
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicrowave assisted batch and continuous flow Suzuki–Miyaura reactions in GVL using a Pd/PiNe biowaste-derived heterogeneous catalyst†
Federica
Valentini
a,
Benedetta
Di Erasmo
a,
Marta
Ciani
a,
Shaomin
Chen
ab,
Yanlong
Gu
 b and
Luigi
Vaccaro
*a
b and
Luigi
Vaccaro
*a
aLaboratory of Green S.O.C. – Dipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Via Elce di Sotto 8, 06123-Perugia, Italy. E-mail: luigi.vaccaro@unipg.it; Web: https://greensoc.chm.unipg.it
bKey Laboratory of Material Chemistry for Energy Conversion and Storage, Ministry of Education, Hubei Key Laboratory of Material Chemistry and Service Failure, School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology, Wuhan 430074, China
First published on 12th March 2024
Abstract
Aiming at the development of alternative and low-impact synthetic pathways, we herein report the exploitation and the simultaneous employment of different tools to improve the overall sustainability of the Suzuki–Miyaura (SM) cross-coupling process with a circular economy approach. For the first time, in this study, we have combined the use of our biowaste-derived heterogeneous catalyst Pd/PiNe with the biomass-derived reaction medium γ-valerolactone (GVL) proving that an optimised protocol can be obtained for the SM process with a significant substrate scope. The microwave irradiation technology highly enhanced the energy efficiency, allowing the synthesis of different biphenyls and reducing the reaction time. In addition, the good efficiency and selectivity of the SM reaction led to further optimisation of the work-up procedure, minimising the waste generation and the E-factor values associated with the process (3.2–9.4). The optimised conditions tolerated the free carboxylic acid group well, realizing the step-economical preparation of the non-steroidal anti-inflammatory analgesic Fenbufen in quantitative yield. Finally, the scale-up of the MW-assisted process was performed in synergy with the optimisation of the continuous flow protocol and the waste minimised synthesis of Fenbufen was achieved.
Introduction
One of the main challenges of modern social, economic and scientific development is the development of more sustainable methods aimed at achieving the main objectives defined by the United Nations plan called “Transforming our world: the 2030 Agenda for Sustainable Development”.1 In this context, the principles of the circular economy (CE) model represent promising guidelines for achieving these objectives.2 In fact, the redesign of products and procedures, aimed at saving resources and minimizing waste, not only contributes to creating new jobs but also reduces the environmental impact of industrial processes and waste management which contribute approximately 9 and 3%, respectively, to the total EU greenhouse gas emission.3In this context, sustainable design plays a crucial role in both chemistry and engineering. However, the optimization and consolidation of alternative and low-impact synthetic pathways are not trivial. Several aspects should be considered, such as the toxicity, safety and environmental profile of raw materials, catalysts, additives and solvents as well as the efficiency and large-scale implementation of the synthetic route designed.4
It is well known that solvents contribute to the major percentage (around 80–90%)5 of wastes produced as they are exploited in different steps (i.e. as a reaction medium, in work-up and purification procedures) and also play a crucial role in the synthetic process.6 For this reason, great efforts have been made in the last decade to replace harmful solvents in all the steps with a particular focus on the use of bio-renewable alternatives.7
Due to the crucial role of cross-coupling reactions in diverse fields, such as pharmaceuticals, optoelectronics, polymer science and many other fields,8 green alternatives have been developed for these processes9 also driven by the definition of strict regulations prohibiting the use of critical solvents.10
Among the C–C coupling processes awarded with the Nobel Prize in 2010,11 the Suzuki–Miyaura (SM) reaction has emerged as a particularly attractive strategy to gain ever-increasing interest.12 Indeed, the SM reaction easily allows the formation of biaryls under mild conditions and with the use of non-toxic boronic acids. For these reasons, the SM cross-coupling represents a key step in several transformations for the total synthesis of highly relevant and valuable products.13
Sherwood et al. have recently studied the influence of green solvents on the SM reaction, concluding the difficulty in unambiguously defining the role of a reaction medium to achieve the best results.14 Anyway, the popularity and the value of the SM reaction in the synthesis of target materials are still related to an intense research activity aimed at the development of green approaches, including solvent selection,15 to improve the sustainability of this widely useful cross-coupling reaction.16
The imperative need to shift towards a more sustainable chemical production can also be summarised by the valuable suggestion given by Varma and Sedghi in a recent review article.15d Anyway, to the best of our knowledge, SM protocols realizing the optimization of reaction conditions for the use of biomass-derived γ-valerolactone (GVL) have not been defined and reported in the literature. GVL has been intriguingly considered to form blending solvent systems by Sherwood under homogeneous catalytic conditions14 but with no relevant success. GVL has recently found use in reducing the viscosity of dihydrolevoglucosenone (Cyrene™).17
As mentioned above, the reaction medium is a crucial parameter, promoting the dissolution of both organic and inorganic reagents. In this context, GVL has already been well demonstrated to be a valid alternative to reprotoxic classic solvents such as DMF and NMP.18
The choice of a renewable reaction medium is additionally crucial when it is also combined with the use of a recoverable and reusable heterogeneous catalytic system. Indeed, the possibility to easily separate the metal from the reaction mixture can reduce the waste at the purification stage, especially in the pharmaceutical and optoelectronics fields where strict regulations limit metal contamination.19 Moreover, the recycling of precious exhaustive metals is of utmost importance.
In the area of heterogeneous metal-based catalysts, porous supports are among the most employed, thanks to their tolerance to different reaction conditions, stability in various solvents and ability to stabilize Pd nanoparticles.20 Although highly engineered and efficient catalysts are available in the literature,21 the employment of renewable waste-derived supports and/or catalysts is crucial in the context of circular economy.22
Moreover, the combination of both renewable and low-impact reaction media and catalyst supports may highly enhance the overall sustainability of the process.15d In this context, we herein demonstrate the efficiency of our waste-derived heterogeneous catalyst in SM cross-coupling in a biomass-derived GVL reaction medium.
The Pd/PiNe catalyst,23 obtained by the careful design of a low-cost heterogeneous support via local urban-waste upcycling, showed comparable catalytic performances to commercially available Pd/C while reducing the metal leaching in solution. It is worth mentioning that this approach well meet the CE requirements, not only proposing an alternative strategy to pine needle urban-waste incineration but also inspiring small-town administrations towards sustainable waste disposal.2
In addition, in this study we considered the use of microwave irradiation-based technology (MW) to improve the process efficiency and reduce the reaction time compared to conventional heating.24 The use of MW in SM reactions has gained increasing interest both under batch25 and flow conditions.26
However, despite the advantages of the MW-based technology, it is worth mentioning that hot spot formation is still one of the main safety issues related to MW irradiation, especially when easily accessible carbon-supported solid catalysts are used.27 Nevertheless, GVL has proven its ability to avoid hot spot formation,28 demonstrating also good efficiency with different catalysts in diverse processes.28,29
Therefore, in this study, we have developed a sustainable and safe protocol to access different biphenyls under the CE and green chemistry guidelines. We have identified the optimal conditions to perform the SM reaction in GVL as the reaction medium under MW irradiation. In addition, we have prepared and employed the biomass-waste derived Pd/PiNe catalyst and optimized the reaction conditions to access active pharmaceutical ingredients (APIs) in high yield. Representatively, Fenbufen, a non-steroidal anti-inflammatory analgesic, can be obtained via different synthetic pathways,30 exploiting SM coupling as the key step. Although in some areas, including the EU, Fenbufen was withdrawn from the market, it is still commercialized in other countries, and strategies for possible repurposing should not be overlooked.31 For the aim of this contribution, including Fenbufen in the substrate scope of the process is of interest. In fact, in its structure different functional groups are present, furnishing a particularly intriguing case study for the versatility and functional group tolerance of the protocol.
Moreover, to consider the limitations related to the scale-up of the MW-based process,26a a continuous flow protocol was efficiently optimized in combination with MW irradiation. In this context, it is worth mentioning that the careful selection of the reaction medium benefited the continuous process of solubilizing both organic and inorganic compounds, thus avoiding the need for reactor regeneration by washing.
Results and discussion
The collected dry pine needle urban waste was initially extracted in a Soxhlet apparatus using a toluene–methanol azeotropic mixture, 95% recovered by distillation. This simple chemical pre-treatment gives the support preparation procedure an added value by enabling the valorisation of the sole lignocellulosic residue, usually underutilized, without wasting the extractive components. Indeed, the importance of the use of essential oils from pine-needles in cosmetics, as flavour additives and in the polymer field is well documented.32Once the pine needle lignocellulosic component was isolated, the PiNe support was prepared following a chemical carbonization method under mild conditions.33 The as-prepared biochar was used without any further modification to immobilize Pd nanoparticles using a polyol method.23 After the addition of H2PdCl4 precursor to the diethylene glycol suspension of PiNe, the reduction step was performed at 130 °C under an Ar atmosphere, affording the final Pd/PiNe heterogeneous catalyst. The described procedure led to small Pd nanoparticles with a size distribution centred at 4.5 nm.23
The metal loading on the biowaste-derived PiNe was measured by MP-AES (microwave plasma atomic emission spectroscopy) analysis (8 wt%).
With the freshly prepared Pd/PiNe catalyst, we started to optimize the SM coupling by selecting 4-bromobenzaldehyde (1a) and phenylboronic acid (2a) as model substrates, employing only 0.5 mol% of our Pd-based catalyst (Table 1). By performing the reaction under conventional heating and using K2CO3 as the base, a conversion of 76% into product 3a was obtained after 16 h in pure GVL as the reaction medium (entry 1, Table 1). Upon addition of different amounts of water to GVL (entries 2–4, Table 1), the conversion of 1a gradually increased to 92% when a mixture of GVL![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1:4) was used (entry 3, Table 1).
:H2O (1:4) was used (entry 3, Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | Reaction medium | Conc. (M) | Base | Convb (%) |
| a Reaction conditions: 1a (0.5 mmol), 2a (1.1 eq.), Pd/PiNe (0.5 mol%), base (1.2 eq.), reaction medium, 100 °C, 16 h. b Determined by GLC analysis; the remaining materials are unreacted 1a and 2a. The yield of 3a is reported in parentheses. c Performed under MW irradiation (MW dynamic mode) in 30 min. d 30% of biphenyl was detected as a byproduct. | ||||
| 1 | GVL | 1 | K2CO3 | 76 (71) |
| 2 | GVL:H2O (4:1) |
1 | K2CO3 | 82 (78) |
| 3 | GVL:H2O (1:4) |
1 | K2CO3 | 92 (88) |
| 4 | GVL:H2O (1:4) |
0.5 | K2CO3 | 74 (70) |
| 5c | GVL | 1 | K2CO3 | 90 (86) |
| 6 | GVL | 1 | DABCO | 35 |
| 7 | GVL | 1 | DBU | Traces |
| 8 | GVL | 1 | TEA | Traces |
| 9c | GVL | 0.7 | TBAAc | >99d (64) |
| 10c | GVL:H2O (5:1) |
0.5 | KOPiv | 91 (86) |
| 11c | GVL | 0.5 | — | Traces |
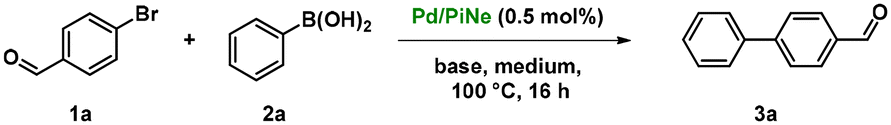
In contrast, by replacing the conventional heating with MW irradiation at a defined temperature, high conversion into 3a was obtained in only 30 minutes using GVL as the sole reaction medium (entry 5, Table 1), also with an energetic efficiency advantage. However, the insolubility of K2CO3 in GVL is an evident limit for a process in flow, and this aspect led us to investigate the use of other organic bases, but only poor results could be obtained (entries 6–8, Table 1).
Still with the goal of defining a continuous flow procedure, we further explored the possibility to reach the homogeneity of the reaction mixture. For this reason, the solubility of different bases in various ratios of the GVL:H2O mixture was investigated. Particular attention was paid to reducing the water excess in order to prevent product precipitation during the process. Indeed, because biphenyls have low solubility in water, their precipitation may complicate the heterogeneous catalyst recovery and the flow scale-up.
Despite K2CO3 and NaOAc being completely soluble in a mixture of GVL:H2O (1:1), under these conditions, phase separation was also observed. Tetrabutylammonium acetate (TBAAc) in GVL and KOPiv in GVL:H2O (5:1) were initially individuated as promising candidates due to their capability of forming clear and homogeneous mixtures in the presence of 1a and 2a.
The latter were tested in the SM coupling as model substrates under MW irradiation (entries 9 and 10, Table 1). Although the use of TBAAc allowed the use of pure GVL and quantitative conversion of 1a, 30% of simple biphenyl was also formed due to the concomitant decarbonylation reaction of 3a. For this reason, KOPiv in a GVL:H2O reaction medium was selected for further optimization, and under these conditions, pure 3a could be obtained. The removal of the base was detrimental, affording mainly triphenylboroxine as a byproduct (entry 11, Table 1).
Keeping in mind the use of fixed MW-irradiation power mode during the flow scale-up, we optimized the reaction conditions in batch using cyclic MW irradiation at a fixed power in the temperature range of 90–120 °C (Table 2).
|
|
|||
|---|---|---|---|
| Entry | MW power | No. of cycles | Convb (%) |
|
a Reaction conditions: 1a (0.5 mmol), 2a (1.1 eq.), Pd/PiNe (0.5 mol%), KOPiv (1.2 eq.), GVL:H2O (1 M), 90–120 °C, MW fixed power (tMW = 2 min; tcool = 1 min).
b Determined by GLC analysis; the remaining materials are unreacted 1a and 2a. The yield of 3a is reported in parentheses.
c Performed using Pd/PiNe recovered from entry 3.
d Commercially available Pd/C (10 wt%) was used.
e GVL:H2O (9:1).
|
|||
| 1 | 150 | 10 | 96 (91) |
| 2 | 100 | 10 | 99 (94) |
| 3 | 100 | 15 | >99 (95) |
| 4c | 100 | 15 | 70 (64) |
| 5 | 50 | 20 | 87 (82) |
| 6 | 50 | 30 | 93 (88) |
| 7 | 50 | 40 | >99 (95) |
| 8d | 50 | 40 | >99 (95) |
| 9e | 50 | 40 | >99 (95) |
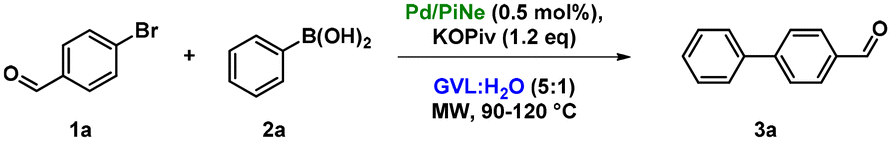
By decreasing the MW irradiation from 150 to 100 W, an increment in 1a conversion was observed after the same 10 cycles (entries 1 and 2, Table 2). The increase in cycles from 10 to 15 (entry 3, Table 2) led to a quantitative formation of the desired product 3a. However, after checking the reusability of the recovered Pd/PiNe under these conditions, a reduced catalytic efficiency during the second run was observed (entry 4, Table 2).
In order to succeed in the conservation of the Pd/PiNe catalytic efficiency as well, the MW irradiation was further reduced to 50 W (entries 5–7, Table 2), affording good results after 40 cycles (entry 7, Table 2). These conditions were also employed with commercially available Pd/C, as a representative of cheap and available palladium systems. In fact, optimum results in terms of efficiency were obtained (entry 8, Table 2). Additionally, the optimization was further assessed by evaluating the metal leaching in solution in both the processes, and with the use of Pd/C, higher Pd leaching was observed compared to Pd/PiNe (77 vs. 14 ppm, respectively). These final performance data of our biowaste-derived Pd-based catalyst are of great interest as they demonstrate the chemical efficiency and sustainability of the newly defined catalyst and conditions.
Ultimately, a further reduction of the water amount in the GVL:H2O mixture (9:1 ratio) led to a quantitative conversion into product 3a (entry 9, Table 2). Under such conditions, the Pd/PiNe catalyst was recovered and reused (see the ESI file for further information†), showing no loss in efficiency (Fig. 1).
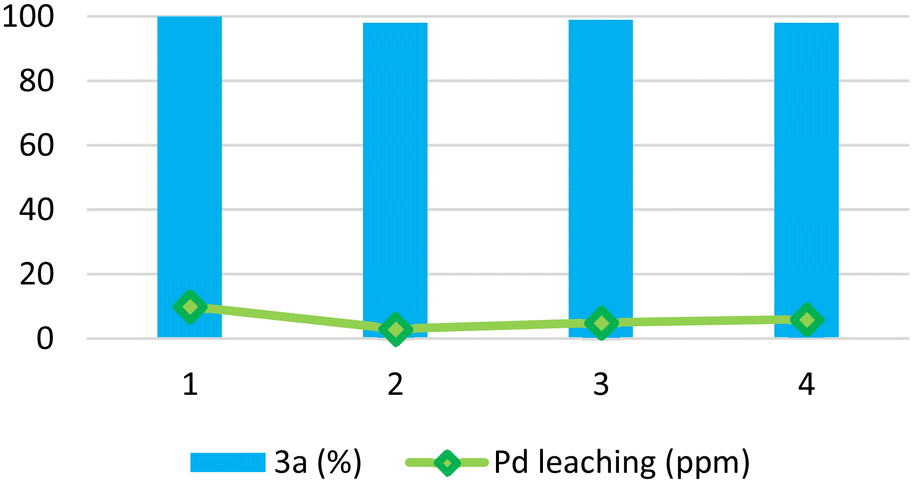 | ||
| Fig. 1 Pd/PiNe recycling and Pd leaching under batch conditions and MW irradiation. Reaction conditions: 1a (0.5 mmol), 2a (1.1 eq.), Pd/PiNe (0.5 mol%), KOPiv (1.2 eq.), 0.5 mL GVL:H2O (9:1), 90–120 °C, 50 W, MW fixed power (tMW = 2 min; tcool = 1 min), and 40 cycles. | ||
Moreover, to demonstrate the stability of our heterogeneous catalyst, we decided to stress the recycle test by performing the recovery and reuse of Pd/PiNe after 30 MW cycles instead of at full conversion (see Figure ESI-1 in the ESI file†). Also, upon reducing the reaction time, Pd/PiNe showed good recyclability.
Given the good catalytic performance of our Pd/PiNe, further efforts were devoted to minimizing the waste generated during the work-up step. At the end of the process, the catalyst was separated from the reaction mixture and washed with pure GVL. Water was removed under vacuum and GVL was recovered by distillation. The solid residue was then washed with water to remove excess 2a and residual KOPiv, affording the pure product 3a in a high isolated yield (95%). The environmental factor (E-factor) associated with this procedure was 14.6 (see the ESI file for further details†). When water is also recovered by distillation (90%), the E-factor was reduced to 70%, leading to a calculated value of 4.3. The E-factor profile34 is shown in Fig. 2 (see the ESI file for further information†).
 | ||
| Fig. 2 E-Factor profile for the model SM reaction between 1a and 2a before (a) and after (b) the recovery of H2O used in product purification. | ||
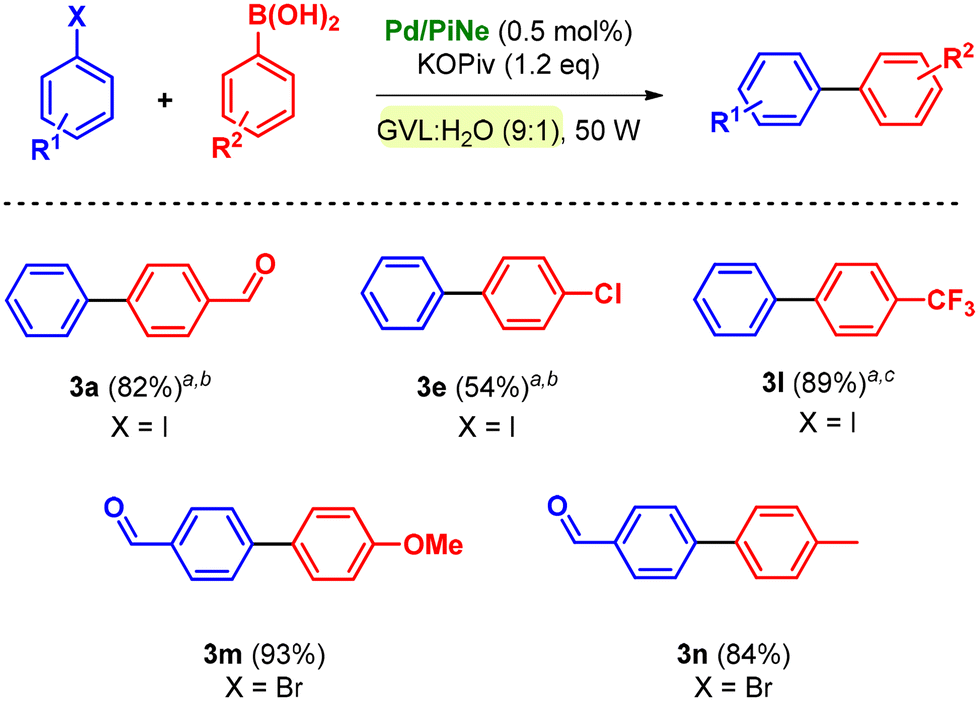
The optimized reaction conditions and the low-impact purification step were then extended to the SM coupling of different aryl halides (1a–i) with phenylboronic acid 2a (Scheme 1).
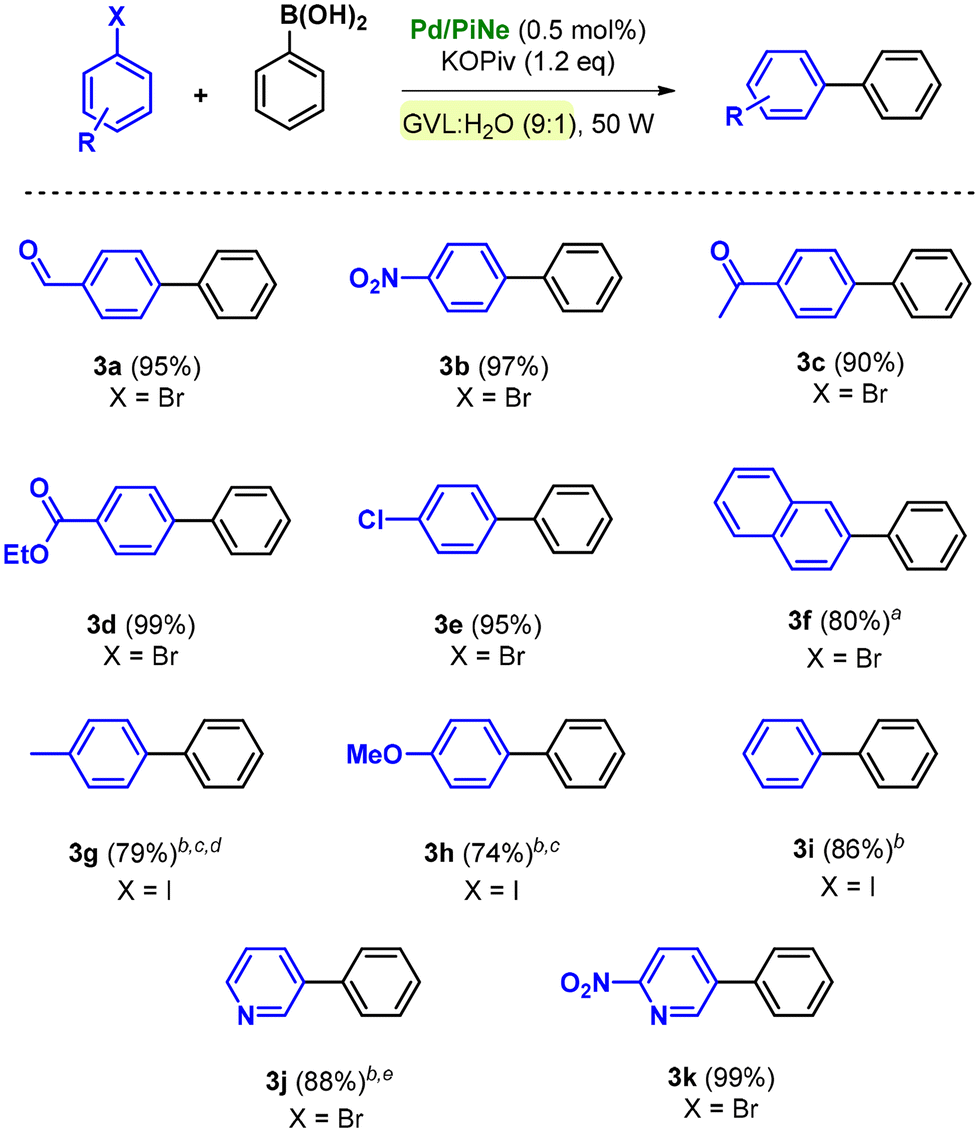 | ||
| Scheme 1 SM coupling between aryl halides and 2a using Pd/PiNe. Reaction conditions: 1 (0.5 mmol), 2a (1.1 eq.), Pd/PiNe (0.5 mol%), KOPiv (1.2 eq.), 0.5 mL GVL:H2O (9:1), 90–120 °C, 50 W, MW fixed power (tMW = 2 min; tcool = 1 min), and 40 cycles. a60 cycles. b100–150 °C. c100 W, 80 cycles. d1 mL GVL:H2O (9:1). e45 cycles. | ||
By using bromoarenes substituted with electron withdrawing groups 1a–e, good to excellent isolated yields (90–99%) were obtained under the optimal reaction conditions. Representatively, when 1c and 1d were replaced with the corresponding aryl chlorides, only traces of the desired products 3c and 3d could be detected.
Product 3f, obtained by the coupling of 2-bromonaphthalene (1f) with 2a, was isolated in 80% yield after increasing the number of cycles to 60.
In contrast, bromoarenes substituted with electron donating groups or unsubstituted bromobenzene poorly reacted under the optimized conditions. The corresponding iodoarenes led to good isolated yields in the range of 74–86% (Scheme 1). In this context, it should be noted that this choice was balanced on both the intrinsic and extrinsic impacts of the haloarene as already quantified and proven.9a
Indeed, the simple replacement of bromides with iodides allowed us to avoid the employment of ligands or extra additives still keeping high the process efficiency.9a Moreover, the synthesis of bromobenzene, compared to iodobenzene as well as the substituted para-methoxy and para-methyl counterparts, exhibits a higher ecological footprint; generally higher yields are associated with lower environmental impact.9a
The efficiency of the Pd/PiNe catalyst under MW irradiation afforded good results also in the SM coupling of N-containing heterocyclic bromides, allowing high isolated yields of arylated pyridines 3j and 3k (Scheme 1). It should be mentioned that for the product 3j, the work-up protocol was modified due to the low melting point of the synthesized compound. The extraction work-up used to isolate the product 3j led to an increase in the E-factor value up to 34 (see the ESI file† for further information and E-factor calculation). However, this value can be reduced by almost four times (7.9) by recovering both the aqueous phase and the heptane used for the extraction.
The conditions were further extended to different substituents such as phenylboronic acids (2b–f), leading always to satisfactory isolated yields (Scheme 2).
 | ||
| Scheme 2 SM coupling under batch conditions using Pd/PiNe. Reaction conditions: 1 (0.5 mmol), 2 (1.1 eq.), Pd/PiNe (0.5 mol%), KOPiv (1.2 eq.), 0.5 mL GVL:H2O (9:1), 90–120 °C, 50 W, MW fixed power (tMW = 2 min; tcool = 1 min), and 40 cycles. a100–150 °C, 100 W. b60 cycles. c80 cycles. | ||
It should be mentioned that the number of cycles and MW irradiation were adjusted based on the substitution of halobenzene rather than phenylboronic acid.
All the products were efficiently isolated following the optimized work-up procedure, achieving high yields and low environmental impact (see the ESI file† for E-factor calculation).
Due to the presence of different functional groups, the access to Fenbufen is of synthetic relevance, and different sustainable alternative technologies have been recently proposed for its production, including the use of mechanochemistry,13i flow-chemistry13a and heterogeneous catalysis.13c
Our newly defined protocol proved to be highly efficient in the synthesis of Fenbufen, achieving almost quantitative yield (Scheme 3) with an associated low waste-generation (see the ESI file for further information†).
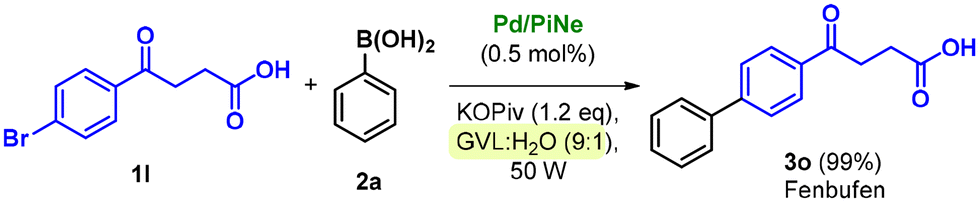 | ||
| Scheme 3 Pd/PiNe catalyses the synthesis of the Fenbufen API in the GVL:H2O reaction medium under MW irradiation. Reaction conditions: 1l (0.5 mmol), 2a (1.1 eq.), Pd/PiNe (0.5 mol%), KOPiv (1.2 eq.), 0.5 mL GVL:H2O (9:1), 90–120 °C, 50 W, MW fixed power (tMW = 2 min; tcool = 1 min), and 40 cycles. | ||
This result is even more important since it is generally not trivial to work directly in the presence of free carboxylic acid groups. However, the high yield obtained well highlights the versatility of the optimized conditions.
Given the impressive results obtained with our waste-derived heterogeneous catalyst, we decided to overcome the scale-up limitations by combining the MW irradiation with the continuous flow protocol.
A 50 cm PTFE tube was packed with 100 mg of Pd/PiNe blended in 2 g of quartz powder (see the ESI file for further details†) and a mixture of 1a, 2a and KOPiv in GVL:H2O was fluxed through the reactor under MW irradiation (Table 3).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Conc. [M] | KOPiv (eq) | GVL:H2O |
MW | Flowb (mL min−1) | Convc (%) |
|
a Reaction conditions: 1a (1 mmol), 2a (1.1 eq.), Pd/PiNe (100 mg), KOPiv, GVL:H2O, MW fixed power.
b Measured at the flow outlet.
c Determined by GLC analysis; the remaining materials are unreacted 1a and 2a.
d Boroxine was detected as a dehydration product.
e Reactor packed with commercially available Pd/C; biphenyl was detected as the main product.
|
||||||
| 1 | 1.00 | 1.2 | 9:1 |
50 | 0.1 | 33 |
| 2 | 0.75 | 1.2 | 9:1 |
50 | 0.1 | 38 |
| 3 | 0.75 | 1.2 | 9:1 |
50 | 0.05 | 58 |
| 4 | 0.50 | 1.2 | 9:1 |
50 | 0.2 | 40 |
| 5 | 0.25 | 1.2 | 9:1 |
50 | 0.2 | 21 |
| 6 | 0.33 | 1.2 | 9:1 |
50 | 0.1 | 56 |
| 7 | 0.33 | 1.2 | 9:1 |
100 | 0.1 | 39d |
| 8 | 0.50 | 1.2 | 9:1 |
75 | 0.1 | 54d |
| 9 | 0.50 | 1.2 | 9:1 |
60 | 0.1 | 63 |
| 10 | 0.50 | 1.2 | 9:1 |
50 | 0.1 | 99 |
| 11 | 0.50 | 1.2 | 9:1 |
40 | 0.1 | 43 |
| 12 | 0.50 | 1.0 | 9:1 |
50 | 0.1 | 61 |
| 13e | 0.50 | 1.2 | 9:1 |
50 | 0.1 | 99 |
| 14 | 0.50 | 1.2 | 5:1 |
50 | 0.1 | 91 |
| 15 | 0.50 | 1.2 | 5:2 |
50 | 0.1 | 65 |
| 16 | 0.50 | 1.2 | 95:5 |
50 | 0.1 | 70 |
Different flow parameters were screened and varied before defining the optimal conditions for the continuous flow protocol. Despite the homogeneity of the selected mixture, a high concentration led to poor conversion into the desired product 3a (entries 1–3, Table 3). At the same time, a high dilution had a detrimental effect on the flow performances (see entries 4 and 5, Table 3).
When the MW irradiation power was increased from 50 to 75 and 100 W (entries 7 and 8, Table 3), boroxine was detected as the byproduct due to the dehydration side reaction of 2a, confirming the irradiation at 50 W as the optimal parameter (entry 10, Table 3). Under these conditions, a quantitative conversion was observed with a flow rate of 0.1 mL min−1.
To further highlight the efficiency and selectivity of our biowaste-derived Pd/PiNe, a commercially available Pd/C catalyst was compared under the optimized flow conditions, leading to the formation of a decarbonylated byproduct as the main product (entry 13, Table 3).
The exploitation of flow technology not only allowed an easy scale-up of the process, being efficient for the continuous conversion of representative 50 mmol of 1a, but also contributed to an increment of the overall sustainability by lowering the E-factor to 3.9. Indeed, the work-up contribution to the E-factor was suppressed, as well as the auxiliary decreased, enhancing the E-kernel from 18%, in the batch process, to 44% (Fig. 3).
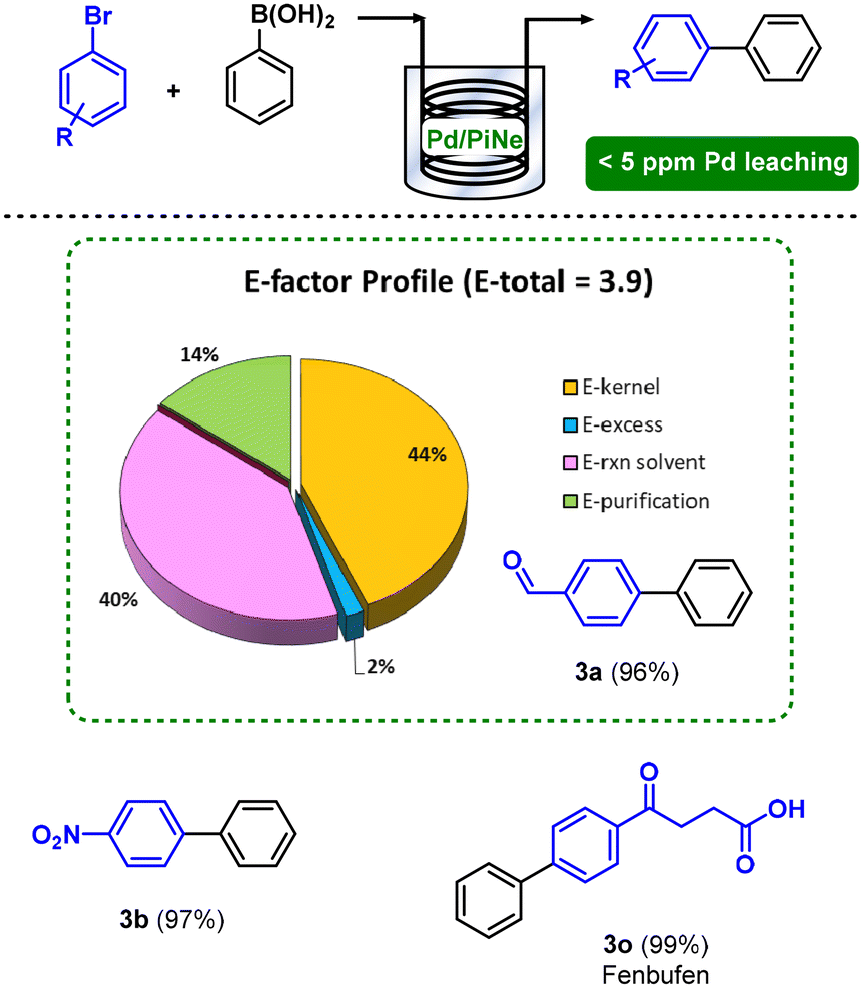 | ||
| Fig. 3 Continuous flow SM coupling between 1 and 2a and the E-factor profile for the synthesis of the product 3a. | ||
The Pd leaching in solution was continuously monitored during the time, showing very good values constantly below 5 ppm.
To confirm the versatility of the flow protocol, representative substrates were selected and fluxed through the same reactor. When 1b was tested, a higher dilution (0.33 M) was employed due to its low solubility. However, product 3b was obtained in a high yield (97%). Finally, Fenbufen API was continuously synthesized, exploiting the possibility of a step-economical access to the API through the synergy between MW irradiation and the flow technology.
To highlight the synergic role of different strategies to improve the overall sustainability of the process, we compared the E-factor values of our protocol with those of selected recent reports employing MW irradiation, whether in batches16h,25 or under flow conditions.26 Attention was directed to those reports specifically dedicated to the design of a sustainable protocol for the SM reaction (see the ESI file for further details†).16 Besides the waste minimisation obtained by our newly reported protocol, it should be noted that many processes combining the continuous flow protocol and MW reported the use of toxic DMF as a reaction medium under highly diluted conditions.
Conclusions
The biowaste-derived Pd/PiNe catalyst and the biomass-derived GVL reaction medium were simultaneously employed to define a sustainable Suzuki–Miyaura cross-coupling process with a circular economy approach. To the best of our knowledge, this represents the first study dedicated to the Suzuki–Miyaura reaction using a heterogeneous catalyst in bioderived GVL as the reaction medium, also reporting a general substrate scope. To further improve the overall sustainability of the process, MW irradiation was exploited to improve the energy efficiency, drastically decreasing the reaction time. With the aim to overcome the scale-up limitations and thus to move towards a continuous flow protocol, the conditions were optimized, balancing the efficiency with the mixture homogeneity using a minimum water content. This was crucial to avoid the precipitation of biphenyl products in the final mixture, thus allowing both an easy separation of the heterogeneous catalyst and a continuous flow protocol without additional regeneration and/or washing steps.The optimized conditions were efficient in the coupling of different substituents; both aryl halides and phenylboronic acids achieved good to excellent isolated yields (54–99%). It should be mentioned that the optimized protocol led also to the definition of a step-economical synthesis of Fenbufen, well tolerating the presence of a free carboxylic acid group and thus affording the desired product in quantitative yield.
Thanks to the good performances of Pd/PiNe, further efforts were devoted to the optimization of the work-up to minimize the waste generation and access low E-factor values (3.2–9.4). The choice of the reaction medium promoted the continuous flow synthesis of biphenyls and further decreased the waste generation associated with the process, enhancing the E-kernel contribution to the total E-factor.
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval to the final version of the manuscript. F. V.: investigation, methodology, data analysis, and manuscript preparation – review; B. D. E., M. C., and S. C.: investigation and manuscript editing; Y. G.: project administration and manuscript preparation – review and editing; L. V.: conceptualization, project administration, and manuscript preparation – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY. We acknowledge the Università degli Studi di Perugia for the support via “Fondo di Ateneo 2022”. The National PhD program in Catalysis coordinated by the University of Perugia is also thanked. This work was also supported by the Innovation and Talent Recruitment Base of New Energy Chemistry and Device (B21003). S. Chen acknowledges the financial support of the China Scholarship Council (CSC).References
- https://sdgs.un.org/2030agenda (accessed 21/11/2023).
- (a) A. Khajuria, V. A. Atienza, S. Chavanich, W. Henning, I. Islam, U. Kral, M. Liu, X. Liu, I. K. Murthy, T. D. T. Oyedotun, P. Verma, G. Xu, X. Zeng and J. Li, Circ. Econ., 2022, 1, 100001 Search PubMed; (b) C. Silvestri, L. Silvestri, A. Forcina, G. Di Bona and D. Falcone, J. Cleaner Prod., 2021, 294, 126137 CrossRef CAS; (c) P. Ghisellini and S. Ulgiati, J. Cleaner Prod., 2020, 243, 118360 CrossRef.
- (a) https://www.europarl.europa.eu/news/en/headlines/economy/20151201STO05603/circular-economy-definition-importance-and-benefits (accessed 21/11/2023); (b) https://www.europarl.europa.eu/news/en/headlines/society/20180301STO98928/greenhouse-gas-emissions-by-country-and-sector-infographic (accessed 22/11/2023).
- F. Ferlin, G. Brufani, G. Rossini and L. Vaccaro, Green Chem., 2023, 25, 7916–7933 RSC.
- (a) C. S. Slater, M. J. Savelski, W. A. Carole and D. J. Constable, Solvent use and waste issues, in Green chemistry in the pharmaceutical industry, 2010, pp. 49–82 Search PubMed; (b) D. J. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- C. Reichardt and T. Welton, Solvents and solvent effects in organic chemistry, John Wiley & Sons, 2011 Search PubMed.
- (a) F. Valentini, G. Brufani, B. Di Erasmo and L. Vaccaro, Curr. Opin. Green Sustainable Chem., 2022, 36, 100634 CrossRef CAS; (b) A. Gevorgyan, K. H. Hopmann and A. Bayer, Green Chem., 2021, 23, 7219–7227 RSC; (c) F. Gao, R. Bai, F. Ferlin, L. Vaccaro, M. Li and Y. Gu, Green Chem., 2020, 22, 6240–6257 RSC; (d) C. J. Clarke, W. C. Tu, O. Levers, A. Bröhl and J. P. Hallet, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed; (e) S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601–1612 RSC; (f) J. H. Clark, T. J. Farmer, A. J. Hunt and J. Sherwood, Int. J. Mol. Sci., 2015, 16, 17101 CrossRef CAS; (g) P. J. Jessop, Green Chem., 2011, 13, 1391–1398 RSC.
- For selected examples see:
(a) B. S. Takale, R. R. Thakore, R. Mallarapu, F. Gallou and B. H. Lipshutz, Org. Process Res. Dev., 2020, 24, 101–105 CrossRef CAS;
(b) C. C. C. JohanssonSeechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed;
(c) P. Devendar, R. Y. Qu, W. M. Kang, B. He and G. F. Yang, J. Agric. Food Chem., 2018, 66, 8914–8934 CrossRef CAS;
(d) J. P. Corbet and G. Mignani, Chem. Rev., 2006, 106, 2651–2710 CrossRef CAS PubMed;
(e) K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4442–4489 CrossRef CAS;
(f) M. Beller, A. Zapf and W. Magerlein, Chem. Eng. Technol., 2001, 24, 575–582 CrossRef CAS.
- (a) J. L. Osorio-Tejada, F. Ferlin, L. Vaccaro and V. Hessel, Green Chem., 2023, 25, 9760–9778 RSC; (b) T. Fantoni, A. Tolomelli and W. Cabri, Catal. Today, 2022, 397, 265–271 CrossRef; (c) T. Fantoni, S. Bernardoni, A. Mattellone, G. Martelli, L. Ferrazzano, P. Cantelmi, D. Corbisiero, A. Tolomelli, W. Cabri, F. Vacondio, F. Ferlenghi, M. Mor and A. Ricci, ChemSusChem, 2021, 14, 2591 CrossRef CAS PubMed; (d) F. Valentini, F. Ferlin, E. Tomarelli, H. Mahmoudi, M. Bagherzadeh, M. Calamante and L. Vaccaro, ChemSusChem, 2021, 14, 3359–3366 CrossRef CAS PubMed; (e) P. Lei, Y. Wang, Y. Mu, Y. Wang, Z. Ma, J. Feng, X. Liu and M. Szostak, ACS Sustainable Chem. Eng., 2021, 9, 14937–14945 CrossRef CAS; (f) S. E. Hooshmand, R. Afshari, D. J. Ramón and R. S. Varma, Green Chem., 2020, 22, 3668–3692 RSC; (g) P. R. Boruah, A. A. Ali, B. Saikia and D. Sarma, Green Chem., 2015, 17, 1442–1445 RSC.
- (a) P. Dutta, A. McGranaghan, I. Keller, Y. Patil, N. Mulholland, V. Murudi, H. Prescher, A. Smith, N. Carson, C. Martin, P. Cox, D. Stierli, M. Boussemghoune, F. Barreteau, J. Cassayrec and E. Godineau, Green Chem., 2022, 24, 3943–3956 RSC; (b) J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC; (c) F. P. Byrne, S. Jin, G. Paggiola, T. H. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Process, 2016, 4, 1–24 CrossRef; (d) A. Marrocchi, A. Facchetti, D. Lanari, C. Petrucci and L. Vaccaro, Energy Environ. Sci., 2016, 9, 763–786 RSC; (e) F. M. Kerton and R. Marriott, Alternative solvents for green chemistry, RSC Green Chemistry Series No 20, Royal Society of Chemistry, Cambridge, 2nd edn, 2013 RSC; (f) R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- https://www.nobelprize.org/prizes/chemistry/2010/summary/ (accessed 4/12/2023).
- For representative examples see: (a) M. Degli Innocenti, T. Schreiner and R. Breinbauer, Adv. Synth. Catal., 2023, 365, 4086 CrossRef CAS; (b) M. Ashraf, M. S. Ahmad, Y. Inomata, N. Ullah, M. N. Tahir and T. Kida, Coord. Chem. Rev., 2023, 476, 214928 CrossRef CAS; (c) M. C. D'Alterio, È. Casals-Cruañas, N. V. Tzouras, G. Talarico, S. P. Nolan and A. Poater, Chem. – Eur. J., 2021, 27, 13481 CrossRef; (d) B. S. Kadu, Catal. Sci. Technol., 2021, 11, 1186–1221 RSC; (e) D. E. Jose, U. S. Kanchana, T. V. Mathew and G. Anilkumar, J. Organomet. Chem., 2020, 927, 121538 CrossRef CAS; (f) I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137–173 CrossRef CAS.
- For representative examples see: (a) Z. Zhang, A. Ohno, H. Takaya and Y. M. A. Yamada, Chem. – Eur. J., 2023, 29, e202300494 CrossRef CAS PubMed; (b) M. Farhang, A. R. Akbarzadeh, M. Rabbani and A. M. Ghadiri, Polyhedron, 2022, 116124 CrossRef CAS; (c) J. Wang, T. Li, Z. Zhao, X. Zhang and W. Pang, Catal. Lett., 2022, 152, 1545–1554 CrossRef CAS; (d) J. Yu, K. S. Iyer and B. H. Lipshutz, Green Chem., 2022, 24, 3640–3643 RSC; (e) P. Orecchia, D. S. Petkova, R. Goetz, F. Rominger, A. S. K. Hashmi and T. Schaub, Green Chem., 2021, 23, 8169–8180 RSC; (f) B. S. Takale, R. R. Thakore, R. Mallarapu, F. Gallou and B. H. Lipshutz, Org. Process Res. Dev., 2020, 24, 101–105 CrossRef CAS; (g) Z. Li, X. Zhang, J. Qin, Z. Tan, M. Han and G. Jin, Org. Process Res. Dev., 2019, 23, 1881–1886 CrossRef CAS; (h) J. Britton and T. F. Jamison, Eur. J. Org. Chem., 2017, 6566–6574 CrossRef CAS; (i) Z. J. Jiang, Z. H. Li, J. B. Yu and W. K. Su, J. Org. Chem., 2016, 81, 10049–10055 CrossRef CAS PubMed; (j) T. N. Glasnova and C. O. Kappe, Adv. Synth. Catal., 2010, 352, 3089–3097 CrossRef.
- J. Sherwood, Beilstein J. Org. Chem., 2020, 16, 1001–1005 CrossRef CAS PubMed.
- For representative examples see: (a) F. Ulusal, E. Erünal and B. Güzel, Inorg. Chim. Acta, 2023, 555, 121127 CrossRef CAS; (b) P. Lei, Y. Mu, Y. Wang, Y. Wang, Z. Ma, J. Feng, X. Liu and M. Szostak, ACS Sustainable Chem. Eng., 2021, 9, 552–559 CrossRef CAS; (c) P. Lei, Y. Ling, J. An, S. P. Nolan and M. Szostak, Adv. Synth. Catal., 2019, 361, 5654 CrossRef CAS; (d) S. E. Hooshmand, B. Heidari, R. Sedghi and R. S. Varma, Green Chem., 2019, 21, 381–405 RSC; (e) P. R. Boruah, A. A. Ali, B. Saikia and D. Sarma, Green Chem., 2015, 17, 1442–1445 RSC; (f) S. D. Ramgren, L. Hie, Y. Ye and N. K. Garg, Org. Lett., 2013, 15, 3950–3953 CrossRef CAS PubMed.
- For representative examples see: (a) C. Palladino, T. Fantoni, L. Ferrazzano, A. Tolomelli and W. Cabri, ACS Sustainable Chem. Eng., 2023, 11, 15994–16004 CrossRef CAS; (b) L. Hosseinoghli, A. Bezaatpour, A. Nuri and M. Amiri, Appl. Organomet. Chem., 2023, e7320 Search PubMed; (c) H. Peng, X. Zhang, V. Papaefthimiou, C. Pham-Huu and V. Ritleng, Green Chem., 2023, 25, 264–279 RSC; (d) X. Yang, C. Wu, W. Su and J. Yu, Eur. J. Org. Chem., 2022, e202101440 CrossRef CAS; (e) Y. Era, J. A. Dennis, S. Wallace and L. E. Horsfall, Green Chem., 2021, 23, 8886–8890 RSC; (f) A. B. Wood, S. Plummer, R. I. Robinson, M. Smith, J. Chang, F. Gallou and B. H. Lipshutz, Green Chem., 2021, 23, 7724–7730 RSC; (g) M. Yousaf, A. F. Zahoor, R. Akhtar, M. Ahmad and S. Naheed, Mol. Diversity, 2020, 24, 821–839 CrossRef CAS PubMed; (h) M. Massaro, S. Riela, G. Lazzara, M. Gruttadauria, S. Milioto and R. Noto, Appl. Organomet. Chem., 2014, 28, 234–238 CrossRef CAS.
- C. Sullivan, Y. Zhang, G. Xu, L. Christianson, F. Luengo, T. Halkoski and P. Gao, Green Chem., 2022, 24, 7184–7193 RSC.
- (a) F. Kerkel, M. Markiewicz, S. Stolte, E. Müller and W. Kunz, Green Chem., 2021, 23, 2962–2976 RSC; (b) V. R. Ravikumar, A. Schröder, S. Köhler, F. A. Çetinel, M. Schmitt, A. Kondrakov, F. Eberle, J.-O. Eichler-Haeske, D. Klein and B. Schmidt-Hansberg, ACS Appl. Energy Mater., 2021, 4, 696–703 CrossRef CAS; (c) G. Strappaveccia, E. Ismalaj, C. Petrucci, D. Lanari, A. Marrocchi, M. Drees, A. Facchetti and L. Vaccaro, Green Chem., 2015, 17, 365–372 RSC.
- (a) C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS; (b) Note for Guidance on Specification Limits for Residues of Metal Catalysts; The European Agency for the Evaluation of Medicinal Products, Evaluation of Medicines for Human Use: London, Dec. 17, 2002; https://www.ema.europa.eu/en/documents/scientific-guideline/noteguidance-specification-limitsresidues-metal-catalysts_en.pdf.
- (a) Y. Gu, S. U. Son, T. Li and B. Tan, Adv. Funct. Mater., 2021, 31, 2008265 CrossRef CAS; (b) R. P. Lopes and D. Astruc, Coord. Chem. Rev., 2021, 426, 213585 CrossRef; (c) M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS; (d) I. C. Gerber and P. Serp, Chem. Rev., 2020, 120, 1250–1349 CrossRef CAS PubMed; (e) M. Zhao, Y. Wu and J.-P. Cao, Appl. Organomet. Chem., 2020, 34, e5539 CrossRef CAS; (f) J. Zhang, S. Lu, Y. Xiang and S. P. Jiang, ChemSusChem, 2020, 13, 2484 CrossRef CAS PubMed; (g) C. Rivera-Cárcamo and P. Serp, ChemCatChem, 2018, 10, 5058 CrossRef; (h) B. Lai, R. Bai and Y. Gu, ACS Sustainable Chem. Eng., 2018, 6, 17076–17086 CrossRef CAS; (i) Y. Cao, S. Mao, M. Li, Y. Chen and Y. Wang, ACS Catal., 2017, 7, 8090–8112 CrossRef CAS; (j) L. He, F. Weniger, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2016, 55, 12582–12594 CrossRef CAS PubMed; (k) S. Sun, R. Bai and Y. Gu, Chem. – Eur. J., 2014, 20, 549–558 CrossRef CAS PubMed; (l) D. S. Su, S. Perathoner and G. Centi, Chem. Rev., 2013, 113, 5782–5816 CrossRef CAS PubMed; (m) F. Rodŕíguez-Reinoso and A. Sepulveda-Escribano, Carbon as Catalyst Support, in Carbon Materials for Catalysis, ed. P. Serp and J. L. Figueiredo, Wiley-VCH, Weinheim, 1st edn, 2008 Search PubMed; (n) H. Marsh and F. R. Reinoso, Activated Carbon, Elsevier, Oxford, 1st edn, 2006 Search PubMed.
- (a) M. Gholinejad, Z. Naghshbandi and C. Nájera, ChemCatChem, 2019, 11, 1792–1823 CrossRef CAS; (b) H. Liu, L. Zhang, N. Wang and D. S. Su, Angew. Chem., Int. Ed., 2014, 53, 12634–12638 CrossRef CAS PubMed; (c) J.-K. Sun and Q. Xu, Energy Environ. Sci., 2014, 7, 2071–2100 RSC; (d) H. Liu, C.-Y. Cao, F.-F. Wei, Y. Jiang, Y.-B. Sun, P.-P. Huang and W.-G. Song, J. Phys. Chem. C, 2013, 117, 21426–21432 CrossRef CAS; (e) X. Xu, Y. Li, Y. Gong, P. Zhang, H. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS PubMed.
- (a) F. Campana, F. Valentini, A. Marrocchi and L. Vaccaro, Biofuel Res. J., 2023, 40, 1989–1998 CrossRef; (b) J. Chopra, V. Dayma, A. Mandal, P. K. Baroliya and D. Maiti, ChemistrySelect, 2022, 7, e202200 CrossRef; (c) H. Hosseinzadeh-Bandbafha, C. Li, X. Chen, W. Peng, M. Aghbashlo, S. S. Lam and M. Tabatabaei, J. Hazard. Mater., 2022, 424, 127636 CrossRef CAS PubMed; (d) C. M. Cova, A. Zuliani, A. R. Puente Santiago, A. Caballero, M. J. Munoz-Batista and R. Luque, J. Mater. Chem. A, 2018, 6, 21516–21523 RSC; (e) A. Zuliani, M. J. Muñoz-Batista and R. Luque, Green Chem., 2018, 20, 3001–3007 RSC; (f) J. A. Bennett, K. Wilson and A. F. Lee, J. Mater. Chem. A, 2016, 4, 3617–3637 RSC.
- (a) F. Valentini, F. Ferlin, S. Lilli, A. Marrocchi, L. Ping, Y. Gu and L. Vaccaro, Green Chem., 2021, 23, 5887–5895 RSC; (b) F. Ferlin, F. Valentini, D. Sciosci, M. Calamante, E. Petricci and L. Vaccaro, ACS Sustainable Chem. Eng., 2021, 9, 12196–12204 CrossRef CAS.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS PubMed.
- For selected representative examples see: (a) A. Y. Khormi, M. Abboud, M. S. Hamdy, M. Eissa and M. R. Shaaban, J. Inorg. Organomet. Polym., 2023, 33, 105–119 CrossRef CAS; (b) A. F. Villamizar-Mogotocoro, S. M. Bonilla-Castañeda and V. V. Kouznetsov, Green Chem., 2022, 24, 7996–8004 RSC; (c) L. Destro, R. Van Melsen, A. Gobbi, A. Terzi, M. Genitoni and A. Zambon, J. Appl. Biosci., 2022, 1, 64–72 CrossRef; (d) S. Supriya, G. S. Ananthnag, V. S. Shetti, B. M. Nagaraja and G. Hegde, Appl. Organomet. Chem., 2020, 34, e5384 CrossRef CAS; (e) V. V. Namboodiri and R. S. Varma, Green Chem., 2001, 3, 146–148 RSC.
- (a) Y. Monguchi, T. Ichikawa, T. Yamada, Y. Sawama and H. Sajiki, Chem. Rec., 2019, 19, 3–14 CrossRef CAS PubMed; (b) V. Konda, J. Rydfjord, J. Savmarker and M. Larhed, Org. Process Res. Dev., 2014, 18, 1413–1418 CrossRef CAS; (c) P. Ohrngren, A. Fardost, F. Russo, J.-S. Schanche, M. Fargrell and M. Larhed, Org. Process Res. Dev., 2012, 16, 1053–1063 CrossRef; (d) P. He, S. J. Haswell, P. D. I. Fletcher, S. M. Kelly and A. Mansfield, Beilstein J. Org. Chem., 2011, 7, 1150–1157 CrossRef CAS PubMed; (e) G. Shore, S. Morin and M. G. Organ, Angew. Chem., Int. Ed., 2006, 45, 2761–2766 CrossRef CAS PubMed; (f) I. R. Baxendale, C. M. Griffiths-Jones, S. V. Ley and G. K. Tranmer, Chem. – Eur. J., 2006, 12, 4407–4416 CrossRef CAS PubMed; (g) P. He, S. J. Haswell and P. D. I. Fletcher, Appl. Catal., A, 2004, 274, 111–114 CrossRef CAS.
- (a) W. Wang, B. Wang, J. Sun, Y. Mao, X. Zhao and Z. Song, RSC Adv., 2016, 6, 52974–52981 RSC; (b) S. Horikoshi, M. Kamata, T. Mitani and N. Serpone, Ind. Eng. Chem. Res., 2014, 53, 14941–14947 CrossRef CAS; (c) S. Horikoshi and N. Serpone, Catal. Sci. Technol., 2014, 4, 1197–1210 RSC; (d) S. Horikoshi, A. Osawa, S. Sakamoto and N. Serpone, Appl. Catal., A, 2013, 460–461, 52–60 CrossRef CAS.
- E. Petricci, C. Risi, F. Ferlin, D. Lanari and L. Vaccaro, Sci. Rep., 2018, 8, 10571 CrossRef PubMed.
- For representative examples see: (a) Z. Jiang, J. Remón, T. Li, V. L. Budarin, J. Fan, C. Hu and J. H. Clark, Cellulose, 2019, 26, 8383–8400 CrossRef CAS; (b) A. Kumar, Y. E. Jad, J. M. Collins, F. Albericio and B. G. de la Torre, ACS Sustainable Chem. Eng., 2018, 6, 8034–8039 CrossRef CAS; (c) S. Tabasso, E. Calcio Gaudino, L. Rinaldi, A. Ledoux, P. Larini and G. Cravotto, New J. Chem., 2017, 41, 9210–9215 RSC; (d) A. S. Amarasekara and M. A. Hasan, Catal. Commun, 2015, 60, 5–7 CrossRef CAS.
- (a) J. K. Mitchell, W. A. Hussain, A. H. Bansode, R. M. O'Connor, D. E. Wise, M. H. Choe and M. Parasram, Org. Lett., 2023, 25, 6517–6521 CrossRef CAS PubMed; (b) X. He, S. Hu, Y. Xiao, L. Yu and W. Duan, Eur. J. Org. Chem., 2022, e202200731 CrossRef CAS; (c) C. Bo, Q. Bu, J. Liu, B. Dai and N. Liu, ACS Sustainable Chem. Eng., 2022, 10, 1822–1828 CrossRef CAS; (d) A. Jorea, D. Ravelli, R. M. Romarowski, S. Marconi, F. Auricchio and M. Fagnoni, ChemSusChem, 2022, 15, e202200898 CrossRef CAS PubMed; (e) D. Zhang, T. Tang, Z. Zhang, L. Le, Z. Xu, H. Lu, Z. Tong, D. Zeng, W.-Y. Wong, S.-F. Yin, A. Ghaderi, N. Kambe and R. Qiu, ACS Catal., 2022, 12(2), 854–867 CrossRef CAS; (f) K. Zheng, G. Xiao, T. Guo, Y. Ding, C. Wang, T.-P. Loh and X. Wu, Org. Lett., 2020, 22, 694–699 CrossRef CAS PubMed; (g) A. Ohno, T. Sato, T. Mase, Y. Uozumi and Y. M. A. Yamada, Adv. Synth. Catal., 2020, 362, 4687 CrossRef CAS.
- (a) B. Wang, Z. Cai, H. Yao, S. Jiao, S. Chen, Z. Yang, W. Huang, Q. Ren, Z. Cao, Y. Chen, L. Zhang and Z. Li, Eur. J. Med. Chem., 2023, 245, 114883 CrossRef CAS PubMed; (b) J. Chandrasekaran and J. Balasubramaniam, Struct. Chem., 2022, 33, 1391–1407 CrossRef CAS PubMed; (c) S. Jardine, S. Anderson, S. Babcock, G. Leung, J. Pan, N. Dhingani, N. Warner, C. Guo, I. Siddiqui, D. Kotlarz, J. J. Dowling, R. A. Melnyk, S. B. Snapper, C. Klein, J. R. Thiagarajah and A. M. Muise, Gastroenterology, 2020, 158, 1000 CrossRef CAS PubMed.
- For representative examples see: (a) A. Koutsaviti, S. Toutoungy, R. Saliba, S. Loupassaki, O. Tzakou, V. Roussis and E. Ioannou, Foods, 2021, 10, 142 CrossRef CAS PubMed; (b) B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS; (c) M. Winnacker, Angew. Chem., Int. Ed., 2018, 57, 14362–14371 CrossRef CAS PubMed; (d) M. Winnacker and J. Sag, Chem. Commun., 2018, 54, 841–844 RSC; (e) H. C. Quilter, M. Hutchby, M. G. Davidson and M. D. Jones, Polym. Chem., 2017, 8, 833–837 RSC; (f) N. A. Kukhta, I. V. Vasilenko and S. V. Kostjuk, Green Chem., 2011, 13, 2362–2364 RSC.
- F. Valentini, E. Cerza, F. Campana, A. Marrocchi and L. Vaccaro, Bioresour. Technol., 2023, 390, 129847 CrossRef CAS PubMed.
- J. Andraos and A. Hent, J. Chem. Educ., 2015, 92, 1820–1830 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General procedures, full characterization of the synthesized compounds and copies of 1H, 13C and 19F NMR spectra. See DOI: https://doi.org/10.1039/d4gc00497c |
| This journal is © The Royal Society of Chemistry 2024 |