoto, S. P. Velagapudi and M. D. Disney, J. Am. Chem. Soc., 2018, 140, 6741–6744 CrossRef CAS PubMed.
- M. G. Costales, H. Aikawa, Y. Li, J. L. Childs-Disney, D. Abegg, D. G. Hoch, S. Pradeep Velagapudi, Y. Nakai, T. Khan, K. W. Wang, I. Yildirim, A. Adibekian, E. T. Wang and M. D. Disney, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 2406–2411 CrossRef CAS PubMed.
- S. Mikutis, M. Rebelo, E. Yankova, M. Gu, C. Tang, A. R. Coelho, M. Yang, M. E. Hazemi, M. P. de Miranda, M. Eleftheriou, M. Robertson, G. S. Vassiliou, D. J. Adams, J. P. Simas, F. Corzana, J. S. Schneekloth, K. Tzelepis and G. J. L. Bernardes, ACS Cent. Sci., 2023, 9, 892–904 CrossRef CAS PubMed.
- S. Mikutis, M. Gu, E. Sendinc, M. E. Hazemi, H. Kiely-Collins, D. Aspris, G. S. Vassiliou, Y. Shi, K. Tzelepis and G. J. L. Bernardes, ACS Cent. Sci., 2020, 6, 2196–2208 CrossRef CAS PubMed.
- C. Martin, M. Bonnet, N. Patino, S. Azoulay, A. Di Giorgio and M. Duca, ChemPlusChem, 2022, 87, e202200250 CrossRef CAS PubMed.
- R. Rozza, P. Janoš, A. Spinello and A. Magistrato, Expert Opin. Drug Discovery, 2022, 17, 1095–1109 CrossRef CAS PubMed.
- C. Cretu, P. Gee, X. Liu, A. Agrawal, T. V. Nguyen, A. K. Ghosh, A. Cook, M. Jurica, N. A. Larsen and V. Pena, Nat. Commun., 2021, 12, 4491 CrossRef CAS PubMed.
- M. Hluchý, P. Gajdušková, I. Ruiz de Los Mozos, M. Rájecký, M. Kluge, B. T. Berger, Z. Slabá, D. Potěšil, E. Weiß, J. Ule, Z. Zdráhal, S. Knapp, K. Paruch, C. C. Friedel and D. Blazek, Nature, 2022, 609, 829–834 CrossRef PubMed.
- R. Chatrikhi, C. F. Feeney, M. J. Pulvino, G. Alachouzos, A. J. MacRae, Z. Falls, S. Rai, W. W. Brennessel, J. L. Jenkins, M. J. Walter, T. A. Graubert, R. Samudrala, M. S. Jurica, A. J. Frontier and C. L. Kielkopf, Cell Chem. Biol., 2021, 28, 1145–1157.e6 CrossRef CAS PubMed.
- R. Rozza, P. Janoš and A. Magistrato, J. Chem. Inf. Model., 2023, 63, 7508–7517 CrossRef CAS PubMed.
- A. Nijhuis, A. Sikka, O. Yogev, L. Herendi, C. Balcells, Y. Ma, E. Poon, C. Eckold, G. N. Valbuena, Y. Xu, Y. Liu, B. M. da Costa, M. Gruet, C. Wickremesinghe, A. Benito, H. Kramer, A. Montoya, D. Carling, E. J. Want, Y. Jamin, L. Chesler and H. C. Keun, Nat. Commun., 2022, 13, 1380 CrossRef CAS PubMed.
- T. B. Faust, H. Yoon, R. P. Nowak, K. A. Donovan, Z. Li, Q. Cai, N. A. Eleuteri, T. Zhang, N. S. Gray and E. S. Fischer, Nat. Chem. Biol., 2020, 16, 7–14 CrossRef CAS PubMed.
- K. Baek and B. A. Schulman, Nat. Chem. Biol., 2020, 16, 2–3 CrossRef CAS PubMed.
- F. Malard, A. Wolter, J. Marquevielle, E. Morvan, F. H. T. Allain and S. Campagne, bioRxiv, 2023, preprint, DOI:10.1101/2023.03.31.535100.
| This journal is © The Royal Society of Chemistry 2024 |