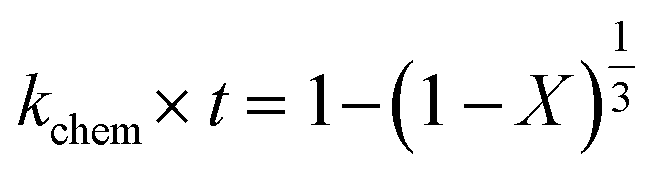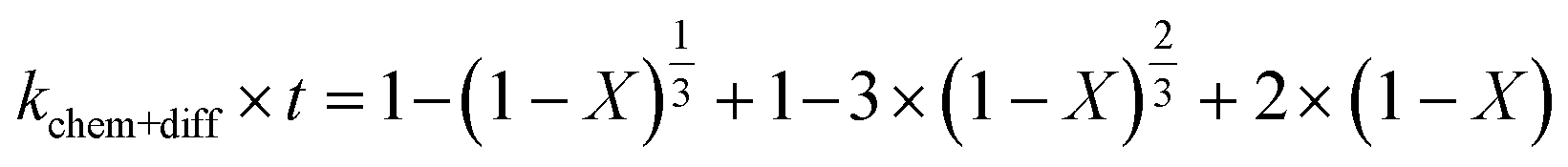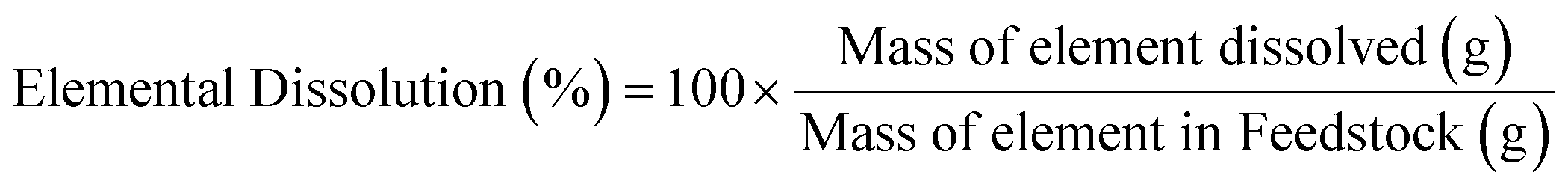DOI:
10.1039/D4IM00025K
(Paper)
Ind. Chem. Mater., 2024, Advance Article
Carboxylic ligands to enhance material recovery from construction waste to produce CaCO3 for carbon utilization†
Received
2nd March 2024
, Accepted 26th April 2024
First published on 3rd May 2024
Abstract
The decarbonization of the built environment is a pressing issue to achieve CO2 reduction targets in the concrete industry. Carbon mineralization of construction and demolition waste (C&DW) is an attractive pathway to capture of CO2 as stable carbonates which can be re-utilized and upcycled in a circularized fashion through the creation of new building blocks. Material recovery from the C&DW is often performed in hydrometallurgical leaching using acidic media; however, this process is often hindered by solubility issues and passivation. To ensure high recoveries of these elements, ligands can be used to enhance dissolution. Carboxylic acids are used in conventional hydrometallurgical mineral processing, such as leaching, floatation, and solvent extraction, and are desired due to their affordability and stability. In this study, we explore the dissolution of waste cement pastes in acidic conditions under the presence of four carboxylic acid ligands: formate, acetate, glutamate, and citrate. The leaching kinetics are categorized and the pseudo-rate constants are established, demonstrating the advantages of these agents to enhance reaction rates in the general order of citrate ⋙ formate > acetate > glutamate > control. The characterization of the post-extraction reactor residue (PERR) revealed a significant increase in Si-content. Finally, the leachate was carbonated to produce calcium carbonate, which was characterized for its use based on morphology and size. Glutamate demonstrated distinct advantages compared to other ligands, with a dual function of not only improving leachability of cement but promoting and stabilizing vaterite during crystallization. Overall, this study motivates the use of sustainable ligands to enhance material recovery during the dissolution of alkaline wastes for carbon mineralization.
Keywords: Carbon mineralization; Material recovery; Leaching; Ligands; Carboxylic acids; Calcium carbonates; Carbon capture utilization and storage.
1 Introduction
1.1 Material recovery from built environment wastes for decarbonization and upcycling potential
The utilization of alkaline wastes rich in Ca and Mg (e.g., steel slag, mine tailings, construction debris, etc.) is posited to contribute to a significant portion of ex situ carbon mineralization as a method of carbon dioxide removal (CDR).1–4 This is especially relevant for the built environment, where 25% of construction and demolition waste (C&DW) is directly landfilled without further processing (U.S.);5 by 2050, estimates report that global C&DW waste could reach 27 billion tonnes.6 On-site sorting and pre-processing of the C&DW will allow for enhanced downstream separation and will ensure different fractions of waste can reach their upcycling centers at lower CO2-footprints.7 Previous studies have demonstrated that the dissolution of waste HCP to recover elements such as Ca, Si, Al, and Fe is a viable method to sequester carbon in the form of calcium carbonates and produce upcycled materials.8,9 This aqueous dissolution and carbonation process could sequester between 100–150 kg CO2 per tonne of C&DW waste; however, this could in fact be lower when considering the distribution, shipping, milling, and comminution carbon impacts.6,9 The replacement of clinker through upcycled materials, such as calcium carbonates and recovered silica residues, could reduce the CO2-burden of cement without compromising the structural integrity of the materials.10 Recent works by Zhao et al. have highlighted the ability for the three anhydrous polymorphs of CaCO3 (vaterite, aragonite, and calcite) produced from the dissolution of waste cement pastes to replace clinker and enhance its hydration during curing.11,12 Additionally, numerous studies have demonstrated that the use of silica-bearing compounds from industrial wastes such as fly ash, steel slag, and other alternatives to fumed silicon could be used as pozzolanic SCMs in new cement mixtures.13–17 The tandem capture of CO2 and production of new built environment materials through hydrometallurgical carbon mineralization processes offers additional advantages which motivates the implementation of carbon capture and utilization (CCU).18
1.2 Carboxylic ligands for enhanced material recovery from alkaline wastes during carbon mineralization
During conventional hydrometallurgical processes involving elemental and metal recovery, complexing agents and ligands are often used to enhance extraction and recoveries.19,20 The use of ligands during aqueous dissolution can also reduce the necessity for pyrometallurgical roasting or smelting, which can lower the overall environmental impact of such processes.21 Classically, cyanide (CN–) has been utilized in gold leaching to form water soluble cyanide salts; however, due to cyanide's environmental toxicity, thiourea (CH4N2S) and thiosulfate (S2O3) are being studied as alternative ligands.22–24 Carboxylic acids and carboxyl derivatives can also be utilized during dissolution to recover valuable metals, such as ethylenediaminetetraacetic (EDTA), oxalic acid, citric acid, malic acid, and nitrilotriacetic acid (NTA).25–27 In certain cases, these organic species are preferable to their inorganic counterparts (e.g., cyanide, ammonia), due to their reduced toxicities. Carboxyl species are also utilized during other hydrometallurgical processes, such as flotation, solvent extraction, and precipitation. Fig. 1 demonstrates the wide variety of uses for these carboxylic species across a wide variety of relevant processes.
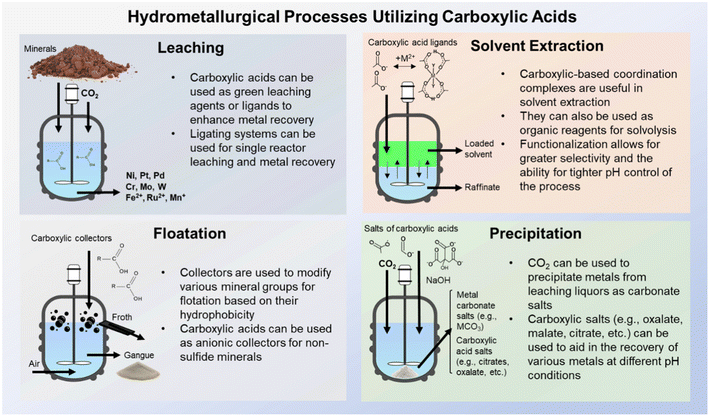 |
| | Fig. 1 Schematic demonstrating classical hydrometallurgical processes which utilize carboxylic chemistry including, but not limited to, leaching, solvent extraction, flotation, and precipitation. | |
Renewed interest into the utilization of CO2 as a green solvent and source of carbonate ions for hydrometallurgical precipitation has occurred, especially in the context of carbon mineralization to capture CO2 itself and to recover energy critical metals, rare earth elements (REEs), and platinum group metals (PGMs) from waste sources.28–31 Carboxylic acids and carboxylic acid derivates can be readily produced through bio-based, electrochemical, and thermochemical processes and are currently utilized in a myriad of industries.32,33 The simplest carboxylic acids, formic and acetic, are mostly produced through reactions of methanol and carbon monoxide, both of which stem from syngas produced from methane reformers.34,35 Formic acid is generally produced from the hydrolysis of methyl formate while acetic acid is produced through the well-known Monsanto or Cativa processes.34 Useful higher order carboxylic acids with large market shares include citric acid, which is mainly produced from fermentation systems using certain fungi, such as Aspergillus niger.36 Certain amino acids also contain valuable carboxylic acid groups, such as glutamic acid, unique as it is one of two amino acids with a negatively charged side chain. Similar to citric acid, it is primarily produced via fermentative bioprocesses.37 Unfortunately, the current carbon footprint of these carboxylic acids is quite high, and thus the net carbon impact of applying them to carbon mineralization methods today is not highly beneficial. However, with the growth of CO2 to chemicals and CO2 utilization, the production of these species in a more renewable fashion could be attainable in the future. For instance, formic acid can be produced via the thermochemical hydrogenation of CO2 or the electrochemical reduction of CO2 in aqueous electrolytes.38,39 Catalysts can also be utilized to convert CO2 into useful molecules, such as cyclic carbonates.40,41 These thermo/electrochemical methods are more popular for lower order carboxylic acids (e.g., formic vs. decanoic acid) due to decreased C–C bond formation required and enthalpy considerations.42
Aqueous dissolution generally occurs at low pHs to ensure protons can hydrolyze metals from the mineral particles via metal oxide (M–O) bond cleavage; moreover, most mineral and industrial wastes are highly alkaline due to the abundance of Mg and Ca hydroxides/carbonate species.43 While there are a variety of previous studies examining the effect of carboxylic ligands, especially citrate, on the recovery of metals from industrial wastes, these are generally limited to electronic residues, spent batteries, slags, fly ash, and certain minerals.44–48 The target elements in these studies include more monetarily valuable species, such as Cu, Pb, Fe, Ni, and even rare earth elements (REEs).46,49–51 There are limited studies in the literature focused on the utilization of carboxylic ligands with respect to Ca-rich heterogenous alkaline wastes, such as hydrated waste cement paste.52 Additionally, these analyses are generally focused only on the chemistry and metal recovery during leaching, and do not consider the full process engineering production of CaCO3 for CCUS applications which follows through multiple process steps. These coordinating molecules not only have interesting properties in terms of leaching, but they may also affect the crystallization of CaCO3 during carbon mineralization downstream as surface modifiers or nucleation inhibitors. Thus, the combined and systematic use of these materials for both leaching and crystallization control within carbon mineralization processes for carbonate formation is both timely and needed.
In this study, we examine the dissolution of waste HCP from the built environment in acidic media in the presence of four representative carboxylic acid salts, formate, acetate, glutamate, and citrate, chosen for their respective industrial relevance and current production market share. The main functionalities which contribute to their differences arise from their protonation potentials at different pHs, as show in Fig. 2. For formate and acetate, one proton is available, as represented by a single pKa being present between 4.2–4.8. Glutamate has two carboxyl groups and one amino group whereas citrate has three carboxyl groups, both species with three available groups for deprotonation. Glutamate can exist as a zwitterion over the majority of the studied pH range. The leaching behavior, kinetics, and reaction rates were analyzed for all four ligands during the dissolution of the waste cement paste. After leaching, the post-extraction reactor residue (PERR) was characterized for its properties and a pH swing was employed to precipitate out non-Ca ions in the solution from pH 3–9. These precipitated metal oxides (PMOs) were characterized for their morphology and elemental composition to assess their potential uses. Finally, the dissolution liquor was reacted with CO2 to produce calcium carbonates and assess the degree of CO2 capture which is afforded by the enhanced material recovery using these carboxylic ligands.
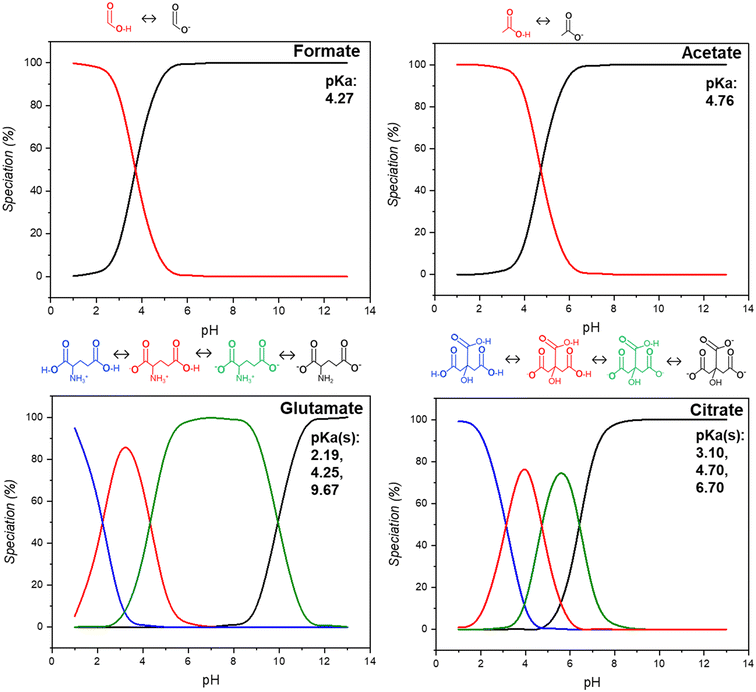 |
| | Fig. 2 Speciation and calculated pKa(s) for salts of formate, acetate, glutamate, and citrate using visual MINTEQ at different pH values. Inserts show the removal of protons from the structures of the molecules at different pKa(s). | |
2 Results and discussion
2.1 Kinetic analysis of ligand-assisted leaching of waste hydrated cement pastes
2.1.1 Leaching kinetics. Fig. 3a–d shows the leaching kinetics over the course of the dissolution experiment for calcium (Fig. 3a), silicon (Fig. 3b), aluminum (Fig. 3c), and iron (Fig. 3d). For Ca removal, the use of the carboxylic ligands, formate, acetate, glutamate, and citrate, greatly enhanced the initial rate of reaction, with citrate reaching near Ca-saturation at approximately 10 min (Fig. 3a). Although these chemicals enhanced the initial rate of Ca extraction, they all reached the same percentage of extraction at 60 min (70–72%). More pronounced extraction behavior was seen in the case of Si, Al, and Fe, where these carboxylic ligands were able to not only enhance the initial rate of extraction, but also the total amount of material extracted. Interestingly, citrate was able to promote the dissolution of up to 4-fold more Si and 95-fold more Fe than HCl alone. Overall, the trend of extraction improvement and material yield was broadly seen as citrate ≫ formate > acetate > glutamate > hydrochloric acid alone. The use of these ligands at low pH conditions (pH 3) required additional HCl addition to ensure full protonation of the ligand (Fig. 2) and bypassing of the buffer regions present. The highest elemental molar recovery per mol of organic ligand was achieved in Ca, followed by Si, then Al and Fe, which tracks with the observed elemental recoveries on a feedstock basis (Table S1†). Compared to Hong et al. (2023), who conducted a similar study with carboxylic ligands during the dissolution of iron slag, a greater increase in the ancillary elements of Si, Fe, Al, was observed herein than during their study.53 Different feedstock compositions, heterogeneities, and crystallinities are likely the result of these differing observations. Additionally, the cooling and quenching rate of slag can have profound impacts on its crystallinity, which in turn can impact waste valorization operations.53,54 Cement paste is hydrated and processed at room temperature (25 °C), yielding different crystallinities which in turn, can yield different leaching potentials of certain elements.55 Increased research is necessary to probe at the effects of carboxylic ligands on a wide variety of alkaline wastes to realize their full potential.
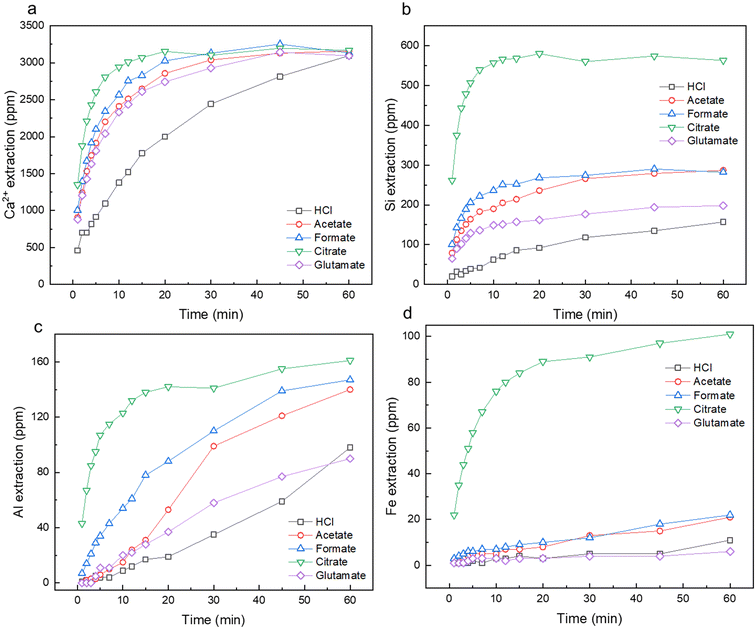 |
| | Fig. 3 Leaching kinetics as measured by ICP-OES for (a) Ca2+; (b) Si; (c) Al; and (d) Fe for HCl, acetate, formate, citrate, and glutamate leaching cases over the course of 60 min extraction. | |
Table 1 shows the calculated calcium-to-silicon (Ca2+/Si) molar ratios at equilibrium for the leaching experiments compared to raw HCP itself. This ratio is an important metric which has been previously used to establish the type of dissolution occurring during the hydrometallurgical processing of mine tailings and Ca and Mg-rich waste minerals.6,9,56,57 In this study, the Ca2+/Si ratio for all cases was higher than the ratio in the starting material, which is attributed to incongruent dissolution of Ca with respect to Si. It is also attributed to the formation of a Si-rich passivation layer on the dissolving particles.9 Incongruent dissolution is widely observed in the aqueous treatment of alkaline materials due to differences in solubility of elemental species, especially Ca with respect to Si. Interestingly, the incongruency of dissolution decreases relative to acid only leaching when carboxylic ligands were used. Citrate had the lowest Ca2+/Si ratio of 5.6 compared to 19.6 in the case of acid only, meaning that more Si was extracted from the cement particles with the use of the ligands. A likely explanation is that increased solvation of silicon and/or the stabilization of pores within the cement matrix during dissolution. Similar behavior has been previously reported in the processing of Mg-rich serpentine waste by Swanson (2014), who utilized oxalate in combination with catechol to stabilize collapsing pores, leading to more congruent dissolution behavior.58 The Si dissolution kinetics (Fig. 3b) also demonstrate that no or limited reprecipitation of the extracted Si is occurring on the timescale of reaction, behavior which was observed by Zhang and Moment (2023) in the dissolution of waste concrete.8 The increased congruency in dissolution afforded by these ligands in the material extraction from waste HCP could be useful to extract as much material as possible.
Table 1 Calcium-to-silicon (Ca2+/Si) ratio at equilibrium after the leaching process had concluded compared to the original hydrated cement paste feedstock
| |
Ca2+/Si ratio |
| HCP feedstock |
2 |
| HCl only |
19.7 |
| Glutamate |
15.6 |
| Acetate |
11.0 |
| Formate |
11.1 |
| Citrate |
5.6 |
2.1.2 Pseudo-rate constants obtained from hydrometallurgical processing. Apparent or pseudo-rate constants were calculated by fitting the kinetic data obtained to three different shrinking-core reaction models.59,60 The shrinking-core reaction model has been widely used to describe the dissolution of alkaline wastes during leaching processes. Recently, these models were utilized by our group to describe the leaching of waste concrete, fly ash, and the same hydrated cement pasted used herein.6,8,61 Generally, the dissolution of these alkaline waste particles can adhere to a diffusion controlled (eqn (1)), chemical controlled (eqn 2), or a combined chemical and diffusion reaction control regime (eqn (3)). In this context, diffusion-controlled processes are limited by the continued diffusion of ions or reactants within or near the cement particles in the aqueous media. To better ascertain details about the reactivity of these ligands with respect to the cement particles, the kinetic data was fit to the three shrinking-core models and the closeness-of-fit (R2) values were established.The early behavior of the leaching data (<10 min) was fit to eqn (1)–(3), which relate the reaction time t (s) and the apparent rate constant kd (s−1) to the elemental extraction X (percent extraction, %) of Ca, Si, Fe, and Al:8
| |
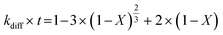 | (1) |
| |
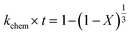 | (2) |
| |
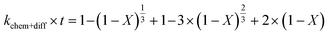 | (3) |
The closeness of fits (R2) from the linearized data fit to all three reaction models is shown in Table 2. Generally, most of the obtained results fit a diffusion-controlled dissolution process, which is consistent with previous studies. It should be noted that this is diffusion-limited reaction regime in reference to the diffusion through and within the solid cement paste and no liquid–liquid dissolution or the diffusion of species within the bulk. Fig. 4a demonstrates an example of this fitting using the diffusion-controlled model (eqn (1)) with the calcium dissolution kinetics over the desired time range of about 420 s (7 min). Shorter time frames are utilized for the kinetic analysis to avoid the saturation region of mass transfer limitations which generally occurs after 10–15 min of dissolution. Ca and Si extraction fit this model the best of all the elements profiled, whereas there are slight deviations in the case of Al and Fe. While part of this could be due to higher error associated with these elements which are present in less than 3% of the starting cement feedstock, it could also be indicative of additional factors controlling the extractive processes of Al and Fe. For the case of citrate, the affinity of iron and aluminum towards citrate is quite high; thus, increased solubility of the formed complexes will have strong implications for the apparent rate constants. In the case of Al, the leaching kinetics suggest that saturation is not reached within the dissolution time tested whereas the high affinity of iron towards citrate and its increased solubility is the main mechanism active in the Fe leaching kinetics. Additional studies using these same ligands with other alkaline wastes (e.g., steel slag, waste to energy ashes, etc.) that contain greater amounts of Al and Fe should be conducted to provide additional mechanistic insights.
Table 2 Best of fit R2 values for the kinetic data obtained for Ca, Si, Fe, and Al in the presence of the four ligands studied. The data was fit to models describing chemical control, diffusion control, and a combination of the two
| Best of fit (R-value) |
| Element of interest |
Ca |
| Ligand |
Control |
Acetate |
Formate |
Citrate |
Glutamate |
| Chemical reaction control |
0.979 |
0.990 |
0.984 |
0.966 |
0.989 |
| Diffusion control |
0.985 |
1.000 |
0.997 |
0.985 |
0.999 |
| Chemical + diffusion control |
0.983 |
0.996 |
0.991 |
0.977 |
0.995 |
| Element of interest |
Si |
| Ligand |
Control |
Acetate |
Formate |
Citrate |
Glutamate |
| Chemical reaction control |
0.891 |
0.968 |
0.959 |
0.934 |
0.958 |
| Diffusion control |
0.913 |
0.991 |
0.983 |
0.963 |
0.978 |
| Chemical + diffusion control |
0.892 |
0.973 |
0.965 |
0.945 |
0.961 |
| Element of interest |
Fe |
| Ligand |
Control |
Acetate |
Formate |
Citrate |
Glutamate |
| Chemical reaction control |
N/A |
0.942 |
0.965 |
0.984 |
0.910 |
| Diffusion control |
N/A |
0.942 |
0.982 |
0.999 |
0.905 |
| Chemical + diffusion control |
N/A |
0.943 |
0.966 |
0.991 |
0.909 |
| Element of interest |
Al |
| Ligand |
Control |
Acetate |
Formate |
Citrate |
Glutamate |
| Chemical reaction control |
0.776 |
0.972 |
0.996 |
0.987 |
0.909 |
| Diffusion control |
0.696 |
0.898 |
0.990 |
0.995 |
0.883 |
| Chemical + diffusion control |
0.773 |
0.969 |
0.999 |
0.993 |
0.908 |
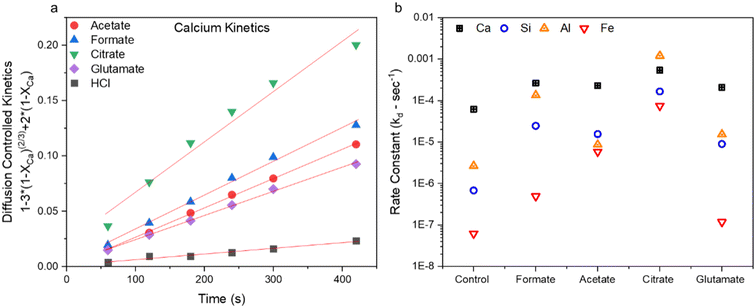 |
| | Fig. 4 (a) Example of the data fitting using the calcium extraction kinetics up to about 7 min for all five test cases using the diffusion-limited model and (b) calculated rate constants (kd) for diffusion-controlled shrinking-core reaction regime Ca, Si, Al, and Fe dissolution from the waste hydrated cement paste (HCP). Control case refers to HCl dissolution only. | |
Fig. 4b shows the rate constants for dissolution as calculated for the five leaching cases for Ca, Si, Al, and Fe. Correspondingly, it can be observed that the increase in rate constants follows a similar trend observed in the kinetic data shown in Fig. 3a–d, where elemental extraction rates for citrate ≫ formate ∼ acetate > glutamate > HCl only. Based on this analysis, it is clear that thermodynamics is dominating the enhancement effects observed by the carboxylic ligands in these reactions. Utilizing these chemicals has altered the apparent solubilities vis-à-vis the observed apparent rates. Calcium extraction rates were more or less similar between the species; however, in the case of citrate Fe rates were increased by 4 orders of magnitude (OOM), Al by 3 OOM, and Si by 2 OOM. Based on kinetic data, apparent rates, and the particle size data (section 3.2) obtained from the samples after the reaction, it is theorized the improvement in the rate of reaction offered for Ca is present at the surface of the dissolving particles, since the Ca extent of extraction is the same for all ligands within the reaction timeframe. Citrate-Ca enhancement could also be a function of 3× greater presence of ligating groups with respect to the other ligands studied. In the case of Si, it seems that there are different Si-ligand species coming to different equilibriums over the course of the dissolution reaction. In reference to the Al and Fe, the ligand enhancement in extraction is likely a result of increased solubilization of these poorly soluble ions, increasing their concentration in the bulk and preventing their reprecipitation of the dissolving particles. Al seems to be pore-controlled, as the kinetics never fully reach saturation. These carboxylic ligands may provide benefits at scale, when mass transfer and solubility limitations can become more pronounced.
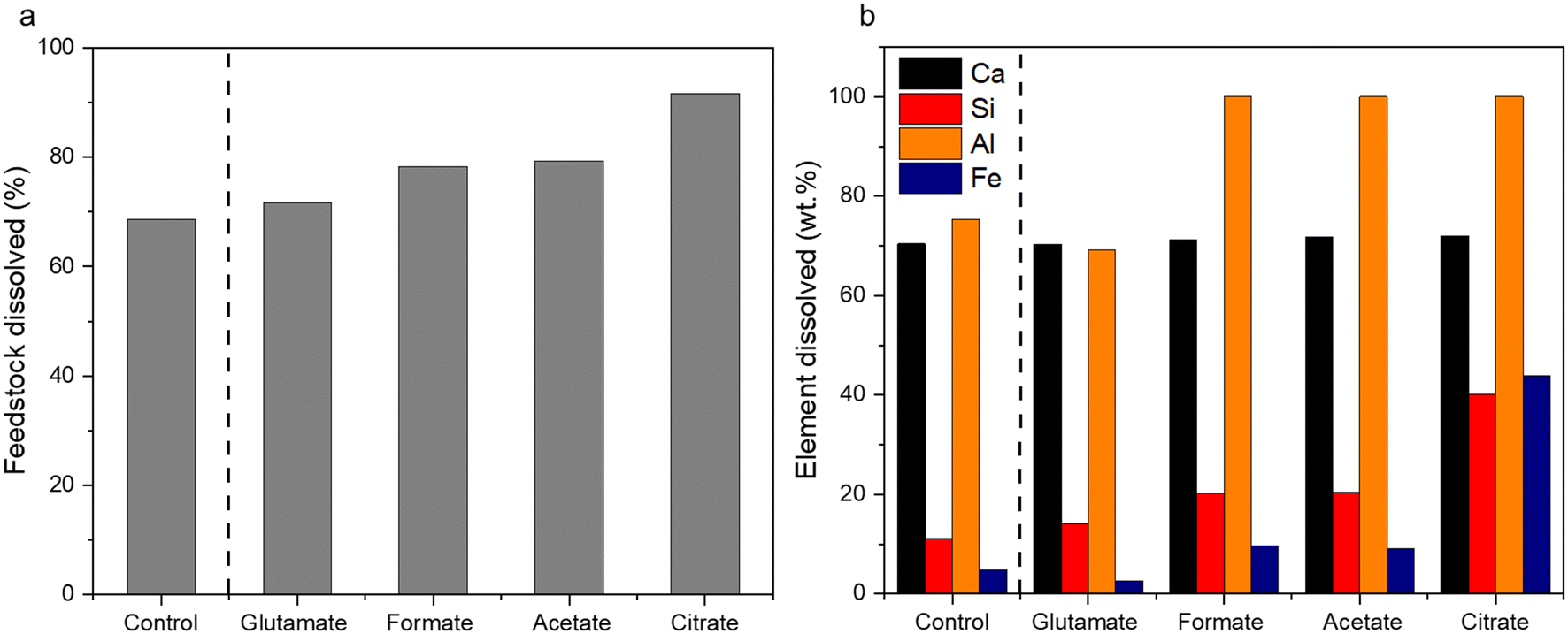
2.2 Characterization of the post-extraction reactor residue (PERR)
The characterization of the unreacted residue accumulated during the single pass leaching provided insights into the dissolution behavior of the hydrometallurgical processing system in addition of the potential downstream uses of these materials. Fig. 5a shows the percentage of feedstock dissolved compared to HCl only and Fig. 5b shows the individual elemental dissolution percentages as a function of the elemental mass of the feedstock itself calculated by eqn (4):| |
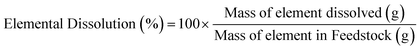 | (4) |
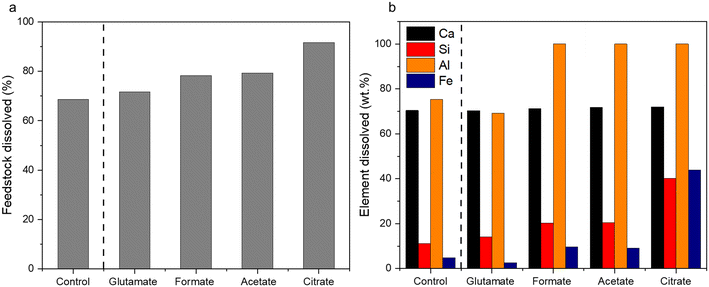 |
| | Fig. 5 (a) Total feedstock dissolved and (b) elemental dissolved mass balance for the carboxylic aid ligands after the hydrometallurgical leaching of waste HCP. | |
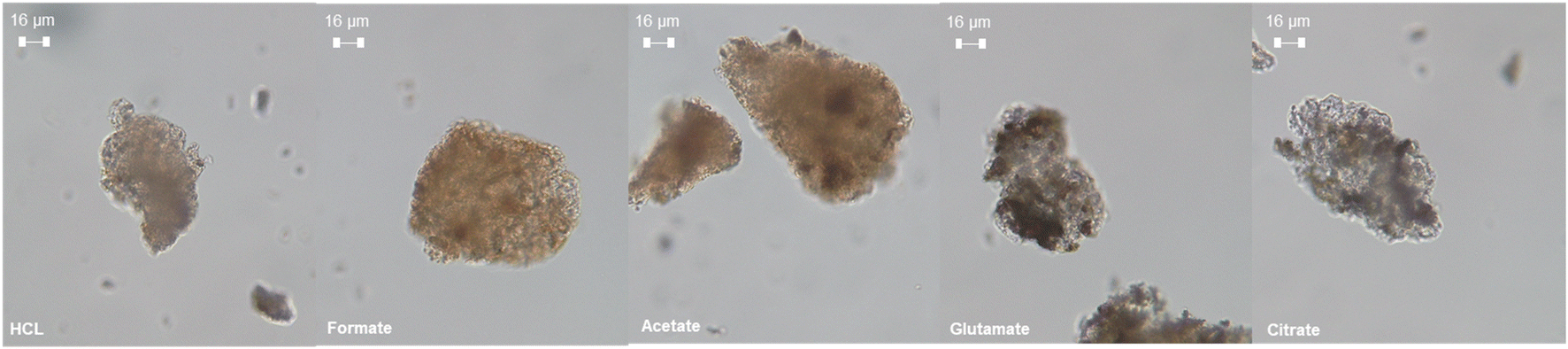
The ligands all enhanced the solubilization of the bulk material itself, with citrate leaving only 8% of the cement remaining in the reactor (Fig. 5a). As demonstrated earlier, the total recovery of calcium was not enhanced much by these salts; however, significant improvements in Fe, Al, and Si total yield were observed, especially in the case of acetate and citrate. XRF analysis of the PERR complemented the mass balance results, showing an increase in SiO2 content and decrease in CaO content for all the samples tested with respect to the original cement itself (Fig. 6). Increased Fe2O3 was observed in the acetate, formate, and glutamate PERR as shown by XRF relative to the acid only. This could be due to the reprecipitation of certain iron species on the undissolved particles during leaching. Visually, the PERR in these three cases looked red compared to the raw feedstock and acid only cases (Fig. S1†), alluding to the presence of iron. Optical microscopy provided additional visual insights into the characteristics of the PERR (Fig. 7 and S2†). In the case of the ligand PERR, the particles appeared smaller and more fragmented that the HCl control (Fig. S2†). The acetate and formate PERR had a red hue due to the presence of iron species, while the glutamate and citrate PERR looked translucent (Fig. 7), likely a result of the high elemental extraction from the dissolved cement matrix.
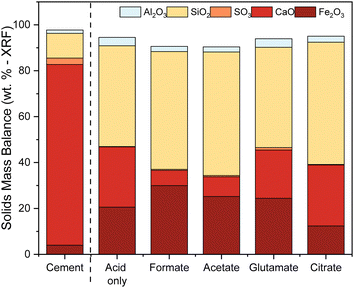 |
| | Fig. 6 XRF solids mass balance of the PERR showing the remainder of the residual elements present after leaching. | |
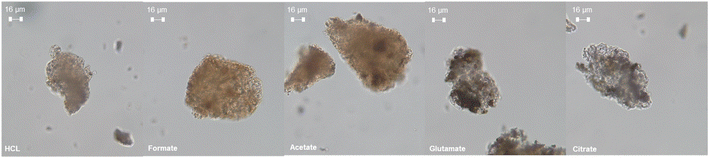 |
| | Fig. 7 Optical microscopy at 40× magnification of the PERR samples after the dissolution process. | |
Sulfur was detected (SO3) in the HCP sample, present mainly as ettringite, which originated from the reaction of gypsum (CaSO4·2H2O) with aluminates in cement.62 No or minimal sulfur was detected in the PERR residues, suggesting that the reaction conditions were adequate to dissolve the ettringite into free sulfate. Previous studies with waste HCP at high slurry densities revealed a reprecipitation of gypsum on the PERR itself, but at low slurry densities sulfate was not observed.6 FTIR data of the PERR revealed similar spectral patterns for all five leaching cases (Fig. 8a). Notably, the disappearance of the OH−1 peak at about 3630 cm−1 from the waste HCP was observed. Additionally, the formation of increased Si–O–Si bonding can be seen through the shift (condensation) of the silanol (Si–O) peak from 950 cm−1 in the raw HCP to about 1030 cm−1 in the PERR samples.63–65 This peak shift has been attributed to the breakage of lower energy Fe–O–Si and Al–O–Si bonds compared to SiO2 bonding, allowing for the condensation of silanol groups after removal of other ions, such as Fe and Al.8 The peak shift in this region is the most pronounced for citrate, which is supported by the observed mass balance and kinetic data. Correspondingly, the particle size data from the PERR (Fig. 8b) shows a similar trend, with particle size reduction following the order of citrate ≫ formate ∼ glutamate ∼ acetate > acid only ≫ raw HCP. The increased reduction in PSD, especially for citrate, supported the observed mass of the residue obtained, with very little remaining.
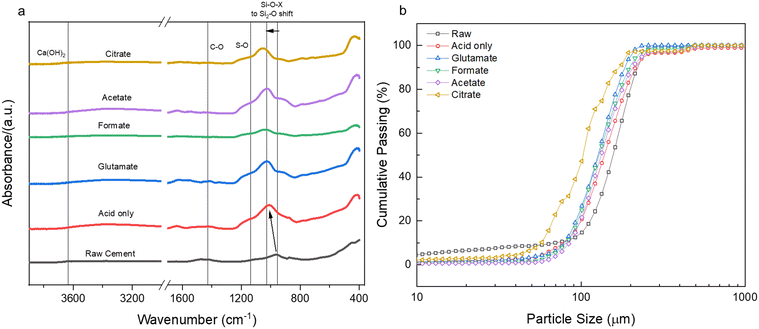 |
| | Fig. 8 (a) FTIR spectral data for the post-extraction reactor residue (PERR) for the five leaching cases studied in comparison to the raw cement paste; (b) cumulative passing particle size distribution (PSD) of the same PERR. | |
2.3 pH-Swing to concentrate ancillary ions present in the leach liquor
Following the dissolution reactions, a pH swing process was employed to precipitate certain species, such as Al, Si, and Fe, which would otherwise contaminate the PCC downstream during carbonation.61 Fig. 9 shows the titration data for the disappearance of Ca, Si, Fe, and Al as a function of increasing pH through NaOH addition. Generally, as the pH increased, the precipitation of Fe and Al species was hindered through the presence of carboxylic ligands. Citrate was the most resistant towards the precipitation of Fe and Al, followed by glutamate, acetate, and formate. The amount of NaOH used for titration also scaled with the potential strength and degree to which the ligand was able to recover material during leaching (Fig. S3†). This is likely due to the increased concentration of ions present in the leaching liquor, especially Si, Al, and Fe, which will require additional base to titrate out. Formate and acetate actually assisted in the precipitation of Si, even at high pHs where it would be thermodynamically more soluble.66
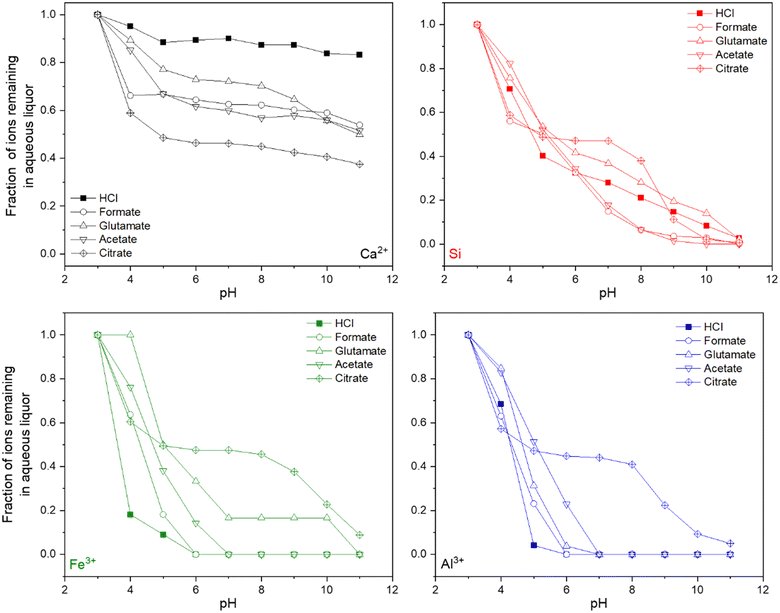 |
| | Fig. 9 ICP-OES data at various pH points for Ca, Si, Fe, and Al during the pH-swing process for different carboxylic salts used during the dissolution of the HCP. | |
Unfortunately, the presence of the carboxylic ligands also allowed for the loss of calcium from the aqueous phase, which was upwards of 60% in the presence of citrate. This behavior could be due to the precipitation of the corresponding calcium salt of the ligand material (i.e., calcium acetate, calcium citrate, etc.) or increased gelation of Si–Fe–Al hydroxides due to higher absolute concentrations (Fig. 3b–d) which precipitate and can trap calcium within via an occlusive process.61 While citrate allows for enhanced material recovery during leaching due to its strength as a chelator, its downside is the subsequent trapping of ions during PMO precipitation. Swinging the pH to as high as 12 could potentially aid in fully precipitating the citrate salts, however a fraction of calcium will also be lost. In a similar study assessing PMO recovery from iron slag leaching, Hong et al. (2023) report similar losses of calcium from the aqueous phase during a pH-swing processing using NaOH;53 thus, sequential PMO redissolution, purification, and precipitation to recover wash out excess calcium might be warranted. This behavior is not seen to the same extent in the cases of formate, acetate, and glutamate, demonstrating that for the production of high-purity carbonates for paper making, food and beverage, and paint applications, other ligands could be more advantageous. However, low-purity carbonates can be produced in high yields even if a PMO-precipitation is not employed and these materials can still have use within the built environment or for long-term carbon storage as carbonates.9 Al, Fe, and Si can act as supplementary cementitious materials (SCMs) and thus there may even be benefits for their incorporation within carbonates for CCU applications.67,68
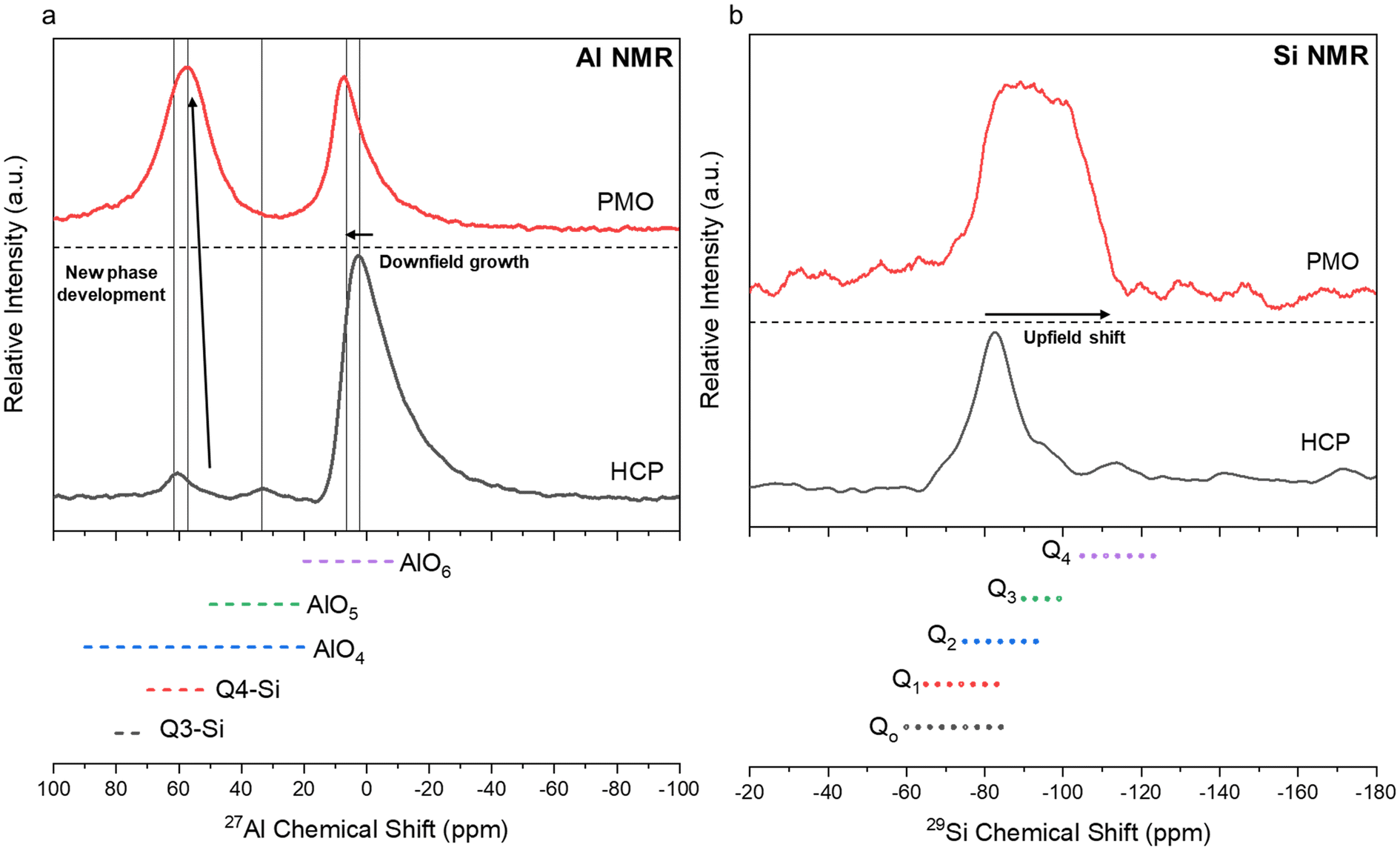
Characterization of the PMO revealed much similarity between the precipitates from each ligand case studied. All of the PMO samples had roughly the same composition of Ca, Al, Si, and Fe present as shown by XRF analysis (Fig. 10a) and were amorphous, as confirmed by Q-PXRD (Fig. 10b). Distinct differences in morphology were observed between the PERR, which after leaching appeared quite porous, and the PMO which looked quite smooth in all cases studied (Fig. S4†). 27Al (Fig. 11a) and 29Si (Fig. 11b) MAS solid-state NMR was performed on the control case PMO to assess its phase differences compared to the raw HCP, specifically in the changes of the Al and Si bonding networks. The raw HCP shows a large peak at 4.9 ppm in the 27Al spectra which could be correlated with the presence of an Al–O5/6 bonding regime, akin to those found in the tricalcium aluminate (celite) and tetracalcium aluminoferrite (felite) clinker phases.69,70 The PMO contained a slightly downshifted version of this same peak (4.9 shifted to 9.9 ppm) and the growth of a peak at 64 ppm, which could be attributed to Al–O4 bonding or the bonding of Al to Q3–Q4 Si structure (Fig. 11a). The bonding of Al with Si during the precipitation with NaOH is a likely outcome of the PMO swing process. In terms of the Si-NMR analysis, a shift from Q0–Q1 silica in the raw HCP towards a broader distribution of Q-structures, with a greater percentage of Q3–Q4 Si bonding in the PMO can be observed (Fig. 11b).6 The presence of the Q0–Q1 structures are indicative of the C–S–H hydrated clinker phases present in the cement itself.8,71 After dissolution and PMO precipitation the growth of Q3–Q4 Si-structures is indicative of the reprecipitation of SiO2 bonding to itself and also other elements, such as Al and Fe.
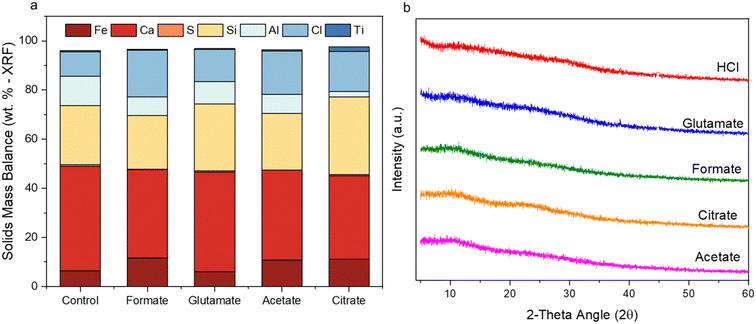 |
| | Fig. 10 (a) Pure elemental balance of the precipitated metal oxides (PMOs) as determined by XRF; (b) PXRD spectral data of the same PMO species for all five cases studied. | |
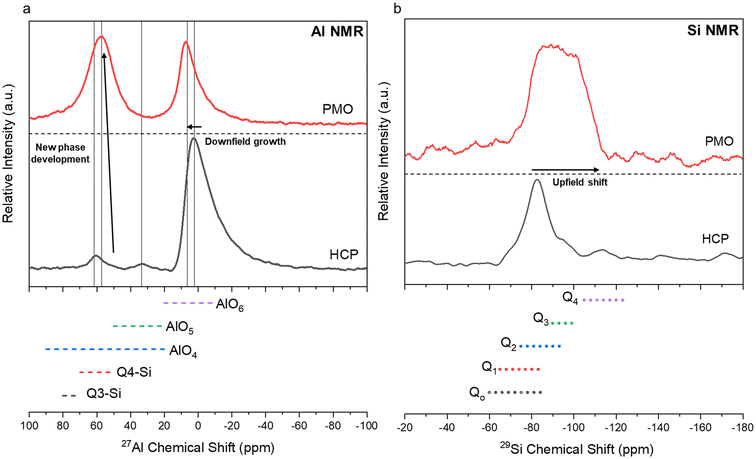 |
| | Fig. 11 27Al (a) and 29Si (b) MAS solid-state nuclear magnetic resonance (NMR) testing of the raw HCP material before leaching and a representative sample of PMO produced during the pH-swing process. The PMO used for the tests was obtained from the HCl only leaching process. | |
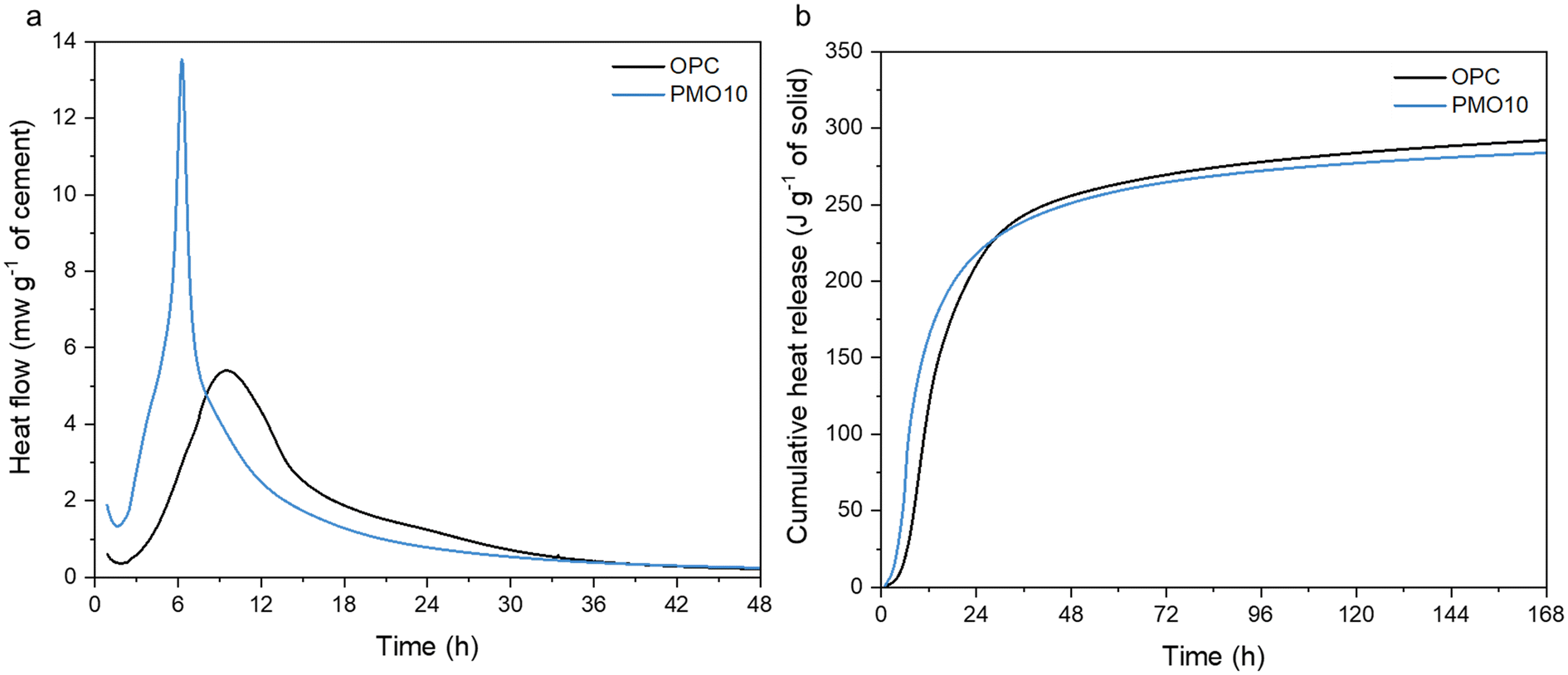
The differences in the structure and phases of the PERR and the PMOs could allow for different uses if they are upcycled within the built environment. These PMOs, which contain calcium, could be useful as pozzolanic materials in new cement hydration.72 The PMOs material is amorphous, which generally means it could react during cement hydration to form new phases.73,74 Cement hydration data is shown in Fig. 12. As observed, the replacement of PMO in OPC considerably enhanced the hydration kinetics of the cement in the first 8 h (Fig. 12a), and also resulted in a similar cumulative heat release as the OPC-only sample (Fig. 12b) over the course of the 168 h curing time. These results indicate that PMO had a strong accelerating effect on cement hydration at the early age, and the amounts of hydrated phases generated after 7 d were quite similar between OPC and PMO10. It can be potentially used as SCMs to replace cement without compromising its engineering properties. In another study, the placement of cement with 10 wt% PMO even improved the compressive strength of cement mortar, similar to commercial SCMs like silica fume.75 This suggests that the PMO materials recovered during the hydrometallurgical processing of alkaline wastes could indeed be used as upcycled materials within the build environment.
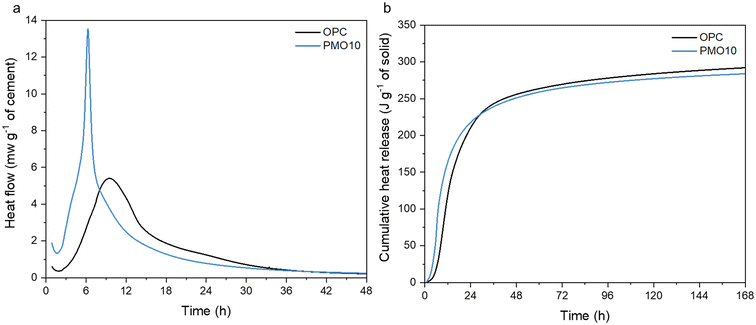 |
| | Fig. 12 (a) Heat flow of OPC and OPC replaced 10 wt% by PMO over 48 hours; (b) cumulative heat release for the same samples over 168 hours of curing and hydration. | |
2.4 Carbonation to produce pure precipitated calcium carbonate (PCC) polymorphs
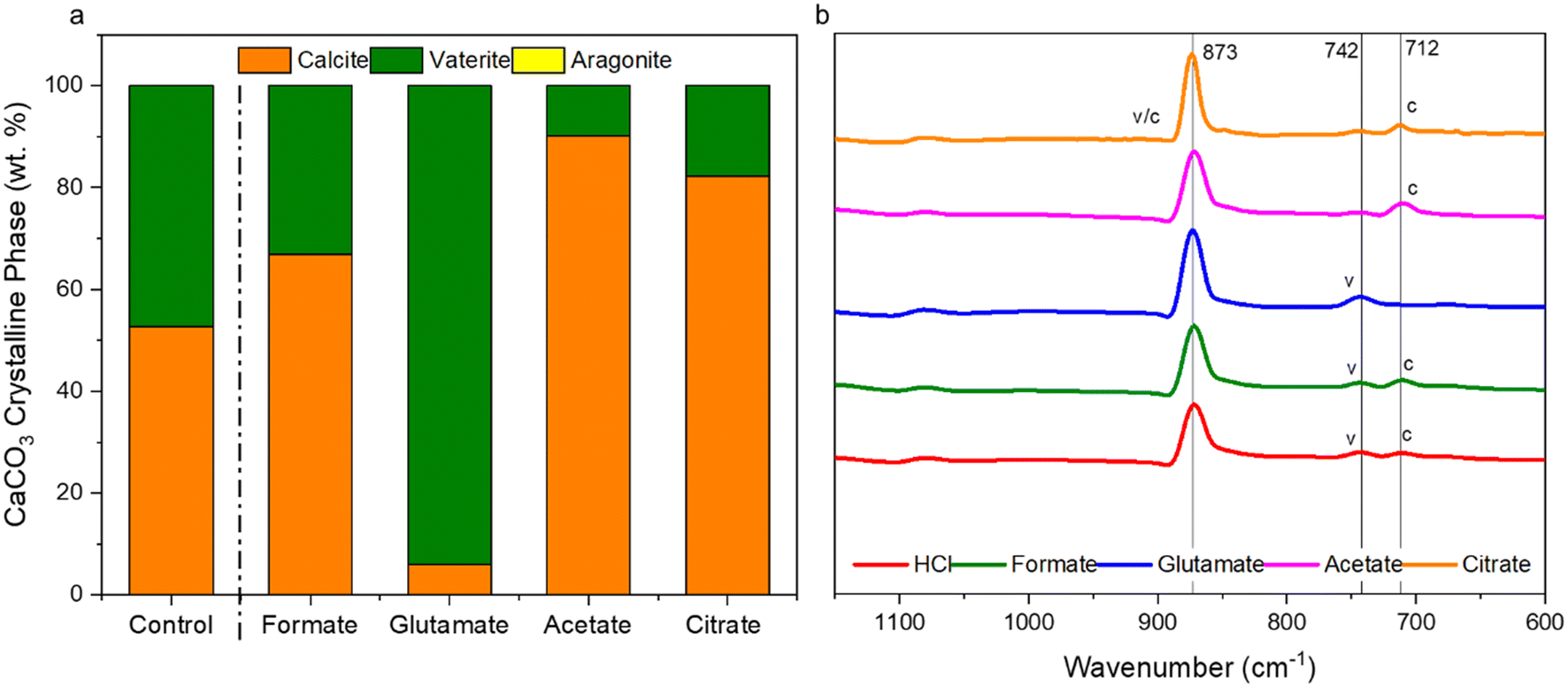
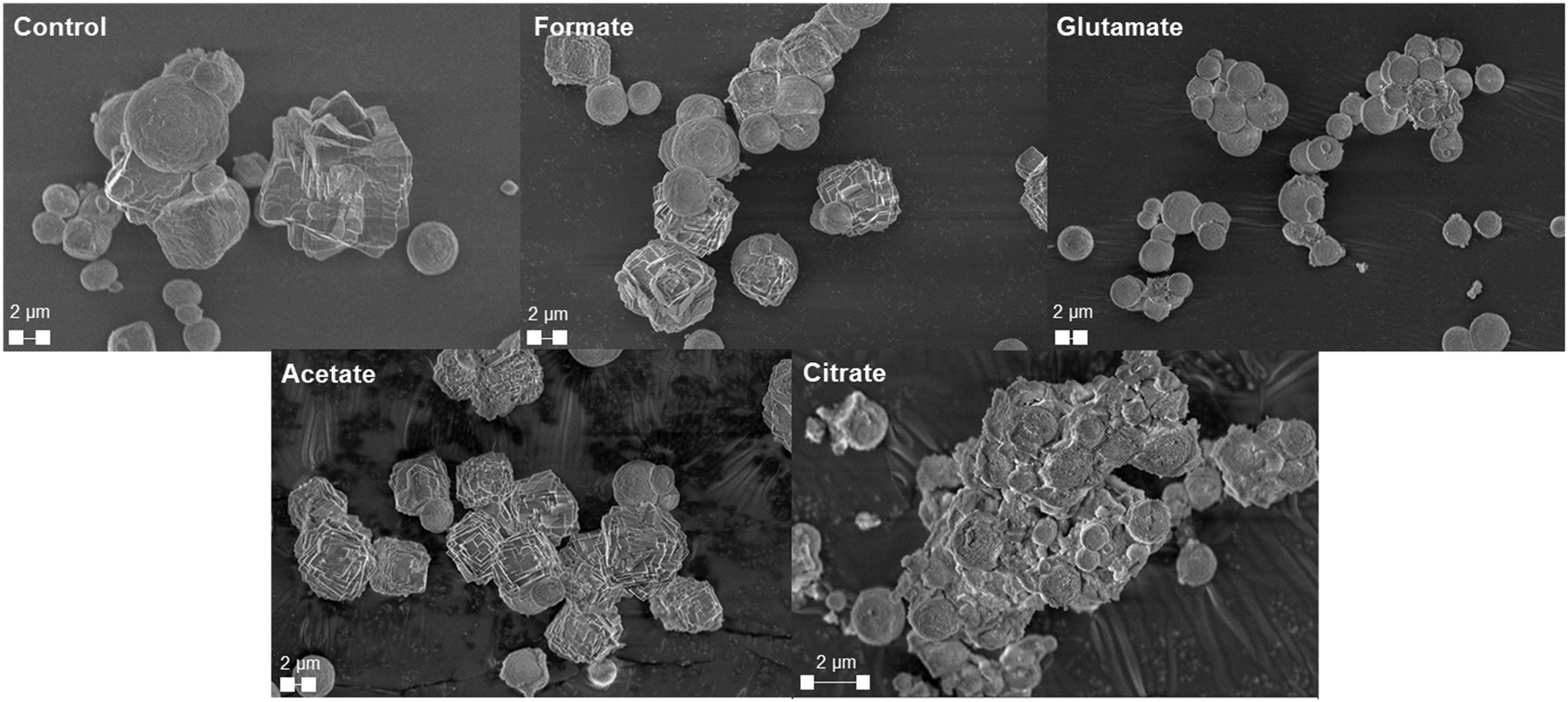
The carbonation of the leachate liquor was performed after PMO removal to produce pure PCC through carbon mineralization. The morphology of the carbonates was analyzed to probe at the various crystalline phases present. Fig. 13a shows the refined Q-PXRD data with the wt% of the crystalline phases of the PCC produced at all five leaching cases. The main anhydrous and most common forms of CaCO3 observed are vaterite, aragonite, and calcite, listed in order of thermodynamic stability.76–78 Calcite is the most widely used polymorph globally and is the main constituent of limestone. However, there is growing interest and research in the production of vaterite and aragonite, the metastable phases of CaCO3, for industrial applications based on their distinct morphologies, with vaterite being spherical and aragonite being needle-like.12,78–80 In this study, the control experiment produced a 50–50 wt% mixture of vaterite and calcite after 30 min carbonation. Due to the relatively dilute amount of Ca present (∼3000 ppm, 0.075 M), the calcite observed is likely a result of the precipitation of vaterite and recrystallization into calcite, which has been reported one of its main synthesis pathways by other researchers.81 The presence of formate, acetate, and citrate seemed to favor the production of calcite over vaterite, and could have decreased the nucleation rate of vaterite itself while promoting that of calcite. Interestingly, glutamate stabilized vaterite and resulted in a vaterite phase purity of the product of almost 97 wt% (Fig. 13a). FTIR analysis confirmed the presence of vaterite in this sample though a peak at 744 cm−1 which corresponds with vaterite CO32− symmetrical vibration (Fig. 13b).82 Calcite peaks in the other samples can be clearly seed at 710–713 cm−1, demonstrating the usefulness of FTIR to fingerprint the polymorphs of CaCO3.82,83 Fig. 14 shows the morphology of the produced carbonates for all the samples obtained during the carbonation reaction, further corroborating the presence of vaterite and calcite. The D50 of all the PCC produced was less than 10 μm (Fig. S5†), however the presence of glutamate and citrate resulted in the greatest degree of particle size reduction, with citrate PCC having a D50 of roughly 3.5 μm. The ability for certain ligands to control the PSD of the PCC during carbonation could be useful if the end use requires smaller carbonates based on size.
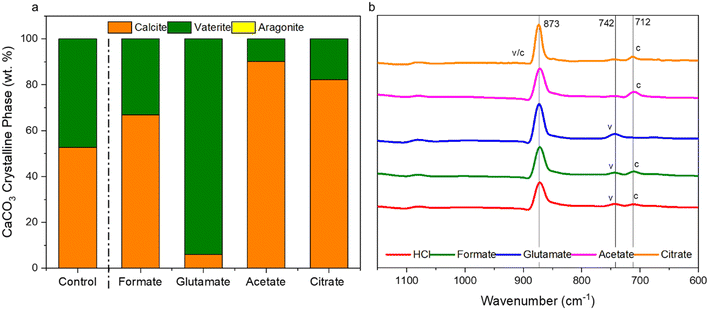 |
| | Fig. 13 (a) Refined Q-PXRD data of the PCC produced showing the polymorphs of CaCO3, vaterite, calcite, or aragonite, present in the samples; (b) FTIR spectral data of the carbonates produced showing characteristic CO32− out-of-plane vibrational bending near 873 cm−1, with vaterite characteristic peaks at 742 cm−1 and calcite in-the-plane bending at 712 cm−1.81–83 Table S2† shows the Rietveld refinement data and Fig. S6† shows the raw PXRD spectral data. | |
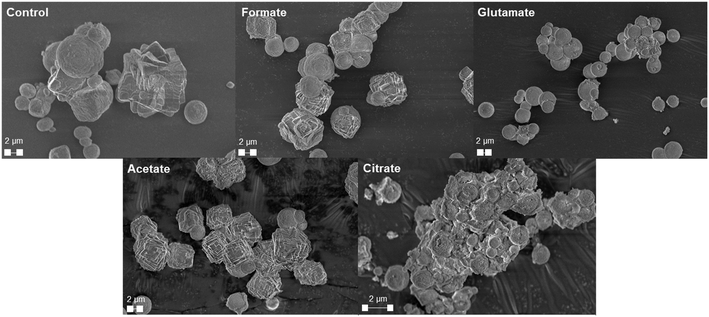 |
| | Fig. 14 SEM micrographs of the produced precipitated calcium carbonates showing the distinct morphologies present for the various ligands utilized during the hydrometallurgical leaching process. | |
The presence of the amino group in glutamate is likely responsible for the stabilization of vaterite, through bonding to the surface of the nucleated vaterite particles and preventing water from dissolving them.78,84,85 Certain amino acids are theorized to affect the crystallization of CaCO3. Finney and coworkers (2020) demonstrated through in silico work that not only is glutamate surface active towards crystallizing carbonates, its affinity for calcium allows organization of cations prior to crystallization to potential template vaterite.86 The suppression of calcite nucleation through the use of inorganic or organic cofactors can be advantageous to prevent calcite templating or seeding of vaterite, the less stable polymorph.87 Previous studies have shown the highest degree of vaterite stability using ammonia or amino-organic additives. Recently, Williams et al. (2023) demonstrated the critical ratio of ammonium salts to calcium which is required to stabilize vaterite during carbon mineralization processes.88 Similar results were also reported by Hood et al. (2014), who showed that of all the amino acids, aspartic and glutamic were able to stabilize vaterite to the best degree, likely due to the presence of the charged amino group which can surface bond to the growing vaterite particles.89 Among the studied carboxylic ligands, glutamate shows promise with two primary advantages over the other chemicals profiled. Firstly, its selectivity towards Ca is high with low yields of Fe and Si. This separation factor is advantageous if high purity PCC is required downstream and could negate the use of the pH-swing process to purify the leachate liquor. Secondly, glutamate shows advantages in surface activity compared to all the other ligands to stabilize vaterite during crystallization of PCC. This dual functionality could motivate its use within carbon mineralization processes for high purity vaterite from alkaline wastes.
Current (Feb. 2024 – Gulf Coast USD) pricing for formic acid is $978 per tonne, acetic acid is $600 per tonne, citric acid is ∼$896 per tonne, and glutamate (monosodium salt) is ∼$700–1000 per tonne utilizing conventional production methods highlighted in section 1.3. In the future, these ligands/commodity chemicals may be synthesized utilizing CO2 captured via DAC or point-source and upgraded through electrochemical, thermochemical, or microbial pathways. The economics of this proposed dissolution process are governed by the cost and recyclability of the ligands. When compared to conventional ex situ carbon mineralization processes, it is clear that the cost to utilize these ligands is higher due to both the ligand spot price and the need for more base (NaOH) to raise the pH during the PMO precipitation step for carbonation (Fig. S3†). However, glutamate requires less base than all the other species, likely due to the fact that the molecule already has one basic group. This further motivates and demonstrates the clear economic and process advantage which glutamate poses, even if the commodity chemical price could be slightly higher than the other molecules. The overall economics of this process could improve if product demand increases and ligand recycling methods are developed. Three distinct products are recovered at each stage of the proposed process: PERR, from the dissolution reactor which is rich is silica, can be reincorporated into new cement to offset OPC;6 PMO, which is recovered during the pH-swing process, can also serve as a clinker replacement; carbonates, which can also be reincorporated into new cement admixtures and replace OPC without compromising the strength of the cement itself.11,12 The carbonate replacement effect has been widely studied, with the improvement in hydration resulting mainly from a “filler effect”, which provides additional sites for nucleation during the hydration and setting of cement.11 The uses of non-traditional calcium carbonate polymorphs, such as vaterite or aragonite, in new built environment materials is also a potential motivation for the production of PCC via ex situ carbon mineralization.12,88,90 The production of circularized and decarbonized materials for the built environment from its own waste can potentially aid in reducing its overall intrinsic carbon intensity, lowering the amount of clinker needed and reducing emissions from cement production.
3 Conclusion
In this work, the dissolution of construction and demolition waste (C&DW) in the form of hydrated cement paste (HCP) was conducted in the presence of four organic carboxylic ligand materials, formate, glutamate, acetate, and citrate. The reactivity of the HCP in the presence of these material was established by kinetic analysis, and the rate constants for Ca, Si, Al, and Fe dissolution and the associated diffusivity of each case was determined. It was observed that not only did these ligands, especially citrate, help with absolute yield of material, they also enhanced the reaction rate of dissolution through their presence. This rate enhancement is theorized to be a result of both increased solubility of the leached elements in addition to more efficient dissolution of the cement into smaller particles. Characterization of the PERR revealed that the residue after leaching was rich is SiO2 with residual CaO present, and the condensation of silanol groups from those bonded to other elements, such as Al and Fe, towards only Si was observed. A pH-swing was employed to cleanse the leaching reaction solution prior to carbonation which revealed the precipitation of Si–Al–Fe rich PMOs. The precipitation of calcium at low pHs was also observed in the presence of the carboxylic acid ligands, with high loss being reported in the case of citrate. New Al phase creation in the PMO and the transition towards higher Si–Q structures was observed through NMR analysis and cement hydration data points towards potential uses of the PMO materials in new cement mixtures. Finally, PCC was produced via mineral carbonation and the morphology, size, and structure of the carbonates was confirmed by PXRD, FTIR, and particle size analysis. Notably, the presence of glutamate was able to produce a carbonate product that was nearly 97 wt% vaterite, likely due to surface active amino group. Future work includes in silico and even in situ techniques (e.g., in-line PAT probe) to more thoroughly establish how these ligands bind to both the dissolving cement particles and the crystalizing carbonates. Overall, this study presents a compelling look into the use of carboxylic acid ligands which can be derived from CO2 to enhance both material recovery and carbon capture during the ex situ carbon mineralization of C&DW for the decarbonization of the built environment.
4 Experimental section
4.1 Materials
The cement paste studied was produced from ordinary Portland Cement in-house and hydrated with a water to cement ratio of 0.4. Hydrated cement samples were crushed, milled, and sieved to a particle size range of 125–210 μm. In order to provide an accurate basis for assessing the degree of elemental extraction, the composition of the hydrated cement paste was assessed by ICP-OES. First, the samples were fused with lithium metaborate and then a solution of this product was analyzed via ICP. The major recoverable elemental composition of the cement was determined to be 44.0 wt% Ca, 14.0 wt% Si, 1.3 wt% Al, and 2.3 wt% Fe.9 The Ca and Si content is much smaller than ordinary Portland cement (OPC), due to the fact that this is hydrated cement paste; thus, to ensure proper material balance the ICP basis is preferred.6 The carboxylic ligands were all sourced from Sigma-Aldrich Chemicals, USA (>99% purity) and were sodium formate (NaCHO2), sodium acetate (NaC2H3O2), sodium glutamate (NaO4NH8C5), and sodium citrate (Na3C6H5O7). CO2 for carbonation was obtained from AirGas (Allentown, PA, USA) at 100% anhydrous. HCl for the leaching experiments was procured at 37 wt% (Sigma-Aldrich Chemicals, USA) and diluted to 5 M prior to use. Stock solutions of 25 wt% (w/v) NaOH (Alfa Aesar, MA, USA) were used for the pH swing process and to maintain the pH within the carbonation reactor during PCC production. Deionized water (MilliQ Grade, Millipore Sigma, USA) was used throughout the entire process, including leaching, washing, and stock solution creation.
4.2 Methods
4.2.1 Leaching reactor design. The dissolution of the waste HCP was carried out in 500 mL ChemGlass (Vineland, NJ, USA) stirred-tank, temperature-controlled reactors in a reaction volume of 300 mL. A slurry-density of 1 wt% (solids/liquid mass ratio) was utilized to properly assess the dissolution kinetics. The reactor was maintained at 25 °C for the duration of the 60 min leaching process which was performed at 900 rpm through overhead agitation. In the case of carboxylic ligand leaching, a 0.05 M solution of the corresponding salt (i.e., formate, acetate, glutamate, and citrate) was made with MilliQ deionized water. This concentration was chosen based on previous studies and preliminary results suggesting an optimal low concentration window of 0–0.1 M for these ligands. The pH was then adjusted to 3 with 5 M HCl prior to leaching in all cases. pH was continually monitored and kept at 3 via HCl addition throughout the entire duration of the experiment. Afterwards, the mixture was quickly filtered through a Buchner apparatus (Whatman, GE, USA) and the post-extraction reactor residue (PERR), or the undissolved portion of the cement matrix, was rinsed with 70% EtOH (v/v) prior to drying overnight in a vacuum oven at 80 °C until the samples reached a constant weight to ensure all water was removed.
4.2.2 pH-Swing process. In an effort to purify the leaching liquor prior to PCC production during carbonation, a pH swing process was employed to precipitate the major ancillary ions present, such as Si, Al, and Fe. The leaching liquor at pH 3 was slowly titrated with 25 wt% (w/v) NaOH towards pH 10, stopping before appreciable amounts of Ca2+ began to precipitate as Ca(OH)2. The system was allowed to equilibrate at each whole-step pH for approximately 30 minutes before moving to the next. At pH 10, the reactor liquors were filtered and washed thoroughly with EtOH prior to drying the precipitated metal oxides (PMOs) in a vacuum oven at 80 °C overnight until the samples reached a constant weight to ensure all water was removed.
4.2.3 Carbonation reaction. The carbonation reaction was carried out in the same 500 mL ChemGlass stirred tank reactor systems at a temperature of 25 °C and a stirring rate of 600 rpm. Carbonation was performed over the course of 30 min and CO2 was sparged into the reactor using a ceramic micro-sparger to ensure adequate solvation and absorption of the CO2 itself. The CO2 flowrate used was 150 mL min−1. The pH of the reactor was maintained at 10 through the continuous addition of NaOH to ensure carbonate ions were present for carbonation. After carbonation finished, the solution was quickly filtered using a Buchner apparatus as described above, and the carbonates were washes with EtOH to ensure removal of trapped water. Finally, the PCC was dried overnight in an 80 °C oven until the samples reached a constant weight to ensure all water was removed prior to characterization.
4.2.4 Characterization techniques. Aqueous species analysis for both the leaching kinetics and the titration during pH swing was performed using an Agilent 5110 inductively coupled plasma optical emissions spectrometer (ICP-OES). Samples were taken and quickly filtered using a 0.22 μm syringe filter to remove any residual debris, prior to being diluted 50 to 100-fold and quenched in 2 wt% HNO3. The following wavelengths were used to track the major elements through the use of the ICP: Ca2+: 422.67 nm; Si: 288.16 nm; Al: 237.31 nm; and Fe: 234.35 nm. To facilitate the titration of the dissolution reactor liquor, a Metrohm Titrando potentiometric titrator was used to dose in the base required to raise the pH of each solution. This also allowed for adequate time to collect samples for precipitation analysis via ICP-OES.The three sets of produced solids, the post-extraction reactor residue (PERR), PMOs, and the PCC, were all subject to rigorous solids characterization techniques. A Panalytical X'pert3 (Malvern) powder X-ray diffractometer (PXRD) with a 3 kW Cu X-ray generator was used to study the crystalline phases present in the solids, especially the presence of the polymorphs of CaCO3, vaterite, calcite, and aragonite, in the PCC. The PXRD data was refined using the open-source material analysis using diffraction (MAUD) software. Relevant crystal structures were obtained from the crystallography open database (COD). A Shimadzu 7200 energy dispersive X-ray fluorescence spectrometer (ED-XRF) was used to analyze the elemental composition of each of these residues, while Nicolet iS50 Fourier transform infrared spectrometer (FTIR) was used to exam the surface bonding structure of the residues, especially in the case of the use of carboxylic ligand materials used during leaching. A Zeiss Sigma VP SEM was used to collect residue morphology images which were sputter-coated for 20 s with AuPd before imaging. A Bruker XFLASH 6-30 energy-dispersive X-ray spectrometer (EDS) was utilized to perform elemental mapping of the samples, especially the PMO, to probe at the homogeneity of the residue composition.
The particle sizes of the residues and PCC were analyzed with a LS 13 320 XR Beckman Coulter particle size analyzer (PSA). Samples were suspended in ethanol prior to injection into the water column to serve as a dispersant over the 90 s measurement time using polarization intensity differential scattering (PIDS) analysis. The precipitated metal oxides (PMOs) were additionally characterized for phase development through solid-state magic angle spinning (MAS) 27Al and 29Si NMR. A Variant direct drive 300 MHz spectrometer equipped with a 3.2 mm chemogenetics broadband MAS probe running in a magnetic field of 7 T was used. The reincorporation and replacement of PMO within cement was tested using isothermal calorimetry to track the hydration of new cement pastes. OPC was replaced 10 wt% by PMO and then mixed together with water to create a final water-to-solid (W/S) ratio of 0.4. After water was loaded, the samples were mixed for 2 min prior to being directly loaded into A TAM Air Isothermal Calorimeter in glass ampules. The temperature was maintained at 25 °C over the course of the 168 h experiment.
Abbreviations
| CCUS | Carbon capture utilization and storage |
| CDR | Carbon dioxide removal |
| PCC | Precipitated calcium carbonate |
| OPC | Ordinary Portland cement |
| HCP | Hydrated cement waste |
| C&DW | Construction and demolition waste |
| DAC | Direct air capture |
Author contributions
J. M. W. and A. J. M. designed and planned this study. J. M. W. performed the primary experiments, data analysis, and wrote the manuscript. D. Z. provided the data and text relating to the hydration of cement pastes with the precipitated metal oxides. N. Z. offered guidance and assistance in the interpretation of kinetic and characterization data. S. K. provided insights and guidance into the cement hydration analysis. S. K. and A. J. M. edited the manuscript and provided guidance.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationship that could have appeared to influence the work reported herein.
Acknowledgements
This work was supported by the New York State Energy Research & Development Authority (NYSERDA, Albany, New York), Agreement Number: 0000185059 and the Lenfest Center for Sustainable Energy (Columbia University, New York, New York). We thank Allen Zheng and Professor Steven Greenbaum (Hunter College, NY, USA) for assisting in collection of the PMO NMR data.
References
- National Academies of Sciences, Engineering, and Medicine, Negative Emissions Technologies and Reliable Sequestration: A Research Agenda, The National Academies Press, Washington, DC, 2019, DOI:10.17226/25259.
- G. Gadikota, J. Matter, P. Kelemen, P. V. Brady and A. H. A. Park, Elucidating the differences in the carbon mineralization behaviors of calcium and magnesium bearing alumino-silicates and magnesium silicates for CO2 storage, Fuel, 2020, 277, 117900 CrossRef CAS.
- C. D. Hills, N. Tripathi and P. J. Carey, Mineralization technology for carbon capture, utilization, and storage, Front. Energy Res., 2020, 8, 553069 Search PubMed.
- G. Gadikota, Carbon mineralization pathways for carbon capture, storage and utilization, Commun. Chem., 2021, 4, 1–5 CrossRef PubMed.
- US EPA, Advancing sustainable materials management: Facts and figures 2018: Assessing trends in material generation, recycling and disposal in the United States, United States Environ. Prot. Agency, 2020, pp. 1–22.
- J. M. Williams, D. Zhao, N. Zhang, A. Zheng, S. G. Greenbaum, S. Kawashima and A. J. Moment, Calcium carbonate and reactive silica recovery from waste cement: The influence of processing parameters on upcycled material properties and carbon intensity, Chem. Eng. J., 2024, 482, 149013 CrossRef CAS.
- J. Soto-Paz, O. Arroyo, L. E. Torres-Guevara, B. A. Parra-Orobio and M. Casallas-Ojeda, The circular economy in the construction and demolition waste management: A comparative analysis in emerging and developed countries, J. Build. Eng., 2023, 78, 107724 CrossRef.
- N. Zhang and A. Moment, Upcycling construction and demolition waste into calcium carbonates: Characterization of leaching kinetics and carbon mineralization conditions, ACS Sustainable Chem. Eng., 2023, 11, 866–879 CrossRef CAS.
- G. Rim, N. Roy, D. Zhao, S. Kawashima, P. Stallworth, S. G. Greenbaum and A.-H. A. Park, CO2 utilization in built environment via the pCO2 swing carbonation of alkaline solid wastes with different mineralogy, Faraday Discuss., 2021, 230, 187–212 RSC.
- M. Fante, F. R. C. Ribeiro, D. B. Nunes, R. C. E. Modolo, C. de S. Kazmierczak, M. Mancio, F. M. W. Tognoli, A. R. G. de Azevedo and M. P. Kulakowski, Behavior of cementitious mixtures with filler carbonate subjected to accelerated carbonation, Case Stud. Constr. Mater., 2022, 17, e01300 Search PubMed.
- D. Zhao, J. M. Williams, Z. Li, A. H. A. Park, A. Radlińska, P. Hou and S. Kawashima, Hydration of cement pastes with calcium carbonate polymorphs, Cem. Concr. Res., 2023, 173, 107270 CrossRef CAS.
- D. Zhao, J. M. Williams, A. H. A. Park and S. Kawashima, Rheology of cement pastes with calcium carbonate polymorphs, Cem. Concr. Res., 2023, 172, 107214 CrossRef CAS.
- T. Strunge, P. Renforth and M. Van der Spek, Towards a business case for CO2 mineralisation in the cement industry, Commun. Earth Environ., 2022, 3, 1–14 CrossRef.
- M. Zajac, J. Skocek, M. Ben Haha and J. Deja, CO2 mineralization methods in cement and concrete industry, Energies, 2022, 15, 1–26 Search PubMed.
- K. A. Knight, P. R. Cunningham and S. A. Miller, Optimizing supplementary cementitious material replacement to minimize the environmental impacts of concrete, Cem. Concr. Compos., 2023, 139, 958–9465 CrossRef.
- F. Farmani, P. Khadiv-Parsi, A. A. Ramezanianpour, B. Bonakdarpour and F. Yazdian, Dual eco-friendly application of silica fume and scoria in cement-based materials through the enhancement of microbially-induced carbonate precipitation, Case Stud. Constr. Mater., 2022, 17, 1–12 Search PubMed.
- H. Sharma, S. K. Sharma, D. K. Ashish, S. K. Adhikary and G. Singh, Effect of various bio-deposition treatment techniques on recycled aggregate and recycled aggregate concrete, J. Build. Eng., 2023, 66, 105868 CrossRef.
- A.-H. A. Park, J. M. Williams, S. J. Friedmann, D. Hanson, S. Kawashima, V. Sick, M. R. Taha and J. Wilcox, Challenges and opportunities for the built environment in a carbon-constrained world for the next 100 years and beyond, Front. Energy Res., 2024, 12, 1388516 CrossRef.
- T. Havlik, Hydrometallurgy: Principles and Applications, Woodhead Publishing, 2008 Search PubMed.
- M. L. Free, Hydrometallurgy: Fundamentals and Applications, Springer Chem, 2nd edn, 2021 Search PubMed.
- H. H. Kellogg, Y. K. Rao and S. W. Marcuson, Pyrometallurgy, Annu. Rev. Phys. Chem., 1976, 27, 87–406 CrossRef.
- K. L. Rees and J. S. J. Van Deventer, The role of metal-cyanide species in leaching gold from a copper concentrate, Miner. Eng., 1999, 12, 877–892 CrossRef CAS.
- J. Li, J. Kou, C. Sun, N. Zhang and H. Zhang, A review of environmentally friendly gold lixiviants: Fundamentals, applications, and commonalities, Miner. Eng., 2023, 197, 108074 CrossRef CAS.
- M. Tanriverdi, H. Mordoǧan and Ü. Ipekoǧlu, Leaching of Ovacık gold ore with cyanide, thiourea and thiosulphate, Miner. Eng., 2005, 18, 363–365 CrossRef CAS.
- R. N. Mutafela, F. Ye, Y. Jani, J. Dutta and W. Hogland, Sustainable extraction of hazardous metals from crystal glass waste using biodegradable chelating agents, J. Mater. Cycles Waste Manage., 2022, 24, 692–701 CrossRef CAS.
- E. Tindanzor, Z. Guo, T. Li, R. Xu, X. Xiao and C. Peng, Leaching and characterization studies of heavy metals in contaminated soil using sequenced reagents of oxalic acid, citric acid, and a copolymer of maleic and acrylic acid instead of ethylenediaminetetraacetic acid, Environ. Sci. Pollut. Res., 2023, 30, 6919–6934 CrossRef CAS PubMed.
- H. Geng, F. Wang, C. Yan, Z. Tian, H. Chen, B. Zhou, R. Yuan and J. Yao, Leaching behavior of metals from iron tailings under varying pH and low-molecular-weight organic acids, J. Hazard. Mater., 2020, 383, 121136 CrossRef CAS PubMed.
- W. J. J. Huijgen and R. N. J. Comans, Carbonation of steel slag for CO2 sequestration: Leaching of products and reaction mechanisms, Environ. Sci. Technol., 2006, 40, 2790–2796 CrossRef CAS PubMed.
- W. J. J. Huijgen, G. J. Witkamp and R. N. J. Comans, Mineral CO2 sequestration by steel slag carbonation, Environ. Sci. Technol., 2005, 39, 9676–9682 CrossRef CAS PubMed.
- P. Kim, A. Anderko, A. Navrotsky and R. E. Riman, Trends in structure and thermodynamic properties of normal rare earth carbonates and rare earth hydroxycarbonates, Miner., 2018, 8, 106 Search PubMed.
- B. Vaziri Hassas, M. Rezaee and S. V. Pisupati, Precipitation of rare earth elements from acid mine drainage by CO2 mineralization process, Chem. Eng. J., 2020, 399, 125716 CrossRef CAS.
- N. Murali, K. Srinivas and B. K. Ahring, Biochemical production and separation of carboxylic acids for biorefinery applications, Fermentation, 2017, 3, 22 CrossRef.
- I. Wender, Reactions of synthesis gas, Fuel Process. Technol., 1996, 48, 189–297 CrossRef CAS.
- W. Ahmad, N. Asadi, P. Aryal, S. Dwivedi, A. Hatwar and A. Tanksale, in Advances in synthesis gas: Methods, technologies and applications, Elsevier, 2022, vol. 3: Syngas products and usages, pp. 199–223 Search PubMed.
- D. A. Bulushev and J. R. H. Ross, Towards sustainable production of formic acid, ChemSusChem, 2018, 11, 821–836 CrossRef CAS PubMed.
- G. Singh Dhillon, S. Kaur Brar, M. Verma and R. Dayal Tyagi, Utilization of different agro-industrial wastes for sustainable bioproduction of citric acid by Aspergillus niger, Biochem. Eng. J., 2011, 54, 83–92 CrossRef.
- R. Kumar, D. Vikramachakravarthi and P. Pal, Production and purification of glutamic acid: A critical review towards process intensification, Chem. Eng. Process., 2014, 81, 59–71 CrossRef CAS.
- M. Aresta and A. Dibenedetto, Utilisation of CO2 as a chemical feedstock: Opportunities and challenges, Dalton Trans., 2007, 2975–2992 RSC.
- C. A. Huff and M. S. Sanford, Catalytic CO2 hydrogenation to formate by a ruthenium pincer complex, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- N. A. Tappe, R. M. Reich, V. D'Elia and F. E. Kühn, Current advances in the catalytic conversion of carbon dioxide by molecular catalysts: An update, Dalton Trans., 2018, 47, 13281–13313 RSC.
- A. Barthel, Y. Saih, M. Gimenez, J. D. A. Pelletier, F. E. Kühn, V. D'Elia and J. M. Basset, Highly integrated CO2 capture and conversion: Direct synthesis of cyclic carbonates from industrial flue gas, Green Chem., 2016, 18, 3116–3123 RSC.
- F. M. Hoffmann, Y. Yang, J. Paul, M. G. White and J. Hrbek, Hydrogenation of carbon dioxide by water: Alkali-promoted synthesis of formate, J. Phys. Chem. Lett., 2010, 1, 2130–2134 CrossRef CAS.
- S. Yadav and A. Mehra, A review on ex situ mineral carbonation, Environ. Sci. Pollut. Res., 2021, 28, 12202–12231 CrossRef CAS PubMed.
- P. N. Olvera-Venegas, L. E. Hernández Cruz and G. T. Lapidus, Leaching of iron oxides from kaolin: Synergistic effect of citrate-thiosulfate and kinetic analysis, Hydrometallurgy, 2017, 171, 16–26 CrossRef CAS.
- Y. Yang, X. Wang, M. Wang, H. Wang and P. Xian, Recovery of iron from red mud by selective leach with oxalic acid, Hydrometallurgy, 2015, 157, 239–245 CrossRef CAS.
- W. Li, N. Wang, F. Lu, H. Chai and H. Gu, Selective separation of aluminum, silicon, and titanium from red mud using oxalic acid leaching, iron precipitation and pH adjustments with calcium carbonate, Hydrometallurgy, 2024, 223, 106221 CrossRef CAS.
- A. Pathak, M. Vinoba and R. Kothari, Emerging role of organic acids in leaching of valuable metals from refinery-spent hydroprocessing catalysts, and potential techno-economic challenges: A review, Crit. Rev. Environ. Sci. Technol., 2021, 51, 1–43 CrossRef CAS.
- E. Gerold, R. Lerchbammer, C. Strnad and H. Antrekowitsch, Towards a sustainable approach using mineral or carboxylic acid to recover lithium from lithium iron phosphate batteries, Hydrometallurgy, 2023, 222, 106187 CrossRef CAS.
- R. Banerjee, A. Mohanty, S. Chakravarty, S. Chakladar and P. Biswas, A single-step process to leach out rare earth elements from coal ash using organic carboxylic acids, Hydrometallurgy, 2021, 201, 105575 CrossRef CAS.
- J. M. Steer and A. J. Griffiths, Investigation of carboxylic acids and non-aqueous solvents for the selective leaching of zinc from blast furnace dust slurry, Hydrometallurgy, 2013, 140, 34–41 CrossRef CAS.
- W. Bao, H. Li and Z. Yi, Selective leaching of steelmaking slag for indirect CO2 mineral sequestration, Ind. Eng. Chem. Res., 2010, 49, 2055–2063 CrossRef CAS.
- E. Mejía, P. Navarro, C. Vargas, J. I. Tobón and W. Osorio, Caracterización de un residuo de construcción y demolición para la obtención de Ca y Si mediante tratamiento con ácido cítrico, DYNA, 2016, 83, 94–101 CrossRef.
- S. Hong, S. Moon, G. Sim and Y. Park, Metal recovery from iron slag via pH swing-assisted carbon mineralization with various organic ligands, J. CO2 Util., 2023, 69, 102418 CrossRef CAS.
- I. Z. Yildirim and M. Prezzi, Chemical, mineralogical, and morphological properties of steel slag, Adv. Civ. Eng., 2011, 2011, 13 Search PubMed.
- G. Artioli and J. W. Bullard, Cement hydration: The role of adsorption and crystal growth, Cryst. Res. Technol., 2013, 48, 903–918 CrossRef CAS.
- G. Rim, A. K. Marchese, P. Stallworth, S. G. Greenbaum and A. H. A. Park, 29Si solid state MAS NMR study on leaching behaviors and chemical stability of different Mg-silicate structures for CO2 sequestration, Chem. Eng. J., 2020, 396, 1–12 CrossRef.
- G. Rim, D. Wang, M. Rayson, G. Brent and A. H. A. Park, Investigation on abrasion versus fragmentation of the Si-rich passivation layer for enhanced carbon mineralization via CO2 partial pressure swing, Ind. Eng. Chem. Res., 2020, 59, 6517–6531 CrossRef CAS.
- E. J. Swanson, PhD thesis, Columbia University, 2014 Search PubMed.
- M. F. Irfan, M. R. Usman and A. Rashid, A detailed statistical study of heterogeneous, homogeneous and nucleation models for dissolution of waste concrete sample for mineral carbonation, Energy, 2018, 158, 580–591 CrossRef CAS.
- S. Y. Pan, T. C. Ling, A. H. A. Park and P. C. Chiang, An overview: Reaction mechanisms and modelling of CO2 utilization via mineralization, Aerosol Air Qual. Res., 2018, 18, 829–848 CrossRef.
- N. Zhang, H. D. Huang, R. Yang, A. Zheng and A. Moment, Aqueous metal ion leaching processes from high sulfur coal fly ash for carbon mineralization: The importance of pH control on cation extraction, carbonate purity, and silicon Q structure, Chem. Eng. J., 2023, 474, 145968 CrossRef CAS.
- I. Jawed and J. Skalny, Alkalies in cement: A review: II. Effects of alkalies on hydration and performance of Portland cement, Cem. Concr. Res., 1978, 8, 37–51 CrossRef CAS.
- M. Handke and W. Mozgawa, Vibrational spectroscopy of the amorphous silicates, Vib. Spectrosc., 1993, 5, 75–84 CrossRef CAS.
- S. Musić, N. Filipović-Vinceković and L. Sekovanić, Precipitation of amorphous SiO2 particles and their properties, Braz. J. Chem. Eng., 2011, 28, 89–94 CrossRef.
- B. Terry, The acid decomposition of silicate minerals part II. Hydrometallurgical applications, Hydrometallurgy, 1983, 10, 151–171 CrossRef CAS.
- D. J. Belton, O. Deschaume and C. C. Perry, An overview of the fundamentals of the chemistry of silica with relevance to biosilicification and technological advances, FEBS J., 2012, 279, 1710–1720 CrossRef CAS PubMed.
- B. Lothenbach, K. Scrivener and R. D. Hooton, Supplementary cementitious materials, Cem. Concr. Res., 2011, 41, 1244–1256 CrossRef CAS.
- S. Park, S. Wu, Z. Liu and S. Pyo, The role of supplementary cementitious materials (Scms) in ultra high performance concrete (uhpc): A review, Materials, 2021, 14, 1–24 Search PubMed.
- X. Pardal, F. Brunet, T. Charpentier, I. Pochard and A. Nonat, 27Al and 29Si solid-state NMR characterization of calcium-aluminosilicate-hydrate, Inorg. Chem., 2012, 51, 1827–1836 CrossRef CAS PubMed.
- B. Walkley and J. L. Provis, Solid-state nuclear magnetic resonance spectroscopy of cements, Mater. Today Adv., 2019, 1, 100007 CrossRef.
- R. A. Hanna, P. J. Barrie, C. R. Cheeseman, C. D. Hills, P. M. Buchler and R. Perry, Solid state 29Si and 27Al NMR and FTIR study of cement pastes containing industrial wastes and organics, Cem. Concr. Res., 1995, 25, 1435–1444 CrossRef CAS.
- J. Moon, S. Bae, K. Celik, S. Yoon, K. H. Kim, K. S. Kim and P. J. M. Monteiro, Characterization of natural pozzolan-based geopolymeric binders, Cem. Concr. Compos., 2014, 53, 97–104 CrossRef CAS.
- M. N. Akhtar, M. Jameel, Z. Ibrahim and N. M. Bunnori, Incorporation of recycled aggregates and silica fume in concrete: an environmental savior-A systematic review, J. Mater. Res. Technol., 2022, 20, 4525–4544 CrossRef CAS.
- C. S. Poon, L. Lam, S. C. Kou, Y. L. Wong and R. Wong, Rate of pozzolanic reaction of metakaolin in high-performance cement pastes, Cem. Concr. Res., 2001, 31, 1301–1306 CrossRef CAS.
- A. Mehta and D. K. Ashish, Silica fume and waste glass in cement concrete production: A review, J. Build. Eng., 2020, 29, 100888 CrossRef.
- O. A. Jimoh, K. S. Ariffin, H. Bin Hussin and A. E. Temitope, Synthesis of precipitated calcium carbonate: A review, Carbonates Evaporites, 2017, 33, 331–346 CrossRef.
- R. Chang, S. Kim, S. Lee, S. Choi, M. Kim and Y. Park, Calcium carbonate precipitation for CO2 storage and utilization: A review of the carbonate crystallization and polymorphism, Front. Energy Res., 2017, 5, 17 CrossRef.
- F. Liendo, M. Arduino, F. A. Deorsola and S. Bensaid, Factors controlling and influencing polymorphism, morphology and size of calcium carbonate synthesized through the carbonation route: A review, Powder Technol., 2022, 398, 117050 CrossRef CAS.
- M. de Beer, F. J. Doucet, J. P. Maree and L. Liebenberg, Synthesis of high-purity precipitated calcium carbonate during the process of recovery of elemental sulphur from gypsum waste, Waste Manage., 2015, 46, 619–627 CrossRef CAS PubMed.
- D. Konopacka-łyskawa, N. Czaplicka, M. Łapiński, B. Kościelska and R. Bray, Precipitation and transformation of vaterite calcium carbonate in the presence of some organic solvents, Materials, 2020, 13, 1–14 CrossRef PubMed.
- R. Chang, D. Choi, M. H. Kim and Y. Park, Tuning crystal polymorphisms and structural investigation of precipitated calcium carbonates for CO2 mineralization, ACS Sustainable Chem. Eng., 2017, 5, 1659–1667 CrossRef CAS.
- N. V. Vagenas, A. Gatsouli and C. G. Kontoyannis, Quantitative analysis of synthetic calcium carbonate polymorphs using FT-IR spectroscopy, Talanta, 2003, 59, 831–836 CrossRef CAS PubMed.
- F. B. Reig, J. V. G. Adelantado and M. C. M. Moya Moreno, FTIR quantitative analysis of calcium carbonate (calcite) and silica (quartz) mixtures using the constant ratio method. Application to geological samples, Talanta, 2002, 58, 811–821 CrossRef CAS PubMed.
- Q. Hu, J. Zhang, H. Teng and U. Becker, Growth process and crystallographic properties of ammonia-induced vaterite, Am. Mineral., 2012, 97, 1437–1445 CrossRef CAS.
- X. Song, C. Weng, Y. Cao, H. Kong and X. Luo, Facile synthesis of pure vaterite using steamed ammonia liquid waste and ammonium carbonate without additives via simple mechanical mixing, Powder Technol., 2021, 386, 361–371 CrossRef CAS.
- A. R. Finney, R. Innocenti Malini, C. L. Freeman and J. H. Harding, Amino acid and oligopeptide effects on calcium carbonate solutions, Cryst. Growth Des., 2020, 20, 3077–3092 CrossRef CAS PubMed.
- Y. Tang, F. Zhang, Z. Cao, W. Jing and Y. Chen, Crystallization of CaCO3 in the presence of sulfate and additives: Experimental and molecular dynamics simulation studies, J. Colloid Interface Sci., 2012, 377, 430–437 CrossRef CAS PubMed.
- J. M. Williams, D. Zhao, S. Moon, S. Kawashima, A. H. A. Park and A. J. Moment, Stabilization of pure vaterite during carbon mineralization: Defining critical activities, additive concentrations, and gas flow conditions for carbon utilization, Cryst. Growth Des., 2023, 23, 8103–8115 CrossRef CAS.
- M. A. Hood, K. Landfester and R. Muñoz-Espí, The role of residue acidity on the stabilization of vaterite by amino acids and oligopeptides, Cryst. Growth Des., 2014, 14, 1077–1085 CrossRef CAS.
- J. M. Williams, D. Zhao, N. Zhang, A. Chin, S. Kawashima and A. J. Moment, Directed synthesis of aragonite through semi- continuous seeded crystallization methods for carbon utilization, CrystEngComm, 2023, 25, 6050–6066 RSC.
|
| This journal is © Institute of Process Engineering of CAS 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Diandian Zhaobc,
Ning Zhang
ab,
Diandian Zhaobc,
Ning Zhang