
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProton conductivity of Li+–H+ exchanged Li7La3Zr2O12 dense membranes prepared by molten long-chain saturated fatty acids†
Akihiro
Ishii
 *a,
Daisuke
Kume
a,
Shoki
Nakayasu
a,
Itaru
Oikawa
a,
Hiroshi
Matsumoto
b,
Hisashi
Kato
b and
Hitoshi
Takamura
a
*a,
Daisuke
Kume
a,
Shoki
Nakayasu
a,
Itaru
Oikawa
a,
Hiroshi
Matsumoto
b,
Hisashi
Kato
b and
Hitoshi
Takamura
a
aDepartment of Materials Science, Graduate School of Engineering, Tohoku University, Sendai, 980-8579, Japan. E-mail: akihiro.ishii.a4@tohoku.ac.jp
bResearch and Development Center, Tohoku Electric Power Co., Inc, Sendai, 980-8550, Japan
First published on 8th January 2024
Abstract
Alkali-proton-exchanged oxides have broad potential as proton-conducting solid-state electrolytes for fuel cells and electrolyzers. However, materials in this class have not been widely explored because of the difficulty in preparing dense-body samples and investigating their electrical conductivities. In this study, using a Li7La3Zr2O12 (LLZ) membrane as a model material, alkali-proton exchange throughout dense-body samples was achieved using molten long-chain saturated fatty acids as proton sources. The 91% of Li+ in the LLZ dense bodies was exchanged with H+ within 15 h using the molten C21H43COOH. This Li+–H+ exchange decreased the conductivity of LLZ by more than 100-fold compared to its pristine state, whereas previous reports have suggested that protons in Li+–H+-exchanged LLZ are mobile. Experimental and theoretical investigations have indicated that most protons in Li+–H+-exchanged LLZ are bound to adjacent oxide ions, and that the proton transfer step of the Grotthuss mechanism is challenging for isolated ZrO6 octahedra. Alkali-proton exchange using molten long-chain saturated fatty acids effectively explores new alkali-proton exchanged materials in which their exchange reactions are rate-determined by a thermally activated diffusion process.
Introduction
Some inorganic materials possess mobile protons in their crystal structure framework at elevated temperatures far above 100 °C and exhibit distinct proton conductivity.1–3 These materials have attracted considerable interest over the last few decades as electrolytes for the production of efficient solid-oxide fuel cells and electrolyzers.4–6 For the practical applications as electrolytes, the proton conducting materials that exhibit the following three characteristics are particularly in demand: (1) high proton conductivity at intermediate temperatures (300–600 °C), (2) high chemical stability against CO2, H2O, and electrode materials, and (3) requiring high temperatures for densification.7 There are two well-known classes of protonic inorganic materials: solid oxyacids that show high proton conductivity but decompose at relatively low temperatures (≈200 °C) as being dehydrated (e.g. CsHSO4 derivatives8–10), and perovskite-type oxide refractories that are stable even in the dehydrated state but show high conductivity only at high temperatures above 600 °C (e.g. BaZrO3–BaCeO3 derivatives11–14). Many efforts have been made to develop thermostable oxyacids (e.g. In-doped SnP2O7,15 NaMg1−xLixHx(PO3)3·yH2O16) or perovskite-type oxides showing high proton conductivity at the intermediate temperatures (e.g. Ba1.01Zr0.399Sc0.590O3-d,17 Ba5Er2Al2ZrO13,18 Ba7Nb4MoO20,19 BaSc0.67O(OH)220). However, the materials that satisfy the three key requirements for electrochemical applications have not yet been developed.Thermodynamically metastable alkali proton-exchanged oxides have gradually gained attention as a new class of proton-conducting materials. They are typically synthesized by preparing alkali-ion-conducting oxides and then immersing them in water or diluted aqueous solutions of sulfuric, nitric, hydrochloric, and carboxylic acids. Alkali-proton-exchanged oxides have the potential to exhibit high proton conductivity at intermediate temperatures, considering that metastable protons may gain high mobility. Indeed, alkali-proton exchanged phosphate-based glass and Li13.9Sr0.1Zn(GeO4)4 show high proton conductivity on the magnitude of 10−3 S cm−1 or higher at the intermediate temperatures.21–23 Unlike perovskite-type oxides, alkali-proton-exchanged oxides suffer less from electronic current leakage under high oxygen potentials because the proton is incorporated by ion exchange, not by the hydration of oxygen vacancies introduced by acceptors.24,25 The alkali-proton exchange technique provides an outstanding possibility for producing a wide variety of proton-conducting oxides with a skeleton of excellent alkali-ion conductors, some of which may show high proton conductivities at intermediate temperatures with reasonable chemical stability.
However, the proton conductivity of alkali-proton-exchanged oxides has not been widely investigated, most likely because of the difficulties in preparing their dense bodies for conductivity measurements. Using water or an acidic aqueous solution for the alkali-proton exchange limits processing temperatures below 100 °C; thus, the thermal acceleration of mass diffusion is limited, and only porous or powder-form oxides are usually applied.26 Sintering of alkali-proton-exchanged oxide powders is also challenging owing to their thermodynamic metastability. Their compaction into gas-leakage-free dense bodies is also unlikely because most oxides exhibit a high elastic modulus.27 Thus, electric conductivity has been reported for only two alkali-proton exchanged oxides: β/β′′-alumina,28–30 and Li5La3M2O12 (M = Nb, Ta, Sn).26,31,32
Some recent reports have achieved the preparation of alkali-proton-exchanged dense oxides using electrochemical techniques. Ishiyama et al. have synthesized the proton-conducting phosphate-based glass by applying DC bias on phosphate-based sodium glass in H2 gas with a Pd electrode and sodium absorption electrode at around 300 °C.21,33,34 A similar electrochemical technique using Ni electrodes has been applied to Li13.9Sr0.1Zn(GeO4)4 and Li2.5Sr0.75Zr1.25(PO4)3 although no direct data on the lithium-proton exchange rate and depth have been reported.22,35 While recognizing that these electrochemical alkali-proton exchange techniques are effective in developing various alkali-proton-exchanged oxides, from an application perspective, electrode deposition and removal processes are problematic. In addition, the electrochemical alkali-proton exchange technique is incompatible with exploring mixed proton–electron conducting electrode materials because of electron current leakage, although they are as crucial as electrodes in electrochemical cells.36–39
In this context, the authors recently proposed a new alkali-proton exchange method that utilizes molten long-chain saturated fatty acids instead of aqueous acids as a proton source and can be applied to a wide variety of dense alkali oxides in principle.40 The molten long-chain saturated fatty acids remain stable in the liquid phase at elevated temperatures well above 100 °C owing to their large molecular weights and strong intermolecular van der Waals and hydrogen bonding. Thus, alkali-proton exchange through dense alkali oxide bodies can be achieved with the aid of thermal acceleration. Their carboxyl branches provide a moderate level of acidity, enabling them to donate protons to alkali oxides without disrupting their crystalline structural frameworks. Saturated hydrocarbon chains are chemically inert, allowing a stable alkali-proton exchange reaction at elevated temperatures for an extended period. Owing to the high hydrophobicity of long hydrocarbon chains, this technique is also beneficial from the viewpoint of avoiding the introduction of bulky hydrated protons (i.e., hydronium) into the oxides,29,41 which leads to the mechanical failure of dense oxide bodies. Thus, the immersion of alkali oxides into molten long-chain saturated fatty acids is a facile technique to prepare various alkali-proton-exchanged oxide dense bodies.
To prove this concept, in this study, a model material of cubic-garnet-type Li7La3Zr2O12 (LLZ) dense bodies was immersed in molten long-chain saturated fatty acids for Li+–H+ exchange. A Li-based oxide was chosen here because exchanging the smaller alkali ion with a proton is suggested to be beneficial for retaining the crystal structure frameworks.42 LLZ is a state-of-the-art Li+-conducting oxide developed by Murugan et al.43 Although LLZ was pronounced to be stable against water in early studies,31,44 Li+–H+ exchange readily takes place on its surface by water exposure, along with minor modification of the crystal structure framework from space group Ia![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) d to I
d to I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d.45–50 LLZ has two types of lithium sites, tetrahedral 24d and interstitialcy octahedral 48g/96h, and protons preferentially substitute the interstitial sites.51,52 Up to about 70% Li+–H+ exchange has been achieved for LLZ powders so far.50,52,53 For highly Li+–H+ exchanged LLZ, Yow et al. and Liu et al. have independently proposed that the proton in LLZ is likely immobile until around 200 °C but turns mobile at higher temperatures.52,54 Orera et al. revealed that protonated LLZ is stable in air at up to 300 °C at least.53 However, the proton conductivity of Li+–H+-exchanged LLZ has not been revealed experimentally, most likely because of the difficulty in preparing dense bodies. In this study, highly Li+–H+-exchanged cubic LLZ dense bodies were prepared using molten long-chain saturated fatty acids, and their electrical conductivities were experimentally determined.
3d.45–50 LLZ has two types of lithium sites, tetrahedral 24d and interstitialcy octahedral 48g/96h, and protons preferentially substitute the interstitial sites.51,52 Up to about 70% Li+–H+ exchange has been achieved for LLZ powders so far.50,52,53 For highly Li+–H+ exchanged LLZ, Yow et al. and Liu et al. have independently proposed that the proton in LLZ is likely immobile until around 200 °C but turns mobile at higher temperatures.52,54 Orera et al. revealed that protonated LLZ is stable in air at up to 300 °C at least.53 However, the proton conductivity of Li+–H+-exchanged LLZ has not been revealed experimentally, most likely because of the difficulty in preparing dense bodies. In this study, highly Li+–H+-exchanged cubic LLZ dense bodies were prepared using molten long-chain saturated fatty acids, and their electrical conductivities were experimentally determined.
Experimental
Synthetic procedures
Cubic garnet-type LLZ dense bodies, nominally Li6.16Al0.28La3Zr2O12, were synthesized by tape-cast with slurries composed of calcined LLZ powders. Doped Al stabilizes the cubic form of LLZ by substitution of the 24d site Li and the introduction of lithium vacancies at the 48g/96h sites, as shown in eqn (1):55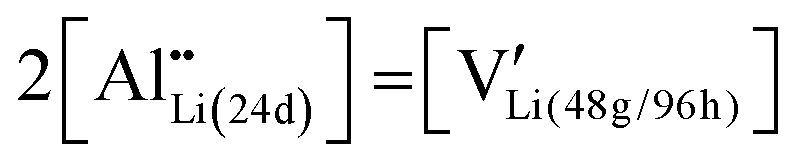 | (1) |
The preparation procedure for the calcined Al-doped LLZ powder is reported in detail in our previous report.27 Raw materials (Li2CO3, Al2O3, La2O3, ZrO2) were ball milled in hexane, then compacted into pellets, and then fired at 950 °C for 12 h in magnesia crucibles. Excess Li (10 mol%) was added to the stoichiometric composition considering sublimation during firing. Hexane inhibited the reactions between water (including air moisture) and the raw materials.5,48,56 The use of alumina crucibles must be prohibited to avoid the contamination of Li-containing specimens with aluminum.57 The calcined pellets were polished on both sides to remove potential impurity phases on their surfaces and then milled again in the same manner. Slurries for tape-casting were prepared as previously described.58 The calcined Al-doped LLZ powder was mixed with a binder (polyvinyl butyral 630), solvent (dehydrated isopropanol and toluene), plasticizer (dibutyl phthalate), dispersant (ethylene glycol), and antifoam (polyoxyethylene (10)-octylphenyl ether) via mixing and deaeration (AR-100, THINKY). The resultant slurries were cast using a doctor blade with a 500 μm gap, dried, and punched out with an 8 mm die. The resultant thin round-shaped Al-doped LLZ green sheets were then heated at 2 °C min−1 to 500 °C and kept for 3 h, and then they were heated at 10 °C min−1 to 1200 °C and sintered for 3 h in the magnesia crucibles.
The immersion of LLZ dense bodies in molten long-chain fatty acids was performed using a homemade quartz vessel, as shown in Fig. 1. The vessel was introduced into a mantle heater stirrer to control the temperature and create a flow of molten fatty acids, which is believed to be beneficial for removing the byproducts of the fatty acid Li salts from the sample surface. Silver rods and mesh suspended the LLZ dense bodies in the middle of the vessel to ensure homogeneous reactions on both sides of the samples. A thermocouple was placed near each sample to achieve precise temperature control. A quartz lid with through-holes for the rods and a thermocouple were placed on the vessel during the reaction to suppress evaporation of the fatty acids. Fatty acids corresponding to a proton content of at least 100 times greater than the lithium content of the LLZ dense bodies were placed in the container. The entire system was placed in a homemade transparent chamber filled with N2 to avoid the oxidative degradation of fatty acids. After the reaction, the samples were rinsed with warm (below 100 °C) cooking oil to remove fatty acid Li salts that potentially remained on the sample surface. Cooking oil is composed of unsaturated hydrophobic carboxylic acids, which blend well with carboxylates and enhance their fluidity. The LLZ dense bodies were then rinsed with acetone using a sonicator.
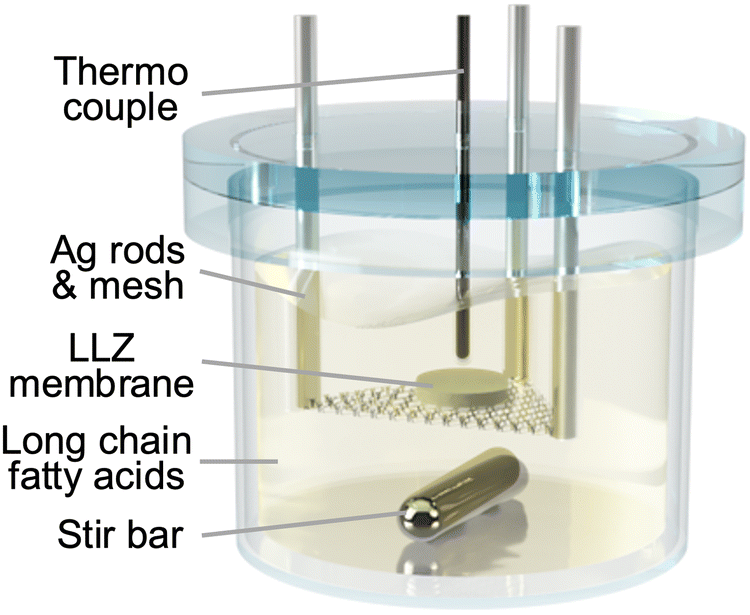 | ||
| Fig. 1 Schematic illustration of the reaction vessel. | ||
Characterization
Crystal and local structures of the samples were determined by X-ray diffraction (XRD, D8 Advance, Bruker AXS) with a Cu-Ka radiation source (λ = 1.5418 Å) and micro-Raman spectroscopy (HR-800, Horiba Jobin Yvon) using a He–Ne laser (λ = 632.8 nm). The lattice constants were refined by the whole powder pattern decomposition with the Pawley method using the TOPAS4 software (Bruker AXS). Cross-sectional Raman analysis of the fracture surfaces of the LLZ dense bodies was also conducted to investigate the Li+–H+ exchange depth. Inductively coupled plasma mass spectrometry (ICP-MS, Agilent 8800, Agilent Technologies) was used for compositional analysis. Sample solution was prepared by heating decomposition at 105 °C for 2h using aqua regia. The composition of the as-sintered LLZ dense bodies was analyzed to be Li: Al: La: Zr equal to 4.29![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.19:3.23:2.00 by mole, suggesting that the concentrations of lithium and aluminum were lower than the nominal composition. The ionic conductivities were measured as functions of temperature using two-probe AC impedance spectroscopy (1260 A, Solartron) in synthetic air. The impedance spectra were recorded in the frequency range of 10 MHz to 0.01 Hz with an AC voltage of 100 mV. Pd electrodes were sputtered onto both sides of the LLZ dense bodies without heating.
:0.19:3.23:2.00 by mole, suggesting that the concentrations of lithium and aluminum were lower than the nominal composition. The ionic conductivities were measured as functions of temperature using two-probe AC impedance spectroscopy (1260 A, Solartron) in synthetic air. The impedance spectra were recorded in the frequency range of 10 MHz to 0.01 Hz with an AC voltage of 100 mV. Pd electrodes were sputtered onto both sides of the LLZ dense bodies without heating.
Theoretical calculation
Theoretical calculations based on density functional theory (DFT) with generalized gradient approximation (GGA) were performed using the Vienna Ab initio Simulation Package (VASP) code.59 An 1 × 1 × 1 cubic LLZ structure with full and partial Li occupation at the 24d and 96h sites (Li(96h)32Li(24d)24La24Zr16O96) was used as the initial structure, following previous studies.60,61 Considering that the simultaneous occupation of neighboring Li atoms at the 96h site is prohibited due to electrostatic repulsion, the 96h sites become equivalent to 48g sites. Al doping was performed by substituting one Li(24d) by Al.62 Considering that the charge compensation (eqn (1)) is accomplished by the closest distance,63,64 Al was introduced as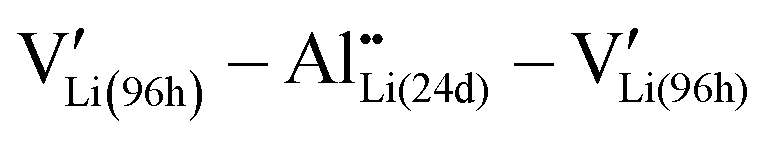 clusters. The cutoff energy was set at 475 eV, and a gamma-point-only calculation was conducted. One hundred crystal structure patterns with random distributions of Li(96h) and protons at Li(96h) sites were calculated, and the most stable structure was considered. VESTA is used for visualization.65
clusters. The cutoff energy was set at 475 eV, and a gamma-point-only calculation was conducted. One hundred crystal structure patterns with random distributions of Li(96h) and protons at Li(96h) sites were calculated, and the most stable structure was considered. VESTA is used for visualization.65
Results and discussion
Li+–H+ exchange of LLZ dense bodies
This study reports the preparation of Li+–H+-exchanged Al-doped LLZ dense bodies via immersion in C21H43COOH (behenic acid). By our preliminary study, C21H43COOH was found to be suitable for steady processing of the ion exchange because of its moderate acidity (dissociation constants around 5 at standard temperature) and high boiling point (>300 °C).40 The expected Li+–H+ exchange reaction is given by eqn (2):| Li6.16Al0.28La3Zr2O12 + yC21H43COOH → HyLi6.16−yAl0.28La3Zr2O12 + yC21H43COOLi | (2) |
LLZ dense bodies were prepared by tape casting to obtain thin samples and achieve uniform Li+–H+ exchange at a high rate. Fig. 2a shows the cross-section of the pristine LLZ dense bodies. It was 100 μm thick and comprised only cubic garnet-type LLZ crystals at the XRD level, as shown in Fig. 2c. The samples were sintered well: their relative density was calculated to be 92 ± 5% by their dimensions and weight. An image analysis supported that the relative density of the sample was 92% (see Fig. S1 in ESI†). Their microstructures and phases were retained after immersion in molten C21H43COOH at 250 °C for 15 h, as shown in Fig. 2b and c. Upon immersion, the XRD peaks shifted to lower angles without any notable shoulder peaks. This indicates that the LLZ lattice expanded by Li+–H+ exchange, as previously reported,45,48,52–54,66 and Li+–H+ exchange took place considerably and uniformly. ICP-MS analysis showed that the composition of the immersed Al-doped LLZ membranes is Li: Al: La: Zr equal to 0.38:0.19:3.14:2.00 by mole, showing that the Li+–H+ exchange rate reached 91%. The lattice constant of the C21H43COOH-immersed LLZ was calculated to be 13.09 Å. This large value also supports a high Li+–H+ exchange rate, as shown in Fig. 3a. Note that the grain boundaries of the surface of the LLZ membranes became clearly visible after the immersion (see Fig. S2 in ESI†), indicating that this process also works as acid etching.
 | ||
| Fig. 2 Cross-sectional SEM images of (a) as-sintered and (b) C21H43COOH-immearsed LLZ dense bodies, and (c) their XRD patterns. | ||
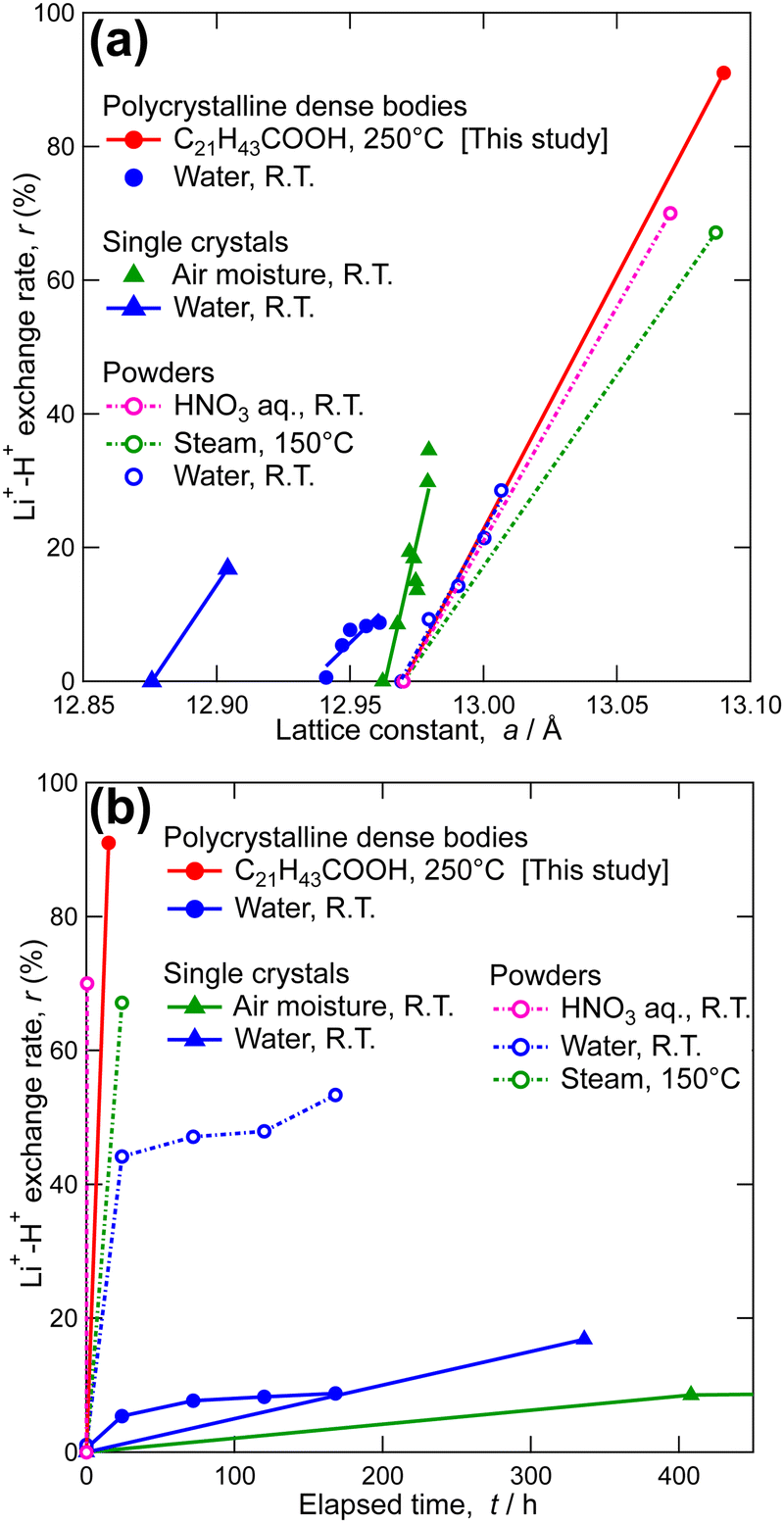 | ||
| Fig. 3 Li+–H+ exchange rate as functions of (a) lattice constant and (b) elapsed time for cubic LLZ in comparison with previously reported data.44,47,51–53,64 | ||
Here, the characteristics of Li+–H+ exchange using molten long-chain saturated fatty acids are presented and compared with those when other proton sources are used. Fig. 3b shows the Li+–H+ exchange rate as a function of reaction time, as reported in this and previous studies.45,52–54,66 Even tepid water achieved approximately 50% Li+–H+ exchange for LLZ powders, while achieving less than 20% exchange for LLZ dense bodies within a reasonable timeframe. Considering that the Li+–H+ exchange reaction is driven by the concentration (activity) gradient and is rate-determined by grain interior diffusion,50,54 enhancement of the reaction kinetics for LLZ dense bodies can be expected using proton sources with higher acidity (e.g., HNO3 aq.) and increasing the reaction temperature (e.g., steam instead of tepid water). Using molten long-chain saturated fatty acids as the proton source achieved both, allowing for a high Li+–H+ exchange rate in a short time, even for the LLZ-dense bodies (91%, 15 h).
The protons of the Li+–H+-exchanged LLZ dense bodies were directly investigated using cross-sectional Raman analysis. Fig. 4a shows the Raman spectra of the fracture cross section of the Al-doped LLZ membranes immersed in C21H43COOH, measured at varying distances from the surface, as schematically depicted in Fig. 4b. The immersed Al-doped LLZ membranes showed peaks at 2850 cm−1, 2900 cm−1, and 3526 cm−1 with no sample location dependence. These frequencies are typical of the stretching vibrations of the OH bonds.67 Meanwhile, the pristine LLZ dense bodies showed no Raman peaks in this range (Fig. S3 in ESI†). This suggests that Li+–H+ exchange occurred uniformly throughout the LLZ dense bodies and that the incorporated protons were located in three different local environments. Given that a lower OH vibration frequency corresponds to stronger OH⋯O hydrogen bonding,67 proton is more mobile when it exhibits the OH vibration peak at a lower frequency.21 Indeed, the vibration frequencies of OH bonding are typically around 2300–3300 cm−1 for oxyacids,21,67 meanwhile, those for perovskite-type oxides are at 3200–3700 cm−1.20,68 For the Li+–H+-exchanged LLZ, the Raman peaks at 2850 cm−1 and 2900 cm−1 are smaller than those at 3526 cm−1, suggesting that the number of mobile protons is negligible at low temperatures.
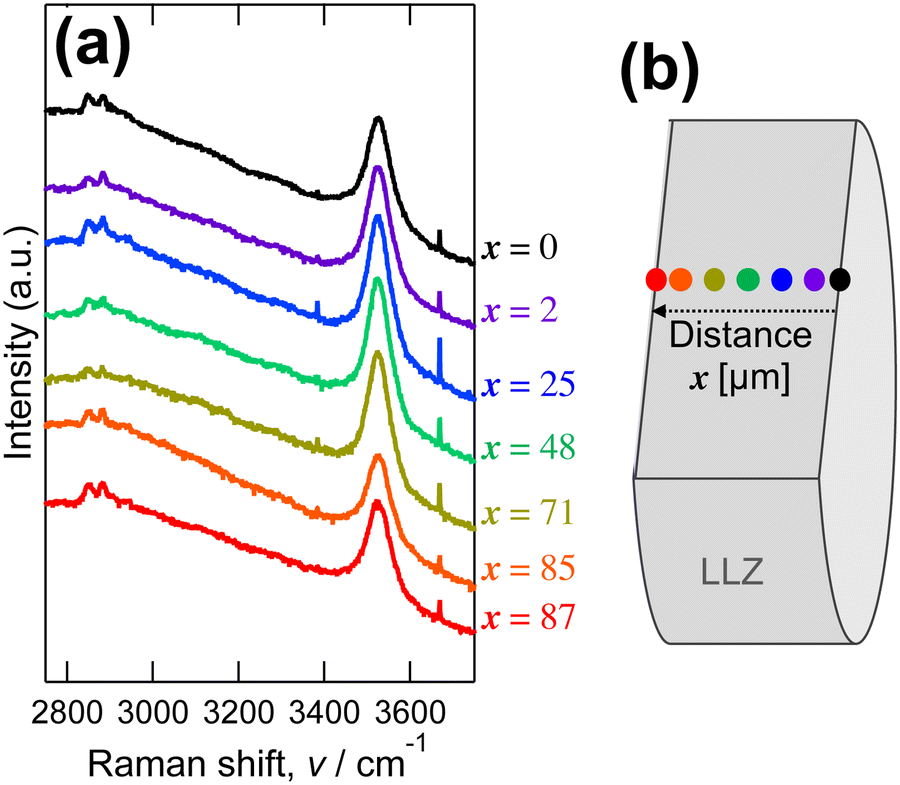 | ||
| Fig. 4 (a) Raman spectra of the fracture surface of Al-doped LLZ dense bodies immersed in C21H43COOH, and (b) schematic illustration of Raman observation location. Spikes at 3384 cm−1 and 3670 cm−1 are attributed to surface adsorbed water. | ||
Electrical conductivity of Li+–H+ exchanged LLZ dense bodies
As demonstrated above, molten long-chain saturated fatty acids were successfully used as proton sources to achieve uniform and high-rate Li+–H+ exchange of the LLZ dense bodies. This enables the experimental determination of the proton conductivity in a cubic garnet-type crystal framework using AC impedance spectroscopy. Distorted semicircles were observed in the high-frequency region of the Nyquist plot (Fig. S4 in ESI†). Considering that the semicircles are attributed to the grain interior and boundary resistance, the total electrical conductivity was calculated by equivalent circuit fitting using a parallel connection of a resistor and a constant phase element, as shown in Fig. 5. In the heating process up to 325 °C, the conductivity increased linearly as increasing temperature in the Arrhenius-type plot, indicating the high uniformity of the Li+–H+ exchange reaction throughout the Al-doped LLZ membrane. Above 325 °C, the conductivity decreased with increasing temperature. This is owing to the decomposition of garnet-type LLZ phase, as previously reported by Orera et al.53 Indeed, XRD after the annealing above 325 °C detected a formation of low crystalline phases (Fig. S5 in ESI†), suggesting that the Li+–H+-exchanged LLZ was decomposed by the dehydration reaction shown in eqn (3.1) or (3.2) at high temperatures.| 2H6.16Al0.28La3Zr2O12 |
| →2La2Zr2O7 + La2O3 + 0.28Al2O3 + 6.16H2O | (3.1) |
| →3La2O3 + 4ZrO2 + 0.28Al2O3 + 6.16H2O | (3.2) |
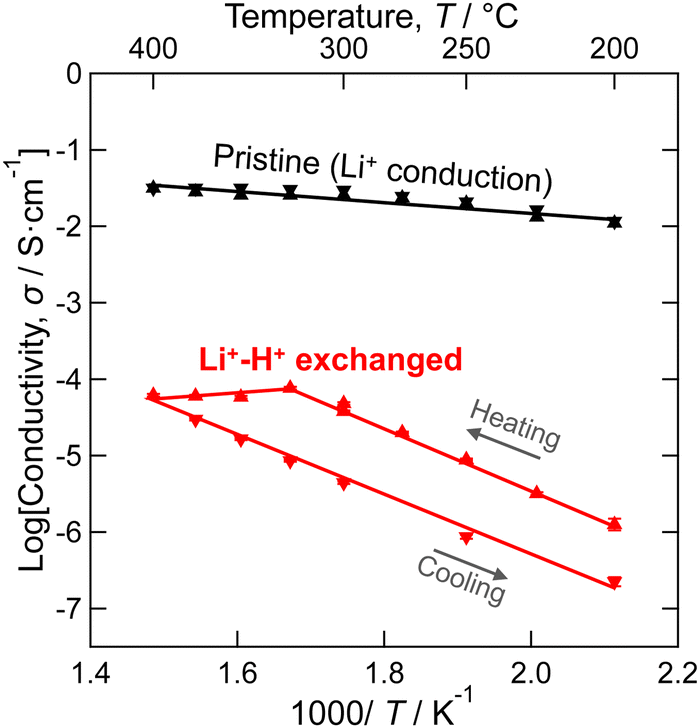 | ||
| Fig. 5 Arrhenius-type plot of total electrical conductivity of the 91% Li+–H+ exchanged LLZ dense bodies measured in heating and subsequent cooling process. Conductivity of the pristine LLZ is also shown for comparison. | ||
Eqn (3.1) and (3.2) suggest the formation of La2O3 and Al2O3, whereas XRD did not detect them. This is likely because their crystallization is limited at these temperatures. Thermogravimetric analysis also supported that the Li+–H+-exchanged LLZ decomposed at this temperature range (see Chapter S2 in ESI†).
As shown in Fig. 5, Li+–H+ exchange decreased the conductivity of LLZ by more than 100-fold. The activation energy for the conduction was also increased from 0.15 eV to 0.80 eV. Meanwhile, protons in highly Li+–H+-exchanged LLZ were previously estimated to be mobile at elevated temperatures.52,54 To clarify the reason for the low proton conductivity, DFT-GGA calculations of Al-doped cubic LLZ were performed. When all Li+ ions were substituted with H+ and the structure relaxed, protons moved from the original Li sites to the vicinity of oxygen (Fig. S6 in ESI†), which is consistent with previous reports.48,52 The distance of OH bond distance in the relaxed structure is shown in Fig. 6a. Most protons are located 0.98 ± 0.005 Å from oxygen. This distance is similar to that in perovskite-type oxides.69 Meanwhile, a few protons are located 0.99 Å or more away from oxygen, showing that their hydrogen bonding is relatively strong. This proton distribution is consistent with the experimental observations made using Raman spectroscopy (Fig. 4). Currently, no relationship can be found between the OH bonding distance and the substituted Li sites or the distance from the Al acceptors.
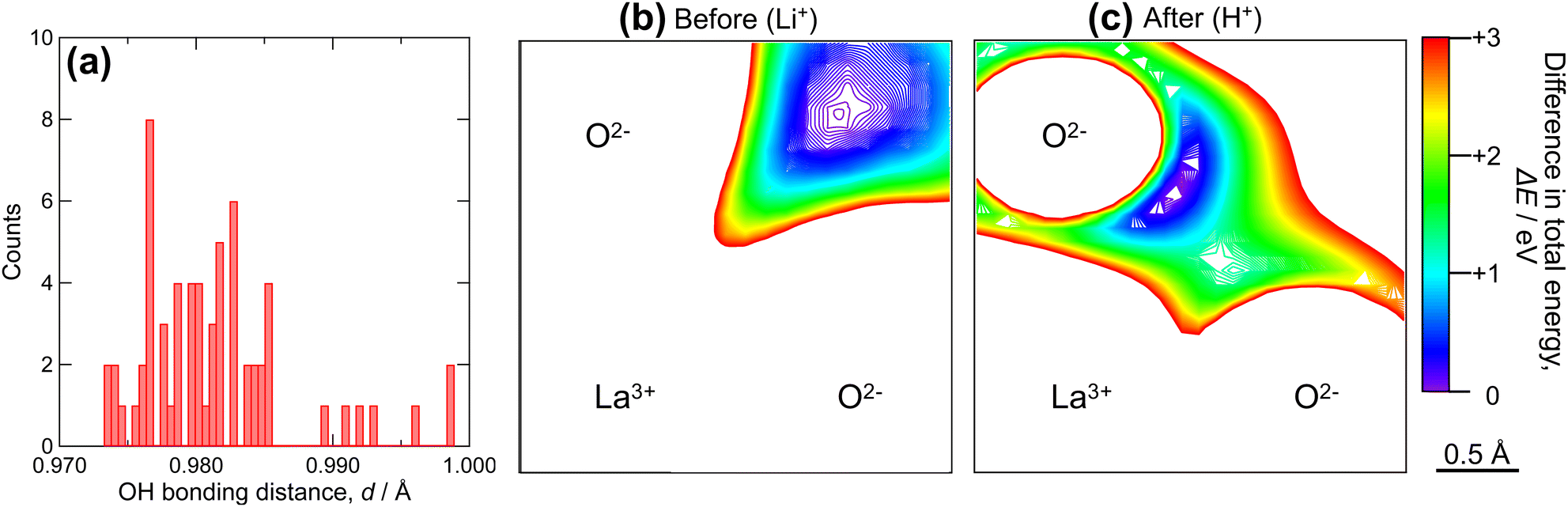 | ||
| Fig. 6 (a) Histogram of OH bonding distance in Li+–H+ exchanged LLZ, and potential energy surface of (b) Li+ and (c) H+ before and after Li+–H+ exchange. | ||
Most protons in Li+–H+-exchanged LLZ are bound to adjacent oxide ions, similar to the perovskite-type oxides; therefore, they are expected to diffuse through a Grotthuss-type two-step mechanism. This mechanism involves proton rotation around oxide ions and proton transfer to the neighboring oxide ions.70 To gain a deeper understanding of the nature of protons in Li+–H+-exchanged LLZ, potential energy surfaces were calculated, as presented in Fig. 6b and c. In these figures, the bluish or violet colors represent the lower potential energy regions. Before the Li+–H+ exchange (Fig. 6b), the low potential energy region for Li+ is relatively broad and spherical, with a diameter of approximately 1 Å. In this situation, Li+ diffusion is expected to occur simply by hopping between the Li sites. Meanwhile, after Li+–H+ exchange (Fig. 6c), the low potential energy region narrowed and took on an arc-like shape centered on the oxide ion. This clearly shows that protons are electrostatically attracted to oxide ions, which is facilitated by their small size; thus, their diffusion occurs via the Grotthuss mechanism. Given that the ZrO6 octahedra are isolated in the garnet-type crystal structure, the proton transfer step of the Grotthuss mechanism is challenging for LLZ, unlike the perovskite-type structure in which they are connected. The difference in ion conduction mechanism before and after Li+–H+ exchange can also be clearly illustrated by bond-valence sum energy (BVSE) calculations. Fig. 7 shows (a) Li+ conduction and (b) H+ conduction pathways of Al-doped LLZ, calculated using the DFT-given crystal structures and BVSE software published by Adams et al.71–73 In Fig. 7a, Li+ is preferentially located at the 24d sites and migrates through the 96h sites, aligning well with the well-known Li+ conduction mechanism of LLZ. Meanwhile, the calculated H+ conduction pathways (Fig. 7b) show spherical motions of H+ around the vertex oxygen of the ZrO6 octahedra. Each spherical motion is connected by conduction paths with a constricted shape, as pointed by a red arrow in Fig. 7b. This picture suggests that the proton transfer step of the Grotthuss mechanism is challenging for LLZ. In this situation, the minor structural modification from Iad to I3d by Li+–H+ exchange most likely has a limited impact on the proton conduction process because the [La3Zr2O12]7− network is preserved through this structural alteration. While previous studies have suggested high proton mobility in Li+–H+-exchanged LLZ,52,54 this study revealed that the actual mobility was low (Fig. 5). This discrepancy is probably because proton rotation is allowed but its transfer is challenging in the LLZ.
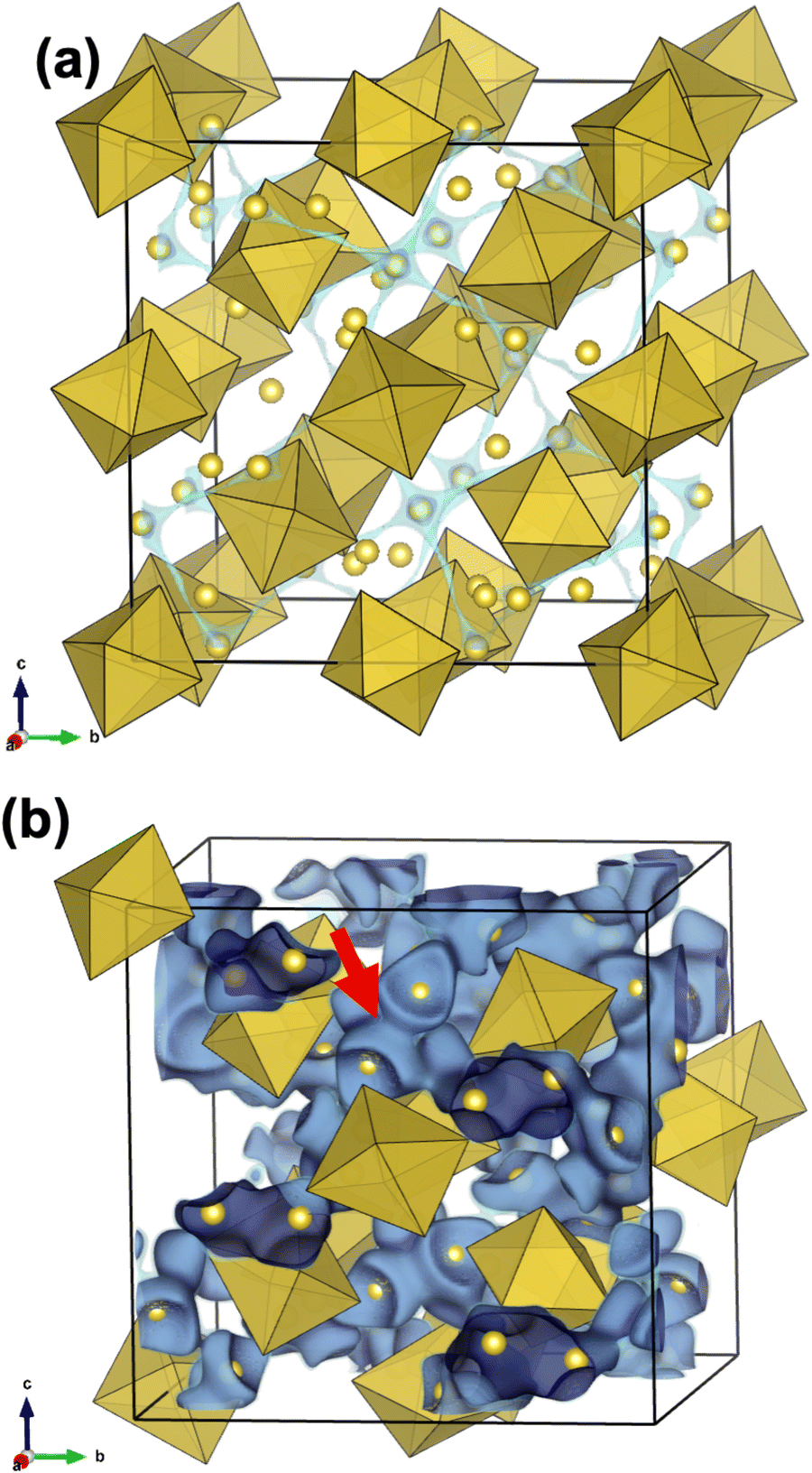 | ||
| Fig. 7 (a) Li+ and (b) H+ conduction pathways of Al-doped LLZ calculated using the DFT-given crystal structures and the BVSE software. Yellow balls and polyhedrons are Li/H and ZrO6, and La and Al are omitted for visibility. | ||
This study demonstrated high-rate Li+–H+ exchange in dense ceramic bodies using molten long-chain saturated fatty acids as a proton source, with LLZ as the model material. However, the crystal structure of LLZ provides low proton conductivity, as mentioned above. Therefore, the authors recently explored Li+–H+ exchange in phosphate-based lithium conductors, which have a structural framework that inherently favors stronger hydrogen bonding than LLZ because of the high covalency of P–O bonding. However, even for NASICON-type doped LiZr2(PO4)3, which shows a high Li+ conductivity comparable to LLZs,74,75 its high-rate Li+–H+ exchange has not been achieved even with molten long-chain saturated fatty acids (see Chapter S3 in ESI†). This indicates that, for the phosphates, not the thermally activated diffusion but the activity gradient determines how the Li+–H+ exchange proceeds. Thus, the alkali-proton exchange method using fatty acids is considered more effective for strongly ionic materials than for materials with covalent characteristics.
Conclusions
Because the exploration of alkali-proton-exchanged materials for proton-conducting solid-state electrolytes has been hindered by the difficulty in preparing dense-body samples, this study developed a new alkali-proton exchange technique using molten long-chain saturated fatty acids as proton sources, which can be applied to dense-body samples. Using molten C21H43COOH and Al-doped Li7La3Zr2O12 dense membranes as the model materials, 91% Li+–H+ exchange was achieved in 15 h. Raman analysis and DFT-GGA calculations suggested that most protons in the Li+–H+-exchanged LLZ were bound to adjacent oxide ions, and the proton transfer step of the Grotthuss mechanism is challenging between isolated ZrO6 octahedra. Consequently, the Li+–H+ exchange decreased the conductivity of LLZ by more than 100-fold compared to its pristine state. Alkali-proton exchange using molten long-chain saturated fatty acids will facilitate the exploration of new alkali-proton-exchanged materials whose exchange reactions are rate-determined by a thermally activated diffusion process.Author contributions
A. I.: conceptualization, writing – original draft, methodology, investigation, validation, data curation, resources, project administration, and funding acquisition. D. K.: methodology, investigation, and data curation. S. N.: validation and investigation. I. O. validation and investigation. H. K.: validation, resources, and project administration. H. M.: validation, resources, and project administration. H. T.: writing – review and editing, supervision, validation, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Murata Science Foundation (Grant number: M21-006). The authors thank Ms Yoko Nakano of the Technical Division of the Graduate School of Engineering, Tohoku University, for technical assistance with the ICP-MS analysis. We thank Mr Youtaro Sakamoto for supplying LLZ for preliminary experiments.References
- S. Mu, H. Huang, A. Ishii, Z. Zhao, M. Zou, P. Kuzbary, F. Peng, K. S. Brinkman, H. Xiao and J. Tong, J. Power Sources Adv., 2020, 4, 100017 CrossRef.
- S. K. Kim, S. H. Hwang, J.-T. Nam and J.-S. Park, Ceram. Int., 2021, 47, 21083–21089 CrossRef CAS.
- Y. Ling, T. Guo, Y. Guo, Y. Yang, Y. Tian, X. Wang, X. Ou and P. Feng, J. Adv. Ceram., 2021, 10, 1052–1060 CrossRef CAS.
- C. K. Vigen and R. Haugsrud, J. Am. Ceram. Soc., 2013, 96, 3812–3820 CrossRef CAS.
- A. Ishii, H. Huang, Y. Meng, S. Mu, J. Gao, J. Lei, F. Peng, H. Xiao, J. Tong and K. S. Brinkman, Mater. Res. Bull., 2021, 143, 111446 CrossRef CAS.
- Z. Zhao, J. Gao, Y. Meng, K. S. Brinkman and J. Tong, Ceram. Int., 2021, 47, 11313–11319 CrossRef CAS.
- C. Duan, J. Huang, N. Sullivan and R. O’Hayre, Appl. Phys. Rev., 2020, 7, 011314 CAS.
- V. G. Ponomareva, K. A. Kovalenko, R. D. Gus’kov, I. N. Bagryantseva, N. F. Uvarov and V. P. Fedin, Solid State Ionics, 2019, 343, 115084 CrossRef CAS.
- C. Dreßler and D. Sebastiani, Phys. Chem. Chem. Phys., 2020, 22, 10738–10752 RSC.
- R. R. Choudhury, R. Chitra, I. P. Makarova, E. V. Selezneva and V. A. Komornikov, Bull. Mater. Sci., 2021, 44, 108 CrossRef CAS.
- S. Mu, Z. Zhao, H. Huang, J. Lei, F. Peng, H. Xiao, K. S. Brinkman and J. (Joshua) Tong, Electrochem. Soc. Interface, 2020, 29, 67 CrossRef CAS.
- I. Oikawa and H. Takamura, Chem. Mater., 2015, 27, 6660–6667 CrossRef CAS.
- H. Takahashi, I. Oikawa and H. Takamura, J. Phys. Chem. C, 2018, 122, 6501–6507 CrossRef CAS.
- J. Lyagaeva, G. Vdovin, L. Hakimova, D. Medvedev, A. Demin and P. Tsiakaras, Electrochim. Acta, 2017, 251, 554–561 CrossRef CAS.
- M. Nagao, T. Kamiya, P. Heo, A. Tomita, T. Hibino and M. Sano, J. Electrochem. Soc., 2006, 153, A1604 CrossRef CAS.
- N. Ueda, J. Nakajima, D. Mori, S. Taminato, N. Imanishi, S. Higashimoto and Y. Matsuda, Mater. Adv., 2021, 2, 6603–6612 RSC.
- J. Hyodo, K. Kitabayashi, K. Hoshino, Y. Okuyama and Y. Yamazaki, Adv. Energy Mater., 2020, 10, 2000213 CrossRef CAS.
- T. Murakami, J. R. Hester and M. Yashima, J. Am. Chem. Soc., 2020, 142, 11653–11657 CrossRef CAS PubMed.
- S. Fop, K. S. McCombie, E. J. Wildman, J. M. S. Skakle, J. T. S. Irvine, P. A. Connor, C. Savaniu, C. Ritter and A. C. Mclaughlin, Nat. Mater., 2020, 19, 752–757 CrossRef CAS PubMed.
- H. Kawamori, I. Oikawa and H. Takamura, Chem. Mater., 2021, 33, 5935–5942 CrossRef CAS.
- T. Ishiyama, T. Yamaguchi and T. Omata, J. Ceram. Soc. Jpn., 2022, 130, 1–9 CrossRef CAS.
- T. Wei, L. A. Zhang, Y. Chen, P. Yang and M. Liu, Chem. Mater., 2017, 29, 1490–1495 CrossRef CAS.
- T. Matsui, T. Ozeki, K. Miyazaki, S. Nagasaka, H. Muroyama, K. Imagawa, Y. Okada and K. Eguchi, J. Mater. Chem. A, 2023, 11, 18207–18212 RSC.
- Y. Matsuzaki, Y. Tachikawa, Y. Baba, K. Sato, H. Iinuma, G. Kojo, H. Matsuo, J. Otomo, H. Matsumoto, S. Taniguchi and K. Sasaki, ECS Trans., 2019, 91, 1009 CrossRef CAS.
- Y. Matsuzaki, Y. Tachikawa, Y. Baba, K. Sato, G. Kojo, H. Matsuo, J. Otomo, H. Matsumoto, S. Taniguchi and K. Sasaki, J. Electrochem. Soc., 2020, 167, 084515 CrossRef CAS.
- C. Galven, J.-L. Fourquet, M.-P. Crosnier-Lopez and F. Le Berre, Chem. Mater., 2011, 23, 1892–1900 CrossRef CAS.
- Y. Sakamoto, A. Ishii, T. Shiratori, I. Oikawa and H. Takamura, Electrochim. Acta, 2023, 457, 142488 CrossRef CAS.
- G. C. Farrington and J. L. Briant, Mater. Res. Bull., 1978, 13, 763–773 CrossRef CAS.
- K. G. Frase, G. C. Farrington and J. O. Thomas, Annu. Rev. Mater. Sci., 1984, 14, 279–295 CrossRef CAS.
- K. G. Frase, J. O. Thomas, A. R. McGhie and G. C. Farrington, J. Solid State Chem., 1986, 62, 297–305 CrossRef CAS.
- L. Truong and V. Thangadurai, Chem. Mater., 2011, 23, 3970–3977 CrossRef CAS.
- L. Truong and V. Thangadurai, Inorg. Chem., 2012, 51, 1222–1224 CrossRef CAS PubMed.
- T. Yamaguchi, S. Tsukuda, T. Ishiyama, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, J. Mater. Chem. A, 2018, 6, 23628–23637 RSC.
- T. Omata, T. Yamaguchi, S. Tsukuda, T. Ishiyama, J. Nishii, T. Yamashita and H. Kawazoe, Phys. Chem. Chem. Phys., 2019, 21, 10744–10749 RSC.
- Z. Lian, F. Chen, X. Song, D. Liao and K. Peng, J. Alloys Compd., 2021, 882, 160696 CrossRef CAS.
- T. Omata, Final Research Report, KAKEN 15K14126, 2017.
- H. Uehara, A. Ishii, I. Oikawa and H. Takamura, Int. J. Hydrogen Energy, 2022, 47, 5577–5584 CrossRef CAS.
- M. Liang, F. He, C. Zhou, Y. Chen, R. Ran, G. Yang, W. Zhou and Z. Shao, Chem. Eng. J., 2021, 420, 127717 CrossRef CAS.
- Y. Cho, M. Ogawa, I. Oikawa, H. L. Tuller and H. Takamura, Chem. Mater., 2019, 31, 2713–2722 CrossRef CAS.
- A. Ishii, H. Takamura and D. Kume, PCT/JP2022/043010, 2022.
- H. Takamura, T. Kagotani, M. Okada and M. Homma, Mater. Trans., 1993, 34, 197–201 CrossRef CAS.
- S. Tsukuda, K. Miyake, T. Yamaguchi, M. Kita, T. Ishiyama, J. Nishii, T. Yamashita, H. Kawazoe and T. Omata, Inorg. Chem., 2017, 56, 13949–13954 CrossRef CAS PubMed.
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- Y. Shimonishi, A. Toda, T. Zhang, A. Hirano, N. Imanishi, O. Yamamoto and Y. Takeda, Solid State Ionics, 2011, 183, 48–53 CrossRef CAS.
- C. Hiebl, D. Young, R. Wagner, H. M. R. Wilkening, G. J. Redhammer and D. Rettenwander, J. Phys. Chem. C, 2019, 123, 1094–1098 CrossRef CAS.
- M. Rosen, R. Ye, M. Mann, S. Lobe, M. Finsterbusch, O. Guillon and D. Fattakhova-Rohlfing, J. Mater. Chem. A, 2021, 9, 4831–4840 RSC.
- H. Nemori, Y. Matsuda, S. Mitsuoka, M. Matsui, O. Yamamoto, Y. Takeda and N. Imanishi, Solid State Ionics, 2015, 282, 7–12 CrossRef CAS.
- R. Kun, F. Langer, M. Delle Piane, S. Ohno, W. G. Zeier, M. Gockeln, L. Colombi Ciacchi, M. Busse and I. Fekete, ACS Appl. Mater. Interfaces, 2018, 10, 37188–37197 CrossRef CAS PubMed.
- R. H. Brugge, A. K. O. Hekselman, A. Cavallaro, F. M. Pesci, R. J. Chater, J. A. Kilner and A. Aguadero, Chem. Mater., 2018, 30, 3704–3713 CrossRef CAS.
- S. Smetaczek, A. Limbeck, V. Zeller, J. Ring, S. Ganschow, D. Rettenwander and J. Fleig, Mater. Adv., 2022, 3, 8760–8770 RSC.
- C. Ma, E. Rangasamy, C. Liang, J. Sakamoto, K. L. More and M. Chi, Angew. Chem., 2015, 127, 131–135 CrossRef.
- X. Liu, Y. Chen, Z. D. Hood, C. Ma, S. Yu, A. Sharafi, H. Wang, K. An, J. Sakamoto, D. J. Siegel, Y. Cheng, N. H. Jalarvo and M. Chi, Energy Environ. Sci., 2019, 12, 945–951 RSC.
- A. Orera, G. Larraz, J. A. Rodríguez-Velamazán, J. Campo and M. L. Sanjuán, Inorg. Chem., 2016, 55, 1324–1332 CrossRef CAS PubMed.
- Z. F. Yow, Y. L. Oh, W. Gu, R. P. Rao and S. Adams, Solid State Ionics, 2016, 292, 122–129 CrossRef CAS.
- D. O. Shin, K. Oh, K. M. Kim, K.-Y. Park, B. Lee, Y.-G. Lee and K. Kang, Sci. Rep., 2015, 5, 18053 CrossRef CAS PubMed.
- A. Ishii, S. Mu, Y. Meng, H. Huang, J. Lei, Y. Li, F. Peng, H. Xiao, J. Tong and K. S. Brinkman, Energy Technol., 2020, 8, 2000364 CrossRef CAS.
- A. Ishii, H. Ishijima, K. Kobayashi, I. Oikawa and H. Takamura, Acta Mater., 2022, 224, 117529 CrossRef CAS.
- Y. Hayamizu, M. Kato and H. Takamura, J. Membr. Sci., 2014, 462, 147–152 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- M. Xu, M. S. Park, J. M. Lee, T. Y. Kim, Y. S. Park and E. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 052301 CrossRef.
- D. Rettenwander, P. Blaha, R. Laskowski, K. Schwarz, P. Bottke, M. Wilkening, C. A. Geiger and G. Amthauer, Chem. Mater., 2014, 26, 2617–2623 CrossRef CAS PubMed.
- C. Li, A. Ishii, L. Roy, D. Hitchcock, Y. Meng and K. Brinkman, J. Mater. Sci., 2020, 55, 16470–16481 CrossRef CAS.
- H. Takahashi, I. Yashima, K. Amezawa, K. Eguchi, H. Matsumoto, H. Takamura and S. Yamaguchi, Chem. Mater., 2017, 29, 1518–1526 CrossRef CAS.
- A. Ishii, I. Oikawa and H. Takamura, J. Appl. Phys., 2019, 126, 215108 CrossRef.
- K. Momma and F. Izumi, J. Appl. Cryst., 2011, 44, 1272–1276 CrossRef CAS.
- G. J. Redhammer, P. Badami, M. Meven, S. Ganschow, S. Berendts, G. Tippelt and D. Rettenwander, ACS Appl. Mater. Interfaces, 2021, 13, 350–359 CrossRef CAS PubMed.
- K. Nakamoto, M. Margoshes and R. E. Rundle, J. Am. Chem. Soc., 1955, 77, 6480–6486 CrossRef CAS.
- J. Liu, Y. Chiba, J. Kawamura and H. Yugami, Solid State Ionics, 2006, 177, 2329–2332 CrossRef CAS.
- M. E. Björketun, P. G. Sundell and G. Wahnström, Faraday Discuss., 2006, 134, 247–265 RSC.
- W. Münch, K.-D. Kreuer, G. Seifert and J. Maier, Solid State Ionics, 2000, 136–137, 183–189 CrossRef.
- H. Chen and S. Adams, IUCrJ, 2017, 4, 614–625 CrossRef CAS PubMed.
- H. Chen, L. L. Wong and S. Adams, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 18–33 CrossRef CAS PubMed.
- L. L. Wong, K. C. Phuah, R. Dai, H. Chen, W. S. Chew and S. Adams, Chem. Mater., 2021, 33, 625–641 CrossRef CAS.
- H. Xie, J. B. Goodenough and Y. Li, J. Power Sources, 2011, 196, 7760–7762 CrossRef CAS.
- M. Harada, H. Takeda, S. Suzuki, K. Nakano, N. Tanibata, M. Nakayama, M. Karasuyama and I. Takeuchi, J. Mater. Chem. A, 2020, 8, 15103–15109 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00901g |
| This journal is © The Royal Society of Chemistry 2024 |