
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in removal of pharmaceutical pollutants in wastewater using metal oxides and carbonaceous materials as photocatalysts: a review†
Suneel Kumar
Srivastava
 *
*
Department of Chemistry, Indian Institute of Technology, Kharagpur-721302, India. E-mail: suneel@chem.iitkgp.ac.in; suneelchemkgp@gmail.com
First published on 31st January 2024
Abstract
The pharmaceuticals industry has played an important role in developing medicines for improving health and quality of life in treating humans and animals around the world. But it is also considered to be one of the sources of pollutants entering deliberately or accidentally into global water bodies causing toxicity that eventually threatens human health, aquatic organisms and environments even at low concentrations. These contaminants are non-biodegradable and cannot be completely removed from various water matrices following conventional treatment methods. In this regard, photodegradation techniques involving modified/unmodified semiconducting materials have attracted a lot of attention as a promising solution in achieving complete antibiotic degradation with the generation of non-toxic by-products. In view of this, the present review article summarizes current research progress in the removal of several emerging contaminants, such as acetaminophen, amoxicillin, sulfamethoxazole, norfloxacin, ibuprofen, ciprofloxacin, tetracycline, diclofenac and atenolol in water. Considerable emphasis has been placed on metal oxides and carbon-based photocatalysts following their modification through doping with metals and non-metals, metal loading, the formation of composites, immobilization and heterostructure/heterojunction approaches. Finally, the review ends with future prospects for nanomaterial-based heterogeneous photocatalysts in the removal of pharmaceutical contaminants from water.
 Suneel Kumar Srivastava | Suneel Kumar Srivastava received his Ph.D degree from the Indian Institute of Technology, Kharagpur in 1984. He is a former Professor in the Department of Chemistry of the same Institute, serving from 1986 to 2021. Dr Srivastava carried out his post-doctoral work as a DAAD Fellow in the Technical University, Karlsruhe (1988–89, 2002, 2006), University of Siegen (1994, 1999), Technical University, Munchen (2009), Leibniz Institute of Polymer Research, Dresden (2013) Germany, and University of Nantes, France (2003, 2007). His research interests are in the field of nondimensional nanomaterials for their application in the fields of energy, environments and polymer nanocomposites. Dr Srivastava has guided 23 Ph.Ds, published about 200 research papers in referred journals, contributed to 16 chapters in books and edited 2 books. |
1 Introduction
Water plays an essential role in sustaining a cherished healthy life for living organisms as well as ecosystems. Therefore, the purity of water remains of utmost concern for the survival of human beings, plants, animals and several other living species in the world. A report presented by UNESCO at the UN 2023 Water Conference revealed the non-availability of safe drinking water for 26% of the global population.1 This problem is also compounded by the presence of several pollutants in water bodies. This contributes to the depletion of fresh water, resulting in an overall water crisis worldwide.2 This adversely affects human health, several other living organisms and sustainable social development. According to an estimate, about 80% of wastewater is discharged globally into the environment without any prior treatment, jeopardizing human health, the ecosystem, and the environment.3 In this regard, dye effluents, heavy metals and pesticides discharged as wastewater from different industries contribute significantly to water pollution.4–12In addition, the wide application of pharmaceuticals in daily life for the treatment of complex diseases is also the major contributor of emerging contaminants, with potential adverse effects on humans and the aquatic environment.13–22 The presence of these pharmaceutical pollutants could lead to cancers, severe bleeding, organ damage, birth defects, reproductive disorders, endocrine disorders, and mild to severe toxic effects in human beings in the global population.14 The toxic effects are also threats to mammals, other organisms, and the ecosystem. Fig. 1 shows the effect of pharmaceuticals in reducing the quality of water.14 The presence of these pharmaceutical pollutants in water through improper disposal, irrigation of crops, and consumption by agriculture, humans, and animals seriously affects the ecosystem.
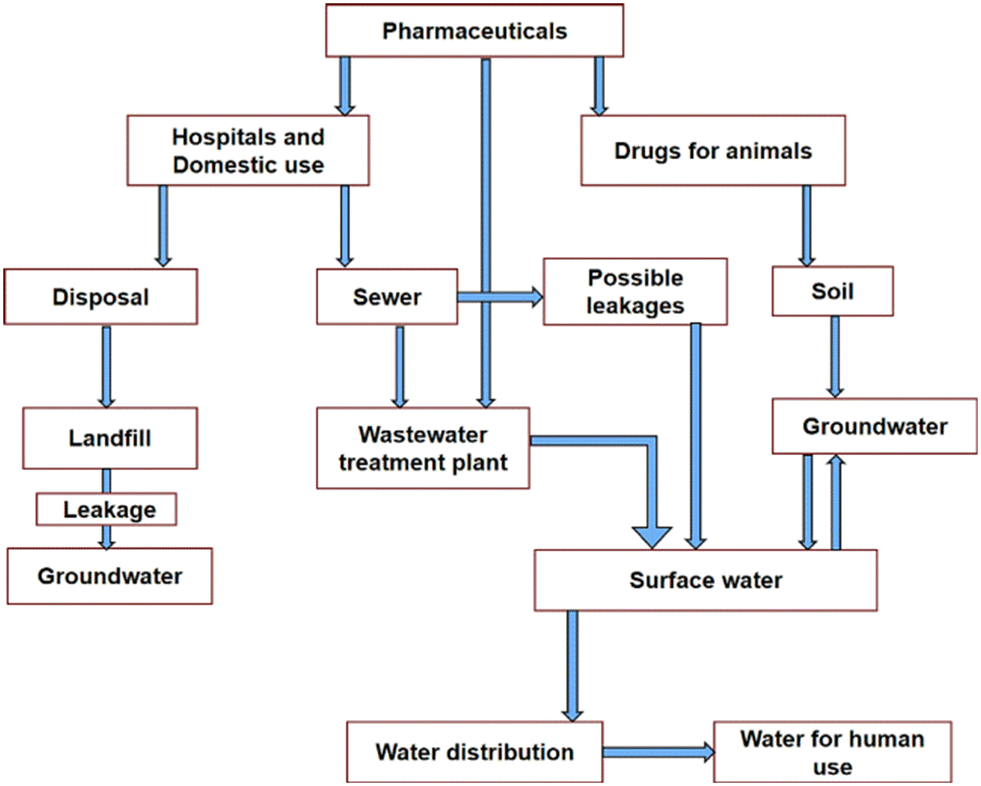 | ||
| Fig. 1 Routes of pharmaceutical contaminants (PCs). Reproduced from ref. 14 with permission from Elsevier (2022). | ||
Further, the accumulation of antibiotic drugs in water can result in the development of antibiotic-resistant bacteria and the dissemination of antibiotic-resistant genes in humans and other living organisms.15,16 According to a recent report, urban wastewater treatment plants are recognized sources for the dissemination of antibiotic resistance in the environment.17 In view of the rising effects of this antibiotic resistance on the global population, the removal of these bioactive molecules from the environment is important to slow down the growth of resistant microorganisms. In addition, antibiotic residues absorbed by plants could interfere with physiological processes, leading to potential ecotoxicological effects.18 These contaminants cannot be completely removed from various water matrices by conventional chemical, physical, flocculation, reverse osmosis or a few other processes, due to the formation of secondary pollutants, high cost, and operational time.19 Therefore, the development of cost-effective, eco-friendly, economical, and effective technologies is urgently needed to remove these emerging contaminants, due to the rising effects of antibiotic resistance in aquatic environments.
Design of the surface and interface plays a promising role in the performance of photocatalysts through maximizing the efficacy of catalysts. Therefore, heterogeneous photocatalysis has been receiving considerable attention as one of the most attractive, low-cost, efficient and outstanding approaches in the degradation of pharmaceutical pollutants.19–55 In this regard, a considerable amount of research interest has focused mostly on TiO2 and to some extent on other semiconducting materials and transition metal oxides as photocatalysts in the degradation of pharmaceutical pollutants in water.23–39 The choice of semiconducting metal oxides as photocatalysts is motivated by the availability of a renewable energy source (solar energy) and the generation of non-toxic degradation products (chemicals and gases). They can be commonly prepared by sol–gel, hydrothermal, solvo-thermal, microwave heating, wet chemical, physical vapour deposition and chemical vapour deposition methods.30 However, the potential of TiO2 and other semiconducting metal oxides could not be harnessed due to the higher rate of recombination of electron–hole pairs and its limited photocatalytic activity under visible light exposure.
Recently, carbonaceous materials have also been reported as promising materials for use in the photocatalytic degradation of antibiotics in water.40–50 This is facilitated by combining these carbon-based materials with other semiconductors, which is considered to be an outstanding approach to enhancing photocatalytic performance. In order to facilitate this, carbonaceous materials with different structures and properties are used as additives in semiconductor materials. This invariably results in enhanced charge separation and visible light activity and is considered the best solution. In addition, semiconducting metal oxides and carbonaceous materials are subjected to doping with metals, non-metals, metal oxides, coupling with noble metal nanoparticles and the formation of composites.36,39,49 Other approaches involving immobilization and the formation of a heterojunction are reported as imperative alternative strategies for achieving enhanced photocatalytic efficiency for these photocatalysts in water treatment.51
According to the available literature, several reviews have been published focusing on metal oxides,23–30 TiO2,31–33 ZnO-based photocatalysts,34 semiconductors,35 doped TiO2,36 hybrids,37 TiO2–carbon dot nanocomposites,38 plasmonic metal–TiO2 composites,39 carbonaceous/carbon-based materials,40,41 g-C3N4,42 MWCNT,43 carbon dots,38,44 activated carbon,45 graphene-based composites,46–48 graphene–TiO2 and doped graphene–TiO2 nanocomposites,49 graphene-based materials,50 and nanomaterial-based heterogeneous photocatalysts51 as photocatalysts for the treatment of wastewater containing pharmaceuticals. Alternatively, several review articles have reported on the photodegradation of antibiotic contaminants in water, such as amoxicillin,21 ibuprofen,22 tetracycline,52,54 ciprofloxacin,53,54 and norfloxacin54 antibiotics in wastewater and several others, which are referred to in section 3. However, there is still a need for an extensive review article in this field, covering in a single window a larger number of pharmaceutical pollutant photocatalysts for their photocatalytic performance.
The present review is focused primarily on the photocatalytic degradation of acetaminophen, amoxicillin, sulfamethoxazole, ibuprofen, norfloxacin, ciprofloxacin, tetracycline, diclofenac, etc. The structure and uses as well as the solubility of these antibiotics in water are provided in Table 1 (ref. 55) and ESI,† respectively. In view of this, the article describes the fundamental properties of semiconducting materials as photocatalysts as well as role of metal oxides, carbon-based materials, and heterojunctions and the immobilization approaches employed and the mechanisms involved in the removal of these pharmaceutical pollutants. Subsequently, the article deals with the removal of the above-mentioned drugs from contaminated water using semiconducting TiO2, ZnO, and many other oxides, their combination with graphitic-carbon nitride (g-C3N4), carbon nanotubes (CNTs), activated carbon (AC), graphene oxide, graphene and graphene quantum dots, doping with metals and nonmetals, the formation of composites, semiconducting materials deposited on certain supports as photocatalysts and a heterojunction approach. It is anticipated that, in the light of this, the current review could be of immense help in identifying cost-effective and efficient photocatalytic methods for the remediation of these pharmaceutical pollutants. In addition, various research gaps, their possible solutions and several future prospects are also provided at the end of this article for the possible enhancement of environmental conservation.
| Pollutant (formula) | Structure | Uses |
|---|---|---|
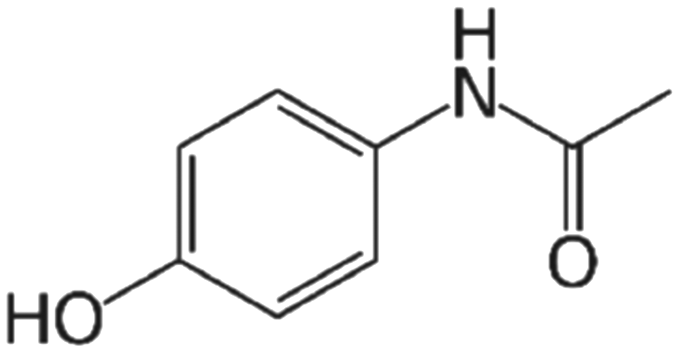
| Acetaminophen (C8H9NO2) |
|
Nonprescription analgesic and antipyretic medication for mild-to-moderate pain and fever |
| Amoxicillin (C16H19N3O5S) |

|
Bacterial infections, and dental abscesses |
| Sulfamethoxazole (C10H11N3O3S) |

|
Used in treatment of a variety of bacterial infections, including those of the urinary, respiratory, and gastrointestinal tracts |
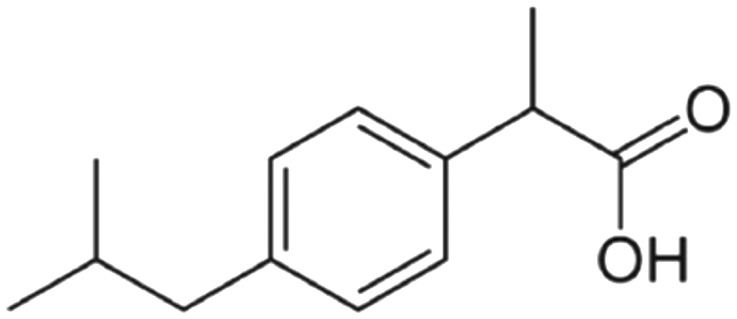
| Ibuprofen (C13H18O2) |
|
Anti-inflammatory; analgesic; antipyretic |
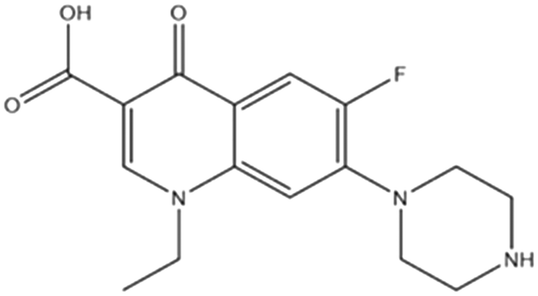
| Norfloxacin (C16H18FN3O3) |
|
In treatment of urinary tract infections and prostatitis |
| Ciprofloxacin (C17H18FN3O3) |

|
Therapy of mild-to-moderate urinary and respiratory tract infections caused by susceptible organisms |
| Tetracycline (C22H24N2O8) |

|
Role as an antimicrobial agent, an antibacterial drug, an antiprotozoal drug, a protein synthesis inhibitor and an Escherichia coli metabolite |
| Diclofenac (C14H11Cl2NO2) |

|
Therapy of chronic forms of arthritis and mild-to-moderate acute pain |
| Atenolol (C14H22N2O3) |

|
As a cardioselective beta-blocker that is widely used in the treatment of hypertension and angina pectoris |
2 Important photocatalysts and their role in the removal of pharmaceutical pollutants
The primary mechanism for the degradation of organic pollutants by a semiconducting material involves irradiating it with light energy in the form of photons (hv) sufficiently greater than the band gap energy of the photocatalyst (Fig. 2 (ref. 37)). Holes (hVB+) and electrons (eCB−) are generated in this manner in the valence band (VB) and the conduction band (CB), respectively. The separated holes reacts with hydroxyl ions (OH−) or water molecules (H2O) to produce hydroxyl radicals (·OH). In addition, the separated electrons reacts with dissolved O2 in water to produce superoxide radicals (·O2−), which upon further reaction, produce ·OH.37,51 Subsequently, the active species generated in this manner react with pharmaceutical pollutants on the surface of the semiconductor catalyst to give H2O, CO2 and other by-products.| Semiconductor + hv → hVB+ + eCB− |
| hVB+ + H2O → H+ + ·OH |
| eCB− + O2 → ·O2− |
| ·O2− + H+ → HO2· |
| HO2· + HO2· → H2O2 + O2 |
| H2O2 + ·O2− → ·OH + OH− + O2 |
| H2O + hVB+ → ·OH + H+ |
| hVB+ + OH− → ·OH |
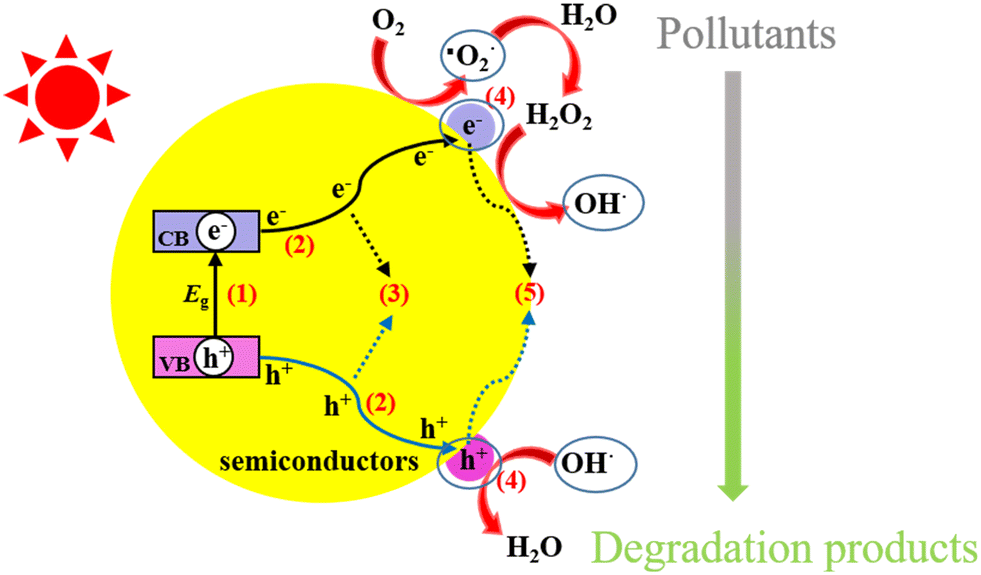 | ||
| Fig. 2 Photocatalytic processes over a heterogeneous photocatalyst. Reproduced from ref. 37 with permission from MDPI (2021). | ||
2.1 Metal oxides
Several semiconductor metal oxides have been used as photocatalysts in the abatement of aqueous pollution due to organic pollutants. From this point of view, TiO2 has received a considerable amount of attention and its choice is mainly guided by its superior photocatalytic degradation efficiency, low processing cost, high environmental stability, nontoxicity, chemical stability, and high oxidizing ability.31–33 However, its wide band gap (∼3–3.2 eV),32 and the fast e−–h+ recombination rate of photogenerated electron–hole pairs in TiO2 limit its applications. Semiconducting ZnO (band gap: 3.37 eV) has been used as another photocatalyst in water treatment as an alternative to TiO2.56 Several other metal oxides (ZrO2, Fe2O3, γ-Fe3O4, SnO2, Mn2O3, WO3, CeO2, CuO, and NiO) have also been investigated as alternatives to TiO2 and ZnO.26 Nano-engineered metal-oxide-based photocatalysts have also attracted a lot of attention in wastewater treatment.57 However, metal oxide catalysts experience similar drawbacks to TiO2. As a consequence, significant developments have taken place in recent years in tailoring these metal oxide photocatalysts. This is achieved by reducing their band gap by the addition of dopants that include both metals and non-metals, such as iron, copper, carbon, nitrogen, platinum and sulfur. In addition, metal sulfides,58 metal ferrites,59 and oxychlorides60 have also been explored as emerging photocatalysts for the removal of pharmaceutical pollutants.Photocatalytic studies have been reported on the performance of semiconductor–metal composites in the removal of several pollutants from water. In this regard, plasmonic composites in combination with various semiconducting photocatalysts have been widely studied for enhancing overall photocatalytic performance.61,62 The improved photocatalytic efficiency is attributed to the surface plasmon resonance effect. In addition, metal nanoparticles can decrease the recombination rate of the photo-induced e−–h+ pairs of the semiconductor material by effective electron trapping in the conduction band. Metal oxide nanocomposites derived from a mixture of two or more oxides or between these oxides and other functional semiconductor materials have also been found to be efficient, economical, and environmentally friendly photocatalysts in water pollutant remediation.63,64
2.2 Carbonaceous materials
The photocatalytic performance of various carbonaceous materials has been receiving more attention for antibiotic removal owing to their intriguing properties and good stability.40,41 The choice of these carbonaceous materials in removing antibiotics is mainly guided by simple and cost-effective synthesis methods, the easy availability of raw materials and their unique physiochemical properties, such as the presence of micropores, mesopores, and macropores, the large number of oxygen-functional groups, high porosity, and high surface area, coupled with good visible-light adsorption ability, chemical stability, excellent electrical conductivity and high intrinsic electron mobility.40 The carbonaceous materials explored for this purpose include carbon dots,38 g-C3N4,42,65 activated carbon45,66 and carbon nanotubes (CNTs).67 Graphene is another carbon-based material composed of a one-atom-thick layer of carbon atoms arranged in a hexagonal lattice.68 It is a semimetal with a small degree of overlap between the valency band and the conduction band.69 This makes graphene a promising candidate for application in photocatalysis. However, the photocatalytic performances and practical applications of carbon-based materials have not been encouraging, due to poor solar-light absorption and the rapid recombination of photogenerated electron–hole pairs.41 Interestingly, combinations of these carbon-based materials with other semiconductor metal oxides have been utilized as promising photocatalysts owing to their notable properties like stability, conductivity, durability and high absorptivity. In addition, carbon-based materials–metal oxide nanocomposites have also enhanced the degradation efficiency of pharmaceuticals by improving the generation of radical species, through improved surface area and light absorption, and reducing the recombination of generated charge carriers.48,692.3 Heterojunction nanocomposites as photocatalysts
A heterojunction is defined as the interface between two layers or regions of different semiconductors with unequal band structures that can result in band alignments. Based on this concept, semiconductor–semiconductor-based heterojunction composites showed excellent improvements in photocatalytic efficiency. This is ascribed to minimized charge carrier recombination, the interface of the heterojunction, superior charge transfer, prolonged charge carrier lifetime, separate active sites, and extended light absorbance characteristics.51 These semiconductor heterojunction photocatalysts are classified into several types: i.e., conventional heterojunctions (type-I, type-II, and type-III), p–n heterojunctions, direct Z-scheme heterojunctions, and S-scheme heterojunctions.70–73 The schematic separation of charges via electron migration from one semiconductor to another in various heterojunction mechanisms is represented in Fig. 3.51 Among these, in a type-I heterojunction, the VB and CB of semiconductor-1 are respectively lower and higher than those of semiconductor-2 (Fig. 3(a)). The photogenerated holes migrate from the VB of semiconductor-1 to the VB of semiconductor-2 accompanied by the transfer of photoelectrons from the CB of semiconductor-1 to the CB of semiconductor-2.52 However, this type-I heterojunction cannot spatially separate e−–h+ pairs and this leads to the accumulation of charge carriers and their accelerated recombination rate. A type-II heterojunction (Fig. 3(b)) involves the transfer of photogenerated holes generated in semiconductor-2 to semiconductor-1, considering the VB of semiconductor-1 to be lower than that of semiconductor-2 on irradiating with light.52 In contrast, photogenerated electrons in the CB of semiconductor-1 can migrate to that of semiconductor-2, if the level of the CB in semiconductor-1 is higher than that of semiconductor-2. It should be noted that the spatial separation of electron–hole pairs can occur in a type-II heterojunction. Furthermore, the structure of a type-III heterojunction is similar to that of a type-II heterojunction; however, charge-carrier separation cannot occur in a type-III heterojunction because the band gaps of both semiconductors do not overlap, since the levels of the VB and CB of both semiconductors are very far apart (Fig. 3(c)). When p-type and n-type semiconductors are combined, a p–n heterojunction can be formed. A space-charge region could be formed at the interface before light irradiation due to diffusion of the majority of charge carriers, leading to a built-in electric field, as shown in Fig. 3(d). In the Z-scheme heterojunction system, the band structure is quite analogous to that of a type-II heterojunction, but the direction of charge transfer is the opposite. The photogenerated electrons from the second semiconductor migrate aggressively to the VB of the first semiconductor and occupy the available holes, while the strongly oxidative holes in the VB of the second semiconductor and strongly reductive electrons in the CB of the first semiconductor take part in the redox reaction (Fig. 3(e)). In a step-scheme (S-scheme) heterojunction, two n-type semiconductors are combined with a staggered band structure similar to a type-II heterojunction (Fig. 3(f)). | ||
| Fig. 3 Schematic illustration of various types of heterojunction: (a) straddling bandgap (type I), (b) staggered bandgap (type II), (c) broken bandgap (type III), (d) p–n type, (e) direct Z-scheme, and (f) S-scheme. Reproduced from ref. 51 with permission from Amer Sci Publ (2023). | ||
2.4 Immobilized photocatalysts
The immobilization of photocatalysts on supports (Fig. 4)51 can maximize the activity of semiconductors by offering a greater number of active sites. The high photocatalytic activity of such immobilized semiconductor photocatalysts is guided by the properties of their semiconductor-active species and the kind of support employed.51 The high catalytic performance of these immobilized photocatalysts originates from impeding the rate of electron–hole pair recombination. The recovery, reusability, and stability issues of a photocatalyst remain challenging after several reaction runs. In this regard, the immobilization of a catalyst on a support facilitates the rapid separation and efficient recycling of the catalyst. This reduces production costs as well as minimizing waste generation, especially in industrial applications compared to conventional pure photocatalysts.74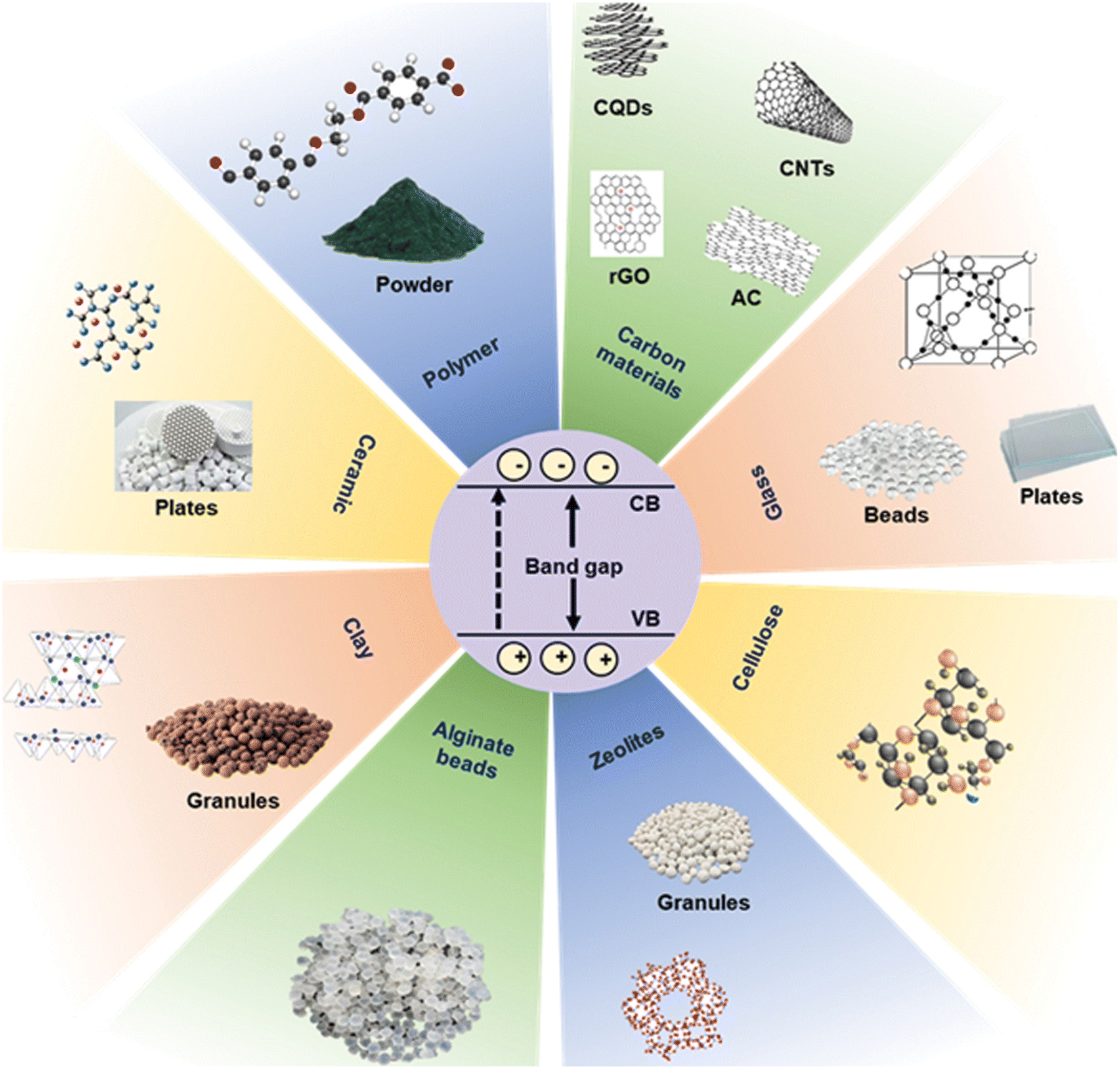 | ||
| Fig. 4 Supporting materials used for the immobilization of photocatalysts. Reproduced from ref. 51 with permission from Amer Sci Publ (2023). | ||
3 Removal of pharmaceutical components using different Photocatalysts
In this review article, we present the use of photocatalysts based on bare metal oxides (TiO2, ZnO and other oxides) and carbon-based materials (graphitic carbon nitride, g-C3N4, carbon nanotubes CNTs, activated carbon AC, and graphene) in the removal of pharmaceutical pollutants from water. In addition, several modification approaches are also highlighted and those involving metal loading, doping with metals and nonmetals, the formation of composites, immobilization and the formation of heterojunctions for this purpose are described below for pharmaceutical pollutants.3.1 Acetaminophen
Acetaminophen (ACT), also known as paracetamol is commonly used all over the world as a painkilling, anti-inflammatory, analgesic, and antipyretic drug.75–78 It is available both as a single-entity formulation and in combination with other medications. The presence of acetaminophen in wastewater, surface water and groundwater can have an adverse effect on living organisms and environmental ecology owing to its oxidative transformation to toxic N-acetyl-p-benzoquinone imine. The stable chemical structure of acetaminophen remains one of the major constraints to its removal through conventional wastewater treatment. Therefore, attention has focused on its removal from aqueous media following a photocatalysis approach, as described below.79–147Zhang et al.82 reported about 95% photocatalytic degradation of acetaminophen in an aqueous solution of TiO2 (1.0 g L−1) after 100 min of irradiation under a 250 W metal halide lamp. This is attributed to direct hole (h+) oxidation and ipso-substitution comprising the main initial steps in the degradation. The photodegradation of paracetamol (20 mg L−1) has been investigated in the presence of nanostructured TiO2 catalysts with a nanotube-type morphology using ultraviolet radiation (λ: 254 nm) and the removal efficiency was found to be 99% after 100 min.83 The photocatalytic degradation of acetaminophen in water has also been reported using ZnO,84 faceted-TiO285 and molecularly imprinted ZnO nanonuts.86
Pd-decorated CuO nanostructured thin film showed enhanced visible-light degradation of acetaminophen.89 The influence of radical trappers revealed no role for ·OH, ·O2− (or 1O2) radicals on the photocatalytic degradation of acetaminophen. The photocatalyst possessed good stability, as indicated by the observed insignificant change in photodegradation even after 5 cycles. According to the available literature, ZnFe2O4 (bandgap: 1.9 eV) is non-toxic and exhibits good photostability.90 Its photocatalytic behaviour is guided by several factors, such as its preparative method, morphology, and the presence of impurities. In view of this, Huerta-Aguilar et al.91 reported the efficient degradation of paracetamol during water treatment using Au nanoparticles grown on ZnFe2O4 as a visible light (200 W halogen lamp, C-type R7s, λ > 400 nm) assisted photocatalyst. TiO2/BN/Pd nanofibers showed significantly enhanced degradation of ACT (>90%), compared to pure TiO2 (20%) after 4 h under visible-light irradiation.92 This was explained on the basis of the good dispersion of Pd nanoparticles on TiO2–BN nanofibers to facilitate the transfer of photoexcited hole carriers and a decrease in photogenerated electron–charge recombination. Reusability studies and recycling tests on the TiO2/BN/Pd photocatalyst indicated its good stability over 5 cycles under UV and visible light.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 103 have also been prepared and examined for the photocatalytic degradation of acetaminophen and paracetamol.
103 have also been prepared and examined for the photocatalytic degradation of acetaminophen and paracetamol.
The degradation of acetaminophen and its reaction mechanism have been investigated in presence of Ag–ZnO104 and La-doped ZnO105 photocatalysts under visible-light irradiation. Abri et al.106 studied the photocatalytic degradation of nizatidine, acetaminophen and levofloxacin over ZnO (1:6) nanostructured photocatalysts under UVB light for 240 min and the findings are displayed in Fig. 5(a). Similar studies on using 1% Ce-doped ZnO produced almost no change in the degradation of acetaminophen and levofloxacin compared to that observed for nizatidine (∼95%), as evidenced from Fig. 5(b). Such different photocatalytic degradation of these pharmaceuticals in the presence of ZnO and 1% Ce–ZnO photocatalysts could be attributed to their chemical structures.
 | ||
| Fig. 5 (a) Photocatalytic degradation of pharmaceuticals over (a) ZnO (1:6) and (b) 1% Ce–ZnO nanostructured photocatalysts [experimental conditions: catalyst dosage: 1 mg mL−1; concentration of pharmaceutical: 5 mg L−1]. Reproduced from ref. 106 with permission from Elsevier (2019). | ||
Kumar et al.107 investigated the photocatalytic degradation of acetophenone by irradiating nitrogen-implanted ZnO nanorod arrays (NRAs) with visible light. It should be noted that an N ion (1 × 1016 ions per cm2) doped ZnO NRA sample (referred to as N–ZnO4) showed maximum degradation efficiency (98.46%) of acetaminophen (20 ppm) in the presence of sunlight under 120 minute duration. The linear variation in ln(C0/C) versus irradiation time followed pseudo-first-order degradation kinetics for acetaminophen. Furthermore, the superior photocatalytic activity of the N–ZnO4 catalyst was inevitable from the high value of its rate constant (0.038 min−1) compared to pristine ZnO NRAs (0.0045 min−1). In addition, further investigations also revealed a more or less unaltered degradation efficiency (98.46% to 97.63%) of N–ZnO4 after five repeated cycles. The findings of the effect of scavengers on the photocatalytic degradation of acetaminophen in the presence of N–ZnO4 showed a decrease in degradation efficiency for acetaminophen (98.4%) in the presence of benzoquinone (BQ 28.52%), EDTA (65.6%) and methanol (98.4%) due to the major role played by O2. The mechanism of acetaminophen degradation on subjecting N-ion-implanted ZnO NRAs to visible light suggested a shifting of the band gap to the visible region.
Magnetic TiO2/Fe3O4 (1.16 g L−1) and TiO2/SiO2/Fe3O4 (1.34 g L−1) nanoparticles degraded acetaminophen, antipyrine, caffeine, and metoprolol pharmaceuticals on illuminating its aqueous solution (pH: 7, ACT concentration: 30 mg L−1).111 TiO2/SiO2/Fe3O4 nanoparticles also showed good reusability, as evidenced within four repeated experiments. Czech and Tyszczuk-Rotko112 explored the visible-light (centered at 500–550 nm) driven photocatalytic removal of acetaminophen from water using MWCNT (1.72 wt%)–TiO2–SiO2 nanocomposites and observed ∼82% efficiency due to the key role played by photogenerated holes. In another study, Fernandes et al.113 selected combinations of Fe2O3 and Fe3O4 nanoparticles due to their easy availability and used them in the photodegradation of acetaminophen under UV-vis irradiation. The total acetaminophen (and caffeine) degradation (20 ppm/150 mL) took place by means of 0.13 g catalyst L−1 solution in 45 min (and 60 min) and it remained almost unaltered over five cycles. A ternary heterogeneous anatase-TiO2 (B) biphasic nanowires/Bi4O5I2 composite exhibited 95% degradation of acetaminophen in 6 min under visible-light irradiation.114 This is ascribed to the multiphase structure, including the synergistic effect of anatase TiO2 and Bi4O5I2. A schematic of the possible charge separation and photocatalytic mechanism of the TiO2–Bi4O5I2 composite under visible-light irradiation is displayed in Fig. 6(a).
 | ||
| Fig. 6 (a) Schematic of the possible charge separation and photocatalytic mechanism of TiO2–Bi4O5I2 composite under visible-light irradiation. Reproduced from ref. 114 with permission from Elsevier (2020). (b) Schematic diagram of charge transfer in the photoexcited TiO2/Fe2O3 core–shell photocatalyst. Reproduced from ref. 117 with permission from Elsevier (2017). | ||
Chau et al.115 synthesized a Cu2O/WO3/TiO2 ternary composite in view of the narrow band gaps of Cu2O (2.20 eV) and 2.70 eV (WO3) guided by their low cost, nontoxicity, chemical stability and strong absorption ability towards visible light. The composite fabricated in this manner produced 92.50% photodegradation of ACT (1 mg L−1) compared to pure TiO2 under 60 min of solar irradiation. This is attributed to the effective separation and low recombination rate of the charge carriers. The produced composite exhibited high reusability for photodegradation with 83% at the fifth cycle of ACT photodegradation. Nanostructured titania supported on activated carbon (AC) has been used to study the effects of photocatalyst dosage, initial solution pH and irradiation (UV) time on the photocatalytic degradation of aqueous acetaminophen.116 Abdel-Wahab et al.117 prepared flower-like core–shell TiO2/Fe2O3 photocatalysts instead of TiO2/Fe3O4 due to the photostability of Fe2O3 compared to Fe3O4 and investigated its activity in the degradation of paracetamol in aqueous solution using a medium-pressure mercury lamp (450 W). These findings indicated increases in the photocatalytic degradation of paracetamol (52.5%) to 87.8% for 50% content of TiO2. This is ascribed to the separation of the photogenerated electron–hole pairs accomplished by coupling the narrow band gap with the wide band gaps of Fe2O3 and TiO2, respectively. A schematic diagram of charge transfer in the photoexcited TiO2/Fe2O3 core–shell photocatalyst is displayed in Fig. 6(b). Jallouli et al.118 used TiO2 nanoparticles and TiO2/cellulosic fiber to carry out the photocatalytic degradation of paracetamol under UV and sunlight irradiation. WO3/TiO2/SiO2119 and TiO2/ZSM-5 (ref. 120) also exhibited enhanced photocatalytic degradation of acetaminophen in contaminated wastewater.
TiO2 immobilized on glass spheres (sunlight)121 and ZnO–polystyrene (UV-LED)122 photocatalysts effectively removed acetaminophen and paracetamol, respectively. The photodegradation of acetaminophen is also reported with zeolite-supported TiO2 and ZnO under UV and sunlight,123 bi-modified titanate nanomaterials (visible light),124 BaTiO3/TiO2 composite (UV-vis),125 and Ag/AgCl@ZIF-8 (visible light).126
Heterostructures comprising α-Fe2O3/g-C3N4130 have been examined for the photocatalytic degradation of acetaminophen. The photocatalytic activity of g-C3N4 combined with UiO-66-NH2 in different proportions (25%-g-C3N4/UiO-66-NH2, 50%-g-C3N4/UiO-66-NH2, 75%-g-C3N4/UiO-66-NH2) was tested for the removal of acetaminophen from an aqueous solution under given experimental conditions ([ACT]: 5 mg L−1, [Cat]: 0.5 g L−1, V: 350 mL).131 The corresponding findings on the temporal evolution of acetaminophen with the different samples and their pseudo-first-order rate constants (kobs) are displayed in Fig. 7(a) and (b). These findings depict complete removal of acetaminophens by the 75%-g-C3N4/UiO-66-NH2 heterostructure in 120 min with a pseudo-first-order rate constant of 2 h−1. It is suggested that incorporation of UiO-66-NH2 in g-C3N4 enhanced the separation of the photogenerated charges. Silica–carbon quantum dots (1 wt%) decorated TiO2 as a sunlight-driven photocatalyst completely removed acetaminophen 33.3% faster than pure TiO2.75 Gupta et al.132 studied the augmented photocatalytic degradation of acetaminophen using hydrothermally treated g-C3N4 and persulfate under LED irradiation.
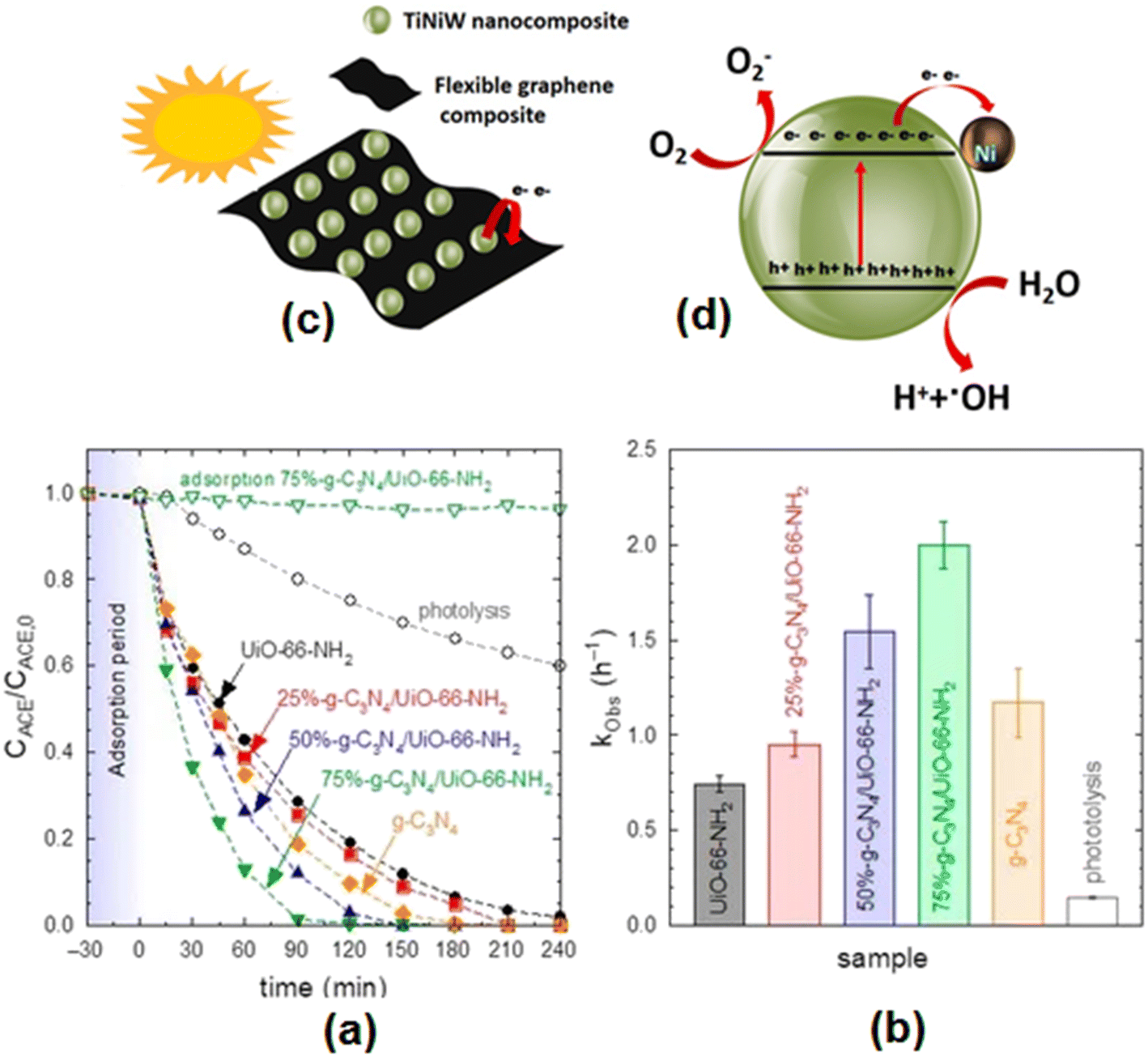 | ||
| Fig. 7 (a) Photocatalytic degradation of acetaminophen with different g-C3N4/UiO-66-NH2 samples. (b) Pseudo-first-order rate constant (kobs) of different g-C3N4/UiO-66-NH2 samples. Experimental conditions: V = 350 mL; T = 20 °C, CACE = 5 mg L−1; CCAT = 0.5 g L−1. Reproduced from ref. 131 with permission from MDPI (2022). (c) Schematic illustration of the TiNiW NPs decorating the surface of the graphene composites and (d) TiNiW nanoparticle showing the possible chemical reactions for the formation of reactive oxygen species that degrade the ACT contaminant. Reproduced from ref. 138 with permission from Elsevier (2021). | ||
A heterojunction magnetic ternary g-C3N4/TiO2–MnFe2O4 halloysite photocatalyst showed about 79.1% removal of acetaminophen (10 ppm) within 90 min under visible light.145 The ternary photocatalyst could be easily recovered by applying an external magnetic field and reused several times without any significant reduction in its catalytic activity. The removal efficiency for acetaminophen under optimum conditions in the presence of a magnetic carbon heterojunction coupled with UV light and peroxymonosulfate was insignificantly reduced from 97.4% even after five consecutive cycles.146 Moradi et al.147 used 0.6 g L−1 of TiO2/graphene/g-C3N4 (60:10:30) Z-type photocatalyst and observed complete degradation of acetaminophen (50 mg L−1) at a pH of 9.0 in 120 min due to a synergistic effect. Their investigations also showed HO· and O2·− radicals to be the dominant species in the degradation of acetaminophen.
Table 2 records the performance data of different photocatalysts on the removal of acetaminophen from wastewater.
| Photocatalyst | Preparative method | ACT | Catalyst dose | pH | Light source | Degradation and time | Rate constant |
|---|---|---|---|---|---|---|---|
| TiO2-rutile76 | Precipitation | 20 ppm | 0.1 g (50 mL) | 9 | Tungsten halogen lamp (400 W), 0.0146 W cm−2 | 68% (60 min) | — |
| TiO2-anatase76 | Thermal precipitation method | 20 ppm | 0.1 g (50 mL) | 9 | Tungsten halogen lamp (400 W), 0.0146 W cm−2 | 60% (60 min) | — |
| ZnO76 | Thermal precipitation method | 20 ppm | 0.1 g (50 mL) | 9 | Tungsten halogen lamp (400 W), (0.0146 W cm−2) | ∼100% (60 min) in 1 h | — |
| TiO2: 80% anatase + 20% rutile (Degussa P25)77 | Commercial | 40 mg L−1 (250 mL) | 2 g L−1 | — | UV lamp (15 W) | 97% (300 min) | — |
| TiO2/Ag (5%)78 | Photodeposition method | 20 μg L−1 (O2: 100 cm3 min−1) | 1 g L−1 | — | UV radiation (365 nm) | 94.50% (240 min) | — |
| TiO279 | Sol–gel method | 50 ppm (750 mL) | 1.33 g L−1 | — | TQ159-ZO lamp (150 W) | ∼50% (180 min) | 0.0056 min−1 |
| TiO280 | Sol–gel method | 35 mg L−1 | 0.15 g | 10 | UV lamp with a wavelength of 256 nm, 1 mW cm−2 | 99% (180 min) | — |
| Solid TiO2 spheres81 | Template-free solvothermal route | 50 mg L−1 | 0.1 g L−1 | — | Mercury lamp (500 W) | 90% (60 min) | 0.075 min−1 |
| Mesoporous TiO2 microspheres81 | Template-free solvothermal route | 50 mg L−1 | 0.1 g L−1 | — | Mercury lamp (500 W) | 94% (60 min) | 0.043 min−1 |
| TiO2 (High Techn. Nano co. Ltd)82 | Commercial | 50 μM | 1.0 g L−1 | 9 | Metal halide lamp (250 W), λ ≥ 365 nm | ∼95% (100 min) | — |
| ZnO powders (Fluka)84 | Commercial (thermally calcined at 100 °C) | 50 mg L−1 | 0.25 g (0.25 L) | — | UV-lamp (315–400 nm), P.D: 0.66 mW cm−2 | ∼97% (240 min) | 0.0136 min−1 |
| ZnO nanonuts86 | Chemical method | 5 × 10−5 M | ∼1.0 mg | 7.2 | UV lamp: 4 mW cm−2, 368 nm | ∼92% (180 min) | 1.32 × 10−2 min−1 |
| TiO2 (Degussa P25)87 | Commercial | 0.3 mg L−1 | 40.5 mg (70 mL) | Neutral | LED lamp – UVA light (15 W), 365 nm | 100% (40 min) | 0.12 min−1 |
| Au–TiO287 | Mixing tempered colloidal solution of au and TiO2 in water | 0.3 mg L−1 | 40.5 mg (70 mL) | Neutral | LED lamp – UVA light (15 W), 365 nm | 100% (32 min) | 0.14 min−1 |
| Au–g-C3N487 | Reflex method | 0.3 mg L−1 | 40.5 mg (70 mL) | 5.9 | Visible light | 100% (25 min) | 0.17 min−1 |
| Ag(1 wt%)/TiO288 | Sonicating mixture of TiO2 and aqueous AgNO3, stirring and irradiating with 450-W ACE lamp for 1 h | 20 mg L−1 | 0.4 g L−1 | 6.3 | Simulated solar light xenon lamp (1000 W), 50.0 mW cm−2 | ∼98% (180 min) | 0.019 min−1 |
| Au(1 wt%)/TiO288 | Sonicating mixture of TiO2 and aqueous H2AuCl6, stirring and irradiating with 450 W ACE lamp for 1 h | 20 mg L−1 | 0.4 g L−1 | 6.3 | Simulated solar light xenon lamp (1000 W), 50.0 mW cm−2 | ∼93% (180 min) | 0.016 min−1 |
| Pt(1 wt%)//TiO288 | Sonicating mixture of TiO2 and aqueous H2AuCl6, stirring and irradiating with 450 W ACE lamp for 1 h | 20 mg L−1 | 0.4 g L−1 | 4.2 | Simulated solar light xenon lamp (1000 W), 50.0 mW cm−2 | ∼100% (180 min) | 0.020 min−1 |
| Pd/CuO89 | Deposition and sputtering | 10 mg L−1 (20 mL) | 15 (l) × 15 (w) × 1 (t) mm film | — | Xenon arc lamp: 150 W, λ > 420 nm | ∼90% (240 min) | 0.796 h−1 |
| TiO2/BN/Pd92 | Electrospinning and atomic layer deposition | 1 mg L−1 (250 mL) | 0.5 g L−1 | 6.8 | Medium-pressure metal halide UV lamp (400 W) | 100% (10 min) | 0.019 min−1 |
| TiO2/BN100/Pd10092 | Electrospinning and atomic layer deposition | 1 mg L−1 (250 mL) | 0.5 g L−1 | 6.8 | 400 W halogen linear lamp (visible irradiation) | 98% (180 min) | 0.28 min−1 |
| C,N-co-doped TiO293 | Peroxo–gel method | 4 mg L−1 | 20 mg | — | UV-light (10 W), λ: 365 nm | 69.31% (120 min) | — |
| C-doped TiO294 | Sol–gel method | 2.0 ppm | 2.0 g L−1 | 7 | Low UV lamp pressure (15 W), 365 nm, 65 W m−2 | 100% (90 min) | 0.0817 min−1 |
| Supported titania-based catalysts (25 wt% mg doping)95 | Industrial petrochemical (source) | 20 mg L−1 | 0.7 g L−1 (25 mL) | 4.3 | UV lamp: 365 nm, 30 W m−2 | 60% (60 min) | — |
| Mercury vapour lamp (125 W), (202 W m−2) | 48.3% (60 min) | — | |||||
| TiO296 | Hydrolysis of Ti isopropoxide (sol–gel method) | 35 mg L−1 | 0.5 g L−1 | 5.5 | UV irradiation: HG500 lamp (30 mW cm−2) | ∼84% (120 min) | 12.4 ± 0.2 × 10−3 min−1 |
| Ta-doped TiO2 (Ti/Ta molar ratio: 1%)96 | Hydrolysis of Ti isopropoxide (sol–gel method) followed by Ta doping through impregnation method | 35 mg L−1 | 0.5 g L−1 | 5.5 | UV irradiation: HG500 lamp (30 mW cm−2) | ∼70% (120 min) | 9.4 ± 0.1 × 10−3 min−1 |
| TiO296 | Hydrolysis of Ti isopropoxide in presence of CH3COOH | 35 mg L−1 | 0.5 g L−1 | 5.5 | UV irradiation: HG500 lamp (30 mW cm−2) | ∼70% (120 min) | 9.3 ± 0.1 × 10−3 min−1 |
| Ta-doped TiO2 (Ti/Ta molar ratio: 1%)96 | Hydrolysis of Ti isopropoxide in presence of CH3COOH followed by ta doping through impregnation method | 35 mg L−1 | 0.5 g L−1 | 5.5 | UV irradiation: HG500 lamp (30 mW cm−2) | ∼73% (60 min) | 10.4 ± 0.1 × 103 min−1 |
| Mesoporous MnOx–TiO297 | Sol–gel method | 25 ppm (150 mL) | 0.1 g L−1 | — | Continuous sonication (20 W) and UVA radiation (160 W m−2) | 26% (180 min) | — |
| IL-Fe-doped TiO2 with Fe to Ti molar ratios (%): 298 | Sol–gel method | 10 mg L−1 (200 mL) | 0.65 g L−1 | 7 | UV lamps | 90.35% (90 min) | 0.25 min−1 |
| Synthetic TiO2 doped with (KAl(SO4)2)99 | Sol–gel method | 0.10 mM | 1.0 g L−1 | 6.9 | Visible light: source (light emitting diodes) with λ > 440 nm | 95% (540 min) | 5.20 × 10−3 min−1 |
| Carbon-self-doped TiO2101 | Sol–gel method (product calcined at 300 °C) | 0.1 mM (500 mL) | 1.0 g L−1 | 6.9 | LEDs (λ > 440 nm) | ∼96% (540 min) | 5.0 × 10−3 min−1 |
| Bi3+(10%)-doped anatase TiO2102 | Hydrolysis method | 104 M (100 mL) | 0.1 g L−1 | 5 | Source: UV-vis, (4 W cm−2) | ∼100% (240 min) | 0.97 h−1 |
| Ba1−xBiFe1−xCuxO3 (x = 0.05)103 | Pechini method | 50 mg L−1 | 0.75 g L−1 | 9 | Metal halide efficacy lamp | 98.1% (120 min) | — |
| Ag/ZnO104 | Chemical method | 5 mg L−1 (500 mL) | 1 g L−1 | 8.5 | Tungsten halogen lamp (300 W) | 90.8% (120 min) | 0.020 min−1 |
| 1.0 wt% La-doped ZnO105 | Precipitation method | 100 mg L−1 (500 mL) | 0.1 g | — | Compact fluorescent lamps: 20 W | 99% (3 h) | — |
| 1% Ce-doped ZnO106 | Hydrothermal method | 5 mg L−1 | 1 mg mL−1 | 6.8 | UV-B mercury lamp (8 W) | 68% (240 min) | 0.0058 min−1 |
| N-Implanted ZnO nanorod array (NRA)107 | ZnO NRAs by two-step process followed by N implantation by low energy ion beam | 20 ppm (5 mL) | 10 × 10 mm aligned ZnO NRA | — | Visible-light irradiation | 98.46% (120 min) | 0.038 min−1 |
| TiO2/SiO2/Fe3O4111 | Ultrasonic-assisted sol–gel method | 30 mg L−1 (400 mL) | 1.34 g L−1 | 7 | Low-pressure mercury lamp: λ: 254 nm, 3.8 × 10−6 Ein L−1 s−1 | ∼97% (300 min) | 1.7 × 109 M−1 s−1 |
| MWCNT (1.72 wt%) TiO2–SiO2112 | Sol–gel method | 10 mg L−1 | — | Nearly neutral | High-pressure mercury lamp, 500–550 nm, 7.31–7.53 mW m−2 | 81.6% (60 min) | 0.0113 min−1 |
| Magnetite–hematite113 | Hydrothermal | 20 mg | 0.13 g L−1 | — | Medium-pressure hg vapour lamp (400 W) | ∼100% (45 min) | — |
| TiO2 (438 mg)–Bi4O5I2114 | In situ calcination method | 3 ppm | 25 mg | — | Xenon lamp with a light filter of 400 nm | ∼95% (6 min) | 0.425 min−1 |
| Cu2O/WO3/TiO2115 | Hydrothermal | 1 mg L−1 (80 mL) | 20 mg | 9 | Solar-light irradiation (source) | 92.5% (60 mL) | 4.42 × 10−2 min−1 |
| Flower-like 50% TiO2/Fe2O3117 | Modified ultrasonic assisted sol–gel method | 50 mg L−1 (50 mL) | 0.1 g L−1 | — | Medium-pressure Hg lamp (450 W) | 87.8% (90 min) | 0.0219 min−1 |
| 3% WO3/TiO2/SiO2119 | Solution method | 10 mg L−1 | 1.0 g L−1 | 9 | Xenon lamp (500 W) without cut-off filter 800 nm cut-off filter (800 nm > λ > 200 nm) | 88% (240 min) | 0.70 h−1 |
| TiO2 (40 wt%) /ZSM-5120 | Sol–gel method | 15 mg L−1 (500 mL) | 1.0 g L−1 | 6.8 | UV lamp (14 W), 254 nm, 0.97 mW cm−2 | 96.6% (180 min) | — |
| 1.1% ZnO/polystyrene122 | Solvent casting method | 12.5 mg L−1 | 25 g (50 mL) | 6.5 | UV light (13 W m−2) | 77% (240 min) | — |
| Bi modified titanate124 | Hydrothermal method | 0.7 mg L−1 | 1.0 g L−1 | 7 | Metal halogen lamp with UV and IR cut-off filters | 88% (180 min) | 12.61 × 10−3 min−1 |
| BaTiO3/TiO2 ratio of 3:1 (w/w)125 |
Grounding followed by drying and calcination | 5 mg L−1 | 1 g L−1 | 7 | Xenon lamp: 500 W (200 nm < λ < 800 nm) | 95% (240 min) | 0.5529 h−1 |
| Ag/AgCl@ZiF-8126 | Stirring method | 1 mg L−1 | 0.5 g L−1 | 5 | Metal halogen lamp (500 W) combined with UV and IR cut-off wave length | 99% (90 min) | 0.0579 min−1 |
| g-C3N4127 | Thermal oxidation etching process | 5 mg L−1 | 0.1 g (250 mL) | — | Solar irradiation (source) | 99% (60 min) | — |
| Exfoliated g-C3N4128 | Thermal synthesis | 25 g dm−3 | 0.9 g | — | UVA lamp: 368 nm, 0.96 mW cm−2 | 41% (120 min) | 4.5 × 10−3 Mol dm−3 min−1 |
| Exfoliated g-C3N4128 | Thermal synthesis | 25 g dm−3 | 0.9 g | — | Visible light lamp (446 nm), an intensity of 8.5 mW cm−2 | 54% (120 min) | — |
| 0.05% ZnO/Ph–g-C3N4129 | Single-step calcination and combustion process | 20 mg L−1 | 1 g L−1 | — | Halogen lamp (500 W) | 90.8% (120 min) | — |
| α-Fe2O3/g-C3N4130 | Dispersion under sonication followed by heating in air | 2.0 mg L−1 (H2O2: 5.0 mM) | 0.1 g L−1 | 5.0 | Xenon lamp: 35.0 W (λ > 420 nm) | 100% (25 min) | 0.134 min−1 |
| g-C3N4(75%)/UiO-66-NH2131 | Hydrothermal method | 5 mg L−1 (350 mL) | 0.5 g L−1 | 4–5 | 9 W lamps, 365 nm | 100% (120 min) | 2.0 h−1 |
| Bi2O3/MnO220 | Room temperature solution phase synthesis | 5 mg L−1 | 1 g L−1 | 6.8 | 200 W LED strip (λ > 420 nm) | 94.3% (120 min) | 0.0202 min−1 |
| TiO2@rGO prepared by using 3 wt% GO133 | Sol–gel method | 50 mg L−1 (25 mL) | 2.0 g L−1 | 5.4 | LED lamps (18 no.) and each of l3 W, λ: 365 nm, 95 μW cm−2 | 100% (50 min) | 0.061 min−1 |
| Calcined ZnFe-LDH/rGO (using 30 mg of GO)135 | Hydrothermal calcined method (using 30 mg GO) | 5 mg L−1 (50 mL) | 25 mg | — | Xenon lamp (500 W), 300 nm cut-off filter | 95% (420 min) | 0.00737 min−1 |
| 5% graphene/TiO2 nanotubes136 | Hydrothermal | 5 mg L−1 (500 mL) | 0.1 g L−1 | 7 | UV lamp (14 W), 254 nm | 96% (180 min) | 00197 min−1 |
| Coal fly ash (CFA)/GO/WO3 NRs137 | Hydrothermal | 5 mg L−1 | 100 mg | — | 250 HW lamp | 86% (180 min) | −0.0116 min−1 |
| Ni@TiO2:W138 | Hydrothermal treatment immobilizing | 25 mg L−1 | 30 mg (100 mL) | 7 | Solar natural irradiation (754 ± 13 W m−2) | 100% (180 min) | 10.7 × 10−3 min−1 |
| Flexible graphene/Ni@TiO2:W138 | TiNiW grown on the surface of graphene | 25 mg L−1 | 30 mg (100 mL) | 7 | Solar natural irradiation (754 ± 13 W m−2) | 86% (180 min) | 8.8 × 10−3 min−1 |
| 1% rGO/BiOBr core/shell139 | Hydrothermal | 5 mg L−1 (30 mL) | — | 5.5–9.5 | Hg/xenon lamp (visible light irradiated with 400 nm cut-off filter), 20 mW cm−2 | 93% (105 min) | 0.006 min−1 |
| rGO–Ag/PANI140 | Mixing reduced GO with polyaniline AgNO3 by vitamin C | 25 mg L−1 | 50 mg | 5 | Visible light | 99.6% (100 min) | — |
| Sr@TiO2 with UiO-66-NH2141 | By carrying out growth of UiO-66-NH2 on SrTiO3 | 5 mg L−1 | 250 mg L−1 (150 mL) | — | Xenon lamp: 600 W m−2 (λ cut-off filter: 320 nm) | ∼94% (240 min) | 0.67 h−1 |
| 15 wt%CeO2/IK–g-C3N4142 | Mixing method | 10 mg L−1 (20 mL) | 2.0 g L−1 | 9 | Visible light lamps (8 W), 465 ± 40 nm | 98% (90 min) | 0.0386 min−1 |
| 5% g-C3N4/TiO2/persulfate143 | Ultrasonic mixing | 5 mg L−1 (100 mL) and PS: 2 mM | 0.331 g L−1 | 7 | Xenon lamp (300 W) with 400 nm cut-off filter | 99.3% (30 min) | 0.181 min−1 |
| CdO–ZnO (0.1:0.2 mole ratio)144 |
Homogeneous co-precipitation | 12 ppm | 1 g L−1 | 6.15 | Halogen lamp (500 W) | 96% (160 min) | 0.05 min−1 |
| Magnetic mesoporous carbon146 | In situ chemical co-precipitation method | 20 mg L−1, PMS: 0.6 mM | 0.12 g L−1 | 6 | UVC lamp – Philips (6 W) with 254 nm cut-off filter | 97.4% (40 min) | — |
| TiO2/graphene/g-C3N4 (60:10:30)147 |
Hydrothermal method | 50 mg L−1 | 0.6 g L−1 | 9 | Xenon lamp (SSL irradiation): 300 W, λ cut-off filter: 420 nm | 100% (120 min) | 2.7 × 10−2 min−1 |
3.2 Amoxicillin
Amoxicillin (AMX) is a widely used semi-synthetic β-lactam and broad-spectrum antibiotic in the treatment of different types of infection for treating both human and animal diseases.148 Therefore, it is possible to find traces of this drug or its degradation products in various aquatic environments in the treated discharge from wastewater treatment plants. Its presence in aquatic animals and humans contributes to toxic effects though the aquatic system due to its structure, high polarity, and water solubility. However, amoxicillin in water is not easy to remove by conventional wastewater treatment processes due to its resistance to biodegradation. Hence, it is necessary to conduct a large amount of research on the treatment and removal of amoxicillin from wastewater using a variety of photocatalysts before discharging it into the natural aquatic environment.149–2163.2.1.1 TiO2. Radosavljević et al.149 applied TiO2 in a nanocrystalline form and compared it with commercial TiO2 to study the photocatalytic degradation of amoxicillin using an Osram Ultra-Vitalux® lamp as the light source. Their findings indicated almost complete degradation of AMX after 210 min for catalyst and AMX concentrations of 2 g dm−3 and 100 mg dm−3, respectively. The UV-mediated photocatalytic degradation of amoxicillin was found to be low (27.6%) in the presence of TiO2 (10–25 nm) compared to cephalexin (63.5%) and tetracycline (100%) under optimal conditions.150 Pereira et al.151 used photoreactors and studied the degradation of amoxicillin in aqueous solution (pH: 7.5) by subjecting it to a solar-driven TiO2 (0.5 g L−1) assisted photocatalytic process. According to this, TiO2/solar UV radiation was able to reduce the antibiotic concentration from 40 to 3.1 mg L−1 after 4.6 kJUV of UV accumulated energy per liter of solution.
The degradation of amoxicillin (10 mg L−1) was also examined under UV and visible irradiation (15 min) and found to be nearly 100% for TiO2 and ZnO (both 0.01 g), respectively.152 Amoxicillin (104 mg L−1) in aqueous solution (pH ∼ 5) was completely degraded under TiO2/UVA (365 nm) in 30 min in the presence of H2O2 (100 mg L−1).153 TiO2-catalyzed photodegradation of amoxicillin (10 mg L−1) was found to be ∼100% under UV irradiation of 30 min duration.154 According to Klauson et al.,155 Degussa P25 TiO2 showed about 83% degradation of AMX (pH: 6.0) after 2 h under solar radiation. Moosavi and Tavakoli156 studied amoxicillin degradation in contaminated water using TiO2 in solar photocatalysis, considering variations in pH, catalyst dose and initial concentration of amoxicillin. These studies showed 84.12% degradation of amoxicillin after 240 min under optimum conditions of pH 9.5, catalyst dose of 1.5 g L−1 and initial concentration of amoxicillin of 17 mg L−1 under 240 min of solar irradiation due to a synergistic effect. In addition, several other studies have also been reported using TiO2,157–159 and supported TiO2160 on the photocatalytic remediation of amoxicillin.
3.2.1.2 ZnO and other metal oxides. The effect of operating variables has been studied on the degradation of amoxicillin (104 mg L−1) in aqueous solution driven by a UV/ZnO photocatalyst prepared by a microwave-assisted gel combustion method, which achieved complete degradation corresponding to a zinc oxide concentration of 0.5 g L−1, irradiation time of 180 min and pH 11.161 The photocatalytic reactions followed pseudo-first-order kinetics with a rate constant of 0.018 min−1. In another study, the photocatalytic removal of amoxicillin (and sulfamethoxazole) was achieved in 6 h from aqueous solutions using ZnO nanoparticles irradiated with UVC irradiation.162 Al-zobai et al.163 reported the recovery of 72.3%, 85.3%, and 100% of amoxicillin under optimum conditions using UV/TiO2, UV/ZnO/TiO2 and UV/ZnO.163 Bi2O3/Fe (3 wt%), successfully synthesized by a microwave-assisted precipitation method, exhibited a degradation efficiency of 76.34% and a degradation rate for amoxicillin of 0.0079 min−1.164
The effect of AMX concentration, WO3 dosage, and pH was studied for the photocatalytic degradation of amoxicillin by solar-driven simulated irradiation.165 These findings revealed the complete removal of AMX under optimal conditions corresponding to an initial AMX concentration of 1.0 μM, catalyst dosage of 0.104 g L−1 and pH 4. Sol–gel-synthesized nano-NiO under optimal conditions efficiently degraded 96% of amoxicillin from pharmaceutical wastewater.166 The photodegradation process was found to follow pseudo-first-order kinetics (k: 0.084 min−1) for an amoxicillin concentration of 25 mg L−1.
A Ce3+-doped TiO2 thin film, prepared using polyethylene glycol as the templating agent, acting as a catalyst succeeded in the removal of amoxicillin under UVA radiation from aqueous solution (pH 6.0).171 It was noted that the removal of amoxicillin increased from 28% to 67% (2 h) in the presence of Ce3+@TiO2, corresponding to a decrease in the initial concentration of amoxicillin from 15.0 to 0.5 mg L−1, respectively. The Ce3+@TiO2 thin film retained its photocatalytic stability more or less unaltered even after 6 cycles. It was suggested that cerium ions trapped the electron and hole pairs in the TiO2 catalyst to form hydroxyl and peroxy radicals that play a significant role in the degradation of amoxicillin. Mn-doped Cu2O nanoparticles synthesized using aloe vera leaf extract exhibited 92% degradation of amoxicillin under sunlight irradiation at pH 9, an initial concentration of amoxicillin of 15 mg L−1, and a photocatalyst dosage of 1 g L−1.172 In all likelihood, Mn doping in Cu2O delays rapid recombination by trapping the photogenerated electrons, accounting for its enhanced photocatalytic performance in amoxicillin degradation.
3.2.4.1 TiO2 nanocomposites. Bergamonti et al.176 studied the photocatalytic activity of TiO2 immobilized on a chitosan scaffold under UV/vis irradiation to examine the degradation of amoxicillin in wastewater under UV-vis irradiation. These findings showed high photodegradation efficiency compared to the direct photolysis of amoxicillin. A TiO2/PAC (powdered activated carbon) mixture in suspension removed 95% amoxicillin in 60 min owing to significant synergy.177 TiO2/zeolite-photocatalysis also presented a feasible methodology for the degradation of the AMX under UV radiation.178 It was noted that a material obtained by acid–alkaline pretreatment and calcination (300 °C) showed the best performance due to its favorable surface structure and TiO2 content.
Pastrana-Martínez and others179 prepared nanodiamond (ND) composites of pristine TiO2 (NDDT) to study its oxidative degradation of amoxicillin soluble in water under near-UV/vis irradiation. Their findings clearly revealed the complete degradation of amoxicillin by NDDT, owing to the generation of holes and better charge separation. In addition, specific surface area, functional groups introduced in ND and the porosity of NDDT compared to bare TiO2 also play an important role in the photocatalytic degradation efficiency of amoxicillin. Li and coworkers180 investigated the effect of Fe3O4 loading in TiO2–Fe3O4 composites, H2O2 concentration, different initial pH and light intensity on the degradation of amoxicillin. The separation showed the following trend towards the degradation of amoxicillin in 100 min under optimum conditions (amoxicillin: 30 mg L−1, UV irradiation: 200 W, [H2O2]: 4.24 mM, pH: 2.84): TiO2/15 wt% Fe3O4 + H2O2 > TiO2/20 wt% Fe3O4 + H2O2 > TiO2/25 wt% Fe3O4 + H2O2 TiO2/10 wt% Fe3O4 + H2O2 > TiO2 + H2O2. It was noted that the presence of H2O2 contributed to oxidation in a photo-Fenton process while the choice of the optimum pH of 2.84 is guided by the scrambling of Fe3+ between OH and H2O2. Furthermore, the reaction rate below 200 W increased remarkably with increasing light intensity due to the generation of electrons and holes. As a consequence, maximum AMX removal efficiency (∼88% in 100 min) was achieved for 0.4 g L−1 of TiO2/15 wt% Fe3O4/H2O2 (6 mM) under optimum conditions corresponding to an initial concentration of amoxicillin of 30 mg L−1 and catalyst loading of 0.4 g L−1. The highest performance for amoxicillin in the presence of TiO2/15 wt% Fe3O4 could be ascribed to the generation of more active ·OH. The proposed mechanism involved the rapid transfer of excited electrons from TiO2 to Fe3O4, reducing h+/e− pair recombination and providing an additional ·OH generation pathway for amoxicillin degradation.
dela Rosa et al.181 studied the degradation and kinetic profiles of amoxicillin using solar/TiO2/Fe2O3/persulfate and the corresponding findings are displayed in Fig. 8(A) and (B), respectively. It was observed that AMX degradation was reduced from 70% (no scavengers) to 39%, 54% and 64% (50 min) in the presence of methanol (MeOH), tert-butanol (t-BuOH) and 1,4-benzoquinone, respectively. Based on the overall findings, arrangements of reactive oxygen species (ROS) for AMX degradation by a solar/TiO2–Fe2O3/PS process follows the order: h+ > SO4·− > HO· > O2·−. The overall amoxicillin degradation can be accounted for by considering the suppression of recombination of charges by the presence of PS as well as the generation of ROS at h+.
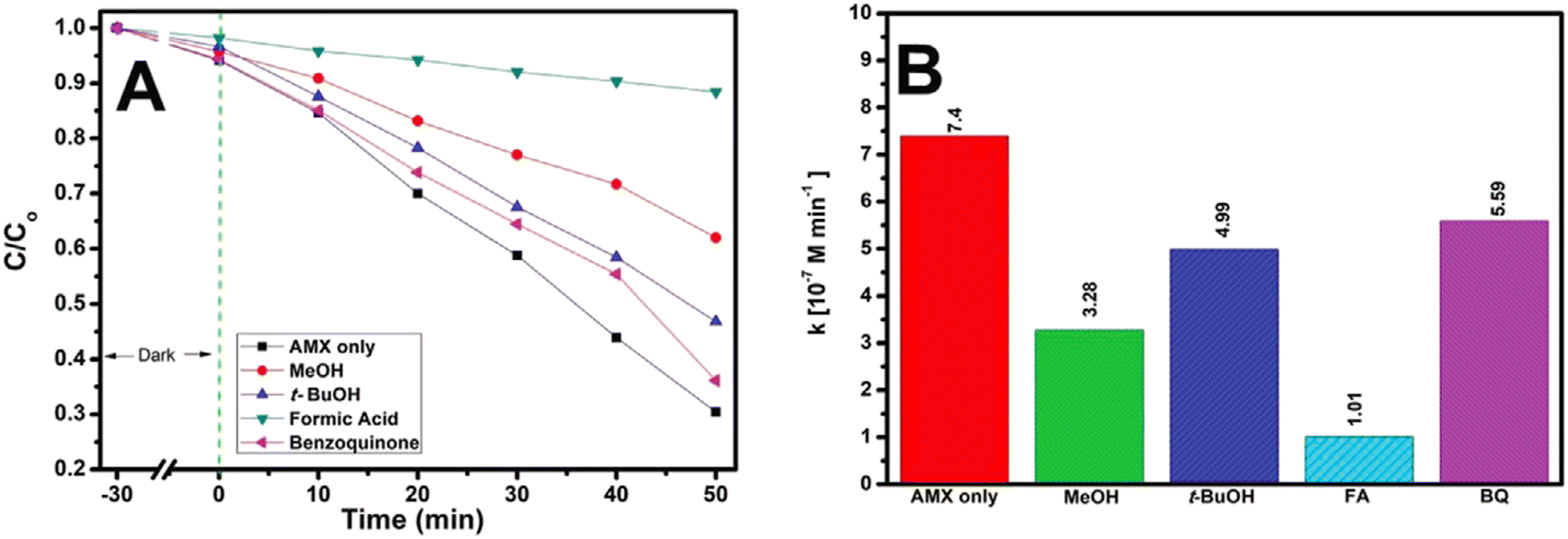 | ||
| Fig. 8 (A) Photocatalytic degradation of AMX under solar irradiation in the presence of scavengers; and (B) corresponding zero-order rate constants (kobs) (experimental conditions: [AMX] = 50 μm; initial pH = 4; [PS] = 334 μm, treatment time, t = 50 min). Reproduced from ref. 181 with permission from Wiley (2021). | ||
TiO2 immobilized on activated carbon fabricated by a high-temperature impregnation method degraded amoxicillin, diclofenac and paracetamol by 100% (120 min), 85% (180 min) and 70% (180 min) in aqueous solution under solar irradiation.182 Li et al.183 reported the photocatalytic degradation of amoxicillin using TiO2 nanoparticles submerged on a porous ceramic membrane. TiO2 immobilized on sand has been used as a catalyst in a solar photocatalytic process for the removal of amoxicillin residues from aqueous solution.184 These findings showed 93.12% degradation of amoxicillin under the optimal conditions of pH 5, 7 5 mg L−1 of TiO2, 400 mg L−1 of H2O2, and 10 mg L−1 of AMX concentration at 150 min irradiation time. Furthermore, the removal of undesirable compounds follows a pseudo-second-order kinetic model. In addition, TiO2/Mg–Al-layered double hydroxide (LDH),185 Ag-ion-exchanged zeolite/TiO2,186 Fe-8-hydroxyquinoline-7-carboxylic/TiO2 flowers187 and TiO2–SiO2188 composites have also been used to remove amoxicillin from aqueous solutions.
3.2.4.2 ZnO-based nanocomposites. Thi et al.189 observed the enhanced photocatalytic activity of ZnO–TiO2 (10%) for the ozonation and perozone degradation of amoxicillin in water under visible-light irradiation. The visible-light-driven MIL-53(Al)/ZnO hierarchical photocatalyst produced 100% removal of amoxicillin corresponding to an initial amoxicillin concentration of 10 mg L−1, solution pH 4.5 and catalyst dose of 1.0 g L−1.190 Recently, Liu and others191 reported significantly high degradation efficiency of amoxicillin (93.10%) in wastewater using Bi2WO6/nano-ZnO (1
:3) after 120 min in comparison to ZnO and Bi2WO6. It is anticipated that the reduction in band gap energy of Bi2WO6/nano-ZnO (1:3) could prevent the recombination of photogenerated charge carriers.
3.2.5.1 g-C3N4-based nanocomposites. Carbon-rich g-C3N4 nanosheet samples were prepared by a combination of 20 g of urea and 60 mg, 90 mg and 120 mg of 1,3,5-cyclohexanetriol as starting materials (referred to as C-CN60, C-CN90 and C-CN120, respectively).192 They included plenty of carbon-rich functionalities and were examined for their photocatalytic activity for amoxicillin degradation under solar and visible light in the aqueous phase and the results are displayed in Fig. 9. The degradation of amoxicillin was found to follow the order: C-CN90 > C-CN60 > C-CN120 > g-C3N4. Photocatalyst C-CN90 showed nearly complete photocatalytic degradation of amoxicillin under solar light and visible light after 150 and 300 minutes, respectively. This has been attributed to the interaction between g-C3N4 and graphited conjugated construction narrowing the band gap and separating photogenerated electron–hole pairs.
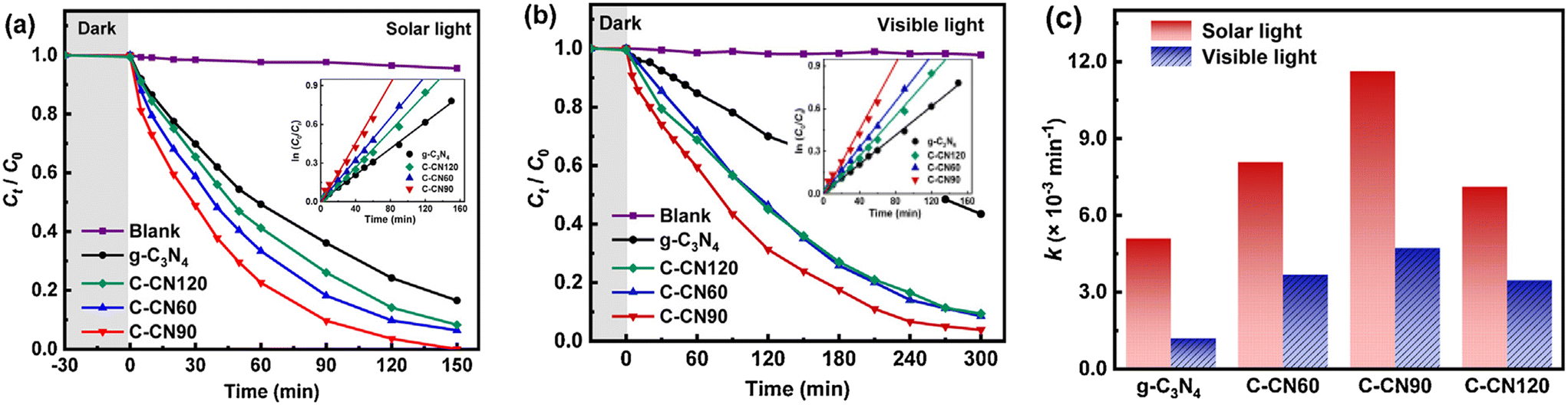 | ||
| Fig. 9 Photocatalytic degradation kinetics of AMX by the synthesized materials under (a) simulated solar light, (b) visible light, and (c) AMX degradation rate constants under solar and visible light. Reproduced from ref. 192 with permission from Elsevier (2021). | ||
Silva et al.193 synthesized metal-free polymeric carbon nitrides using melamine (CN-M), thiourea (CN-T) and their 1:1 mixture (CN-1M:1T) as precursors in a Teflon reactor comprising 25 mL of deionized water followed by heating of the products at 550 °C for 30 min. Their investigations revealed 100% degradation of AMX for CN-T followed by CN-M (65%) and CN-1M:1T (56%) after 48 h of visible-light exposure. The superior performance of CN-T was found to be directly related to the greater number of defects present in its structure, that can help in the separation of electron–hole pairs. An Ag/g-C3N4/ZnO nanorod (0.08 g L−1) nanocomposite has also acted as an efficient photocatalyst in the photocatalytic degradation of amoxicillin of high concentration (40 mg L−1) irradiated by visible light.194 V2O5-nanodot-decorated laminar C3N4 degraded amoxicillin under solar light, exhibiting 91.3% removal efficiency.195 It is suggested that such a V2O5/C3N4 S-scheme structure provides an internal electron channel at the interface and maintains the active sites with high potentials for the photodegradation of amoxicillin. Mesoporous g-C3N4/persulfate exhibited 99% degradation of AMX under visible-light irradiation within 60 min at pH 7 due to a synergistic effect.196 Graphitic-carbon–CuO–ZnO nanocomposites exhibited 49% efficiency in the photocatalytic degradation of amoxicillin under direct sunlight and followed pseudo-first-order kinetics.197 α-Fe2O3/g-C3N4,198 mesoporous g-C3N4,199 and CQDs/K2Ti6O13200 photocatalysts have also been reported in the photocatalytic degradation of amoxicillin.
3.2.5.2 Graphene-based nanocomposites. Changotra et al.201 prepared nanocomposites of varying FeS2 to GO weight to study the degradation of amoxicillin as a function of different parameters, such as solution pH value, optimal doses of H2O2 and catalyst, stability of the catalyst, and leaching effect of the catalyst, under optimal solar-Fenton treatment. These investigations showed the complete degradation of amoxicillin (∼99%) by FeS2/GO (4
:3) in 180 min owing to the synergistic coupling of FeS2 and GO under the optimal conditions of [amoxicillin]init conc 25 mg L−1, [FeS/GO] 0.75 g L−1, 12 mM [H2O2] and pH 5. Further, HO· acted as dominant reactive species and no toxic secondary products were produced in the amoxicillin degradation. The photocatalytic degradation efficiency for amoxicillin by TiO2 nanoparticles loaded on graphene oxide under UV light was found to be >99% at pH 6, catalyst dose of 0.4 g L−1, amoxicillin concentration of 50 mg L−1 and intensity of 36 W (Fig. 10(a–d)).202
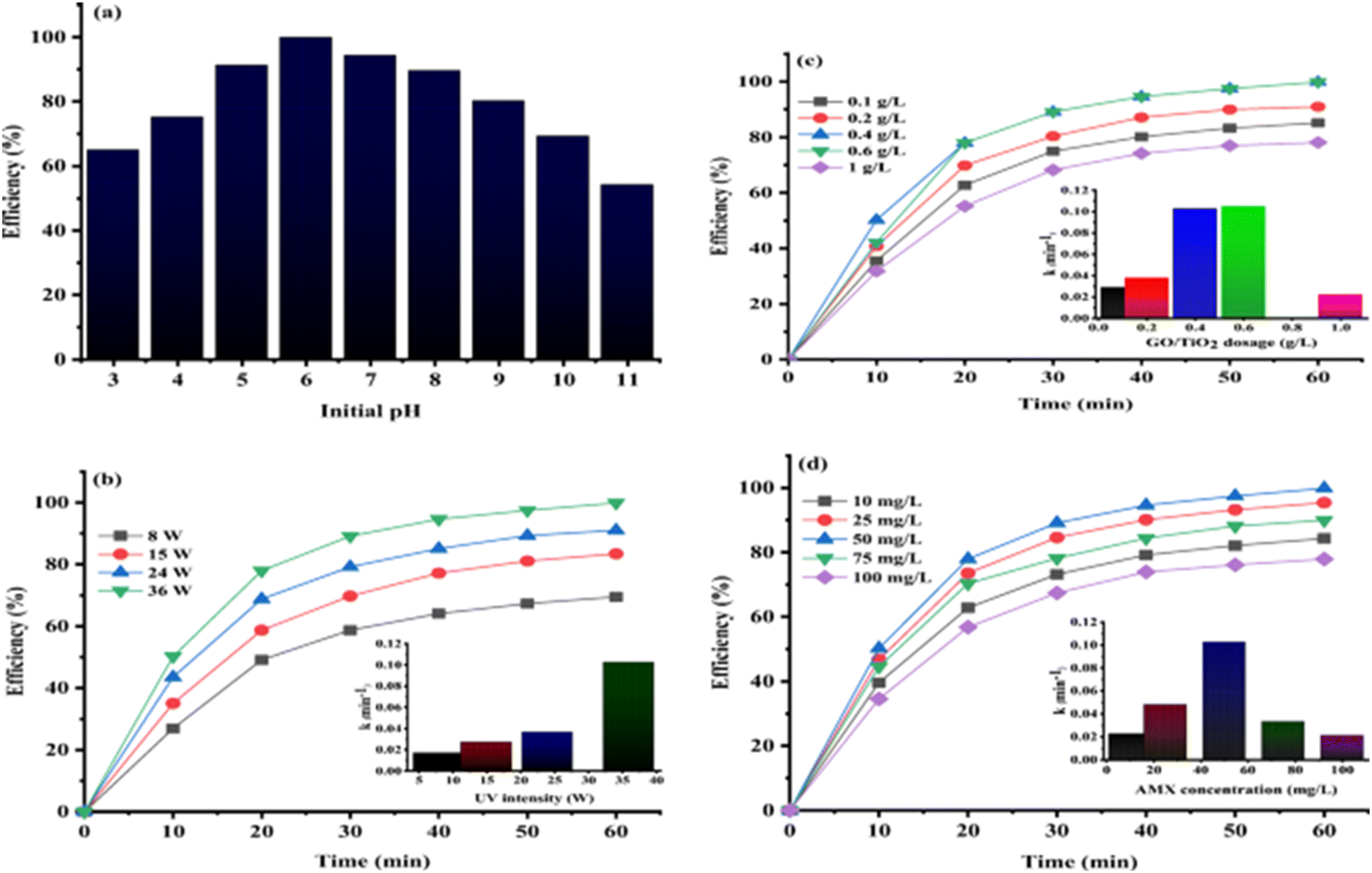 | ||
| Fig. 10 The effect of different operational factors on AMX photocatalytic degradation and kinetic constant (a–d). Reproduced from ref. 202 with permission from Springer (2021). | ||
According to Song and others,203 KBrO3 added to graphene–TiO2 nanotubes achieved 100% photodegradation of amoxicillin under UVA-light irradiation. It is suggested that KBrO3 prevents electron–hole recombination and has a direct role as an oxidant in the degradation of amoxicillin. A visible-light-driven MIL-68(In)–NH2/graphene oxide (GO) composite photocatalyst (0.6 g L−1) exhibited 93% degradation (120 min) of amoxicillin in aqueous solution of pH 5 compared to pure MIL-68(In)–NH2.204 It is suggested that MIL-68(In)–NH2/GO acted as an electron transporter for suppressing photogenerated carrier recombination and also acted as a sensitizer for enhancing visible-light absorption. The proposed mechanism suggested that h+ and ·O2− are active species. In another study, a 2D/3D g-C3N4/BiVO4 hybrid photocatalyst decorated with rGO (1.2 wt%) degraded amoxicillin by 91.9% under optimized conditions with visible-light illumination.205
El-Fawal et al.209 observed the better performance of an AgFeO2–graphene/Cu2(BTC)3 MOF heterojunction compared to AgFeO2/graphene and AgFeO2/Cu2(BTC)3 binary photocatalysts in achieving about 97% removal of amoxicillin and diclofenac after 150 min under sunlight irradiation, which exhibited excellent stability up to four cycles. Based on these findings, a direct Z-scheme heterojunction mechanism has been proposed for the separation of photo-induced charge carriers at the interface of these photocatalysts. The enhanced photocatalytic activity of the tertiary heterojunction photocatalyst was mainly attributed to its superiority for light absorption (up to 650 nm) with high photostability, accelerated e−/h+ pair separation and increased lifetime of photogenerated charges. The heterojunction p-ZnO/CuO (50:50 wt%) assisted photocatalytic process removed amoxicillin (initial concentration: 50 mg L−1) from water (pH: 11) almost completely on exposure to solar irradiation for 4 h.210 The degradation of amoxicillin followed pseudo-first-order kinetics (k: 9.95 × 10−3 min−1).
Gao et al.211 deposited Ag nanoparticles on the surface of a TiO2/mesoporous g-C3N4 heterojunction and used it in the photocatalytic removal of amoxicillin under visible light. A photocatalyst fabricated in this manner achieved higher degradation efficiency for amoxicillin than a TiO2/mesoporous-g-C3N4 heterojunction, mesoporous-C3N4, or bulk-g-C3N4. Such photoactivity of an Ag/TiO2/M–g-C3N4 catalyst has been assigned to the synergistic effect accounting for the effective transfer of electrons and inhibition of electron–hole recombination. The effectiveness of this photocatalyst was also tested for the removal of amoxicillin in real situations. A WO3/Ag3VO4 Z-scheme heterojunction with enhanced separation efficiency of electron–hole and surface area was deposited on rGO and used as a photocatalyst in the degradation of amoxicillin under irradiation by visible light.212 The amoxicillin photocatalytic degradation followed the following order on irradiating it with visible light: Ag3VO4/WO3/r-GO (∼96%) > Ag3VO4/WO3 (∼37%) > WO3 > Ag3VO4 (∼32%). It is suggested that the presence of rGO, by increasing the surface area in Ag3VO4/WO3/rGO, facilitates amoxicillin adsorption and electron transfer for charge separation of Ag3VO4/WO3.
Investigations have also been made on the photodegradation of amoxicillin via a magnetic TiO2–graphene oxide–Fe3O4 composite213 and Pd nanoparticles anchored to anatase TiO2.214 Hajipour et al.215 fabricated heterojunctions of TiO2/CuO, adopting the surface modification of TiO2 with CuO, and investigated its application in the photocatalytic degradation of amoxicillin in wastewater. It should be noted that TiO2/CuO (7.5%) showed reduced photo-activity compared to a TiO2/CuO (10%) photocatalyst, which could be attributed to the partial blockage of the active sites in the TiO2 nanoparticles, In another study, a novel nanophotocatalyst of CuO nanoparticles and ZnO nanorods anchored on thermally-exfoliated g-C3N4 nanosheets established the complete removal of amoxicillin corresponding to a catalytic dosage of 0.9 g L−1 and pH 7.0 within 120 min under simulated sunlight illumination.216 Subsequently, a double Z-scheme mechanism as well as a tentative pathway were proposed in detail.
Table 3 records the performance data of different photocatalysts on the removal of amoxicillin from wastewater.
| Photocatalyst | Method of preparation | AMX | Catalyst dose | pH | Light source details | Degradation (time) | Rate constant |
|---|---|---|---|---|---|---|---|
| TiO2 nanoparticles (US3490)150 | Commercial | 15 mg L−1 | 2 g L−1 | 5 | UV lamp (18 W) | 27.6% (15 min) | — |
| ZnO nanoparticles (US3590)150 | Commercial | 15 mg L−1 | 2 g L−1 | 5 | UV lamp (18 W) | 48.6% (15 min) | — |
| GO–Fe3O4150 | Ultrasonic mixing followed by reflexing | 15 mg L−1 | 2 g L−1 | — | Lamp (UV): 18 W | 87.1% (15 min) | — |
| TiO2 (P25 Degussa)152 | Commercial | 10 mg L−1 (20 mL) | 0.01 g | — | UV | 100% (15 min) | 4.33 × 10−1 min−1 |
| TiO2 (P25 Degussa)152 | Commercial | 10 mg L−1 (20 mL) | 0.01 g | — | Visible | 99% (15 min) | — |
| ZnO (Hoechst)152 | Commercial | 10 mg L−1 (20 mL) | 0.01 g | — | UV | 98% (15 min) | 3.03 × 10−1 min−1 |
| ZnO (Hoechst)152 | Commercial | 10 mg L−1 (20 mL) | 0.01 g | — | Visible | 99% (15 min) | — |
| TiO2 (Fluka)153 | Commercial | 104 mg L−1 (500 mL) | 1.0 g L−1 | 11 | UV lamp: 6 W (365 nm) | ∼71% (300 min) | 0.007 min−1 |
| TiO2 (H2O2: 100 m L−1)153 | Commercial | 104 mg L−1 (500 mL) | 1.0 g L−1 | 5 | UV lamp: 6 W (365 nm) | 100% (20 min) | — |
| TiO2 (P25 Degussa)154 | Commercial | 0.01 g | 10 mg L−1 (20 mL) | — | UV lamp | 100% (30 min) | 0.433 min−1 |
| TiO2 (Degussa P25)155 | Commercial | 25 mg L−1 | 1 g L−1, slurry | 6 | Solar light (16 mW cm−2) | ∼83% (120 min) | — |
| Carbon (32%) doped TiO2 (Degussa P25)155 | Commercial | 25 mg L−1 | 1 g L−1, slurry | 6 | Solar light (16 mW cm−2) | ∼73% (120 min) | — |
| Fe (2.2%) doped TiO2 (Degussa P25)155 | Commercial | 25 mg L−1 | 1 g L−1, slurry | 6 | Solar light (16 mW cm−2) | ∼75% (120 min) | — |
| TiO2 (sigma Aldrich)156 | Commercial | 1.5 g L−1 | 17 mg L−1 | 9.5 | Solar irradiation | 84.12% (240 min) | — |
| ZnO162 | Microwave assisted gel combustion method | 10 mg L−1 (200 mL) | 0.25 g L−1 | 10 | UVC lamp (30 W) | 100% (5 h) | 0.014 min−1 |
| WO3 (sigma Aldrich)165 | Commercial | 1.0 μM | 0.104 g L−1 | 4 | Xenon lamp (300 W) | 99.99% (180 min) | 2.908 × 10−2 min−1 |
| NiO166 | Sol–gel method | 25 mg L−1 | 0.2 g L−1 | — | Low mercury lamp (15 W) | ∼96% (120 min) | 0.084 min−1 |
| Cu (4.54 mg g−1) doped TiO2169 | Photoreduction method | 10 mg L−1 | 40 mg | 6 | Wolfram lamp as visible light source | ∼90% (24 h) | 4 × 10−4 min−1 |
| Fe3+ doped TiO2170 | Sol–gel method | 10 mg L−1 | 90 mg L−1 | 11 | UV lamp of C type, 125 W, 247 nm | Synthetic water: 99.14% (120 min), pharmaceutical water: 88.92% (120 min) | — |
| Mn-doped Cu2O172 | Green synthesis | 15 mg L−1 (100 mL) | 1 g L−1 | 9 | Sunlight irradiation (900 W m−2) | 92% (180 min) | 0.073 min−1 |
| La–Ce (1 wt%) TiO2173 | Sonochemical-assisted synthesis | 10 mg L−1 (100 mL) | Appropriate amount | — | Halogen lamp (500 W) | 75.7% (?) | — |
| Ag/ZnO175 | Conventional method | 5 mg L−1 | 0.15 g L−1 | 5 | UVA, 365 nm | 93.76% (120 min) | 0.073 min−1 |
| TiO2/chitosan176 | 3D printing | 0.1 mM (40 mL) | 15 layers (AMX/TiO2 molar ratio: 1/100) | 6.7 | Medium-pressure Hg vapour water jacket lamp (UV-vis), 125 W, 300–800 nm, 3.5 mW cm−2 | ∼95% (2 h) | 0.57 × 10−2 min−1 |
| TiO2/PAC177 | Suspension method | 15 mg L−1 | TiO2: 1 g L−1, PAC: 0.1 g L−1 | 6.5 | UV-vis (540 W m−2) | 90–97% (60 min) | 0.034 min−1 |
| TiO2/zeolite178 | Modified reported method | 30 mg L−1 (100 mL) | 2 g L−1 | 4.05 | Medium-pressure Hg lamp (47 W) with λ ≤ 290 nm cut-off | 88% (240 min) | — |
| Functionalized nanodiamond-TiO2179 | Liquid phase deposition | 0.1 mM (7.5 mL) | 1 g L−1 | — | Medium-pressure hg vapor lamp | 100% (60 min) | 83.3 × 10−3 min−1 |
| TiO2-15 wt% Fe3O4180 | Hydrothermal | 30 mg L−1, (H2O2: 24 mM) | 0.4 g L−1 | 2.84 | Low-pressure mercury vapor lamp: 100 W, 1200 mW cm−2 | ∼88% (100 min) | — |
| TiO2@α-Fe2O3 film (PS: 334 μm)181 | Spin coating | 50 μm | — | 4 | Xenon lamp (450 W) | 70% (50 min) | 7.4 × 10−7 M min−1 |
| TiO2 immobilized on activated carbon182 | High-temperature impregnation method | 50 mg L−1 (4 L) | 1.2 g L−1 | 10 | Solar irradiation | 100% (120 min) | 0.037 min−1 |
| TiO2–sand184 | Sol–gel dip-coating | 10 mg L−1, H2O2, 400 mg L−1 | 75 mg L−1 | 5 | Solar irradiation | 93.12% (150 min) | 0.0175 min−1 |
| TiO2/Mg–Fe-LDH185 | Direct co-precipitation method | 30 mg L−1 | 2 g L−1 | 11 | UVA light (λmax: 365 nm) | ∼100% (240 min) | — |
| TiO2/Mg–Al-LDH185 | Direct co-precipitation method | 30 mg L−1 | 2 g L−1 | 5.5 | UVA light (λmax: 365 nm) | ∼95% (240 min) | — |
| Ag/zeolite/TiO2186 | Liquid ion-exchange method | One g L−1 (15 mL) | 0.01 g | 6.7 | High-pressure Hg lamp (400 W), 120 mW cm−2 | ∼25% (75 min) | — |
| TiO2(80%)–SiO2(20%)188 | Sol–gel method | 20 mg L−1 (100 mL) | 4 g L−1 | 5 | Hg lamp – UVA (15 W), 365 nm | 88% (150 min) | 0.0014 min−1 |
| MIL-53 (Al)/ZnO190 | Hydrothermal/chemical conditions followed | 10 mg L−1 | 1.0 g L−1 | 4.5 | Metal halide lamp: 400 W, 510 nm | 100% (60 min) | — |
| g-C3N4193 | Heating of aq. Thiourea in Teflon reactor | 30 mg | 50 mg L−1 (10 mL) | pH ∼ 6 | Visible light: 150 W, 16 mW cm−2 | 100% (48 h) | 0.088 h−1 |
| Ag/g-C3N4/ZnO nanorods194 | Dispersion method | 40 mg L−1 | 0.08 g L−1 (60 mL) | — | Solar simulator lamp: 300 W (λ ≥ 420 nm) | 41.36% (180 min) | 0.01017 min−1 |
| V2O5/C3N4195 | Heating powdered NH4VO3/g-C3N4 mixture | 20 mg L−1 | 0.5 g L−1 | 7 | Simulated sunlight | ∼91% (120 min) | 0.0268 min−1 |
| α-Fe2O3 (5%)/g-C3N4198 | Solution method | 20 mg L−1 | 0.02 g (60 mL) | Neutral | Solar simulator (300 W) with cut-off filter (λ > 420 nm) | 46% (180 min) | 40.20 × 10−4 min−1 |
| Mesoporous g-C3N4199 | Template-free method | 2 mg L−1 | 100 g L−1 (100 mL) | 9 | Xenon lamp: 300 W (λ > 420 nm) | 90% (60 min) | 0.036 min−1 |
| CQDs modified K2Ti6O13 nanotubes200 | Hydrothermal method combined with calcination | 1 mg L−1 (50 mL) | 0.2 g L−1 | 6 | Light-emitting diode, 10 mW cm−2, 365 nm | 100% (90 min) | 0.0495 min−1 |
| GO/TiO2202 | Chemical hydrothermal method | 50 mg L−1 (100 mL) | 0.4 g L−1 | 6 | UV light (36 W) | 99.84% (60 min) | 0.105 min−1 |
| Graphene@TiO2 nanotube/KBrO3 (0.20 g L−1)203 | Reaction under autoclave | 5 mg L−1 | — | — | Light: UVA lamp: 19 W, λ = 369 nm | 96.94% (180 min) | 0.0186 min−1 |
| MIL-68(In)–NH2/GrO204 | Dispersion method | 20 ppm (200 mL) | 0.6 g L−1 | 5 | Xenon lamp (300 W) with 420 nm cut-off filter | 93% (120 min) | 0.0187 min−1 |
| 1.2 wt% rGO@g-C3N4/BiVO4205 | Wet impregnation method | 10 mg L−1 (100 mL) | 0.1 g (100 mL) | — | Halogen lamp (500 W) | 91.9% (180 min) | 0.0023 min−1 |
| InVO4/Ag/g-C3N4206 | Hydrothermal | 10 ppm | 0.5 g L−1 | — | Visible light (30 W bulb) | >99% (420 min) | — |
| CuI/FePO4207 | Reflux-assisted co-precipitation technique | 20 mg L−1 (50 mL) | 50 mg | — | Visible light (400 W) | 90% (120 min) | — |
| Mesoporous SnO2/g-C3N4208 | Green modified technique | 10 ppm (40 mL) | 10 mg | — | Xenon lamp: 300 W with a cut-off filter (λ > 400 nm) | 92.1% (80 min) | — |
| AgFeO2–graphene/Cu2(BTC)3 MOF209 | In situ solvothermal impregnation | 5 mg L−1 | 5 g L−1 (50 mL) | 8 | Halogen lamp 500 W, 420–600 nm | 97% (150 min) | (6.4–8.7) × 10−2 min−1 |
| p-CuO/n-ZnO (50:50 wt%)210 | Chemical route | 50 mg L−1 | 0.5 g L−1 | 11 | Sunlight (109 mW cm−2) | >87% (240 min) | 9.95 × 10−3 min−1 |
| 1.94 wt% Ag/TiO2/mesoporous g-C3N4211 | Photodeposition means | 5 ppm (0.1 L) | 0.1 g | — | Xe lamp: 300 W (λ > 420 nm) | 99% (60 min) | 0.0614 min−1 |
| WO3/Ag3VO4/rGO212 | Multiple steps | 20 ppm | 0.5 g L−1 | — | LED lamp (220 V, 30 W) | ∼96% (420 min) | — |
| CuO and ZnO co-anchored on g-C3N4216 | Via isoelectric point-mediated annealing | 60 mg L−1 | 0.9 g L−1 | 7.0 | Xenon lamp (250 W) simulated sunlight | 100% (120 min) | 0.0269 min−1 |
3.3 Sulfamethoxazole
Sulfamethoxazole is used to treat a wide variety of bacterial infections, including those of the urinary, respiratory, and gastrointestinal tracts.217 However, it has been frequently detected in wastewater and surface water in aquatic environments due to its extensive consumption, excretion and disposal. Therefore, several investigations have been made by many researchers focusing on the biodegradation of sulfamethoxazole during wastewater treatment following photocatalytic degradation of sulfamethoxazole in water using a variety of photocatalysts.218–2913.3.1.1 TiO2. The photodegradation of sulfonamides has been studied in the UV/TiO2 system to study the effects of pH and salinity on sulfamethoxazole concentration and total organic carbon (TOC) during the removal of sulfonamides in a UV/TiO2 system.219 The photodegradation and mineralization rates of sulfonamides in the UV/TiO2 system satisfied pseudo-first-order kinetics. A TiO2 suspension has been used as a catalyst in a sunset solar simulator to examine the degradation of sulfamethoxazole in real municipal wastewater treatment plant effluent.220 It was inferred that hydrogen peroxide can be highly recommended for working with TiO2 at low concentrations. The photocatalytic degradation of sulfamethoxazole in surface and drinking water in the absence and presence of UV (265 nm) involving TiO2 nanoparticles after 60 minutes follow the order: UV (∼100%) > anatase TiO2 (∼92%) > rutile and commercial TiO2 (∼90%).221 The effects of different UV-LED (UVA, UVB, and UVC) wavelengths were studied in carrying out the photocatalytic decomposition of sulfamethoxazole by TiO2.222 These findings showed complete decomposition within 1 h by TiO2/UVC under the conditions of TiO2: 0.5 g L−1, natural pH, and initial concentration of sulfamethoxazole: 20 mg L−1. Sulfamethoxazole in an aqueous suspension of TiO2 (0.5 g L−1) showed 82% degradation of sulfamethoxazole under UV irradiation.223 In another study, the removal efficiency for the photocatalytic degradation of sulfamethoxazole (20 mg L−1) in aqueous solution (pH: 3) by TiO2 (0.08 g L−1) as a photocatalyst was found to be 96.5% in 60 min under UV light.224 In addition, investigations have also been reported on the degradation of sulfamethoxazole using TiO2,225–227 biochar-supported TiO2228 and immobilized TiO2229–231 as photocatalysts.
3.3.1.2 ZnO. ZnO nanoparticles prepared by a microwave-assisted gel combustion synthesis method showed complete removal of amoxicillin (and sulfamethoxazole) from contaminated water in six hours under UVC irradiation.162 It was inferred that the photocatalytic removal followed the Langmuir–Hinshelwood model in the range of concentration of 5–20 mg L−1. Mirzaei et al.232 achieved ∼97% removal of sulfamethoxazole by a zinc oxide photocatalyst in the presence of fluoride ions (F–ZnO) after 30 min of reaction illuminated by UV irradiation under optimum conditions and followed pseudo-first-order kinetics (k: 0.099 min−1). The hydrothermally synthesized ZnO at 200 °C for 8 h at pH 7.5 reached 84% removal of sulfamethoxazole after 60 min under UVA irradiation.233 In addition, TiO2 and WO3 nanoparticles have also been utilized in the removal of sulfamethoxazole by its photocatalytic degradation.234
TiO2 nanotube arrays (TNAs), TiO2 nanowires on nanotube arrays (TNWs/TNAs), Au-nanoparticle-decorated TNAs, and TNWs/TNAs efficiently degraded sulfamethazine amoxicillin, ampicillin, doxycycline, oxytetracycline, lincomycin, vancomycin and sulfamethoxazole irradiated in water under UV-vis and visible light.174 Among these, the Au–TNWs/TNAs photocatalyst showed the highest activity towards the degradation of all the antibiotics due to synergistic and surface plasmonic effects. In another study, Cu–TiO2 (at low mass ratios of 0.016–0.063 wt%) produced nearly complete degradation of sulfamethoxazole by visible light at pH 5.2 for a 4 mg L−1 initial concentration of sulfamethoxazole.237 Further studies revealed the highly stable photoactivity of Cu–TiO2, as evident from experiments comprising at least 4 cycles. Au, Ag, Cu, Au–Ag and Au–Cu nanoparticles deposited on TiO2 showed increased photocatalytic activity for the photocatalytic degradation of sulfamethoxazole using UVC light.238
F,Pt-co-doped photocatalysts have also been employed in photocatalytic degradation using direct solar light.241 Fluoride ions and Pt in the TiO2 lattice were chosen in order to control the growth of the photocatalytically active anatase phase and to introduce new energy levels between the valence and conductive bands of TiO2 to narrow its band gap. These findings demonstrated degradation of sulfamethoxazole under direct solar light and a solar simulator corresponding to about >93% (90 min) and 58% (360 min), respectively. An iodine (I)–potassium (K)–C3N4 photocatalyst removed nearly 100% of sulfamethoxazole within 45 min under visible-light irradiation.242 N,Cu-co-doped TiO2 decorated on SWCNTs demonstrated total removal of sulfamethoxazole under a pH of 6.0, catalyst dosage of 0.8 g L−1, light intensity of 200 W, US power of 200 W, and initial sulfamethoxazole concentration of 60 mg L−1 in 60 min.243
Ag metal has been used as a co-dopant in P-doped g-C3N4 in order to overcome its poor photocatalytic performance.244 The investigations of Ag (nano)–P-co-doped@g-C3N4 (Ag–P@UCN) as a photocatalyst in visible light followed the trend in the removal of sulfamethoxazole in water: Ag(nano)–P@g-C3N4 (>99%) > P-doped g-C3N4 (68%) > g-C3N4 (47%). The presence of silver nanoparticles Ag(nano)–P@g-C3N4 enhanced light absorption and also acted as photogenerated electron traps, thereby enabling the effective separation of electron and hole pairs. A mechanism has also been proposed for the degradation of sulfamethoxazole in presence of an Ag–P@UCN photocatalyst. In another study, multi-homojunction gradient-nitrogen-doped TiO2 exhibited enhanced performance in the removal of sulfamethoxazole from water compared to pristine TiO2 and non-gradient-doped TiO2 under simulated solar-light irradiation.245 Zammit et al.246 examined the removal of sulfamethoxazole using a cerium-doped zinc oxide (Ce–ZnO) photocatalyst and its comparison with ZnO and benchmark TiO2–P25 in immobilized form on a metallic support and found Ce–ZnO to be most effective under UVA irradiation. In another study,247 Zn (10 wt%)–TiO2/pBC (pretreated biochar) was investigated for the photodegradation of sulfamethoxazole under visible-light irradiation and a comparison with TiO2/pBC and TiO2 after 3 h took the following order: Zn–TiO2/pBC (80.81%) > TiO2/pBC (59.05%) > TiO2 (50.07%).
A composite comprising titania nanoparticles/activated carbon prepared by calcination at 400 °C exhibited much better performance in the removal of sulfamethoxazole from deionized water and seawater.252 Clay–TiO2 nanocomposites prepared via biomass-assisted synthesis showed fast degradation of sulfamethoxazole (>90%) in 30 min under sunlight.253 An LDH–TiO2 (10%) nanocomposite has been developed, keeping in view its possible reusability and regeneration after subjection to UVA radiation, to carry out the degradation of sulfamethoxazole.254 These findings established almost complete degradation after 360 min of UVA irradiation, corresponding to initial sulfamethoxazole concentration of 20 mg L−1, pH 10 and LDH–TiO2 catalyst loading of 50 mg. Recycling and reusability studies were also conducted by dissolving a mass of 50 mg of LDH–TiO2 in sulfamethoxazole (concentration: 20 mg L−1) and pH 10, irradiated for 8 h under UVA. Further investigations revealed no significant variation in sulfamethoxazole degradation efficiency from the first cycle (100%) to the fifth cycle (90.5%).
According to Długosz et al.,255 a floating TiO2-expanded perlite (referred to as EP-TiO2-773: where 773 is the calcination temperature in °C) photocatalyst enhanced the photodegradation of sulfamethoxazole in the aqueous medium over a wide range of pH values on irradiation from the near-UV spectral region. However, the fastest decrease in the concentration of sulfamethoxazole was observed for the system irradiated at pH 10. The degradation of sulfamethoxazole followed pseudo-first-order kinetics in accordance with the Langmuir–Hinshelwood model. Their findings also suggested the key role of hydroxyl radical formation in the degradation of sulfamethoxazole. Noroozi et al.256 synthesized copper doped TiO2 decorated with carbon quantum dots (CQDs) and observed its excellent performance in the degradation of SMX during 60 minute time under optimum conditions corresponding to initial SMX concentration, catalyst dosage, pH, visible light intensity and CQDs ratio in the composites of 20 mg L−1, 0.8 g L−1, 6, 75 Wm−2 and 4 wt% respectively. The photocatalytic degradation of sulfamethoxazole was found to be guided by a pseudo-first-order kinetic model with HO· and O2·− as active species. Poly(ethylene terephthalate)–TiO2,257 BiVO4/SrTiO3,258 CuOx–BiVO4259 and TiO2@CuCo2O4260 were also used for the photocatalytic degradation of sulfamethoxazole.
3.3.5.1 MWCNT-based composites. WO3–MWCNT composites with different amounts of functionalized MWCNTs were prepared by a hydrothermal method (named WT-2, WT-4 and WT-8), and SMX degradation was studied under visible-light irradiation.261Fig. 11(a) shows the highest efficiency of 73.3% within 3 h for WT-8; however, WT-4 with efficiency of 65.2% was preferred due to its better dispersion in water. Further studies on SMX (10 mg L−1) degradation at different catalyst dosages of WT-4 in Fig. 11(b) showed its maximum efficiency (88.5%) corresponding to a loading of 2.00 g L−1. A possible degradation mechanism highlighting the role of O2− and OH· radicals during the photocatalytic process has also been proposed and is displayed in Fig. 11(c). Awfa et al.262 reported ∼60% photodegradation of sulfamethoxazole by magnetic carbon nanotube–TiO2 composites. Martini et al.263 observed almost complete reduction of toxicity using photocatalytic ozonation with H2O2 and Fe/CNT.
 | ||
| Fig. 11 (a) SMX degradation under visible-light irradiation by WO3, WT-2, WT-4 and WT-8. Conditions: catalyst: 0.50 g L−1, SM: 10 mg L−1. (b) SMX degradation by WT-4 at different catalyst dosage (0.25, 0.50, 1.00 and 2.00 g L−1). Conditions: SMX: 10 mg L−1. (c) Schematic illustration of the proposed mechanism for the enhanced degradation of SMX by WO3-CNT composites under visible-light irradiation. Reproduced from ref. 261 with permission from Elsevier (2018). | ||
3.3.5.2 g-C3N4-based composites. An Ag (5%)/P–g-C3N4 composite synthesized by thermal polymerization combined with a photodeposition method completely degraded sulfamethoxazole within 20 min under visible-light irradiation.264 This is attributed to the formation of holes and superoxide radicals acting as dominant active species. In addition, the surface plasmon resonance effect (Ag) and the formation of a Schottky barrier on the Ag/P–g-C3N4 interface could facilitate the enhanced generation of electrons/holes as well as accounting for the recombination of photogenerated electron–hole pairs. A magnetic ZnO@g-C3N4 composite under optimum conditions removed 90.4% of sulfamethoxazole after 60 min.265 In addition, core–shell g-C3N4@ZnO,266 peroxymonosulfate (PMS)/g-C3N4267 and Ag/g-C3N4268 have also been reported in the photocatalytic degradation of sulfamethoxazole.
3.3.5.3 Graphene-based composites. Visible-light-derived rGO–WO3 composites showed 98% removal of sulfamethoxazole within 3 hours.269 In another study, Ag@Ag2O–graphene nanocomposites comprising variable graphene concentrations (1.7, 2.5, and 3.4 wt%) were prepared to study the degradation of sulfamethoxazole under simulated solar light (λ > 280 nm) and visible-light irradiation (λ > 400 nm), including the stability of the photocatalyst and the mechanism of photocatalytic degradation.270 These findings indicated higher activity and comparable stability for the first and second cycles in an Ag@Ag2O–graphene photocatalyst loaded with 2.5 wt% graphene. Possible charge transfer processes were suggested to take place under visible-light irradiation, and holes were major active species for Ag@Ag2O–graphene photocatalytic degradation while Ag0 acted as an electron capture center. Lin et al.271 observed 92% degradation of sulfamethoxazole after subjecting an immobilized TiO2–reduced graphene oxide (rGO) nanocomposite on optical fibers to 180 min of UV irradiation. A visible-light-driven Cu2O/rGO photocatalyst successfully degraded sulfamethoxazole.272
Nawaz et al.273 used graphene oxide and titanium dioxide in combination with sodium alginate to synthesize a reduced graphene oxide–TiO2/sodium alginate (rGOT/SA) aerogel. They observed more than 99% removal of these contaminants taking place within 45–90 min under UVA light, corresponding to an optimal mass ratio of TiO2 nanoparticles with respect to graphene oxide of 2:1 in an rGOT/sodium alginate aerogel in the presence of 1 wt% sodium alginate solution. Zhou et al.274 investigated the photocatalytic decomposition of SMX by Ag3PO4, Ag3PO4–graphene and Ag/Ag3PO4–graphene under simulated solar-light irradiation. They observed that the photocatalytic activities of Ag3PO4–graphene and Ag/Ag3PO4–graphene were no better than pure Ag3PO4. However, these studies indicated the enhanced structural stability of Ag/Ag3PO4–graphene, which would be more practical in real treatment processes.
 | ||
| Fig. 12 (a) Degradation of SMX by WCN-8 at various pH values under visible light: Conditions: catalyst = 0.5 g L−1, SMX = 10 mg L−1. (b) Degradation of SMX by WCN-8 at different catalyst dosages under visible light: Conditions: SMX: 10 mg L−1, no pH adjustment. (c) Schematic illustration of SMX photodegradation process over WCN composites under visible-light irradiation. Reproduced from ref. 275 with permission from RSC (2017). | ||
In another study, Ag2S/Bi2S3/g-C3N4 heterojunctions exhibited 97.4% degradation of sulfamethoxazole in 90 min in aqueous solution under visible light.277 The stable hierarchical Fe2O3/Co3O4 heterojunction on nickel foam exhibited enhanced photocatalytic degradation of sulfamethoxazole.278 The photocatalyst was also studied to evaluate its effectiveness in surface water, hospital wastewater, and wastewater treatment. A magnetic quaternary BiOCl/g-C3N4/Cu2O/Fe3O4 nano-heterojunction exhibited 99.5% photodegradation of sulfamethoxazole (100 μM) in 60 and 120 min under visible and natural sunlight, respectively.279 Photocatalysts comprising graphene-supported p–n heterojunction rGO@Cu2O/BiVO4 composites with different Cu2O loadings (l, 5, 10, 15 and 20 wt%) were prepared to study their photocatalytic degradation activity for sulfamethoxazole oxidation under LED light at neutral pH.280 All the composites were found to be effective in sulfamethoxazole oxidation owing to the electrical conductivity of rGO and the p–n heterojunction between Cu2O and BiVO4.
Zhang et al.281 evaluated the performance of a Bi2WO6/TiO2 heterojunction for photocatalytic ozonation degradation of sulfamethoxazole under simulated sunlight. They attained 97.1% removal rate of sulfamethoxazole corresponding to a catalyst dosage of 0.2 g L−1, ozone concentration of 1.5 mg L−1, sulfamethoxazole concentration of 10 mg L−1 and pH 5.25. These studies also established excellent recyclability and stability, as evidenced through 5 cycle experiments. They also proposed a new Z-scheme transfer pathway for electrons and a degradation mechanism. A direct Z-scheme MIL-53(Co/Fe)/10 wt% MoS2 heterojunction composite photocatalyst displayed 99% removal of sulfamethoxazole (10 mg L−1) in aqueous solution (pH: 6) following visible-light-driven activation of peroxymonosulfate (initial concentration: solution 0.2 g L−1).282 Bi2O3/C3N4/TiO2@C quaternary hybrids (fabricated by a hydrothermal and calcination two-step method) exhibited high photocatalytic activity, degrading 100% sulfamethoxazole (SMZ, 5 mg L−1) within 100 min under visible-light irradiation.283 These investigations further revealed the photocatalytic degradation rates of SMZ by a Bi2O3/C3N4/TiO2@C junction to be 5.12, 2.87, and 1.35 times higher than those with Bi2O3/C3N4, C3N4/TiO2@C, and Bi2O3/TiO2@C junctions, respectively.
Ren et al.284 examined Ag (0.5, 1 and 2 wt%) nanoparticles/g-C3N4/Bi3TaO7 as Z-scheme photocatalysts prepared by combining hydrothermal and photodeposition for visible-light-driven performance in the degradation of sulfamethoxazole. It should be noted that the removal efficiency for sulfamethoxazole by Ag (1 wt%)/g-C3N4/Bi3TaO7 was found to be about 98% after 25 min and adopted the following order compared to g-C3N4, Bi3TaO7, g-C3N4–Bi3TaO7 and other Ag/g-C3N4/Bi3TaO7 composites: Ag (1 wt%)/g-C3N4/Bi3TaO7 > Ag (2 wt%)/g-C3N4/Bi3TaO7 > Ag (0.5 wt%)/g-C3N4/Bi3TaO7 > g-C3N4/Bi3TaO7 > g-C3N4 > Bi3TaO7. Such improved performance of Ag (1 wt%)/g-C3N4/Bi3TaO7 is attributed to the effective separation/transfer of photo-excited electrons and holes. In another study, an in situ prepared Ag3PO4/Bi4Ti3O12-20% heterojunction composite photocatalyst under visible-light irradiation exhibited much better photocatalytic activity in degrading sulfamethoxazole and stability compared to Ag3PO4 or pure Bi4Ti3O12.285 This is attributed to the formation of a direct Z-scheme improving the stability and activity of the Ag3PO4/Bi4Ti3O12 composite.
An Ag2O–KNbO3 (0.15Ag–Nb) composite fabricated by an in situ deposition method exhibited improved degradation of sulfamethoxazole under visible-light irradiation compared to the corresponding pure KNbO3 and Ag2O.286 The apparent rate constant of the composite was found to be 0.40 and 8 times those of KNbO3 and Ag2O, respectively. According to these studies, a type-I heterojunction formed between KNbO3 and Ag2O significantly enhanced the separation of photo-induced holes and electrons and accounted for sulfamethoxazole degradation. The rate constant value of the visible-light-driven optimal 0D/1D AgI/MoO3 (0.13 min−1) Z-scheme heterojunction photocatalyst in sulfamethoxazole degradation was found to be ∼22.4 times and 32.5 times those of MoO3 (0.0058 min−1) and AgI (0.0040 min−1), respectively.287 In addition, Z-scheme Ag3PO4/g-C3N4,288 Fe3O4–ZnO@g-C3N4,289 CeO2/g-C3N4 (CeO2: 5% mass ratio)290 and S-scheme-based N–SrTiO3/NH4V4O10291 photocatalysts have also been evaluated for the removal of sulfamethoxazole from water.
Table 4 records the performance data of different photocatalysts on the removal of sulfamethoxazole in wastewater.
| Photocatalyst | Preparative method | SMX | Catalyst dose | pH | Light source and other details | Degradation/removal (time) | Rate constant |
|---|---|---|---|---|---|---|---|
| TiO2: mainly of anatase (80%), (P25 Degussa)219 | Commercial | 20 mg L−1 | 1 g L−1 | 5 | Xenon lamp: 400 W (200 nm < λ < 700 nm) | 96% (180 min) | 0.026 min−1 |
| TiO2, P-25 Degussa222 | Commercial | 20 mg L−1 | 0.5 g L−1 | Natural | UV lamp equipped with UV C (260 nm) | 100% (180 min) | — |
| TiO2 Degussa P25223 | Commercial | 100 mgL−1 | 1.0 g L−1 | 5 | Xenon lamp (1000 W) with λcut-off < 290 nm | 88% (360 min) | 0.054 min−1 |
| TiO2 Merck224 | Commercial | 20 mg L−1 | 0.08 g L−1 | 3 | Low-pressure mercury vapour lamp (15 W) | 96.5% (60 min) | — |
| Biochar supported TiO2228 | Sol–gel method | 10 mg L−1 (0.1 L) | 0.5 g | 4 | UV lamp-UVC (15 W), λ: 254 nm | 91% (6 h) | — |
| TiO2 immobilized on glass spheres229 | By dip coating on glass | 100 μg L−1 | 0.335 g L−1 | 7.82 | Solar UV radiation (λ < 400 nm) | 100% (120 min) | 0.030 min−1 (first cycle) |
| F–ZnO232 | Commercial | 1 mM (NH4F: 2.505 mM) | 1.48 g L−1 | 4.7 | UVC lamp: 10 W | 97% (30 min) | 0.099 min−1 |
| ZnO233 | Hydrothermal | 10 mg L−1 | 200 mg L−1 | 7.5 | UVA lamp | 84% (60 min) | 0.030 min−1 |
| TiO2 nanoparticles (sigma-Aldrich)234 | Commercial | 50 mg L−1 | 500 mg L−1 | 4 | UV lamp | 100% (90 min) | 0.0356 min−1 |
| WO3 commercial (sigma-Aldrich)234 | Commercial | 25 mg L−1 | 750 mg L−1 | 3 | UV lamp | 100% (90 min) | 0.0093 min−1 |
| Pd/TiO2 (1%)236 | UV-reduction | 1 mg L−1 | ∼50 mg L−1 | — | Natural sunlight | 100% (10 min) | 52.1 ± 5.1 × 10−2 min−1 |
| Pt/TiO2 (1%)236 | UV-reduction | 1 mg L−1 | ∼50 mg L−1 | — | Natural sunlight | ∼90% (10 min) | 7.6 ± 501 × 10−2 min−1 |
| Cu (0.045 wt%)–TiO2237 | Microwave assisted impregnation method | 4 mg L−1 (20 mL) | 1 g L−1 | 5.2 | Lamps: 8 W, 77 mW cm−2 | 100% (120 min) | 0.0506 min−1 |
| TiO2 Evonik P25238 | Sol–gel procedure | 30 mg L−1 | 0.5 g L−1 | — | UVC | 100% (90 min) | 0.046 min−1 |
| TiO2 Evonik P25238 | Sol–gel procedure | 30 mg L−1 | 0.5 g L−1 | — | Simulated solar light | 100% (240 min) | 0.022 min−1 |
| 1.5% au/TiO2238 | Deposition precipitation method | 30 mg L−1 | 0.5 g L−1 | — | UVC light (254 nm) | 100% (90 min) | 0.071 min−1 |
| 1.5% au/TiO2238 | Deposition precipitation method | 30 mg L−1 | 0.5 g L−1 | — | Simulated solar light | 100% (180 min) | 0.039 min−1 |
| 1.5% Ag/TiO2238 | Deposition precipitation method | 30 mg L−1 | 0.5 g L−1 | — | UVC light (254 nm) | 100% (45 min) | 0.201 min−1 |
| Simulated solar light | 100% (240 min) | 0.027 min−1 | |||||
| 1.0% Cu/TiO2238 | Deposition precipitation method | 30 mg L−1 | 0.5 g L−1 | — | UVC light (254 nm) | 100% (90 min) | 0.186 min−1 |
| Simulated solar light | 100% (240 min) | 0.028 min−1 | |||||
| Au–Ag/TiO2238 | Deposition precipitation method | 30 mg L−1 | 0.5 g L−1 | — | UVC light (254 nm) | 100% (45 min) | 0.143 min−1 |
| Simulated solar light | 100% (240 min) | 0.025 min−1 | |||||
| Au–Cu/TiO2238 | Deposition precipitation method | 30 mg L−1 | 0.5 g L−1 | — | UVC light (254 nm) | 100% (45 min) | 0.145 min−1 |
| Simulated solar light | 100% (240 min) | 0.026 min−1 | |||||
| Fe-doped Titania (Fe/Ti molar ratio: 0.04%)239 | Co-precipitation method | 234 mg L−1 | 1 g L−1 | Natural pH | Xenon ozone free lamp (100 W) | 95% (120 min) | 29 × 10−3 min−1 |
| F–Pd co-doped TiO2240 | Microwave assisted hydrothermal method | 30 mg L−1 | 1 g L−1 | — | Sunlight | 98.4% (40 min) | — |
| F–Pd co-doped TiO2240 | Microwave assisted hydrothermal method | 30 mg L−1 | 1 g L−1 | — | Solar simulator | 98.5% (220 min) | — |
| F–Pt co doped TiO2241 | Microwave assisted hydrothermal method | 20 mg L−1 (50 mL) | 50 mg | ∼5.1 | Solar light | >93% (90 min) | — |
| F–Pt co-doped TiO2241 | Microwave assisted hydrothermal method | 20 mg L−1 (50 mL) | 50 mg | ∼5.1 | Simulated solar light | ∼58% (360 min) | — |
| N–Cu co doped TiO2@f-SWCNT243 | Sol–gel method | 60 mg L−1 | 0.8 g L−1 | 6 | Xenon lamp (200 W) | 100% (60 min) | 0.0512 min−1 |
| Ag,P-co-doped g-C3N4244 | Pyrolysis method | 5 mg L−1 | 1000 mg L−1 | 9.0 | Visible lamps (8 W each), λ: 465 ± 40 nm | >99% (30 min) | 2.06 × 10−1 min−1 |
| Ce-doped ZnO246 | Spray coating | 6.332 μg L−1 | Catalyst immobilized on 11.5 cm dia discs of area 104 cm2 | 6.28 | UVA lamp (36 W) | — | 1.09 × 10−2 min−1 |
| Zn–TiO2/pBC247 | Modified sol–gel method | 10 mg L−1 (160 mL) | 0.2 g | 5.03 | Xenon lamp (50 W) with 420 nm cut-off filter | 80.8% (180 min) | 0.0085 min−1 |
| Fe3O4/ZnO, (H2O2:100 mg L−1)248 | Polyol-mediated preparation | 100 μg L−1 (20 mL) | 200 mg L−1 | 7 | UVA lamp (15 W), λ: 365 nm, 4 mW cm−2 | ∼100% (240 min) | — |
| Bi2O3–TiO2/PAC251 | Two-stage calcination method | 20 mg L−1 (250 mL) | 0.05 g | 11 | Solar light–xenon arc lamp (300 W) | ∼100% (30 min) | 0.159 min−1 |
| LDH–TiO2254 | Impregnation process | 20 mg L−1 (100 mL) | 50 mg | 10 | UVA lamp (λ: 300–400 nm, 300 W) | 100% (360 min) | — |
| Poly(ethylene terephthalate)-10% TiO2257 | Solvent casting method | 1 mg L−1 (100 mL) | 50 mg L−1 | — | Xenon lamp (simulated solar light): 1.5 kW, 500 W m−2 | 98% (360 min) | 0.015 min−1 |
| BiVO4/SrTiO3 (1%)258 | Self-template method under hydrothermal condition | 10 mg L−1 (50 mL) | 0.05 g | — | Xenon lamp (500 W) | 91% (60 min) | — |
| 0.75 CuOx–BiVO4259 | Polyol-reduction method | 0.5 mg L−1 | 500 mg L−1 (persulfate: 100 mg L−1) | — | Simulated solar light | 100% (30 min) | 0.0991 min−1 |
| WO3–MWCNT261 | Hydrothermal method | 10 mg L−1 | 2.0 g L−1 | — | Solar simulator–xenon arc lamp (300 W), 420–630 nm | 88.5% (180 min) | — |
| Magnetic ZnO@g-C3N4265 | In situ growth | 30 mg L−1 (1000 mL) | 0.65 g L−1 | 5.6 | UVC lamp (10 W) | 90.4% (60 min) | 0.0384 min−1 |
| 5 wt% Ag/g-C3N4268 | Photo-reduction method | 10 μM (100 mL) | 5 mg | Natural pH | Xenon lamp (300 W) with a 400 nm cut-off filter | 97.5% (60 min) | — |
| rGO–WO3269 | Hydrothermal method | 10 mg L−1 | 1.0 g L−1 | No pH adjustment | Xenon arc lamp: 200 W (420–630 nm) | >98% (180 min) | 1.607 h−1 |
| Ag@Ag2O-2.5 wt% graphene270 | Precipitation method | 1 mg L−1 | 0.05 g L−1 | — | Xenon lamp (300 W) with a cut-off filter (λ > 280 nm), 37.7 mW cm−2 | ∼100% (90 min) | 0.038 min−1 |
| Immobilized TiO2-2.7% rGO271 | Polymer assisted hydrothermal deposition method | 5 mg L−1 | Bundle of thirty 10 cm photocatalyst-coated SOF (25 mL) placed on a petri disc | 6 | High-pressure UV mercury vapor lamp (160 W) | 92% (180 min) | 0.757 h−1 |
| Cu2O/rGO-80 (80 refers amount of GO (mg) used in preparation of rGO)272 | Wet chemical method | 5 mg L−1 (80 mL) | 20 mg | — | Xe lamp: 300 W (420 nm cut-off filter) | 50% (120 min) | 0.00525 min−1 |
| rGO–TiO2/sodium alginate (1:3)273 |
Hydrothermal method | 10 ppm (200 mL) | 0.5 g L−1 | Neutral | High-pressure mercury lamp (100 W) | >99% (45–90 min) | 0.108 min−1 |
| WO3–g-C3N4 (referred as WCN-8)275 | Hydrothermal method | 10 mg L−1 | 1.0 g L−1 | No pH adjustment | Xenon arc lamp (300 W), 420–630 nm | 91.7% (240 min) | — |
| Ce0.8Gd0.2O2−δ/TiO2276 | Modified Pechini method | 25 mg L−1 (300 mL) | 30 mg | — | Mercury lamp (15 W) | 97% (120 min) | 0.2959 mg−1 min−1 |
| Ag2S/Bi2S3/g-C3N4277 | Hydrothermal | 20 mg L−1 | 0.25 mg mL−1 | 7 | Xenon lamp (visible light): 300 W | 97.3% (90 min) | 0.0642 min−1 |
| BiOCl/g-C3N4/Cu2O/Fe3O4279 | Co-precipitation method | 100 μM | 0.2 g L−1 | 6.5 | Xenon lamp | 99.5% (60 min) | 0.0543 min−1 |
| rGO@Cu2O/BiVO4280 | Solution method | 0.5 mg L−1 | 100 mg (250 ml) | 7 | LED light (30 W) | ∼98.5% (270 min) | — |
| Bi2WO6/TiO2281 | Hydrothermal method | 10 mg L−1, [ozone]: 1.5 mg L−1 | 0.2 g L−1 | 5.25 | Simulated sunlight | 97.1% (180 min) | 1.83 × 10−2 min−1 |
| MIL-53(Co/Fe)/10 wt% MoS2282 | Hydrothermal through in situ growth | 10 mg L−1, (peroxymonosulfate: 0.2 g L−1) | 0.01 g L−1 | 6 | Visible light | 99% (60 min) | — |
| Bi2O3/C3N4/TiO2@C283 | Hydrothermal and calcination | 5 mg L−1 | 1 g L−1 | 5 | Visible light | 100% (100 min) | — |
| Ag (1 wt%)/g-C3N4/Bi3TaO7284 | Photo deposition method | 5 mg L−1 | 25 mg (50 mL) | — | Xenon lamp (300 W) | 98% (25 min) | 0.1499 min−1 |
| Ag3PO4/Bi4Ti3O12-20%285 | In situ growth method | 5 ppm (50 mL) | — | — | Xenon lamp: 300 W (λ > 400 nm) | ∼77% (40 min) | 0.0372 min−1 |
| Ag2O–KNbO3 (Ag–Nb molar ratio: 0.15/1)286 | In situ growth | 5 ppm | 0.3 mg mL−1 | — | Visible-light irradiation | 91% (40 min) | 0.0603 min−1 |
| 97.9% Ag3PO4/2.1% g-C3N4288 | In situ precipitation method | 1 mg L−1 (100 mL) | 5 mg | Neutral pH | Xenon lamp (300 W), λ > 400 nm, 138.7 mW cm−2 | ∼99% (90 min) | 0.063 min−1 |
| Fe3O4–ZnO@g-C3N4289 | In situ growth | 30 mg L−1 | 0.5 g L−1 | 7 | UVC lamp (10 W) | 95% (90 min) | 0.0351 min−1 |
3.4 Ibuprofen
Ibuprofen (IPF) is a drug belonging to the class of propanoic acid derivatives and is extensively used in the treatment of fever, pain in human beings, inflammatory disorders, muscle problems, including migraines, rheumatoid arthritis, analgesic and painful menstrual periods.22,292 It is slightly soluble in water, stable, is eliminated from the body through urine and does not undergo biodegradation. As a result, it can be found in water samples of different origins originating from municipal wastewater treatment plant effluents, groundwater through leaching and natural water and cannot be treated through conventional wastewater treatments. The presence of ibuprofen even in low concentration through water affects the reproduction of aquatic animals, including the photosynthesis of aquatic plants. Ibuprofen can leach into ground water and soil in daily life. In view of this, several studies have been made using metal oxide and graphitic material related photocatalysts to make wastewater free from ibuprofen.293–357The photodegradation of ibuprofen has been tested as a function of catalyst type (TiO2 and ZnO), loading (50–500 mg L−1), initial drug concentration (10, 40, 80 mg L−1) and wavelength (200–600 nm) of irradiation.297 The photocatalytic efficiency was found to be greater than 90% in 15 min under UVA and visible-light irradiation corresponding to an initial concentration of ibuprofen of 10 mg L−1 and amount of photocatalysts (TiO2 and ZnO) of 100 mg L−1. These findings also indicated over 90% conversion of the drug within 8 min with k-values of 0.382 and 0.326 min−1 under UVA for TiO2 and ZnO, respectively, and it correspondingly decreased to 0.199 and 0.144 min−1 under visible light. Tanveer and others298 used UV and solar irradiation to compare the photocatalytic degradation of ibuprofen in water using TiO2 and ZnO. A much higher rate of degradation was observed in UV for TiO2 (99%) compared to ZnO (86%) after 15 min compared to solar degradation.
The degradation of ibuprofen using a heterogeneous ZnO photocatalyst irradiated with UVC achieved 82.97% removal efficiency within a reaction time of 95 min under optimized conditions (pH: 6.7, ZnO loading: 583 mg L−1, initial IBP concentration: 1.5 mg L−1, humic acid concentration: 54 mg L−1).299 The reactive species responsible for oxidizing ibuprofen were found to be h+, O2·−, H2O2, and ·OH. In another experiment, ZnO–Ce (0.50 g L−1) showed 60% removal of ibuprofen (20 ppm) under acidic conditions after 120 min under UVC irradiation.300 Holes played a vital role in the degradation process of ibuprofen and it displayed good degradation activity even after 3 cycles under UV light. Hexagonal α-Fe2O3 flakes have removed up to 80% of ibuprofen in a combination of adsorption treatment followed by UV (265 nm) irradiation.301 TiO2 immobilized on glass coupled with simulated solar irradiation also eliminated ibuprofen and its derivatives.302 Investigations on the photocatalytic activity of TiO2,303,304 ZnO,304,305 and ZnO membrane306 have also been reported in the remediation of water from ibuprofen.
:1 wt. ratio demonstrated complete degradation of ibuprofen under visible light within 5 h in contaminated water.308
Bi (0.25 wt%) and Ni (0.5 wt%) doped TiO2 photocatalysts synthesized by a sol–gel method under irradiation of solar light for 6 h achieved degradation of ibuprofen by 89% and 78% repectively.309 The degradation of ibuprofen followed kinetics in accordance with the Langmuir–Hinshelwood model. In addition, La3+-doped TiO2 monolith,310 Cu-doped LaFeO3,311 Cu2O-doped TiO2 nanotube arrays,312 C,N-co-doped mesoporous TiO2313 and TiO2 co-doping with urea and functionalized CNT314 photocatalysts also displayed enhanced photocatalytic degradation of ibuprofen in aqueous solution.
Ag and Fe3O4 co-modified WO3−x (Ag/Fe3O4/WO3−x) composites were fabricated by hydrothermal and photodeposition processes and showed almost complete photocatalytic-Fenton degradation of ibuprofen (and diclofenac), as evident from (Fig. 13(a) and (b)).322 This is attributed to the surface plasmon resonance effect of Ag, separation of photogenerated carriers and heterostructures of Ag/Fe3O4/WO3−x. In addition, the possibility of absorption of light greatly improving the photocatalytic-Fenton degradation efficiency cannot be ruled out. The fabricated Ag/Fe3O4/WO3−x also exhibited good photocatalytic-Fenton stability in the photodegradation of ibuprofen (and diclofenac), as indicated by the almost unchanged degradation rate of the antibiotic in (Fig. 13(c) and (d)). The degradation and charge transfer mechanism involved in the removal of the ibuprofen and diclofenac have also been proposed and are displayed in Fig. 13(e).
 | ||
| Fig. 13 Photocatalytic-Fenton degradation of (a) ibuprofen and (b) diclofenac by Fe3O4, WO3–x, Fe3O4/WO3–x, and Ag/Fe3O4/WO3–x samples. (c and d) Corresponding recycling study and stability of Ag/Fe3O4/WO3−x. (e) Schematic illustration of the possible catalytic degradation mechanism and charge transfer of Ag/Fe3O4/WO3–x under light irradiation (modified image). Reproduced from ref. 322 with permission from ACS (2021). | ||
Lenzi et al.323 showed that the photocatalytic degradation of ibuprofen (10 ppm) solution (pH: 7) by 0.3 g L−1 of Ag/ZnO/CoFe2O4 (5 wt%) exhibited removal efficiencies of 80% and 47% under artificial and solar radiation, respectively. These studies also confirmed the recovery and reuse of the catalyst after 3 cycles without significant loss of catalytic activity. Visible-light-driven mesoporous hierarchical BiOBr/Fe3O4@SiO2 (dose: 1 g L−1) photocatalyst degraded ibuprofen (initial concentration: 2 mg L−1) almost completely in 60 min.324 Further studies have shown BiOBr/Fe3O4@SiO2 maintaining its initial photocatalytic activity (∼80%) even after five cycles. In another study, a magnetically separable Fe3O4–SiO2-coated TiO2 composite demonstrated excellent photocatalytic activity.325 An immobilized TiO2/ZnO-sensitized copper(II) phthalocyanine heterostructure displayed about 80% degradation of ibuprofen (initial conc.: 5 mg L−1) after 4 h of irradiation under 365 nm UV.326 The studies revealed a small decline in the IBF degradation (77%) after the 5th cycle. PANI-coated WO3@TiO2,327 polyacrylonitrile (PAN)–MWCNT/TiO2–NH2,328 TiO2 nanoparticles and C-nanofiber-modified magnetic Fe3O4 nanospheres (TiO2@Fe3O4@C-NF),329 carbon dots/Fe3O4@carbon sphere pomegranate-like composites,330 PVDF–ZnO/Ag2CO3/Ag2O,331 and PAN–MWCNT nanofiber crosslinked TiO2–NH2 nanoparticles332 have also been examined for their photodegradation performance for ibuprofen.
Acidified g-C3N4/polyaniline/rGO@biochar (0.5 mg L−1) nano-assemblies degraded ibuprofen (20 mg L−1) to the extent of 98.4% in 50 min under exposure to visible light.336 Such significant performance is attributed to multiple reasons, such as highly separated charges, enhanced visible absorption and diffusion. The major reactive species in the degradation process for ibuprofen involved hydroxyl and superoxide radical anions. Akbarzadeh et al.337 explored the photodegradation of ibuprofen solution in the presence of a hydrothermally fabricated g-C3N4/Ag/AgCl/BiVO4 microflower composite as photocatalyst under visible light and compared its performance with BiVO4, g-C3N4/BiVO4 and Ag/AgCl/BiVO4. These findings revealed remarkably enhanced degradation efficiency of g-C3N4/Ag/AgCl/BiVO4 (94.7%) compared to g-C3N4 (6.5%), BiVO4 (11.4%), g-C3N4/BiVO4 (68.6%), or Ag/AgCl/BiVO4 (88.3%) in 1 h corresponding to a photocatalyst dosage of 0.25 g L−1 and initial concentration of 2 mg L−1. The reduced band gap energy and recombination rate of the g-C3N4/Ag/AgCl/BiVO4 photocatalyst are ascribed to charge transfer along the heterojunction. The photocatalytic degradation performance of IPF increases with the (121)/(040) XRD plane intensity ratio of BiVO4, Ag/AgCl/BiVO4, g-C3N4/BiVO4 and g-C3N4/Ag/AgCl/BiVO4 and is found to be in good agreement with the photoluminescence findings.
A hierarchical assembly of Ag (7%)/g-C3N4/kaolinite composite fabricated following an in situ calcination and photodeposition process exhibited 99.9% degradation of ibuprofen (k: 0.01128 min−1) after 5 h under visible-light irradiation compared to g-C3N4, g-C3N4/kaolinite and Ag/g-C3N4.338 This outcome is due to the stronger adsorption property, efficient separation and transfer of electron–hole pairs. In addition, the presence of monodispersed Ag nanoparticles in the g-C3N4/kaolinite sheets led to more active sites, accounting for this. The efficient photocatalytic degradation of ibuprofen has also been reported in aqueous solution using graphene quantum dots/AgVO3 nanoribbons,339 g-C3N4/MIL-68(In)–NH2 composites,340 graphene oxide and TiO2 heterostructures doped with F,341 reduced-graphene-oxide–TiO2/sodium alginate 3-dimensional structure aerogel273 and Fe3O4/graphene/S-doped g-C3N4342 also exhibited enhanced visible-light photocatalytic activity for the degradation of ibuprofen.
:4 mol ratio) by a hydrothermal reaction. Such an assembly of 2D/2D heterojunctions removed 96.1% ibuprofen under visible-light irradiation within 60 min due to a synergistic effect.
Kumar and others345 synthesized a magnetically recyclable direct-contact Z-scheme g-C3N4/TiO2/Fe3O4@SiO2 heterojunction nanophotocatalyst and recorded 97% removal of ibuprofen solution (pH: 3) after 15 min under irradiation by visible light (∼330 W m−2). Such excellent performance of a magnetically recyclable direct-contact Z-scheme nanophotocatalyst was attributed to the low recombination rate of photogenerated e− and h+. Visible-light-assisted persulfate activation by an SnS2 (0.5%)/MIL-88B(Fe) Z-scheme heterojunction achieved 100% removal of ibuprofen in 120 min.346 This was found to be 54 and 4 times higher than SnS2 and SnS2 (0.5%)/MIL-88B(Fe), respectively. Such findings could be ascribed to the structure and crystallinity of the photocatalysts. In another reported study, an optimized Z-scheme based 1D/2D FeV3O8/g-C3N4 composite comprising 10% FeV3O8 achieved a maximum degradation rate for ibuprofen of 95% at 85 min under visible-light irradiation.347 Kinetic studies established that the rate constant is 4 times that of g-C3N4 nanosheets. However, the presence of 30% FeV3O8 in g-C3N4 decreased the degradation efficiency to 52.8%.
Heterostructure g-C3N4/Bi2WO6/rGO nanocomposites prepared by microwave- assisted treatment for 120 min in a hydrothermal method undertook the maximum photocatalytic degradation of ibuprofen (93.9%) under visible-light illumination.348 In addition, g-C3N4@NiO/Ni@MIl-101,349 Bi5O7I–MoO3,350 AgSCN/Ag3PO4/C3N4,351 N–TiO2@SiO2@Fe3O4,352 g-C3N4/CQDs/CdIn2S4,353 direct Z-scheme Co3O4/BiOI,354 a double Z-scheme system of α-SnWO4/UiO-66(NH2)/g-C3N4,355 CdS/Fe3O4/TiO2356 and Ag2CO3/Ag2O/ZnO357 heterojunctions also exhibited excellent photocatalytic degradation of ibuprofen.
Table 5 records the performance data of different photocatalysts on the removal of ibuprofen from wastewater.
| Photocatalyst | Preparation method | IPF | Catalyst dose | pH | Light source | % degradation | Rate constant |
|---|---|---|---|---|---|---|---|
| TiO2 Degussa P25 (80% anatase and 20% rutile)294 | Commercial | 213 mg L−1 | 2.5 g L−1 | 5.0–5.3 | UV-LEDs (10 W), 365 nm, 375 W m−2 | 100% (5 min) | 24 × 10−3 min−1 |
| TiO2 nanoparticles (Degussa P25)295 | Commercial | 5 μg mL−1 (50 mL) | 134.5 mg | 5.5 | UV light: 15 W, 365 nm | 100% (10 mi) | 1.0 min−1 |
| TiO2 (Vetec, 98% of purity)296 | Commercial | 10−4 M (100 mL) | 0.03 g | 5 | Mercury lamp (125 W) | 100% (5 min) | — |
| TiO2 P-25 Degussa (75:25 w/w mixture of anatase:rutile)297 |
Commercial | 10 mg L−1 | 100 mg L−1 | 4 | UVA | ∼100% (18 min) | 0.382 min−1 |
| ZnO Sigma Aldrich297 | Commercial | 10 mg L−1 | 100 mg L−1 | 4 | UVA | ∼100% (18 min) | 0.326 min−1 |
| TiO2 P-25 Degussa (75:25 w/w mixture of anatase:rutile)297 |
Commercial | 10 mg L−1 | 100 mg L−1 | 4 | Visible | ∼94% (18 min) | 0.199 min−1 |
| ZnO Sigma Aldrich297 | Commercial | 10 mg L−1 | 100 mg L−1 | 4 | Visible | ∼90% (18 min) | 0.144 min−1 |
| TiO2 (Sigma-Aldrich)298 | Commercial | 20 mg L−1 | 1.5 g L−1 | 3 | UV lamp (40 W) | 99% (15 min) | 0.54 min−1 |
| ZnO (Sigma-Aldrich)298 | Commercial | 20 mg L−1 | 1.0 g L−1 | 7 | UV lamp (40 W) | 86% (15 min) | 0.31 min−1 |
| ZnO (Nano pars Spadana)299 | Commercial | 5 mg L−1 (humic acid: 50 mg L−1) | 500 mg L−1 | 7 | 125 W medium-pressure Hg lamp (UVC) | 98% (100 min) | — |
| ZnO–Ce300 | Precipitation method | 20 ppm | 0.5 g L−1 | 3 | UV light: 125 W Hg without bulb | 60% (120 min) | 6.86 × 10−3 min−1 |
| ZnO–Ce; H2O2: 0.5 m mole per L300 | Precipitation method | 20 ppm | 0.5 g L−1 | 3 | UV light: 125 W Hg without bulb | 70% (120 min) | — |
| TiO2 (Degussa P25) dispersed powder302 | Commercial | 25 mg L−1 | 0.2 g L−1 | 4.5 | Solar simulator exposed to xenon lamp irradiation | ∼95% (150 min) | 0.2378 mg L−1 min−1 (zero order), 0.0251 min−1 (first order), 0.0034 L mg−1 min−1 (second order) |
| TiO2 immobilized on the active coated glass302 | Chemical vapour deposition | 25 mg L−1 | 0.2 g L−1 | 4.5 | Solar simulator and exposed to xenon lamp irradiation | 100% (1480 min) | 0.0124 mg L−1 min−1 (zero order), 0.0012 min−1 (first order), 0.0001 L mg−1 min−1 (second order) |
| TiO2 Degussa (P-25)303 | Commercial | 4 mg L−1 | 20 mg L−1 | 7.8 | 125 W Hg vapor lamp, 10.75 mW cm−2 | >98% (30 min) | — |
| TiO2 Degussa P25 (ref. 304) | Commercial | 5 mg dm−3 | 50 mg dm−3 | — | Mercury lamp (150 W), λ < 300 nm | ∼89% (60 min) | 0.0425 min−1 |
| ZnO Degussa P25 (ref. 304) | Commercial | 1 mg dm−3 | 50 mg dm−3 | — | Mercury lamp (150 W), λ < 300 nm | 60% (30 min) | 0.0328 min−1 |
| ZnO nanoparticles305 | Chemical method | 60 ppm | 10 mg L−1 | — | Four UV-vis solarium lamps (60 W) | 24% (180 min) | 0.055 min−1 |
| PVDF- ZnO/Ag2CO3/Ag2O membrane306 | Casting solution using wet phase inversion method | 10 ppm (300 mL) | 1.96 wt% (membrane area: 12.56 cm2) | — | White light-emitting diode lamp (λ > 400 nm, 100 W) | 49.96% (180 min) | — |
| N,S-co-doped TiO2 nanoparticles307 | Sol–gel and hydrothermal methods | 5 mg L−1 (50 mL) | 2.0 g L−1 | 6 | Simulated solar radiation: 350 W xenon lamp | 85% (90 min) | 0.062 min−1 |
| C–N–S co-doped TiO2308 | Thermal treatment method | 20 ppm (200 mL) | 0.5 g L−1 | — | LED lamp (λmax: 420 nm, 1 mW cm−2) | ∼100% (300 min) | 0.021 min−1 |
| Bi (0.25 wt%) doped TiO2309 | Sol–gel method | 25 ppm | 2 g L−1 | 6 | UV (36 W, 254 nm) | 89% (360 min) | 0.0064 min−1 |
| Ni (0.5 wt%) doped TiO2309 | Sol–gel method | 25 ppm | 2 g L−1 | 6 | UV (36 W, 254 nm) | 78% (360 min) | 0.0046 min−1 |
| La3+(2%)-doped TiO2 monolith310 | Sol–gel method | 50 mg L−1 (70 mL) | 0.1 g | 5 | Sunlight | 96.9% (150 min) | 2.2 × 10−2 min−1 |
| C,N-co-doped mesoporous TiO2313 | Hydrothermal method | 20 ppm (220 mL) | 0.5 g L−1 | — | High-pressure Hg lamp (150 W), λmax: 254 nm | 98.9% (120 min) | 0.0377 min−1 |
| C,N-doped mesoporous TiO2313 | Hydrothermal method | 20 ppm (220 mL) | 0.5 g L−1 | — | LED lamp (visible light, λmax: 420 nm, 1 mW cm−2) | 100% (120 min) | 0.0207 min−1 |
| N doped CNT COOH/TiO2 (anatase/rutile: 20/80)314 | Hydrothermal | 5 mg L−1 ppm | 400 mg L−1 | Natural pH | LED light: 240 W, 40 mW cm−2 and 410 nm | 85–86% (120 min) | 4.45 × 10−3–1.22 × 10−2 min−1 |
| Activated carbon impregnated with TiO2316 | Sol–gel method | 25 mg L−1 (20 mL) | 1.6 g L−1 | 4.3 | UV lamp: 15 W, 254 nm | 92% (240 min) | — |
| Fe3O4@MIL-53(Fe)318 | Calcination (400 °C) | 10 mg L−1 (50 mL), H2O2 (20 mM) | 0.4 g L−1 | — | Xenon lamp (500 W with 420 nm cut-off filter) | 99% (60 min) | 4.71 × 10−2 min−1 |
| Fe3O4/Bi2WO6319 | Two-step approach | 10 mg L−1 (70 mL) | 70 mg | 4.7 | Solar light | >80% (120 min) | 0.0144 min−1 |
| Ag/Fe3O4/WO3−x/H2O2 (10 mM)322 | Simultaneous calcination | 10 mg L−1 (30 mL) | 30 mg | — | Xenon lamp (500 W) with optical filter (λ ≥ 420 nm) | ∼100% (90 min) | — |
| Ag/ZnO/CoFe2O4323 | Coating CoFe2O4 with ag/ZnO using Pechini method | 10 ppm | 0.3 g L−1 | 7 | UV light (125 W medium-pressure Hg lamp) | 80% (60 min) | 0.03905 min−1 |
| BiOBr/Fe3O4@SiO2324 | Solvothermal | 2 mg L−1 | 1 g L−1 (50 ml) | 7 | Fluorescent lamp (visible light) | ∼99% (60 min) | 0.08 min−1 |
| TiO2/ZnO/copper phthalocyanine (CuPc)326 | Multiple steps | 5 mg L−1 (50 mL) | Film | 6.5 | Hg lamp with 365 nm cut-off filter, 1,2 W cm−2 | 80% (240 min) | 0.42 h−1 |
| PAN–MWCNT/TiO2–NH2328 | Electrospinning | 5 mg L−1 (100 mL) | 15 mg L−1 | 2 | UVA lamp (315–400 nm) of 40 W | ∼100% (120 min) | — |
| Carbon dots/Fe3O4@carbon sphere (in presence of persulfate)330 | Solvothermal method | 50 μmol L−1 | 0.3 g L−1 | — | Xenon lamp (350 W) with a glass filter (λ > 420 nm) | 96% (120 min) | — |
| PAN–MWCNT/TiO2–NH2 composite nanofibers332 | Multiple steps | 5 mg L−1 (100 mL) | 15 mg | 2 | Xenon lamp (125 W) with cut-off filter (λ > 400 nm), 0.1 W cm−2 | 100% (210 min) | — |
| g-C3N4333 | Polycondensation | 20 mg L−1 (200 mL) | 200 mg | 5.5 | Xenon lamp (35 W) | 20% (4 h) | — |
| Reduced graphene oxide–HoVO4–TiO2335 | Hydrothermal | 10 mg L−1 | 40 mg L−1 | 7 | Tungsten lamp (150 W), (λ > 4900 nm) | ∼96% (60 min) | — |
| g-C3N/ag/AgCl/BiVO4337 | Hydrothermal | 2 mg (50 mL) | 0.25 g L−1 | 4 | Visible light | 94.7% (60 min) | — |
| Ag (7%)/g-C3N4/kaolinite338 | Two steps | 5 ppm (50 mL) | 50 mg | — | Xenon lamp (500 W with 400 nm cut-off filter) | 99.9% (300 min) | 0.01128 min−1 |
| Graphene quantum dots (3 wt%)/AgVO3339 | Hydrothermal | 10 mg L−1 (50 mL) | 0.01 g | — | Xenon lamp (350 W with λ > 420 nm) | ∼100% (180 min) | 0.1678 min−1 |
| g-C3N4 (10 wt%)/MIL-68(In)–NH2 composites340 | In situ solvothermal assisted by ultrasonication | 20 mg L−1 | 0.15 g | 4 | Xenon lamp (300 W with λ > 420 nm) | 93% (120 min) | 0.01739 min−1 |
| Graphene oxide/TiO2 doped with F (BrO3− 100 μg L−1)341 | Hydrothermal | 100 μg L−1 | 0.05 g L−1 | 5.2 | Low-pressure Hg lamp (10 W), (26 μW cm−2) | ∼100% (60 min) | 0.4504 min−1 |
| rGO–TiO2/sodium alginate273 | Hydrothermal | 10 ppm (200 mL) | 0.5 g L−1 | 7 | High-pressure Hg lamp (100 W), (13.5 W m−2) | ∼100% (90 min) | 0.047 min−1 |
| TiO2/5% g-C3N4343 | Solvothermal | 5 mg L−1 | 50 mg | 7 | Xenon lamp (259 W) | ∼90% (60 min) | 0.03833 min−1 |
| g-C3N4/Bi2WO6 (1:4 molar ratio)344 |
Hydrothermal | 25 μM | 0.2 g L−1 | — | Xenon lamp (300 W) with 420 nm cut-off filter | ∼96.1% (60 min) | 0.062 min−1 |
| g-C3N4/TiO2/Fe3O4@SiO2345 | Sol–gel method | 2 mg L−1 (50 mL) | 50 mg | 7 | Visible light, 330 W m−2 | 97% (15 min) | — |
| FeV3O8 (10%)/g-C3N4347 | Dispersion, grinding and calcination | 10 ppm (30 mL) | 10 mg | — | Xenon lamp (300 W) with UV cut-off filter (λ: 420 nm) | 95% (85 min) | 0.03 min−1 |
| g-C3N4/Bi2WO6/rGO348 | Microwave assisted hydrothermal preparation | 5 mg L−1 | 1.0 g L−1 | 4.3 | Xenon lamp (300 W), λ > 420 nm | 93% (240 min) | 0.011 min−1 |
| g-C3N4/Bi2WO6/rGO348 | Microwave assisted hydrothermal preparation | 5 mg L−1 | 1.0 g L−1 | 4.3 | Sunlight | 98.6% (240 min) | — |
| AgSCN/Ag3PO4/C3N4 (molar % of AgSCN: 11.3)351 | Precipitation reaction | 5 mg L−1 (100 mL) | 50 mg | — | Sunlight (500 W halide lamp) | 91% (6 min) | 0.46 min−1 |
| N–TiO2@SiO2@Fe3O4352 | Sol–gel method | 2 mg L−1 (50 mL) | 50 mg | — | Fluorescent lamps (9 W), 320 μW cm−2 | 94% (300 min) | — |
| g-C3N4/CQDs/CdIn2S4353 | Hydrothermal | 80 mg L−1 (100 mL) | 0.1 g | — | 300 W xenon lamp with 420 nm cut-off filter, 200 mW cm−2 | 91% (60 min) | — |
| Co3O4/BiOI (1:2)354 |
Solvothermal | 10 ppm (50 mL) | 40 mg | 11.3 | 60 W LED lamp with 420 nm cut-off filter | 93.87% (60 min) | 0.0945 min−1 |
| α-SnWO4/UiO-66(NH2)/g-C3N4355 | Solvothermal | 10 mg L−1 (100 mL) | 50 mg | — | Simulated sunlight using high-pressure 300 W xenon lamp | 95.5% (120 min) | 0.017 min−1 |
3.5 Norfloxacin
Norfloxacin (NOR) is an effective antibacterial agent of the fluoroquinolone family and is widely used as a drug in clinical treatments for bacterial infections of urinary, biliary, and respiratory tracts, and gastrointestinal and skin infections.358–360 Norfloxacin has frequently been detected in municipal/wastewater treatment plants, is difficult to biodegrade and is predicted to be a potential risk to human beings and the environment. Therefore, it is considered a potential threat to the water environment and human health.361–422The photocatalytic degradation of norfloxacin (and ciprofloxacin) was found to be 90–93% under optimized conditions in B and Ce doped TiO2, irradiated by sunlight.373 Bi3+ and Fe2+ ion doped ZnO showed significant photocatalytic degradation of norfloxacin with the addition of HSO5− under solar irradiation and followed pseudo-first-order kinetics.374 The co-doped ZnO exhibited a lower band gap, which accounted for the increased absorption of solar irradiation and reduced electron and hole recombination, which facilitated high norfloxacin degradation compared to undoped ZnO. Fe-doped CeO2 exhibited about 95% photocatalytic degradation of norfloxacin in aqueous solution (pH: 8.0) within 180 min corresponding to an initial norfloxacin concentration of 2.5 mg L−1 and catalyst dose of 0.1 g L−1.375 An Ag-doped TiO2/CFA (coal fly ash) photocatalyst has also been used to monitor the photocatalytic degradation of norfloxacin.376
:SiO2 ratios of 0, 0.25, 1.0 and 5.0). Subsequent investigations on the removal of norfloxacin revealed the better photocatalytic activity of 1.0TiO2/SBA-15 hybrid material in achieving 96.6% degradation of norfloxacin in 150 min under UV-light irradiation. Fe-complex/TiO2 composites comprising [FeII(dpbpy)2 (H2O)2]/TiO2, [FeII(dpbpy)(phen)2]/TiO2 and [FeII(dpbpy)(bpy)2]/TiO2 (dpbpy: 2,2′-bipyridine-4,4′-diphosphoric acid, phen: 1,10-phenanthroline, bpy: 2,2-bipyridyl) photocatalysts exhibited 98.5% degradation of norfloxacin in water under visible-light irradiation after 3 h.379 Further, the photocatalytic performance and cyclic stability of these composites were found to be much better than those of pure TiO2 or P25. An Ag2O/TiO2-zeolite composite fabricated through a modified sol–gel method exhibited high performance in the decomposition of norfloxacin under simulated solar-light illumination.380 This is a consequence of the narrow band gap of the photocatalyst, its enhanced light absorbance ability in the visible region and high charge separation efficiency.
FeVO4/Fe2TiO5 (2:1) synthesized via a one-pot hydrothermal method exhibited high photocatalytic activity and excellent stability for the removal of norfloxacin in aqueous solution under visible-light irradiation.381 This is ascribed to the synergistic effect of photogenerated electron–holes with radical OH· and h+. MIL-101(Fe)–NH2 immobilized on an α-Al2O3 sheet has also been investigated for effective norfloxacin elimination via a photo-Fenton process.382 Ag/AgCl–CeO2 composite photocatalysts fabricated by in situ interspersal of AgCl on CeO2 and subsequent photoreduction of AgCl to Ag exhibited enhanced photocatalytic activity in the photodegradation of norfloxacin under visible-light irradiation.383Fig. 14(a) shows the highest degradation efficiency (91%) for norfloxacin achieved by sample Ag/AgCl–CeO2 composites with an Ag mass ratio of 13.94 wt% (denoted AC-3) within 90 min under visible-light irradiation. It is also apparent from Fig. 14(b) and (c) that the photodegradation process followed a pseudo-first-order kinetic model with the highest rate constant (0.02279 min−1) for AC-3 compared to CeO2, Ag/AgCl, Ag/CeO2 and other AC composites. Fig. 14(d) shows the time-dependent UV-vis spectra of NOF solution for the AC-3 sample. ZnO/ZnS@biochar,384 ZnFe2O4/hydroxyapatite–Sn2+,385 (BiO)2CO3–Bi–TiO2,386 and Ag/AgCl/Ag2MoO4387 composites have also been reported as promising photocatalysts in the degradation of norfloxacin in water under UV irradiation.
 | ||
| Fig. 14 (a) Photocatalytic degradation NOF curves; (b) kinetic curves of NOF degradation; (c) apparent rate constants for the degradation of NOF; (d) time-dependent UV–vis spectra of NOF solution for AC-3 sample (Ag/AgCl–CeO2). Reproduced from ref. 383 with permission from Elsevier (2017). | ||
3.5.5.1 g-C3N4-based composites. Fei et al.388 investigated the photocatalytic degradation of norfloxacin in the presence of a sunlight-driven mesoporous g-C3N4. The results showed 90% decomposition of norfloxacin in 1.5 h under simulated sunlight irradiation. Co/g-C3N4, Co/g-C3N4/H2O2 and Co/g-C3N4/PMS composite photocatalysts exhibited better performance compared to pure g-C3N4 in the photocatalytic degradation of norfloxacin under visible-light irradiation.389 The optimization and variations of different parameters have been used to study the photocatalytic degradation of norfloxacin in the presence of ZnO/g-C3N4/Fe3O4 under visible light.390 These findings indicated a removal rate of norfloxacin greater than 90% in 120 min for a catalyst concentration of 1.43 g L−1, solution pH 7.12 and norfloxacin concentration of <8.61 mg L−1. Shuttle-like CeO2/g-C3N4 combined with persulfate391 and NiWO4 nanorods anchored on g-C3N4 nanosheets392 also exhibited enhanced degradation of norfloxacin under visible light.
3.5.5.2 Graphene-based composites. A TiO2/Bi2WO6/rGO (0.5%) photocatalyst attained about 87.79% removal of norfloxacin in water under visible-light irradiation after 60 min and was found to be superior to its individual components under optimal conditions.393 Such enhanced catalytic activity of TiO2/Bi2WO6/rGO arises due to the ligand–metal electron transfer mechanism. According to Zhao et al.,394 an rGO/Bi2WO6 composite exhibited outstanding photocatalytic activity for norfloxacin degradation in an aquatic environment under visible-light irradiation, as evident from the time-dependent-UV spectrum and time-dependent-HPLC spectrum displayed in Fig. 15(a) and (b), respectively. Fig. 15(c) and (d) indicate about 87.49% degradation of norfloxacin within 180 min compared to Bi2WO6, under visible-light irradiation. Additional investigations revealed ·OH and e− playing dominant roles in the photocatalytic degradation of norfloxacin. N-doped TiO2/graphene exhibited enhanced photocatalytic degradation under UV-light irradiation.395 It is suggested that graphene acts as an efficient “electron pump”, thereby promoting the separation of carriers to account for the observed photodegradation.
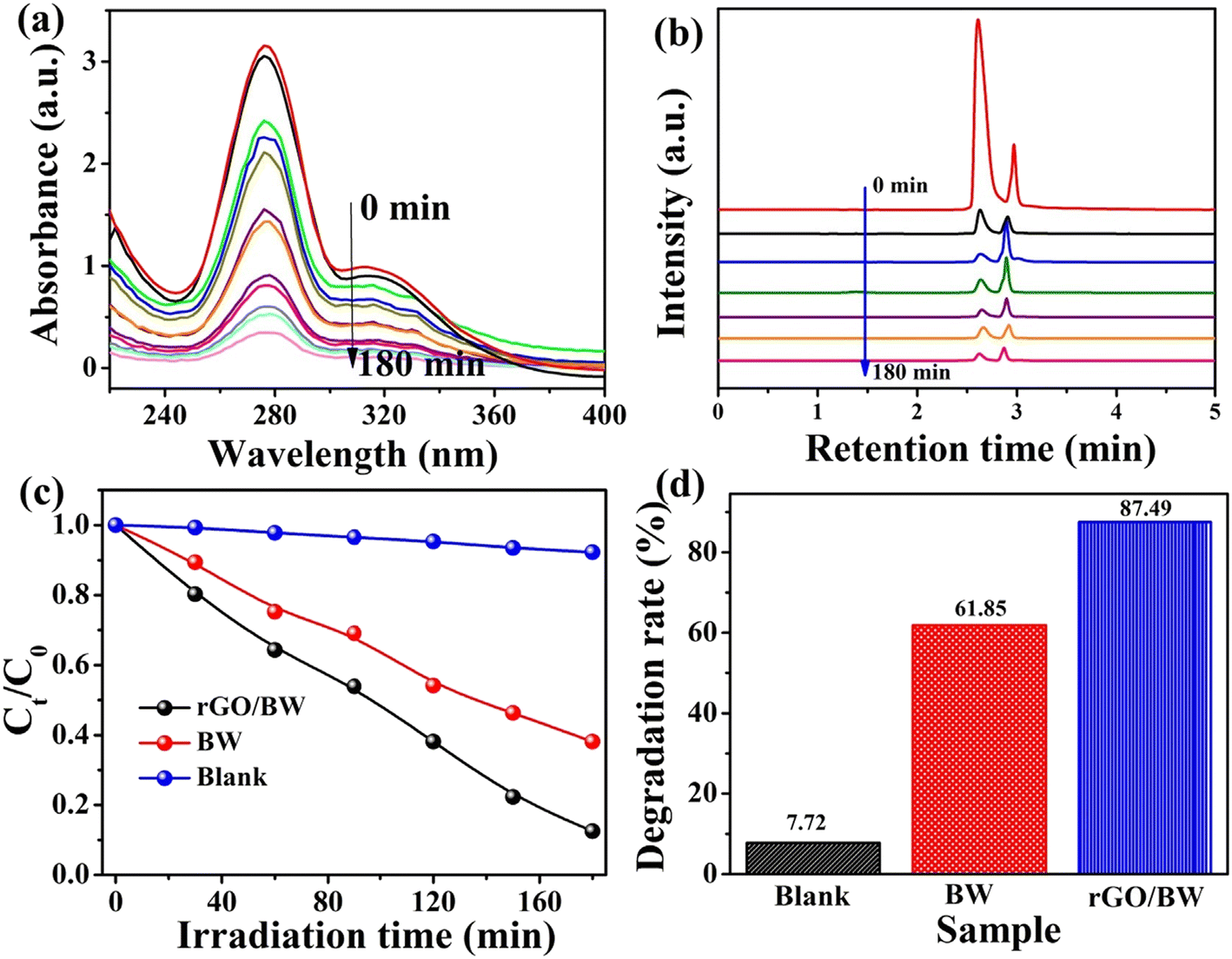 | ||
| Fig. 15 (a) The time-dependent UV spectrum, (b) the time-dependent-HPLC spectrum, (c) the photodegradation curve, and (d) photocatalytic degradation rate of norfloxacin. Reproduced from ref. 394 with permission from Elsevier (2021). | ||
Wu et al.396 reported a UV-assisted nitrogen-doped reduced graphene oxide/Fe3O4 composite by a simple hydrothermal–co-precipitation method and investigated the degradation of norfloxacin with activated peroxodisulfate. These findings demonstrated 100% degradation efficiency of norfloxacin (pH: 3.0) within 13 min due to an excellent synergistic effect at m(NGO–Fe3O4):m(PDS) of 4:1, and concentrations of NOR and S2O82− of 100 mg L−1 and 1 mM, respectively. According to this, in situ generated ·OH was considered to be the main active free radical. rGO-coupled manganese oxynitride,397 immobilized Ag3PO4/GO on 3D nickel foam398 and γ-Fe2O3-MIL-53(Fe)–GO399 photocatalysts also displayed efficient degradation of norfloxacin.
Z-Scheme ternary heterojunctions comprising phosphate-doped BiVO4/graphene quantum dots/P-doped g-C3N4 (BVP/GQDs/PCN) produced an 86.3% degradation rate for norfloxacin under visible light.405 Such an excellent performance of the photocatalyst is guided by interfacial charge transfer efficiency and a broadened visible-light response range compared to binary type-II heterojunction phosphate-doped BiVO4/PCN. CoWO4 nanoparticles assembled with g-C3N4 nanosheets fabricated by a hydrothermal method showed 3.18 and 2.69 times higher photocatalytic degradation of norfloxacin under visible light compared to g-C3N4 and CoWO4, respectively.406 Such enhanced performance of CoWO4/g-C3N4 is attributed to the synergism between CoWO4 and g-C3N4 inhibiting the fast recombination of photogenerated electron–hole pairs. Investigations involving radical scavengers suggested that ·OH rather than O2−˙ plays a dominant role in the degradation of norfloxacin. Fig. 16 shows the possible mechanism responsible for the photodegradation of norfloxacin by this synthesized CoWO4/g-C3N4, a phenomenon driven through a Z-scheme mechanistic pathway.
 | ||
| Fig. 16 Schematic illustration of possible Z-scheme photocatalytic mechanism. Reproduced from ref. 406 with permission from Elsevier (2019). | ||
A Bi2Sn2O7/heated perylene diimide (PDIH) Z-scheme heterojunction photocatalyst reached 98.71% degradation of norfloxacin in 90 min under visible light.407 The apparent rate constant of norfloxacin was found to be 3.65 and 20 times those of PDIH and Bi2Sn2O7, respectively. The fabricated Bi2Sn2O7/PDIH heterojunction catalyst also facilitated the separation of charge carriers and preserved the redox capability. In another study, piezo-photocatalytic degradation of norfloxacin by the S-scheme heterojunction BaTiO3/TiO2 was found to be 91.7% (60 min) with a rate constant of 43 × 10−3 min−1.408 Free radical trapping investigations indicated h+ and ·OH to be the main active species in the degradation process. The heterojunction also showed excellent stability and cyclability, as evident after 5 cycles. An LaFeO3/g-C3N4 heterojunction showed 95% photocatalytic degradation of norfloxacin under visible light in 180 min, which was found to 9.32 times higher than pristine g-C3N4.409 Zhang et al.410 prepared an optimized AgBr (3%)/LaNiO3 (30%)/g-C3N4 (100%) dual Z-scheme composite system via ultrasound-assisted hydrothermal method considering energy band matching and observed 92% photodegradation of norfloxacin within two hours under visible light owing to a synergistic effect. These studies also indicated an almost unaltered photodegradation rate (>90%) even after six cycles.
Ag3PO4/CNTs exhibited an efficiency of about 93% for the photoelectrocatalytic degradation of NOR within 30 min.411 This is explained based on the Z-scheme mechanism that significantly promoted the separation of electron–hole pairs. Further, h+ and ·O2− made a major contribution to the degradation process to oxidize NOR. An oxygen-vacancy-rich CuWO4/BiOCl composite exhibited excellent photocatalytic degradation of norfloxacin (96.69%) in 120 min under a 300 W xenon lamp due to a Z-scheme structure compared with pure CuWO4 and oxygen-vacancy-rich BiOCl.412 A dual Z-scheme mechanism has been proposed for Ag (0.3 wt%)@BiPO4/BiOBr/BiFeO3 that enabled 98.1% and 99.1% degradation of norfloxacin (20 mg L−1) in 90 min and in less than 45 min under visible and UV light exposure, respectively.413 It is suggested that the synergistic effects of ternary nanoheterostructures heterojunctions, electron capture and the surface plasmon resonance effect of Ag lead to such high photocatalytic activity. Immobilized Z-scheme CdS/Au/TiO2 nanobelts displayed 64.67% (60 min) degradation of norfloxacin under xenon-light-simulated sunlight irradiation which was ascribed to the synergistic effect.414
The formation of an S-scheme in the heterojunction of a photocatalyst facilitates the separation of photogenerated electron–hole pairs and reduces the recombination of charge carriers. In view of this, an S-scheme heterojunction comprising N–ZnO/g-C3N4 prepared by calcining ZIF-L/g-C3N4 in a mass ratio of 15% showed more than 90% degradation of norfloxacin in 90 min under a visible system.415 The corresponding rate constant was 4.15 times and 4.65 times higher than g-C3N4 and N–ZnO, respectively. The effective light capture capacity and migration and separation of carriers accounted for such behavior. Further, holes and superoxide radicals are reported to be the active species in the photodegradation of norfloxacin. The degradation rate of norfloxacin on a 10% g-C3N4/Bi8(CrO4)O11 heterojunction photocatalyst is about 1.38 and 2.33 times higher than that of pure Bi8(CrO4)O11 and g-C3N4, respectively.416
Efficient photocatalytic performance for norfloxacin degradation has also been reported in chitosan/TiO2@g-C3N4,417 AgI/MFeO3/g-C3N4 (M: Y, Gd, La),418 Bi2Sn2O7/g-C3N4,419 Ag/graphitic carbon nitride quantum dots (CNQDs)/g-C3N4,420 BiOBr/iron oxides,421 and CdS QDs/CaFe2O4@ZnFe2O4422 photocatalysts.
Table 6 records the performance data of different photocatalysts on the removal of norfloxacin from wastewater.
| Photocatalyst | Preparation | NOR | Catalyst dose | pH | Light type | Degradation (time) | Rate constant |
|---|---|---|---|---|---|---|---|
| TiO2−x361 | Combustion method | 100 μM L−1 | 0.1 g L−1 | 7 | Xenon lamp: 300 W (>400 nm) | ∼100% (240 min) | 0.0361 min−1 |
| Cu2O363 | Hydrothermal | 20 mg L−1, (50 mL) | 50 mg | — | Xenon lamp (500 W) | 79.87% (210 min) | 0.0081 min−1 |
| Bi2WO6 with [Fe3+]: 0.3 mmol L−1364 | Ultrasonic spray pyrolysis | 0.0313 mM L−1 (100 mL) | 0.5 g L−1 | 9 | Xenon lamp: 300 W | 89.7% (20 min) | 0.1006 min−1 |
| TiO2/Ti film with exposed {001} facets (HF: 0.02 M)365 | Hydrothermal | 10 mg L−1 | — | 2.62 | Low-pressure mercury lamp (10 W), λmax: 254 nm | 70.5% (90 min) | 0.0504 min−1 |
| ZnO nanoflowers366 | Sol–gel method | 10 mg L−1 | 0.1 g L−1 | 11 | Fluorescent lamp: 8 W (0.55 mW cm−2) | ∼72% (100 min) | 3.93 × 10−2 min−1 |
| Triangular Ag nanoplates coated ZnO nanoflowers366 | Sol–gel method | 10 mg L−1 | 1.0 g L−1 | 11 | Fluorescent lamp (8 W), 0.55 mW cm−2 | ∼97% (100 min) | 3.93 × 10−2 min−1 |
| Triangular Ag nanoplates coated ZnO nanoflowers367 | Hydrothermal method and dual-reduction method | 10 ppm (3 mL) | — | — | Fluorescent lamp (8 W), 0.55 mW cm−2 | 92.2% (270 min) | 9.2 × 10−3 min−1 |
| Prussian blue doped CeO2 (ratio: 10%) with H2O2: 9 mM368 | Physical and chemical loading approaches | 16 mg L−1 (50 mL) | 0.6 g L−1 | 6 | W fluorescent lamp (0.55 mW cm−2) | 88.93% (30 min) | — |
| N doped TiO2370 | Hydrothermal method | 6.03 mg L−1 | 0.54 g L−1 | 6.37 | Xenon lamp (300 W), 350–780 nm, 150 mW cm−2 | 99.53% (30 min) | — |
| Al (1 Mol%)-doped TiO2 nanoflakes371 | Solvothermal | 2 × 10−4 M | 15 mg (50 ml) | 10.1 | Visible light | 93% (120 min) | 0.0143 min−1 |
| C–TiO2372 | Solution phase carbonization method | 0.0094 mM | 0.2 g L−1 | Neutral | Low-pressure mercury lamps (420 nm) | ∼100% (70 min) | 5.44 × 10−4 [NFX]0-1 + 0.10 [C–TiO2] − 1.99 × 10−2 min−1 |
| Bi3+ and Fe2+ doped ZnO374 | Sol–gel method | 10.0 mg L−1 | 1.0 g L−1 | 8 | Xenon lamp (300 W), 45.2 mW cm−2 | 80% (120 min) | — |
| Bi3+ and Fe2+ doped ZnO (0.2 mM HSO5−)374 | Sol–gel method | 10.0 mg L−1 | 1.0 g L−1 | 8 | Xenon lamp (300 W), 45.2 mW cm−2 | 99% (120 min) | 9.8 × 109 M−1 s−1 (·OH), 9.0 × 109 M−1 s−1 (SO4·−) |
| [FeII(dpbpy)(phen)2]/TiO2379 | Hydrothermal | 0.313 mM | 1 g L−1 | 5 | Xenon lamp (300 W), λ > 420 nm, 140 mW cm−2 | 98.5% (180 min) | 0.0412 min−1 |
| Ag2O/TiO2–zeolite380 | Sol–gel method | 5 mg L−1 (100 mL) | 50 mg | — | Xenon lamp (35 W), 6.7 mW cm−2 | 98.7% (60 min) | — |
| FeVO4/Fe2TiO5 (2:1)381 |
One-pot hydrothermal method | 10 mg L−1 (50 mL) | 0.05 g | — | 500 W Xe lamp | 95% (30 min) | — |
| Ag/AgCl–CeO2 (Ag mass ratio: 13.94 wt%)383 | Via urea hydrolysis and calcination | 10 mg L−1 (50 mL) | 30 mg | — | Xe lamp: 300 W (equipped with a UV cut-off filter) | 91% (90 min) | 0.02279 min−1 |
| ZnO/ZnS@biochar (ZnSO4/poplar sawdust ratio: 1:1)384 |
Impregnation-roasting method | 0.025 g L−1 (50 mL) | 0.5 g L−1 | 7 | UV-light | 95% (180 min) | 0.021 min−1 |
| Ag/AgCl/Ag2MoO4387 | In situ photoreduction | 10 mg L−1 (50 mL) | 30 mg | — | Xenon lamp: 300 W, (λ > 420 nm) | ∼65% (90 min) | — |
| ZnO/g-C3N4–Fe3O4390 | Hydrothermal | 8.61 mg L−1 | 1.43 g L−1 | 7.12 | Xenon lamp with 280 nm UV filter | >90% (120) min | 0.0117 min−1 |
| CeO2/g-C3N4 (mass ratio of CeO2 to g-C3N4:5 and PS: 5 mM)391 | Mixing method | 10 mg L−1 (50 mL) | 0.05 g | 2 | 150 W high-pressure xenon lamp with cut-off λ of 420 nm | 88.6% (60 min) | 0.03573 min−1 |
| NiWO4 nanorods/g-C3N4392 | Hydrothermal followed by sonication | 10 mg L−1 | 50 mg (100 mL) | — | W lamp (visible light), 150 mW cm−2 | 97% (60 min) | 0.0547 min−1 |
| rGO/Bi2WO6394 | Hydrothermal | 10 mg mL−1 (100 mL) | 50 mg | — | Xenon lamp (300 W) | 87.79% (180 min) | — |
| N–TiO2/graphene395 | Three-step method | 30 mg L−1 (20 mL) | — | — | Mercury lamp (250 W), 365 nm | 50% (160 min) | 0.0051 min−1 |
| N-doped rGO/Fe3O4 [m(N–GO–Fe3O4):m(peroxodisulfate) = 4:1]396 |
Hydrothermal-co-precipitation | 100 mg L−1, S2O82−: 1 mM | 1 g L−1 | 3 | UV lamp: 15 W, 254 nm, 44 μW cm−2 | 100% (13 min) | 0.238 min−1 |
| Ni foam supported Ag3PO4/GO (16.78 wt%)398 | Dip-coating | 15 mg L−1 (120 mL) | — | — | Xenon lamp (250 W) with 400 nm cut-off filter, 100 mW cm−2 | 83.68% (100 min) | 0.426 min−1 |
| γ-Fe2O3-MIL-53(Fe)–GO399 | Multiple steps | 10 mg L−1 | 20 mg | — | 500 W Xe lamp (100 mW cm−2), (420 nm cut-off filter) | 92.8% (90 min) | — |
| Ni-doped ZnO/MWCNTs400 | Dispersion method | 100 mgL−1 (100 mL) | — | 6.8 | UV | 100% (200 min) | — |
| Visible | 100% (160 min) | ||||||
| Bi contained glass–ceramic401 | Multiple steps | 20 mg L−1 (20 mL) | 20 mg | — | UV-vis–NIR | ∼53% (180 min) | 6.76 × 10−3 min−1 |
| Bi contained glass–ceramic401 | Multiple steps | 20 mg L−1 (20 mL) | 20 mg | — | Visible | ∼35% (180 min) | 2.52 × 10−3 min−1 |
| Bi contained glass–ceramic401 | Multiple steps | 20 mg L−1 (20 mL) | 20 mg | — | UV | ∼52% (180 min) | 4.05 × 10−3 min−1 |
| LaOCl/LDH404 | Precipitation method | 10 mg L−1 (50 mL) | 20 mg | 7 | Xenon lamp: 300 W | 85% (80 min) | 0.014 min−1 |
| Phosphate-doped BiVO4/graphene quantum dots/P-doped g-C3N4405 | Hydrothermal | 20 mg L−1 (50 mL) | 50 mg | 9.6 | Xenon lamp (300 W) with a 420 nm cut-off filter | 86.3% (120 min) | 0.0148 min−1 |
| CoWO4/g-C3N4406 | Hydrothermal method, followed by ultrasonication | 10 mg L−1 (100 mL) | 50 mg | — | 250 W halogen lamps (visible light) | 91% (80 min) | 0.0283 s−1 |
| LaFeOx/g-C3N4409 | Ultrasound assisted hydrothermal method | 20 mg (100 mL) | 20 mg L−1 | — | Xenon lamp with 420 nm cut-off filter | 95% (180 min) | 0.01371 min−1 |
| 3 wt% AgBr/30 wt% LaNiO3/100% g-C3N4410 | Ultrasound-assisted hydrothermal method | 20 mg L−1 (100 mL) | 20 mg | 7 | Xenon lamp (500 W) with a 420 nm cut-off filter | 92% (120 min) | 0.01790 min−1 |
| 0.3 wt% ag@BiPO4/BiOBr/BiFeO3413 | Precipitation-wet impregnation-photo deposition method | 20 mg L−1 | 0.3 g | 7.3 | Visible | 98.1% (90 min) | 0.04123 min−1 |
| 0.3 wt% ag@BiPO4/BiOBr/BiFeO3413 | Precipitation-wet impregnation-photo deposition method | 20 mg L−1 | 0.3 g | 7.3 | UV | 99.1% (45 min) | 0.07023 min−1 |
| Immobilized CdS/au/TiO2414 | Multiple steps | 5 mg L−1 (35 mL) | 4 cm3 | — | Xenon lamp (35 W) | 64.67% (60 min) | 0.018 min−1 |
| AgI/LaFeO3/g-C3N4418 | Ultrasound-assisted hydrothermal approach | 20 mg L−1 (100 ml) | 0.2 g | — | Xenon lamp (500 W), (40 mW cm−2) | 95% (180 min) | 0.0188 min−1 |
| 20% Bi2Sn2O7/g-C3N4419 | Ultrasound-assisted hydrothermal method | 20 mg L−1 (100 mL) | 0.02 g | — | 500 W xenon lamp with a UV cut-off filter | 94% (180 min) | 0.01261 min−1 |
| BiOBr/iron oxides421 | In situ co-precipitation method | 10 mg L−1 (50 mL) | 0.5 g | ∼7 | 800 W xenon lamp with 420-nm cut-off filter | 99.8% (90 min) | ∼0.076 min−1 |
3.6 Ciprofloxacin
Ciprofloxacin (CIP) is a synthetic antimicrobial agent of the fluoroquinolone class and considered to be a very promising and efficacious drug for use in the treatment of various community-acquired and nosocomial infections.360,423,424 It is not easily biodegradable and is considered a potential risk to human health. The presence of ciprofloxacin in water acts as pollutant and can be removed by means of a photocatalytic approach.425–5243.6.1.1 TiO2. The photocatalytic degradation of ciprofloxacin as a micropollutant in water has been receiving considerable attention in the presence of metal oxides. Zeng et al.424 used carbon-dot-doped TiO2 to investigate the kinetics, mechanism and pathway following heterogeneous photocatalytic ozonation degradation of ciprofloxacin. It was noted that 1.0 wt% introduction of carbon dots enhanced the degradation of CIP by 91.1% compared to pristine TiO2 (64%) in 30 min. Several studies have been made on ciprofloxacin degradation using commercial TiO2 as a photocatalyst irradiated with simulated solar light,425,426 artificial sunlight,426 simulated sunlight427 and UVA/LED428 and UVC radiation.429 TiO2 nanoparticles irradiated with UVA light demonstrated removal of ciprofloxacin (300 μg L−1) from water in less than 6 minutes.430 The hydrothermally synthesized mesoporous TiO2 exhibited 96% photocatalytic degradation of ciprofloxacin hydrochloride (CIP·HCl) under artificial sunlight compared to that prepared by calcination of a titanium glycolate precursor and subsequent hydrothermal-calcination.431 This is ascribed to the higher electron–hole separation and charge transfer capability.
Li et al.432 fabricated 3D tripyramid TiO2 (TP-TiO2) architectures and rod-like morphology of TiO2 (RL-TiO2) and studied their application in the photocatalytic degradation of ciprofloxacin hydrochloride under UV-vis-light irradiation. They observed relatively superior removal efficiency (90% within 60 min) for ciprofloxacin and its significantly higher rate constants in the presence of TP-TiO2 compared to RL-TiO2. This is ascribed to the key role played by superoxide radicals and photogenic holes in the degradation of ciprofloxacin. Usman et al.433 used TiO2 nanoparticles (50 mg) in the ∼91% degradation of ciprofloxacin aqueous solution (pH: 5.5) on irradiation by a white mercury UV lamp for 5 hours.
3.6.1.2 ZnO and other oxides. ZnO (125 nm) is found to be a very effective photocatalyst in removing 300 μg L−1 ciprofloxacin from aqueous solution treated by UVA in less than 6 minutes.430 ZnO nanoparticles prepared by a chemical precipitation method on irradiation with UV light (365 nm) for 60 min degraded ciprofloxacin (∼48%) in aqueous solution (pH: 10) and also followed pseudo-first-order kinetics (∼0.00437 min−1).434 ZnO nanoparticles synthesized by a sol–gel method were used to examine the degradation of ciprofloxacin in contaminated water under UVC light.435 These findings showed complete photodegradation in 140 minutes corresponding to an initial concentration of ciprofloxacin of 10 mg L−1, pH 5, ZnO loading of 0.15 g L−1 and irradiation time of 140 min. According to Ulyankina et al.,436 UVA-irradiated ZnO nanoparticles synthesized by a pulse alternating current electrochemical method reached 93.6% removal efficiency in 30 min under optimal conditions (initial CIP concentration: 5 mg L−1, pH: 6.5, catalyst dosage: 0.5 g L−1, UV light intensity: 2.0 mW cm−2). Such performance of ZnO nanoparticles is attributed to their higher surface area and increased charge carrier separation compared to commercial ZnO. In another study, ZnO nanoparticles prepared by chemical precipitation immobilized on a glass plate showed 69.5% degradation efficiency for an aqueous solution (pH: 6.8) of ciprofloxacin (10 mg L−1) under UVC irradiation (180 min).437 A ZnO nanostructure prepared by a pyrolysis method achieved 95.5% ciprofloxacin degradation in 60 min under visible light.438
A ZnO nanotube photocatalyst on irradiation with the terrestrial solar spectrum showed about 2.9 times faster degradation of ciprofloxacin compared to TiO2 Degussa P25.439 The flower-like ZnO architectures assembled with nanorods displayed 96% efficiency (240 min) for the degradation of ciprofloxacin (initial conc.: 0.015 μM) in aqueous solution under a UV lamp as a light source.440 Finčur et al.441 undertook comparative studies by examining the photocatalytic properties of TiO2, ZnO and MgO nanopowders prepared by a sol–gel method in the removal of ciprofloxacin from water under UV/simulated sunlight. The corresponding efficiencies of 93.4%, 86.9% and 59.6% suggested TiO2 to be most efficient nanopowder for this. The photocatalytic activity of CdO nanoparticles synthesized via a green route imparted 95% degradation of ciprofloxacin in aqueous media under sunlight (60 minutes).442 In another work, ZnO nanorod irradiated with UV lamp recorded 92% degradation of ciprofloxacin in 60 minutes.443
TiO2 modified with monometallic and bimetallic nanoparticles comprising 1.5%-Au/TiO2, 1.5%-Ag/TiO2, 1.0%-Cu/TiO2, 1%Au–0.5%Ag/TiO2 and 1.0%Au–0.5% Cu/TiO2 were fabricated by a deposition–precipitation method and used as photocatalysts in the degradation of ciprofloxacin in pure water under UVC-light irradiation.447 These investigations revealed 100% degradation of ciprofloxacin for all these modified TiO2 catalysts corresponding to 60, 30, 60, 90 and 45 min, respectively. This is ascribed to the lower recombination of the hole–electron pairs arising from the electron trap effect by metal nanoparticles.
A ZnO-modified g-C3N4 photocatalyst removed 93.8% ciprofloxacin from water, corresponding to an amount of 0.05 g L−1 and pH value of 8.452 Further studies have shown the degradation rate of ciprofloxacin by ZnO-doped g-C3N4 to be 4.9 times faster than that of undoped g-C3N4. The photocatalyst also exhibited high reusability, as evident from 89.8% efficiency after 3 cycles. Boron-doped TiO2 and cerium-doped TiO2 demonstrated about 90–93% photocatalytic degradation of ciprofloxacin and norfloxacin under solar light.373 Such enhanced photocatalytic activity was explained on the basis of the narrowed band gap and electron–hole separation. In addition, metal-doped metal oxides, such as Fe0/TiO2,453 Fe-doped ZnO454 Zn-doped Cu2O,455 and Cu-doped ZnO,456 have also been successfully reported in the photodegradation of ciprofloxacin.
Several investigations have also been reported on co-doped metal oxides for their applications as photocatalysts in the removal of ciprofloxacin from water. According to Nguyen and others,457 the UV-visible-light-driven photocatalytic degradation of ciprofloxacin hydrochloride (30 mg L−1) by N,S-co-doped TiO2 exhibited a removal efficiency of 78.7% at pH 5.5 for a catalyst dose of 0.05 g. The synthesized N,C-co-doped TiO2 under optimum conditions demonstrated the highest photocatalytic activity in the removal of ciprofloxacin in water under visible light.458 It was concluded that photogenerated holes and superoxide radicals play an active role in the degradation of ciprofloxacin. ZnO nanowires doped with copper and cerium oxides displayed 88.9% removal of ciprofloxacin under UV irradiation.459
Teixeira et al.466 made an assessment of the optimization and reusability of Fe3O4/SiO2/TiO2 magnetic photocatalytic particles in the degradation of ciprofloxacin. These studies have shown 95% degradation of ciprofloxacin (pH: 5.5) after 90 min under UV with no significant loss even after five uses. Ternary core–shell Fe3O4/SiO2/TiO2 nanocomposite photocatalysts showed good synergistic properties on the removal efficiency for ciprofloxacin under UVA-light irradiation.467 The photocatalytic degradation of ciprofloxacin hydrochloride by Ag–SrTiO3/TiO2 composite nanostructures under simulated sunlight resulted in 97.6% degradation of ciprofloxacin due to an increase in the carriers and separation between electron–hole pairs.468
Metal oxide/hydroxyapatite,469 CuFe2O4@methyl cellulose,470 TiO2-modified Bi2MoO6471 and Ag2O/Ag2CO3/MWNTs472 have also been examined successfully as composite photocatalysts for the enhancement of ciprofloxacin degradation in water under UV, UVC and visible light, respectively.
3.6.5.1 g-C3N4 and carbon-dot-based composites. Hernández-Uresti et al.333 used polymeric g-C3N4 powder and observed 60% degradation of ciprofloxacin in aqueous solution (pH: 5.5) after 240 min under UV-vis irradiation. Recent studies on exfoliated g-C3N4 (2 g L−1) showed 78% degradation of ciprofloxacin (20 ppm) irradiated under solar light for 1 h.473 In another finding, a 3D g-C3N4/TiO2/kaolinite heterogeneous composite displayed ∼92% degradation efficiency for ciprofloxacin in 240 min under visible-light irradiation.474 This is ascribed to the larger surface area and the availability of more reactive sites, and the efficient separation and longer lifetimes of photogenerated electron–hole pairs. Chuaicham et al.475 observed 98% decomposition of ciprofloxacin (10 mg L−1) within 120 min after irradiation with visible light of a Zn–Cr layered double oxide/fly ash composite photocatalyst in aqueous conditions. The formation of new electronic levels accounted for such enhanced photocatalytic performance. In situ synthesized 3D g-C3N4/La–N–TiO2 also showed complete degradation of ciprofloxacin (5 mg L−1 starting concentration) at a pH of about 6.5 in about 60 min under exposure to simulated solar light.476 Carbon dots/Bi4O5Br2477 nanocomposites also displayed improved visible-light photocatalytic degradation of ciprofloxacin.
3.6.5.2 Composites of graphene oxide and graphene. Graphene oxide and reduced graphene have been used to fabricate binary and ternary composites and they have been used as photocatalysts in the removal of ciprofloxacin from water. Sponza et al.478 prepared nano-GO–Fe3O4 nanocomposites by adding water-dispersed Fe3O4 nanoparticles to an aqueous solution of GO. This irradiated with sunlight produced 80% removal efficiency for ciprofloxacin in water under optimum conditions (initial conc. of ciprofloxacin: 1 mg L−1, original pH: 6.5, nano-GO/M concentration: 2 g L−1, irradiation time: 250 min). ZnO-particle-coated carboxyl-enriched GO (ZnO@cGO) degraded almost 100% ciprofloxacin in water (pH: 7) within about 5 min under visible irradiation (initial concentration of CIP: 25 μg mL−1, catalyst: 0.5 mg mL−1).479 It was concluded that degradation of ciprofloxacin depends mainly on O2− and h+. An rGO-supported BiVO4/TiO2 heterostructure nanocomposite achieved 80.5% degradation rate for ciprofloxacin in acidic ambient (pH: 5) within 150 min, 2.06 times higher than BiVO4/TiO2.480 A nanostructured ZnO–CdO incorporated rGO photocatalyst showed degradation of ciprofloxacin of around 99.28% in 75 min under UV light.481 This is attributed to the effective separation of charge carriers consequential on the production of more reactive oxygen species after incorporation of rGO nanosheets with ZnO–CdO.
The performance of ZnAl mixed metal-oxide (MMO)/rGOx (x: wt% of rGO) composites was tested and compared with ZnAl MMO and pure ZnAl MMO in the photodegradation of ciprofloxacin hydrochloride in aqueous solution under visible light.482 It was found to show the following order of photodegradation efficiency at the end of 2 h of irradiation time: ZnAl MMO/rGO20 (∼90.58%) > ZnAl LDH/rGO20 (∼67.74)% > ZnAl MMO (50.96%) > ZnAl LDH (36.47%). Such enhanced performance of the ZnAl MMO/rGO20 photocatalyst has been ascribed to the synergistic effect of the heterogeneous structure. The degradation mechanism of ciprofloxacin has been clearly explained based on the heterostructure that accounts for efficient charge separation and inhibition of the recombination of photogenerated carriers. It is believed that O2· radicals and h+ predominantly contribute to the degradation of ciprofloxacin. TiO2 (64.3 wt%)-pillared multilayer graphene nanocomposites showed better photodegradation efficiency of 78% than TiO2 (42%) under light-emitting diode irradiation for 150 min.483 The photodegradation followed pseudo-first-order kinetics with the rate constant of graphene/TiO2 composite about 3.89 times that of pristine TiO2. The graphene/TiO2 composite also exhibited high stability and reusability even after five consecutive photocatalytic cycles. Urus et al.484 used a GO@Fe3O4@TiO2-type core@shell@shell nanohybrid (10 mg) as a catalyst to remove 91.5% of ciprofloxacin (10 ppm) from water solution (pH: 7) after 240 min. In addition, the photocatalytic removal of ciprofloxacin has also been evaluated using 3D-structured flower-like bismuth tungstate/magnetic graphene nanoplates485 and Ag2CrO4/Ag/BiFeO3@rGO photocatalysts.486
Huo et al.487 synthesized an N-doped ZnO/CdS/graphene oxide ternary composite via a two-step method and tested its photocatalytic activity in the degradation of ciprofloxacin hydrochloride under visible light and compared it with pure CdS, N–ZnO, and N–ZnO:CdS (2:1, 1:1, 1:2, 1:3). The highest degradation rate of about 86% was shown for the 2:1 molar ratio of N–ZnO and CdS. This is explained in terms of heterostructure and the contribution from GO in N–ZnO/CdS promoting photogenerated electron transfer and suppressing the recombination of electron–hole pairs. The proposed schematic suggested that charge transfer and holes played a major role in the photocatalytic system.
Magnetic g-C3N4/MnFe2O4/graphene composites have been examined for the photocatalytic degradation of ciprofloxacin in the presence of persulfate as an oxidant under visible-light irradiation.491 Graphene-layer-anchored TiO2/g-C3N4 showed enhanced photocatalytic performance (degradation rate: 61.7%, k: 0.01675 min−1) under visible light compared to graphene-layer-anchored TiO2, g-C3N4 and g-C3N4/TiO2.492 This is explained on the basis of accumulation of g-C3N4 electrons with high reduction capability and TiO2 holes with high oxidation capability. Enhanced photocatalytic activity has also been displayed by a visible-light-driven mesoporous TiO2@g-C3N4 hollow core@shell heterojunction in the degradation of ciprofloxacin.493
A heterostructure comprising Ag nanoparticles deposited on the surface of ZnO nanoplates and Fe2O3 nanorods exhibited superior solar-light-driven photocatalytic activity in ciprofloxacin degradation (76.4%) under optimized conditions (initial ciprofloxacin concentration: 10 mg L−1; pH 4; catalyst loading: 0.3 g L−1).494 The e−, h+, ·OH and ·O2− played important roles as reactive species in the photocatalytic degradation process. The efficient separation of charge carriers and migration of e−/h+ across the heterostructure interface accounted for this. Zhao et al.495 achieved 95.6% removal of ciprofloxacin under visible-light irradiation for 40 min by a ternary Mn2O3/Mn3O4/MnO2 (molar ratio of 3:1:2) valence state heterojunction with dual heterostructures under visible light. Such a performance is derived from its enhanced surface area, light absorption and charge separation of the Mn2O3/Mn3O4/MnO2 heterostructure. Further studies established that holes and superoxide radicals play an important role in the degradation of ciprofloxacin. Other studies comprising a unique 2D/3D/2D rGO (3%)/Fe2O3 (4%)/g-C3N4 heterojunction showed almost 100% degradation of ciprofloxacin (pH: 7) compared to pristine g-C3N4 nanosheets under visible-light irradiation for 40 minutes.496 Such photocatalytic properties of a heterojunction nanocomposite system are accounted for in terms of enhanced charge migration and separation.
Chen et al.497 noted the enhanced degradation of ciprofloxacin over Bi2O3/(BiO)2CO3 heterojunctions compared to pristine (BiO)2CO3 and Bi2O3 in the presence of simulated solar light. The decay process for ciprofloxacin followed pseudo-first-order kinetics with the rate constant increasing with decreasing concentration of CIP. In addition, CdS/BiOBr,498 Cu2O/Cu2(PO4)(OH),499 Sm-doped g-C3N4/Ti3C2MXene,500 CeO2/La2O3/TiO2,501 g-C3N4/NH2-MIL-88B(Fe)502 and a polypyrrole-sensitized ZnFe2O4/g-C3N4 n–n heterojunction503 have also displayed enhanced photocatalytic degradation of ciprofloxacin.
Costa et al.504 observed ∼98% photodegradation of ciprofloxacin (initial concentration: 5 ppm) at neutral pH in the presence of a Z-scheme TiO2/SnO2 nanostructure photocatalyst. These findings also revealed the active role of oxygen singlets, holes, and superoxide radicals as the main species in the photodegradation of ciprofloxacin. Li et al.505 prepared an oxygen-vacancy-rich TiO2/Ta3N5 composite by a solvothermal method and used it as a direct Z-scheme heterojunction photocatalyst. They observed 95.7% (90 min) degradation rate of ciprofloxacin hydrochloride under visible-light irradiation. It was suggested that oxygen vacancies form an intermediate energy level in TiO2 that accounts for the separation of photogenerated electrons and holes. In addition, the formation of a Z-scheme energy band structure by oxygen-vacancy-rich TiO2 and Ta3N5 is likely to enable more photogenerated carriers to participate in the photocatalytic reaction. This was also inevitable from the excellent photocatalytic degradation of ciprofloxacin delivered by an oxygen-vacancy-rich TiO2/Ta3N5 composite under visible light. CeO2/ZnO nanocomposites prepared by a co-precipitation method displayed twice the activity in the photocatalytic degradation of ciprofloxacin compared to undoped ZnO and was ten times more active than pristine CeO2.506 Such enhanced formation of a Z-scheme heterojunction is attributed to the migration of photo-excited electrons from the conduction band of ZnO to the valence band of CeO2.
N-doped carbon quantum dot (NCQD)-decorated Bi2O2CO3 heterojunction nanosheets exhibited remarkably enhanced photocatalytic activities for ciprofloxacin photodegradation mediated by radiation in the ultraviolet to near-infrared region.507 It is suggested that NCQDs act as photosensitizers (hole reservoirs) to harvest solar light and a type-II heterojunction facilitates efficient charge carrier separation to account for this. The mechanisms and pathways of ciprofloxacin degradation mediated by different lights were also discussed. N-doped carbon dots (NCDs) decorated onto a Bi2MoO6/g-C3N4 (BMCN) nanocomposite photodegraded ciprofloxacin by 98% (30 min) under visible-light irradiation.508 It is proposed that NCDs play a role as a mediator to transfer electrons from the conduction band to the valence band of Bi2MoO6 and g-C3N4, respectively. The findings also revealed ·OH and ·O2− radicals acting as the dominant reactive species. The photocatalyst also displayed good stability and reusability after five consecutive cycles of ciprofloxacin photodegradation.
A Z-scheme involving a TiO2 nanorod/g-C3N4 (30 wt%) nanosheet nanocomposite showed 93.4% degradation of ciprofloxacin (initial concentration: 15 mmol L−1) aqueous solution (pH: 6.3) under simulated sunlight irradiation in 60 min.509 It was also concluded that h+ and ·OH played a major role in the degradation of ciprofloxacin. In another study, a biochar@ZnFe2O4/BiOBr Z-scheme heterojunction photocatalyst prepared by a solvothermal method under visible-light irradiation (λ > 420 nm) showed no significant degradation efficiency for ciprofloxacin (65.26%).510 Wen et al.511 fabricated CeO2–Ag/AgBr composite photocatalysts with a Z-scheme configuration by following the in situ interspersal of AgBr on CeO2 and subsequently studied the photodegradation of ciprofloxacin under visible-light irradiation (Fig. 17(a)). According to this, CeO2 itself has almost no ability to degrade ciprofloxacin, though it can be partly eliminated in the presence of pristine Ag/AgBr. However, CIP concentration decreased further to some extent for CeO2 decorated with Ag/AgBr in CeO2–Ag/AgBr composites with 21.26 wt% of Ag (denoted CAB-21.26) exhibiting the most pronounced photocatalytic activity. This is ascribed to the accelerated interfacial charge transfer process and the improved separation of the photogenerated electron–hole pairs. Furthermore, the kinetic behavior followed pseudo-first-order kinetics and exhibited higher k-values for the CeO2–Ag/AgBr hybrids (Fig. 17(b)). Another Z-scheme-based AgBr/Ag/Bi2WO6 heterostructure achieved 57% (5 h) photocatalytic degradation of ciprofloxacin under visible-light irradiation in pure water.512 Such a performance was ascribed to the synergistic effect of the AgBr/Ag/Bi2WO6 heterostructure compared to its single components.
 | ||
| Fig. 17 (a) Photocatalytic degradation CIP curves and (b) apparent rate constants for the degradation of CIP solution for a CAB-21.26 sample. Reproduced from ref. 511 with permission from Elsevier (2018). | ||
Z-Scheme-guided g-C3N4/Bi2WO6,513 Fe3O4/Bi2WO6,514 g-C3N4/Ti3C2/MXene/black phosphorus,515 g-C3N4/Ag3PO4/chitosan,516 Ag/AgVO3/g-C3N4,517 CeO2/Co3O4 p–n hetrojunctions,518 Bi nanodots/2D Bi3NbO7 nanosheets,519 Bi2WO6/Ta3N5,520 g-C3N4@Cs0.33WO3,521 ZnO/SnS2,522 g-C3N4/rGO/WO3,523 and CuS/BiVO4524 have also displayed enhanced photocatalytic degradation of ciprofloxacin.
Table 7 records the performance data of different photocatalysts on the removal of norfloxacin from wastewater.
| Photocatalyst | Preparative method | CIPa/CIP·HClb | Catalyst dose | pH | Light source | Degradation (time) | Rate constant |
|---|---|---|---|---|---|---|---|
| P25 TiO2 (anatase:rutile = 80:20), [H2O2]: 82.5 mg L−1425 |
Commercial | 0.030 mmol L−1a (500 mL) |
0.5 g L−1 | 6 | Simulated solar irradiation, 800 W xenon lamp | ∼100% (90 min) | 0.022 min−1 |
| Degussa P-25 TiO2 (80:20% w/w anatase-to-rutile)426 | Commercial | 100 mg L−1b |
1 g L−1 | 9 | Simulated solar irradiation (850 W cm−2) | ∼100% (160 min) | 0.108 min−1 |
| Degussa P-25 TiO2 (80:20% w/w anatase-to-rutile)428 |
Commercial | 20 mg L−1a (100 mL) |
100 mg L−1 | 6.0 | UVA/LED lamp (3 W), 10 mW cm−2, λ > 365 nm | — | 0.2217 ± 0.0179 min−1 |
| TiO2 (80% anatase and 20% rutile) immobilized on glass plates429 | Multiple steps | 60 μmol L−1a (500 mL) |
TiO2 (7.5 g L−1) | 9 | UVC lamp: 15 W 254 nm | ∼98% (120 min) | ∼25 × 10−3 min−1 |
| TiO2 P25 and ZnO430 | Commercial | 300 μg L−1a (50 mL) |
1 g L−1 | — | UVA (1.6 to 1.7 mW cm−2) | 100% (6 min) | — |
| Mesoporous TiO2 nanoparticles431 | Hydrothermal | 160 mg L−1b (40 ml) |
0.01 g | — | Xenon lamp (500 W), 200–1000 nm | 96.05% (360 min) | 0.45 min−1 |
| 3D tripyramid TiO2 architectures432 | Hydrothermal method | 32.6 μMa (50 mL) | 5 mg | — | UV-vis light | 90% (60 min) | 4.03 × 10−2 min−1 |
| ZnO nanoparticles434 | Chemical precipitation method | 4 mg L−1a (3 mL) |
20 mg L−1 | 10 | Xenon lamp (365 nm) | ∼48% (60 min) | 0.0043 ± 0.003 min−1 |
| ZnO nanoparticles435 | Sol–gel method | 10 mg L−1a |
0.15 g L−1 | 5 | Low-pressure mercury-vapour lamps (9 W) | 100% (140 min) | 0.032 min−1 |
| Nano-ZnO436 | Pulse electrochemical synthesis | 5 mg L−1a |
0.5 g L−1 | 6.5 | UV light (2.0 mW cm−2) | 93.6% (30 min) | — |
| Immobilized ZnO nanoparticles437 | Heat attachment method | 10 mg L−1a |
14 × 14 × 5 cm3 | 6.8 | UV lamp (15 W, 42 W m−2) | 69.5% (180 min) | ∼0.008 min−1 |
| ZnO nanotubes439 | Modified published protocol | 2 × 10−5 mol L−1a (0.4 L) |
14 mg | 8.0 | 300 W xenon lamp with AM1.5 filter (1000 W m−2) | 12% (120 min) | 9.61 × 10−4 min−1 |
| Flower-like ZnO440 | Thermionic vacuum arc | 0.015 μMa | ZnO deposited on 2 × 2 cm2 (Si wafer) | — | UV lamp, 1 W m−2, 253.7 nm | 96% (240 min) | 14.8 × 10−3 min−1 |
| TiO2441 (NH4)2S2O8: 0.125 mM | Sol–gel method | 0.05 mMa | 0.5 mg mL−1 | — | High-pressure Hg lamp (125 W), 1.4 × 10−2 W cm−2 in UV region | 93.4% (60 min) | — |
| ZnO441 | Sol–gel method | 0.05 mMa | 0.5 g L−1 | — | High-pressure Hg lamp (125 W) in UV region, 1.4 × 10−2 W cm−2 | 86.9% (60 min) | — |
| CdO442 | Green approach | 10 ppma (50 mL) | 50 mg | — | Sunlight | 95% (60 min) | 0.04722 min−1 |
| ZnO–Ag-Graphite443 | Hydrothermal method | 5 mg L−1a (50 mL) |
0.3 g L−1 | — | 24 W UV lamp, λ: 254 nm | 98% (60 min) | 0.05983 min−1 |
| 2.5% N-1.5% Fe–TiO2444 | Hydrothermal method | 20 mg L−1a (300 mL) |
0.3 g | — | LED illumination source | 70% (360 min) | 5.52 × 10−3 min−1 |
| Ag nanoparticles@TiO2445 | Sonicating TiO2 and aq. AgNO3 + aq. Na2CO3 | 1.0 mMa (100 mL) | 1.0 mg L−1 | 7 | UV light (120 W Hg lamp) | 85.21% (14500 s) |
1.53 mM s−1 |
| Ag nanoparticles@TiO2445 | Sonicating TiO2 and aq. AgNO3 + aq. Na2CO3 | 1.0 mMa (100 mL) | 1.0 mg L−1 | 7 | Sunlight | 75.58% (14500 s) |
1.210 mM s−1 |
| Mesoporous Cu (0.1 wt%)@TiO2446 | Reduction method | 40 mg L−1b (40 mL) |
0.01 g | — | 500 W xenon lamp (sunlight) | ∼100% (3 h) | 1.16 h−1 |
| 1.5%-Au/TiO2447 | Deposition–precipitation method | 30 mg L−1a (250 mL) |
0.5 g L−1 | — | UVC light irradiation (15 W low-pressure Hg lamp, 254 nm 44 W m−2) | 100% (60 min) | 0.06 min−1 |
| 1.5%-Ag/TiO2447 | Deposition–precipitation method | 30 mg L−1a (250 mL) |
0.5 g L−1 | — | UVC light irradiation (15 W low-pressure Hg lamp, 254 nm 44 W m−2) | 100% (30 min) | 0.117 min−1 |
| 1.0%-Cu/TiO2447 | Deposition–precipitation method | 30 mg L−1a (250 mL) |
0.5 g L−1 | — | UVC light irradiation (15 W low-pressure Hg lamp, 254 nm 44 W m−2) | 100% (60 min) | 0.072 min−1 |
| 1% Au–0.5% Ag/TiO2447 | Deposition–precipitation method | 30 mg L−1a (250 mL) |
0.5 g L−1 | — | UVC light irradiation (15 W low-pressure Hg lamp, 254 nm 44 W m−2) | 100% (90 min) | 0.053 min−1 |
| 1.0% Au–0.5% Cu/TiO2447 | Deposition–precipitation method | 30 mg L−1a (250 mL) |
0.5 g L−1 | — | UVC light irradiation (15 W low-pressure Hg lamp, 254 nm 44 W m−2) | 100% (45 min) | 0.099 min−1 |
| N (12.9%) doped–TiO2 nanorice particles448 | Hydrothermal method | 20 ppma | 0.3 g L−1 | 5.5 | UVA lamps: 20 W, 365 nm, 0.493 mW cm−2 | 94.29% (240 min) | — |
| N doped–TiO2 (N/Ti wt ratio:0.34%) immobilized on glass spheres449 | Sol–gel method followed by immobilization | 20 mg L−1a (20 mL) |
3 g L−1 | — | Xenon lamp: 500 W and λ > 420 nm | 93.5% (90 min) | 0.02859 min−1 |
| P-doped TiO2 (using 50 mg NaH2PO2)450 | Heat treatment under flowing NH3 | 5 ppma (50 mL) | 25 mg | — | Visible-light irradiation | 100% (60 min) | 0.065 min−1 |
| Polyaniline doped ZrO2451 | In situ oxi. Polym. | 4 × 10−5 Ma (100 mL) | 30 mg | — | UV-light irradiation (λ > 400 nm) | 96.6% (120 min) | — |
| TiO2/Fe0453 | Liquid-phase reduction process | 30 mg L−1a |
1.0 g L−1 | 3.0 | UV-lamp: 10 W, 254 nm, 2.0 W m−2 | 94.6% (60 min) | — |
| Fe doped ZnO nanoparticles454 | Precipitation route | 5 mg L−1b |
150 mg L−1 | 9 | Sunlight, 650 W m−2, 80000 ± 3000 lux |
∼80% (210 min) | — |
| Zn-doped Cu2O (by adding 0.05 g of ZnCl2)455 | Solvothermal method | 20 mg L−1a (50 mL) |
30 mg | — | 500 W metal halide lamp, λ < 400 nm filter | 94.6% (240 min) | 0.0038 min−1 |
| N-S-doped TiO2457 | Sol–gel method | 30 ppma | 0.05 mg | 5.5 | Halogen lamp: 500 W (360–780 nm) | 78.7% (220 min) | 0.0065 min−1 |
| Graphitized mesoporous carbon–TiO2460 | Extended resorcinol-formaldehyde method | 15 mg L−1a (200 mL) |
70 mg | — | 14 W UV lamp, 254 nm | 100% (120 min) | 0.102 min−1 |
| Mo/co oxides461 | Sol–gel method | 10 mg L−1a |
1 g L−1 | 4 | Sunlight | 56.3% (180 min) | 7.9 × 10−2 min−1 |
| TiOF2/TiO2462 | Hydrothermal (160 °C) | 20 mg L−1b (50 mL) |
50 mg | — | Xenon lamp: 300 W with a UV-cut-off filter (420 nm) | ∼95% (90 min) | 0.034 min−1 |
| Core–shell 3D γ-Fe2O3@ZnO464 | Hydrothermal-sintering and atomic layer deposition | 10 mg L−1a (100 mL) |
0.5 g L−1 | 5.8 | Xenon lamp (300 W) | 92.5% (60 min) | 0.0419 min−1 |
| rGO–BiVO4–ZnO465 | Hydrothermal method | 4 × 10−5 Ma (100 mL) | 30 mg | — | W lamp (150 mW cm−2), (λ < 400 nm) | 98.4% (60 min) | — |
| Fe3O4/SiO2/TiO466 | Sol–gel synthesis (calcined at 600 °C) | 5 mg L−1a |
1 g L−1 | 5.5 | UV irradiation, (365 nm, 1.6 mW cm−2) | 95% (90 min) | 0.032 min−1 |
| Core–shell Fe3O4/SiO2/TiO2(100 °C)467 | Microwave-assisted synthesis | 10 mg dm−3a (100 cm3) |
50 mg | 6.5 | UVA lamp (365 nm) | 94.0% (120 min) | 0.0158 min−1 |
| Ag–SrTiO3/TiO2468 | Hydrothermal/photoreduction | 20 mg L−1b (50 mL) |
20 mg | — | 300 W xenon lamp | 97.6% (60 min) | 0.070 min−1 |
| TiO2/hap (with 40% by wt% of oxide:Hap)469 | Soft chemical method | 20 ppma (100 mL) | 2 g L−1 | — | HPK 125 W lamp- UV light | 100% (15 min) | — |
| ZnO/HAp (with 40% by wt% of oxide:Hap)469 | Soft chemical method | 20 mg L−1a |
2 g L−1 | — | HPK 125 W lamp-UV light | 100% (20 min) | — |
| CuFe2O4@methyl cellulose470 | Microwave-assisted method | 3 mg L−1a |
0.2 g | 7 | UVC lamps (low pressure, 6 W, Philips) | 72.87% (90 min): real sample | 0.902 min−1 |
| TiO2/Bi2MoO6 (TiO2 content: 0.41 wt%)471 | Solvothermal–calcination process | 10 mg L−1a (50 mL) |
30 mg | — | Xenon lamp 350 W with a UV cut-off filter | 88% (150 min) | ∼8 × 10−3 min−1 |
| Ag2O/Ag2CO3/MWNTs472 | Calcination (10 min) | 10 mg L−1a (100 mL) |
0.05 g | — | Xenon lamp: 300 W (visible light) | 76% (60 min) | — |
| g-C3N4333 | Polycondensation of melamine | 10 mg L−1a |
200 mg (200 mL) | — | Xenon lamp (35 W): UV-vis radiation source | 60% (240 min) | 4 × 10−5 s−1 |
| Exfoliated g-C3N4473 | Green route | 20 ppma | 1 gL−1 | — | Solar-light irradiation | 78% (60 min) | 23 × 10−3 min−1 |
| g-C3N4/TiO2/kaolinite474 | Sol–gel method/chemical stripping/self-assembly | 10 ppma (100 mL) | 0.2 g | — | Xenon lamp (90 mW cm−2 with 400 nm cut-off filter) | ∼92% (240 min) | 0.00813 min−1 |
| Zn–Cr LDH/fly ash (molar ratio = 2:1)475 |
Coprecipitation method followed by dispersion method | 10 ppma (50 mL) | 1.0 g L−1 | Xenon lamp (500 W) with UV cut-off filter | ∼98% (150 min) | — | |
| g-C3N4/La–N–TiO2476 | In situ synthetic method | 10 mg L−1a |
0.75 g L−1 | ∼6.5 | Xenon lamp; (300 W), λ > 420 nm | 96.8% (60 min) | — |
| Nano graphene oxide–magnetite478 | Mixing and dispersion | 1 mg L−1a |
2 g L−1 | 6.5 | Sunlight irradiation at 80 W power | 80% (250 min) | — |
| ZnO–CdO/rGO481 | Refluxing method | 10 mg L−1a (50 mL) |
10 mg | 7 | UV light, 800 W xenon lamp with 420-nm cut-off filter | 99.28% (75 min) | — |
| ZnAl mixed metal oxides/rGO482 | Hydrothermal combined with calcination | 10 mg L−1a (50 ml) |
10 mg | — | 800 W xenon lamp with 420 nm cut-off | 90.58% (120 min) | 0.01893 min−1 |
| TiO2 (64.3 wt%)-pillared multilayer graphene (35.7 wt%)483 | Hydrothermal | 15 mg L−1a (40 mL) |
20 mg | 5.8 | LED lamp (5 W), λ > 420 nm | 78% (150 min) | 0.99111 min−1 |
| GO@Fe3O4@TiO2484 | In situ method | 10 ppma (100 mL) | 10 mg | 7–8 | Solar simulator: 300 W | 91.5% (240 min) | 0.0079 min−1 |
| Ag2CrO4/Ag/BiFeO3@8% wt ratio of rGO486 | Dispersion method | 10 mg L−1a |
0.2 mg mL−1 | 7 | Xenon lamp (300 W) with 400 nm cut-off filter, 450 mW cm−2 | 96% (60 min) | 0.0638 min−1 |
| N–ZnO/CdS/GO487 | Hydrothermal | 15 mg L−1a (100 mL) |
50 mg | — | Xenon lamp (300 W) with λ > 420 nm | 86% (60 min) | — |
| 0.6Ag3PO4/TiO2 nanotube arrays (600 °C)488 | In situ growth method | 10 mg L−1a (40 mL) |
40 mg | — | Xenon lamp (300 W), 200 mW cm−2 | 85.3% (60 min) | 0.02499 min−1 |
| P-doped ultrathin g-C3N4/BiVO4489 | Impregnated process | 10 mg L−1a |
1 g L−1 | 6.72 | Visible-light irradiation (λ > 420 nm) | 92.6% (120 min) | 0.0203 min−1 |
| ZnO–Ag2O/porous g-C3N4490 | Hydrothermal | 20 mg L−1a (100 mL) |
50 mg | — | W lamp (500 W), λ ≥ 420 nm | 97.4% (48 min) | 0.057 min−1 |
| Graphene layers anchored TiO2/g-C3N4492 | In situ calcination method using 40 g of Ti3C2 | 3 mg L−1a (100 mL) |
60 mg | — | Xenon lamp (300 W), λ > 400 nm, 300 mW cm−2 | 61.7% (60 min) | 0.01675 min−1 |
| Ag/Fe2O3/ZnO494 | Ultrasonic-assisted hydrothermal method | 10 mg L−1a (100 mL) |
0.3 g L−1 | 4 | Solar illumination | 76.4% (210 min) | 0.3036 h−1 |
| Mn2O3/Mn3O4/MnO2495 | Hydrothermal and in situ method | 10 mg L−1a (120 mL) |
0.2 g L−1 | 7 | Xenon lamp (300 W), 900 mW cm−2 | 95.6% (40 min) | — |
| rGO/Fe2O3/g-C3N4496 | Embedding approach | 50 mg L−1a |
100 mg | 7 | Halogen lamp: 500 W | ∼100% (40 min) | 1.0878 min−1 |
| Bi2O3/(BiO)2CO3497 | Hydrothermal/calcination | 10 mg L−1a |
0.5 g L−1 (100 mL) | 7 | Xenon lamp: 300 W, 0.641 W cm−2 | 93.4% (30 min) | 0.476 min−1 |
| CdS/BiOBr-1:3498 |
Solvothermal route | 10 mg L−1a (200 mL) |
50 mg | 7 | Sunlight | 99.1% (240 min) | 0.00692 min−1 |
| Cu2O/Cu2(PO4)(OH)499 | Reflex method | 20 mg L−1a (100 mL) |
100 mg | — | Direct sunlight irradiation | ∼98% (120 min) | — |
| CeO2/La2O3/TiO2501 | Sol–gel followed by calcination | 10 ppma (50 mL) | 50 mg | 6–7 | Visible light using tungsten lamp (300 W cm−2) | 100% (120 min) | — |
| TiO2/SnO2504 | Hydrothermal and ion exchange | 2.5 × 10−3 g L−1a |
2.5 × 10−3 g | Neutral | UVC lamps with 35 W each (253 nm) | 92.8% (120 min) | 22.4 × 10−3 min−1 |
| CeO2/ZnO506 | Co-precipitation method | 15 mg L−1a (100 mL) |
0.25 g L−1 | 3.2 | 200 W mercury-xenon lamp with 365 nm filter | ∼60% (60 min) | 0.0130 min−1 |
| 5 wt% N-doped carbon quantum dots decorated Bi2O2CO3507 | Hydrothermal method | 10 mg L−1a (80 mL) |
40 mg | — | UV-vis light | 91.1% (60 min) | ∼0.0325 min−1 |
| Visible | 92.8% (60 min) | ∼0.02 min−1 | |||||
| Bi2MoO6/g-C3N4508 | Hydrothermal method | 5 mg L−1a |
1.0 g L−1 | 8 | Visible lamps (77 mW cm−2) | 98% (30 min) | 0.12 min−1 |
| TiO2 nanorod/30 wt% g-C3N4 nanosheets509 | Mixing followed by ultrasonication | 15 μmol L−1a (50 mL) |
10 mg | 6.3 | Xenon lamp: 500 W | 93.4% (60 min) | 0.0381 min−1 |
| 5 wt% biochar@ZnFe2O4/BiOBr510 | Solvothermal/photodeposition/precipitation | 15 mg L−1a |
50 mg (100 mL) | — | Xenon lamp: 300 W | 65.26% (60 min) | — |
| CeO2-21.26 wt% Ag/AgBr511 | In situ | 10 mg L−1a (50 mL) |
50 mg | — | Xenon lamp (300 W) with a UV cut-off filter | 93.05% (120 min) | 0.02011 min−1 |
| Bi2WO6/ag/AgBr512 | Precipitation followed by dispersion | 30 mg L−1a (250 mL) |
125 mg | — | Phillips lamp (50 W), λ = 380–800 nm | 57% (5 h) | — |
| g-C3N4/Bi2WO6513 | Solvothermal and grind calcination method | 15 mg L−1a (100 mL) |
0.1 g | — | Xenon lamp:300 W, λ < 400 nm | 98% (120 min) | — |
| Fe3O4/Bi2WO6 (4% iron content)514 | Hydrothermal method | 10 mg L−1a (100 mL) |
30 mg | — | Visible-light irradiation (λ > 420 nm) | ∼99.7% (15 min) | — |
| g-C3N4/Ti3C2 MXene/black P515 | Calcination process | 20 mg L−1a (100 mL) |
20 mg | — | Xenon lamp: 300 W, λ > 420 nm | >99% (60 min) | 0.048 min−1 |
| g-C3N4/Ag3PO4/chitosan516 | Multiple steps | 20 mg L−1a |
2.0 mg | 7 | Visible light | 90.34% (60 min) | 0.01771 min−1 |
| 0.5 wt% Ag/AgVO3/g-C3N4517 | Wet-impregnation method | 10 ppma (100 mL) | 0.1 g | — | Halogen lamp (500 W): visible light | 82.6% (120 min) | — |
| Bi (7%) nanodots/Bi3NbO7 nanosheets519 | Two-step wet chemical reaction | 10 mg L−1a (100 mL) |
50 mg | — | Xenon lamp: 300 W with 400 nm cut-off filter | 86% (120 min) | 0.01427 min−1 |
| Bi2WO6/Ta3N5 (1.0/1 mole ratio)520 | Electrospinning–calcination–solvothermal route | 20 mg L−1a (100 mL) |
40 mg | 3 | Xenon lamp: 300 W with a cut-off filter (λ > 400 nm), 97 mW cm−2 | 81.1% (120 min) | 0.0105 min−1 |
| g-C3N4@Cs0.33WO3521 | Solvothermal | 20 ppm | 20 mg (100 mL) | 3 | Xenon lamp (500 W), λ: 230–2500 nm, 0.25 W cm−2 | 97% (145 min) | 14.9 × 10−3 min−1 |
| g-C3N4/rGO/WO3523 | Photo reduction method | 20 mg L−1a (50 mL) |
10 mg | — | High-pressure xenon arc lamp with 400 nm cut-off filter and 100 mW cm−2 | 85% (180 min) | — |
| CuS/BiVO4 (mass ratio: 7%)524 | In situ | 10 mg L−1a (100 mL) |
100 mg | — | Xenon lamp (300 W) with a 420 nm cut-off filter | 86.7% (90 min) | 0.02151 min−1 |
3.7 Tetracycline
Tetracycline (TC) is invariably used as an antibiotic against different bacterial infections, such as urinary tract infections, acne, gonorrhea, chlamydia, mycoplasma, rickettsia, cholera, brucellosis, plague and syphilis.52 It finds extensive application in the medical field, for veterinary purposes, and as a feed additive in the agricultural sector. However, extensive applications of tetracycline mean its presence in surface water, groundwater, wastewater, domestic wastewater and other source-related environments, causing a serious threat to the environment. Therefore, several approaches have been made to develop a highly efficient approach to remove antibiotics by a photocatalysis approach.525–6623.7.1.1 TiO2. Several investigations have been reported using TiO2 as a photocatalyst in water treatment for the removal of tetracycline. According to Palominos et al.,527 an aqueous suspension of TiO2 has been used to facilitate the photocatalytic oxidation of tetracycline on irradiation with simulated solar light. Studies indicated the rapid degradation of tetracycline, undergoing 100% completion after 15 min under optimum conditions (tetracycline: 20 mg L−1, TiO2: 1.5 g L−1, pH: 8.7). The mechanism of photocatalytic tetracycline oxidation involved active roles for holes and OH radicals. The nanosized TiO2 achieved more than 95% removal of tetracycline within 40 min under UV irradiation for a tetracycline concentration of 40 mg L−1 and catalyst dose of 1000 mg L−1.528 Safari et al.529 also used nanosized TiO2 (1.0 g L−1) to study the degradation kinetics of a tetracycline hydrochloride (TC·HCl) aqueous solution (55 mg L−1, pH: 5) under ultraviolet irradiation. They observed 100% degradation after 30 min on adding H2O2 (100 mg L−1) compared to 91.4% degradation after 90 min for TiO2/UV. The photocatalytic degradation of tetracycline over commercial TiO2–P25 showed 94.8% (120 min) removal efficiency under visible light (λ = 700 nm).530 Recently, a crosslinking method has been followed for immobilizing TiO2 (P25) nanoparticles in chitosan film, which showed promising photocatalytic activity in the purification of water containing tetracycline hydrochloride under UV irradiation.531 The stability and reusability of this composite film in four consecutive cycles revealed a significant decrease in removal efficiency after the second run, from 87% to 57%. Tetracycline hydrochloride degradation has also been studied using a green and low-cost approach, involving the preparation of immobilized titania samples by depositing two successive TiO2 layers on two different commercial supports.532
3.7.1.2 ZnO and other oxides. Palominos et al.527 carried out the photocatalytic oxidation of tetracycline in an aqueous suspension containing ZnO and found its performance comparable to TiO2 (∼100% degradation) under simulated solar light. According to the suggested mechanism, the contribution towards photocatalytic tetracycline oxidation on ZnO is mainly guided by hydroxyl radicals. UV-irradiated ZnO/peroxymonosulfate has shown about 95.6% degradation of tetracycline (10 mg L−1, pH: 7) in 90 min compared to UV/ZnO (50.14%), attributed to the formation of SO4·−.533 In addition, HSO5− acts as an electron acceptor and inhibits electron–hole pair recombination, thereby allowing the formation of more ·OH radicals. Iron oxide nanoparticles,534 nanospherical α-Fe2O3 supported on 12-tungstosilicic acid,535 SnO2 hollow microspheres,536 polyaniline coated on magnetic MoO3537 and BiFeO3538 have also been studied in the photocatalytic degradation of tetracycline aqueous solutions.
Liu et al.542 studied the photoactivity of an Au–ZnO nanomotor system based on vertically aligned ZnO in the photocatalytic degradation of tetracycline as a function of different photocatalysts, UV light intensity and cycling tests, as presented in Fig. 18(a)–(c), respectively. The findings revealed that the respective degradation rates of tetracycline within 30 min and rate constants corresponding to pseudo-first-order kinetics follow the order: Au–ZnO nanorod motors (Au–ZnO-M): 99.3% > Au–ZnO nanorod array (Au–ZnO-A): 95.5% > ZnO (86.5%), and k ‘Au–ZnO nanorod motors (k (Au–ZnO-M): 0.1451 min−1 > k (Au–ZnO-A): 0.1120 min−1 > k (ZnO): 0.0542 min−1). It was suggested that the Au layer in the Au–ZnO heterojunction nanoarrays acted as an electron reservoir to facilitate charge separation, thereby lowering the possibility of photogenerated carrier recombination. A possible photocatalytic mechanism for the photocatalytic degradation of tetracycline by Au–ZnO nanomotors under UV-light irradiation is displayed in Fig. 18(d). According to this, electrons in Au could react with O2 to form ·O2−, accounting for the degradation of tetracycline. In contrast, h+ in the VB of ZnO could directly degrade tetracycline to a stable product.
 | ||
| Fig. 18 Photocatalytic degradation of TC. (a) Dynamic curves of different photocatalysts (initial conditions: 40 mg L−1, TC, 0.2 g L−1 photocatalyst, and 350 mW cm−2 UV light). (b) The impact of UV light intensity (initial conditions: 40 mg L−1 TC and 0.2 g L−1 Au–ZnO nanomotors). (c) Cycling tests (initial conditions: 30 mg L−1 TC: 0.2 g L−1, Au–ZnO nanomotors, and 350 mW cm−2 UV light). (d) Proposed photocatalytic mechanism for TC degradation. Reproduced from ref. 542 with permission from RSC (2022). | ||
3.7.3.1 Doped TiO2 and ZnO. Several studies have been carried out on the performance of doped TiO2 and ZnO photocatalysts and subsequently used in the removal of tetracycline from water.543–546 Red mud and modified red mud originating from industrial solid waste discharged from the aluminum industry have been investigated as low-cost, effective photocatalysts under irradiated visible light.543 Xu et al.544 developed a C-doped TiO2–polymethylsilsesquioxane (PMSQ) aerogel followed by thermal treatment at 400 °C in air. They used it to achieve 98% removal of tetracycline hydrochloride from aqueous solution in 180 min and ascribed it to enhanced charge separation. In another study, hydrothermally prepared carbon (3 wt%)-doped TiO2 with metal (Ni/Co/Cu) nitrate hydroxide was used as a nanocomposite photocatalyst.545 The photocatalytic activity of this catalyst displayed 97% removal of tetracycline hydrochloride within 60 min. TiO2 doped with acetylene black,546 N-doped TiO2/diatomite,547 P-doped carbon nitride tubes combined with peroxydisulfate (PDS),548 N,S-doped TiO2 and N,S-doped ZnO modified chitosan,549 and C,N,S-tri-doped TiO2550 photocatalysts have also been investigated for the degradation of tetracycline.
Metal-doped photocatalysts have also received a lot of attention for their application in the photocatalytic degradation of tetracycline in aqueous solution. Nb-doped ZnO (Nb:Zn molar ratio: 1:1) showed 93.2% degradation efficiency for tetracycline in 180 min under visible light and also possessed superior recyclability and stability.525 Zhang et al.551 fabricated Ag-doped TiO2 (Ag+:Ti4+ mole ratio: 0, 0.5, 1.0, 2.0, 3.0, and 5.0%) hollow microspheres following an applied hydrothermal process by a template-free method. It was noted that Ag-doped TiO2 (Ag+:Ti4+ mole ratio: 3.0%) exhibited maximum removal of tetracycline hydrochloride following first-order kinetics with OH· and h+ playing an active role. Ce (2%)-doped TiO2/halloysite nanotubes and Ce (2%)–TiO2/halloysite nanotubes enabled about 78% tetracycline removal within 60 min under visible-light irradiation.552 TiO2 composite nanofibers doped with CuO were also studied for the photocatalytic degradation of pharmaceutical wastewater.553 Bembibre et al.554 used Ca-doped ZnO nanoparticles in the removal of tetracycline under a visible-light-driven sonocatalytic process.
3.7.3.2 Doped graphitic materials. Doped graphitic materials have attracted a lot of attention as photocatalysts in the removal of tetracycline from water.555 Nitrogen-self-doped g-C3N4 nanosheets prepared by a combination of N-self-doping and thermal exfoliation showed higher photocatalytic activity for tetracycline degradation than bulk g-C3N4, N-self-doped g-C3N4 or g-C3N4 nanosheets.556 This is attributed to the enlarged visible-light absorption ability, reduced recombination and prolonged lifetime of photogenerated charge carriers. Chen et al.557 reported the removal of tetracycline hydrochloride from wastewater (pH: 5) using an S–g-C3N4/PTFE membrane under irradiated visible light. These findings indicated 98.1% photocatalytic degradation corresponding to an initial concentration of tetracycline hydrochloride of 10 mg L−1, catalyst dosage of 1 g L−1, and S–g-C3N4 loading of 50 mg. Further, the S–g-C3N4/PTFE membrane displayed good recovery performance and photocatalytic stability. Ba (2%)-doped g-C3N4 demonstrated significant influence on the photocatalytic activity owing to its low band gap and the effective separation of photo-induced e−–h+.558
Er-doped g-C3N4,559 Cd-doped g-C3N4,560 S-doped carbon quantum dot loaded hollow tubular g-C3N4,561 single-atom Ni,S-co-coped g-C3N4,562 nitrogen defect/boron dopant engineered tubular g-C3N4,563 Ag–g-C3N4,564 Bi-nanoparticle-decorated g-C3N4 nanosheets (10 wt%),565 Co-doped TiO2rGO,566 rGO-doped ZnAlTi-LDH,567 and graphene oxide/magnetite/cerium-doped titania568 photocatalysts also acted as efficient photocatalysts in the degradation of tetracycline.
The degradation of tetracycline in water has been investigated on TiO2 decorated on magnetically activated carbon as a function of different parameters under ultraviolet and ultrasound irradiation.573 These findings revealed 93% tetracycline removal at the end of 180 min under optimum conditions corresponding to an optimum intensity of 70 W US power, pH 6.0, catalyst loading of 0.4 g L−1, and initial concentration of tetracycline of 30 mg L−1. ZnO rod-activated carbon fiber,574 Fe3O4/FeP,575 spatially confined Fe2O3 in hierarchical SiO2@TiO2 hollow spheres,576 La-enriched titania–zirconia oxide,577 Ni(OH)2-decorated TiO2,578 IO–TiO2–CdS,579 and WO2.72/ZnIn2S4580 have also been demonstrated as efficient photocatalysts for the removal of tetracycline from water.
Wang et al.581 converted harmful algae into bio-nanohybrid materials by immobilizing Microcystis aeruginosa cells onto PAN–TiO2/Ag hybrid nanofibers. They observed about 96% degradation efficiency for tetracycline hydrochloride under visible light compared to PAN/TiO2/Ag nanofiber (77%) and M. aeruginosa (49%) due to a synergistic effect. It is suggested that enhanced degradation in M. aeruginosa/PAN–TiO2/Ag could be caused by algae facilitating the effective separation of photogenerated electron–holes on TiO2. The presence of ZnO, carbonaceous layers and Ag nanoparticles improved the optical absorption property in the Ag/ZnO/C structure, resulting in 95.8% (35 min) and 90.6% (280 min) degradation of tetracycline hydrochloride under UV- and visible-light irradiation, respectively.582 This is ascribed to efficient photogenerated electron separation and transportation and an increase in the active reaction sites. According to Wei et al.,583 an SiO2–TiO2–C (nC:nTi mol ratio: 3.5) aerogel composite displayed 80.01% degradation efficiency for tetracycline hydrochloride within 180 min under visible light and also retained its high stability and reusability. ·O2− and ·OH were considered as the active species responsible for the photocatalytic degradation of tetracycline. In addition, ternary chitosan comprising chitosan–TiO2–ZnO over graphene,584 palygorskite-supported Cu2O/TiO2,585 CuO/Fe2O3,586 ZnO@zeolitic imidazolate,587 and bimetallic oxide/carbon588 have also been tested for the photocatalytic degradation of tetracycline in water.
3.7.5.1 g-C3N4. Insufficient sunlight usage, low surface area and rapid charge recombination of electron and hole pairs are a major hinderance contributing towards the low photocatalytic performance of g-C3N4.65 As a result, several investigations have been made into the photodegradation of tetracycline using g-C3N4 and its composites. Hernández-Uresti et al.333 prepared a graphite-like C3N4 photocatalyst by the polycondensation of a melamine precursor and observed the following trend for the photodegradation of four different pharmaceuticals in aqueous solution under UV-vis irradiation: tetracycline > ciprofloxacin > salicylic acid > ibuprofen. The active species responsible for the degradation of tetracycline were considered to be photogenerated holes, OH radicals and H2O2. Self-assembly-based g-C3N4 nanoflakes showed up to 70% removal efficiency for tetracycline (20 ppm) within 180 min under light irradiation (420 nm).589
Shi et al.590 studied the degradation performance of tetracycline in real water systems by metal-free g-C3N4 microspheres under various conditions through visible-light catalysis and PMS activation synergy. According to this, the rate constant values for the degradation of tetracycline by photocatalysis, Fenton-like catalysis, and photo-Fenton-like catalysis are 0.013, 0.025, and 0.028 min−1, respectively. The observed superior degradation performance of photo-Fenton-like catalysis is attributed to the synergetic effect between PMS activation and photocatalysis. In another study, Wang and others591 used graphitic carbon nitride microspheres and recorded 80.54% degradation efficiency for the removal of tetracycline hydrochloride under visible-light irradiation for 2 h, corresponding to a photocatalyst dose of 1.0 g L−1, initial concentration of tetracycline hydrochloride solution of 10 mg L−1 and initial pH 7. Porous g-C3N4,592 GQDs/g-C3N4,593 S-doped graphitic carbon nitride,594 hexagonal BN/g-C3N4,595 poly-o-phenylenediamine (POPD)/g-C3N4,596 and N–CNT/mesoporous g-C3N4597 photocatalysts have also been evaluated for the removal of tetracycline.
Jiang et al.598 studied the degradation of tetracycline in aqueous solution using P and S doped g-C3N4 under visible light (λ ≥ 420 nm) and showed higher photocatalytic than bare g-C3N4 or single-doped g-C3N4. According to this, P and S doping in g-C3N4 inhibited the recombination of electron–hole pairs and facilitated the efficient separation of photogenerated charges. The h+ and ·O2− were the dominant active species responsible for the degradation of tetracycline. Porous g-C3N4/TiO2 (g-C3N4:TiO2 mass ratio: 12:1) photocatalysts removed 88.43% of tetracycline from aqueous solution under a xenon lamp for 90 min, which was ascribed to the synergistic effect.599 In another study, a ZrO2-embedded MoS2/g-C3N4 nanocomposite exhibited 94.8% tetracycline degradation in aqueous solution in 90 min under visible light owing to the dual charge-transfer channel between the layers of MoS2/g-C3N4 and ZrO2 nanoparticles.600 Poly-N-isopropylacrylamide (PNIPAM)/Fe3O4/g-C3N4 prepared by a hydrothermal method and thermal photoinitiation under visible-light irradiation decomposed tetracycline into harmless small molecules.601 The catalytic activity remain more or less unaltered even after 5 repeated uses and could be easily separated. In addition, CDs/g-C3N4/BiPO4,602 ZnO/N-doped g-C3N4,603 and g-C3N4/H3PW12O40/TiO2604 exhibited enhanced photocatalytic degradation performance for tetracycline under visible light.
3.7.5.2 Graphene. Binary and ternary graphitic composite materials have been reported as photocatalysts in the removal of tetracycline from aqueous solution.605–621 According to Ren et al.,605 a red mud/graphene oxide (mass ratio: 93
:7) composite attained the best degradation rate for tetracycline (79.8%) compared to raw red mud under visible light within 80 min owing to its enhanced specific surface area, light absorption and charge separation. Porous hydroxyapatite (Hap) hollow microspheres as a source of cheap and green photocatalysts have been harnessed in fabricating rGO/Hap composites.606 Investigations revealed significantly enhanced photocatalytic activity of rGO (1.5 wt%)/Hap in tetracycline degradation (92.1%, 30 min) under a xenon lamp (300 W) for full-spectrum irradiation. This is explained on the basis of the photogenerated electrons accumulating at rGO (acting as an electron acceptor) that could interact with O2 to form ·O2−. In addition, separated positive holes in the VB of porous hollow Hap (acting as an electron donor) microspheres directly participate in the oxidation of tetracycline.
Heteropoly acid (H3PMo8W4O40)/graphene oxide nanocomposites based on UiO-66 have been synthesized following an in situ growth hydrothermal method and tested as photocatalysts in tetracycline photodegradation under visible-light irradiation.607 The photocatalytic degradation efficiency for tetracycline was found to be significantly higher (95%: 120 min) compared to GO or heteropoly acid. An Fe3O4/GO/ZnO magnetic nanocomposite showed 74% (100 min) degradation of tetracycline hydrochloride under visible-light irradiation.608 This is explained on the basis of ZnO and Fe3O4/GO in Fe3O4/GO/ZnO contributing to the generation of the electron–hole pairs under visible light and promoting the transfer of photogenerated electrons, respectively. In another study, graphene quantum dot decorated ZnO–ZnF2O4 nanocage ternary composites, prepared by a one-step deposition method exhibited superior performance in the degradation of tetracycline hydrochloride under visible light compared to ZnO or ZnO–ZnFe2O4.609 According to Chakraborty et al.,610 an rGO–ZnTe (1:1) photocatalyst facilitated the degradation of tetracycline due to a synergistic effect. It is suggested that the 2D wrinkled surface of rGO contributes in minimizing the recombination probabilities of photoinduced electron–hole pairs. N-doped TiO2 nanoparticles deposited on rGO exhibited more pronounced photodegradation activity for tetracycline hydrochloride than pure TiO2 or N-doped TiO2.611 Subsequent studies on the reusability of N-doped TiO2/rGO also established the stability of the composite photocatalyst.
Kumar et al.612 fabricated ZnO quantum dots (1.5 wt%)/rGO by a hydrothermal method and observed 68% removal of tetracycline from wastewater (pH: 5) after 120 min under visible light. Fe3O4/g-C3N4/rGO exhibited 86.7% degradation rate of tetracycline hydrochloride, following pseudo-second-order kinetics.613 The proposed mechanism suggested ·O2− and ·OH radicals as the most reactive species in the photocatalytic degradation of tetracycline. Ghoreishian et al.614 reported sonophotocatalytic degradation of tetracycline using a flower-like rGO/CdWO4 composite under simulated visible-light irradiation. These findings under optical conditions (pH: 5.7, initial concentration of tetracycline: 13.54 mg L−1, catalyst dosage: 0.216 g L−1, time: 60 min) revealed its photocatalytic catalytic activity to be 1.5 and 3 times higher than that of commercial nano-ZnO and TiO2, respectively.
Interfacial growth of a TiO2–rGO composite by the Pickering emulsion approach showed 94% removal efficiency for tetracycline hydrochloride (10 ppm) after 40 min under the visible light.615 Such significant enhancement in the photocatalytic efficiency of TiO2–rGO is ascribed to its 2D sandwich-like structure. Porous hollow hydroxyapatite microspheres decorated with rGO,616 rGO–CdS,617 rGO–CdS/ZnS,72 Ag/TiO2 nanosheets/rGO,618 Ag/TiO2 nanosheets,619 Ag/TiO2 nanosheets–rGO,620 and TiO2/rGO/activated carbon621 have also been harnessed as photocatalysts in the degradation of tetracycline in aqueous solution.
Ti0.7Sn0.3O2/g-C3N4 (mass ratio: 10 wt%) achieved 83% degradation of tetracycline hydrochloride in 40 min under irradiated visible light.627 This is explained on the basis of an S-scheme between Ti0.7Sn0.3O2 and g-C3N4 to increase and transport photogenerated charges. The ultrasonic-assisted precipitation method has been used to fabricate a ZnO (20 wt%)/GO (2 wt%)/Ag3PO4 heterojunction and it has been used as a photocatalyst in the elimination of tetracycline hydrochloride from wastewater.628 These findings showed 96.32% (75 min) degradation under visible light corresponding to initial concentration of tetracycline of 30 mg L−1 and catalyst dose of 1.0 g L−1.
The degradation rate of tetracycline was found to be about 10 times higher in a g-C3N4/C/Fe3O4 ternary nanocomposite compared to its individual or binary components under simulated solar light.629 The degradation process followed a first-order kinetics model with a much higher apparent rate constant for g-C3N4/C/Fe3O4 (0.0063 min−1) compared to g-C3N4 (0.0029 min−1) or carbon (0.0003 min−1). The photoinduced h+ and ·O2− free radicals are suggested to act as the main active components in the degradation. The enhanced activity of g-C3N4/C/Fe3O4 in tetracycline degradation is attributed to heterojunction formation and is due to the effective separation of the photocarriers. In addition, the introduction of C into g-C3N4/C/Fe3O4 facilitates an enhancement of the optical response range and effective electron transfer.
Liao et al.630 examined the utility of a core–shell BiFeO3/TiO2 heterostructure with a p–n heterojunction as a photocatalyst prepared by forming nanospheres of TiO2 on BiFeO3 (nanocubes) in tetracycline degradation under visible-light irradiation. The findings indicated much higher degradation efficiency of BiFeO3/TiO2 (72.2%) compared to BiFeO3 (64.9%) and TiO2 (38.3%) after 180 min of visible illumination. A BiFeO3/TiO2 p–n heterojunction photocatalyst showed superior degradation efficiency for tetracycline due to its enlarged specific surface area and higher sensitivity to visible light, improved separation and transfer efficiency of photoelectron–hole pairs and a synergistic effect. Fiber-shaped Ag2O/Ta3N5 p–n heterojunctions designed as efficient photocatalysts showed enhanced photocatalytic activity with good stability in photocatalytic activity for tetracycline under visible light (λ > 400 nm) due to the synergistic effect.631 It is anticipated that photogenerated holes and superoxide radicals played prominent roles in the photocatalytic process.
Chen and others632 fabricated an α-Bi2O3/g-C3N4 heterostructure modified by plasmonic metallic Bi and oxygen vacancies and observed a remarkably high degradation rate for tetracycline (90.2%) under visible light after 180 min. Such enhancement is attributed to the formation of a p–n junction arising from a combination of n-type (g-C3N4) and p-type (α-Bi2O3) semiconductors, which is beneficial in a ternary photocatalyst. It is suggested that Bi nanoparticles and the presence of oxygen vacancies favor the consumption and separation of the photogenerated electrons and holes in the ternary heterojunction photocatalyst. Several other heterojunction photocatalysts, such as C3N4@MnFe2O4–rGO,491 BiVO4/TiO2/rGO,633 porous g-C3N4/AgBr/rGO,634 C3N4-supported WO3/BiOCl,635 BiOI/exfoliated C3N4,636 CuO@ZnO,637 ZnO/SnO2,638 Cu2O–TiO2,639 MoS2/Ag/g-C3N4,640 g-C3N4/ZrO2−x,641 and needle SnO2 nanoparticles anchored on exfoliated g-C3N4642 have also shown enhancement and stability in the degradation of tetracycline.
N-doped ZnO–MoS2 binary heterojunctions have been fabricated by a hydrothermal method and used to study its photocatalytic activity for the degradation of tetracycline under visible-light irradiation.643Fig. 19(a)–(d) show corresponding findings based on variations in the degradation of TC with time, corresponding ln(C/C0) vs. time plots, a histogram showing a comparative degradation rate (%) of TC under visible light illumination and a bar graph showing the values of rate constants for all the photocatalysts. It should be noted that photocatalytic degradation of tetracycline followed pseudo-first-order kinetics. In addition, fabricated semiconductor heterojunctions demonstrated enhanced performance for the degradation of tetracycline due to the synergistic effect. Furthermore, the enhanced photostability of the photocatalyst over three cycles for a period of 360 min is ascribed to the transfer of holes from the valence band of N-doped ZnO to the valence band of MoS2.
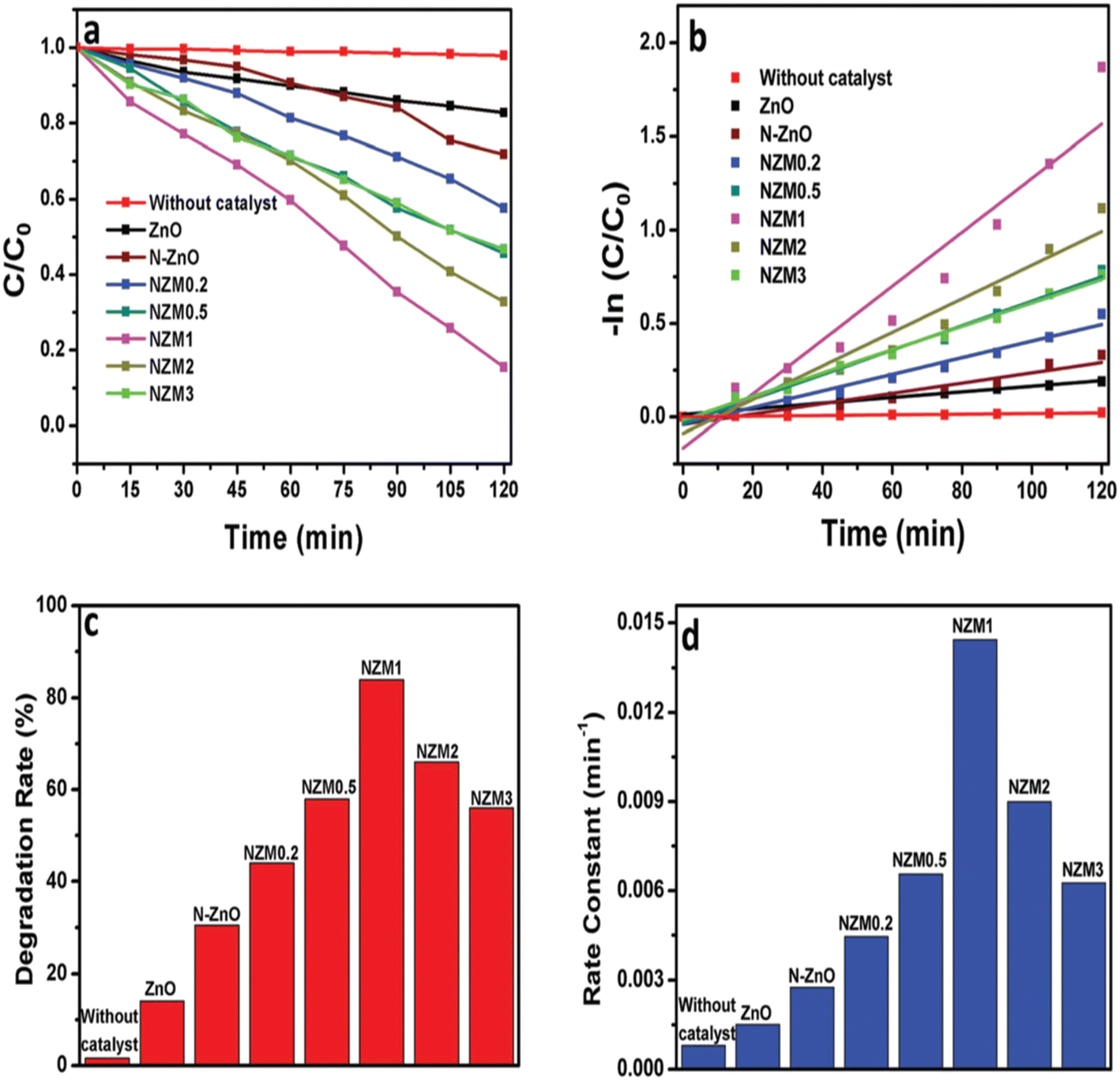 | ||
| Fig. 19 (a) Kinetic curves for the degradation of TC, (b) ln(C/C0) vs. time curve for the degradation of TC, (c) a histogram showing a comparative degradation rate (%) of TC under visible light illumination and (d) a bar graph showing the values of rate constants for all the photocatalysts (N-doped ZnO nanorods loaded 0.2, 0.5, 1, 2 and 3 wt% of with MoS2 nanoflowers (MNF) are referred to as NZM0.2, NZM0.5, NZM1, NZM2, and NZM3, respectively). Reproduced from ref. 643 with permission from RSC (2017). | ||
A novel type-II Bi2W2O9/g-C3N4 heterojunction has been fabricated and studied for its photocatalytic performance in the removal of tetracycline under simulated solar irradiation and it was compared with Bi2W2O9 and g-C3N4, as displayed in Fig. 20(a).72 It is inferred that Bi2W2O9/g-C3N4 yields high photodegradation (∼95%) compared to the degradation observed for pristine g-C3N4 (75%) or Bi2W2O9 (∼60%). This is attributed to the Bi2W2O9 semiconductor acting as a trap for photogenerated holes and electrons. A photocatalytic mechanism has also been proposed for the Bi2W2O9/g-C3N4 system in Fig. 20(b).
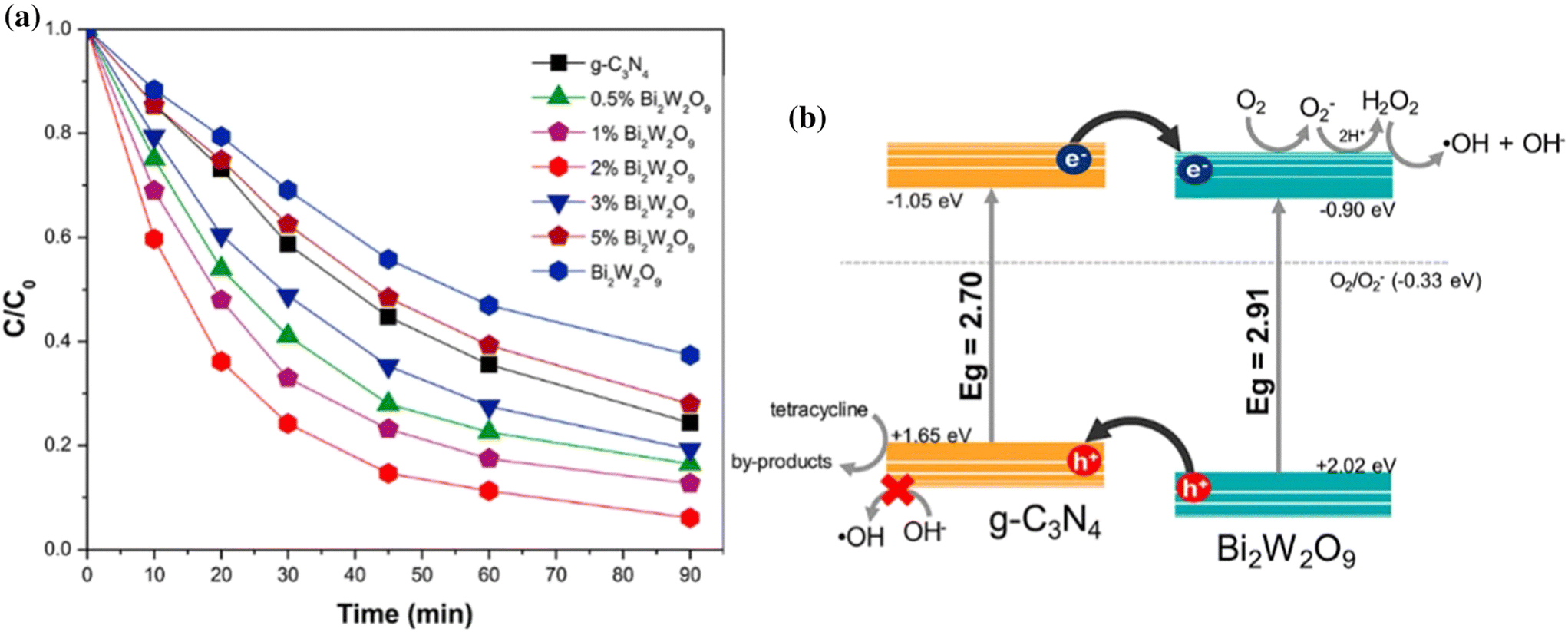 | ||
| Fig. 20 (a) Photocatalytic degradation of tetracycline antibiotic (C0 = 10 mg L−1, pH = 4.89) as a function of irradiation time over Bi2W2O9, g-C3N4 and Bi2W2O9/g-C3N4 samples. (b) Proposed photocatalytic mechanism for the Bi2W2O9/g-C3N4 system under solar-like irradiation. Reproduced from ref. 72 with permission of Elsevier (2020). | ||
Z-scheme WO3/g-C3N4 composite hollow microspheres fabricated by an in situ hydrolysis and polymerization process showed an enhanced degradation rate towards tetracycline hydrochloride (82% in 120 min) under visible-light irradiation.644 The enhanced separation of photoinduced electrons and holes and the synergistic effect of g-C3N4 and WO3 are considered to be a few reasons for this. In addition, the presence of hollow cavities could enable trapping of the incident photons and facilitate availability of more electrons and holes in the photocatalytic process. In another study, a Z-scheme mesoporous Sn3O4/g-C3N4 heterostructure exhibited superior photocatalytic performance in degrading tetracycline hydrochloride present in water.645 A possible photocatalytic reaction mechanism has also been examined in detail for this. In another study, BiOI/g-C3N4/CeO2 (3 wt%) photocatalyst possessed the best photocatalytic activity for degradation of tetracycline (91.6%) under visible-light irradiation.646 It is anticipated that CeO2/g-C3N4 and BiOI/g-C3N4 catalysts block the recombination of photoinduced electron–hole pairs through the formation of a heterojunction.
Dai et al.73in situ prepared 3D-20% polyaniline/perylene diimide (PANI/PDI) and found the degradation rate for tetracycline under visible-light irradiation in a static system, by 15.3 times and 17.0 times those of pure PDI and PANI, respectively. The main reactive species in the degradation of tetracycline comprised superoxide radicals, hydrogen peroxide and holes. Fig. 21(a) and (b) schematically show the electron–hole pair separation process and TC degradation mechanism of a 3D 20%-PANI/PDI heterojunction under visible-light irradiation. Scanning electron microscopy images of 3D PANI/PDI in Fig. 21(c and d) indicate a significant decrease in size after the dissolution/assembly process and the PDI are uniformly/orderly dispersed in the 3D network structure of PANI.
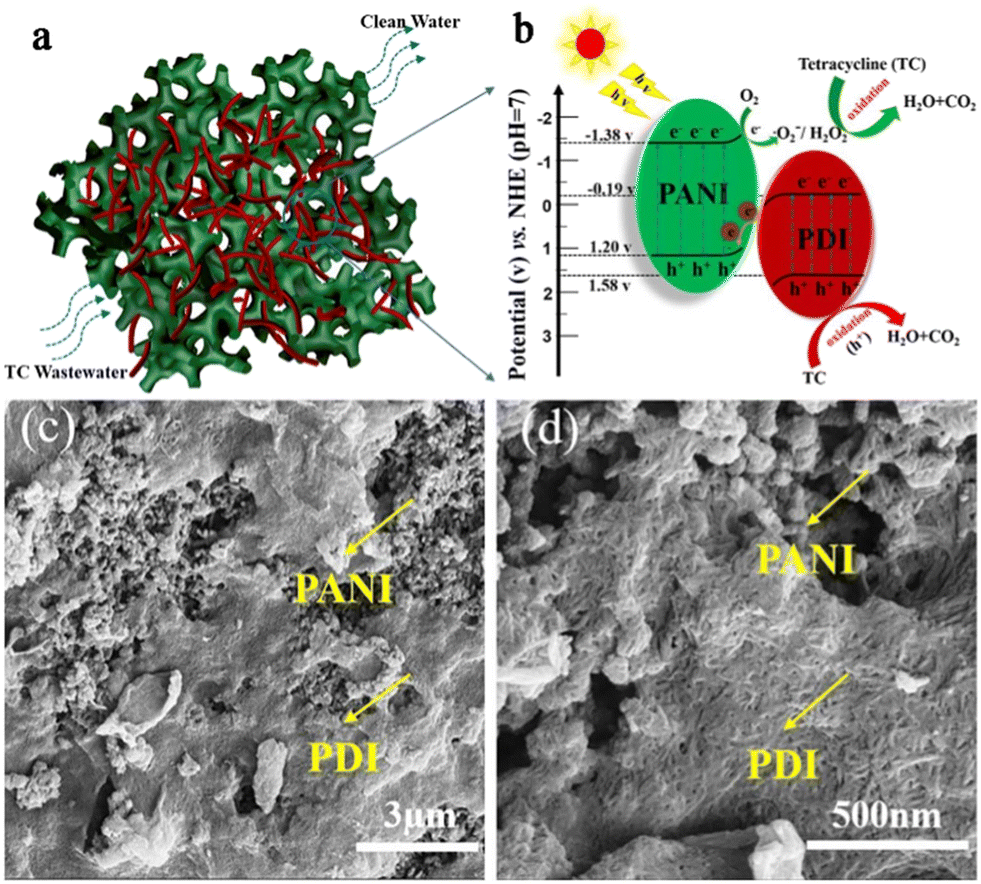 | ||
| Fig. 21 (a) Morphological structure of PANI/PDI. (b) Photocatalytic mechanism of PANI/PDI heterojunction photocatalysts under visible-light irradiation: direct Z-scheme heterojunction mechanism. (c and d) Scanning electron microscopy images of 3D PANI/PDI. (Modified) Reproduced from ref. 73 with permission of Elsevier (2020). | ||
In addition, TiO2−x/ultra thin g-C3N4/TiO2−x,647 K-doped g-C3N4/TiO2/CdS,648 γ-Fe2O3 nanospheres anchored on g-C3N4,649 CQDs/g-C3N4,650 Ag3PO4/MIL-88A(Fe),651 BiOBr/MoS2/GO,652 g-C3N4/MnO2/GO,653 BiVO4@polypyrrole/g-C3N4,654 AgI/BiOBr/rGO,655 graphene-bridged Ag3PO4/Ag/BiVO4,656 g-C3N4 nanoparticles/WO3 hollow microspheres,657 CuIn2S2/g-C3N4,658 Ag3PO4/g-C3N4/ZnO,659 g-C3N4 nanosheet/Ag3PO4/α-Bi2O3,660 LaNiO3-modified C3N4661 and ultrafine TiO2 nanoparticle modified g-C3N4662 heterojunction photocatalysts have also been harnessed in the removal of tetracycline in water.
Table 8 records the performance data of different photocatalysts used in the removal of tetracycline from water.
| Photocatalyst | Preparative method | CIPa/CIP·HClb | Catalyst dose | pH | Light source | Degradation and time | Rate constant |
|---|---|---|---|---|---|---|---|
| TiO2526 | Solvothermal | 50 mg L−1a (100 mL) |
30 mg | 6.0 | Visible light LED 50 W | ∼25% (12 min) | ∼0.03 min−1 |
| TiO2 (P25 Degussa)527 | Commercial | 20 mg L−1a (100 L) |
1.5 g L−1 | 8.7 | Xenon lamp, 250 Wm−2, 300–800 nm | 100% (15 min) | — |
| ZnO (Sigma Aldrich)527 | Comme rcial | 20 mg L−1a (100 mL) |
1.0 g L−1 | 11 | Xenon lamp, 250 Wm−2, 300–800 nm | 100% (15 min) | — |
| Nanosized TiO2 (P25) with 70% anatase and 30% rutile528 | Commercial | 40 mg L−1b (40 ml) |
1000 mg L−1 | 9 | Medium-pressure mercury lamp (UV), λ < 290 nm 525 μW cm−2 | 95% (60 min) | — |
| Nanosized TiO2 with 80% anatase and 20% rutile in presence of H2O2 (100 mg L−1)529 | Commercial | 55 mg L−1b (250 mL) |
1 g L−1 | 5 | UV lamp: 18 W, λ: 254 nm, 2500 μW cm−2 | 100% (30 min) | 7.25 × 10−3 min−1 |
| TiO2–P25 (80% anatase and 20% rutile) in presence of H2O2 (100 mg L−1)530 | Commercial | 10 mg L−1a (100 mL) |
0.2 g L−1 | — | Xenon lamp 300 W, 20 mW cm−2, λ: 350 nm | 94.8% (120 min) | ∼3.8 × 10−2 min−1 |
| TiO2 (P25) immobilized in chitosan531 | Dispersion method | 30 mg L−1b |
0.12 g | 4 | UV lamp (30 W), λmax: 360 nm | 87% (360 min) | 0.025 min−1 |
| Nanometric and immobilized TiO2532 | Modified sol–gel method (product calcined at 400 °C) | 35 ppmb | 300 mg | — | Jelosil HG500 UV lamp, 30 mW cm−2 | 90% (35 min) | 56 ± 2 × 10−3 |
| ZnO nanoparticles (peroxy monosulfate:2 mM)533 | Biosynthesis | 10 mg L−1b |
2 g L−1 | 7.0 | Low-pressure UV lamp (6 W), λ: 254–258 nm | 95.6% (90 min) | 0.018 min−1 |
| Iron oxide nanoparticles534 | Co-precipitation | 83 μMb (10 mL) | 10 mg | 7 | Hg quartz lamp (280 W), λ: 180 nm to 623 nm | 40% (60 min) | 0.0092 min−1 |
| Nanospherical α-Fe2O3 supported on 12-tungstosilicic acid (H2O2: 0.1 ppm/250 ml)535 | Solid state dispersion | 30 ppma | 150 ppm | 8 | Hg lamp (15 W), λ: 254 nm | 97.39% (50 min) | 0.0098 min−1 |
| SnO2 hollow microspheres (SnCl2·2H2O:Na3C6H5O7·2H2O mole ratio = 1:4)536 |
Hydrothermal method | 50 mg L−1b (40 ml) |
50 mg | — | Hg lamp, λ: 365 nm | 76% (140 min) | 0.00861 min−1 |
| BiFeO3 (in presence of H2O2: 9.8 mM)538 | Calcination of gel formed from bi and Fe nitrates at 600 °C | 40 mg L−1a |
2 g L−1 | 4 | Hg lamp (300 W), λ = 365 nm | 100% (210 min) | 0.02650 min−1 |
| Nb doped TiO2 (Nb:Zn molar ratio of 1:1)525 |
Green synthesis | 150 mg L−1a (100 mL) |
0.25 g L−1 | 7 | 250 W xenon arc lamp with a 420 nm cut-off filter | 91. 5% (180 min) | 7.3 × 10−3 min−1 |
| Au–TiO2 (0.3 g)/PVDF539 | Three-step synthesis strategy | 20 mla solution | 0.1 g | — | Xenon lamp (300 W), λ < 420 nm | 75% (120 min) | 0.01212 min−1 |
| C-doped TiO2 (in PMSQ)544 | Multiple steps | 10 mg L−1b (50 mL) |
0.5 g | 7 | W halogen lamp (100 W) with filter (λ > 420 nm) | 98% (180 min) | — |
| TiO2/acetylene black with PS: 3 mM L−1546 | Mixing method | 30 mg L−1b (100 mL) |
0.5 g L−1 | 4.1 | LED lamp (30 W), λ: 400–780 nm, 50 W m−2 | 93.3% (120 min) | 2.2 × 10−2 min−1 |
| N doped TiO2 diatomite547 | Mixing followed by calcination | 20 mg L−1b |
5 g L−1 | 6 | Xenon lamp (150 W), λ < 400 nm | 91% (300 min) | — |
| P doped carbon nitride tube (peroxydisulfate: 1.0 g L−1)548 | Hydrothermal calcination | 20 mg L−1a (100 mL) |
0.3 g L−1 | 4.59 | Xenon lamp (300 W) with a cut-off filter of λ: 400 nm, 180 mW cm−2 | 96.4% (60 min) | 0.0492 min−1 |
| Chitosan modified N,S-doped TiO2549 | Sol–gel-hydrothermal method | 10 mg L−1a (100 mL) |
0.6 g L−1 | 8.2 | LED lamp: 18 W | 91% (20 min) | 0.048 min−1 |
| C,N,S-tri-doped TiO2550 | Sol–gel method (thiourea-to-Ti molar ratio of 0.05:1 and calcined at 450) |
5.0 mg L−1a |
0.5 g L−1 | 9 | Solar stimulator equipped with xenon arc lamp (150 W), λ < 420 nm | 98% (180 min) | 24.6 × 10−3 min−1 |
| Ag-doped TiO2 (Ag+ to Ti4+molar ratio: 3.0%)551 | Template-free route (hydrothermal) | 30 mg L−1b |
100 mg | — | Xenon lamp (300 W) and (λ > 420 nm) | ∼88% (30 min) | 6.77 × 10−2 min−1 |
| Ce (2%)–TiO2/halloysite nanotubes552 | Modified sol–gel method | 20 mg L−1a (100 mL) |
50 mg | — | Xenon lamp (300 W), λ > 420 nm | 78% (60 min) | — |
| TiO2 composite nanofibers doped with CuO553 | Electrospinning technique | 100 ppma | 1.0 g L−1 | Neutral | Xenon lamp (400 W) | 71% (60 min) | — |
| N self-doped g-C3N4556 | Combination of N self-doping and thermal exfoliation process | 10 mg L−1a (100 mL) |
0.5 g L−1 | — | Xenon lamp (300 W), λ > 350 nm | 89.14% (60 min) | — |
| S–g-C3N4/PTFE membrane557 | Ultrasonic device method | 10 mg L−1b |
50 mg | 5 | 300 W xenon light irradiation with a 420 nm cut-off filter | 98.1% (120 min) | 0.03348 min−1 |
| Ba (2%)-doped g-C3N4558 | Facial thermal condensation method | 20 mg L−1a (50 mL) |
50 mg | 10 | Xenon lamp (150 W) with 400 nm cut-off filter | 91.94% (120 min) | 0.0175 min−1 |
| Er (0.0035 g)-doped g-C3N4559 | Calcination | 25 mg L−1a (50 ml) |
25 mg | 4 | Xenon lamp (35 W) | ∼90% (90 min) | 0.0204 min−1 |
| Cd (4.6 wt%) doped g-C3N4560 | Thermal polymerization method | 10 mg L−1a |
0.8 g L−1 | 5 | Xenon lamp (300 W) with λ > 420 nm | 98.1% (60 min) | — |
| S-doped CQDs loaded hollow tubular g-C3N4561 | Ultrasonic assisted synthesis strategy | 20 mg L−1a |
1 g L−1 | — | Xenon lamp (300 W), 0.33100 W cm−2 | 82.67% (60 min) | 0.0293 min−1 |
| Ni–S co-coped g-C3N4562 | Thermal polymerization followed by calcination | 10 ppmb (30 mL) | 5 mg | 5 | Xenon lamp (300 W), 100 W cm−2 | 91.77% (60 min) | 0.031 min−1 |
| Ag doped g-C3N4564 | Heating melamine and urea mixture | 20 mg L−1a (50 mL) |
0.1 g | 7 | Solar light | 96.8% (120 min) | — |
| Bi nanoparticle-decorated g-C3N4 nanosheet (10 wt%)565 | Ultrasound-assisted electrostatic self-assembly method | 10 mg L−1b |
40 mg | 7 | Lamp (300 W), λ: 420–780 nm | 90.7% (70 min) | — |
| Co (0.20 wt%) doped TiO2/rGO566 | One-pot hydrothermal method | 30 mg L−1b (100 mL) |
100 mg | — | Halogen lamp: 500 W (400 nm cut-off filter) | 60% (180 min) | — |
| Magnetic graphene oxide–Ce (10% mass ratio) doped titania568 | Dispersion method | 25 mg L−1a (100 mL) |
50 mg | — | Xenon lamp: 300 W, 400 nm cut-off filter | 82.92% (180 min) | 0.03005 min−1 |
| MoS2 (20 wt%)/TiO2571 | Hydrothermal route | 10 mg L−1a |
100 mg | 5.5 | Metal halide lamp: 400 W (UV-vis light source) | 95% (100 min) | 0.0276 min−1 |
| ZnO/γ-Fe2O3572 | Microwave-assisted solution method | 30 mg L−1a (20 mL) |
0.5 mg L−1 | 6.7 | Halogen lamp, 100 mW cm−2 | 88.52% (150 min) | 0.01321 min−1 |
| Magnetic activated C@TiO2573 | Impregnation method | 10 mg L−1a |
0.4 g L−1 | 6 | UVC lamp (40 W), (λ: 254 nm) | ∼100% 180 min | 0.19 min−1 |
| ZnO rod-activated carbon fiber (ACF)574 | Microwave | 40 mg L−1a (150 mL) |
One piece (5.5 cm of ZnO rod-ACF) | 8 | UV lamp (20 W), λ: 8365 nm | >99% (60 min) | — |
| Fe3O4/FeP (molar ratios of Fe:P at 1:6)575 |
Hydrothermal synthesis and partial phosphating annealing method | 50 mg L−1b (40 mL) |
20 mg | — | Xenon lamp (1000 W) | 88% (180 min) | 0.00984 min−1 |
| Hierarchical hollow SiO2–Fe2O3@TiO2576 | Dispersion/in situ polymerization/sol–gel approach | 10 mg L−1a (50 mL) |
0.2 mg mL−1 | 3–7 | Simulated solar-light irradiation | 100% (140 min) | — |
| Fe2O3 in hierarchical SiO2@TiO2 hollow sphere576 | Dispersion/in situ polymerization/sol–gel | 10 mg L−1a (50 mL) |
0.2 mg mL−1 | — | Natural sunlight irradiation | 100% (80 min) | — |
| La–TiO2–ZrO2577 | Sol–gel process | 10 mg L−1a (100 mL) |
0.35 mg L−1 | 5 | UV lamp | 100% (120 min) | 0.0359 min−1 |
| Ni(OH)2 decorated rutile TiO2578 | Deposition of Ni(OH)2 on hydrothermally prepared TiO2 nanorods using 0.2 M TiCl4 | 100 mg L−1a (20 mL) |
200 mg | — | 200 W Hg xenon lamp, λ < 420 nm (cut-off filter) | 76% (150 min) | 0.0090 min−1 |
| 3D IO–TiO2–CdS579 | Hydrothermal synthesis | 30 mg L−1b |
30 mg | — | Xenon lamp (λ < 420 nm) | >99% (20 min) | — |
| 1D/2D WO2.72/ZnIn2S4580 | Hydrothermal reaction | 50 mg L−1b (300 mL) |
30 mg | — | Xenon lamp: 300 W (λ > 420 nm) | 97.3% (60 min) | — |
| PAN/TiO2/Ag nanofiber581 | Immobilizing M. aeruginosa cells onto PAN/TiO2/Ag | 20 mg L−1b (500 mL) |
1 g L−1 | 6 | Halogen lamp: 500 W (λ < 420 nm) | 96% (240 min) | 5.62 × 10−3 min−1 |
| Ag/ZnO/C582 | Calcination and photodeposition route | 20 mg L−1b (100 mL) |
100 mg | — | Xenon lamp: 500 W, λ > 400 nm | 81% (280 min) | — |
| Ag/ZnO/C582 | Calcination and photodeposition route | 20 mg L−1b (100 mL) |
100 mg | — | UV lamp: 250 W, λmax: 365 nm | 95.8% (35 min) | — |
| SiO2–TiO2–C (nc:nTi: 3.5)583 |
Sol–gel method | 10 mg L−1b (50 mL) |
— | — | Visible light (λ > 420 nm) | 80.31% (180 min) | 0.00831 min−1 |
| Chitosan–TiO2–ZnO584 | Sol–gel and ultrasound-assisted method | 20 mg L−1a |
0.5 g L−1 | 4 | UV | 97.2% (180 min) | — |
| Palygorskite-supported Cu2O/TiO2585 | Liquid phase reduction method | 30 mg L−1b (50 mL) |
1.0 mg | 8.7 | Xenon lamp: 500 W | 88.81% (240 min) | 0.0129 min−1 |
| CuO/Fe2O3586 | Green synthesis | 20 mg L−1a |
40 mg | 7 | UV irradiation | 88% (80 min) | 0.048 min−1 |
| Zr0.3Ti/C588 | Calcination | 10 mg L−1a |
— | — | Xenon lamp (300 W) | 98% (30 min) | 0.84 L Mol−1 min−1 |
| Polymeric g-C3N4333 | Polycondensation | 20 mg L−1a (200 mL) |
200 mg | 5.5 | Xenon lamp (35 W) | 86% (240 min) | — |
| g-C3N4 nanoflakes589 | Thermal condensation followed by heat treatment | 20 ppma | — | — | LED (6 W), λ: 365 nm | 70% (180 min) | — |
| Self-assembled g-C3N4 microsphere591 | Supramolecular self-assembly with post-heating treatment | 10 mg L−1b |
1.0 g L−1 | 7 | Xenon lamp: 500 W (λ > 420 nm) | 80.54% (120 min) | — |
| Porous g-C3N4592 | Calcination of bulk g-C3N4 | 20 mg L−1b (50 mL) |
30 mg | 9 | Xenon lamp (300 W) with UV cut-off filter 420 nm | 91.8% (60 min) | — |
| 0.5 wt% GQDs/g-C3N4593 | Electrostatic interaction method | 20 mg L−1b |
25 mg | — | 300 W xenon arc lamp, λ > 400 nm | ∼67% (120 min) | — |
| S-doped graphitic carbon nitride594 | Thermal induction copolymerization | 30 mg L−1a |
0.01 g L−1 | 4 | Solar light | 93.8% (60 min) | — |
| h-BN (2.0 mg)/g-C3N4595 | In situ method | 10 mg L−1a (100 mL) |
1.0 g L−1 | — | Xenon lamp (300 W, λ > 400 nm) | 79.7% (60 min) | 0.02775 min−1 |
| POPD/g-C3N4596 | Suspension polymerization | 10 mg L−1b (50 mL) |
0.5 g L−1 | — | Xenon lamp (300 W) | 86.0% (120 min) | — |
| N doped CNT/mpg-C3N4597 | Thermal polycondensation | 20 mg L−1b |
1.0 g L−1 | — | Xenon lamp (300 W) | 67.1% (240 min) | — |
| P,S-doped g-C3N4 (hexachloro triphosphazene: 50 mg)598 | In situ thermal copolymerization | 10 mg L−1a |
1.0 g L−1 | — | 300 W xenon lamp, λ > 420 nm | 85.85% (60 min) | 0.03823 min−1 |
| Porous g-C3N4/TiO2 nanoparticles599 | Reaction carried out under autoclave | 10 mg L−1a (70 mL) |
70 mg | 5 | Xenon lamp irradiation | 88.43% (90 min) | — |
| ZrO2 nanoparticles@MoS2/g-C3N4600 | Multiple steps | 20 mg L−1a (100 mL) |
50 mg | 3 | Xenon lamp (300 W), λ > 420 nm | 94.8% (90 min) | 0.0230 min−1 |
| PNIPAM/Fe3O4/g-C3N4601 | Thermal photoinitiation technology | 20 mg L−1a (100 mL) |
0. 1 g | — | Xenon lamp (300 W): visible light | ∼78% (120 min) | — |
| CDs doped g-C3N4//BiPO4602 | Hydrothermal method | 10 mg L−1b |
1 g L−1 | 4 | Xenon lamp (500 W), under visible light | 75.50% (220 min) | 0.0005 min−1 |
| 20% ZnO/N doped g-C3N4603 | Self-assembled method through electrostatic attraction | 20 mgL−1b (200 mL) |
0.1 mg L−1 | — | Xenon lamp (300 W) under visible light | 81.3% (15 min) | 0.1016 min−1 |
| Red mud modified with graphene oxide (mass ratio: 93:7)605 |
Ultrasonic mixing | 10 mg L−1a |
50 mg | 6.9 | Xenon lamp (300 W), λ > 420 nm | 79.8% (80 min) | 0.02011 min−1 |
| rGO (1.5 wt%) hydroxyapatite microsphere606 | Hydrothermal method | 60 mg L−1a |
1.0 g L−1 | 5 | Xenon lamp (300 W) with full spectrum irradiation | 92.1% (30 min) | 0.1816 min−1 |
| Heteropoly acid/GO/UiO-66607 | In situ growth hydrothermal method | 20 ppma (50 mL) | 0.02 g | 7 | Hg lamp 500 W (λ > 400 nm) | 95% (120 min) | — |
| Fe3O4/GO/ZnO608 | Dispersion, followed by hydrothermal treatment | 50 mg L−1b |
1 mg L−1 | — | Under simulated light irradiation (intensity: 1 kW m−2) | 74% (100 min) | 14 × 10−3 min−1 |
| GQDs (1 mL)/ZnO–ZrFe2O4609 | One-step deposition process | 20 mg L−1b |
20 mg (50 mL) | — | Xenon lamp (500 W) coupled with 420 nm cut-off filter | ∼90% (27 min) | 0.08809 min− |
| rGO/ZnTe (1:1)610 |
Single-pot one-step solvothermal process | 10 mg L−1a |
100 mg (50 mL) | — | Solar simulator (AM 1.5, 100 mW cm−2) | ∼70% (40 min) | 0.033 min−1 |
| N doped–TiO2/rGO611 | Photoreduction method | 10 mg L−1b |
50 mg | — | Xenon arc lamp (300 W) with 400 nm cut-off filter | 98% (60 min) | 0.05655 min−1 |
| 1.5 w% ZnO quantum dots/rGO612 | Precipitation and hydrothermal methods | 20 ppma (50 ml) | 50 mg L−1 | 5 | Non halogen lamps (24 V, 250 W) | 68% (120 min) | 0.00961 min−1 |
| Fe3O4/g-C3N4/rGO613 | Ultrasonic dispersion | 20 mg L−1b (100 ml L−1) |
0.1 g | 7 | Xenon lamp (500 W) | 86.7% (60 min) | 0.0306 min−1 |
| rGO/CdWO4614 | Heating method | 13.54 mg L−1a |
0.216 g L−1 | 5.7 | Simulated solar light | 100% (60 min) | 0.0693 min−1 |
| rGO/CdS617 | Solvothermal | 0.08 mmolea (40 ml) | 40 mg | — | Solar light | 83.25% (16 min) | 0.13 min−1 |
| Ag/TiO2/rGO618 | Ultrasonic impregnation assisted photoreduction strategy | 20 mg L−1a (50 mL) |
1 g L−1 | 7 | Hg lamp (300 W), λ < 400 nm | ∼100% (60 min) | 0.1578 min−1 |
| TiO2/rGO/activated carbon621 | Hydrothermal method | 5 × 10−4 Mb | 2.0 g L−1 | — | Xenon lamp (solar simulator) | ∼95% (100 min) | 0.0286 min−1 |
| Core–shell g-C3N4@Co–TiO2622 | Electrospinning approach/thermal polymerization | 20 mg L−1b (10 mL) |
2 × 2 cm2 membrane | 7 | Xenon lamp (300 W), λ > 420 nm, 50 mW cm−2 | 90.8% (60 min) | 0.038 min−1 |
| Hierarchical 2% Au–g-C3N4–ZnO623 | In situ preparation of g-C3N4 ZnO nanorods on g-C3N4 nanosheets and the deposition of Au nanoparticles | 50 mg L−1a (50 mL) |
10 mg | 9.3 | Xenon lamp | 74.7% (30 min) | 3.998 × 10−2 min−1 |
| Mesoporous TiO2-modified ZnO QDs immobilized on LLDPE624 | Casting method | 40 mg L−1a (100 mL) |
— | 9 | Fluorescent lamp: 48 W | 89.5% (90 min) | 0.01312 min−1 |
| 7% CuO/g-C3N4625 | Dispersion method | 50 mgb (1000 mL) | 0.2 g | — | Xenon lamp (500 W), λ > 365 nm | 55% (60 min) | 0.014 min−1 |
| ZnO globular (15 wt%)/g-C3N4626 | In situ growth | 20 mg L−1a (100 mL) |
20 mg | — | PLS-SXE300 (300 W), L.I: 9.6 W m−2, 400–780 nm | 78.4% (50 min) | — |
| ZnO (20 wt%)/GO (2 wt%)/Ag3PO4628 | Ultrasonic-assisted precipitation method | 30 mg L−1b (50 mL) |
1.0 g L−1 | 6 | Visible lamp: 65 W | 96.32% | — |
| g-C3N4/C/Fe3O4629 | Sonication and in situ precipitation technique | 10 mg L−1a (40 mL) |
10 mg | — | Xenon lamp (500 W) | 96.4% (120 min) | 0.0292 min−1 |
| Core–shell BiFeO3/TiO2630 | Hydrolysis and precipitation method | 20 mg L−1a (300 mL) |
1 g L−1 | 5 | UV light | 67.9% (180 min) | — |
| Core–shell BiFeO3/TiO2630 | Hydrolysis and precipitation method | 20 mg L−1a (300 mL) |
1 g L−1 | 5 | Visible light | 72.2% (180 min) | — |
| Fiber-shaped Ag2O/Ta3N5 (molar ratios: 0.3/1)631 | Electrospinning–calcination–nitridation method, followed by in situ anchoring of Ag2O deposition | 10 mg L−1a (80 mL) |
— | — | Xenon lamp (300 W), λ > 400 nm | 78.3% (60 min) | 0.0079 min−1 |
| BiVO4/TiO2/rGO633 | Reaction under Teflon reactor | 10 μg L−1a |
— | — | Xenon lamp (1000 W), λ > 420 nm | 96.2% (120 min) | 0.02613 min−1 |
| g-C3N4/AgBr/rGO634 | Mixing followed by heating | 20 mg L−1a |
0.05 g (100 mL) | — | Xenon lamp (250 W) | 78.4% (90 min) | — |
| C3N4@MnFe2O4–rGO491 | Impregnation approach | 20 mg L−1a (50 mL) + PS |
50 mg | — | Xenon lamp (300 W) with 400 nm cut-off filter | 94.5% (60 min) | 0.0337 min−1 |
| BiOl/exfoliated C3N4 (mass ratio: 0.4)636 | Combination of thermal exfoliation and chemical precipitation | 20 mg L−1a (50 mL) |
1.0 g L−1 | 6 | Xenon lamp (500 W) with 420 nm cut-off filter | 86% (30 min) | 0.0705 min−1 |
| 3.0 wt% CuO@ZnO637 | One-pot method | 20 ppma | 1.5 g L−1 | — | Xenon lamp (300 W), λcutoff: 420 nm, 45.2 mW cm−2 | 100% (45 min) | 113.50 × 10−3 min−1 |
| ZnO/5 wt% SnO2638 | Solvothermal process | 1 g L−1b (100 mL) |
60 mg | — | Xenon lamp (300 W), λ: 420–780 nm | ∼90% (60 min) | 0.0385 min−1 |
| Cu2O–TiO2639 | Surfactant-free preparation method (TiO2:Cu2O = 0.1; 0.2; 0.3) |
50 mga (100 mL) | 30 mg | — | Xenon lamp (300 W) | 91% (60 min) | 0.0432 min−1 |
| 10 wt% MoS2/Ag/g-C3N4640 | Ag deposition and MoS2 coupling is applied co-modify g-C3N4 nanosheets | 20 mg L−1a (50 mL) |
10 mg | 5.5 | Xenon lamp (300 W), λ > 420 nm | 90.1% (30 min) | 0.0507 min−1 |
| g-C3N4 (7.1 wt%)/ZrO2−x641 | Anodic oxidation and thermal deposition method (0.06 g melamine taken) | 10 ppmb (5 mL) | 2 mg | — | Xenon lamp (300 W), λ > 420 nm | 90.6% (60 min) | 0.0474 min−1 |
| 3 wt% needle SnO2 needle nanoparticles anchored on exfoliated g-C3N4642 | Hydrothermal method | 30 mg L−1a (100 ml) |
50 mg | — | Xenon lamp (250 W) with a cut-of filter of 420 nm | 95.90% (120 min) | 0.0205 min−1 |
| N-doped ZnO nanorods–MoS2 nanoflowers (1 wt% MoS2 loaded in N–ZnO)643 | Hydrothermal strategy | 0.01 g L−1a |
25 mg (50 mL) | — | CFL lamp (45 W), λ ≥ 420 nm | 84% (120 min) | 14.43 × 10−3 min−1 |
| WO3/g-C3N4644 | In situ hydrolysis and polymerization process | 25 mg L−1b (100 mL) |
50 mg | — | Xenon lamp (300 W) with a 420 nm cut-off filter | 82% (120 min) | 0.0164 min−1 |
| Sn3O4/g-C3N4 (with load ratio of 3%)645 | Two-step hydrothermal process | 10 mg L−1b (100 mL) |
50 mg | — | Xenon lamp (500 W) | 72.2% (120 min) | 0.0108 min−1 |
| BiOI/g-C3N4/CeO2 (3 wt%)646 | Calcination and hydrothermal treatment | 20 mg L−1a (30 mL) |
50 mg | — | Xenon lamp (300 W), λ > 420 nm | 91.6% (120 min) | 0.0205 min−1 |
| TiO2−x/g-C3N4647 | Grinding and in situ reduction | 10 mg L−1b (50 mL) |
50 mg | 9 | Xeon-lamp (300 W), λ > 420 nm | 87.7% (90 min) | 31.7 × 10−3 min−1 |
| K doped g-C3N4/TiO2/CdS648 | Hydrothermal method | 20 mg L−1a (50 mL) |
50 mg | — | Xenon lamp (300 W), λ > 420 nm | 94.2% (30 min) | 0.08554 min−1 |
| γ-Fe2O3 nanospheres (5%) anchored on g-C3N4649 | Anchoring mesoporous γ-Fe2O3 nanospheres on g-C3N4 nanosheet surface | 10 mg L−1b (100 mL) |
50 mg | — | Xenon light source (500 W) with 420 nm cut-off filter | 73.8% (120 min) | 0.0134 min−1 |
| 0.50 wt% CQDs/g-C3N4650 | Low-temperature process | 10 mg L−1b (100 mL) |
50 mg | — | Xenon lamp (250 W) with 420 nm UV-cut-off filter | 78.6% (210 min) | ∼0.0065 min−1 |
| BiOBr/MoS2/graphene oxide652 | Hydrolysis method | 10 mg L−1b |
25 mg | — | Xenon lamp (300 W) with 380-nm cut-off filter | >98% (40 min) | 0.04277 min−1 |
| g-C3N4/MnO2/GO653 | Wet-chemical method | 10 mg L−1b (100 mL) |
0.5 g L−1 | 6 | Xenon lamp (300 W) with a 420 nm filter | 91.4% (90 min) | — |
| BiVO4@Polypyrrole/g-C3N4654 | Dispersion method | 30 mg L−1a (50 mL) |
30 mg | — | Xenon lamp (300 W), λ > 420 nm | 90% (120 min) | — |
| AgI/BiOBr/rGO655 | Solvothermal method followed by in situ precipitation | 20 mg L−1a (100 mL) |
50 mg | — | Xenon lamp (5.00 W), simulated sunlight | 94.2% (80 min) | 0.018 min−1 |
| Ag/Ag3PO4/BiVO4/rGO656 | In situ deposition method followed by photo-reduction | 10 mg L−1a |
0.5 g L−1 | 6.75 | Xenon lamp (300 W) | 94.96% (60 min) | — |
| g-C3N4/WO3657 | Dispersion method | 10 mg L−1b (100 ml) |
40 mg | — | Xenon lamp (500 W), 320–780 nm, 100 mW cm−2 | 79.8% (180 min) | — |
| (50%)CuIn2S4/g-C3N4658 | Synthesis under autoclave | 20 mg L−1a (100 mL) |
0.5 g L−1 | — | Xenon lamp (300 W), λ > 420 nm cut-off filter | 83.7% (60 min) | 0.02583 min−1 |
| TiO2/g-C3N4662 | Co-annealing process | 20 mg L−1b (40 mL) |
250 mg | 7 | Xenon lamp (150 W) | 99.40% (120 min) | 3.70 × 10−4 min−1 |
3.8 Diclofenac
Diclofenac (DCF), an important non-steroidal anti-inflammatory drug, finds multifaceted applications as a painkiller primarily for dysmenorrhea, rheumatoid arthritis and inflammation.663,664 The intake of diclofenac even at low levels by humans and other living organisms is reported to have an adverse biochemical effect. The solubility and high polarity of diclofenac in water and lower degradability account for its water pollution. Further, it can accumulate in food chains owing to its migration through the aquatic medium (surface water, drinking water, underground water) in food chains. In view of this, the following photocatalytic methods have been used in the removal of diclofenac from water.665–748ZnO showed highly active photodegradation of diclofenac sodium in aqueous solution under UV lamp irradiation compared to solar radiation.674 Mimouni et al.675 investigated the effect of heat treatment on the photocatalytic activity of α-Fe2O3 nanoparticles towards diclofenac elimination. The findings in Fig. 22(a) and (b) show the highest degradation for α-Fe2O3 (calcinated at 300 °C) and the value of the degradation rate constant corresponds to 0.060 min−1. The generation of extremely active OH· radicals is responsible for the total photodegradation of DCF, as schematically described in Fig. 22(c). Meroni et al.676 achieved 70% degradation of diclofenac (25 ppm) by a piezo-enhanced sonophotocatalytic approach based on ZnO (0.1 g L−1) subjected to UV-light irradiation for 360 min. In addition, ZnO modified with rare earth elements (Ce, Yb) and Fe,677 NixZn1−x Fe2O4 (x = 0, 0.3, 0.7),678 cobalt ferrite,679 MgO,680 and WO3681 photocatalysts have also been investigated for the removal of diclofenac from aqueous solution.
 | ||
| Fig. 22 (a) Conversion plots for photodegradation of DCF in the presence of α-Fe2O3 calcinated at different temperatures. (b) The degradation rate of different samples at 120 min. (c) Schematic presentation on the generation of OH· radicals in α-Fe2O3. Reproduced from ref. 675 with permission from Springer (2022). | ||
The photocatalytic performance of a sodium diclofenac solution (pH: 6.5) in F-doped (20 wt%) ZnO under simulated solar radiation indicated the complete degradation of diclofenac sodium of concentration: 10 mg L−1 under the optimized experimental conditions (ZnO–F concentration: 1 g L−1).688 The enhanced photocatalytic activity of F-doped TiO2 is ascribed to the reduction in the recombination rate of electron–hole pairs. In another similar study, fluorine (0.25, 0.5 and 1 at%)-doped ZnO nano- and meso-crystalline ZnO showed high rates of diclofenac degradation in water compared to bare ZnO.689 Chaudhari and others690 used a sol–gel method to prepare Mn/CeO2, Cu/CeO2 Ag/CeO2 (metal semiconductors) and Agl/CeO2 (an n–p semiconductor–semiconductor) by doping with Mn, Cu, Ag and AgI, respectively. Further investigations have been made to compare their photocatalytic degradation for diclofenac sodium in water under the same optimal conditions (pH: 7, diclofenac concentration: 10 ppm) within 90 min exposure to UV light. It is noted that AgI-doped CeO2 (1 g L−1) exhibited higher degradation of diclofenac sodium solution (95%) compared to Mn/CeO2, Cu/CeO2 or Ag/CeO2, such enhancement in the photocatalytic activity of AgI/CeO2 is attributed to its larger surface area and charge separation efficiency.
In addition, Ce@TiO2,691 granular activated carbon modified with N-doped TiO2,692 C,N-co-doped TiO2,693 Ce,Mn-co-doped TiO2,694 N,S-co-doped carbon quantum dots/TiO2,695 TiO2 doped with B, F, N, P,696 and S,N,C-tri-doped TiO2697 photocatalysts have been investigated for the removal of diclofenac from aqueous solution.
:1) synthesized by a hydrothermal method was the most effective catalyst in the photocatalytic removal of diclofenac under visible-light irradiation compared to pure TiO2.698 The composite catalyst successfully degraded diclofenac almost completely in 270 min corresponding to pH 5, initial diclofenac concentration of 25 mg L−1 and catalyst concentration of 0.6 g L−1. Subsequent studies showed the catalyst retained 80% catalyst efficiency after four consecutive reaction cycles. N-doped WO3/TiO2 synthesized by a sol–gel method enhanced the degradation of diclofenac sodium using simulated solar light owing to the synergistic effect and narrowing of the bandgap.699 The visible-light-irradiated photocatalytic degradation of diclofenac sodium using ZnO–WO3 has shown better catalytic activity than bare ZnO.700 These studies revealed ZnO–WO3 (Zn:W mole ratio: ≈10:1) exhibiting ∼76% degradation efficiency at a given pH (6), DCF diclofenac concentration (20 mg L−1) and catalyst loading (0.8 g L−1).
Cordero-García et al.701 studied the effect of carbon doping on WO3/TiO2 on the photocatalytic degradation of diclofenac sodium and observed its higher photocatalytic activity compared to WO3/TiO2 and TiO2. Hydroxyapatite/TiO2 (dose: 4 g L−1) in water degraded DCF (initial concentration: 5 ppm) by 95% in 24 h on irradiating it with simulated solar light.702 According to Sun et al.,703 the intensity of UV irradiation plays a more significant role in the significant removal of diclofenac by a nano-TiO2/diatomite composite in a photocatalytic reactor. According to this, diclofenac degraded completely at 30 min under higher UV irradiation intensity at a flux of 3.0 L h−1. A visible-light-responsive TiO2/Ag3PO4 (10:1) nanocomposite immobilized in a spherical polymeric matrix showed almost complete removal of diclofenac (k: 0.018 min−1) in 120 min corresponding to initial drug concentration of 20 mg L−1 bead loading of 10 g L−1, and reaction volume of 0.8 L.704 The ·OH radical and h+ are reported to be the primary reactive oxygen species in the photodegradation of diclofenac.
An Ag–Ag2O/reduced TiO2 nanophotocatalyst demonstrated 99.8% degradation of diclofenac after 50 min of visible irradiation.705 This is attributed to the effective charge separation, enhanced visible light absorbance and localized SPR of nanocrystalline Ag0. Silvestri et al.706 synthesized PPy–ZnO (25:1) via a polymerization method and studied the degradation of DCF under simulated solar light. In this regard, the composite catalyst (1 g L−1) facilitated 81% (60 min) degradation of diclofenac (10 mg L−1) with h+ the main reactive species involved in the reaction. This performance is ascribed to the mesoporous structure, superior surface area and reduced band gap of PPy–ZnO. According to Das et al.,707 a titania–zirconia (Zr/Ti mass ratio of 11.8 wt%) composite catalyst exhibited a reasonably higher removal of DCF (∼92.41%) compared to the anatase form of titania without zirconia.
Attempts have been made to eliminate diclofenac sodium from wastewater through the photocatalytic degradation of hydrothermally prepared TiO2–SnO2 (Ti–Sn molar ratio: 1:1, 5:1, 10:1, 20:1 and 30:1) under various operating conditions.708 The results indicated the TiO2–SnO2 catalyst with a molar ratio of 20:1 to be the most effective photocatalyst compared to the other binary composites. The catalyst achieved complete degradation of diclofenac under optimum conditions comprising initial drug concentration of 20 mg L−1, catalyst loading of 0.8 g L−1 and pH 5. The photocatalyst also displayed excellent repeatability and better stability over repeated reaction cycles. Fe3O4/TixOy/activated carbon,709 Fe3O4 (nanosphere)/Bi2S3 (nanorod)/BiOBr (nanosheet)710 TiO2@ZnFe2O4/Pd,711 nanotubular titanium dioxide–polyethersulfone (PES) membrane,712 Al2O3–Nd2O3,713 and TiO2–zeolite714 based photocatalysts have also been evaluated for the photocatalytic degradation of diclofenac.
3.8.5.1 g-C3N4 and its composites. Carbon quantum dot (CQD)-modified porous g-C3N4 (dose: 200 mg L−1) synthesized using 20 mL of CQD stock solution showed almost complete degradation of diclofenac solution (pH: 9) of an initial concentration of 10 mg L−1 in 12 min under visible light.715 This is attributed to the tuning of the band structure and enhanced separation of charge carriers. The studies also suggested DCF degradation to be dominated by a photosensitization-like mechanism. The CQD/g-C3N4 photocatalyst also exhibited excellent reusability, as evident from studies in the 5th cycle (>90%). Pd quantum dots (1 wt%) deposited on g-C3N4 (dose: 0.5 g L−1) achieved 100% removal of diclofenac solution (initial concentration: 1 mg L−1, pH: 7) within 15 min under solar light.716 The rate constant (0.72 min−1) was found to be 8 times higher than that of g-C3N4. Such enhanced photocatalytic activity has been explained based on its narrowed bandgap, reduction in the recombination of photogenerated charge carriers and availability of a photosensitization-like electron transfer pathway.
Graphite-like C3N4-modified Ag3PO4 nanoparticles exhibited highly enhanced photocatalytic activity under visible-light irradiation owing to the synergistic effect.717 This is mainly ascribed to the matching band potentials between Ag3PO4 and g-C3N4, effectively suppressing recombination of electron–hole pairs and promoting their separation efficiency. Diclofenac sodium and ibuprofen (5 mg L−1) achieved complete degradation (180 min) in the presence of carbon microspheres (dia: 0.9–1.9 μm) supported on an anatase phase of TiO2 (mass ratio TiO2 to C microspheres: 2) heterostructure photocatalyst under solar light.718 Further studies revealed the high performance of the photocatalyst even after five successive cycles (80%) as evident from the findings in the first cycle (94%).
Hu et al.719 fabricated eco-friendly 2D heterojunction photocatalyst composites (BCCNT) comprising C-doped supramolecule based g-C3N4 (BCCN) layers and TiO2 nanoparticles and corresponding findings are displayed in Fig. 23(a). It should be noted that degradation of diclofenac solution (10 mg L−1, initial pH: 5.05) by 1 g L−1 of 30% C-doped supramolecular based g-C3N4 (BCCNT) reached 98.92% within 30 min under LED lamp illumination owing to ·O2− and h+ as the main active species. Further investigations established that the degradation kinetics of DCF fitted the pseudo-first-order equation (Fig. 23(b)) with an apparent reaction rate constant (kapp: 0.1796 min−1) about 29.4 times higher than BCCN (0.0061 min−1). A possible mechanism for the photodegradation of DCF under LED lamp irradiation is also displayed in Fig. 23(c).
 | ||
| Fig. 23 (a) Ct/C0versus time plots of different photocatalysts. (b) Respective kinetic curves (inset) and apparent reaction rate constants of diclofenac (conditions: [DCF]0 = 10 mg L−1, [Catal.] =1 g L−1, no pH adjustment and pHinitial = 5.05) and (c) possible mechanism for the photodegradation of DCF and CBZ under LED lamp irradiation over 30% BCCNT composites. Reproduced from ref. 719 with permission from Elsevier (2019). | ||
An AgI/gC3N4 (AgI molar mass ratio: 45%) composite photocatalyst exhibited almost complete degradation of diclofenac sodium in 6 min under visible-light irradiation compared to AgI and g-C3N4.720 The reaction rate constant value of AgI/gC3N4 (k: 0.561 min−1) was found to be ∼12.5 and 43.2 times higher than those achieved by AgI (0.045 min−1) and g-C3N4 (0.013 min−1). The photocatalytic degradation of diclofenac was guided by photogenerated holes and superoxide anion radicals as the main reactive species. Such enhanced photocatalytic activity of AgI/g-C3N4 is ascribed to the heterojunction between g-C3N4 and AgI that facilitated interfacial charge transfer and prevented the recombination of electron–hole pairs. Ag/g-C3N4 (mass ratio of Ag: 54%) heterostructure photocatalysts prepared by photodeposition under ambient conditions showed complete degradation of DCF compared to g-C3N4 under visible-light irradiation and followed pseudo-first-order kinetics. The rate constant was k = 0.0429 min−1.721 The rate constant of diclofenac degradation over Ag/g-C3N4 was almost 3.1 times higher than that of pure g-C3N4. Further investigations also revealed generated holes as the main reactive species in diclofenac degradation and also established the excellent stability of Ag/g-C3N4. CNT–Ni@TiO2:W nanoparticles722 and C3N4/NH2-MIL-125 (ref. 723) have also shown remarkable performance in the removal of diclofenac present in water.
3.8.5.2 Graphene composites. The removal of diclofenac (and amoxicillin) has been reported by maltodextrin/reduced graphene and maltodextrin/reduced graphene/copper oxide nanocomposites.724 Kovacic et al.725 fabricated S-doped TiO2/rGO by a one-pot solvothermal method to study the removal of diclofenac sodium in aqueous medium (pH 4) under simulated solar irradiation. These findings revealed strong dependence on rGO loading of the photocatalytic performance of S–TiO2/rGO in the degradation of DCF. Accordingly, 5 wt% rGO in TiO2 showed improved diclofenac photocatalytic activity compared to bare TiO2 owing to the effective photogenerated charge separation, as inferred from a photoluminescence study. John et al.726 investigated sunlight-mediated removal of diclofenac sodium from water (25 mg L−1) using TiO2–reduced graphene oxide (75 mg L−1) and persulfate (20 mg L−1). They achieved an efficiency of more than 98% within 30 min under sunlight illumination. The diclofenac degradation followed the Langmuir–Hinshelwood mechanism and pseudo-first-order kinetics with a pseudo-first-order rate constant (99.4 × 103 min−1) about twice that of TiO2–rGO (50.9 × 10−3 min−1). A hydrothermally synthesized BiOCl–GO composite showed 100% and 47.88% removal of DCF from solution (25 mg L−1) under UV light and visible spectrum solar light, respectively.727 Li et al.728 also used a hydrothermal method to synthesize an Ag–BiOI–rGO nanocomposite. They observed the complete removal of diclofenac (10.0 mg mL−1) by 5 mol% Ag–BiOI–rGO (5 wt%) in 80 min under visible-light irradiation compared to pure BiOI, Ag–BiOI or BiOI–rGO photocatalysts (50 mg in 50 mL). This is attributed to the enhanced charge separation and reduced recombination of photogenerated charge carriers due to Ag and rGO in BiOCl. Other studies reported ∼93% decomposition of diclofenac sodium (25 mg L−1) solution (pH: 6) within 6 min by cubic Ag/AgBr/GO (0.030 g) on illumination with sunlight.729 It is suggested that the large surface area of the catalyst as well as the superior charge separation and transfer efficiency accounted for this. UV-light-assisted activation of persulfate by rGO–Cu3BiS3 (30 mg) reportedly achieved 81% degradation of DCF in 60 min.730 An AgFeO2–graphene/Cu2(BTC)3 MOF heterojunction has also been studied under sunlight for the degradation of diclofenac in aqueous solution.209
The optimal BiOCl/CuBi2O4 exhibited a 90% degradation rate for aqueous DCF in 60 min under visible-light irradiation.739 The degradation followed pseudo-first-order kinetics (k: 0.03539 min−1), much higher than CuBi2O4 (k: 0.00139 min−1) or BiOCl (k: 0.00319 min−1). Such enhanced photocatalytic performance of BiOCl/CuBi2O4 is most likely to be due to the upgraded charge separation and transfer caused by the formation of an S-scheme heterojunction and the presence of oxygen vacancies. Chen et al.740 investigated the photocatalytic performance and mechanism of a Z-scheme CuBi2O4/Ag3PO4 photocatalyst in the degradation of diclofenac sodium under visible-light irradiation. Studies have also been reported on Z-scheme CuBi2O4/Ag3PO4 to study the effects of pH, H2O2, and S2O82− on the visible-light-driven degradation of diclofenac sodium.741
Visible-light-driven TiO2/g-C3N4 achieved maximum degradation efficiency (93.49%) for the removal of diclofenac sodium from aqueous solution (5 ppm) and the process followed pseudo-first-order kinetics.742 Such a Z-scheme photocatalyst successfully prevents the fast recombination of electron–hole pairs. Elangovan and others743 prepared a TiO2–CdS heterojunction following a two-step hydrothermal treatment. Subsequent use of this as a photocatalyst achieved 86% diclofenac degradation within 4 h under visible-light irradiation. It was suggested that the direct Z-scheme heterojunction structure accounts for the direct charge transfer between heterojunction catalysts. Investigations of a TiO2–CdS photocatalyst in five successive reaction cycles established its appreciable photochemical stability and reusability. ZnSnO3/Bi2WO6,744 Ag3PO4/g-C3N4,745 V2O5-B-doped g-C3N4,746 MoS2/Cd0.9Zn0.1S747 and MoO3@ZrO2748 photocatalysts have also shown enhanced degradation of diclofenac and diclofenac sodium.
Table 9 records the data on the performance of metal oxides and carbonaceous materials based photocatalyst in the removal of diclofenac from water under optimum conditions.
| Photocatalysts | Preparative method | DCF | Catalyst dose | pH | Light source | Degradation and time | Rate constant |
|---|---|---|---|---|---|---|---|
| TiO2665 |
Sol–gel method | 5 ppm (100 mL) | 50 mg | 6 | Xenon arc lamp, 300 W, 70 mW cm−2, λcut-off: 420 nm | ∼80% (120 min) | — |
| TiO2665 |
Sol–gel method | 5 ppm (100 mL) | 50 mg | 6 | Natural sunlight | ∼72% (120 min) | — |
| TiO2P25666 | Commercial | 2 mg L−1 | 200 mg L−1 | — | Blacklight Philips TLK05 (40 W), 290–400 nm | 100% (60 min) | ∼0.09 min−1 |
| TiO2SG666 | Commercial | 2 mg L−1 | 200 mg L−1 | — | Blacklight Philips TLK05 (40 W), 290–400 nm | 100% (30 min) | ∼0.13 min−1 |
| TiO2 aerogel P25 (Degussa)667 | Commercial | 5 mg L−1 (100 mL) | 0.2 g L−1 | — | 125 W black light fluorescent lamp: 300–420 nm | 100% (80 min) | 4.24 × 10−2 min−1 |
| TiO2 nano thin film on glass slide669 | Chemical bath deposition | 10 ppm | 25 × 75 mm deposited film | 2 | UV lamp | 26% (12 min) | — |
| TiO2 immobilized on glass670 | Solution method | 0.5 mg L−1 | Film of area 144 cm2 | 6.2–7.2 | UVA lamp: 15 W (300–400 nm) | ∼100% (26 h) | 0.15 h−1 |
| ZnO (Merck)671 | Commercial | 300 mg L−1 | 1.0 g L−1 | 4 | UV | 90.7% (180 min) | 0.0144 min−1 |
| ZnO (Merck)671 | Commercial | 300 mg L−1 | 1.0 g L−1 | 4 | Solar | 56.5% (190 min) | 0.0044 min−1 |
| V2O5 (Merck)671 | Commercial | 300 mg L−1 | 1.0 g L−1 | 4 | UV | ∼100% (180 min) | 0.0196 min−1 |
| V2O5 (Merck)671 | Commercial | 300 mg L−1 | 1.0 g L−1 | 4 | Solar | ∼100% (180 min) | 0.0141 min−1 |
| TiO2 immobilized on activated carbon182 | Temperature impregnation method | 50 mg L−1 (4 L) | 1.2 g L−1 | 10 | Solar irradiation | ∼85% (180 min) | 0.010 min−1 |
| Degussa P25 TiO2 (75% A:25% R)/H2O2: 1.4 mM672 | Commercial | 5 mg L−1 | 250 mg L−1 | — | UVA lamp (9 W lamp) | ∼99.5% (60 min) | — |
| TiO2 (anatase and rutile)673 | Commercial | 20 ppm (5 L) | 0.3 g L−1 | 4 | UV lamp: 250 W | 80.25% (120 min) | 0.0152 min−1 |
| TiO2 (anatase and rutile)/H2O2: 0.3 g L−1673 |
Commercial | 20 ppm (5 L) | 0.3 g L−1 | 4 | UV lamp: 250 W | 95.7% (120 min) | 0.0273 min−1 |
| ZnO674 | Commercial | 30 μM | 0.25 g L−1 | 3 | UV lamp: 40 W, 254 nm | 95% (5 min) | 0.403 min−1 |
| α-Fe2O3 nanoparticles (calcinated at 300 °C)675 | Drying followed by heat treatment | 15 mg L−1 (100 mL) | 1 g L−1 | — | UVC lamp: 15 W, 254 nm | 96% (120 min) | 0.04 min−1 |
| MgO nanoparticles680 | Direct precipitation method | 10 mg L−1 | 0.1 g | 6.5 | UV light source (254 nm) | 100% (60 min) | 0.1191 min−1 |
| TiO2–Pd683 | Sol–gel method | 50 g L−1 (0.20 L) | 1 g L−1 | 5 | UV light source (15 W), 300–400 nm | 100% (120 min) | ∼0.05 min−1 |
| TiO2–Ag683 | Sol–gel method | 50 mg L−1 (0.20 L) | 1 g L−1 | 5 | UV light source (15 W), 300–400 nm | 100% (120 min) | ∼0.04 min−1 |
| Ag/Ag2O/WO3 (H2O2: 1 × 10−4 mM)684 | Deposition/hydrothermal | 0.006 g (100 mL) | 0.1 g | 12 | Mercury lamp (160 W), λ ≥ 400 nm | 85% (60 min) | 32.0 × 10−3 min−1 |
| C-doped TiO2 (anatase phase)686 | Microwave digestion method | 50 μg L−1 | 250 mg L−1 | 7.5 | High-pressure W visible lamp (150 W), λ > 400 nm, 8000 lx | ∼100% (150 min) | 0.0334 min−1 |
| Mg (25 wt%)-doped SiO2687 | Mixing of Mg/SiO2 with MgCl2 | 20 mg L−1 (25 mL) | 0.7 g L−1 | 4.3 | UV light | 55% (60 min) | — |
| Mg (25 wt%)-doped SiO2687 | Mixing of Mg/SiO2 with MgCl2 | 20 mg L−1 (25 mL) | 0.7 g L−1 | 4.3 | Visible light | 48% (60 min) | — |
| F (0.25)-doped ZnO nano689 | Hydrothermal approach | 10 mg L−1 (100 mL) | 1.0 g L−1 | — | UV-LEDs strip: 10 W, 365 nm | 85% (30 min) and ∼99% (180 min) | 0.06 min−1 |
| Mn doped CeO2690 | Sol–gel | 10 ppm | 1.0 g L−1 | 7 | Mercury vapour lamp (125 W) with cut-off wavelength of 455 nm | 48% (60 min) | — |
| Cu doped CeO2690 | Sol–gel | 10 ppm | 1.0 g L−1 | 7 | Mercury vapour lamp (125 W) with cut-off wavelength of 470 nm | 50% (60 min) | — |
| Ag doped CeO2690 | Sol–gel | 10 ppm | 1.0 g L−1 | 7 | Mercury vapour lamp (125 W) with cut-off wavelength of 510 nm | 57% (60 min) | — |
| AgI/CeO2690 | Sol–gel | 10 ppm | 1.0 g L−1 | — | Mercury vapour lamp (125 W) with cut-off wavelength of 460 nm | 88% (60 min) | 1.758 × 104 L Mol−1 min−1 |
| Ce@TiO2691 | Precipitation method | 5 μM (100 mL) | 75 mg | — | UV light | ∼100% (80 min) | — |
| 1% Ce–0.6% Mn/TiO2694 | Sol–gel method | 10 mg L−1 | 50 mg L−1 | 6 | UV lamp: 30 W, λ: 254 nm | 94% (240 min) | 0.012 min−1 |
| N,S co-doped-CQDs/TiO2695 | Via in situ phase inversion method | 10 ppm (200 mL) | 1.5 g (25 cm2 membrane area) | — | Visible-light irradiation (λ > 400 nm) | 62.3% (150 min) | — |
| UV light (λ < 380 nm) | ∼55% (150 min) | — | |||||
| B (5 wt%) doped TiO2696 | Sol–gel method | 15 mg dm−3 | 250 mg dm−3 | — | UV lamp | ∼30% (120 min) | 0.0035 min−1 |
| P (5 wt%) doped TiO2696 | Sol–gel method | 15 mg dm−3 | 250 mg dm−3 | — | UV lamp | ∼24% (120 min) | 0.0019 min−1 |
| F (5 wt%) doped TiO2696 | Sol–gel method | 15 mg dm−3 | 250 mg dm−3 | — | UV lamp | ∼27% (120 min) | 0.0021 min−1 |
| C–S–N-tri-doped TiO2 (thiourea/Ti molar ratio: 0.2:1)697 |
Sonochemical method | 25 mg L−1 (50 mL) | 0.05 g L−1 | Neutral pH | Sunlight | 76.48% (90 min) | 0.0632 min−1 |
| TiO2–WO3 (10:1 molar ratio)698 |
Hydrothermal method | 25 mg L−1 (100 mL) | 0.6 g L−1 | 5 | Metal halide lamp, 400 W, visible light | 100% (210 min) | — |
| Hydroxyapatite–TiO2702 | Annealing of Ti salt and hydroxyapatite | 5 mg L−1 (50 mL) | 4 g L−1 | — | UV lamp, λ: 365 nm, 1.80 mW cm−2 | 95% (24 h) | — |
| Nano TiO2/diatomite703 | Hydrolysis, precipitation and roasting of diatomite and TiCl4 | 400 μg L−1 | 0.5 g L−1 | — | UV lamps: 16 W, 254 nm, 1.17 mW cm−2 | 100% (30 min) | — |
| Immobilized (12 wt% TiO2)/Ag3PO4 (10:1)704 |
Sol–gel method | 20 mg L−1 | 10 g L−1 (beads), 0.8 L | — | Visible light source | ∼90% (120 min) | 0.018 min−1 |
| 4.25-Ag–Ag2O/r-TiO2-0.130705 | One-step solution reduction strategy | 5 mg·L−1 (100 mL) | 30 mg | — | Visible light | 100% (50 min) | 0.04767 min−1 |
| PPy:ZnO (25:1)706 |
Via polymerization method | 10 mg L−1 (100 mL) | 1 g L−1 | 6 | Xenon lamp (250–800 nm) | 81% (60 min) | 0.986 min−1 |
| TiO2–SnO2 (molar ratio: 20 to 1)708 | Hydrothermal method | 20 mg L−1 | 0.8 g L−1 | 5 | UV lamp | 100% (300 min) | 0.0147 min−1 |
| Fe3O4/Bi2S3/BiOBr (with Bi2S3 mass ratio of 4%)710 | One-pot solvothermal | 10 mg L−1 (50 mL) | 0.03 g L−1 | 5 | LED lamp (50 W), 475 nm | 93.81% (40 min) | 0.0527 min−1 |
| TiO2@ZnFe2O4/Pd711 | Photodeposition technique | 10 mg L−1 | 0.03 g L−1 | 4 | Solar light | 84.87% (120 min) | 0.0172 min−1 |
| Nanotubular TiO2-PES712 | Via anodization of TiO2 nanotubes on polyethersulfone membrane | 5 mg L−1 | Circular membranes (Dia: 47 mm) | — | UVA sunlamp (7.6 mW cm−2) | ∼94% (240 min) | 9.96 × 10−3 min−1 |
| Al2O3-(15%) Nd2O3713 | Sol–gel method | 80 ppm | 200 mg (200 mL) | — | UV lamp, 254 nm, 4400 μW cm−2 | >92.0% (40 min) | 9.5 × 10−2 min−1 |
| CQDs (50 mL) modified g-C3N4715 | Mixing method | 10 mg L−1 (50 mL) | 200 mg L−1 | 9 | Xenon arc lamp (300 W) with UV cut-off filter (λ ≥ 400 nm), 150 ± 5 mW cm−2 | 100% (12 min) | 0.47 min−1 |
| TiO2–carbon microspheres (CMS) with Ti:CMS molar ratio = 2718 |
Solvothermal treatment | 5 mg L−1 (50 mL) | 250 mg L−1 | 6.0 | Xenon lamp (500 W m−2). With light correction filter (λ ≤ 350 nm) | 100% (180 min) | — |
| 30% TiO2-hybridize C-doped based g-C3N4719 | In situ method | 10 mg L−1 (100 mL) | 1 g L−1 | 5.05 | LED lamp: 50 W, 380–780 nm | 98.92% (30 min) | 0.1796 min−1 |
| AgI/g-C3N4 (molar ratio of AgI: 45%)720 | Deposition–precipitation method | 1 mg L−1 (100 mL) | 10 mg | — | Xenon lamp (300 W), λ ≥ 400 nm, 100 mW cm−2 | 100% (6 min) | 0.561 min−1 |
| Ag modified g-C3N4 (mass ratio of Ag: 54%)721 | Photodeposition | 100 mg L−1 (100 mL) | 10 mg | — | Xenon lamp: 300 W with cut-off filter (λ ≥ 400 nm), 100 mW cm−2 | ∼100% (120 min) | 0.0429 min−1 |
| TiO2–rGO in presence of persulfate726 | Solvothermal treatment (using 5 wt% GO) | 25 mg L−1 (50 mL), (persulfate:20 mg L−1) | 75 mg L−1 | 4 | Sunlight (1.25 × 106 lx) | >98% (30 min) | 99.4 × 10−3 min−1 |
| BiOCl–GO727 | One-pot hydrothermal method | 25 mg L−1 (100 mL) | 1 g L−1 | 5 | Visible spectrum solar light (17.38 mW cm−2) | 47.88% (180 min) | — |
| 5 Mol% Ag–BiOI–rGO 5 wt%728 | Hydrothermal strategy | 10.0 μg mL−1 (50 mL) | 50 mg | — | Halogen lamp: 300 W | 100% (80 min) | 0.026 min−1 |
| Ag/AgBr/GO729 | Sonochemical route | 25 mg L−1 (25 mL) | 0.030 g | 6.2 | Sunlight irradiation | ∼93% (6 min) | — |
| rGO–Cu3BiS3 (15%)/PS (5 mM)730 | Solvothermal process | 10 mg L−1 (50 mL) | 30 mg | — | UV LED light (15 W) | 85% (60 min) | 3.8 × 10−2 min−1 |
| Co3O4/WO3 (annealed)731 | Dispersion method | 15 ppm (50 ml) | 30 mg | 6.8 | Mercury lamp (80 W) with cut-off of 420 nm | 90.8% (180 min) | 0.1412 min−1 |
| Fe3O4@SrTiO3/Bi4O5I2732 | In situ hydrothermal route | 10 mg L−1 | 0.3 mg mL−1 | 6 | Xenon lamp (300 W) | 98.4% (90 min) | 0.06214 min−1 |
| N,S co-doped TiO2@MoS2735 | Hydrothermal method | 0.15 mg L−1 | 0.98 g L−1 | 5.5 | Visible LED light irradiation | 98% (150 min) | 0.002 min−1 |
| S–B-co-doped g-C3N4 nanotubes–MnO2 (PMS: 0.06 mM)736 | Hydrothermal | 20 mg L−1 | 0.5 g L−1 | 7 | Visible light (8 × 8 W), 460 nm | 99% (10 min) | — |
| Pt–TiO2–Nb2O5738 | Multiple steps | 12.5 mg L−1 (100 mL) | 0.5 g L−1 | — | UV-LED | 100% (20 min) | 0.446 min−1 |
| BiOCl/CuBi2O4 (mass ratio: 40%)739 | Solvothermal process | 50 mg L−1 (40 mL) | 1 mg mL−1 | — | Xenon lamp (300 W), λ > 420 nm | ∼90% (60 min) | 0.03539 min−1 |
| CuBi2O4/Ag3PO4 (1:1)740 |
Combination of hydrothermal and in situ deposition | 10 mg L−1 (50 mL) | 0.025 g | — | Xenon lamp (300 W) with cut-off filter at λ ≥ 400 nm | ∼90% (120 min) | 0.0143 min−1 |
| CuBi2O4/Ag3PO4 (mass ratio of 3:7)741 |
Hydrothermal synthesis and in situ deposition method | 10 mg L−1 | 25 mg (50 mL) | 4.42 | Xenon lamp (300 W), λ > 400 nm | 82% (60 min) | 0.0072 minc |
| CuBi2O4/Ag3PO4 (mass ratio of 3:7)/S2O82−: 1–06 mM741 |
Hydrothermal synthesis and in situ deposition method | 10 mg L−1 | 25 mg (50 mL) | 4.42 | Xenon lamp (300 W), λ > 400 nm | 100% (60 min) | 0.0272 min−1 |
| CuBi2O4/Ag3PO4 (mass ratio of 3:7)/H2O2: 1 mM741 |
Hydrothermal synthesis and in situ deposition method | 10 mg L−1 | 25 mg (50 mL) | 4.42 | Xenon lamp (300 W), λ > 400 nm | 98.40% (60 min) | 0.0162 min−1 |
| TiO2/g-C3N4742 | Wet impregnation method | 5 ppm | 0.3 g | 5 | W halogen lamp (1000 W) | 93.49% (90 min) | 0.0324 min−1 |
| Ag3PO4/g-C3N4 (30%)745 | Deposition–precipitation method | 1 mg L−1 (100 mL) | 0.1 g L−1 | — | Xenon lamp (300 W) with filter (λ ≥ 400 nm) | ∼100% (12 min) | 0.453 min−1 |
| 50% V2O5–g-C3N4 (molar ratio: 30%)746 | Mixing method | 10 mg L−1 | 0.2 mg mL−1 | >7 | Monochromatic blue lamps (8 W), 465 ± 40 nm | 100% (<105 min) | ∼0.53 min−1 |
| MoS2/Cd0.9Zn0.1S747 | One-step hydrothermal method | 20 μM (50 mL) | 25 mg | — | Xenon lamp (300 W) with 420 nm cut-off filter | 86% (30 min) | — |
3.9 Atenolol
Atenolol (ATL) belongs to the group of β-blockers and is extensively used in the treatment of cardiovascular diseases, such as hypertension, coronary arterial disease and cardiac arrhythmia.749 As a result, it has been widely detected in sewage effluent, surface water and wastewater treatment plants on its release into the environment through urban discharges. Atenolol can prevent the growth of human embryonic cells and is toxic to water species. Therefore, it is essential to develop simple and cost-effective technologies for the effective removal of ATL in wastewater before release into natural water.750–780Among the different commercial TiO2 catalysts, TiO2 (Degussa P25) aqueous suspensions (250 mg L−1) delivered 80% photocatalytic conversion of atenolol (10 mg L−1) under irradiation by a 1 kW Xe-OP lamp in 120 min.755 TiO2 (Degussa P25) has been tested for the removal by degradation of atenolol, acetaminophen, sulfamethoxazole in hospital wastewater.756 Rimoldi et al.757 evaluated the degradation of tetracycline hydrochloride, paracetamol, caffeine and atenolol, both as individual pollutants and in mixtures, using UV and simulated-solar-mediated TiO2. According to Ponkshe and Thakur,758 degradation of atenolol (2 × 10−4 M) using different commercially available TiO2 (0.03 g L−1) as photocatalysts in a 100 mL reaction solution (natural pH) under UV light for 120 min followed the order: Aeroxide TiO2 P25 (94%) > TiO2 Hombikat UV 100 (68%) > Merck TiO2 (60%) > TiO2 Kronoclean 7000 (45%). Rogé et al.759 prepared ZnO nanowires by metal organic chemical vapor deposition and investigated their photocatalytic activity in a solution containing atenolol and sulfadimidine under low-power 365 nm UV light (2.28 mW cm−2). The corresponding pseudo-first-order rate constants in these pollutants were found to be 6.5 × 10−3 and 2.3 × 10−3 min−1. Several other studies also reported the photocatalytic degradation of atenolol in aqueous solution using Degussa TiO2 P25 suspension,760 TiO2,761 TiO2/salicylaldehyde–NH2-MIL-101(Cr)762 and ZnO.763
Atenolol has been removed from domestic wastewater effluent using green-synthesized Fe (0–5%)-doped TiO2 (Fe–TiO2) under visible-light irradiation.765 These findings showed 85% removal of atenolol in the presence of Fe (2 wt%)–TiO2 after 105 min at solution pH 9, initial atenolol concentration of 10 mg L−1 and catalyst dose of 1.25 g L−1. The degradation of atenolol by visible-light-activated Fe–TiO2 was attributed to the cleavage of the ether bond, hydroxylation of the aromatic ring and oxidation of amine moieties. Alternatively, the enhanced photocatalytic activity for atenolol by Fe-doped TiO2 due to the reduced band gap of TiO2 cannot be ruled out.
Ag–TiO2 (Ag/Ti molar ratio: 2%) microtubes showed enhanced degradation of atenolol under UV-light irradiation (λ: 365 nm, power: 0.111 mW cm−2).766 Further investigations revealed Ag acting as a good photogenerated electron acceptor for photocatalysis. Cobalt-doped TiO2 nanoparticles (dose: 2.0 g L−1) exhibited about 90% photodegradation (ATL: 15 mg L−1, H2O2: 2.0 mL, pH: 2) of atenolol in 40 min under UV irradiation.767 The photodegradation of atenolol followed first-order kinetics, and the process involved the formation of hydroxyl free radicals and superoxide oxygen anions as active species.
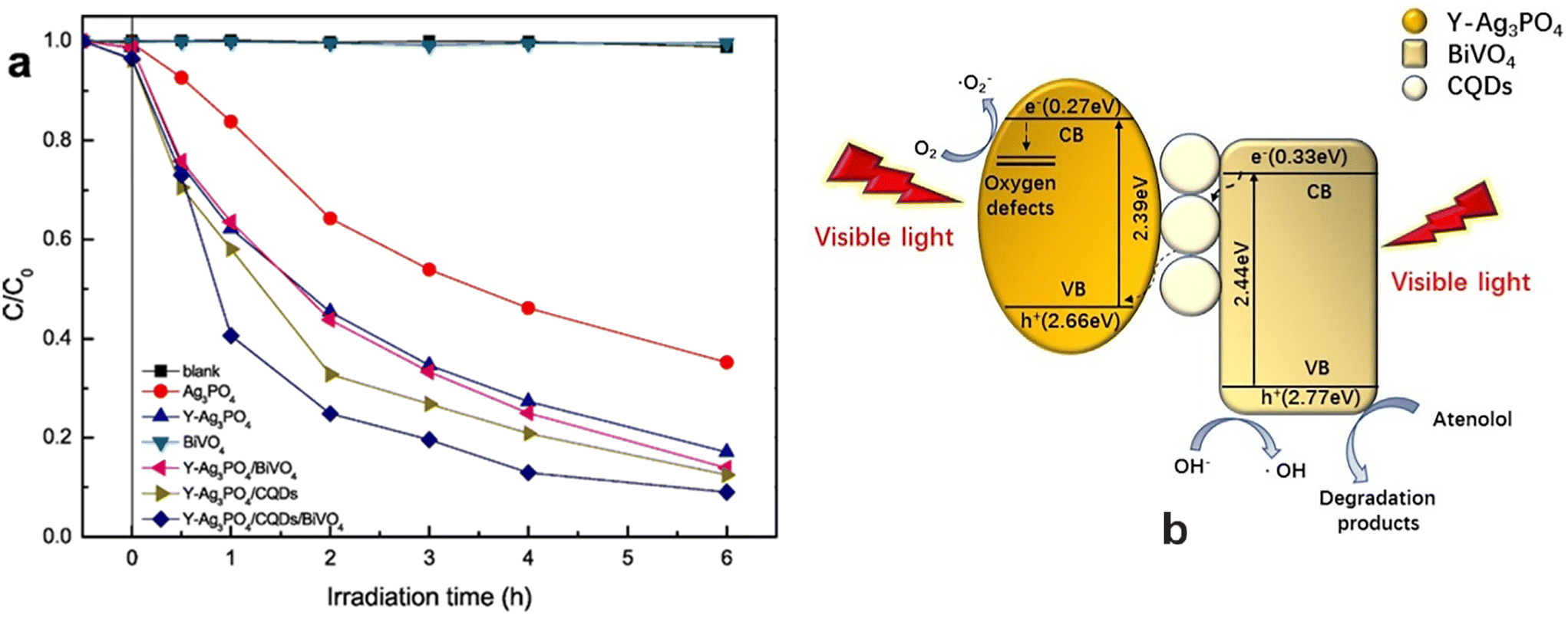 | ||
| Fig. 24 (a) Photocatalytic activities of photocatalysts for atenolol degradation of Ag3PO4, Y–Ag3PO4, BiVO4, Y–Ag3PO4/BiVO4, Y–Ag3PO4/CQDs and Y–Ag3PO4/CQDs/BiVO4 for the degradation of atenolol and (b) Z-scheme photocatalysis mechanism for atenolol degradation by Y–Ag3PO4/CQDs/BiVO4. Reproduced from ref. 779 with permission from Elsevier (2020). | ||
Table 10 records data on the performance of different photocatalysts on removal of diclofenac from water under optimum conditions.
| Photocatalyst | Preparation method | ATL | Catalyst dose | pH | Light source | Degradation (time) | Rate constant |
|---|---|---|---|---|---|---|---|
| TiO2: mixed phase (source: Shandong Xiya Chemical Co)754 | Commercial | 18.77 μM | 2 g L−1 | 7.6 | UV-lamp (365 nm) and I0: 774 mW cm−2 | 100% (60 min) | 0.064 min−1 |
| TiO2 (75% A + 25% R) Degussa P25755 | Commercial | 10 mg L−1 | 250 mg L−1 | 8 | Xenon-OP lamp (1 kW), I0: 272.3 W m−2 | 80% (120 min) | — |
| Degussa P25756 | Commercial | 10 mg L−1 | 1.0 g L−1 | — | Natural solar irradiation | 100% (400 kJ m−2) | — |
| Degussa TiO2 P25758 | Commercial | 37.6 mM | 2.0 g L−1 (25 mL) | 6.8 | High-pressure Hg lamp (125 W), 365 nm, 31.3 mW m−2 | ∼100% (60 min) | 0.0570 min−1 |
| Degussa P25 TiO2760 |
Commercial | 37.6 μM | 2.0 g L−1 | 7 | UV light | 100% (60 min) | — |
| TiO2 immobilized on the clinoptilolite nano particles support762 | Dispersion method | 10 mg L−1 (25 mL) | 1.5 g L−1 | — | UV lamp (80 W) | 75% (60 min) | — |
| TiO2 immobilized on Salicylaldehyde-NH2-MIL 101 (Cr) support762 | Dispersion method | 10 mg L−1 (25 mL) | 1.5 g L−1 | — | Xenon lamp (100 W) | 82% (60 min) | — |
| ZnO nanoparticles763 | Synthetic method | 20 mg L−1 | 10 mg L−1 | 7 | 9 W UVC lamp | 100% (120 min) | — |
| Fe–TiO2764 |
Green method | 5 mg L−1 (100 mL) | 1005 mg L−1 | 8 | Xenon arc lamp, 300 W, λ: 650 nm | 71.2% (98 min) | — |
| Ag–TiO2764 |
Green method | 5 mg L−1 (100 mL) | 1065 mg L−1 | 8 | Xenon arc lamp, 300 W, λ: 650 nm | 65.7% (120 min) | — |
| Ag–ZnO microtubes104 | Solution method | 5 mg L−1 | 1 g L−1 | 8.5 | W halogen lamp (300 W) | 70.2% (120 min) | 0.01 min−1 |
| Fe–TiO2765 |
Green synthesis | 10 mg L−1 | 1.25 g L−1 | 9 | 300 W halogen lamp | 85% (105 min) | 0.013 min−1 |
| Ag–TiO2 microtubes (Ag/Ti molar ratio: 2%)/O3766 |
Calcination | 20 mg L−1 | 0.2 g | 9.11 | Medium-pressure Hg lamp: 365 nm and 0.111 mW cm−2 | 92.23% (9 min) | 0.3275 min−1 |
| Co doped-TiO2 (H2O2: 2.0 mL L−1)767 | Mixing followed by calcination | 15 mg L−1 | 2.0 g L−1 | 2 | UV (200 nm) | 90% (40 min) | 0.059 min−1, 1.75 × 10−4 g mg−1 min−1 |
| Bi2O3/TiO2768 |
Solvothermal method | 10 mg L−1 | 400 mg L−1 | 7 | UVC (visible-light irradiation) | 68.92% (60 min) | — |
| TiO2/zeolites769 | Solid-state dispersion method | 50 mg L−1 | 1 g L−1 (40 mL) | 6.5 | Lamp (Osram Vitalux (300 W)) | ∼94% (70 min) | 0.132 ± 0.001 min−1 |
| TiO2@Fe3O4770 |
Mixing method | 10 ppm | 0.75 g L−1 | 5.5 | Low-pressure Hg vapour lamp (UVC. l: 280 nm) | 43.7% (30 min) | — |
| Fe3O4@AgCl770 | Mixing method | 10 ppm | 0.75 g L−1 | 5.5 | Low-pressure Hg vapour lamp (UVC. l: 280 nm) | 66% (30 min) | — |
| Fe3O4@TiO2770 |
Mixing method | 10 ppm | 0.75 g L−1 | 5.5 | Low-pressure Hg vapour lamp (UVC. l: 280 nm) | 66% (30 min) | — |
| BiOCl@Fe3O4 with [PS]: 1.0 mM771 | Precipitation process | 2.5 mg L−1 | 0.1 g L−1 | 6.5 | Xenon lamp (simulated sunlight): 500 W | ∼99% (60 min) | (5.34–6.04) × 10−2 min−1 |
| Graphene oxide–TiO2773 |
Hydrothermal | 25 ppm | 1.5 g L−1 (150 mL) | 6 | 1000 W xenon arc lamp, 750 mW cm−2 | 72% (60 min) | — |
| Y–Ag3PO4/CQDs/BiVO4779 |
Mixing method | 10 mM (50 mL) | 5 mg photocatalyst | — | 250 W xenon lamp with UV cut-off filter, λ > 420 nm | 90.9% (6 h) | 0.50 h−1 |
4 Future scope and perspectives
Pharmaceutical pollutants found in water supplies through human and animal consumption of antibiotics, antipyretics, analgesics, etc. are considered potential hazards to the environment, humans and aquatic life.781 However, conventional wastewater treatment methods are ineffective in eliminating them completely. In view of this, the photocatalytic degradation of these pharmaceutical pollutants using semiconducting materials is considered an effective method.An efficient semiconducting material acting as an efficient photocatalyst is guided by enhanced visible-light absorption, facilitating charge carrier migration and a reduced recombination rate. In view of this, TiO2, WO3, ZnO, Fe2O3, CdS, MoS2etc. are widely used photocatalysts for the photodegradation of pharmaceutical pollutants in water.23–39 However, the large band gaps of photogenerated charge carriers, i.e. rapid recombination rate (i.e., short lifetimes) of photogenerated charge carriers, instability in an aqueous medium, reusability of the photocatalyst and poor absorption ability for visible light, are a few drawbacks that limit the practical applications of metal oxide as photocatalysts. Therefore, increasing attention has been focused on achieving the effective separation of photogenerated charge carriers, improvements in the visible-light response and other factors782 through designing and constructing advanced light energy harvesting assemblies for environmental remediation.783 This problem has been overcome by modifying semiconducting metal oxides through doping, composite formation, immobilizing semiconducting materials on supports and heterojunction formation for the removal of drugs from contaminated water. In addition, the combination of these semiconducting metal oxides with carbon-based materials, such as activated carbon, biochar, carbon nanotubes, carbon dots, g-C3N4 and graphene, has also attracted a lot of attention in the removal of pharmaceutical pollutants present in wastewater. However, there are still several research gaps in the removal of antibiotics by photocatalysts. These future challenges are described below.
The expensive precursors used in the synthesis of metal oxides limit their large-scale application. Therefore, it is desirable to realize the simple, facile, affordable, low-cost synthesis of photocatalysts. The specific surface area,782 crystallite size,784 size, shape and overall structure785 of photocatalysts play important roles in the photocatalytic activity of emerging pollutants. This needs to be correlated with light trapping, charge separation and pollutant adsorption ability parameters under optimized operational conditions.
Carbon-based materials have also attracted significant interest in recent years due to their unique physicochemical, optical and electrical properties following band-gap tuning, composite formation and heterojunction construction, etc.40–50 The enhanced photo-efficiency of the corresponding nanocomposites is ascribed to improvement in visible-range absorption, fast charge carrier migration and reduced recombination rate. However, their choices are limited to batch experiments at the laboratory scale rather than the pilot scale. As a result, there is a gap between on-going research and its application.
The literature revealed considerable interest in investigating the photocatalytic degradation of individual pharmaceutical pollutants in water. However, wastewater could contain complex pollutant mixtures, including other organic and inorganic species originating from heavy metals, dyes, personal care products, pesticides and other sources.757,786–794 This can affect the degradation process for pharmaceutical pollutants through interference and matrix effects. Therefore, attention also needs to be focused on developing photocatalysts capable of simultaneously removing pharmaceuticals even in the presence of other pollutants/interfering substances in the wastewater. Recovery, reusability, and stability remain other issues in the development of high-performing photocatalysts in wastewater treatment. Toxicity assessment is considered to be one of important parameters in the treatment of wastewater by photocatalysis.795 This could be ascribed to the formation of carcinogenic secondary metabolites due to the incomplete mineralization of targeted contaminants.
Nanomaterial-based photocatalysts have shown great promise due to their superior adsorptive and photocatalytic properties in the removal of pharmaceutical pollutants.51,57 In this regard, leaching of toxic components could adversely affect the quality of the water environment. This aspect remains a matter of great concern and as a consequence, extensive investigations are needed to fully understand the role of various photocatalyst nanoparticles and their toxicity risks in aquatic environments.796,797 Therefore, it remains challenging to recover and separate the nanoparticle-based photocatalysts invariably used in water treatment. Recently, this difficulty has been overcome by immobilizing the photocatalysts on various support materials. Therefore, in the future innovations will be needed for effective, eco-friendly, and sustainable immobilization techniques for the separation/recovery and reuse of photocatalytic materials. Existing research has also invariably focused on laboratory-scale photocatalysis in the degradation of emerging pharmaceutical pollutants without much implementation in real water systems. More studies need to be focused at the pilot and industrial scale levels for its commercialization. The fabrication of economical, environmentally friendly and effective photocatalysts taking into account many of these aspects remains a major challenge in this field.
5 Conclusions
Antibiotics have been invariably used in different fields, such as the medical field, agriculture, and veterinary medicine for the purpose of killing or preventing bacterial growth. However, the presence of these pharmaceutical pollutants on entering surface water and groundwater are a potential threat to human and marine lives and need to be eliminated. Considering this, various conventional processes have been developed for the removal of these pharmaceutical pollutants. However, their choice is limited due to their high cost as well as incomplete elimination of contaminants from the contaminated water.In view of this, the current review highlights recent advances in the applications of different photocatalysts to the removal of emerging pharmaceutical pollutants in wastewater. As a result, the performance of several metal oxides, carbonaceous materials, composites including surface modification, doping with metals/nonmetals, heterojunction formation, and immobilization using support materials, homo- or hetero-materials composed of two or more inorganic phases, inorganic semiconductors coupled with carbon-based materials, inorganic semiconductors hybridized with 2D materials as excellent photocatalysts have been reviewed to find out the optimum removal efficiency for the pollutants (acetaminophen, amoxicillin, sulfamethoxazole, acetaminophen, norfloxacin, ciprofloxacin, tetracycline, diclofenac and atenolol) in water. However, secondary pollution produced by the formation of by-products during the photocatalytic process, leaching of dopants/active components of the photocatalysts, and the generation of excess CO2 during the photocatalysis process are additional challenges that need to be addressed in future. Further, most of these findings are reported on the laboratory scale, and real-world and industrial-scale applications have yet to be fully realized. The further development of low-cost, robust photocatalysts utilizing semiconductors and renewable visible/solar light to solve both the world crises of energy supply and environmental pollution remains a pressing demand for industrial application.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author began working on this review article while still a Professor in the Department of Chemistry, Indian Institute of Technology, Kharagpur and thus, remains very appreciative for making this effort possible. Author also expresses his heartfelt thanks to Professor Ashok Kumar Gupta, Department of Civil Engineering, Indian Institute of Technology, Kharagpur and his research scholars Brahma Gupta, Vishal Parida, Adarsh Singh and Akash Rawat for the valuable interactions. The table of content figure was drawn with the help of Biorender and acknowledged. Author also thanks Dr. Kunal Manna and Dr. Ayon Karmakar for their help in arranging copyright permissions and the Figures, respectively.References
- https://www.unesco.org/en/articles/imminent-risk-global-water-crisis-warns-un-world-water-development-report-2023 .
- I. Bashir, F. A. Lone, R. A. Bhat, S. A. Mir, Z. A. Dar and S. A. Dar, in Bioremediation and biotechnology sustainable approaches to pollution degradation, ed. K. R. Hakeem, R. A. Bhat and H. Qadri, Springer, 2020, pp. 1–26, DOI:10.1007/978-3-030-35691-0_1.
- https://reliefweb.int/report/world/wastewater-resource-may-2022 .
- S. Kundu and A. K. Gupta, Chem. Eng. J., 2006, 122, 93–106 CrossRef CAS.
- S. Dutta, B. Gupta, S. K. Srivastava and A. K. Gupta, Mater. Adv., 2021, 2, 4497–4531 RSC.
- S. Dutta, S. K. Srivastava and A. K. Gupta, Mater. Adv., 2021, 2, 2431 RSC.
- S. Senapati, S. K. Srivastava and S. B. Singh, Nanoscale, 2012, 4, 6604–6612 RSC.
- P. A. Kobielska, A. J. Howarth, O. K. Farha and S. Nayak, Coord. Chem. Rev., 2018, 358, 92–102 CrossRef CAS.
- S. Ayoob and A. K. Gupta, Crit. Rev. Environ. Sci. Technol., 2006, 36, 433–487 CrossRef CAS.
- A. Singh, D. Saidulu, A. K. Gupta and V. Kubsad, J. Environ. Chem. Eng., 2022, 10, 109012 CrossRef CAS.
- B. Gupta, R. S. Ambekar, R. M. Tromer, P. S. Ghosal, R. Sinha, A. Majumder, P. Kumbhakar, P. M. Ajayan, D. S. Galvao, A. K. Gupta and C. S. Tiwary, RSC Adv., 2021, 11, 19788–19796 RSC.
- B. S. Rathi, P. S. Kumar and D.-V. N. Vo, Sci. Total Environ., 2021, 797, 149134 CrossRef CAS PubMed.
- M. Hejna, D. Kapuścińska and A. Aksmann, Int. J. Environ. Res. Public Health, 2022, 19, 7717 CrossRef CAS PubMed.
- K. Samal, S. Mahapatra and M. H. Ali, Energy Nexus, 2022, 6, 100076 CrossRef CAS.
- A. C. Duarte, S. Rodrigues, A. Afonso, A. Nogueira and P. Coutinho, Pharmaceuticals, 2022, 15, 393 CrossRef CAS PubMed.
- T. Zhang, K. Lv, Q. Lu, L. Wang and X. Liu, J. Environ. Sci., 2021, 104, 415–429 CrossRef CAS PubMed.
- J. O'Neill, Tackling Drug-Resistant Infections Globally: Final Report and Recommendations, 2016, Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf.
- S. I. Polianciuc, A. E. Gurzău, B. Kiss, M. G. Ştefan and F. Loghin, Med. Pharm. Rep., 2020, 93, 231–240 Search PubMed.
- V. Vinayagam, S. Murugan, R. Kumaresan, M. Narayanan, M. Sillanpää, D. V. N. Vo, O. S. Kushwaha, P. Jenis, P. Potdar and S. Gadiya, Chemosphere, 2022, 300, 134597 CrossRef CAS PubMed.
- V. K. Parida, S. K. Srivastava, S. Chowdhury and A. K. Gupta, Chem. Eng. J., 2023, 472, 144969 CrossRef CAS.
- A. A. Aryee, R. Han and L. Ou, J. Cleaner Prod., 2022, 368, 133140 CrossRef CAS.
- B. Sruthi, S. Janani and S. S. Khan, Sep. Purif. Technol., 2021, 279, 119709 CrossRef.
- D. Kanakaraju, B. D. Glass and M. Oelgemoller, Environ. Chem. Lett., 2014, 12, 27–47 CrossRef CAS.
- M. Minella, D. Fabbri, P. Calza and C. Minero, Curr. Opin. Green Sustainable Chem., 2017, 6, 11–17 CrossRef.
- R. Karuppannan, S. Mohan and T.-O. Do, in Nanostructured materials for environmental applications, ed. S. Balakumar, V. Keller and M. V. Shankar, Springer Nature Switzerland AG, 2021, pp. 85–112 Search PubMed.
- A. Krishnan, A. Swarnalal, D. Das, M. Krishnan, V. S. Saji and S. M. A. Shibli, J. Environ. Sci., 2024, 139, 389–417 CrossRef PubMed.
- N. Roy, S. A. Alex, N. Chandrasekaran, A. Mukherjee and K. Kannabiran, J. Environ. Chem. Eng., 2021, 9, 104796 CrossRef CAS.
- R. Gusain, K. Gupta, P. Joshi and O. P. Khatri, Adv. Colloid Interface Sci., 2019, 272, 102009 CrossRef CAS PubMed.
- S. Gautam, H. Agrawal, M. Thakur, A. Akbari, H. Sharda, R. Kaur and M. Amini, J. Environ. Chem. Eng., 2020, 8, 103726 CrossRef CAS.
- T. Velempini, E. Prabakaran and K. Pillay, Mater. Today Chem., 2021, 19, 100380 CrossRef CAS.
- R. Krakowiak, J. Musial, P. Bakun, M. Spychała, B. Czarczynska-Goslinska, D. T. Mlynarczyk, T. Koczorowski, L. Sobotta, B. Stanisz and T. Goslinski, Appl. Sci., 2021, 11, 8674 CrossRef CAS.
- K. P. Gopinath, N. V. Madhav, A. Krishnan, R. Malolan and G. Rangarajan, J. Environ. Manage., 2020, 270, 110906 CrossRef CAS PubMed.
- D. Chen, Y. Cheng, N. Zhou, P. Chen, Y. Wang, K. Li, S. Huo, P. Cheng, P. Peng, R. Zhang, L. Wang, H. Liu, Y. Liu and R. Ruan, J. Cleaner Prod., 2020, 268, 121725 CrossRef CAS.
- A. Mirzaei, Z. Chen, F. Haghighat and L. Yerushalmi, Sustain. Cities Soc., 2016, 27, 407–418 CrossRef.
- P. Kar, K. Shukla, P. Jain, G. Sathiyan and R. K. Gupta, Nano Mater. Sci., 2021, 3, 25–46 CrossRef CAS.
- K. S. Varma, R. J. Tayade, K. J. Shah, P. A. Joshi, A. D. Shukla and V. G. Gandhi, Water-Energy Nexus, 2020, 3, 46–61 CrossRef.
- G. Ren, H. Han, Y. Wang, S. Liu, J. Zhao, X. Meng and Z. Li, Nanomaterials, 2021, 11, 1804 CrossRef CAS PubMed.
- R. M. S. Sendão, J. C. G. Esteves da Silva and L. P. da Silva, Chemosphere, 2022, 301, 134731 CrossRef PubMed.
- L. G. Devi and R. Kavitha, Appl. Surf. Sci., 2016, 360, 601–622 CrossRef.
- J. Ge, Y. Zhang and S.-J. Park, Materials, 2019, 12, 1916 CrossRef CAS PubMed.
- R. Ma, Y. Xue, Q. Ma, Y. Chen, S. Yuan and J. Fan, Nanomaterials, 2022, 12, 4045 CrossRef CAS PubMed.
- T. H. Pham, N. M. Viet, P. T. T. Hoai, S. H. Jung and T. Y. Kim, Environ. Res., 2023, 231, 116246 CrossRef CAS PubMed.
- K. K. Gangu, S. Maddila and S. B. Jonnalagadda, Sci. Total Environ., 2019, 646, 1398–1412 CrossRef CAS PubMed.
- R. B. González-González, A. Sharma, R. Parra-Saldívar, R. A. Ramirez-Mendoza, M. Bilal and H. M. N. Iqbal, J. Hazard. Mater., 2022, 423, 127145 CrossRef PubMed.
- M. Abdullah, J. Iqbal, M. S. U. Rehman, U. Khalid, F. Mateen, S. N. Arshad, A. G. Al-Sehemi, H. Algarni, O. A. Al-Hartomy and T. Fazal, Chemosphere, 2023, 317, 137834 CrossRef CAS PubMed.
- M.-F. Li, Y.-G. Liu, G.-M. Zeng, N. Liu and S.-B. Liu, Chemosphere, 2019, 226, 360–380 CrossRef CAS PubMed.
- M. Minale, Z. Gu, A. Guadie, D. M. Kabtamu, Y. Li and X. Wang, J. Environ. Manage., 2020, 276, 111310 CrossRef CAS PubMed.
- O. C. Olatunde and D. C. Onwudiwe, Int. J. Environ. Res. Public Health, 2021, 18, 1529 CrossRef CAS PubMed.
- B. A. Bhanvase, T. P. Shende and S. H. Sonawane, Environ. Technol. Rev., 2017, 6, 1–14 CrossRef CAS.
- W. W. Anku, E. M. Kiarii, R. Sharma, G. M. Joshi, S. K. Shukla and P. P. Govender, in A New Generation Material Graphene: Applications in Water Technology, ed. M. Naushad, Springer International Publishing AG, part of Springer Nature, 2019, pp. 187–208 Search PubMed.
- V. K. Parida, S. K. Srivastava, A. K. Gupta and A. Rawat, Mater. Express, 2023, 13, 1–38 CrossRef CAS.
- A. Abbasnia, A. Zarei, M. Yeganeh, H. R. Sobhi, M. Cholami and A. Esrafili, Inorg. Chem. Commun., 2022, 145, 109959 CrossRef CAS.
- S. S. Imam, R. Adnan and N. H. M. Kaus, Toxicol. Environ. Chem., 2018, 100, 518–539 CrossRef.
- S. Shurbaje, P. T. Huong and T. M. Altahtamounti, Catalysts, 2021, 11, 437 CrossRef.
- https://pubchem.ncbi.nlm.nih.gov/ .
- Z. L. Wang, J. Phys.: Condens.Matter, 2004, 16, R829–R858 CrossRef CAS.
- A. Pattnaik, J. N. Sahu, A. K. Poonia and P. Ghosh, Chem. Eng. Res. Des., 2023, 190, 667–686 CrossRef CAS.
- Z. Wu, Z. Xiong and B. Lai, Environmental Functional Materials, 2022, 1, 298–315 CrossRef.
- Y. Wang, Z. Lu, Z. Zhu, X. Zhao, N. Gao, D. Wang, Z. Hua, Y. Yan, P. Huoa and M. Song, RSC Adv., 2016, 6, 51877–51887 RSC.
- S. Heidari, M. Haghighi and M. Shabani, J. Cleaner Prod., 2020, 259, 120679 CrossRef CAS.
- P. Wang, B. Huang, Y. Dai and M.-H. Whangbo, Phys. Chem. Chem. Phys., 2012, 14, 9813–9825 RSC.
- A. Kumar, P. Choudhary, A. Kumar, P. H. C. Camargo and V. Krishnan, Small, 2022, 18, 2101638 CrossRef CAS PubMed.
- E. S. Araújo, M. F. G. Pereira, G. M. G. da Silva, G. F. Tavares, C. Y. B. Oliveira and P. M. Faia, Toxics, 2023, 11, 658 CrossRef PubMed.
- A. Moharana, A. Kumar, A. Thakur, D.-V. N. Vo, A. Sharma and D. Kumar, in Nanostructured photocatalysts from Fundamental to practical applications, 2021, pp. 145–167, DOI:10.1016/B978-0-12-823007-7.00010-9.
- S. Biswas and A. Pal, Catalysts, 2023, 13, 925 CrossRef CAS.
- B. Samir, N. Bouazizi, P. N. Fotsing, J. Cosme, V. Marquis, G. L. Dotto, F. L. Derf and J. Vieillard, Appl. Sci., 2023, 13, 8074 CrossRef CAS.
- S. Kurwadkar, T. V. Hoang, K. Malwade, S. R. Kanel, W. F. Harper Jr and G. Struckhof, Nanotechnol. Environ. Eng., 2019, 4, 12 CrossRef CAS.
- Y. Tang, X. Liu, C. Ma, M. Zhou, P. Huo, L. Yu, J. Pan, W. Shia and Y. Yan, New J. Chem., 2015, 39, 5150–5160 RSC.
- B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 075404 CrossRef.
- Y. Ren, D. Zeng and W.-J. Ong, Chin. J. Catal., 2019, 40, 289–319 CrossRef CAS.
- X. He, T. Kai and P. Ding, Environ. Chem. Lett., 2021, 19, 4563–4601 CrossRef CAS PubMed.
- S. Obregón, M. A. Ruíz-Gómez, V. Rodríguez-González, A. Vázquez and D. B. Hernández-Uresti, Mater. Sci. Semicond. Process., 2020, 113, 105056 CrossRef.
- W. Dai, L. Jiang, J. Wang, Y. Pu, Y. Zhu, Y. Wang and B. Xiao, Chem. Eng. J., 2020, 397, 125476 CrossRef CAS.
- M. Miceli, P. Frontera, A. Macario and A. Malara, Catalysts, 2021, 11, 591 CrossRef CAS.
- V. Wongso, H. K. Chung, N. S. Sambudi, S. Sufian, B. Abdullah, M. D. H. Wirzal and W. L. Ang, J. Photochem. Photobiol., A, 2020, 394, 112436 CrossRef CAS.
- A. H. Zyoud, A. Zubi, S. Hejjawi, S. H. Zyoud, M. H. Helal, S. H. Zyoud, N. Qamhieh, A. R. Hajamohideen and H. S. Hilal, J. Environ. Chem. Eng., 2020, 8, 104038 CrossRef CAS.
- C. A. Aguilar, C. Montalvo, J. G. Ceron and E. Moctezuma, Int. J. Environ. Res., 2011, 5, 1071–1078 CAS.
- C. A. Aguilar, C. Montalvo, B. B. Zermeño, R. M. Cerón, J. G. Cerón, F. Anguebes and M. A. Ramírez, Int. J. Environ. Sci. Technol., 2019, 16, 843–852 CrossRef CAS.
- R. Trujillano, V. Rives and I. García, Molecules, 2022, 27, 2904 CrossRef CAS PubMed.
- A. Marizcal-Barba, J. A. Sanchez-Burgos, V. Zamora-Gasga and A. Perez Larios, Photochem, 2022, 2, 225–236 CrossRef CAS.
- C. J. Lin, W. T. Yang, C.-Y. Chou and S. Y. H. Liou, Chemosphere, 2016, 152, 490–495 CrossRef CAS PubMed.
- X. Zhang, F. Wu, X. W. Wu, P. Chen and N. Deng, Materials, 2008, 157, 300–307 CAS.
- S. A. Lozano-Morales, G. Morales, M. Á. L. Zavala, A. Arce-Sarria and F. Machuca-Martínez, Processes, 2019, 7, 319 CrossRef CAS.
- N. Kaneva, A. Bojinova, K. Papazova, D. Dimitrov, K. Zaharieva, Z. Cherkezova-Zheleva and A. Eliyas, Arch. Pharmacal Res., 2016, 39, 1418–1425 CrossRef CAS PubMed.
- R. Katal, M. H. D. A. Farahani and H. Jiangyong, Sep. Purif. Technol., 2020, 230, 115859 CrossRef CAS.
- M. Cantarella, A. D. Mauro, A. Gulino, L. Spitaleri, G. Nicotra, V. Privitera and G. Impellizzeri, Appl. Catal., B, 2018, 238, 509–517 CrossRef CAS.
- M. Jiménez-Salcedo, M. Monge and M. T. Tena, Photochem. Photobiol. Sci., 2022, 21, 337–347 CrossRef PubMed.
- O. Nasr, O. Mohamed, A.-S. Al-Shirbini and A.-M. Abdel-Wahab, J. Photochem. Photobiol., A, 2019, 374, 185–193 CrossRef CAS.
- R. Katal, S. M. Panah, M. Saeedikhani, M. Kosari, C. C. Sheng, O. S. Leong, G. Xiao and H. Jiangyong, Adv. Mater. Interfaces, 2018, 5, 1801440 CrossRef.
- T. H. Yu, W. Y. Cheng, K.-J. Chao and S. Y. Lu, Nanoscale, 2013, 5, 7356–7360 RSC.
- C. A. Huerta-Aguilar, A. A. Ramírez-Alejandre, P. Thangarasu, J. A. Arenas-Alatorre, I. A. Reyes-Dominguez and M. de la L. Corea, Catal. Sci. Technol., 2019, 9, 3066 RSC.
- S. Sayegh, F. Tanos, A. Nada, G. Lesage, F. Zaviska, E. Petit, V. Rouessac, I. Iatsunskyi, E. Coy, R. Viter, D. Damberga, M. Weber, A. Razzouk, J. Stephan and M. Bechelany, Dalton Trans., 2022, 51, 2674–2695 RSC.
- V. Y. Safitri, A. Santoni, D. V. Wellia, K. Khoiriah and S. Safni, Mol. Ther., 2017, 12, 189–195 CAS.
- Y. A. Shaban and H. M. Fallata, Res. Chem. Intermed., 2019, 45, 2529–2547 CrossRef CAS.
- W. L. da Silva, M. A. Lansarin, J. H. Z. dos Santos and F. Silveira, Water Sci. Technol., 2016, 74, 2370–2383 CrossRef PubMed.
- L. Rimoldi, D. Meroni, E. Falletta, A. M. Ferretti, A. Gervasini, G. Cappelletti and S. Ardizzone, Appl. Surf. Sci., 2017, 424, 198–205 CrossRef CAS.
- Z. Khani, D. Schieppati, C. L. Bianchi and D. C. Boffito, Catalysts, 2019, 9, 949 CrossRef CAS.
- A. M. S. Asadi and M. Malakootian, J. Mater. Sci.: Mater. Electron., 2019, 30, 14878–14889 CrossRef.
- M. L. P. Dalida, K. M. S. Amer, C.-C. Su and M.-C. Lu, Environ. Sci. Pollut. Res., 2014, 21, 1208–1216 CrossRef CAS PubMed.
- M. Abid, E. Makhoul, F. Tanos, I. F. Iatsunskyi, E. Coy, G. Lesage, M. Cretin, D. Cornu, A. B. H. Amara and M. Bechelany, Membranes, 2023, 13, 204 CrossRef CAS PubMed.
- M. D. G. de Luna, J. C.-T. Lin, M. J. N. Gotostos and M.-C. Lu, Sustainable Environ. Res., 2016, 26, 161–167 CrossRef CAS.
- A. Alzamly, F. Hamed, T. Ramachandran, M. Bakiro, M. Bakiro, S. H. Ahmed, S. Mansour, S. Salem, K. Abdul al, N. S. Al Kaabi, M. Meetani and A. Khalee, J. Water Reuse Desalin., 2019, 9, 31–46 CrossRef CAS.
- H. A. Abbas, R. A. Nasr, R. N. Vannier and T. S. Jamil, J. Environ. Sci., 2022, 112, 331–342 CrossRef CAS PubMed.
- B. Ramasamy, J. Jeyadharmarajan and P. Chinnaiyan, Environ. Sci. Pollut. Res., 2021, 28, 39637–39647 CrossRef CAS PubMed.
- V. H.-T. Thi and B.-K. Lee, Mater. Res. Bull., 2017, 96, 171–182 CrossRef CAS.
- R. A. Abri, F. A. Marzouqi, A. T. Kuvarega, M. A. Meetani, S. M. Z. Al Kindy, S. Karthikeyan, Y. Kim and R. Selvaraj, J. Photochem. Photobiol., A, 2019, 384, 112065 CrossRef.
- D. R. Kumar, K. S. Ranjith, Y. Haldorai, A. Kandasami and R. T. R. Kumar, ACS Omega, 2019, 4, 11973–11979 CrossRef PubMed.
- O. F. S. Khasawneh, P. Palaniandy, M. Ahmadipour, H. Mohammadi, M. Razak and B. Hamdan, J. Environ. Chem. Eng., 2021, 9, 104921 CrossRef CAS.
- O. F. S. Khasawneh, P. Palaniandy and L. P. Teng, MethodsX, 2019, 6, 2735–2743 CrossRef PubMed.
- H. Shahabi, A. Allahrasani and A. Naghizadeh, Desalin. Water Treat., 2019, 149, 164–170 CrossRef CAS.
- P. M. Álvareza, J. Jaramillo, F. López-Piñero and P. K. Plucinski, Appl. Catal., B, 2010, 100, 338–345 CrossRef.
- B. Czech and K. Tyszczuk-Rotko, Sep. Purif. Technol., 2018, 206, 343–355 CrossRef CAS.
- T. A. Fernandes, S. G. Mendo, L. P. Ferreira, N. R. Neng, M. C. Oliveira, A. Gil, M. D. Carvalho, O. C. Monteiro, J. M. F. Nogueira and M. J. Calhorda, Environ. Sci. Pollut. Res., 2021, 28, 17228–17243 CrossRef CAS PubMed.
- R. Mu, Y. Ao, T. Wu, C. Wang and P. Wang, J. Hazard. Mater., 2020, 382, 121083 CrossRef CAS PubMed.
- J. H. F. Chau, C. W. Lai, B. F. Leo, J. C. Juan and M. R. Johan, Catal. Commun., 2022, 163, 106396 CrossRef CAS.
- S. Basha, D. Keane, K. Nolan, M. Oelgemöller, J. Lawler, J. M. Tobin and A. Morrissey, Environ. Sci. Pollut. Res., 2015, 22, 2219–2230 CrossRef CAS PubMed.
- A.-M. Abdel-Wahab, A.-S. Al-Shirbini, O. Mohamed and O. Nasr, J. Photochem. Photobiol., A, 2017, 347, 186–198 CrossRef CAS.
- N. Jallouli, K. Elghniji, H. Trabelsi and M. Ksibi, Arabian J. Chem., 2017, 10, S3640–S3645 CrossRef CAS.
- L. Yanyan, T. A. Kurniawan, Z. Ying, A. B. Albadarin and G. Walker, J. Mol. Liq., 2017, 243, 761–770 CrossRef.
- C.-T. Chang, J.-J. Wang, T. Ouyang, Q. Zhang and Y.-H. Jing, Mater. Sci. Eng., B, 2015, 196, 53–60 CrossRef CAS.
- N. Miranda-García, S. Suárez, M. Ignacio Maldonado, S. Malato and B. Sánchez, Catal. Today, 2014, 230, 27–34 CrossRef.
- V. Vaiano, M. Matarangolo and O. Sacco, Chem. Eng. J., 2018, 350, 703–713 CrossRef CAS.
- S. Behravesh, N. Mirghaffari, A. A. Alemrajabi, F. Davar and M. Soleimani, Environ. Sci. Pollut. Res., 2020, 27, 26929–26942 CrossRef CAS PubMed.
- G. Fan, H. Peng, J. Zhang, X. Zheng, G. Zhu, S. Wang and L. Hong, Catal. Sci. Technol., 2018, 8, 5906–5919 RSC.
- T. A. Kurniawan, L. Yanyan, T. Ouyang, A. B. Albadarin and G. Walker, Mater. Sci. Semicond. Process., 2018, 73, 42–50 CrossRef CAS.
- G. Fan, X. Zheng, J. Luo, H. Peng, H. Lin, M. Bao, L. Hong and J. Zhou, Chem. Eng. J., 2018, 351, 782–790 CrossRef CAS.
- F. Al Marzouqi, R. Selvaraj and Y. Kim, Mater. Res. Express, 2019, 6, 125538 CrossRef CAS.
- A. Smýkalová, B. Sokolová, K. Foniok, V. Matejka and P. Praus, Nanomaterials, 2019, 9, 1194 CrossRef PubMed.
- M. Z. A. Warshagha and M. Muneer, Int. J. Environ. Anal. Chem., 2022, 102, 6339–6358 CrossRef.
- M. Kohantorabi, G. Moussavi, P. Oulego and S. Giannakis, Sep. Purif. Technol., 2022, 299, 121723 CrossRef CAS.
- R. R. Solís, M. A. Quintana, M. Á. Martín-Lara, A. Pérez, M. Calero and M. J. Muñoz-Batista, Int. J. Mol. Sci., 2022, 23, 12871 CrossRef PubMed.
- S. Gupta, J. Gandhi, S. Kokate, L. G. Raikar, V. G. Kopuri and H. Prakash, Heliyon, 2023, 9, e16450 CrossRef CAS PubMed.
- A. H. C. Khavar, G. Moussavi and A. R. Mahjoub, Appl. Surf. Sci., 2018, 440, 963–973 CrossRef.
- B. Palas, G. Ersöz and S. Atalay, Chem. Eng. Sci., 2021, 242, 116593 CrossRef CAS.
- J. Zhu, Z. Zhu, H. Zhang, H. Lu, W. Zhang, Y. Qiu, L. Zhuc and S. Küppers, Appl. Catal., B, 2018, 225, 550–562 CrossRef CAS.
- H. Tao, X. Liang, Q. Zhang and C.-T. Chang, Appl. Surf. Sci., 2015, 324, 258–264 CrossRef CAS.
- E. C. Umejuru, E. Prabakaran and K. Pillay, ACS Omega, 2021, 6, 11155–11172 CrossRef CAS PubMed.
- C. Gomez-Solis, R. Mendoza, J. F. Rios-Orihuela, G. Robledo-Trujillo, L. A. Diaz-Torres, J. Oliva and V. Rodriguez-Gonzalez, J. Environ. Manage., 2021, 290, 112665 CrossRef CAS PubMed.
- L. Allagui, B. Chouchene, T. Gries, G. Medjahdi, E. Girot, X. Framboisier, A. B. H. Amara, K. Balan and R. Schneider, Appl. Surf. Sci., 2019, 490, 580–591 CrossRef CAS.
- S. Sravya, D. R. Devi, N. Belachew, K. Eswara Rao and K. Basavaiah, RSC Adv., 2021, 11, 12030 RSC.
- Y. L. Wang, M. Penas-Garzon, J. J. Rodriguez, J. Bedia and C. Belver, Chem. Eng. J., 2022, 446, 137229 CrossRef CAS.
- L. K. B. Paragas, V. D. Dang, R. S. Sahu, S. Garcia-Segura, M. D. G. de Luna, J. A. I. Pimentel and R.-A. Doong, Sep. Purif. Technol., 2021, 272, 117567 CrossRef CAS.
- X. Du, X. Bai, L. Xu, L. Yang and P. Jin, Chem. Eng. J., 2020, 384, 123245 CrossRef CAS.
- M. Z. A. Warshagha and M. Muneer, Environ. Nanotechnol., Monit. Manage., 2022, 18, 100728 CAS.
- Z. Ren, Z. Zhao, Z. Yang, B. Cheng and X. Yang, Nano, 2021, 16, 2150100 CrossRef CAS.
- M. Noorisepehr, K. Ghadirinejad, B. Kakavandi, A. R. A. Esfahani and A. Asadi, Chemosphere, 2019, 232, 140–151 CrossRef CAS PubMed.
- S. Moradi, A. A. Isari, F. Hayati, R. Rezaei, R. R. Kalantary and B. Kakavandi, Chem. Eng. J., 2021, 414, 128618 CrossRef CAS.
- I. Gozlan, A. Rotstein and D. Avisar, Chemosphere, 2013, 91, 985–992 CrossRef CAS PubMed.
- K. D. Radosavljević, A. V. Golubović, M. M. Radišić, A. R. Mladenović, D. Ž. Mijin and S. D. Petrović, Chem. Ind. Chem. Eng. Q., 2017, 23, 187–195 CrossRef.
- M. Fazilati, Desalin. Water Treat., 2019, 169, 222–231 CrossRef CAS.
- J. H. O. S. Pereira, A. C. Reis, O. C. Nunes, M. T. Borges, V. J. P. Vilar and R. A. R. Boaventura, Environ. Sci. Pollut. Res., 2014, 21, 1292–1303 CrossRef CAS PubMed.
- B. Ambrosetti, L. Campanella and R. Palmisano, J. Environ. Sci. Eng. A, 2015, 4, 273–281 Search PubMed.
- E. S. Elmolla and M. Chaudhuri, Desalination, 2010, 252, 46–52 CrossRef CAS.
- R. Palmisano, L. Campanella and B. Ambrosetti, J. Environ. Anal. Chem., 2015, 2, 1000143 Search PubMed.
- D. Klauson, J. Babkina, K. Stepanova, M. Krichevskaya and S. Preis, Catal. Today, 2010, 151, 39–45 CrossRef CAS.
- F. S. Moosavi and T. Tavakoli, Environ. Sci. Pollut. Res., 2016, 23, 23262–23270 CrossRef CAS PubMed.
- D. Dimitrakopoulou, I. Rethemiotaki, Z. Frontistis, N. P. Xekoukoulotakis, D. Venieri and D. Mantzavinos, Environ. Manage., 2012, 98, 168–174 CAS.
- N. Ellepola and G. Rubasinghege, Environments, 2022, 9, 77 CrossRef PubMed.
- A. F. Alkaim and A. M. Aljobree, Int. J. Adv. Sci. Technol., 2020, 29, 5480–5487 Search PubMed.
- F. Moosavi, C. Cheng, T. T. Gheinani, M. Traore, A. Kanaev and M. Nikravech, Chem. Eng. Trans., 2019, 73, 175–180 Search PubMed.
- E. S. Elmolla and M. Chaudhuri, J. Hazard. Mater., 2010, 173, 445–449 CrossRef CAS PubMed.
- S. Pourmoslemi, A. Mohammadi, F. Kobarfard and N. Assi, Desalin. Water Treat., 2016, 57, 14774–14784 CrossRef CAS.
- K. M. M. Al-zobai, A. A. Hassan and N. O. Kariem, Solid State Technol., 2020, 63, 1–9 Search PubMed.
- B. A. Utami, H. Sutanto, I. Alkian, F. Sa'Adah and E. Hidayanto, Cogent Eng., 2022, 9, 2119534 CrossRef.
- T. T. Nguyen, S. N. Nam, J. Son and J. Oh, Sci. Rep., 2019, 9, 9349 CrossRef PubMed.
- D. Balarak and F. K. Mostafapour, Indones. J. Chem., 2019, 19, 211–218 CrossRef CAS.
- R. Mohammadi, B. Massoumi and M. Rabani, Int. J. Photoenergy, 2012, 514856, DOI:10.1155/2012/514856.
- R. Mohammadi, B. Massoumi and H. Eskandarloo, Desalin. Water Treat., 2015, 53, 1995–2004 CrossRef CAS.
- E. T. Wahyuni, P. Y. Yulikayani and N. H. Aprilita, J. Mater. Environ. Sci., 2020, 11, 670–683 CAS.
- N. Olama, M. Dehghani and M. Malakootian, Appl. Water Sci., 2018, 8, 97 CrossRef.
- Lalliansanga, D. Tiwari, S.-M. Lee and D.-J. Kim, Environ. Res., 2022, 210, 112914 CrossRef CAS PubMed.
- Y. T. Gaim, S. M. Yimanuh and Z. G. Kidanu, J. Compos. Sci., 2022, 6, 317 CrossRef CAS.
- M. F. R. Samsudin, F. Maeght, N. Kamarudin and S. Sufian, Malays. J. Microsc., 2020, 16, 37–43 Search PubMed.
- T. C. M. V. Do, D. Q. Nguyen, K. T. Nguyen and P. H. Le, Materials, 2019, 12, 2434 CrossRef CAS PubMed.
- M. R. Rezaei, M. H. Sayadi and N. Ravankhah, Journal of Natural Environment, 2021, 74, 331–344 Search PubMed.
- L. Bergamonti, C. Bergonzi, C. Graiff, P. P. Lottici, R. Bettini and L. Elviri, Water Res., 2019, 163, 114841 CrossRef CAS PubMed.
- S. Moles, J. Berges, M. P. Ormad, M. J. Nieto-Monge, J. Gómez and R. Mosteo, Environ. Sci. Pollut. Res., 2021, 28, 24167–24179 CrossRef CAS PubMed.
- D. Kanakaraju, J. Kockler, C. A. Motti, B. D. Glass and M. Oelgemöller, Appl. Catal., B, 2015, 166–167, 45–55 CrossRef CAS.
- L. M. Pastrana-Martínez, S. Morales-Torres, S. A. C. Carabineiro, J. G. Buijnsters, J. L. Figueiredo, A. M. T. Silva and J. L. Faria, Appl. Surf. Sci., 2018, 458, 839–848 CrossRef.
- Q. Li, H. Kong, R. Jia, J. Shao and Y. He, RSC Adv., 2019, 9, 12538 RSC.
- F. M. dela Rosa, J. Papac, S. Garcia-Ballesteros, M. Kovačić, Z. Katančić, H. Kušić and A. L. Božić, Adv. Sustainable Syst., 2021, 5, 2100119 CrossRef CAS.
- M. G. Alalm, A. Tawfik and A. Ookawara, J. Environ. Chem. Eng., 2016, 4, 1929–1937 CrossRef.
- Q. Li, R. Jia, J. Shao and Y. He, J. Cleaner Prod., 2019, 209, 755–761 CrossRef CAS.
- F. A. Sulaiman and A. I. Alwared, J. Ecol. Eng., 2022, 23, 293–304 CrossRef.
- A. Aissani, M. Kameche and K. Benabbou, Inorg. Nano-Met. Chem., 2022, 52, 1197–1207 CrossRef CAS.
- N. Torkian, A. Bahrami, A. Hosseini-Abari, M. M. Momeni, M. Abdolkarimi Mahabadi, A. Bayat, P. Hajipour, H. A. Rourani, M. S. Abbasi, S. Torkian, Y. Wen, M. Y. Mehr and A. Hojjati-Najafabadi, Environ. Res., 2022, 207, 112157 CrossRef CAS PubMed.
- Y. Chen, X. Zhang, L. Wang, X. Cheng and Q. Shang, Chem. Eng. J., 2020, 402, 126260 CrossRef CAS.
- S. Rani, A. Garg and N. Singh, Toxicol. Environ. Chem., 2021, 103, 137–153 CrossRef CAS.
- T. D. N. Thi, L. H. Nguyen, X. H. Nguyen, H. V. Phung, T. H. T. Vinh, P. V. Viet, N. V. Thai, H. N. Le, D. T. Pham, H. T. Van, L. H. T. Thi, T. D. P. Thi, T. L. Minh, H. H. P. Quang, H. P. N. Vu, T. T. Duc and H. M. Nguyen, Mater. Sci. Semicond. Process., 2022, 142, 106456 CrossRef.
- A. Fawzy, H. Mahanna and M. Mossad, Environ. Sci. Pollut. Res., 2022, 29, 68532–68546 CrossRef CAS PubMed.
- H. Liu, L. Wang, S. Wei, Y. Wu, Y. Zheng, F. Yuan and J. Hou, Opt. Mater., 2022, 123, 111835 CrossRef CAS.
- D. Huang, X. Sun, Y. Liu, H. Ji, W. Liu, C.-C. Wang, W. Ma and Z. Cai, Chin. Chem. Lett., 2021, 32, 2787–2791 CrossRef CAS.
- I. F. Silva, I. F. Teixeira, R. D. F. Rios, G. M. do Nascimento, I. Binatti, H. F. V. Victória, K. Krambrock, L. A. Cury, A. P. C. Teixeira and H. O. Stumpf, J. Hazard. Mater., 2021, 401, 123713 CrossRef CAS PubMed.
- N. Q. Thang, A. Sabbah, L.-C. Chen, K.-H. Chen, C. M. Thi and P. V. Viet, Chemosphere, 2021, 282, 130971 CrossRef CAS PubMed.
- S. Le, C. Zhu, Y. Cao, P. Wang, Q. Liu, H. Zhou, C. Chen, S. Wang and X. Duan, Appl. Catal., B, 2022, 303, 120903 CrossRef CAS.
- M. Dou, J. Wang, B. Gao, Z. Ma and X. Huang, Chem. Eng. J., 2020, 394, 124899 CrossRef CAS.
- E. G. Shankar, S. Billa, A. B. V. Kiran Kumar and J. S. Yu, Ceram. Int., 2021, 47, 30572–30583 CrossRef CAS.
- V. V. Pham, T. K. Truong, L. V. Hai, H. P. P. La, H. T. Nguyen, V. Q. Lam, H. D. Tong, Q. Nguyen, A. Sabbah, K.-H. Chen, S.-J. You and T. M. Cao, ACS Appl. Nano Mater., 2022, 5, 4506–4514 CrossRef CAS.
- M. Dou, J. Wang, B. Gao, C. Xu and F. Yang, Chem. Eng. J., 2020, 383, 123134 CrossRef CAS.
- Q. Chen, L. Chen, J. Qi, Y. Tong, Y. Lv, C. Xu, J. Ni and W. Liu, Chin. Chem. Lett., 2019, 30, 1214–1218 CrossRef CAS.
- R. Changotra, Q. He and A. Dhir, Chem. Eng. J., 2022, 442, 136201 CrossRef CAS.
- D. Balarak, N. Mengelizadeh, P. Rajiv and K. Chandrika, Environ. Sci. Pollut. Res., 2021, 28, 49743–49754 CrossRef CAS PubMed.
- J. Song, Z. Xu, W. Liu and C.-T. Chang, Mater. Sci. Semicond. Process., 2016, 52, 32–37 CrossRef CAS.
- C. Yang, X. You, J. Cheng, H. Zheng and Y. Chen, Appl. Catal., B, 2017, 200, 673–680 CrossRef CAS.
- M. F. R. Samsudin and S. Sufian, J. Mol. Liq., 2020, 314, 113530 CrossRef CAS.
- D. V. Thuan, T. L. Nguyen, H. H. P. Thi, N. T. Thanh, S. Ghotekar, A. K. Sharma, M. T. Binh, T. T. Nga, T.-D. Pham and D. P. Cam, Opt. Mater., 2022, 123, 111885 CrossRef.
- F. Beshkar, A. Al-Nayili, O. Amiri, M. Salavati-Niasari and M. Mousavi-Kamazani, J. Alloys Compd., 2022, 892, 162176 CrossRef CAS.
- M. R. S. Nivetha, J. V. Kumar, J. S. Ajarem, A. A. Allam, V. Manikandan, R. Arulmozhi and N. Abirami, Environ. Res., 2022, 209, 112809 CrossRef CAS PubMed.
- E. M. El-Fawal, S. A. Younis and T. Zaki, J. Photochem. Photobiol., A, 2020, 401, 112746 CrossRef CAS.
- Y. Belaissa, D. Nibou, A. A. Assadi, B. Bellal and M. Trari, J. Taiwan Inst. Chem. Eng., 2016, 68, 254–265 CrossRef CAS.
- B. Gao, J. Wang, M. Dou, C. Xu and X. Huang, Environ. Sci. Pollut. Res., 2020, 27, 7025–7039 CrossRef CAS PubMed.
- M. H. T. Tung, T. T. T. Phuong, D. M. N. Tram, D. M. The, N. V. N. Mai, T. T. T. Hien, L. T. C. Nhung, N. T. T. Binh, C. V. Hoang, D. N. Nhiem, T.-D. Pham and N. T. D. Cam, Diamond Relat. Mater., 2022, 121, 108788 CrossRef CAS.
- Q. Li, H. Kong, P. Li, J. Shao and Y. He, J. Hazard. Mater., 2019, 373, 437–446 CrossRef CAS PubMed.
- K. H. Leong, H. Y. Chu, S. Ibrahi and P. Saravanan, Beilstein J. Nanotechnol., 2015, 6, 428–437 CrossRef PubMed.
- P. Hajipour, A. Eslami, A. Bahrami, A. Hosseini-Abari, F. Y. Saber, R. Mohammadi and M. Y. Mehr, Ceram. Int., 2021, 47, 33875–33885 CrossRef CAS.
- M. Moradi, F. Hasanvandian, A. A. Isari, F. Hayati, B. Kakavandi and S. R. Setayesh, Appl. Catal., B, 2021, 285, 119838 CrossRef CAS.
- G. Prasannamedha and P. S. Kumar, J. Cleaner Prod., 2020, 250, 119553 CrossRef CAS.
- J. Musial, D. T. Mlynarczyk and B. J. Stanisz, Sci. Total Environ., 2023, 856, 159122 CrossRef CAS PubMed.
- C. H. Wu, C. Y. Kuo, C. D. Dong, C. W. Chen and Y. L. Lin, Water Sci. Technol., 2019, 79, 349–355 CrossRef CAS PubMed.
- L. Prieto-Rodriguez, S. Miralles-Cuevas, I. Oller, P. Fernández-Ibánez, A. Agüera, J. Blanco and S. Malato, Appl. Catal., B, 2012, 128, 119–125 CrossRef CAS.
- A. Hu, X. Zhang, D. Luong, K. D. Oakes, M. R. Servos, R. Liang, S. Kurdi, P. Peng and Y. Zhou, Waste Biomass Valorization, 2012, 3, 443–449 CrossRef CAS.
- M. R. Eskandarian, H. Choi, M. Fazli and M. H. Rasoulifard, Chem. Eng. J., 2016, 300, 414–422 CrossRef CAS.
- M. N. Abellán, B. Bayarri, J. Giménez and J. Costa, Appl. Catal., B, 2007, 74, 233–241 CrossRef.
- D. Balarak and H. Azarpira, Int. J. ChemTech Res., 2016, 9, 731–738 CAS.
- J. Carbajo, M. Jiménez, S. Miralles, S. Malato, M. Faraldos and A. Bahamonde, Chem. Eng. J., 2016, 291, 64–73 CrossRef CAS.
- A. Kutuzova, T. Dontsova and W. Kwapinski, Catalysts, 2021, 11, 728 CrossRef CAS.
- O. Porcar-Santos, A. Cruz-Alcalde, N. López-Vinent, D. Zanganas and C. Sans, Sci. Total Environ., 2020, 736, 139605 CrossRef CAS PubMed.
- J. R. Kim and E. Kan, J. Environ. Manage., 2016, 180, 94–101 CrossRef CAS PubMed.
- N. Miranda-Garcia, S. Suarez, B. Sanchez, J. M. Coronado, S. Alato and M. I. Maldonsado, Appl. Catal., B, 2011, 103, 294–301 CrossRef CAS.
- S. Ramasundaram, M. G. Seid, J. W. Choe, E.-J. Kim, Y. C. Chung, K. Cho, C. Lee and S. W. Hong, Chem. Eng. J., 2016, 306, 344–351 CrossRef CAS.
- M. J. Arlos, M. M. Hatat-Fraile, R. Liang, L. M. Bragg, N. Y. Zhou, S. A. Andrews and M. R. Servos, Water Res., 2016, 101, 351–361 CrossRef CAS PubMed.
- A. Mirzaei, L. Yerushalmi, Z. Chen, F. Haghighat and J. Guo, Water Res., 2018, 132, 241–251 CrossRef CAS PubMed.
- T. Makropoulou, I. Kortidis, K. Davididou, D. E. Motaung and E. Chatzisymeon, J. Water Process. Eng., 2020, 36, 101299 CrossRef.
- F. Beheshti, R. M. A. Tehrani and A. Khadir, Int. J. Environ. Sci. Technol., 2019, 16, 7987–7996 CrossRef CAS.
- A. Tiwari, A. Shukla, Lalliansanga, D. Tiwari and S. M. Lee, J. Environ. Manage., 2018, 220, 96–108 CrossRef CAS PubMed.
- E. Borowska, J. F. Gomes, R. C. Martins, R. M. Quinta-Ferreira, H. Horn and M. Gmurek, Catalysts, 2019, 9, 500 CrossRef CAS.
- L.-F. Chiang and R. A. Doong, Sep. Purif. Technol., 2015, 156, 1003–1010 CrossRef CAS.
- R. Zanella, E. Avella, R. M. Ramírez-Zamora, F. Castillón-Barraza and J. C. D. Álvarez, Environ. Technol., 2018, 39, 2353–2364 CrossRef CAS PubMed.
- A. Tsiampalis, Z. Frontistis, V. Binas, G. Kiriakidis and D. Mantzavinos, Catalysts, 2019, 9, 612 CrossRef CAS.
- M. Jahdi, S. B. Mishra, E. N. Nxumalo, S. D. Mhlanga and A. K. Mishra, Appl. Catal., B, 2020, 267, 118716 CrossRef CAS.
- M. Jahdi, S. B. Mishra, E. N. Nxumalo, S. D. Mhlanga and A. K. Mishra, RSC Adv., 2020, 10, 27662 RSC.
- L. K. B. Paragas, M. D. G. de Luna and R.-A. Doong, Chemosphere, 2018, 210, 1099–1107 CrossRef CAS PubMed.
- A. A. Isari, F. Hayati, B. Kakavandi, M. Rostami, M. Motevassel and E. Dehghanifard, Chem. Eng. J., 2020, 392, 123685 CrossRef CAS.
- T.-B. Nguyena, C. P. Huang, R. Doong, C.-W. Chen and C.-D. Dong, Chem. Eng. J., 2020, 384, 123383 CrossRef.
- A. Mirzaei, M. Eddah, S. Roualdès, D. Ma and M. Chaker, Chem. Eng. J., 2021, 422, 130507 CrossRef CAS.
- I. Zammit, V. Vaiano, A. R. Ribeiro, A. M. T. Silva, C. M. Manaia and L. Rizzo, Catalysts, 2019, 9, 222 CrossRef.
- X. Xie, S. Li, H. Zhang, Z. Wang and H. Huang, Sci. Total Environ., 2019, 659, 529–539 CrossRef CAS PubMed.
- L. Fernández, M. Gamallo, M. A. González-Gómez, C. Vázquez-Vázquez, J. Rivas, M. Pintado and M. T. Moreira, J. Environ. Manage., 2019, 237, 595–608 CrossRef PubMed.
- V. Polliotto, F. R. Pomilla, V. Maurino, G. Marcì, A. B. Prevot, R. Nisticò, G. Magnacca, M. C. Paganini, L. Ponce Robles, L. Perez and S. Malato, Catal. Today, 2019, 328, 164–171 CrossRef CAS.
- N. T. T. Nguyen, A. Q. K. Nguyen, M. S. Kim, C. Lee, S. Kim and J. Kim, Sep. Purif. Technol., 2021, 267, 118610 CrossRef CAS.
- N. Wang, X. Li, Y. Yang, Z. Zhou, Y. Shang and X. Zhuang, J. Water Process. Eng., 2020, 36, 101335 CrossRef.
- N. Rioja, P. Benguria, F. J. Peñas and S. Zorita, Environ. Sci. Pollut. Res., 2014, 21, 11168–11177 CrossRef CAS PubMed.
- M. O. Alfred, M. O. Omorogie, O. Bodede, R. Moodley, A. Ogunlaja, O. G. Adeyemi, C. Gunter, A. Trubert, H. Eckert, L. D. A. Silva, A. S. S. de Camargo, A. de J. Motheo, S. M. Clarke and E. I. Unuabonah, Chem. Eng. J., 2020, 398, 125544 CrossRef.
- E. H. Mourid, E. M. El Mouchtari, L. El Mersly, L. Benaziz, S. Rafqah and M. Lakraimi, J. Photochem. Photobiol., A, 2020, 396, 112530 CrossRef CAS.
- M. Długosz, P. Zmudzki, A. Kwiecień, K. Szczubiałka and J. Krzek, J. Hazard. Mater., 2015, 298, 146–153 CrossRef PubMed.
- R. Noroozi, M. Gholami, M. Farzadkia and R. R. Kalantary, Environ. Sci. Pollut. Res., 2022, 29, 56403–56418 CrossRef CAS PubMed.
- N. Malesic-Eleftheriadou, E. Evgenidou, G. Z. Kyzas, D. N. Bikiaris and D. A. Lambropoulou, Chemosphere, 2019, 234, 746–755 CrossRef CAS PubMed.
- J. Li, F. Wang, L. Meng, M. Han, Y. Guo and C. Sun, J. Colloid Interface Sci., 2017, 485, 116–122 CrossRef CAS PubMed.
- K. Kouvelis, A. A. Kampioti, A. Petala and Z. Frontistis, Catalysts, 2022, 12, 882 CrossRef CAS.
- O. Mertah, A. Gomez-Aviles, A. Kherbeche, C. Belver and J. Bedia, J. Environ. Chem. Eng., 2022, 10, 108438 CrossRef CAS.
- W. Zhu, Z. Li, C. He, S. Faqian and Y. Zhou, J. Alloys Compd., 2018, 754, 153–162 CrossRef CAS.
- D. Awfa, M. Ateia, M. Fujii and C. Yoshimura, J. Water Process. Eng., 2019, 31, 100836 CrossRef.
- J. Martini, C. A. Orge, J. L. Faria, M. F. R. Pereira and O. S. G. P. Soares, Appl. Sci., 2019, 9, 2652 CrossRef CAS.
- M. Chen, C. Guo, S. Hou, L. Wu, J. Lv, C. Hu, Y. Zhang and J. Xu, J. Hazard. Mater., 2019, 366, 219–228 CrossRef CAS PubMed.
- A. Mirzaei, L. Yerushalmi, Z. Chen and F. Haghighat, J. Hazard. Mater., 2018, 359, 516–526 CrossRef CAS PubMed.
- G. K. Teye, J. Huang, Y. Li, K. Li, L. Chen and W. K. Darkwah, Nanomaterials, 2021, 11, 2609 CrossRef CAS PubMed.
- X. Ao, Z. Li and H. Zhang, J. Cleaner Prod., 2022, 356, 131822 CrossRef CAS.
- Y. Song, J. Qi, J. Tian, S. Gao and F. Cui, Chem. Eng. J., 2018, 341, 547–555 CrossRef CAS.
- W. Zhu, F. Sun, R. Goei and Y. Zhou, Appl. Catal., B, 2017, 207, 93–102 CrossRef CAS.
- L. Zhou, G. Zou and G. L. Deng, Catalysts, 2018, 8, 272 CrossRef.
- L. Lin, H. Wang and P. Xu, Chem. Eng. J., 2017, 310, 389–398 CrossRef CAS.
- S.-H. Liu, Y.-S. Wei and J. S. Lu, Chemosphere, 2016, 154, 118–123 CrossRef CAS PubMed.
- M. Nawaz, A. A. Khan, A. Hussain, J. Jang, H.-Y. Jung and D. S. Lee, Chemosphere, 2020, 261, 127702 CrossRef CAS PubMed.
- L. Zhou, O. G. Alvarez, C. S. Mazon, L. Chen, H. Deng and M. Sui, Catal. Sci. Technol., 2016, 6, 5972 RSC.
- W. Zhu, F. Sun, R. Goei and Y. Zhou, Catal. Sci. Technol., 2017, 7, 2591–2600 RSC.
- M. H. de M. Rodrigues, P. A. R. de Sousa, K. C. M. Borges, L. de M. Coelho, R. F. Gonçalves, M. D. Teodoro, F. da V. Motta, R. M. do Nascimento and M. G. Júnior, J. Alloys Compd., 2019, 808, 151711 CrossRef.
- A. Kumar, G. Sharma, M. Naushad, Z. A. Alothman and P. Dhiman, Earth Syst. Environ., 2022, 6, 141–156 CrossRef.
- J. Qiu, C. Yue, W. Zheng, F. Liu and J. Zhu, Chin. Chem. Lett., 2021, 32, 3431–3434 CrossRef CAS.
- A. Kumar, A. Kumar, G. Sharma, A. H. Al-Muhtaseb, M. Naushad, A. A. Ghfar and F. J. Stadler, Chem. Eng. J., 2018, 334, 462–478 CrossRef CAS.
- Z. Huang, X. Dai, Z. Huang, T. Wang, L. Cui, J. Ye and P. Wu, Chemosphere, 2019, 221, 824–833 CrossRef CAS PubMed.
- L. Zhang, G. Meng, B. Liu and X. Ge, J. Mol. Liq., 2022, 360, 119427 CrossRef CAS.
- D. Roy, S. Niyogi and S. De, Process Saf. Environ. Prot., 2022, 161, 723–738 CrossRef CAS.
- T. Ke, S. Shen, K. Yang and D. Lin, Appl. Surf. Sci., 2022, 580, 152302 CrossRef CAS.
- M. Ren, Y. Ao, P. Wang and C. Wang, Chem. Eng. J., 2019, 378, 122122 CrossRef CAS.
- C. Liu, J. Xu, J. Niu, M. Chen and Y. Zhou, Sep. Purif. Technol., 2020, 241, 116622 CrossRef CAS.
- C. Liu, J. Xu, X. Du, Q. Li, Y. Fu and M. Chen, Opt. Mater., 2021, 112, 110742 CrossRef CAS.
- J. Xu, J. Chen, Y. Ao and P. Wang, Chin. Chem. Lett., 2021, 32, 3226–3230 CrossRef CAS.
- L. Zhou, W. Zhang, L. Chen and H. Deng, J. Colloid Interface Sci., 2017, 487, 410–417 CrossRef CAS PubMed.
- A. Mirzaei, Z. Chen, F. Haghighat and L. Yerushalm, Chemosphere, 2018, 205, 463–474 CrossRef CAS PubMed.
- G. Liu, H. Wang, D. Chen, C. Dai, Z. Zhang and Y. Feng, Sep. Purif. Technol., 2020, 237, 116329 CrossRef CAS.
- Y. Zhang, Y. Li and Y. Yuan, J. Colloid Interface Sci., 2023, 645, 860–869 CrossRef CAS PubMed.
- E. Brillas, Chemosphere, 2022, 286, 131849 CrossRef CAS PubMed.
- J. Jan-Roblero and J. A. Cruz-Maya, Molecules, 2023, 28, 2097 CrossRef CAS PubMed.
- N. Jallouli, L. M. Pastrana-Martinez, A. R. Ribeiro, N. F. F. Moreira, J. L. Faria, O. Hentati, A. M. T. Silva and M. Ksibi, Chem. Eng. J., 2018, 334, 976–984 CrossRef CAS.
- M. Jiménez-Salcedo, M. Monge and M. T. Tena, Chemosphere, 2019, 215, 605–618 CrossRef PubMed.
- M. O. Miranda, W. E. C. Cavalcanti, F. F. Barbosa, J. A. de Sousa, F. I. da Silva, S. B. C. Pergher and T. P. Braga, RSC Adv., 2021, 11, 27720 RSC.
- I. Georgaki, E. Vasilaki and N. Katsarakis, Am. J. Anal. Chem., 2014, 5, 518–534 CrossRef.
- M. Tanveer, G. T. Guyer and G. Abbas, Water Environ. Res., 2019, 91, 822–829 CrossRef CAS PubMed.
- N. Rastkari, A. Eslami, S. Nasseri, E. Piroti and A. Asadi, Pol. J. Environ. Stud., 2017, 26, 785–794 CrossRef CAS PubMed.
- A. S. Sá, R. P. Feitosa, L. Honório, R. Peña-Garcia, L. C. Almeida, J. S. Dias, L. P. Brazuna, T. G. Tabuti, E. R. Triboni, J. A. Osajima and E. C. da Silva-Filho, Materials, 2021, 14, 5891 CrossRef PubMed.
- M. Ulfa, D. Prasetyoko, H. Bahruji and R. E. Nugraha, Materials, 2021, 14, 6779 CrossRef CAS PubMed.
- S. Khalaf, J. H. Shoqeir, F. Lelario, S. A. Bufo, R. Karamanand and L. Scrano, Catalysts, 2020, 10, 560 CrossRef CAS.
- F. S. Braz, M. R. A. Silva, F. S. Silva, S. J. Andrade, A. L. Fonseca and M. M. Kondo, J. Environ. Prot., 2014, 5, 620–626 CrossRef.
- J. Bohdziewicz, E. Kudlek and M. Dudziak, Desalin. Water Treat., 2016, 57, 1552–1563 CrossRef CAS.
- J. Choina, A. Bagabas, C. Fisher, G. U. Flechsig, H. Kosslick, A. Alshammari and A. Schulz, Catal. Today, 2015, 241, 47–54 CrossRef CAS.
- N. Rosman, W. N. W. Salleh, J. Jaafar, Z. Harun, F. Aziz and A. F. Ismail, Catalysts, 2022, 12, 209 CrossRef CAS.
- A. Eslami, M. M. Amini, A. R. Yazdanbakhsh, A. Mohseni-Bandpei, A. A. Safari and A. Asadi, J. Chem. Technol. Biotechnol., 2016, 91, 2693–2704 CrossRef CAS.
- T. M. Khedr, S. M. El-Sheikh, A. Hakki, A. A. Ismail, W. A. Badawy and D. W. Bahnemann, J. Photochem. Photobiol., A, 2017, 346, 530–540 CrossRef CAS.
- V. Bhatia and A. Dhir, J. Environ. Chem. Eng., 2016, 4, 1267–1273 CrossRef CAS.
- A. Jraba, Z. Anna and E. Elaloui, J. Mater. Sci.: Mater. Electron., 2020, 31, 1072–1083 CrossRef CAS.
- R. Pelosato, V. Carrara and I. N. Sora, Chem. Eng. Trans., 2019, 73, 181–186 Search PubMed.
- Q. Sun, Y.-P. Peng, H. Chen, K.-L. Chang, Y.-N. Qiu and S.-W. Lai, J. Hazard. Mater., 2016, 319, 121–129 CrossRef CAS PubMed.
- S. M. El-Sheikh, T. M. Khedr, A. Hakki, A. A. Ismail, W. A. Badawy and D. W. Bahnemann, Sep. Purif. Technol., 2017, 173, 258–268 CrossRef CAS.
- C. Yuan, C.-H. Hung, H.-W. Li and W.-H. Chang, Chemosphere, 2016, 155, 471–478 CrossRef CAS PubMed.
- L. Lin, W. Jiang, M. Bechelany, M. Nasr, J. Jarvis, T. Schaub, R. R. Sapkota, P. Miele, H. Wang and P. Xu, Chemosphere, 2019, 220, 921–929 CrossRef CAS PubMed.
- Y. Gu, J. Yperman, R. Carleer, J. D'Haen, J. Maggen, S. Vanderheyden, K. Vanreppelen and R. M. Garcia, Chemosphere, 2019, 217, 724–731 CrossRef CAS PubMed.
- M. Mohadesi, A. Gouran and K. Seifi, Environ. Sci. Pollut. Res., 2022, 29, 34338–34348 CrossRef CAS PubMed.
- N. Liu, J. Wang, J. Wu, Z. Li, W. Huang, Y. Zheng, J. Lei, X. Zhang and L. Tang, Mater. Res. Bull., 2020, 132, 111000 CrossRef CAS.
- T. R. Bastami, A. Ahmadpour and F. A. Hekmatikar, J. Ind. Eng. Chem., 2017, 51, 244–254 CrossRef.
- C. S. L. Fung, M. Khan, A. Kumar and I. M. C. Lo, Sep. Purif. Technol., 2019, 216, 102–114 CrossRef CAS.
- M. P. Villavicencio, A. E. Morales, M. de L. R. Peralta, M. Sánchez-Cantú, L. R. Blanco, E. C. Anota, J. H. C. García and F. Tzompantzi, Catal. Lett., 2020, 150, 2385–2399 CrossRef.
- J. Liao, Y. Xu, Y. Zhao, C.-C. Wang and C. Ge, ACS Appl. Nano Mater., 2021, 4, 1898–1905 CrossRef CAS.
- G. G. Lenzi, M. F. Lopes, D. I. Andrade, J. S. Napoli, A. Parolin, Y. B. Fávaro, M. E. K. Fuziki, L. N. B. de Almeida, T. G. Josué, D. T. Dias and A. M. Tusset, Water Sci. Technol., 2021, 84, 2158–2179 CrossRef CAS PubMed.
- M. Khan, C. S. L. Fung, A. Kumar and I. M. C. Lo, J. Hazard. Mater., 2019, 365, 733–743 CrossRef CAS PubMed.
- K. Kang, M. Jang, M. Cui, P. Qiu, B. Park, S. A. Snyder and J. Khim, J. Mol. Catal. A: Chem., 2014, 390, 178–186 CrossRef CAS.
- C. B. Anucha, I. Altin, E. Bacaksiz, I. Degirmencioglu, T. Kucukomeroglu, S. Yilmaz and V. N. Stathopoulos, Separations, 2021, 8, 24 CrossRef.
- S. Sambaza, A. Maity and K. Pillay, J. Saudi Chem. Soc., 2022, 26, 101563 CrossRef CAS.
- A. Mohamed, A. Salama, W. S. Nasser and A. Uheida, Environ. Sci. Eur., 2018, 30, 47 CrossRef PubMed.
- E. Yilmaz, S. Salem, G. Sarp, S. Aydin, K. Sahin, I. R. Korkmaz and D. Yuvali, Talanta, 2020, 213, 120813 CrossRef CAS PubMed.
- B.-T. Zhang, Q. Wang, Y. Zhang, Y. Teng and M. Fan, Sep. Purif. Technol., 2020, 242, 116820 CrossRef CAS.
- N. Rosman, W. N. W. Salleh, N. A. M. Razali, S. Z. Ahmad, N. H. Ismail, F. Aziz, Z. Harun, A. F. Ismail and N. Yuso, Mater. Today: Proc., 2021, 42, 69–74 CAS.
- A. Uheida, A. Mohamed, M. Belaqziz and W. S. Nasser, Sep. Purif. Technol., 2019, 212, 110–118 CrossRef CAS.
- D. Hernández-Uresti, A. Vázquez, D. Sanchez-Martinez and S. Obregón, J. Photochem. Photobiol., A, 2016, 324, 47–52 CrossRef.
- X. Wang, J. Luo, Y. Huang, J. Mei and Y. Chen, Environ. Sci.: Water Res. Technol., 2021, 7, 610 RSC.
- A. Raja, P. Rajasekaran, K. Selvakumar, M. Arivanandhan, S. A. Bahadur and M. S. Swaminathan, Appl. Surf. Sci., 2020, 513, 145803 CrossRef CAS.
- A. Kumar, G. Sharma, M. Naushad, A. H. Al-Muhtaseb, A. Kumar, I. Hira, T. Ahamad, A. A. Ghfar and F. J. Stadler, J. Environ. Manage., 2019, 231, 1164–1175 CrossRef CAS PubMed.
- R. Akbarzadeh, C. S. L. Fung, R. A. Rather and I. M. C. Lo, Chem. Eng. J., 2018, 341, 248–261 CrossRef CAS.
- Z. Sun, X. Zhang, X. Dong, X. Liu, Y. Tan, F. Yuan, S. Zheng and C. Li, J. Materiomics, 2020, 6, 582–592 CrossRef.
- Z.-D. Lei, J.-J. Wang, L. Wang, X.-Y. Yang, G. Xu and L. Tang, J. Hazard. Mater., 2016, 312, 298–306 CrossRef CAS PubMed.
- W. Cao, Y. Yuan, C. Yang, S. Wu and J. Cheng, Chem. Eng. J., 2020, 391, 123608 CrossRef CAS.
- Y. Zhang, J. Li and H. Liu, J. Nanomater., 2020, 6094984, DOI:10.1155/2020/6094984.
- L. Wang, Q. Sun, Y. Dou, Z. Zhang, T. Yan and Y. Li, J. Hazard. Mater., 2021, 413, 125288 CrossRef CAS PubMed.
- X. Chen, X. Li, J. Yang, Q. Sun, Y. Yang and X. Wu, Int. J. Hydrogen Energy, 2018, 43, 13284–13293 CrossRef CAS.
- J. Wang, L. Tang, G. Zeng, Y. Deng, Y. Liu, L. Wang, Y. Zhou, Z. Guo, J. Wang and C. Zhang, Appl. Catal., B, 2017, 209, 285–294 CrossRef CAS.
- A. Kumar, M. Khan, X. Zeng and I. M. C. Lo, Chem. Eng. J., 2018, 353, 645–656 CrossRef CAS.
- N. Liu, F. Fei, W. Dai, J. Lei, F. Bi, B. Wang, G. Quan, X. Zhang and L. Tang, J. Colloid Interface Sci., 2022, 625, 965–977 CrossRef CAS PubMed.
- S. Mao, C. Liu, M. Xia, F. Wang and X. Ju, Catal. Sci. Technol., 2021, 11, 3466–3480 RSC.
- S.-H. Liu and W.-T. Tang, Sci. Total Environ., 2020, 731, 139172 CrossRef CAS PubMed.
- Y. Cong, Y. Li, X. Wang, X. Wei, L. Che and S.-W. Lv, Sep. Purif. Technol., 2022, 297, 121531 CrossRef CAS.
- P. J. Mafa, U. S. Swana, D. Liu, J. Gui, B. B. Mamba and A. T. Kuvarega, Colloids Surf., A, 2021, 612, 126004 CrossRef CAS.
- J. Zhang, L. Xin, L. Qianwen, L. Yuqian, G. Liu and X. Shi, Ceram. Int., 2020, 46, 106–113 CrossRef CAS.
- A. Kumar, M. Khan, L. Fang and I. M. C. Lo, J. Hazard. Mater., 2019, 370, 108–116 CrossRef CAS PubMed.
- M. Liang, Z. Zhang, R. Long, Y. Wang, Y. Yu and Y. Pei, Environ. Pollut., 2020, 259, 113770 CrossRef CAS PubMed.
- M. E. Malefane, U. Feleni, P. J. Mafa and A. T. Kuvarega, Appl. Surf. Sci., 2020, 514, 145940 CrossRef CAS.
- Q. Wei, S. Xiong, W. Li, C. Jin, Y. Chen, L. Hou, Z. Wu, Z. Pan, Q. He, Y. Wang and D. Tang, J. Alloys Compd., 2021, 885, 160984 CrossRef CAS.
- A. Zhou, L. Liao, X. Wu, K. Yang, C. Li, W. Chen and P. Xie, Sep. Purif. Technol., 2020, 250, 117241 CrossRef CAS.
- N. Rosman, W. N. W. Salleh, M. A. Mohamed, Z. Harun, A. F. Ismail and F. Aziz, Sep. Purif. Technol., 2020, 251, 117391 CrossRef CAS.
- L. Chierentin and H. R. N. Salgado, Crit. Rev. Anal. Chem., 2016, 46, 22–39 CrossRef CAS PubMed.
- B. Holmes, R. N. Brogden and D. M. Richards, Drugs, 1985, 30, 482–513 CrossRef CAS PubMed.
- A. J. Schaeffer, Eur. Urol., 1990, 17, 19–23 CrossRef PubMed.
- H. Yang, L. Mei, P. Wang, J. Genereux, Y. Wang, B. Yi, C. Au, L. Dang and P. Feng, RSC Adv., 2017, 7, 45721 RSC.
- M. M. Haque and M. Muneer, J. Hazard. Mater., 2007, 145, 51–57 CrossRef CAS PubMed.
- Z. Liu, X. Yu, F. Yang, J. Nie, N. Zhao, J. Li and B. Yao, IOP Conf. Ser. Earth Environ. Sci., 2020, 453, 012084 CrossRef.
- M. Chen, Y. Huang and W. Chu, Chin. J. Catal., 2019, 40, 673–680 CrossRef CAS.
- M. Sayed, L. A. Shah, J. A. Khan, N. S. Shah, J. Nisar, H. M. Khan, P. Zhang and A. R. Khan, J. Phys. Chem. A, 2016, 120, 9916–9931 CrossRef CAS PubMed.
- S.-L. Zhou, S. Zhang, F. Liu, J.-J. Liu, J.-J. Xue, D.-J. Yang and C.-T. Chang, J. Photochem. Photobiol., A, 2016, 328, 97–104 CrossRef CAS.
- S. Zhang, S.-L. Zhou, J.-J. Liu, D.-J. Yang, J.-J. Xue and C.-T. Chang, J. Nanosci. Nanotechnol., 2018, 18, 4087–4092 CrossRef CAS PubMed.
- R. Xiao, Y. Zhang, S. Wang, H. Zhu, H. Song, G. Chen, H. Lin, J. Zhang and J. Xiang, Environ. Sci. Pollut. Res., 2021, 28, 69301–69313 CrossRef CAS PubMed.
- S. S. Mohtar, F. Aziz, A. F. Ismail, N. S. Sambudi, H. Abdullah, A. N. Rosli and B. Ohtani, Catalysts, 2021, 11, 1160 CrossRef CAS.
- X. Jin, X. Zhou, P. Sun, S. Lin, W. Cao, Z. Li and W. Liu, Chemosphere, 2019, 237, 124433 CrossRef CAS PubMed.
- R. Kaushik, P. K. Samal and A. Halder, ACS Appl. Nano Mater., 2019, 2, 7898–7909 CrossRef CAS.
- M. Chen and W. Chu, J. Hazard. Mater., 2012, 219, 183–189 CrossRef PubMed.
- M. Manasa, P. R. Chandewar and H. Mahalingam, Catal. Today, 2021, 375, 522–536 CrossRef CAS.
- N. S. Shah, J. A. Khan, M. Sayed, Z. U. H. Khan, A. D. Rizwan, N. Muhammad, G. Boczkaj, B. Murtaza, M. Imran, H. M. Khan and G. Zaman, Chem. Eng. J., 2018, 351, 841–855 CrossRef CAS.
- Y. Hsu, J. Thomas, C. T. Chang and C. M. Ma, J. Nanosci. Nanotechnol., 2021, 21, 3099–3106 CrossRef CAS PubMed.
- B. P. Patil and R. V. Jayaram, Catal. Green Chem. Eng., 2021, 4, 51–63 CrossRef CAS.
- P. Garcia-Muñoz, N. P. Zussblatt, G. Pliego, J. A. Zazo, F. Fresno, B. F. Chmelka and J. A. Casas, J. Environ. Manage., 2019, 238, 243–250 CrossRef PubMed.
- N. T. T. Trang, T. Q. Vinh, H. V. Giang, N. S. Mai, N. T. Dong, P. T. Linh, N. V. Hoang and N. M. Tan, Vietnam J. Sci. Technol., 2020, 58, 13–19 CrossRef.
- X. Wang, Y. Sun, L. Yang, Q. Shang, D. Wang, T. Guo and Y. Guo, Sci. Total Environ., 2019, 656, 1010–1020 CrossRef CAS PubMed.
- J. Gou, Q. Ma, X. Deng, Y. Cui, H. Zhang, X. Cheng, X. Li, M. Xie and Q. Cheng, Chem. Eng. J., 2017, 308, 818–826 CrossRef CAS.
- J. Li, M. Han, Y. Guo, F. Wang and C. Sun, Chem. Eng. J., 2016, 298, 300–308 CrossRef CAS.
- Q. Zhao, C.-C. Wang and P. Wang, Chin. Chem. Lett., 2022, 33, 4828–4833 CrossRef CAS.
- X.-J. Wen, C.-G. Niu, D.-W. Huang, L. Zhang, C. Liang and G.-M. Zeng, J. Catal., 2017, 355, 73–86 CrossRef CAS.
- W. Liu, T. He, Y. Wang, G. Ning, Z. Xu and X. Chen, Sci. Rep., 2020, 10, 11903 CrossRef CAS PubMed.
- L. Bharali, J. Kalita, S. S. Dhar and N. S. Moyon, ChemistrySelect, 2022, 7, e202203487 CrossRef CAS.
- C.-H. Zhang, X.-B. Zhao, Y.-J. Li and D.-W. Sun, Yingyong Huaxue, 2021, 38, 99–106 CAS.
- Z. Jiao, J. Zhang, Z. Liu and Z. Ma, J. Photochem. Photobiol., A, 2019, 371, 67–75 CrossRef CAS.
- W. Y. Fei, W. F. Liang, L. J. Hua, X. Z. Jie, S. Y. Han, Z. Q. Xin, Y. Kun, L. W. Ying, L. Jin and L. G. Guang, Zhongguo Huanjing Kexue, 2018, 38, 1346–1355 Search PubMed.
- W. Zhang, Z. Bian, X. Xin, L. Wang, X. Geng and H. Wang, Chemosphere, 2021, 262, 127955 CrossRef CAS PubMed.
- Q. Chen, Y. Hao, Z. Song, M. Liu, D. Chen, B. Zhu, J. Chen and Z. Chen, Ecotoxicol. Environ. Saf., 2021, 225, 112742 CrossRef CAS PubMed.
- W. Liu, J. Zhou and J. Yao, Ecotoxicol. Environ. Saf., 2020, 190, 110062 CrossRef CAS PubMed.
- S. L. Prabavathi and V. Muthuraj, Colloids Surf., A, 2019, 567, 43–54 CrossRef.
- L. Ma, J. Duan, B. Ji, Y. Liu, C. Li, C. Li, W. Zhao and Z. Yang, J. Alloys Compd., 2021, 869, 158679 CrossRef CAS.
- Y. Zhao, X. Liang, X. Hu and J. Fan, J. Colloid Interface Sci., 2021, 589, 336–346 CrossRef CAS PubMed.
- W. Zhao, J. Duan, B. Ji, L. Ma and Z. Yang, J. Environ. Chem. Eng., 2018, 8, 102206 CrossRef.
- J. Wu, J. Bai, Z. Wang, Z. Liu, Y. Mao, B. Liu and X. Zhu, Environ. Technol., 2022, 43, 95–106 CrossRef CAS PubMed.
- H. Mohan, S. Vadivel, P. M. Sathya, M. Fujii, G. H. Ha, G. Kim and T. Shin, ACS Appl. Energy Mater., 2022, 5, 12851–12859 CrossRef CAS.
- B. Ji, W. Zhao, J. Duan, L. Fu, L. Ma and Z. Yang, RSC Adv., 2020, 10, 4427–4435 RSC.
- Q. Wu, Y. Liu, H. Jing, H. Yu, Y. Lu, M. Huo and H. Huo, Chem. Eng. J., 2020, 390, 124615 CrossRef CAS.
- S. Wen, M. Chen and H. Cao, Int. J. Electrochem. Sci., 2021, 16, 210733, DOI:10.20964/2021.07.63.
- G. Li, S. Huang, N. Zhu, H. Yuan, D. Ge and Y. Wei, Chem. Eng. J., 2021, 421, 127852 CrossRef CAS.
- J. Guo, C.-H. Shen, J. Sun, X.-J. Xu, X.-Y. Li, Z.-H. Fei, Z.-T. Liu and X.-J. Wen, Sep. Purif. Technol., 2021, 259, 118109 CrossRef CAS.
- S. Subudhi, L. Paramanik, S. Sultana, S. Mansingh, P. Mohapatra and K. Parida, J. Colloid Interface Sci., 2020, 568, 89–105 CrossRef CAS PubMed.
- W. Zhang, Y. Meng, Y. Liu, H. Shen, Z. Ni, S. Xia, W. Han, Y. Li and H. Tang, J. Environ. Chem. Eng., 2022, 10, 107812 CrossRef CAS.
- M. Wang, H. Yu, P. Wang, Z. Chi, Z. Zhang, B. Dong, H. Dong, K. Yu and H. Yu, Sep. Purif. Technol., 2021, 274, 118692 CrossRef CAS.
- S. L. Prabavathi, K. Govindan, K. Saravanakumar, A. Jang and V. Muthuraj, J. Ind. Eng. Chem., 2019, 80, 558–567 CrossRef CAS.
- N. Yin, H. Chen, X. Yuan, Y. Zhang, M. Zhang, J. Guo, Y. Zhang, L. Qiao, M. Liu and K. Song, J. Hazard. Mater., 2022, 436, 129317 CrossRef CAS PubMed.
- Z. Zhao, Q. Ling, Z. Li, K. Yan, C. Ding, P. Chen, L. Yang, Z. Sun and M. Zhang, Sep. Purif. Technol., 2023, 308, 122928 CrossRef CAS.
- J. Jiang and Y. Li, Crystals, 2021, 11, 1173 CrossRef CAS.
- J. Zhang, Z. Zhu, J. Jiang and H. Li, Molecules, 2020, 25, 3706 CrossRef CAS PubMed.
- Y. Lv, H. Liu, D. Jin, H. Yang, D. He, Z. Zhang, Y. Zhang, J. Qu and Y.-N. Zhang, Chem. Eng. J., 2022, 429, 132092 CrossRef CAS.
- L. Chang, Y. Pu, P. Jing, J. Ji, X. Wei, B. Cao, Y. Yu, S. Xu and H. Xie, Surf. Interfaces, 2022, 31, 102010 CrossRef CAS.
- A. Kumar, S. K. Sharma, G. Sharma, A. H. Al-Muhtaseb, M. Naushad, A. A. Ghfar and F. J. Stadler, J. Hazard. Mater., 2019, 364, 429–440 CrossRef CAS PubMed.
- J. Li, Z. Xia, D. Ma, G. Liu, N. Song, D. Xiang, Y. Xin, G. Zhang and Q. Chen, J. Colloid Interface Sci., 2021, 586, 243–256 CrossRef CAS PubMed.
- C. Zhang, M. Jia, Z. Xu, W. Xiong, Z. Yang, J. Cao, H. Peng, H. Xu, Y. Xiang and Y. Jing, Chem. Eng. J., 2022, 430, 132652 CrossRef CAS.
- X. Gu, T. Chen, J. Lei, Y. Yang, X. Zheng, S. Zhang, Q. Zhu, X. Fu, S. Meng and S. Chen, Chin. J. Catal., 2022, 43, 2569–2580 CrossRef CAS.
- Y. Liu, R. Chen, F. Wu, W. Gan, L. Chen, M. Zhang and Z. Sun, Sep. Purif. Technol., 2023, 324, 124572 CrossRef.
- J. Zhang, Z. Zhu, J. Jiang and H. Li, Catalysts, 2020, 10, 373 CrossRef CAS.
- Z. Zhu, H. Xia, H. Li and S. Han, Inorganics, 2022, 10, 131 CrossRef CAS.
- C. Li, T. Sun, G. Yi, D. Zhang, Y. Zhang, X. Lin, J. Liu, Z. Shi and Q. Lin, Colloids Surf., A, 2023, 662, 131001 CrossRef CAS.
- C. Guo, S. Gao, J. Lv, S. Hou, Y. Zhang and J. Xu, Appl. Catal., B, 2017, 205, 68–77 CrossRef CAS.
- A. Behera, D. Kandi, S. Sahoo and K. Parida, J. Phys. Chem. C, 2019, 123, 17112–17126 CrossRef CAS.
- P. C. Sharma, A. Jain, S. Jain, R. Pahwa and M. S. Yar, J. Enzyme Inhib. Med. Chem., 2010, 25, 577–589 CrossRef CAS PubMed.
- Y. Zeng, D. Chen, T. Chen, M. Cai, Q. Zhang, Z. Xie, R. Li, Z. Xiao, G. Liu and W. Lv, Chemosphere, 2019, 227, 198–206 CrossRef CAS PubMed.
- W. Li, C. Guo, B. Su and J. Xu, J. Chem. Technol. Biotechnol., 2012, 87, 643–650 CrossRef CAS.
- R. Shetty, G. Kothari, A. Tambe, B. D. Kulkarni and S. P. Kamble, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2016, 55, 16–22 Search PubMed.
- T. A. Gad-Allah, M. E. M. Ali and M. I. Badawy, J. Hazard. Mater., 2011, 186, 751–755 CrossRef CAS PubMed.
- S. Li and J. Hu, Water Res., 2018, 132, 320–330 CrossRef CAS PubMed.
- A. Salma, S. Thoröe-Boveleth, T. C. Schmidt and J. Tuerk, J. Hazard. Mater., 2016, 313, 49–59 CrossRef CAS PubMed.
- A. R. Silva, P. M. Martins, S. Teixeira, S. A. C. Carabineiro, K. Kuehn, G. Cuniberti, M. M. Alves, S. Lanceros-Mendezagh and L. Pereira, RSC Adv., 2016, 6, 95494–95503 RSC.
- Y. Gan, Y. Wei, J. Xiong and G. Cheng, Chem. Eng. J., 2018, 349, 1–16 CrossRef CAS.
- Y. Li, Y. Fu and M. Zhu, Appl. Catal., B, 2020, 260, 118149 CrossRef CAS.
- M. R. Usman, A. Prasasti, S. Fajriyah, A. W. Marita, S. Islamiah, A. N. Firdaus, A. R. Noviyanti and D. R. Eddy, Bull. Chem. React. Eng. Catal., 2021, 16, 752–762 CrossRef CAS.
- M. El-Kemary, H. El-Shamy and I. El-Mehasseb, J. Lumin., 2010, 130, 2327–2331 CrossRef CAS.
- M. Eskandari, N. Goudarzi and S. G. Moussavi, Water Environ. J., 2018, 32, 58–66 CrossRef CAS.
- A. Ulyankina, T. Molodtsova, M. Gorshenkov, I. Leontyev, D. Zhigunov, E. Konstantinova, T. Lastovina, J. Tolasz, J. Henych, N. Licciardello, G. Cuniberti and N. Smirnova, J. Water Process. Eng., 2021, 40, 101809 CrossRef.
- S. Aghdasi and M. Shokri, Iran. J. Catal., 2016, 6, 481–487 CAS.
- F. Nekouei and S. Nekouei, Sci. Total Environ., 2017, 601–602, 508–517 CrossRef CAS PubMed.
- C. Bojer, J. Schöbel, T. Martin, M. Ertl, H. Schmalz and J. Breu, Appl. Catal., B, 2017, 204, 561–565 CrossRef CAS.
- V. Tiron, M. A. Ciolan, G. Bulai, G. Mihalache, F. D. Lipsa and R. Jijie, Nanomaterials, 2022, 12, 2193 CrossRef CAS PubMed.
- N. Finčur, P. Sfîrloagă, P. Putnik, V. Despotović, M. Lazarević, M. Uzelac, B. Abramović, P. Vlazan, C. Ianăs, T. Alapi, M. Náfrádi, I. Maksimović, M. Putnik-Delić and D. Š. Merkulov, Nanomaterials, 2021, 11, 215 CrossRef PubMed.
- M. Mahjoore, A. Aryafar and M. Honarmand, Journal of Mining and Engineering, 2022, 13, 155–164 Search PubMed.
- T. S. Roy, S. Akter, M. R. Fahim, M. A. Gafur and T. Ferdous, Heliyon, 2023, 9, e13130 CrossRef CAS PubMed.
- T. Suwannaruang, J. P. Hildebrand, D. H. Taffa, M. Wark, K. Kamonsuangkasem, P. Chirawatkul and K. Wantala, J. Photochem. Photobiol., A, 2020, 391, 112371 CrossRef CAS.
- C. A. Huerta-Aguilar, Y. S. García Gutiérrez and P. Thangarasu, Chem. Eng. J., 2020, 394, 124286 CrossRef CAS.
- Y. Gan, M. Zhang, J. Xiong, J. Zhu, W. Li, C. Zhang and G. Cheng, J. Taiwan Inst. Chem. Eng., 2019, 96, 229–242 CrossRef CAS.
- J. C. Durán-Álvarez, E. Avella, R. M. Ramírez-Zamora and R. Zanella, Catal. Today, 2016, 266, 175–187 CrossRef.
- T. Suwannaruang, P. Kidkhunthod, N. Chanlek, S. Soontaranon and K. Wantala, Appl. Surf. Sci., 2019, 478, 1–14 CrossRef CAS.
- X. Xing, Z. Du, J. Zhuang and D. Wang, J. Photochem. Photobiol., A, 2018, 359, 23–32 CrossRef CAS.
- X. Feng, P. Wang, J. Hou, J. Qian, Y. Ao and C. Wang, J. Hazard. Mater., 2018, 351, 196–205 CrossRef CAS PubMed.
- S. Vijayalakshmi, E. Kumar, P. S. Venkatesh and A. Raja, Ionics, 2020, 26, 1507–1513 CrossRef CAS.
- D. V. Thuan, T. B. H. Nguyen, T. H. Pham, J. Kim, T. T. Hien Chu, M. V. Nguyen, K. D. Nguyen, W. A. Al-onazi and M. S. Elshikh, Chemosphere, 2022, 308, 136408 CrossRef PubMed.
- Z.-H. Diao, X.-R. Xu, D. Jiang, J.-J. Liu, L.-J. Kong, G. Li, L.-Z. Zuo and Q.-H. Wu, Chem. Eng. J., 2017, 315, 167–176 CrossRef CAS.
- S. Das, S. Ghosh, A. J. Misra, A. J. Tamhankar, A. Mishra, C. S. Lundborg and S. K. Tripathy, Int. J. Environ. Res. Public Health, 2018, 15, 2440 CrossRef CAS PubMed.
- X. Yu, J. Zhang, J. Zhang, J. Niu, J. Zhao, Y. Wei and B. Yao, Chem. Eng. J., 2019, 374, 316–327 CrossRef CAS.
- A. Tofanello, E. Belleti, A. M. M. Brito and I. L. Nantes-Cardoso, Mater. Res., 2021, 24 DOI:10.1590/1980-5373-mr-2021-0198.
- L. T. Nguyen, H. T. Nguyen, T. D. Pham, T. D. Tran, H. T. Chu, H. T. Dang and B. V. Bruggen, Top. Catal., 2020, 63, 985–995 CrossRef CAS.
- X. Huang, W. Yang, G. Zhang, L. Yan, Y. Zhang, A. Jiang, H. Xu, M. Zhou, Z. Liu, H. Tang and D. D. Dionysiou, Catal. Today, 2021, 361, 11–16 CrossRef CAS.
- M. Mirzai, F. Akhlaghian and F. Rahmani, Water Environ. J., 2020, 34, 420–431 CrossRef CAS.
- X. Zheng, S. Xu, Y. Wang, X. Sun, Y. Gao and B. Gao, J. Colloid Interface Sci., 2018, 527, 202–213 CrossRef CAS PubMed.
- A. Alam, U. Rahman, Z. U. Rahman, S. A. Khan, Z. Shah, K. Shaheen, H. Suo, M. N. Qureshi, S. B. Khan, E. M. Bakhsh and K. Akhtar, J. Mater. Sci.: Mater. Electron., 2022, 33, 4255–4267 CrossRef CAS.
- Z. Liu, X. Liu, Q. Lu, Q. Wang and Z. Ma, J. Taiwan Inst. Chem. Eng., 2019, 96, 214–222 CrossRef CAS.
- A. Hassani, A. Khataee and S. Karacaa, J. Mol. Catal. A: Chem., 2015, 409, 149–161 CrossRef CAS.
- N. Li, J. Zhang, Y. Tian, J. Zhao, J. Zhang and W. Zuo, Chem. Eng. J., 2017, 308, 377–385 CrossRef CAS.
- A. Raja, P. Rajasekaran, K. Selvakumar, M. Arunpandian, K. Kaviyarasu, S. Asath Bahadur and M. Swaminathan, Sep. Purif. Technol., 2020, 233, 115996 CrossRef CAS.
- S. Teixeira, H. Mora, L.-M. Blasse, P. M. Martins, S. A. C. Carabineiro, S. Lanceros-Méndez, K. Kühn and G. Cuniberti, J. Photochem. Photobiol., A, 2017, 345, 27–35 CrossRef CAS.
- I. Gabelica, L. Ćurković, V. Mandić, I. Panžić, D. Ljubas and K. Zadro, Catalysts, 2021, 11, 1136 CrossRef CAS.
- Z. Liu and Z. Ma, Mater. Res. Bull., 2019, 118, 110492 CrossRef CAS.
- A. J. Labrag, C. E. Bekkali, I. Es-Saidi, H. Bouyarmane, A. Laghzizil, M. Khamar and D. Robert, E3S Web Conf., 2020, 150, 02006 CrossRef.
- F. Tamaddon, A. Nasiri and G. Yazdanpanah, MethodsX, 2020, 7, 100764 CrossRef CAS PubMed.
- Z. Liu, J. Tian, D. Zeng, C. Yu, W. Huang, K. Yang, X. Liu and H. Liu, Mater. Res. Bull., 2019, 112, 336–345 CrossRef CAS.
- H. Wang, J. Li, P. Huo, Y. Yan and Q. Guan, Appl. Surf. Sci., 2016, 366, 1–8 CrossRef CAS.
- S. P. Pattnaik, A. Behera, S. Martha, R. Acharya and K. Parida, J. Mater. Sci., 2019, 54, 5726–5742 CrossRef CAS.
- C. Li, Z. Sun, W. Zhang, C. Yu and S. Zheng, Appl. Catal., B, 2018, 220, 272–282 CrossRef CAS.
- C. Chuaicham, T. Inoue, V. Balakumar, Q. Tian, B. Ohtani and K. Sasaki, J. Environ. Chem. Eng., 2022, 10, 10697 Search PubMed.
- Y. Yu, K. Liu, Y. Zhang, X. Xing and H. Li, Int. J. Environ. Res. Public Health, 2022, 19, 4793 CrossRef CAS PubMed.
- Y. Zhang, H. Pan and F. Zhang, Mater. Lett., 2019, 251, 114–117 CrossRef CAS.
- D. T. Sponza and P. Koyuncuoglu, Clin. Microbiol. Infect., 2019, 4, 1–10 Search PubMed.
- Y. He, S. Han, G. Zhao, J. Luo, C. Jia, Y. Chen, Q. Liu, J. Gao, C. Wang and J. Wang, J. Environ. Chem. Eng., 2022, 10, 107829 CrossRef CAS.
- M. Shi, W. Li, Q. Wang, H. Xu, Y. Zhao, G. He, Q. Meng and H. Chen, Opt. Mater., 2021, 122, 111726 CrossRef CAS.
- S. Kumar, R. D. Kaushik and P. Purohit, J. Hazard. Mater., 2022, 424, 127332 CrossRef CAS PubMed.
- J. Ni, J. Xue, J. Shen, G. He and H. Chen, Appl. Surf. Sci., 2018, 441, 599–606 CrossRef CAS.
- X.-F. Zeng, J.-S. Wang, Y.-N. Zhao, W.-L. Zhang and M.-H. Wang, Int. J. Miner., Metall. Mater., 2021, 28, 503–510 CrossRef CAS.
- S. Urus, M. Caylar, H. Eskalen and S. Ozganm, J. Mater. Sci.: Mater. Electron., 2022, 33, 4314–4329 CrossRef CAS.
- X. Hou, J. Liu, W. Guo, S. Li, Y. Guo, Y. Shi and C. Zhang, Catal. Commun., 2019, 121, 27–31 CrossRef CAS.
- A. Kumar, G. Sharma, M. Naushad, T. Ahamad, R. C. Veses and F. J. Stadler, Chem. Eng. J., 2019, 370, 148–165 CrossRef CAS.
- P. Huo, M. Zhou, Y. Tang, X. Liu, C. Ma, L. Yu and Y. Yan, J. Alloys Compd., 2016, 670, 198–209 CrossRef CAS.
- J. Du, S. Ma, Y. Yan, K. Li, F. Zhao and J. Zhou, Colloids Surf., A, 2019, 572, 237–249 CrossRef CAS.
- Y. Deng, L. Tang, C. Feng, G. Zeng, J. Wang, Y. Zhou, Y. Liu, B. Peng and H. Feng, J. Hazard. Mater., 2018, 344, 758–769 CrossRef CAS PubMed.
- X. Rong, F. Qiu, Z. Jiang, J. Rong, J. Pan, T. Zhang and D. Yang, Chem. Eng. Res. Des., 2016, 111, 253–261 CrossRef CAS.
- X. Wang, A. Wang and J. Ma, J. Hazard. Mater., 2017, 336, 81–92 CrossRef CAS PubMed.
- Z. Wu, Y. Liang, X. Yuan, D. Zou, J. Fang, L. Jiang, J. Zhang, H. Yang and Z. Xiao, Chem. Eng. J., 2020, 394, 124921 CrossRef CAS.
- N. Guo, Y. Zeng, H. Li, X. Xu, H. Yu and X. Han, J. Hazard. Mater., 2018, 353, 80–88 CrossRef CAS PubMed.
- A. Kaur, W. A. Anderson, S. Tanvir and S. K. Kansal, J. Colloid Interface Sci., 2019, 557, 236–253 CrossRef CAS PubMed.
- J. Zhao, Z. Zhao, N. Li, J. Nan, R. Yu and J. Du, Chem. Eng. J., 2018, 353, 805–813 CrossRef CAS.
- S. Shanavas, S. M. Roopan, A. Priyadharsan, D. Devipriya, S. Jayapandi, R. Acevedo and P. M. Anbarasan, Appl. Catal., B, 2019, 255, 117758 CrossRef CAS.
- M. Chen, J. Yao, Y. Huang, H. Gong and W. Chu, Chem. Eng. J., 2018, 334, 453–461 CrossRef CAS.
- T. Senasu, S. Nijpanich, S. Juabrum, N. Chanlek and S. Nanan, Appl. Surf. Sci., 2021, 567, 150850 CrossRef CAS.
- F. Beshkar, M. Salavati-Niasari and O. Amiri, Ind. Eng. Chem. Res., 2021, 60, 9578–9591 CrossRef CAS.
- M. Yu, H. Liang, R. Zhan, L. Xu and J. Niu, Chin. Chem. Lett., 2021, 32, 2155–2158 CrossRef CAS.
- A. S. Prasad, S. N. Kumar, M. A. Maheswari and D. Prabhakaran, Bull. Mater. Sci., 2021, 44, 99 CrossRef CAS.
- J. Yi, K. Wu, H. Wu, J. Guo, L. Zhang, H. Li and J. Lim, Nano, 2021, 16, 2150063 CrossRef CAS.
- K. K. Das, S. Patnaik, S. Mansingh, A. Behera, A. Mohanty, C. Acharya and K. M. Parida, J. Colloid Interface Sci., 2020, 561, 551–567 CrossRef CAS PubMed.
- L. N. Costa, F. X. Nobre, A. O. Lobo and J. M. E. de Matos, Environ. Nanotechnol., Monit. Manage., 2021, 16, 100466 CAS.
- M. Li, J. Zhang, L. Wang, X. Cheng, X. Gao, Y. Wang, G. Zhang, Y. Qi, H. Zhai, R. Guan and Z. Zhao, Appl. Surf. Sci., 2022, 583, 152516 CrossRef CAS.
- L. Wolski, K. Grzelak, M. Muńko, M. Frankowski, T. Grzyb and G. Nowaczyk, Appl. Surf. Sci., 2021, 563, 150338 CrossRef CAS.
- X. Hu, H. Zhao, Y. Liang, F. Chen, J. Li and R. Chen, Chemosphere, 2021, 264, 128434 CrossRef CAS PubMed.
- V. D. Dang, J. A. Jr, T. Annadurai, T. A. N. Bui, H. L. Tran, L.-Y. Lin and R.-A. Doong, Chem. Eng. J., 2021, 422, 130103 CrossRef CAS.
- K. Hu, R. Li, C. Ye, A. Wang, W. Wei, D. Hu, R. Qiu and K. Yan, J. Cleaner Prod., 2020, 253, 120055 CrossRef CAS.
- M. Chen, Y. Dai, J. Guo, H. Yang, D. Liu and Y. Zhai, Appl. Surf. Sci., 2019, 493, 1361–1367 CrossRef CAS.
- X.-J. Wen, C.-G. Niu, L. Zhang, C. Liang, H. Guo and G.-M. Zeng, J. Catal., 2018, 358, 141–154 CrossRef CAS.
- J. C. Durán-Álvarez, M. Méndez-Galván, L. Lartundo-Rojas, M. Rodríguez-Varela, D. Ramírez-Ortega, D. Guerrero-Araque and R. Zanella, Top. Catal., 2019, 62, 1011–1025 CrossRef.
- J. Mao, B. Hong, J. Wei, J. Xi, Y. Han, H. Jin, D. Jin, X. Peng, J. Li, Y. Yang, J. Gong, H. Ge and X. Wang, ChemistrySelect, 2019, 4, 13716–13723 CrossRef CAS.
- B. Zhu, D. Song, T. Jia, W. Sun, D. Wang, L. Wang, J. Guo, L. Jin, L. Zhang and H. Tao, ACS Omega, 2021, 6, 1647–1656 CrossRef CAS PubMed.
- Y. Zhou, M. Yu, H. Liang, J. Chen, L. Xu and J. Niu, Appl. Catal., B, 2021, 291, 120105 CrossRef CAS.
- N. S. Alhokbany, R. Mousa, M. Naushad, S. M. Alshehri and T. Ahamad, Int. J. Biol. Macromol., 2020, 164, 3864–3872 CrossRef CAS PubMed.
- M. F. R. Samsudin, C. Frebillot, Y. Kaddoury, S. Sufian and W.-J. Ong, J. Environ. Manage., 2020, 270, 110803 CrossRef CAS PubMed.
- C.-H. Shen, X.-J. Wen, Z.-H. Fei, Z.-T. Liu and Q.-M. Mu, Chem. Eng. J., 2020, 391, 123612 CrossRef CAS.
- K. Wang, Y. Li, G. Zhang, J. Li and X. Wu, Appl. Catal., B, 2019, 240, 39–49 CrossRef CAS.
- S. Li, J. Chen, S. Hu, H. Wang, W. Jiang and X. Chen, Chem. Eng. J., 2020, 402, 126165 CrossRef CAS.
- A. A. Tessema, C.-M. Wu and K. G. Motora, ACS Omega, 2022, 7, 38475–38486 CrossRef CAS PubMed.
- A. B. Makama, A. Salmiaton, E. B. Saion, T. S. Y. Choong and N. Abdullah, J. Nanomater., 2015, 108297, DOI:10.1155/2015/108297.
- N. Lu, P. Wang, Y. Su, H. Yu, N. Liu and X. Quan, Chemosphere, 2019, 215, 444–453 CrossRef CAS PubMed.
- C. Lai, M. Zhang, B. Li, D. Huang, G. Zeng, L. Qin, X. Liu, H. Yi, M. Cheng, L. Li, Z. Chen and L. Chen, Chem. Eng. J., 2019, 358, 891–902 CrossRef CAS.
- T. H. A. Nguyen, V. T. Le, V.-D. Doan, A. V. Tran, V. C. Nguyen, A.-T. Nguyen and Y. Vasseghian, Mater. Lett., 2022, 308, 131129 CrossRef CAS.
- K. Wang, L. Liang, Y. Zheng, H. Li, X. Niu, D. Zhang and H. Fan, New J. Chem., 2021, 45, 16168–16178 RSC.
- R. A. Palominos, M. A. Mondaca, A. Giraldo, G. Penuela, M. Perez-Moya and H. D. Mansilla, Catal. Today, 2009, 144, 100–105 CrossRef CAS.
- X.-D. Zhu, Y.-J. Wang, R.-J. Sun and D.-M. Zhou, Chemosphere, 2013, 92, 925–932 CrossRef CAS PubMed.
- G. H. Safari, M. Hoseini, M. Seyedsalehi, H. Kamani, J. Jaafari and A. H. Mahvi, Int. J. Environ. Sci. Technol., 2015, 12, 603–616 CrossRef CAS.
- S. Wu, H. Hu, Y. Lin, J. Zhang and Y. H. Hu, Chem. Eng. J., 2020, 382, 122842 CrossRef CAS.
- T. Ikhlef-Taguelmimt, A. Hamiche, I. Yahiaoui, T. Bendellali, H. Lebik-Elhadi, H. A. Amar and F. Aissani-Benissad, Water Sci. Technol., 2020, 82, 1570–1578 CrossRef CAS PubMed.
- L. Rimoldi, D. Meroni, G. Cappelletti and S. Ardizzone, Catal. Today, 2017, 281, 38–44 CrossRef CAS.
- M. Malakootian, S. N. Asadzadeh, M. Mehdipootarr and D. Kalantar-Neyestanaki, Desalin. Water Treat., 2021, 222, 302–312 CrossRef CAS.
- S. J. Olusegun, G. Larrea, M. Osial, K. Jackowska and P. Krysinski, Catalysts, 2021, 11, 1243 CrossRef CAS.
- M. Saghi and K. Mahanpoor, Int. J. Ind. Chem., 2017, 8, 297–313 CrossRef CAS.
- H. Shi, Q. Wu, L. Jiang, L. Wang, M. Huang, B. Han, Z. Yu and Y. Zuo, Int. J. Electrochem. Sci., 2020, 15, 1539–1547 CrossRef CAS.
- S. K. Fanourakis, S. Q. Barroga, R. A. Mathew, J. Pena-Bahamonde, S. M. Louie, J. V. D. Perez and D. F. Rodrigues, J. Environ. Chem. Eng., 2022, 10, 107635 CrossRef CAS.
- Y. Jiang, C. Xing, Y. Chen, J. Shi and S. Wang, Environ. Sci. Pollut. Res., 2022, 29, 57656–57668 CrossRef CAS PubMed.
- M. Yan, Y. Wu and X. Liu, J. Alloys Compd., 2021, 855, 157548 CrossRef CAS.
- O. Linnik, E. Manuilov, S. Snegir, N. Smirnova and A. Eremenko, J. Adv. Oxid. Technol., 2009, 12, 265–270 CAS.
- S. Zhao, Y. Yang, R. Lu, Y. Wang, Y. Lu, R. D. Rodriguez, E. Sheremet and J. Chen, J. Environ. Chem. Eng., 2022, 10, 107107 CrossRef CAS.
- M. Liu, J. Jiang, H. Tan, B. Chen, J. Ou, H. Wang, J. Sun, L. Liu, F. Wang, J. Gao, C. Liu, F. Peng, Y. Liu and Y. Tu, Nanoscale, 2022, 14, 12804–12813 RSC.
- W. Shi, H. Ren, M. Li, K. Xu, Y. Shu, C. Yan and Y. Tang, Chem. Eng. J., 2020, 382, 122876 CrossRef CAS.
- H. Xu, Y. Dingi, S. H. Yang, H. Zhang, X. Wang, J. Yuan, M. Long, Q. Zhao and Y. Liu, Bull. Mater. Sci., 2021, 44, 226 CrossRef CAS.
- K. Pandi, M. Preeyanghaa, V. Vinesh, J. Madhavan and B. Neppolian, Environ. Res., 2022, 207, 112188 CrossRef CAS PubMed.
- T. Zhang, Y. Liu, Y. Rao, X. Li, D. Yuan, S. Tang and Q. Zhao, Chem. Eng. J., 2020, 384, 123350 CrossRef CAS.
- Y. Chen and K. Liu, Chem. Eng. J., 2016, 302, 682–696 CrossRef CAS.
- K. Zheng, J. Chen, X. Gao, X. Cao, S. Wu and J. Su, Water Sci. Technol., 2021, 84, 1919–1929 CrossRef CAS PubMed.
- N. Farhadian, R. Akbarzadeh, M. Pirsaheb, T. C. Jen, Y. Fakhri and A. Asadi, Int. J. Biol. Macromol., 2019, 132, 360–373 CrossRef CAS PubMed.
- P. Wang, P.-S. Yap and T.-T. Lim, Appl. Catal., A, 2011, 399, 252–261 CrossRef CAS.
- J. Zhang, X. Li, M. Peng, Y. Tang, A. Ke, W. Gan, X. Fu and H. Hao, Mater. Res. Express, 2018, 5, 065008 CrossRef.
- H. Wang, D. Wu, X. Li and P. Huo, J. Mater. Sci.: Mater. Electron., 2019, 30, 19126–19136 CrossRef CAS.
- A. Moradi, F. Rahmani and M. Khamforoush, Iran. J. Polym. Sci. Technol., 2021, 33, 465–478 Search PubMed.
- A. Bembibre, M. Benamara, M. Hjiri, E. Gómez, H. R. Alamri, R. Dhahri and A. Serrà, Chem. Eng. J., 2022, 427, 132006 CrossRef CAS.
- D. S. Pattanayak, D. Pal, J. Mishra, C. Thakur and K. L. Wasewar, Environ. Sci. Pollut. Res., 2023, 30, 24919–24926 CrossRef CAS PubMed.
- L. Jiang, X. Yuan, G. Zeng, J. Liang, Z. Wu, H. Yu, D. Mo, H. Wang, Z. Xiao and C. Zhou, J. Colloid Interface Sci., 2019, 536, 17–29 CrossRef CAS PubMed.
- S. Chen, F. Liu, R. Cui, B. Zhu and X. You, Water Cycle, 2022, 3, 8–17 CrossRef.
- T. S. Bui, P. Bansal, B.-K. Lee, T. Mahvelati-Shamsabadi and T. Soltani, Appl. Surf. Sci., 2019, 506, 144184 CrossRef.
- G. Li, B. Wang, J. Zhang, R. Wang and H. Liu, Chem. Eng. J., 2020, 391, 123500 CrossRef CAS.
- Y. Zhao, H. Qin, Z. Wang, H. Wang, Y. He, Q. Tian, Q. Luo and P. Xu, Environ. Sci. Pollut. Res., 2022, 29, 74062–74080 CrossRef CAS PubMed.
- W. Wang, Z. Zeng, G. Zheng, C. Zhang, R. Xiao, C. Zhou, W. Xiong, Y. Yang, L. Lei, Y. Liu, D. Huang, M. C. Y. Cheng, Y. Fu, H. Luo and Y. Zhou, Chem. Eng. J., 2019, 378, 122132 CrossRef CAS.
- J. Xu, Y. Chen, M. Chen, J. Wang and L. Wang, Chem. Eng. J., 2022, 442, 136208 CrossRef CAS.
- L. Chen, Y. Wang, S. Cheng, X. Zhao, J. Zhang, Z. Ao, C. Zhao, B. Li, S. Wang, S. Wang and H. Sun, Appl. Catal., B, 2022, 303, 120932 CrossRef CAS.
- N. L. M. Tri, J. Kim, B. L. Giang, T. M. A. Tahtamouni, P. T. Huong, C. Lee, N. M. Viet and D. Q. Trung, J. Ind. Eng. Chem., 2019, 80, 597–605 CrossRef.
- D. Jia, Y. Zhang, X. Zhang, P. Feng, L. Yang, R. Ning, H. Pan and Y. Miao, Environ. Sci.: Nano, 2021, 8, 415–431 RSC.
- S. J. Alyani, A. E. Pirbazari, F. E. Khalilsaraei, N. A. Kolur and N. Gilani, J. Alloys Compd., 2019, 799, 169–182 CrossRef.
- J. Ye, J. Liu, Z. Huang, S. Wu, X. Dai, L. Zhang and L. Cui, Chemosphere, 2019, 227, 505–513 CrossRef CAS PubMed.
- M. Cao, P. Wang, Y. Ao, C. Wang, J. Hou and J. Qian, J. Colloid Interface Sci., 2016, 467, 129–139 CrossRef CAS PubMed.
- J. Liu, H. Chen, C. Zhu, S. Han, J. Li, S. She and X. Wu, J. Colloid Interface Sci., 2022, 622, 526–538 CrossRef CAS PubMed.
- N. Belhoucheta, B. Hamdia, H. Chenchounid and Y. Bessekhoua, J. Photochem. Photobiol., A, 2019, 372, 196–205 CrossRef.
- Y. M. Hunge, A. A. Yadav, S.-W. Kang and H. Kim, J. Colloid Interface Sci., 2022, 606, 454–463 CrossRef CAS PubMed.
- P. Semeraro, S. Bettini, S. Sawalha, S. Pal, A. Licciulli, F. Marzo, N. Lovergine, L. Valli and G. Giancane, Nanomaterials, 2020, 10, 1458 CrossRef CAS PubMed.
- B. Kakavandi, N. Bahari, R. R. Kalantary and E. D. Fard, Ultrason. Sonochem., 2019, 55, 75–85 CrossRef CAS PubMed.
- V. H. T. Thi and B.-K. Lee, J. Hazard. Mater., 2017, 324, 329–339 CrossRef PubMed.
- H. Shi, Q. Wu, X. Yang, Y. Zuo, H. Yang, R. Zhang, Y. Zhang, Y. Fan, X. Du and L. Jiang, Int. J. Electrochem. Sci., 2021, 16, 210314, DOI:10.20964/2021.03.70.
- S. Zhang, J. Yi, J. Chen, Z. Yin, T. Tang, W. Wei, S. Cao and H. Xu, Chem. Eng. J., 2020, 380, 122583 CrossRef CAS.
- E. Weidner, K. S. Ciesielczyk, D. Moszynsky, T. Jesionowski and F. Ciesielczyk, Environ. Technol. Innovation, 2021, 24, 102016 CrossRef CAS.
- S. Leong, D. Li, K. Hapgood, X. Zhang and H. Wang, Appl. Catal., B, 2016, 198, 224–233 CrossRef CAS.
- C. Lv, X. Lan, L. Wang, X. Dai, M. Zhang, J. Cui, S. Yuan, S. Wang and J. Shi, Environ. Technol., 2021, 42, 377–387 CrossRef CAS PubMed.
- W. Chen, L. Chang, S.-B. Ren, Z.-C. He, G.-B. Huang and X.-H. Liu, J. Hazard. Mater., 2020, 384, 121308 CrossRef CAS PubMed.
- L. Wang, C. Zhang, R. Cheng, J. Ali, Z. Wang, G. Mailhot and G. Pan, Catalysts, 2018, 8, 628 CrossRef.
- J. Xue, S. Ma, Y. Zhou, Z. Zhang and P. Jiang, RSC Adv., 2015, 5, 18832–18840 RSC.
- J. Wei, P. Zhu and P. Chen, Materials, 2022, 15, 1963 CrossRef CAS PubMed.
- H. A. Patehkhor, M. Fattahi and M. R. Khosravi-Nikou, Sci. Rep., 2021, 11, 24177 CrossRef PubMed.
- Y. Shi, Z. Yang, B. Wang, H. An, Z. Chen and H. Cui, Appl. Clay Sci., 2016, 119, 311–320 CrossRef CAS.
- S. Kaushal, A. Kumar, H. Bains and P. P. Singh, Environ. Sci. Pollut. Res., 2023, 30, 37092–37104 CrossRef CAS PubMed.
- X. Zhang, H. Wang, M. Gao, P. Zhao, W. Xia, R. Yang, Y. Huang, L. Wang, M. Liu, T. Wei, L. Wang, R. Yao, X. Li and Z. Fan, Chemosphere, 2022, 294, 133782 CrossRef CAS PubMed.
- Y. Wang, S. Zhang, Y. Ge, C. Wang, J. Hu and H. Liu, Acta Phys.-Chim. Sin., 2020, 36, 1905083 Search PubMed.
- C. Hu, Z.-T. Liu, P.-C. Yang, Y.-X. Ding, K.-Y. A. Lin and B.-S. Nguven, J. Taiwan Inst. Chem. Eng., 2021, 123, 219–227 CrossRef CAS.
- H. Shi, Y. He, Y. Li, T. He and P. Luo, Sep. Purif. Technol., 2022, 280, 119864 CrossRef CAS.
- D. Wang, S. Li and Q. Feng, J. Mater. Sci.: Mater. Electron., 2018, 29, 9380–9386 CrossRef CAS.
- J. Zhang and Z. Ma, Chin. J. Chem. Eng., 2018, 26, 753–760 CrossRef CAS.
- J. Liu, H. Xu, Y. Xu, Y. Song, J. Lian, Y. Zhao, L. Wang, L. Huang, H. Ji and H. Li, Appl. Catal., B, 2017, 207, 429–437 CrossRef CAS.
- V. T. Quyen, H. J. Kim, J. T. Kim, L. T. T. Ha, P. T. Huong, D. M. Thanh, N. M. Viet and P. Q. Thang, Sol. Energy, 2021, 214, 288–293 CrossRef.
- L. Jiang, X. Yuan, G. Zeng, Z. Wu, J. Liang, X. Chen, L. Leng, H. Wang and H. Wang, Appl. Catal., B, 2018, 221, 715–725 CrossRef CAS.
- X. Chen, L. Liu, Y. Zhao, J. Zhang, D. Li, B. Hu and X. Hai, ChemistrySelect, 2017, 2, 9256–9260 CrossRef CAS.
- J. Liu, Y. Song, H. Xu, X. Zhu, J. Lian, Y. Xu, Y. Zhao, L. Huang, H. Ji and H. Li, J. Colloid Interface Sci., 2017, 494, 38–46 CrossRef CAS PubMed.
- L. Jiang, X.-Z. Yuan, G. Zeng, X. Chen, Z. Wu, J. Liang, J. Zhang, H. Wang and H. Wang, ACS Sustainable Chem. Eng., 2017, 5, 5831–5841 CrossRef CAS.
- R. Chen, X. Wang and L. Zhu, Research Square, 2022, preprint, DOI:10.21203/rs.3.rs-1172368/v1.
- E. Vijayakumar, M. G. Raj, M. G. Narendran, R. Preetha, R. Mohankumar, B. Neppolian and A. J. Bosco, ACS Omega, 2022, 7, 5079–5095 CrossRef CAS PubMed.
- X. Tang, L. Ni, J. Han and Y. Wang, Chin. J. Catal., 2017, 38, 447–457 CrossRef CAS.
- W. Qian, W. Hu, Z. Jiang, Y. Wu, Z. Li, Z. Diao and M. Li, Catalysts, 2022, 12, 774 CrossRef CAS.
- F. Wang, Z. Zhu and J. Guo, RSC Adv., 2021, 11, 35663–35672 RSC.
- H. Shi, T. Zhao, J. Wang, Y. Wang, Z. Chen, B. Liu, H. Ji, W. Wang, G. Zhang and Y. Li, J. Alloys Compd., 2021, 860, 157924 CrossRef CAS.
- H.-J. Ren, Y.-B. Tang, W.-l. Shi, F.-Y. Chen and Y.-S. Xu, New J. Chem., 2019, 43, 19172–19179 RSC.
- R. Zou, T. Xu, X. Lei, Q. Wu and S. Xue, Solid State Sci., 2020, 99, 106067 CrossRef CAS.
- A. Maridiroosi, A. R. Mahjoub and H. Fakhr, Int. J. Chem. Biomol. Sci., 2020, 14, 31–36, DOI:10.5281/zenodo.3669194.
- D. Qiao, Z. Li, J. Duan and X. He, Chem. Eng. J., 2020, 400, 125952 CrossRef CAS.
- B. Li, X. Yu, X. Yu, R. Du, L. Liu and Y. Zhang, Appl. Surf. Sci., 2019, 478, 991–997 CrossRef CAS.
- K. Chakraborty, T. Pal and S. Ghosh, ACS Appl. Nano Mater., 2018, 1, 3137–3144 CrossRef CAS.
- X. Tang, Z. Wang and Y. Wang, Chem. Phys. Lett., 2018, 691, 408–414 CrossRef CAS.
- K. V. A. Kumar, B. Lakshminarayana, D. Suryakala and C. Subrahmanyam, RSC Adv., 2020, 10, 20494–20503 RSC.
- J. Shan, X. Wu, C. Li, J. Hu, Z. Zhang, H. Liu, P. Xia and X. Huang, Research Square, 2022, preprint, DOI:10.21203/rs.3.rs-1721079/v1.
- S. M. Ghoreishian, G. S. R. Raju, E. Pavitra, C. H. Kwak, Y.-K. Han and Y. S. Huh, Appl. Surf. Sci., 2019, 489, 110–122 CrossRef CAS.
- S. Zhang, J. Xu, J. Hu, O. Cui and H. Liu, Langmuir, 2017, 33, 5015–5024 CrossRef CAS PubMed.
- R. Zou, T. Xu, X. Lei, Q. Wu and S. Xue, Solid State Sci., 2020, 99, 106067 CrossRef CAS.
- S. K. Ibrahim, T. Pal and S. Ghosh, AIP Conf. Proc., 2019, 2115, 030188, DOI:10.1063/1.5113027.
- Y. Hou, S. Pu, Q. Shi, S. Mandal, H. Ma, S. Xue, G. Cai and Y. Bai, J. Taiwan Inst. Chem. Eng., 2019, 104, 139–150 CrossRef CAS.
- F.-S. Tabatabai-Yazdi, A. E. Pirbazari, F. E. Khalilsaraei, N. A. Kolur and N. Gilani, Water Environ. Res., 2020, 92, 662–676 CrossRef CAS PubMed.
- F.-S. Tabatabai-Yazdi, A. E. Pirbazari, F. E. K. Saraei and N. Gilani, Phys. B, 2021, 608, 412869 CrossRef CAS.
- L.-L. Qu, N. Wang, Y.-Y. Li, D.-D. Bao, G.-H. Yang and H.-T. Li, J. Mater. Sci., 2017, 52, 8311–8320 CrossRef CAS.
- J. Song, X. Wu, M. Zhang, C. Liu, J. Yu, G. Sun, Y. Si and B. Ding, Chem. Eng. J., 2020, 379, 122269 CrossRef CAS.
- L. Huang, D. Bao, J. Li and X. Jiang, Appl. Surf. Sci., 2021, 555, 149696 CrossRef CAS.
- A. Iqbal, U. Saidu, F. Adam, S. Sreekantan, N. Yahaya, M. N. Ahmad, R. J. Ramalingam and L. D. Wilson, Molecules, 2021, 26, 2509 CrossRef CAS PubMed.
- Y. Zhao, A. Zada, Y. Yang, J. Pan, Y. Wang, Z. Yan, Z. Xu and K. Qi, Front. Chem., 2021, 9, 797738 CrossRef CAS PubMed.
- H. Jingyu, Y. Ran, L. Zhaohui, S. Yuanqiang, Q. Lingbo and A. N. Kani, Solid State Sci., 2019, 92, 60–67 CrossRef.
- B. Guo, B. Liu, C. Wang, Y. Wang, S. Yin, M. S. Javed and W. Han, J. Environ. Chem. Eng., 2022, 10, 107118 CrossRef CAS.
- P. Zhu, M. Duan, R. Wang, J. Xu, P. Zou and H. Jia, Colloids Surf., A, 2020, 602, 125118 CrossRef CAS.
- J. Gu, H. Jia, S. Ma, Z. Ye, J. Pan, R. Dong, Y. Zong and J. Xue, ACS Omega, 2020, 5, 30980–30988 CrossRef CAS PubMed.
- X. Liao, T.-T. Li, H.-T. Ren, Z. Mao, X. Zhang, J.-H. Lin and C.-W. Lou, Ceram. Int., 2021, 47, 10786–10795 CrossRef CAS.
- S. Li, S. Hu, K. Xu, W. Jiang, Y. Liu, Z. Leng and J. Liu, J. Colloid Interface Sci., 2017, 504, 561–569 CrossRef CAS PubMed.
- D. Chen, S. Wu, J. Fang, S. Lu, G. Y. Zhou, W. Feng, F. Yang, Y. Chen and Z. Q. Fang, Sep. Purif. Technol., 2018, 193, 232–241 CrossRef CAS.
- W. Wang, Q. Han, Z. Zhu, L. Zhang, S. Zhong and B. Liu, Adv. Powder Technol., 2019, 30, 1882–1896 CrossRef CAS.
- Y. Zhou, J. Li, C. Liu, P. Huo and H. Wang, Appl. Surf. Sci., 2018, 458, 586–596 CrossRef CAS.
- Z. Liu, A. Zhang, Y. Liu, Y. Fu and Y. Du, Colloids Surf., A, 2022, 643, 128818 CrossRef CAS.
- H. Liu, W. Huo, T. C. Zhang, L. Ouyang and S. Yuan, Mater. Today Chem., 2022, 23, 100729 CrossRef CAS.
- M. Al-Haddad, A. Shawky and I. A. Mkhalid, J. Taiwan Inst. Chem. Eng., 2021, 123, 284–292 CrossRef CAS.
- H. M. Lwin, W. Zhan, S. Song, F. Jia and J. Zhou, Chem. Phys. Lett., 2019, 736, 136806 CrossRef CAS.
- X. Zhang, D. Han, M. Dai, K. Chen, Z. Han, Y. Fan, Y. He, D. Han and L. Niu, Catal. Sci. Technol., 2021, 11, 6248 RSC.
- C. Jin, J. Kang, Z. Li, M. Wang, Z. Wu and Y. Xie, Appl. Surf. Sci., 2020, 514, 146076 CrossRef CAS.
- Q. Chen, W. Yang, J. Zhu, L. Fu, D. Li and L. Zhou, J. Hazard. Mater., 2020, 384, 121275 CrossRef CAS PubMed.
- A. O. Oluwole and O. S. Olatunji, Environ. Sci. Eur., 2022, 34, 5 CrossRef CAS.
- S. Kumar, V. Sharma, K. Bhattacharyya and V. Krishnan, Mater. Chem. Front., 2017, 1, 1093–1106 RSC.
- T. Xiao, Z. Tang, Y. Yang, L. Tang, Y. Zhou and Z. Zou, Appl. Catal., B, 2018, 220, 417–428 CrossRef CAS.
- C. Li, S. Yu, H. Dong, C. Liu, H. Wu, H. Che and G. Chen, Appl. Catal., B, 2018, 238, 284–293 CrossRef CAS.
- X. Jiang, S. Lai, W. Xu, J. Fang, X. Chen, J. Beiyuan, X. Zhou, K. Lin, J. Liu and G. Guan, J. Alloys Compd., 2019, 809, 151804 CrossRef CAS.
- J. Ni, W. Wang, D. Liu, Q. Zhu, J. Jia, J. Tian, Z. Li, X. Wang and Z. Xing, J. Hazard. Mater., 2021, 408, 124432 CrossRef CAS PubMed.
- Y. Liu, J. Tian, L. Wei, Q. Wang, C. Wang, Z. Xing, X. Li, W. Yang and C. Yang, Sep. Purif. Technol., 2021, 257, 117976 CrossRef CAS.
- C. Li, S. Yu, H. Che, X. Zhang, J. Han, Y. Mao, Y. Wang, C. Liu and H. Dong, ACS Sustainable Chem. Eng., 2018, 6, 16437–16447 CrossRef CAS.
- Y. Hong, Y. Meng, G. Zhang, B. Yin, Y. Zhao, W. Shi and C. Li, Sep. Purif. Technol., 2016, 171, 229–237 CrossRef CAS.
- Z. Yang, X. Xia, L. Shao, L. Wang and Y. Liu, Chem. Eng. J., 2021, 410, 128454 CrossRef CAS.
- Y. Li, Z. Lai, Z. Huang, H. Wang, C. Zhao, G. Ruan and F. Du, Appl. Surf. Sci., 2021, 550, 149342 CrossRef CAS.
- C. Du, Z. Zhang, S. Tan, G. Yu, H. Chen, L. Zhou, L. Yu, Y. Su, Y. Zhang, F. Deng and S. Wang, Environ. Res., 2021, 200, 111427 CrossRef CAS PubMed.
- L. Yan, W. Li, Q. Zhao, Z. Zhu, C. Hu and B. Liu, Colloids Surf., A, 2021, 524, 126783 CrossRef.
- J. Chen, X. Xiao, Y. Wang, M. Lu and X. Zeng, J. Alloys Compd., 2019, 800, 88–98 CrossRef CAS.
- F. Chen, Q. Yang, X. Li, G. Zeng, D. Wang, C. Niu, J. Zhao, H. An, T. Xie and Y. Deng, Appl. Catal., B, 2017, 200, 330–342 CrossRef CAS.
- H. Jing, R. Ou, H. Yu, Y. Zhao, Y. Lu, M. Huo, H. Huo and X. Wang, Sep. Purif. Technol., 2021, 255, 117646 CrossRef CAS.
- F. Guo, W. Shi, M. Li, Y. Shi and H. Wen, Sep. Purif. Technol., 2019, 210, 608–615 CrossRef CAS.
- P. Zhu, M. Hu, M. Duan, L. Xie and M. Zhao, J. Alloys Compd., 2020, 840, 155714 CrossRef CAS.
- Z. Zhang, X. Xue and X. Chen, Dalton Trans., 2022, 51, 8015–8027 RSC.
- X. Zhou, Y. Chen, C. Li, L. Zhang, X. Zhang, X. Ning, L. Zhan and J. Luo, Sep. Purif. Technol., 2019, 211, 179–188 CrossRef CAS.
- B. Zhang, X. He, X. Ma, Q. Chen, G. Liu, Y. Zhou, D. Ma, C. Cui, J. Ma and Y. Xin, Sep. Purif. Technol., 2020, 247, 116932 CrossRef CAS.
- L. Lonappan, S. K. Brar, R. K. Das, M. Verma and R. Y. Surampalli, Environ. Int., 2016, 96, 127–138 CrossRef CAS PubMed.
- A. S. Mestre and A. P. Carvalho, Molecules, 2019, 24, 3702, DOI:10.3390/molecules24203702.
- K. C. Hui, W. L. Ang, W. Z. N. Yahya and N. S. Sambudi, Chemosphere, 2022, 290, 133377 CrossRef CAS PubMed.
- D. Fabbri, M. J. López-Muñoz, A. Daniele, C. Medana and P. Calza, Photochem. Photobiol. Sci., 2019, 18, 845–852 CrossRef CAS PubMed.
- L. Rizza, S. Meric, D. Kassinos, M. Guida, F. Russo and V. Belgiorno, Water Res., 2009, 43, 979–988 CrossRef PubMed.
- W. Z. Khan, I. Najeeb, S. Ishtiaque and S. Jabeen, International Organization of Scientific Research (IOSR) Journal of Engineering, 2016, 6, 36–46 Search PubMed.
- A. C. Khraibet, N. J. Imran, H. M. Majeed, M. A. Ehmood, G. S. Hasoon, Z. F. Nathim, A. K. Alwan and A. S. A. Alsada, J. Genet. Environ. Resour. Conserv., 2022, 10, 41–48 Search PubMed.
- U. Schulze-Hennings, I. Brückner, W. Gebhardt, M. Groteklaes, S. P. Blöβ, M. Wett, V. Linnemann, D. Montag and J. Pinnekamp, Water Environ. J., 2017, 31, 508–514 CrossRef CAS.
- M. Baniamer, A. Almasi and S. Sharifnia, Iran. J. Chem. Chem. Eng., 2018, 15, 3–16 Search PubMed.
- A. Achilleos, E. Hapeshi, N. P. Xekoukoulotakis, D. Mantzavinos and D. Fatta-Kassinos, Chem. Eng. J., 2010, 161, 53–59 CrossRef CAS.
- M. V. Bagal and P. R. Gogate, Ultrason. Sonochem., 2014, 21, 1035–1043 CrossRef CAS PubMed.
- M. Tanveer, G. Tezcanli, M. T. Sadiq, S. M. Kazmi, N. Noshad, G. Abbas and A. Ali, Eng. Proc., 2022, 12, 76 Search PubMed.
- I. Mimouni, A. Bouziani, Y. Naciri, M. Boujnah, M. Alaoui, E. Belghiti and M. E. Azzouzi, Environ. Sci. Pollut. Res., 2022, 29, 7984–7996 CrossRef CAS PubMed.
- D. Meroni, C. L. Bianchi, D. C. Boffito, G. Cerrato, A. Bruni, M. A. Sartirana and E. Falletta, Ultrason. Sonochem., 2021, 75, 105615 CrossRef CAS PubMed.
- I. Berruti, N. P. F. Gonçalves, P. Calza, M. C. Paganini, I. Oller and M. I. Polo-López, Chemosphere, 2022, 303, 135017 CrossRef CAS PubMed.
- H. Mohan, P. M. Sathya, S. Vadivel, G. H. Ha, H. S. Oh, G. Kim, K.-K. Seralathan and T. Shin, Chemosphere, 2022, 301, 134699 CrossRef CAS PubMed.
- M. V. Gerbaldo, S. G. Marchetti, V. R. Elías, S. N. Mendieta and M. E. Crivello, Chem. Eng. Res. Des., 2021, 166, 237–247 CrossRef CAS.
- M. A. H. Karim and B. K. Aziz, React. Kinet., Mech. Catal., 2022, 135, 1113–1124 CrossRef CAS.
- A. Rey, E. Mena, A. M. Chavez, F. J. Beltron and F. Medina, Chem. Eng. Sci., 2015, 126, 80–90 CrossRef CAS.
- H. Chakhtouna, H. Benzeid, N. Zari, A. E. K. Qaiss and R. Bouhfid, Environ. Sci. Pollut. Res., 2021, 28, 44638–44666 CrossRef CAS PubMed.
- M. R. Espino-Estevez, C. Fernandez-Rodríguez, O. M. Gonzalez-Díaz, J. Arana, J. P. Espinos, J. A. Ortega-Mendez and J. M. Dona-Rodríguez, Chem. Eng. J., 2016, 298, 82–95 CrossRef CAS.
- E. Torad, E. H. Ismail, M. M. Mohamed and M. M. H. Khalil, Mater. Res. Bull., 2021, 137, 111193 CrossRef CAS.
- V.-H. Nguyena, Q. B. Tran, X. C. Nguyen, L. T. Hai, T. T. T. Ho, M. Shokouhimehr, D.-V. N. Vo, S. S. Lami, V.-H. Nguyena, Q. B. Tranc, X. C. Nguyenc, L. T. Hai, T. T. T. Ho, M. Shokouhimehr, D.-V. N. Vo, S. S. Lam, H. P. Nguyenj, C. T. Hoang, Q. V. Ly, W. Peng, S. Y. Kim, T. V. Tunge and Q. V. Lec, Process Saf. Environ. Prot., 2020, 142, 229–237 CrossRef.
- A. Surenjan, B. Sambandam, T. Pradeep and L. Philip, J. Environ. Chem. Eng., 2017, 5, 757–767 CrossRef CAS.
- I. Tbessi, M. Benito, J. Llorca, E. Molins, S. Sayadi and W. Najjar, J. Alloys Compd., 2019, 779, 314–325 CrossRef CAS.
- L. Rueda-Salaya, A. Hernández-Ramírez, L. Hinojosa-Reyes, J. L. Guzmán-Mar, M. Villanueva-Rodríguez and E. Sánchez-Cervantes, J. Photochem. Photobiol., A, 2020, 391, 112364 CrossRef CAS.
- G. Vitiello, G. Iervolino, C. Imparato, I. Rea, F. Borbone, L. D. Stefano, A. Aronne and V. Vaiano, Sci. Total Environ., 2021, 762, 143066 CrossRef CAS PubMed.
- S. M. Chaudhari, O. S. Gonsalves and R. Nemade, Mater. Res. Bull., 2021, 143, 111463 CrossRef CAS.
- M. Thiruppathi, P. S. Kumar, P. Devendran, C. Ramalingan, M. Swaminathan and E. R. Nagarajan, J. Alloys Compd., 2018, 735, 728–734 CrossRef CAS.
- P. Apopei, C. Orha, M. I. Popescu, C. Lazau, F. Manea, C. Catrinescu and C. Teodosiu, Process Saf. Environ. Prot., 2020, 138, 324–336 CrossRef CAS.
- W. Buda and B. Czech, Water Sci. Technol., 2013, 68, 1322 CrossRef CAS PubMed.
- I. Tbessi, M. Benito, E. Molins, J. Liorca, A. Touati, S. Sayadi and W. Najjar, Solid State Sci., 2019, 88, 20–28 CrossRef CAS.
- W. S. Koe, W. C. Chong, Y. L. Pang, C. H. Koo, M. Ebrahim and A. W. Mohammad, J. Water Process. Eng., 2020, 33, 101068 CrossRef.
- A. Gil, A. M. García, M. Fernández, M. A. Vicente, B. González-Rodríguez, V. Rives and S. A. Korili, J. Ind. Eng. Chem., 2017, 53, 183–191 CrossRef CAS.
- S. Ramandi, M. H. Entezari and N. Ghows, Ultrason. Sonochem., 2017, 38, 234–245 CrossRef CAS PubMed.
- E. Mugunthan, M. B. Saidutta and P. E. Jagadeeshbabu, Environ. Nanotechnol., Monit. Manage., 2018, 10, 322–330 Search PubMed.
- A. Cordero-García, G. T. Palomino, L. Hinojosa-Reyes, J. L. Guzmán-Mar, L. MayaTeviño and A. Hernández-Ramírez, Environ. Sci. Pollut. Res., 2017, 24, 4613–4624 CrossRef PubMed.
- E. Mugunthan, M. B. Saidutta and P. E. Jagadeeshbabu, J. Photochem. Photobiol., A, 2019, 383, 111993 CrossRef CAS.
- A. Cordero-García, J. L. Guzmán-Mar, L. Hinojosa-Reyes, E. Ruiz-Ruiz and A. Hernández-Ramírez, Ceram. Int., 2016, 42, 9796–9803 CrossRef.
- S. Murgolo, I. S. Moreira, C. Piccirillo, P. M. L. Castro, G. Ventrella, C. Cocozza and G. Mascolo, Materials, 2018, 11, 1779 CrossRef PubMed.
- W. Sun, H. Chu, B. Dong, D. Cao and S. Zheng, Int. J. Electrochem. Sci., 2014, 9, 4566–4573 CrossRef.
- C. T. Mehmood, Z. Zhong, H. Zhou, C. Zhang and Y. Xiao, RSC Adv., 2020, 10, 36349–36362 RSC.
- Y. Cui, Q. Ma, X. Deng, Q. Meng, X. Cheng, M. Xie, X. Li, Q. Cheng and H. Liu, Appl. Catal., B, 2017, 206, 136–145 CrossRef CAS.
- S. Silvestri, C. D. Ferreira, V. Oliveira, J. M. T. B. Varejão, J. A. Labrincha and D. M. Tobaldi, J. Photochem. Photobiol., A, 2019, 375, 261–269 CrossRef CAS.
- L. Das, S. K. Barodia, S. Sengupta and J. K. Basu, Int. J. Environ. Sci. Technol., 2015, 12, 317–326 CrossRef CAS.
- E. Mugunthan, M. B. Saidutta and P. E. Jagadeeshbabu, Environ. Technol., 2019, 40, 929–941 CrossRef CAS PubMed.
- E. I. Moreno-Valencia, S. P. Paredes-Carrera, J. C. Sánchez-Ochoa, S. O. Flores-Valle and J. R. Avendaño-Gómez, Mater. Res. Express, 2017, 4, 115026 CrossRef.
- S. Li, Z. Wang, X. Zhang, J. Zhao, Z. Hu, Z. Wang and X. Xie, Chem. Eng. J., 2019, 378, 122169 CrossRef CAS.
- N. Ahmadpour, M. H. Sayadi, S. Sobhani and M. Hajiani, J. Environ. Manage., 2020, 271, 110964 CrossRef CAS PubMed.
- K. Fischer, M. Kühnert, R. Gläser and A. Schulze, RSC Adv., 2015, 5, 16340–16348 RSC.
- J. E. Casillas, J. Campa-Molina, F. Tzompantzi, G. G. C. Arízaga, A. López-Gaona, S. Ulloa-Godínez, M. E. Cano and A. Barrera, Materials, 2020, 13, 1345 CrossRef CAS PubMed.
- S. Salaeh, D. J. Perisic, M. Biosic, H. Kusic, S. Babic, U. Lavrencic Stangar, D. D. Dionysiou and A. L. Bozic, Chem. Eng. J., 2016, 304, 289–302 CrossRef CAS.
- W. Liu, Y. Li, F. Liu, W. Jiang, D. Zhang and J. Liang, Water Res., 2019, 151, 8–19 CrossRef CAS PubMed.
- X. Liu, F. Li, Y. Liu, P. Li, L. Chen, B. Li, T. Qian and W. Liu, J. Environ. Chem. Eng., 2022, 10, 107545 CrossRef CAS.
- Z. Xiu, H. Bo, Y. Wu and X. Hao, Appl. Surf. Sci., 2014, 289, 394–399 CrossRef CAS.
- M. Penas-Garzon, W. H. M. Abdelraheem, C. Belver, J. J. Rodriguez, J. Bedia and D. D. Dionysiou, Sep. Purif. Technol., 2021, 275, 119169 CrossRef CAS.
- Z. Hu, X. Cai, Z. Wang, S. Li, Z. Wang and X. Xie, J. Hazard. Mater., 2019, 380, 120812 CrossRef CAS PubMed.
- W. Zhang, L. Zhou, J. Shi and H. Deng, J. Colloid Interface Sci., 2017, 496, 167–176 CrossRef CAS PubMed.
- W. Zhang, L. Zhou and H. Deng, J. Mol. Catal. A: Chem., 2016, 423, 270–276 CrossRef CAS.
- E. Valadez-Renteria, R. Perez-Gonzalez, C. Gomez-Solis, L. A. Diaz-Torres, A. Encinas, J. Oliva and V. Rodriguez-Gonzalez, J. Environ. Sci., 2023, 126, 575–589 CrossRef CAS PubMed.
- V. Muelas-Ramos, M. J. Sampaio, C. G. Silva, J. Bedia, J. J. Rodriguez, J. L. Faria and C. Belver, J. Hazard. Mater., 2021, 416, 126199 CrossRef CAS PubMed.
- O. Moradi, H. Alizadeh and S. Sedaghat, Chemosphere, 2022, 299, 134435 CrossRef CAS PubMed.
- M. Kovacic, K. Perovic, J. Papac, A. Tomic, L. Matoh, B.
![[Z with combining breve]](https://www.rsc.org/images/entities/char_005a_0306.gif) ener, T. Brodar, I. Capan, A. K. Surca, H. Kuši, U. L. Štangar and A. L. Božic, Materials, 2020, 13, 1621 CrossRef CAS PubMed.
ener, T. Brodar, I. Capan, A. K. Surca, H. Kuši, U. L. Štangar and A. L. Božic, Materials, 2020, 13, 1621 CrossRef CAS PubMed. - D. John, J. Jose, S. G. Bhat and V. S. Achari, Heliyon, 2021, 7, e07451 CrossRef CAS PubMed.
- J. Rashid, S. Oooim, R. Kumar, M. A. Barakat, B. Akram, N. Hussain, H. B. Bin and M. Xu, Sci. Rep., 2020, 10, 14191 CrossRef CAS PubMed.
- W. Li, R. Yu, M. Li, N. Guo, H. Yu and Y. Yu, Chemosphere, 2019, 218, 966–973 CrossRef CAS PubMed.
- A. Esmaeili and M. H. Entezari, RSC Adv., 2015, 5, 97027 RSC.
- O. C. Olatunde and D. C. Onwudiwe, Results Chem., 2022, 4, 100273 CrossRef CAS.
- M. E. Malefane, U. Feleni and A. T. Kuvarega, J. Environ. Chem. Eng., 2020, 8, 103560 CrossRef CAS.
- A. Kumar, S. K. Sharma, A. Kumar, G. Sharma, N. Almasoud, T. S. Alomar, M. Naushad, Z. A. Alothman and F. J. Stadler, J. Cleaner Prod., 2021, 315, 128137 CrossRef CAS.
- L. Zhang, L. Liu, X. Zhang, C. Ge and J. Liao, Adv. Energy Sustainability Res., 2022, 3, 2200138 CrossRef CAS.
- D. Kulhary and S. Singh, ChemistrySelect, 2022, 7, e202201964 CrossRef CAS.
- M. Irandost, R. Akbarzadeh, M. Pirsaheb, A. Asadi, P. Mohammadi and M. Sillanpää, J. Mol. Liq., 2019, 291, 111342 CrossRef CAS.
- M. D. Nguyen, T. B. Nguyen, L. H. Tran, T. G. Nguyen, I. Fatimah, E. P. Kuncoro and R.-A. Doong, Chem. Eng. J., 2023, 452, 139249 CrossRef.
- N. Aghababaei, M. Abdouss, H. Hosseini-Monfared and F. Ghanbari, Journal of Water Process Engineering, 2023, 53, 103702 CrossRef.
- O. Sacco, J. J. Murcia, A. E. Lara, M. Hernández-Laverde, H. Rojas, J. A. Navío, M. C. Hidalgo and V. Vaiano, Mater. Sci. Semicond. Process., 2020, 107, 104839 CrossRef CAS.
- S. Wu, X. Yu, J. Zhang, Y. Zhang, Y. Zhu and M. Zhu, Chem. Eng. J., 2021, 411, 128555 CrossRef CAS.
- X. Chen, C. Yu, R. Zhu, N. Li, J. Chen, S. Li, W. Xia, S. Xu, H. Wang and X. Chen, Nanomaterials, 2019, 9, 959 CrossRef CAS PubMed.
- X. Chen, C. Yu, R. Zhu, N. Li, J. Chen, Q. Lin, S. Xu, X. Chen and H. Wang, Sci. Total Environ., 2020, 711, 134643 CrossRef CAS PubMed.
- P. John, K. Johari, G. S. Nirmala, A. Appusamy and M. Thanabalan, Environ. Technol. Innovation, 2020, 22, 101412 CrossRef.
- M. Elangovan, S. M. Bharathaiyengar and J. P. Ettiyappan, Environ. Sci. Pollut. Res., 2021, 28, 18186–18200 CrossRef CAS PubMed.
- N. Li, W. Fu, G. Sun, M. Shi, M. Wu, W. Shen, Q. Li and J. Ma, Appl. Organomet. Chem., 2023, 37, e7170 CrossRef CAS.
- W. Zhang, L. Zhou, J. Shi and H. Deng, Catalysts, 2018, 8, 45 CrossRef.
- A. N. Oliveros, J. A. I. Pimentel, M. D. G. de Luna, S. Garcia-Segura, R. R. M. Abarca and R.-A. Doong, Chem. Eng. J., 2021, 403, 126213 CrossRef CAS.
- M. Cai, R. Li, Z. Xie, J. Huang, Y. Zeng, Q. Zhang, H. Liu, W. Lv and G. Liu, Appl. Catal., B, 2019, 259, 118033 CrossRef CAS.
- G. A. Ashraf, R. T. Rasool, R. U. Rasool, M. F. Saleem, J. Ali, D. Ghernaout, M. Hassan, A. M. Aljuwayid, M. A. Habila and H. Guo, J. Water Process Eng., 2023, 51, 103435 CrossRef.
- M. V. Krishna, G. Madhavi, N. F. Idris, S. A. M. Idris and L. R. K. Chowdary, Arabian J. Chem., 2019, 12, 1290–1297 CrossRef.
- F. Pomati, S. Castiglioni, E. Zuccato, R. Fanelli, D. Vigetti, C. Rossetti and D. Calamari, Environ. Sci. Technol., 2006, 40, 2442 CrossRef CAS PubMed.
- Z. Bensaadi, N. Yeddou-Mezenner, M. Trari and F. Medjene, J. Environ. Chem. Eng., 2014, 2, 1371–1377 CrossRef CAS.
- H. Yang, T. An, G. Y. Li, W. Song, W. J. Cooper, H. Luo and X. Guo, J. Hazard. Mater., 2010, 179, 834–839 CrossRef CAS PubMed.
- E. Hapeshi, A. Achilleos, M. I. Vasquez, C. Michael, N. P. Xekoukoulotakis, D. Mantzavinos and D. Kassinos, Water Res., 2010, 44, 1737–1746 CrossRef CAS PubMed.
- Z. Ran, L. Wang, Y. Fang, C. Ma and S. Li, Catalysts, 2019, 9, 876 CrossRef CAS.
- L. A. Ioannou, E. Hapeshi, M. I. Vasquez, D. Mantzavinos and D. Fatta-Kassinos, Sol. Energy, 2011, 85, 1915–1926 CrossRef CAS.
- D. A. Pino-Sandoval, L. Hinojosa-Reyes, J. L. Guzmán-Mar, J. C. Murillo-Sierra and A. Hernandez-Ramirez, Water, Air, Soil Pollut., 2022, 233, 17, DOI:10.1007/s11270-021-05484-7.
- L. Rimoldi, D. Meroni, E. Falletta, V. Pifferi, L. Falciola, G. Cappelletti and S. Ardizzone, Photochem. Photobiol. Sci., 2017, 16, 60 CrossRef CAS PubMed.
- A. Ponkshe and P. Thakur, Mater. Today: Proc., 2019, 18, 1162–1175 CAS.
- V. Rogé, C. Guignard, G. Lamblin, F. Laporte, I. Fechete, F. Garin, A. Dinia and D. Lenoble, Catal. Today, 2018, 306, 215–222 CrossRef.
- Y. Ji, L. Zhou, C. Ferronato, X. Yang, A. Salvador, C. Zeng and J.-M. Chovelon, J. Photochem. Photobiol., A, 2013, 254, 35–44 CrossRef CAS.
- C. Medana, P. Calza, F. Carbone, E. Pelizzetti, H. Hidaka and C. Baiocchi, Rapid Commun. Mass Spectrom., 2009, 23, 206 CrossRef CAS.
- Z. Mehrabadi and H. Faghihian, J. Photochem. Photobiol., A, 2018, 356, 102–111 CrossRef CAS.
- M. Eskandari, N. Goudarzi, M. A. Chamjangali and S. G. Moussavi, Journal of Sabzevar University of Medical Science, 2020, 27, 131–141 Search PubMed.
- B. Ramasamy, J. Jeyanthi and P. Chinnaiyan, Environ. Nanotechnol., Monit. Manage., 2023, 19, 100779 CAS.
- R. Bhuvaneswari, J. Jeyanthi and S. M. Kumar, Optik, 2021, 239, 166658 CrossRef.
- Y. Ling, G. Liao, Y. Xie, J. Yin, J. Huang, W. Feng and L. Li, J. Photochem. Photobiol., A, 2016, 329, 280–286 CrossRef CAS.
- Z. A. Alothman, A. Y. Badjah, O. M. Alharbi and I. Ali, Environ. Sci. Pollut. Res., 2021, 28, 7423–7430 CrossRef CAS PubMed.
- B. Bina, A. Fatehizadeh, E. Taheri, M. Heydari, M. Darvishmotevalli and A. Bazmeh, Int. J. Environ. Anal. Chem., 2022, 1–12, DOI:10.1080/03067319.2022.2085045.
- S. Stojanović, M. Vranješ, Z. Šaponjić, V. Rac, V. Rakić, L. Ignjatović and L. Damjanović-Vasilić, Int. J. Environ. Sci. Technol., 2023, 20, 1–16 CrossRef.
- R. Corchero, R. Rodil, A. Soto and E. Rodil, Nanomaterials, 2021, 11, 411 CrossRef CAS PubMed.
- Y. Shi, H. Chen, Y. Wu and W. Dong, Environ. Sci. Pollut. Res., 2017, 25, 693–703 CrossRef PubMed.
- V. Pistková, M. Tasbihi, M. Vávrová and U. L. Stangar, J. Photochem. Photobiol., A, 2015, 305, 19–28 CrossRef.
- V. Bhatia, G. Malekshoar, A. Dhir and A. K. Ray, J. Photochem. Photobiol., A, 2017, 332, 182–187 CrossRef CAS.
- V. Bhatia, K. Manoli, A. Dhir and A. K. Ray, Can. J. Chem. Eng., 2020, 98, 1767–1775 CrossRef CAS.
- V. Bhatia, A. Dhar and A. K. Ray, J. Photochem. Photobiol., A, 2021, 409, 113136 CrossRef CAS.
- N. F. F. Moreira, M. J. Sampaio, A. R. Ribeiro, C. G. Silva, J. L. Faria and A. M. T. Silva, Appl. Catal., B, 2019, 248, 184–192 CrossRef CAS.
- Y. Zhang, S. Yang, L. Wei, X. Zhao and X. Lu, Environ. Technol., 2024, 45, 972–987 CrossRef CAS PubMed.
- A. Kumar, A. Kumar, G. Sharma, M. Naushad, T. Ahamad and F. J. Stadler, Chemosphere, 2018, 209, 457–469 CrossRef CAS PubMed.
- Y. Wang, J. Niu, X. Gao and Y. Zhang, Appl. Surf. Sci., 2020, 533, 147458 CrossRef.
- M. R. Rajeshwari, A. Syed, A. H. Bahkali, A. M. Elgorban, M. K. Rahiman, R. S. Varma and S. S. Khan, J. Ind. Eng. Chem., 2022, 115, 402–415 CrossRef CAS.
- A. H. Shah and M. A. Rather, Adv. Nano Res., 2021, 10, 397–414 Search PubMed.
- S. Sun, X. Yu, Q. Yang, Z. Yang and S. Liang, Nanoscale Adv., 2019, 1, 34–63 RSC.
- Photocatalytic Functional Materials for Environmental Remediation, ed. A. Pandikumar and K. Jothivenkatachalam, 2019, Wiley Search PubMed.
- A. B. D. Nandiyanto, R. Zaen and R. Oktiani, Arabian J. Chem., 2020, 13, 1283–1296 CrossRef CAS.
- Z. Cui, L. Zhang, Y. Xue, Y. Feng, M. Wang, J. Chen, B. Ji, C. Wang and Y. Xue, Int. J. Miner., Metall. Mater., 2022, 29, 2221–2231 CrossRef CAS.
- Z.-H. Diao, J.-C. Jin, M.-Y. Zou, H. Liu, J.-Q. Qin, X.-H. Zhou, W. Qian, P.-R. Guo, L.-J. Kong and W. Chu, Sep. Purif. Technol., 2021, 278, 119620 CrossRef.
- L. Han, B. Li, H. Wen, Y. Guo and Z. Lin, J. Mater. Sci. Technol., 2021, 70, 176–184 CrossRef CAS.
- E. Omrani, A. Ahmadpour, M. Heravi and T. R. Bastami, J. Water Process Eng., 2022, 47, 102581 CrossRef.
- A. Majumder, A. K. Gupta and M. Sillanpää, Colloids Surf., A, 2022, 648, 129250 CrossRef CAS.
- S. Das, M. Sanjay, S. Kumar, S. Sarkar, C. S. Tiwary and S. Chowdhury, Chem. Eng. J., 2023, 476, 146719 CrossRef CAS.
- T. Zhu, J. Yang, Y. Shen, T. Liang, S. Fan, S. Zhang, Z. Yu, S. Wang and Y. Hou, Colloids Surf., A, 2024, 681, 132802 CrossRef CAS.
- M. Wang, B. Liang, S. Lin, Z. Zhao, H. He, X. Li and S. Liang, Appl. Surf. Sci., 2024, 657, 15979 Search PubMed.
- C. Yang, X. Zhang, Z. Li, Z. Wang, J. Shi, H. Xie, T. Li and Y. Zhou, Mater. Res. Bull., 2024, 174, 112709 CrossRef CAS.
- Y. Xia, H. Liu, F. Sun, B. Yue, X. Wang, F. Guo, Y. Zhu, H. Yu, G. Liu, W. Yu and X. Dong, J. Cleaner Prod., 2024, 434, 140445 CrossRef CAS.
- J. J. Rueda-Marquez, I. Levchuk, P. F. Ibañez and M. Sillanpää, J. Cleaner Prod., 2020, 258, 120694 CrossRef CAS.
- C. Blaise, F. Gagné, J. F. Férard and P. Eullaffroy, Environ. Toxicol., 2008, 23, 591–598 CrossRef CAS PubMed.
- N. B. Turan, H. S. Erkan, G. O. Engin and M. S. Bilgili, Process Saf. Environ. Prot., 2019, 130, 238–249 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3lf00142c |
| This journal is © The Royal Society of Chemistry 2024 |