 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe unexpected fast polymerization during the synthesis of a glycolated polythiophene
Abdulrahman
Bakry†
 a,
Preeti
Yadav†
a,
Shin-Ya (Emerson)
Chen
b and
Christine K.
Luscombe
*a
a,
Preeti
Yadav†
a,
Shin-Ya (Emerson)
Chen
b and
Christine K.
Luscombe
*a
api-Conjugated Polymers Unit, Okinawa Institute of Science and Technology Graduate University, Kunigami-gun, Okinawa 9040495, Japan. E-mail: christine.luscombe@oist.jp
bMolecular Engineering and Sciences Institute, University of Washington, Seattle, WA 98195, USA
First published on 8th September 2023
Abstract
Conjugated polymers with ethylene glycol side chains are emerging as ideal materials for bioelectronics, particularly for application in organic electrochemical transistors (OECTs). To improve the OECT device performance, it is important to develop an efficient synthetic strategy that will provide access to novel high-performing materials besides focusing on molecular design. While a lot of efforts are being devoted to designing of new polymers by modifying the glycol side chains, understanding how their nature affects the polymerization kinetics and eventually the polymer structure and properties is not known. In this work, we have studied the influence of the content of the ethylene glycol side chain and its linkage on the formation of the active Grignard monomer species upon Grignard metathesis in three thiophene derivatives. A strong dependence of the monomer's concentration on polymerization was noted in our study indicating that for synthesizing P3MEEMT, a high-performing OECT material, by Kumada catalyst transfer polymerization (KCTP) a minimum of 0.15 M monomer is needed. Furthermore, kinetic studies by GPC show uncontrolled polymerization behavior contrary to the controlled chain growth characteristics of the KCTP.
1. Introduction
The interfacing of biological systems with micro and nano-electronic systems has opened new frontiers in life science research bringing a whole new insight into medical diagnosis and health monitoring.1 In recent years, organic electrochemical transistors (OECTs) have emerged as a key technology dominating bioelectronic research due to their efficient transduction of biochemical signals into electronic signals.2–4 OECTs have been successfully employed as miniaturized sensors for metabolite detection,5 neural interfacing,6 and neuromorphic computing.7 Similar to other transistors, an OECT is a three-terminal device comprising a source, drain, and gate electrode, however, it is the electrochemical gating of OECTs through an electrolyte that drives the movement of ions into and out of the channel material thereby transducing ionic signals into electrical signals.3,8 This means that the channel between the source and drain electrode of OECTs requires materials exhibiting mixed ionic–electronic conduction properties.9 Hence, to achieve a high-performing OECT device, carrier mobility, and ion transport of the polymer should be balanced in such a way that maximum transconductance (figure of merit for OECTs)10 is achieved along with good operational stability in ambient and aqueous environments and fast kinetics.11Among the various classes of mixed conductors developed in recent years for OECTs, organic semiconductors particularly conjugated polymers functionalized with ethylene glycol side chains have been of particular interest demonstrating high OECT performance due to improved volumetric capacitance (rivaling PEDOT) and operation in accumulation mode, facile synthetic tunability, and excellent enzymatic biocompatibility.11–14 Given the intricate relationship between ionic and electronic conduction, several molecular design strategies have been explored for improving the device performance of ethylene glycol functionalized polymers including the use of ethylene glycol with different chain lengths,15,16 mixed alkyl ethylene glycol chains,17,18 and modifying of the polymer backbone.19,20 One such polymer developed by our group is poly(3-{[2-(2-methoxyethoxy)ethoxy]methyl}thiophene-2,5-diyl) (P3MEEMT)21 demonstrating figure of merit comparable to state-of-the-art OECT channel materials. Considering P3MEEMT as a model system, several such polymers have been developed by varying the ethylene glycol content of glycol side chains and the position of oxygen atoms.15,16,22,23 Despite various aspects of the molecular design strategy investigated for improving the performance of these materials, the molecular design and synthesis for OECT channel material still lack a coherent strategy. Therefore, the advent of simple and efficient synthetic methods that will enable the synthesis of such types of polymers with high regioregularity and precisely defined molecular weight will be crucial in advancing the field of OECTs.
Synthesis of conjugated polymers by Kumada catalyst transfer polymerization (KCTP) remains the best approach for obtaining polymers with controlled molecular weight, narrow dispersity, and well-defined end groups.24,25 Since KCTP polymerization proceeds via the active Grignard monomer formed from Grignard metathesis with a dihaloderivative, conditions for active monomer formation and its polymerization are the deciding factor for polymer structure and molecular weight. Despite being a simple and efficient approach, the challenge in utilizing KCTP for OECT channel material synthesis arises from the presence of polar side groups in these systems. For example, the hygroscopic nature of ethylene glycol side chains can interfere with the formation of the active Grignard monomer leading to loss of polymerization control or total reaction failure. Furthermore, apart from the length and content of the oligoethylene glycol side chain, the position of oxygen atoms can have a strong impact on the formation of active monomer species and polymerization kinetics. This can cause a serious batch-to-batch reproducibility issue.
Though many oligoethylene glycol functionalized polythiophene derivatives prepared by KCTP for OECT applications have appeared in the literature in recent years,15–18 no such report throws light on the mechanistic aspects of their polymerization. Therefore, in order to develop a KCTP method that is capable of producing ethylene glycol-substituted conjugated polymers in a reproducible manner, a detailed understanding of the polymerization mechanism, particularly the role of reaction parameters, is essential for developing high-performing OECT channel materials.
In this article, we sought to elucidate the mechanistic influences of the ethylene glycol side chain on the monomer's reactivity towards the Grignard metathesis step and eventually the polymerization. We report how the content and length of the ethylene glycol side chain, and the proximity of oxygen atoms influence the formation of the active Grignard monomer. We also show that polymerization in such molecular systems is sensitive to the monomer's concentration. Furthermore, we provide evidence that the KCTP for P3MEEMT is extremely fast and follows a non-controlled pathway contrary to the controlled synthesis of poly(3-hexylthiophene).
2. Results and discussion
The mechanistic aspects of KCTP for polymerization of 3-alkyl-substituted thiophene have been well studied, but no such study is available for the ethylene-glycol functionalized polythiophene derivatives. Therefore, we aim to understand both the formation of the active Grignard monomer and its polymerization to focus on developing the appropriate reaction conditions that would serve as a guide to the synthesis of such polymeric systems for OECT applications. Fig. 1 shows the chemical structures of monomers 2,5-dibromo-3-((2-(2-methoxyethoxy)ethoxy)methyl)thiophene (3MEEMT) (1), 2,5-dibromo-3-((heptyloxy)methyl)thiophene (3PAAT) (2), and 2,5-dibromo-3-(4-(2-(2-methoxyethoxy)ethoxy)butyl)thiophene (3MEEBT) (3) used in this study. It is important to mention here that while all three monomers were investigated for the Grignard metathesis step, polymerization studies were carried out with only monomer 1 (3MEEMT).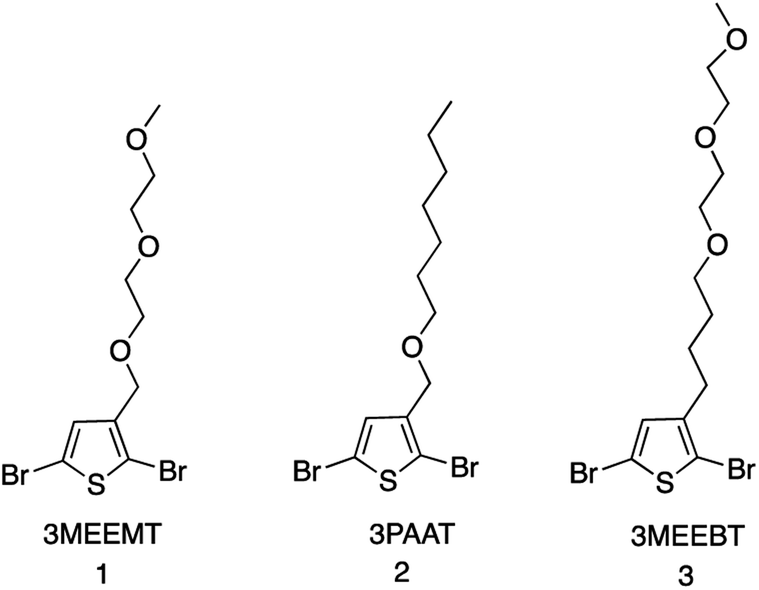 | ||
| Fig. 1 Chemical structures of monomers for the Grignard metathesis reaction. | ||
2.1 Grignard metathesis reaction
For KCTP, the active Grignard monomer is obtained in situ from the dihalomonomer via Grignard metathesis with a suitable Grignard reagent as shown in Scheme 1. The quantitative formation of only one isomer upon Mg/Br exchange of 3MEEMT with iPrMgCl at the 5-position is preferable. However, as shown in the results summarized in Table 1, the selectivity is greatly reduced in 3MEEMT producing two regioisomers A and B in the ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 with the undesirable monomer being the major product, with 15% monomer unexchanged, contrary to the 4:1 ratio obtained in 2,5-dibromo-3-hexylthiophene.26 The preferential formation of isomer B intrigued us to investigate if reducing the oxygen content in the side chain and/or moving it away from the thiophene core would improve the regioselectivity for the Mg/Br exchange in such systems. A significant effect in the formation of two regioisomers was noticed upon Grignard metathesis. A Mg/Br exchange in monomer 3MEEBT (3) produces a 3:1 ratio of isomers A and B when the three oxygen atoms are moved further from the thiophene unit, while the presence of one oxygen in the side chain of monomer 3PAAT (2) leads to the formation of both the isomers in 1:1 ratio on Grignard metathesis with iPrMgCl. The preferential formation of isomer B over A in 3MEEMT may result from coordination between the neighboring oxygen atom of the glycol side chain and the exchange reagent similar to the complexity-induced proximity effect in aromatic ortho-lithiation.27,28 Given the hygroscopic nature of the ethylene glycol chain which can interfere with the Grignard metathesis step, we used a water scavenger29 prior to the addition of the turbo Grignard reagent. Though the water scavenger did not affect the regioselectivity, it led to the failure of polymerization. Furthermore, the ratio of the isomers A and B in 3MEEMT did not change significantly (1:1.8) when iPrMgCl was replaced by the turbo Grignard reagent iPrMgCl·LiCl however the presence of LiCl instead pushed the Mg/Br exchange to completion. Therefore, the turbo Grignard reagent was chosen for the Grignard metathesis for polymerization.
:2 with the undesirable monomer being the major product, with 15% monomer unexchanged, contrary to the 4:1 ratio obtained in 2,5-dibromo-3-hexylthiophene.26 The preferential formation of isomer B intrigued us to investigate if reducing the oxygen content in the side chain and/or moving it away from the thiophene core would improve the regioselectivity for the Mg/Br exchange in such systems. A significant effect in the formation of two regioisomers was noticed upon Grignard metathesis. A Mg/Br exchange in monomer 3MEEBT (3) produces a 3:1 ratio of isomers A and B when the three oxygen atoms are moved further from the thiophene unit, while the presence of one oxygen in the side chain of monomer 3PAAT (2) leads to the formation of both the isomers in 1:1 ratio on Grignard metathesis with iPrMgCl. The preferential formation of isomer B over A in 3MEEMT may result from coordination between the neighboring oxygen atom of the glycol side chain and the exchange reagent similar to the complexity-induced proximity effect in aromatic ortho-lithiation.27,28 Given the hygroscopic nature of the ethylene glycol chain which can interfere with the Grignard metathesis step, we used a water scavenger29 prior to the addition of the turbo Grignard reagent. Though the water scavenger did not affect the regioselectivity, it led to the failure of polymerization. Furthermore, the ratio of the isomers A and B in 3MEEMT did not change significantly (1:1.8) when iPrMgCl was replaced by the turbo Grignard reagent iPrMgCl·LiCl however the presence of LiCl instead pushed the Mg/Br exchange to completion. Therefore, the turbo Grignard reagent was chosen for the Grignard metathesis for polymerization.
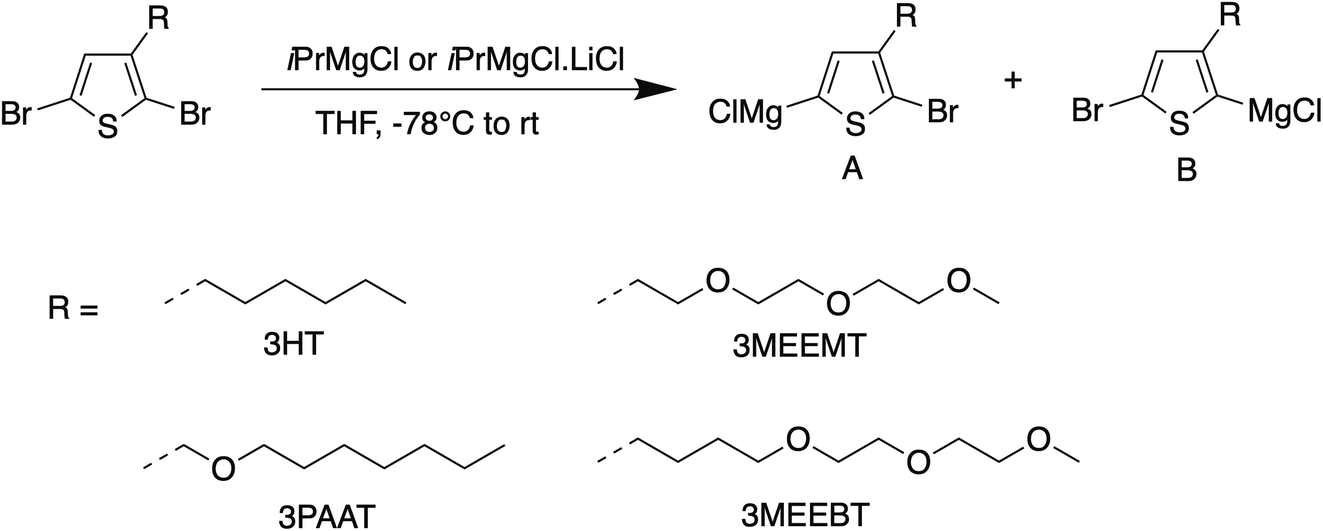 | ||
| Scheme 1 Grignard metathesis of monomers 1–3. | ||
2.2 Polymerization studies
After developing the appropriate conditions for the Grignard metathesis step, we focused on screening the polymerization conditions. All the polymerization studies were carried out at room temperature using Ni(dppe)Cl2 catalyst in a 1 mol% ratio to the 3MEEMT monomer (Scheme 2). We found that the polymerization of 3MEEMT via KCTP was affected by monomer concentration. A preliminary study with four different concentrations of the monomer 0.3, 0.15, 0.07, and 0.03 M revealed that a minimum monomer concentration of 0.15 M was needed to obtain the polymer P3MEEMT with the lower concentrations of 0.03 and 0.07 M affording no polymer highlighting the sensitivity of the polymerization to the monomer's concentration. Based on this observation, polymerization was carried out with 0.3 M monomer concentration and the polymerization kinetics was investigated by GPC by withdrawing a 0.2 mL aliquot from the reaction mixture at t = 1, 2, 5, and 10 minutes (Table 2). A concentration-dependent study for P3HT synthesis available in the literature30 with three different monomer concentrations, 0.1, 0.25, 0.5 M, indicated that for 0.1 and 0.25 M an increase in Mn was noted during the first 20 minutes of the polymerization followed by no significant change during the 1 hour course of the polymerization. While for 0.5 M, Mn increased significantly during the first 10 minutes. Most of the P3HT syntheses in literature are performed using 0.1 M monomer concentration.31,32 A higher minimum monomer concentration required for P3MEEMT compared to P3HT may reflect the lower amount of the desired Grignard monomer produced in the initial Mg/Br exchange.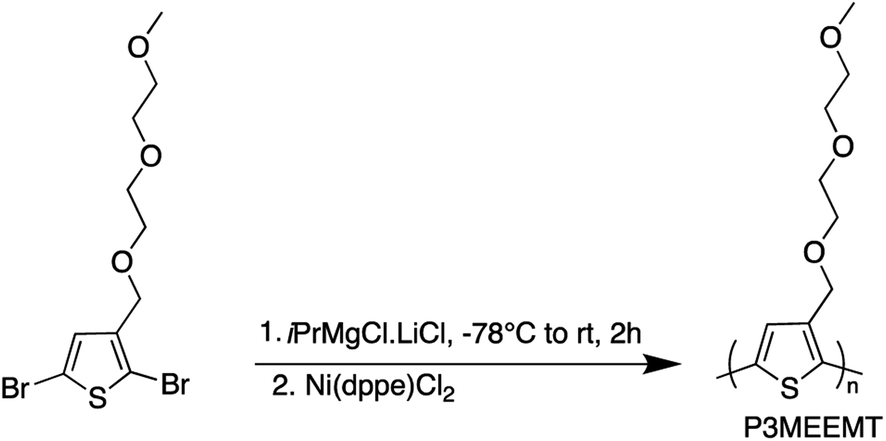 | ||
| Scheme 2 Synthesis of P3MEEMT by KCTP. | ||
| Time (min) | M n (g mol−1) | M w (g mol−1) | Đ |
|---|---|---|---|
| 1 | 18200 |
38000 |
2.0 |
| 2 | 13200 |
30000 |
2.3 |
| 5 | 10300 |
15700 |
1.5 |
| 10 | 10000 |
16400 |
1.6 |
Analysis of GPC data indicates that polymerization is extremely fast with no change or even a decline in the number average molecular weight (Mn) after 1 minute contrary to our and others' common practice where the polymerization is left for 1–4 hours.16,21,22 Insoluble material starts to appear after 10 minutes. GPC of the soluble fraction of the polymer obtained after 10 minutes indicates a polymer with Mn of 10000 g mol−1. The polymerization is significantly faster than that of P3HT where 1 hour is often needed.33 A characteristic of KCTP is the ability to control the molecular weight of the product by altering the monomer-to-catalyst ratio. Based on this, and if KCTP was operating, we would expect a polymer with an Mn of 7000 g mol−1 to form. However, this Mn is not achieved and the dispersities are high suggesting an uncontrolled polymerization.
Further studies are required to elucidate the origin of this extremely fast polymerization. We speculate that the polymerization kinetics observed in the synthesis of P3MEEMT may result from either the coordination of oxygen atoms in the ethylene glycol side chain with Li and/or Mg, or due to a radical-mediated path.27,34 Also, a literature report by Hu and co-workers mention that an electron-rich nucleophile facilitates Kumada coupling.35 Elucidating the origin of the fast polymerization is a subject of future work.
3. Conclusions
In conclusion, we have shown that both the content of the ethylene glycol chain and the proximity of the oxygen atom play a decisive role in the formation of the active Grignard monomer obtained upon the Grignard metathesis with glycolated thiophene derivatives. Furthermore, the use of the turbo-Grignard reagent assists the Mg/Br exchange towards completion without affecting the ratio of regioisomers formed during the reaction. In addition, it was found that the polymerization is sensitive to monomer concentration needing at least 0.15 M of the monomer. We further show that the polymerization of P3MEEMT is extremely fast indicating uncontrolled chain growth which may have been associated with either the coordination of Li and/or Mg by oxygen of the side chain or a radical-mediated polymerization path leading to loss of polymerization control. Further studies are currently being carried out to understand the mechanism involved in the polymerization.4. Experimental section
4.1 Materials and methods
All the chemicals were purchased from either Sigma-Aldrich or Tokyo Chemical Industries and used as received. Deuterated solvents were obtained from EURISO-TOP. Trimethoxybenzene was used as an internal standard for 1H NMR studies. Anhydrous dichloromethane (DCM) was dried and deoxygenated on a Glass Contour solvent purification system. Anhydrous tetrahydrofuran (THF) was purchased from Sigma-Aldrich and used as received. All the reactions were performed using standard Schlenk techniques under a dry nitrogen atmosphere. All the reaction flasks were dried overnight in an oven at 120 °C before use. 1H NMR spectra were recorded on either a Bruker 400 or 500 MHz spectrometer using CDCl3 or CD2Cl2 solvents. The synthesized polymers were characterized with size exclusion chromatography using a Malvern Viscotek TDA 305 GPC equipped with a UV detector. Molecular weight was determined from GPC using THF as eluent at a temperature of 40 °C and a flow rate of 1 mL min−1 and molecular weights were determined relative to polystyrene standards.4.2 Monomer synthesis
Monomers 1 and 2 were synthesized by following the procedure reported in the literature.21,22 For measuring the ratio of regioisomers formed during Grignard metathesis, 0.2 mL of the aliquot was taken from the reaction mixture and added to a vial containing 1.0 mL of chloroform and quenched with 0.5 M HCl. The organic layer was dried over anhydrous sodium sulfate followed by evaporation of the solvent in vacuo. 0.6 mL of CDCl3/CD2Cl2 was added to each dried vial and analyzed by 1H NMR.4.3 Polymer synthesis
Polymerization of 3MEEMT was carried out by following the procedure reported in the literature.21,22 2,5-Dibromo-3-((2-(2-methoxyethoxy)ethoxy)methyl)thiophene (225.0 mg, 0.602 mmol) and the reference standard, 1,3,5-trimethoxybenzene (5.0 mg, 0.03 mmol) were degassed for 30 min with N2. 2 mL of THF was added, then the flask was cooled to −78 °C and iPrMgCl·LiCl (87.0 mg, 0.46 mL, 0.602 mmol, 1.3 M) was added dropwise over 10 min and the reaction was allowed to proceed for 2 h at room temperature. Ni(dppe)Cl2 (3.0 mg, 0.006 mmol, 1 mol%) was added in one portion, and the polymerization was allowed to continue for 24 h at room temperature. To quench the polymerization, HCl (0.5 mL, 5.0 M) was added to the reaction, and the polymer was precipitated into 150 mL of MeOH. The polymer was purified using successive Soxhlet extractions using hexanes, and MeOH, and then collected with CHCl3.The obtained polymer was again precipitated into MeOH, filtered to be collected as a powder, and dried overnight in a vacuum oven. 1H NMR (400 MHz, CD2Cl2) δ 7.22 (s, 1H), 4.58 (s, 2H), 3.64 (d, J = 5.5 Hz, 2H), 3.62–3.57 (m, 2H), 3.54 (dd, J = 5.8, 3.5 Hz, 3H), 3.45–3.41 (m, 2H), 3.24 (d, J = 1.3 Hz, 4H).
For polymerization kinetics, 0.2 mL of the aliquot was taken from the reaction mixture at the time intervals of 1, 2, 5 and 10 minutes and added to a vial containing 1.0 mL of chloroform and quenched with 0.5 M HCl. The organic layer was dried over anhydrous sodium sulfate followed by evaporation of the solvent in vacuo. The solid residue was dried under vacuum at 60 °C overnight and GPC was recorded.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This was supported by funding from the Okinawa Institute of Science and Technology and JSPS KAKENHI Grant Number JP22K20547.References
- Y. Fang, L. Meng, A. Prominski, E. N. Schaumann, M. Seebald and B. Tian, Chem. Soc. Rev., 2020, 49, 7978–8035 RSC.
- J. Rivnay, R. M. Owens and G. G. Malliaras, Chem. Mater., 2014, 26, 679 CrossRef CAS.
- T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379 CrossRef CAS PubMed.
- A. Marks, S. Griggs, N. Gasparini and M. Moser, Adv. Mater. Interfaces, 2022, 9, 2102039 CrossRef CAS.
- A. M. Pappa, D. Ohayon, A. Giovannitti, I. P. Maria, A. Savva, I. Uguz, J. Rivnay, I. McCulloch, R. M. Owens and S. Inal, Sci. Adv., 2018, 4, eaat0911 CrossRef.
- D. Khodagholy, T. Doublet, P. Quilichini, M. Gurfinkel, P. Leleux, A. Ghestem, E. Ismailova, T. Hervé, S. Sanaur, C. Bernard and G. G. Malliaras, Nat. Commun., 2013, 4, 1575 CrossRef PubMed.
- A. Melianas, T. J. Quill, G. LeCroy, Y. Tuchman, H. V. Loo, S. T. Keene and A. Giovannitti, Sci. Adv., 2020, 6, eabb2958 CrossRef CAS PubMed.
- J. Rivnay, S. Inal, A. Salleo, R. M. Owens, M. Berggren and G. G. Malliaras, Nat. Rev. Mater., 2018, 3, 17086 CrossRef CAS.
- X. Wu, Q. Liu, A. Surendran, S. E. Bottle, P. Sonar and W. L. Leong, Adv. Electron. Mater., 2021, 7, 2000701 CrossRef CAS.
- S. Inal, J. Rivnay, P. Leleux, M. Ferro, M. Ramuz, J. C. Brendel, M. M. Schmidt, M. Thelakkat and G. G. Malliaras, Adv. Mater., 2014, 26, 7450–7455 CrossRef CAS PubMed.
- M. Moser, J. F. Ponder Jr, A. Wadsworth, A. Giovannitti and I. McCulloch, Adv. Funct. Mater., 2018, 1807033 Search PubMed.
- N. A. Kukhta, A. Marks and C. K. Luscombe, Chem. Rev., 2022, 122, 4325–4355 CrossRef CAS PubMed.
- P. Li and T. Lei, J. Polym. Sci., 2022, 60, 377–392 CrossRef CAS.
- J. H. Kim, S.-M. Kim, G. Kim and M.-H. Yoon, Macromol. Biosci., 2020, 20, 2000211 CrossRef CAS PubMed.
- B. T. DiTullio, L. R. Savagian, O. Bardagot, M. De Keersmaecker, A. M. Österholm, N. Banerji and J. R. Reynolds, J. Am. Chem. Soc., 2023, 145, 122–134 CrossRef CAS PubMed.
- P. Schmode, A. Savva, R. Kahl, D. Ohayon, F. Meichsner, O. Dolynchuk, T. Thurn-Albrecht, S. Inal and M. Thelakkat, ACS Appl. Mater. Interfaces, 2020, 12, 13029–13039 CrossRef CAS.
- E. Tan, J. Kim, K. Stewart, C. Pitsalidis, S. Kwon, N. Siemons, J. Kim, Y. Jiang, J. M. Frost, D. Pearce, J. E. Tyrrell, J. Nelson, R. M. Owens, Y.-H. Kim and J.-S. Kim, Adv. Mater., 2022, 34, 2202574 CrossRef CAS.
- I. P. Maria, B. D. Paulsen, A. Savva, D. Ohayon, R. Wu, R. Hallani, A. Basu, W. Du, T. D. Anthopoulos, S. Inal, J. Rivnay, I. McCulloch and A. Giovannitti, Adv. Funct. Mater., 2021, 31, 2008718 CrossRef CAS.
- K. Feng, W. Shan, J. Wang, J.-W. Lee, W. Yang, W. Wu, Y. Wang, B. J. Kim, X. Guo and H. Guo, Adv. Mater., 2022, 34, 2201340 CrossRef CAS.
- I. P. Maria, S. Griggs, R. B. Rashid, B. D. Paulsen, J. Surgailis, K. Thorley, V. N. Le, G. T. Harrison, C. Combe, R. Hallani, A. Giovannitti, A. F. Paterson, S. Inal, J. Rivnay and I. McCulloch, Chem. Mater., 2022, 34(19), 8593–8602 CrossRef CAS.
- L. Q. Flagg, C. G. Bischak, J. W. Onorato, R. B. Rashid, C. K. Luscombe and D. S. Ginger, J. Am. Chem. Soc., 2019, 141, 4345–4354 CrossRef CAS.
- S. E. Chen, L. Q. Flagg, J. W. Onorato, L. J. Richter, J. Guo, C. K. Luscombe and D. S. Ginger, J. Mater. Chem. A, 2022, 10, 10738–10749 RSC.
- B. X. Dong, C. Nowak, J. W. Onorato, T. Ma, J. Niklas, O. G. Poluektov, G. Grocke, M. F. DiTusa, F. A. Escobedo, C. K. Luscombe, P. F. Nealey and S. N. Patel, Chem. Mater., 2021, 33, 741–753 CrossRef CAS.
- S. Cheng, R. Zhao and D. S. Seferos, Acc. Chem. Res., 2021, 54(22), 4203–4214 CrossRef CAS.
- R. H. Lohwasser and M. Thelakkat, Macromolecules, 2011, 44, 3388–3397 CrossRef CAS.
- R. S. Loewe, S. M. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250–253 CrossRef CAS.
- S. Vandeleene, K. Van den Bergh, T. Verbiest and G. Koeckelberghs, Macromolecules, 2008, 41, 5123–5131 CrossRef CAS.
- A. Desaintjean, T. Haupt, L. J. Bole, N. R. Judge, E. Hevia and P. Knochel, Angew. Chem., Int. Ed., 2021, 60, 1513–1518 CrossRef CAS.
- S. Cheng, S. Ye, C. N. Apte, A. K. Rudin and D. S. Seferos, ACS Macro Lett., 2021, 10, 697–701 CrossRef CAS PubMed.
- B. C. Achord and J. W. Rawlins, Macromolecules, 2009, 42, 8634–8639 CrossRef CAS.
- D. S. Dissanayake, E. Sheina, M. C. Biewer, R. D. McCullough and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 79–82 CrossRef CAS.
- J.-P. Lamps and J.-M. Catala, Macromolecules, 2011, 44, 7962–7968 CrossRef CAS.
- J. Buenaflor, P. Sommerville, H. Qian and C. Luscombe, Macromol. Chem. Phys., 2020, 221, 1900363 CrossRef CAS.
- G. Manolikakes and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 205–209 CrossRef CAS.
- O. Vechorkin, V. Proust and X. Hu, J. Am. Chem. Soc., 2009, 131, 9756–9766 CrossRef CAS.
Footnote |
| † Contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2024 |