
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Amine functionalized surface frustrated Lewis pairs boost CO2 photocatalysis†‡
Qinhui
Guan
a,
Chengzhe
Ni
a,
Tingjiang
Yan
 *ab,
Na
Li
*ac,
Lu
Wang
d,
Zhe
Lu
d,
Weiguang
Ran
a,
Yipin
Zhang
a,
Wenjuan
Li
a,
Lulu
Zhang
a,
Dapeng
Zhang
a,
Baibiao
Huang
c and
Geoffrey A.
Ozin
*e
*ab,
Na
Li
*ac,
Lu
Wang
d,
Zhe
Lu
d,
Weiguang
Ran
a,
Yipin
Zhang
a,
Wenjuan
Li
a,
Lulu
Zhang
a,
Dapeng
Zhang
a,
Baibiao
Huang
c and
Geoffrey A.
Ozin
*e
aKey Laboratory of Catalytic Conversion and Clean Energy in Universities of Shandong Province, School of Chemistry and Chemical Engineering, Qufu Normal University, Qufu, 273165, P.R. China. E-mail: tingjiangn@163.com; lina20201130@163.com
bCollege of Chemistry and Chemical Engineering, Shaanxi University of Science and Technology, Xi’an, 710021, P.R. China
cState Key Laboratory of Crystal Materials, Shandong University, Jinan, 250100, P.R. China
dSchool of Science and Engineering, The Chinese University of Hong Kong, Shenzhen, 518172, Shenzhen, Guangdong, P.R. China
eMaterials Chemistry and Nanochemistry Research Group, Solar Fuels Cluster, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, ON M5S 3H6, Canada. E-mail: gozin@chem.utoronto.ca
First published on 9th January 2024
Abstract
The archetype surface frustrated Lewis pair (SFLP) that facilitates CO2 photocatalytic hydrogenation to methanol and carbon monoxide, is an InOH⋯In site positioned in the surface of a nanoscale indium oxide hydroxide, denoted In2O3−x(OH)y. Proximal Lewis acid In(III) and Lewis base InOH of this genre serve as surface active sites that enable the photochemical heterolytic H2 dissociation and reduction of CO2 to the mentioned products. The conversion rate enabled by light has been found to far exceed that enabled by heat. Efforts to enhance the CO2 photocatalytic performance of the SFLP have involved modifications of the Lewis acidity and basicity through isomorphic substitution of In(III) with Bi(III) and changes in the population of oxygen vacancies through control of oxide non-stoichiometry. Replacement of the Lewis base hydroxide InOH by the stronger Lewis base amine InNH2 heretofore remains unexplored. The strategy described herein to explore this opportunity begins with the synthesis of In2O3−x(EDA)y. This new material is proven to contain an InNH2⋯In SFLP and its CO2 photocatalytic performance is demonstrated to outperform that of its In2O3−x(OH)y progenitor. Tailored Lewis acidity and basicity surfaces bring CO2 photocatalysis another step closer to the vision of solar CO2 refineries.
Broader contextIn heterogeneous catalysis the surface frustrated Lewis pair, SFLP, has played a central role in the hydrogenation of CO2 to commodity chemicals exemplified by syngas, methanol, methane, and higher hydrocarbons. Notably, all prior reports of SFLP in CO2 catalysis involve a Lewis acid metal M site proximal to a Lewis base hydroxide MOH site, described as M⋯MOH. The thermochemical and photochemical activity of the SFLP is determined by its electronic ground (thermocatalysis) and excited (photocatalysis) state Lewis acidity and basicity. To this end, the acidity of the Lewis site has previously been investigated by modifying the effective charge on the metal site. This has been curated by changes in the ionic radius, oxidation state, and electronegativity of the metal. This goal has been achieved in the work described herein by switching the Lewis basic hydroxide site M⋯MOH to a stronger Lewis basic amine M⋯MNH2, a change found to boost photocatalytic CO2 hydrogenation performance, one more step to the vision of CO2 refineries powered by light. |
Introduction
Photocatalytic conversion of CO2 into valuable chemical fuels has emerged as a potential solution to address both energy and environmental concerns. However, breaking the highly stable C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in CO2 molecules remains a challenging task. Previous studies have shown that surface oxygen vacancies (OVs) on metal oxides exhibit high catalytic activity and can effectively activate and split CO2.1–3 The thermodynamically metastable OVs, which are formed through hydrogen reduction or hydroxyl dehydration, facilitate the incorporation of CO2 oxygen atoms into the oxide sublattice, enabling subsequent CO2 hydrogenation. More recently, heterogeneous catalysts with solid surface frustrated Lewis pair (SFLP) have been developed for various significant catalytic reactions, including CO2 hydrogenation, hydroamination, and methane conversion.4,5 SFLP consists of Lewis acidic and Lewis basic sites in close proximity, preventing their interaction and forming Lewis acid–base pair that synergistically activates small molecules such as H2, CO2, and CH4. The existing solid SFLP systems can be categorized into two types: isolated OVs systems and OH–OVs systems.6–11 Isolated OVs systems, represented by CeO2−x, exhibit a high density of surface OVs, which leads to adjacent metal atoms with unsaturated coordination; thus, creating independent Lewis acidic and basic sites.12 On the other hand, OH–OVs systems typically involve a surface metal site with unsaturated coordination (Lewis acid) located near oxygen vacancies and a surface hydroxide site (Lewis base). Examples of such systems include defect-laden hydroxylated oxides like In2O3−x(OH)y, TiO2−x(OH)y, and CoGeO2(OH)2.7,13,14 However, achieving a balanced concentration between OVs and hydroxide sites in this manner is challenging. Although our recent research demonstrated that the reactivity of Lewis acidic sites can be controlled through the replacement of In2O3 with single-site Bi3+ ion, maximizing the reactivity of SFLP by simultaneously creating high-level Lewis acidic and basic sites remains a significant challenge.15
O bonds in CO2 molecules remains a challenging task. Previous studies have shown that surface oxygen vacancies (OVs) on metal oxides exhibit high catalytic activity and can effectively activate and split CO2.1–3 The thermodynamically metastable OVs, which are formed through hydrogen reduction or hydroxyl dehydration, facilitate the incorporation of CO2 oxygen atoms into the oxide sublattice, enabling subsequent CO2 hydrogenation. More recently, heterogeneous catalysts with solid surface frustrated Lewis pair (SFLP) have been developed for various significant catalytic reactions, including CO2 hydrogenation, hydroamination, and methane conversion.4,5 SFLP consists of Lewis acidic and Lewis basic sites in close proximity, preventing their interaction and forming Lewis acid–base pair that synergistically activates small molecules such as H2, CO2, and CH4. The existing solid SFLP systems can be categorized into two types: isolated OVs systems and OH–OVs systems.6–11 Isolated OVs systems, represented by CeO2−x, exhibit a high density of surface OVs, which leads to adjacent metal atoms with unsaturated coordination; thus, creating independent Lewis acidic and basic sites.12 On the other hand, OH–OVs systems typically involve a surface metal site with unsaturated coordination (Lewis acid) located near oxygen vacancies and a surface hydroxide site (Lewis base). Examples of such systems include defect-laden hydroxylated oxides like In2O3−x(OH)y, TiO2−x(OH)y, and CoGeO2(OH)2.7,13,14 However, achieving a balanced concentration between OVs and hydroxide sites in this manner is challenging. Although our recent research demonstrated that the reactivity of Lewis acidic sites can be controlled through the replacement of In2O3 with single-site Bi3+ ion, maximizing the reactivity of SFLP by simultaneously creating high-level Lewis acidic and basic sites remains a significant challenge.15
Incorporating amine functional groups has been widely employed to enhance CO2 capture efficiency in CO2 photocatalysis. Amine functionalization of various photocatalysts, such as oxides, sulfides, and graphitic carbon nitride, enables substantial CO2 adsorption through acid–base interactions.16–18 The adsorbed amine groups on photocatalysts can activate CO2 molecules by forming carbamate intermediates, resulting in a significant distortion of the original linear CO bond and reducing the energy barrier for C–O bond formation.19 The previous work by Lu et al. demonstrated that amine groups can act as Lewis basic sites, coupling with Lewis acidic sites (e.g., Au surface) to form solid-molecule SFLP. This SFLP configuration enables the activation of H2 and subsequent hydrogenation of imines and nitriles.20 In contrast to the aforementioned SFLP types, amine-functionalized SFLP systems circumvent the reliance on hydroxyl groups to generate OVs, potentially maximizing the reactivity of SFLP. Moreover, the amine group possesses higher electronegativity than the hydroxyl group, suggesting its potential as a more active Lewis basic site. Consequently, grafting amine groups onto defect-laden oxides by replacing hydroxyl groups could result in the formation of NH2–OVs systems, offering enhanced reactivity and providing strong Lewis acid and base sites to facilitate CO2 adsorption and activation, thereby improving photocatalytic CO2 reduction performance.
In this study, we present a one-pot solvothermal approach for synthesizing amine-functionalized In2O3 catalysts for gas-phase CO2 hydrogenation photocatalysis. Extensive characterization techniques, coupled with density functional theory (DFT) calculations, are employed to gain valuable insights into the formation of InNH2⋯In SFLP systems, the surface and electronic nature of the active sites, and the identification of key intermediates during photocatalytic CO2 hydrogenation. The findings of this study provide a foundation for sustainable chemical fuel production through the engineering of molecule-functionalized metal oxide-based heterogeneous CO2 photocatalysis.
Results and discussion
A facile one-pot solvothermal method was employed to synthesize amine-functionalized In2O3 nanocrystals (E-IO) using In(NO3)3 as the indium precursor and ethylenediamine (EDA) as both the reaction medium and functionalizing agent. As illustrated in Fig. 1A, EDA initially chelates with indium ions in the precursor solution, forming Inn(EDA)m3+ complexes. These complexes then react with OH− ions, derived from the hydrolysis of EDA, resulting in the generation of Inn(EDA)m−x(OH)(3−x)+x intermediates. These intermediates gradually dissociate and condense, forming small In2O3 nuclei with EDA attached to the surface through hydrogen bonding by single coordination according to the previous reports.21,22 The newly formed In2O3 nuclei tend to rapidly attach together, minimizing surface energy. Subsequently, they undergo further rearrangement and crystallization through a “self-assembly” aggregation growth mechanism.23,24 SEM and TEM images of the amine-functionalized In2O3 nanocrystals are shown in Fig. 1B and C, respectively. These images reveal a predominantly spherical morphology with an average diameter of approximately 30 nm. The In2O3 nanospheres are composed of small agglomerated nanoparticles with average diameters around 6.50 nm (Fig. 1D). HRTEM analysis in Fig. 1E shows well-defined lattice fringes with an interplanar distance of 0.293 nm, corresponding to the (222) facet of the cubic structure of In2O3. The SAED pattern displayed in Fig. 1F exhibits multiple rings composed of individual spots, indicating the polycrystalline nature of the amine-functionalized In2O3 nanocrystals. EDS mapping of the nanospheres confirms the homogeneous distribution of In, O, and N elements in the In2O3 nanocrystals (Fig. 1G). The atomic content of N in the E-IO sample was determined to be approximately 5.51 wt% (Fig. S1, ESI‡).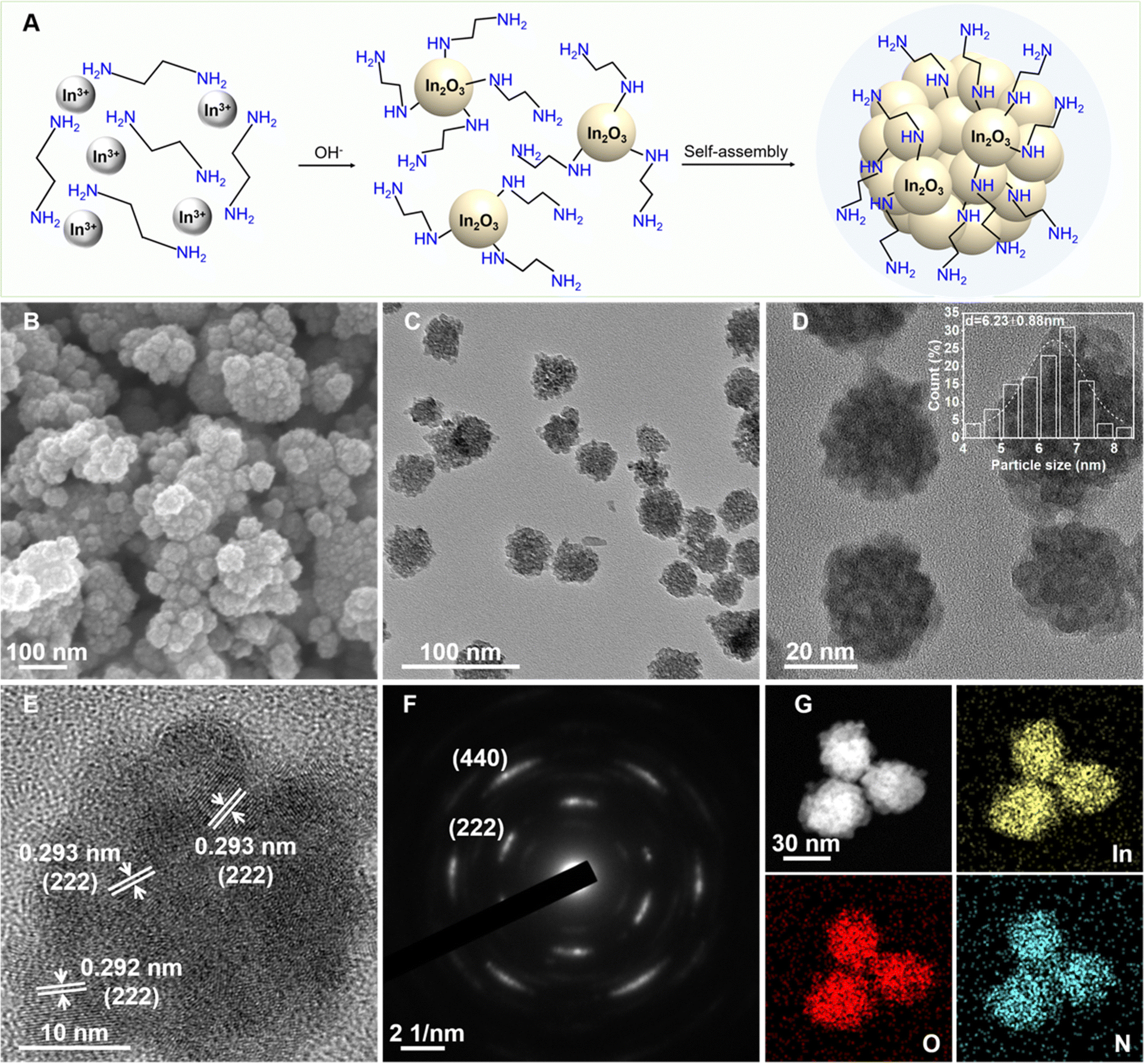 | ||
| Fig. 1 Synthesis and morphology of amine-functionalized In2O3 nanocrystals. (A) Schematic of the synthetic route. (B) SEM images. (C)–(E) TEM images, particle size distribution (inset) and HRTEM images. (F) SAED pattern. (G) EDS mapping. | ||
To gain a deeper understanding of the role played by amine groups in the active In2O3-based photocatalysts, we synthesized indium hydroxide via a hydrothermal method (Scheme S1, ESI‡), where only water was used as the reactant solvent. The resulting indium hydroxide was then calcined to produce In2O3 with a fresh surface.25 TEM images of the hydrothermally synthesized In2O3 sample (H-IO) (Fig. S2A–C, ESI‡) reveal that it is a cube-shaped nanoparticle with an edge length exceeding 400 nm. The significant difference in particle size between E-IO and H-IO makes it challenging to determine whether the variation in photocatalytic activity is attributable to particle size or surface amine groups. To directly compare the In2O3 samples with and without amine functionalization, we further synthesized In2O3 (Si-IO) with smaller particle size using SiO2 nanospheres as templates in an aqueous solution. The resulting In2O3 sample consists of small agglomerated nanorods with a diameter of approximately 10 nm (Fig. S2D–F, ESI‡), which is comparable in particle size to the E-IO sample.
Fig. S3 (ESI‡) presents the XRD patterns of the In2O3 samples with and without amine functionalization. The patterns of all three samples exhibit similar diffraction peaks that can be assigned to the cubic phase of indium oxide (c-In2O3) (JCPDS no. 71-2195). The crystallite sizes estimated using the Scherrer equation are 6.41 nm for E-IO, 29.95 nm for H-IO, and 9.47 nm for Si-IO (Table S1, ESI‡). Nitrogen adsorption/desorption isotherms of the three samples are shown in Fig. S4 (ESI‡), revealing the presence of mesopores in all In2O3 samples. The specific surface area follows the order of Si-IO (94.97 m2 g−1) > E-IO (50.54 m2 g−1) > H-IO (15.67 m2 g−1). Although a larger surface area may facilitate CO2 adsorption, the subsequent photocatalytic results indicate that surface area is not the primary factor contributing to the improved photocatalytic activity in CO2 hydrogenation.
To confirm the successful functionalization of amine groups on the surface of In2O3 nanocrystals, several characterizations, including FTIR, TG-MS, XPS, and NMR were conducted on the collected samples. In the FTIR spectra of the three In2O3 samples (Fig. S5, ESI‡), four intense peaks centered at 602, 565, 538, and 428 cm−1 are observed, corresponding to the vibration peaks of In–O bonds in cubic In2O3.26 The peak at 1383 cm−1 corresponds to the stretching vibration of OH bonds, with the H-IO sample exhibiting the strongest OH peak due to the abundance of surface OH groups. Notably, the E-IO sample displays several new peaks centered at 3241, 2947, 2890, 1561, 1495, 1343, 1270, 1178 and 1113 cm−1 (Fig. 2A). According to previous studies and the measured IR spectra of EDA reference, the peaks at 3241 and 1561 cm−1 correspond to the symmetric bending vibration of NH,27,28 while the peaks at 1343, 1270, 1178 and 1113 cm−1 can be attributed to the stretching vibration of C–N.29–32 Additionally, peaks centered at 2947, 2890 and 1495 cm−1 can be assigned to CH2 vibrational bands.27,28,32 TG-MS analysis was further performed to confirm the presence of amine groups in the E-IO samples. It is evident that E-IO exhibits significant weight loss during thermal treatment in the temperature range of 30 to 520 °C, compared to the other two samples (Fig. S6, ESI‡). This weight loss can be attributed to the release of ethylenediamine molecules bonded to the surface of E-IO upon thermal treatment. The TG-MS data (Fig. 2B) reveal that the E-IO sample has a substantial amount of physically and chemically absorbed H2O on the surface, which is desorbed at around 150 and 280 °C, respectively. Physically absorbed CO2 is completely removed at approximately 200 °C. Additionally, the observed CO2 in the range of 200–400 °C can be attributed to the decomposition of amine groups, which is supported by the presence of a mass spectrometer signal for NO with m/z value of 30 starting from 245 °C.33–35 Of note, the additional Raman (Fig. S7, ESI‡) and wide-range N 1s XPS (Fig. S8, ESI‡) results can exclude the possibility of the generation of NO deriving from salt anion (NO3−). Furthermore, according to the previous reports shown in Table S2 (ESI‡), the N 1s XPS spectrum of E-IO exhibits three distinct peaks: O–In–N bonding at 399.55 eV,36 C–N bonding at 398.30 eV and coordinately-bound NH groups at 400.39 eV (Fig. 2C).37,38 What's more, as shown in Fig. S9 and S10 (ESI‡), the three peaks detected in the C 1s XPS spectrum of E-IO can be attributed to the C–C bonding (284.60 eV), C–N bonding (285.98 eV) and O–CO bonding (288.83 eV), respectively,39,40 and the In 3d5/2 and In 3d1/2 peaks of E-IO are conventionally deconvoluted to In–N bonding (443.59 eV and 251.18 eV), and In–O bonding (444.17 eV and 451.73 eV).41 Through these results, the existence state of EDA on the surface of E-IO can be confirmed: the amine group at one end of EDA is coordinated with the indium atom of In2O3, while the amine group at the other end exists in an uncoordinated NH2 group state. Solid-state NMR experiment can further provide strong evidence for the successful grafting of EDA molecular onto the catalyst surface. As shown in Fig. 2D, the 13C magic angle spinning nuclear magnetic resonance (13C MAS NMR) spectrum for the E-IO presents the characteristic carbon resonance signals at 39.1 ppm, which can be assigned to the C atom in CH2 groups arising from EDA molecular. Moreover, an additional resonance peak at 167.0 ppm is clearly observed and should be attributed to the C atom in carbamate groups that are formed by the binding of NH2 groups with CO2 in the air.42–44 Collectively, all these results confirm the successful grafting of amine groups onto the surface of E-IO nanoparticles during the solvothermal process.
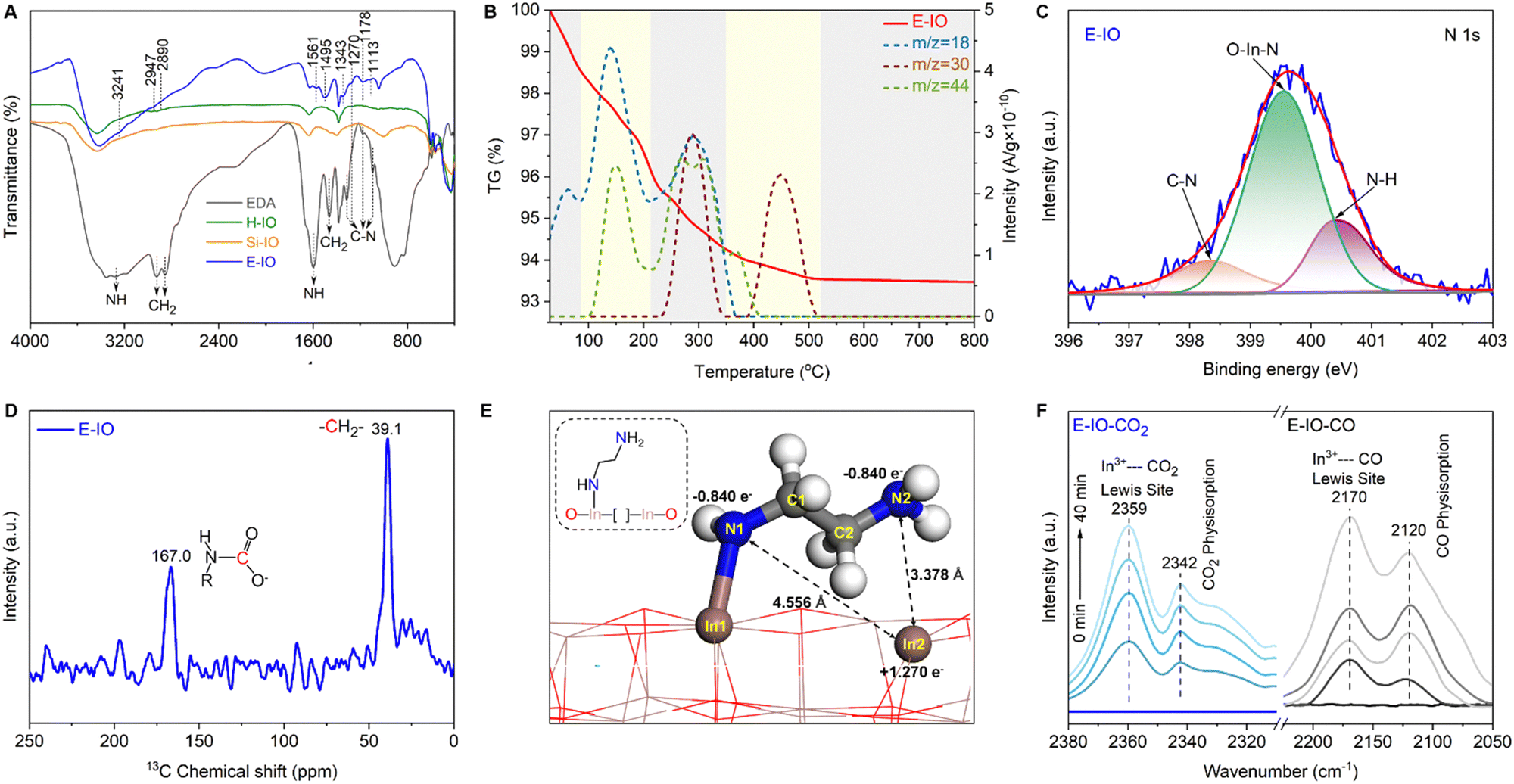 | ||
| Fig. 2 Characterization of samples (A) FTIR spectra of H-IO, Si-IO and E-IO. (B) TGA-MS curves of E-IO in air condition. (C) High resolution N 1s XPS spectra of E-IO. (D) 13C MAS NMR spectra of E-IO at room temperature. (E) Optimized structure of the grafting state InNH2⋯In. (F) In situ DRIFTS spectra of the adsorption of CO2 (left) and CO (right) on the E-IO surface at 298 K. | ||
Ethylenediamine was employed as both an appropriate amine-modifying reagent and a facilitator for generating a significant quantity of oxygen vacancies during the synthesis of amine-functionalized In2O3 nanocrystals. The presence and semi-quantitative analysis of oxygen vacancies were determined using XPS and EPR techniques. Fig. S11A–C (ESI‡) shows the high-resolution XPS spectra of the O 1s region in the H-IO, Si-IO, and E-IO samples. The spectra exhibit three distinct peaks at binding energies of 529.35, 530.86, and 531.81 eV, corresponding to lattice oxides, oxygen vacancies, and hydroxyl groups, respectively.9,45 Notably, E-IO displays a significantly higher oxygen vacancy concentration of 34.43% compared to H-IO (20.68%) and Si-IO (18.15%). Additionally, the binding energy of the In 3d core level in E-IO exhibits a noticeable shift toward lower binding energy (Fig. S11D, ESI‡), indicating an unsaturated state of indium in the amine-functionalized In2O3 nanocrystals and a higher surrounding electron density. This observation further confirms the presence of a substantial number of oxygen vacancies in the amine-functionalized In2O3 sample. EPR spectroscopy is an effective semi-quantitative characterization method for evaluating oxygen vacancies. As depicted in Fig. S12 (ESI‡), all three samples exhibit an EPR signal related to oxygen vacancies, appearing at the g-value of 2.003.46 Notably, E-IO displays the strongest EPR signal, indicating that the presence of ethylenediamine promotes the formation of OVs during the solvothermal synthesis. Consequently, based on the XPS and EPR findings, it can be concluded that the amine-functionalized In2O3 sample possesses a substantial number of surface oxygen vacancies. As a higher population of oxygen vacancies corresponds to a lower oxygen coordination number around the In(III) sites, the coordinately unsaturated In(III) becomes more Lewis acidic. Furthermore, the substitution of hydroxyl groups with amine groups, which possess stronger electronegativity in the amine-functionalized In2O3 nanocrystals, makes them more Lewis basic. Consequently, the amine-functionalized In2O3 (E-IO) should have intensified Lewis acid and Lewis base sites, showing promising potential in enhancing photocatalytic performance for CO2 reduction.
Density functional theory (DFT) slab calculations were conducted to validate the SFLP property achieved by substituting EDA for hydroxide groups on the In2O3−x(OH)y surface. Previous studies have demonstrated that SFLP, consisting of an unsaturated In atom as the Lewis acid site and a hydroxide group as the Lewis base site, can be constructed by removing a surface oxygen atom and introducing a hydroxide group on the (110) facet of cubic In2O3.15,47,48 Herein, we investigate the optimized structure of the substitution of EDA at the hydroxide group site. As shown in Fig. 2E, two kinds of surface configurations, In2–N1 and In2–N2, are identified on the amine-functionalized surface. The In2–N1 and In2–N2 with respective distances of 4.556 Å and 3.378 Å fall in the domain of solid SFLP. The Bader charge calculations indicate that the related In2, N1 and N2 pairs have atomic local charges of +1.27e, −0.84e and −0.84e, respectively. Compared with In2–N1 configuration, In2–N2 with a shorter distance may deliver a higher capability to activate reactant molecules.49
The existence of unsaturated In atom as Lewis acid site is essential for the construction of InNH2⋯In SFLP, where the NH2 in the InNH2⋯In SFLP systems refers to the non-coordinated NH2 group of EDA, while the other NH2 group being coordinated with the In atom in In2O3. To prove this, in situ diffuse reflectance infrared Fourier-transform spectroscopy (DRIFTS) of adsorbed CO and CO2 at 298 K were investigated, respectively. After thermal treatment of the E-IO at 423 K for 2 h, saturated doses of CO or CO2 were introduced into IR cells. The spectrum (Fig. 2F, right) of adsorbed CO on E-IO shows a diagnostic peak centered at 2170 cm−1, which can be assigned to the C–O stretching vibration of CO interacting with the coordinatively unsaturated In sites.50–52 In addition, in the adsorption spectrum of CO2 (Fig. 2F, left), the diagnostic peak at 2359 cm−1 can be clearly observed and this peak is usually ascribed to the linear adduct of CO2 coordinated to Lewis acidic sites.53,54 Moreover, the diagnostic peaks at 2120 and 2342 cm−1 can be attributed to the physical adsorption of CO and CO2, respectively. These two spectroscopic characterizations clearly highlight that CO/CO2 can be able to interact with unsaturated In atoms that act as Lewis acid sites, providing strong evidence for the presence of grafting state InNH2⋯In.
The catalytic performance of CO2 hydrogenation was assessed in a flow reactor under reaction temperatures of 200, 250, and 300 °C with and without simulated solar light irradiation, using a gas mixture of CO2 and H2 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio, within a miniaturized photothermal catalytic microreactor system (Fig. S13, ESI‡). All three In2O3 samples (E-IO, Si-IO, H-IO) exhibited catalytic activity for the reverse water gas shift (RWGS) reaction to produce CO and were capable of synthesizing CH3OH, although the production rate and selectivity of the products varied among the catalysts. At 200 °C, regardless of light irradiation, all In2O3 samples showed negligible CO activity, while the E-IO sample exhibited significantly higher CH3OH activity compared to the other catalysts (Fig. S14 and S15, ESI‡). When the reaction temperature was increased to 250 °C, the Si-IO and H-IO catalysts displayed poor CO and CH3OH production rates similar to those observed at 200 °C (Fig. 3A and B). Remarkably, the amine-functionalized E-IO catalyst showed a substantial improvement in both CO and CH3OH formation under these reaction conditions. Specifically, at this temperature, E-IO demonstrated CO production rates of 248.07 μmol gcat−1 h−1 in the dark and 390.74 μmol gcat−1 h−1 under light irradiation, with a photo-enhancement rate of 158%. A significant photo-enhancement rate of approximately 217% was also observed for CH3OH on E-IO. The solar-powered CH3OH production rate of 59.02 μmol gcat−1 h−1 was more than 15.4 higher than that of the reference cubic In2O3 catalyst (Si-IO). This photo-enhancement activity can be attributed to the lower activation energy of the photocatalytic process compared to the thermochemical process.15 Furthermore, when the reaction temperature was further increased to 300 °C (Fig. S13, ESI‡), although both H-IO and Si-IO samples exhibited improved activities for CO and CH3OH production, the E-IO sample still exhibited the best catalytic performance among the three catalysts. In particular, under light irradiation, the CO production rate on E-IO reached a high value of 1814.73 μmol gcat−1 h−1, which is nearly 2.3 times higher than that of H-IO and Si-IO, and comparable to the reported rhombohedral polymorph of In2O3.15 However, due to the exothermic nature of the CH3OH production reaction, the production rate of CH3OH on E-IO decreased to 37.57 and 41.06 μmol gcat−1 h−1 under dark and light conditions, respectively, showing almost no photo-enhancement at this high reaction temperature. The estimated turnover number (TON) of the three samples shows a similar activity trend towards CO and CH3OH production (Fig. S15, ESI‡), further demonstrating that the amine functionalization of In2O3 can significantly enhance the performance of photocatalytic CO2 hydrogenation.
:3 ratio, within a miniaturized photothermal catalytic microreactor system (Fig. S13, ESI‡). All three In2O3 samples (E-IO, Si-IO, H-IO) exhibited catalytic activity for the reverse water gas shift (RWGS) reaction to produce CO and were capable of synthesizing CH3OH, although the production rate and selectivity of the products varied among the catalysts. At 200 °C, regardless of light irradiation, all In2O3 samples showed negligible CO activity, while the E-IO sample exhibited significantly higher CH3OH activity compared to the other catalysts (Fig. S14 and S15, ESI‡). When the reaction temperature was increased to 250 °C, the Si-IO and H-IO catalysts displayed poor CO and CH3OH production rates similar to those observed at 200 °C (Fig. 3A and B). Remarkably, the amine-functionalized E-IO catalyst showed a substantial improvement in both CO and CH3OH formation under these reaction conditions. Specifically, at this temperature, E-IO demonstrated CO production rates of 248.07 μmol gcat−1 h−1 in the dark and 390.74 μmol gcat−1 h−1 under light irradiation, with a photo-enhancement rate of 158%. A significant photo-enhancement rate of approximately 217% was also observed for CH3OH on E-IO. The solar-powered CH3OH production rate of 59.02 μmol gcat−1 h−1 was more than 15.4 higher than that of the reference cubic In2O3 catalyst (Si-IO). This photo-enhancement activity can be attributed to the lower activation energy of the photocatalytic process compared to the thermochemical process.15 Furthermore, when the reaction temperature was further increased to 300 °C (Fig. S13, ESI‡), although both H-IO and Si-IO samples exhibited improved activities for CO and CH3OH production, the E-IO sample still exhibited the best catalytic performance among the three catalysts. In particular, under light irradiation, the CO production rate on E-IO reached a high value of 1814.73 μmol gcat−1 h−1, which is nearly 2.3 times higher than that of H-IO and Si-IO, and comparable to the reported rhombohedral polymorph of In2O3.15 However, due to the exothermic nature of the CH3OH production reaction, the production rate of CH3OH on E-IO decreased to 37.57 and 41.06 μmol gcat−1 h−1 under dark and light conditions, respectively, showing almost no photo-enhancement at this high reaction temperature. The estimated turnover number (TON) of the three samples shows a similar activity trend towards CO and CH3OH production (Fig. S15, ESI‡), further demonstrating that the amine functionalization of In2O3 can significantly enhance the performance of photocatalytic CO2 hydrogenation.
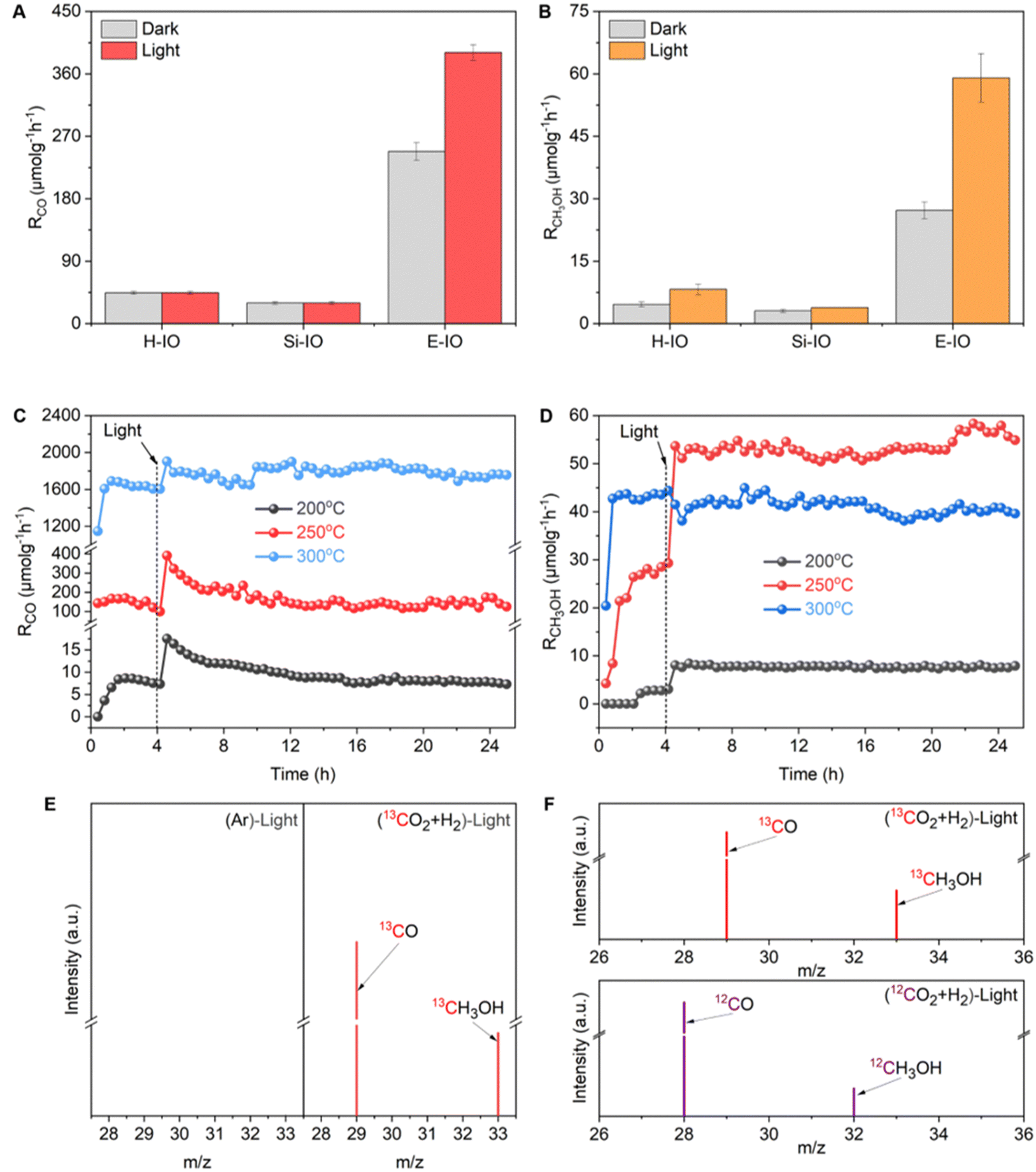 | ||
| Fig. 3 CO2 hydrogenation performance (A) CO production rate at 250 °C with and without solar irradiation. (B) CH3OH production rate at 250 °C with and without solar irradiation. (C) Long-term (20 h) CO production over E-IO at different reaction temperatures with solar irradiation. (D) Long-term (20 h) CH3OH production over E-IO at different reaction temperatures with solar irradiation. (E) GC-MS data from Ar and 13CO2/H2 atmosphere under irradiation condition at 250 °C. (F) 13CH3OH and 13CO obtained from 13CO2 hydrogenation, and 12CH3OH and 12CO as a reference. | ||
Based on the catalytic results, it can be concluded that the amine functionalization of In2O3 significantly enhances the performance of CO2 hydrogenation, exhibiting remarkable photo-enhancement in both CO and CH3OH production. In light of the superior catalytic performance of E-IO, we conducted long-term stability tests under atmospheric pressure conditions at 200, 250, and 300 °C. Prior to the long-term light irradiation testing, a 4 hour thermal catalytic process was performed to establish a steady-state activity. As depicted in Fig. 3C and D, the E-IO catalyst demonstrated excellent photocatalytic stability for both CO and CH3OH production. Notably, optimal CO production and CH3OH formation were achieved at 300 and 250 °C, respectively. After 20 h of continuous light irradiation testing, no significant decrease in production rate was observed, with a CO rate of 1787.18 μmol gcat−1 h−1 and a CH3OH rate of 53.38 μmol gcat−1 h−1. Furthermore, the selectivity of CH3OH was evaluated throughout the tests (Fig. S16, ESI‡). The results revealed a CH3OH selectivity of approximately 49%, 30%, and 2% at 200, 250, and 300 °C under light irradiation, respectively. Notably, the E-IO catalyst exhibited both a high CH3OH production rate (53.38 μmol gcat−1 h−1) and excellent CH3OH selectivity (30%) at 250 °C reaction conditions, demonstrating outstanding overall performance compared to reported indium oxide catalysts.9 Moreover, the spent E-IO catalyst after 20 h of reactions at 250 °C showed no significant changes in crystal phase, surface chemical groups, and chemical oxidation states, confirming its structural stability (Fig. S17 and S18, ESI‡). It should also be noted that when the E-IO catalyst suffered from catalytic reaction at a higher temperature (300 °C), the surface coordinated EDA molecules disappeared (Fig. 2B and Fig. S18c, ESI‡) and the InNH2⋯In SFLP was destroyed, which then contributes to a remarkably decreased photocatalytic activity.
To substantiate that the CO and CH3OH products were from the photocatalytic reaction, we conducted two parallel control experiments: one was using high-purity Ar instead of CO2, and the other was using carbon-13-labeled carbon dioxide (13CO2). Fig. 3E demonstrated that no product was observed in the Ar condition, indicating that both carbon products were exclusively derived from CO2 reduction. When the reaction atmosphere changed into 13CO2 and H2, obvious M/S signals of 13CO (m/z = 29) and 13CH3OH (m/z = 33) products were detected, supporting the conclusion that the products were generated via CO2 reduction. As compared, under 12CO2 and H2 atmosphere, unequivocal signals of 12CO and 12CH3OH products were detected (Fig. 3F). All these results collectively confirm that the carbon source in the CO2 hydrogenation originates from the CO2, rather than the decomposition of EDA molecules grafted on the catalyst surface. To further reflect the strong chemical interaction of EDA molecule grafting on the In2O3 surface, we also prepared amine-modified In2O3 samples via the impregnation method. As shown in FTIR spectra (Fig. S19, ESI‡), there exhibited nearly absent characteristic absorption peaks related to EDA on the catalyst surface, indicating efficient removal of surface-grafted EDA molecules during the washing process. To maintain the residual of the EDA molecules, the impregnated catalyst without washing was also prepared and the characteristic absorption peaks related to EDA can be clearly detected. However, the photocatalytic CO2 hydrogenation performance of all the impregnated amine-modified In2O3 showed almost the same as the pristine In2O3 samples (Fig. S20, ESI‡), suggesting the minimal effect of physical impregnation.
Fig. 4A illustrates the UV-visible DRS spectra and corresponding bandgaps of the H-IO, Si-IO, and E-IO catalysts. All samples exhibit solar light absorption in the ultraviolet region (wavelengths below 440 nm). Notably, E-IO demonstrates significantly stronger UV light absorption, approximately double that of the other two samples. This observation suggests an increase in surface charge of the indium oxide material due to the introduction of surface amine groups. Furthermore, by employing the Kubelka–Munk equation and Mott–Schottky analysis (Fig. S21, ESI‡), we calculated the band gap (Eg) and conduction band (CB) of the samples, resulting in the band diagrams (Fig. 4B).55 It is important to note that In2O3−x(EDA)y (E-IO) exhibits a more moderate band gap energy with a value of 3.04 eV, compared to In2O3−x(OH)y (H-IO with a band gap energy of 2.80 eV and Si-IO with an Eg of 2.90 eV), as well as pristine cubic indium oxide (with an Eg of 3.6 eV as a direct semiconductor).56 This indicates that E-IO enhances light absorption while avoiding excessive recombination of electron–hole pairs caused by a narrow band gap, thereby improving the transfer efficiency of photogenerated charge carriers. Moreover, E-IO exhibits a more negative conduction band at approximately −0.759 eV compared to the other two samples, indicating a better reduction potential that facilitates CO2 photocatalytic reduction.
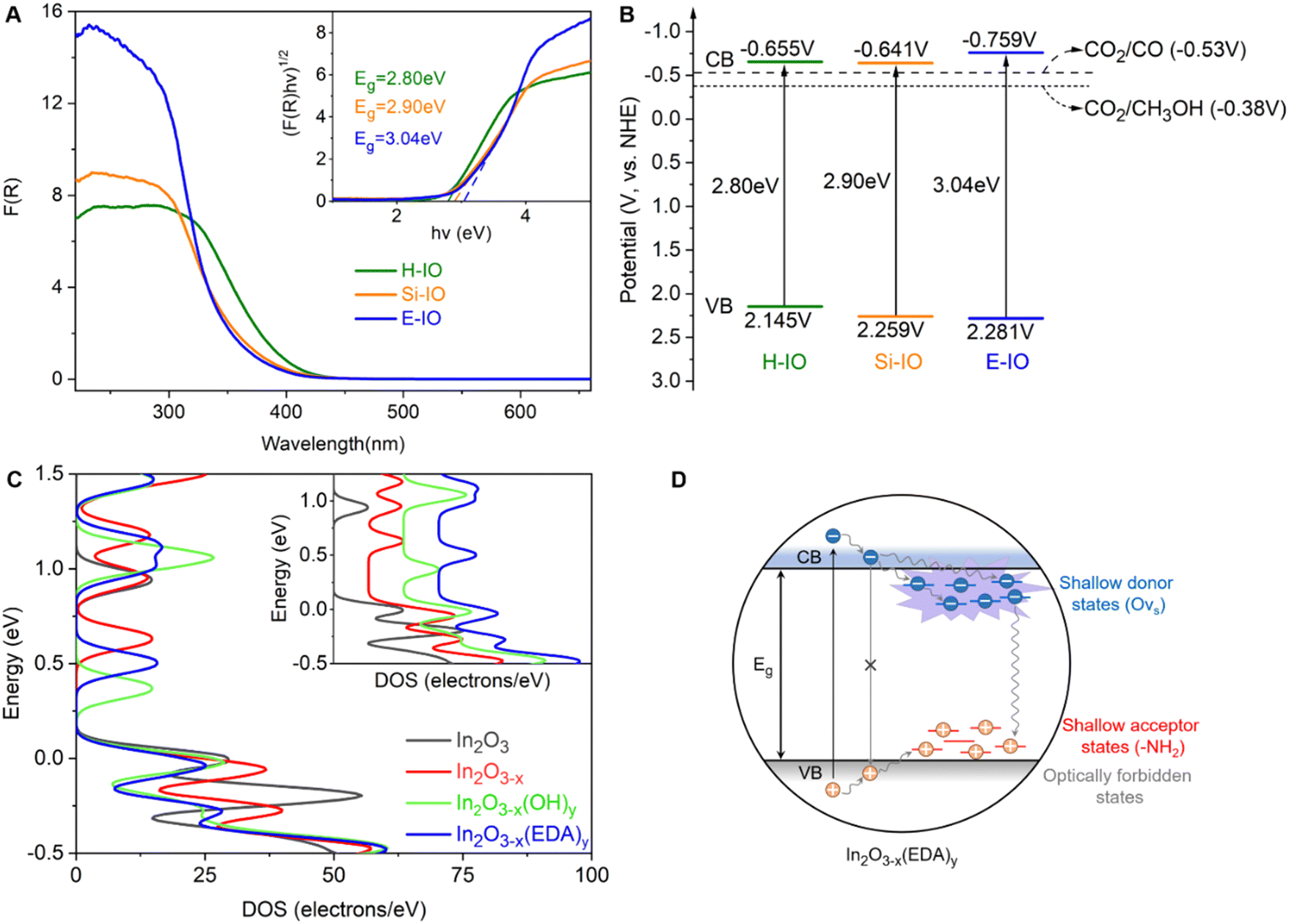 | ||
| Fig. 4 Photoelectrochemical characterizations (A) DRS and corresponding Tauc plots (inset) of H-IO, Si-IO, and E-IO. (B) Band diagrams. (C) The total DOS for In2O3, In2O3−x, In2O3−x(OH)y, and In2O3−x(EDA)y (110) surface, expanded view of the DOS in the conduction band region (inset). (D) A schematic illustration of charge carrier recombination pathways in In2O3−x(EDA)y nanoparticles. | ||
To investigate the photogenerated charge transfer mechanism, the room-temperature photoluminescence (PL) spectra of the H-IO, Si-IO, and E-IO samples are presented in Fig. S22A (ESI‡). All three samples display a green emission peak centered around 460 nm. However, E-IO exhibits a weaker PL emission peak compared to the other two samples. This can be attributed to the fact that amine groups and oxygen vacancies, similar to hydroxide and oxygen vacancies, act as traps, creating more moderate mid-gap states. These mid-gap states effectively capture photo-excited electrons and inhibit electron–hole recombination, leading to a reduction in PL emission intensity. Furthermore, Fig. S22B and C (ESI‡) demonstrate that E-IO exhibits the highest photocurrent density and lowest impedance, indicating superior charge separation and transfer efficiency. These photoelectrochemical characterizations substantiate that the construction of InNH2⋯In SFLP can enhance the transport of photogenerated charges, thereby improving the photocatalytic performance of amine-functionalized In2O3.
To gain a deeper understanding of how surface-grafted NH2 groups and oxygen vacancies affect the electronic band structure of amine-functionalized In2O3, we conducted density of states (DOS) calculations and partial density of states (PDOS) for four types of surfaces: pristine In2O3, In2O3−x, In2O3−x(OH)y, and In2O3−x(EDA)y, using hybrid DFT calculations (Fig. 4C). On the surface of In2O3−x with only oxygen vacancies, new states near the original conduction band were observed, closer to the Fermi level compared to the defect-free In2O3 surface. These states were fully occupied, indicating an increase in electron surface charge due to the donor properties of oxygen vacancies. However, the introduction of oxygen vacancies also pushed the valence band further away from the Fermi level, somewhat diminishing the overall advantage of introducing an oxygen vacancy. Fortunately, these drawbacks can be effectively mitigated when oxygen vacancies are introduced together with amine or hydroxyl groups. Similar to the hydroxyl group, the amine group induced the accumulation of p electrons at the edge of the valence band and shifted the Fermi level toward the edge, exhibiting typical acceptor properties (Fig. S23, ESI‡). The simplified schematic in Fig. 4D illustrates that in the In2O3−x(EDA)y specimen, oxygen vacancies create shallow donor states below the conduction band edge, while surface amine groups act as shallow acceptor states above the valence band edge. The presence of surface amine groups and oxygen vacancies alters the relaxation dynamics of charge carriers, enhancing non-radiative relaxation processes and facilitating the separation of electron–hole pairs, thereby improving the catalytic efficiency of photogenerated electrons in reduction reactions. These findings align with the results obtained from the DRS and PL experiments discussed earlier.
Besides the electronic structure and charge transfer efficiency, the enhanced Lewis acid–base pairs in amine-functionalized In2O3 are also expected to improve the adsorption and activation capacity for CO2. To investigate the CO2 uptake ability, we recorded the CO2 adsorption profiles on H-IO, Si-IO, and E-IO catalysts. As shown in Fig. 5A, E-IO exhibits the highest adsorption capacity of 5.72 cm3 g−1 for CO2, which is 2.44 and 2.86 times higher than that of H-IO and Si-IO, respectively. It is evident that the superior CO2 adsorption capacity of E-IO cannot be attributed solely to its surface area, as E-IO has a moderate surface area (50.54 m2 g−1) compared to the other catalysts. Considering the significant enhancement of Lewis acid–base pairs due to NH2 substitution, the improved CO2 adsorption capacity can be mainly attributed to the strong chemical interaction between CO2 molecules and Lewis basic NH2 sites, as well as the abundance of oxygen vacancies. To gain further insight into the effect of NH2 substitution on the interaction between CO2 and the catalysts, we conducted CO2 temperature-programmed desorption (CO2-TPD) tests. As depicted in Fig. 5B, the TPD peak intensity is higher for the amine-functionalized In2O3 compared to the other catalysts. Notably, the TPD profile of E-IO can be divided into three regions: a low-temperature region (100–200 °C), a medium-temperature region (200–360 °C), and a high-temperature region (360–550 °C). The weak desorption peak at 146 °C corresponds to physically adsorbed CO2. The significant desorption peak observed at 289 °C is attributed to the desorption of CO2 that is bound to NH2 groups, forming carbamate (H2NCOO−) species.57,58 The additional peak at 324 °C can be associated with the chemical desorption of bent CO2δ− species, which is related to oxygen vacancies.59,60 Furthermore, the desorption peaks at higher temperatures (449 and 495 °C) are primarily attributed to the decomposition of surface carbonates and bicarbonates.15,61
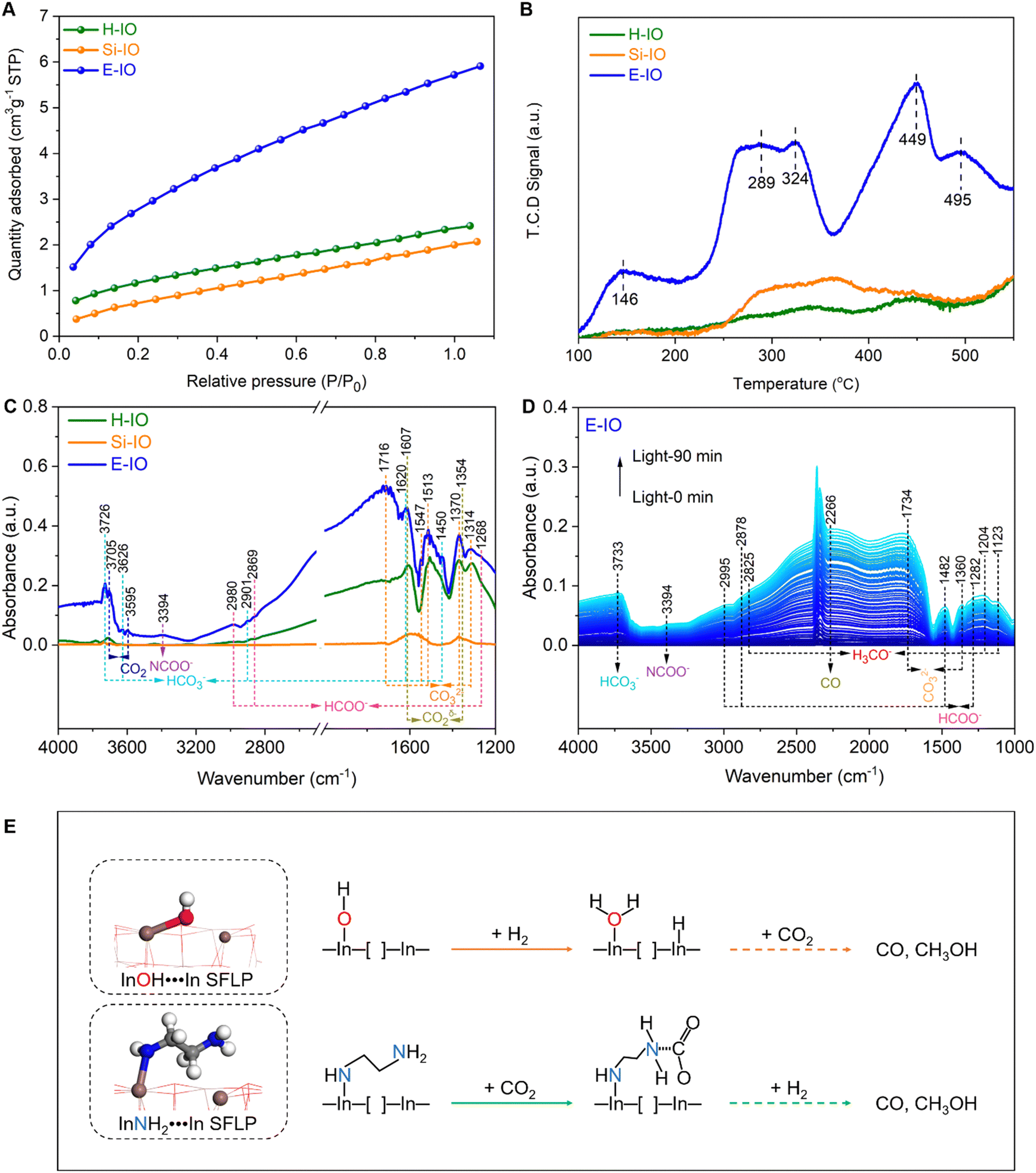 | ||
| Fig. 5 CO2 adsorption and activation on H-IO, Si-IO, and E-IO (A) CO2 adsorption isotherms. (B) CO2-TPD profiles. (C) In situ DRIFTS spectra of the adsorption of CO2 at 40 minutes. (D) In situ DRIFTS spectra of surface species under reaction conditions on E-IO with light. (E) The traditional InOH⋯In SFLP photocatalytic CO2 hydrogenation mechanism (top) and InNH2⋯In SFLP photocatalytic CO2 hydrogenation mechanism (bottom). | ||
In situ DRIFTS experiments were conducted to investigate the adsorption of CO2. It is evident that E-IO exhibits superior adsorption capacity for CO2 compared to H-IO and Si-IO, resulting in the formation of a greater number of intermediate species through bonding interactions (Fig. S24, ESI‡). The transient evolution of surface species during CO2 adsorption on H-IO, Si-IO, and E-IO samples is depicted in Fig. 5C. In the case of E-IO, the observed features at 3394 cm−1 can be attributed to the NCOO− skeletal vibration and N–H stretching of carbamate formation, indicating that surface amine groups adsorb and activate CO2 by forming carbamates.62,63 The peaks at 1716, 1513, 1370, and 1314 cm−1 correspond to the asymmetric and symmetric OCO stretching modes of carbonates (CO32−), and the appearance of four peaks at 3726, 3626, 2901, 1620 and 1450 cm−1 indicates the formation of bicarbonate species (HCO3−).15,64,65 Peaks at 1607 and 1356 cm−1 represent the asymmetric and symmetric stretching modes, respectively, and can be attributed to the bent CO2δ− species adsorbed at oxygen vacancy sites. Peaks at 3705 and 3595 cm−1 correspond to gaseous CO2 fingerprint modes.66 Additionally, peaks at 2980, 2869, and 1268 cm−1 can be attributed to the asymmetric and symmetric OCO stretching vibrations of adsorbed bidentate formate (HCOO−) species.45,67,68 Although some formate species may also be observed on H-IO and Si-IO, their peak intensities are weaker compared to E-IO. These collective results indicate that amine-functionalized In2O3 exhibits strong CO2 adsorption, bonding, and activation abilities due to the presence of grafted NH2 groups and abundant oxygen vacancies.
To gain a deeper understanding of the enhanced reactivity facilitated by InNH2⋯In SFLP. In situ DRIFTS experiments were conducted under reaction conditions (CO2 + H2, 250 °C) to identify reaction intermediates and elucidate the photocatalytic mechanism involved in the CO2 hydrogenation process. Fig. 5D shows the identified surface species on E-IO under reaction conditions with different irradiation times. Vibration absorption peaks of HCOO− were detected at 2995, 2878, 1482, and 1282 cm−1, which have been previously identified as intermediate species in the generation of CO and CH3OH.62,67 The vibration absorption peak of H3CO− species, another key intermediate in methanol formation, was indicated by the diagnostic mode at 2825 cm−1, which corresponds to the CH3 stretching modes.15,68 Additionally, the peaks at 1204 and 1123 cm−1 correspond to the CO stretching mode of bridged methoxide species. The peak at 2266 cm−1 is attributed to CO, indicating the formation of CO on the surface of E-IO.69 In contrast, only vibration absorption peaks of CO and a small amount of HCOO− were detected on the surfaces of H-IO and Si-IO (Fig. S25, ESI‡). These results demonstrate that In2O3−x(EDA)y catalysts can effectively promote the formation of intermediate species HCOO− and H3CO− during the photocatalytic hydrogenation reaction, leading to an enhanced production of CH3OH and CO.
Based on the comprehensive analysis, a plausible catalytic mechanism for CO2 hydrogenation on In2O3−x(EDA)y with robust InNH2⋯In SFLP can be proposed in Fig. 5E. The traditional InOH⋯In SFLP with moderate Lewis acid and base sites, can facilitate the heterolysis of H2, in which the proton bound to the hydroxide Lewis base and hydride bound to the coordinately unsaturated indium, and subsequently react with CO2 to generate CO and CH3OH. As for InNH2⋯In SFLP, the amine group Lewis base provides a robust adsorption site for CO2 molecules, preferentially favouring the activation of CO2 into carbamate (NHCOO−) intermediates. Moreover, the unsaturated In atom acting as Lewis acid can also provide an active site to activate and bond with CO2 molecules. This synergetic Lewis acid–base enhancement in the activation ability towards CO2 molecules effectively promotes a continuous multi-electron hydrogenation and reduction process, leading to the formation of crucial intermediates and ultimately the production of CO and CH3OH.
Conclusions
In summary, this study successfully synthesized amine-functionalized In2O3 (In2O3−x(EDA)y) nanoparticles, which possess robust InNH2⋯In SFLP, using a one-step solvothermal method. In comparison to conventional InOH⋯In, the InNH2⋯In SFLP demonstrates superior performance in the photocatalytic hydrogenation of CO2. This enhanced activity can be attributed to the strong adsorption, binding, and activation capabilities of the Lewis acid–base pairs present in InNH2⋯In SFLP. Additionally, the existence of moderate shallow states facilitates the separation of electron–hole pairs, thereby enhancing the reactivity of the Lewis acid–base sites. By leveraging the unique properties of surface InNH2⋯In SFLP, it becomes possible to design efficient and sustainable indium oxide catalysts for the conversion of CO2 into valuable products. This research contributes a molecule-functionalized engineering strategy for the development and optimization of heterogeneous photocatalysts, showcasing one more step towards future solar CO2 refineries.Author contributions
T. Y., N. L. and G. A. O. conceived and designed the experiments. Q. G. and C. N. conducted the catalysts preparation and the catalysts testing flow experiments for CO2 hydrogenation. W. R. conducted and analyzed the DFT calculations. Q. G., C. N., Y. Z., W. L., L. Z. and D. Z. performed the structural characterizations and the in situ DRIFTS. L. W. and Z. L. performed the carbon-13-labeled carbon dioxide test. T. Y., B. H., and G. A. O. conceived the project and co-wrote the manuscript. The manuscript was written through collective contributions from all authors. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. Y. is thankful for the financial support from the National Natural Science Foundation of China (22172086 and 22105117), Taishan Scholars Program of Shandong Province (no. tsqn202103064), the major basic research project of Shandong Province (ZR2021ZD06), Natural Science Foundation of Shandong Province (ZR2022MB078, ZR2020QE053 and ZR2021QB041). G. A. O. thanks the Natural Sciences and Engineering Council of Canada (NSERC) for support of this research. L. W. acknowledges the financial support from the National Natural Science Foundation of China (Grants no. 52102311).Notes and references
- L. Li, W. Liu, R. Chen, S. Shang, X. Zhang, H. Wang, H. Zhang, B. Ye and Y. Xie, Angew. Chem., Int. Ed., 2022, 61, e202214490 CrossRef CAS PubMed.
- Z. Shi, Q. Tan and D. Wu, Ind. Eng. Chem. Res., 2021, 60, 3532–3542 CrossRef CAS.
- Z. Huang, L. Liu, S. Qi, S. Zhang, Y. Qu and C.-R. Chang, ACS Catal., 2017, 8, 546–554 CrossRef.
- L. B. Hoch, T. E. Wood, P. G. O’Brien, K. Liao, L. M. Reyes, C. A. Mims and G. A. Ozin, Adv. Sci., 2014, 1, 1400013 CrossRef.
- S. Zhang, Z. Xia, Y. Zou, F. Cao, Y. Liu, Y. Ma and Y. Qu, J. Am. Chem. Soc., 2019, 141, 11353–11357 CrossRef CAS PubMed.
- K. K. Ghuman, L. B. Hoch, T. E. Wood, C. Mims, C. V. Singh and G. A. Ozin, ACS Catal., 2016, 6, 5764–5770 CrossRef CAS.
- L. Wang, T. Yan, W. S. Rui Song, Y. Dong, J. Guo, Z. Zhang, X. Wang and G. A. Ozin, Angew. Chem., Int. Ed., 2019, 58, 9501–9505 CrossRef CAS PubMed.
- H. Wang, J. Jia, L. Wang, K. Butler, R. Song, G. Casillas, N. P. K. Le He, D. D. Perovic, L. Jing, A. Walsh, R. Dittmeyer and G. A. Ozin, Adv. Sci., 2019, 6, 1902170 CrossRef CAS.
- L. Wang, M. Ghoussoub, H. Wang, Y. Shao, W. Sun, A. A. Tountas, T. E. Wood, H. Li, J. Y. Y. Loh, Y. Dong, M. Xia, Y. Li, S. Wang, J. Jia, C. Qiu, C. Qian, N. P. Kherani, L. He, X. Zhang and G. A. Ozin, Joule, 2018, 2, 1369–1381 CrossRef CAS.
- Q. Wang, Z. Miao, Y. Zhang, T. Yan, L. Meng and X. Wang, ACS Catal., 2022, 12, 4016–4025 CrossRef CAS.
- Y. Xie, J. Chen, X. Wu, J. Wen, R. Zhao, Z. Li, G. Tian, Q. Zhang, P. Ning and J. Hao, ACS Catal., 2022, 12, 10587–10602 CrossRef CAS.
- S. Zhang, Z. Huang, Y. Ma, W. Gao, J. Li, F. Cao, L. Li, C. Chang and Y. Qu, Nat. Commun., 2017, 8, 15266–15276 CrossRef CAS PubMed.
- Z. Li, C. Mao, Q. Pei, P. N. Duchesne, T. He, X. Zhang, M. Xia, J. Wang, L. Wang, R. Song, A. A. Jelle, D. M. Meira, Q. Ge, K. K. Ghuman, L. He and G. A. Ozin, Nat. Commun., 2022, 13, 7205–7215 CrossRef CAS.
- X. Wang, L. Lu, B. Wang, Z. Xu, Z. Xin, S. Yan, Z. Geng and Z. Zou, Adv. Funct. Mater., 2018, 28, 1804191 CrossRef.
- T. Yan, N. Li, L. Wang, W. Ran, P. N. Duchesne, L. Wan, N. T. Nguyen, L. Wang, M. Xia and G. A. Ozin, Nat. Commun., 2020, 11, 6095–6104 CrossRef CAS.
- Y. Liao, S. Cao, Y. Yuan, Q. Gu, Z. Zhang and C. Xue, Chem. – Eur. J., 2014, 20, 10220–10222 CrossRef CAS PubMed.
- K. M. Cho, K. H. Kim, K. Park, C. Kim, S. Kim, A. Al-Saggaf, I. Gereige and H.-T. Jung, ACS Catal., 2017, 7, 7064–7069 CrossRef CAS.
- Q. Huang, J. Yu, S. Cao, C. Cui and B. Cheng, Appl. Surf. Sci., 2015, 358, 350–355 CrossRef CAS.
- A. Lo and F. Taghipour, J. Mater. Chem. A, 2021, 9, 26430–26453 RSC.
- G. Lu, P. Zhang, D. Sun, L. Wang, K. Zhou, Z. Wang and G. Guo, Chem. Sci., 2014, 5, 1082–1090 RSC.
- C. Kim, H. S. Cho, S. Chang, S. J. Cho and M. Choi, Energy Environ. Sci., 2016, 9, 1803–1811 RSC.
- L. Mafra, T. Čendak, S. Schneider, P. V. Wiper, J. Pires, J. R. B. Gomes and M. L. Pinto, Chem. Eng. J., 2018, 336, 612–621 CrossRef CAS.
- Y.-D. Li, H.-W. Liao, Y. Ding, Y.-T. Qian, L. Yang and G.-E. Zhou, Chem. Mater., 1998, 10, 2301–2303 CrossRef CAS.
- M. Vatanparast and M. T. Taghizadeh, J. Mater. Sci.: Mater. Electron., 2015, 27, 54–63 CrossRef.
- T. Yan, X. Wang, J. Long, H. Lin, R. Yuan, W. Dai, Z. Li and X. Fu, New J. Chem., 2008, 32, 1843–1846 RSC.
- Y. Shen, X. Zhong, J. Zhang, T. Li, S. Zhao, B. Cui, D. Wei, Y. Zhang and K. Wei, Appl. Surf. Sci., 2019, 498, 143873 CrossRef CAS.
- X. Yan, L. Zhang, Y. Zhang, G. Yang and Z. Yan, Ind. Eng. Chem. Res., 2011, 50, 3220–3226 CrossRef CAS.
- U. Tumuluri, M. Isenberg, H.-S. Tan and S. S. C. Chuang, Langmuir, 2014, 30, 7405–7413 CrossRef CAS.
- M. Shanbedi, S. Z. Heris, M. Baniadam, A. Amiri and M. Maghrebi, Ind. Eng. Chem. Res., 2012, 51, 1423–1428 CrossRef CAS.
- C. Li, T. Zhao, A. Yang and F. Liu, ACS Omega, 2021, 6, 34027–34034 CrossRef CAS PubMed.
- T. Li, P. Huang, T. Liao, J. Guo, X. Yu, B. Han, L. Peng, Y. Zhu and Y. Zhang, Environ. Sci. Pollut. Res. Int., 2019, 26, 33269–33280 CrossRef CAS PubMed.
- P. Zhang, Y. Lu, M. Fan, P. Jiang, Y. Bao, X. Gao and J. Xia, Prog. Org. Coat., 2020, 147, 105811 CrossRef CAS.
- K. M. Parida and D. Rath, J. Mol. Catal. A: Chem., 2009, 310, 93–100 CrossRef CAS.
- X. Zhang, H. Qin, X. Zheng and W. Wu, Mater. Res. Bull., 2013, 48, 3981–3986 CrossRef CAS.
- F. Zheng, D. N. Tran, B. J. Busche, G. E. Fryxell, R. S. Addleman, T. S. Zemanian and C. L. Aardahl, Ind. Eng. Chem. Res., 2005, 44, 3099–3105 CrossRef CAS.
- M. Yao, B. Sun, N. Wang, W. Hu and S. Komarneni, Appl. Surf. Sci., 2019, 480, 655–664 CrossRef CAS.
- M. Tabbal, P. Merel, S. Moisa, M. Chaker, E. Gat, A. Ricard, M. Moisan and S. Gujrathi, Surf. Coat. Technol., 1998, 98, 1092–1096 CrossRef CAS.
- Z. Ren, X. Xu, X. Wang, B. Gao, Q. Yue, W. Song, L. Zhang and H. Wang, J. Colloid Interface Sci., 2016, 468, 313–323 CrossRef CAS PubMed.
- N. Murugesan, S. Suresh, M. Kandasamy, S. Murugesan and S. K. Kumar, Ceram. Int., 2020, 46, 27897–27902 CrossRef CAS.
- J. L. Hueso, J. P. Espinos, A. Caballero, J. Cotrino and A. R. Gonzalez-Elipe, Carbon, 2007, 45, 89–96 CrossRef CAS.
- S. Ilhom, A. Mohammad, D. Shukla, J. Grasso, B. G. Willis, A. K. Okyay and N. Biyikli, RSC Adv., 2020, 10, 27357–27368 RSC.
- R. Vieira, I. Marin-Montesinos, J. O. Pereira, R. Fonseca, M. Ilkaeva, M. Sardo and L. Mafra, J. Phys. Chem. C, 2021, 125, 14797–14806 CrossRef CAS.
- G. Gatti, D. Costenaro, C. Vittoni, G. Paul, V. Crocellà, E. Mangano, S. Brandani, S. Bordiga, M. Cossi, L. Marchese and C. Bisio, Phys. Chem. Chem. Phys., 2017, 19, 14114–14128 RSC.
- G. Gatti, C. Vittoni, D. Costenaro, G. Paul, E. Mangano, S. Brandani, L. Marchese and C. Bisio, Phys. Chem. Chem. Phys., 2017, 19, 29449–29460 RSC.
- T. Yan, L. Wang, Y. Liang, M. Makaremi, T. E. Wood, Y. Dai, B. Huang, A. A. Jelle, Y. Dong and G. A. Ozin, Nat. Commun., 2019, 10, 2521–2530 CrossRef PubMed.
- Y. Qi, L. Song, S. Ouyang, X. Liang, S. Ning, Q. Zhang and J. Ye, Adv. Mater., 2020, 32, 1903915 CrossRef CAS PubMed.
- J. Ye, C. Liu, D. Mei and Q. Ge, ACS Catal., 2013, 3, 1296–1306 CrossRef CAS.
- A. Posada-Borbón and H. Grönbeck, ACS Catal., 2021, 11, 9996–10006 CrossRef.
- L. L. Zeonjuk, N. Vankova, A. Mavrandonakis, T. Heine, G.-V. Rçschenthaler and J. Eicher, Chem. – Eur. J., 2013, 19, 17413–17424 CrossRef CAS PubMed.
- S. Kim, D. C. Sorescu, O. Byl, J. John and T. Yates, J. Phys. Chem. B, 2006, 110, 4742–4750 CrossRef CAS PubMed.
- A. Vimont, J.-M. Goupil, J.-C. Lavalley, M. Daturi, S. Surblé, C. Serre, F. Millange, G. Férey and N. Audebrand, J. Am. Chem. Soc., 2006, 128, 3218–3227 CrossRef CAS PubMed.
- C. P. Cabello, G. Berlier, G. Magnacca, P. Rumori and G. T. Palomino, CrystEngComm, 2015, 17, 430–437 RSC.
- A. Comès, S. Fiorilli and C. Aprile, J. CO2 Util., 2020, 37, 213–221 CrossRef.
- P. L. Llewellyn, S. Bourrelly, C. Serre, A. Vimont, M. Daturi, L. Hamon, G. D. Weireld, J.-S. Chang, D.-Y. Hong, Y. K. Hwang, S. H. Jhung and G. R. Férey, Langmuir, 2008, 24, 7245–7250 CrossRef CAS PubMed.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef CAS.
- X. Meng, C. Peng, J. Jia, P. Liu, Y. Men and Y. Pan, J. CO2 Util., 2022, 55, 101844 CrossRef CAS.
- X. Zheng, L. Shen, X. Chen, X. Zheng, C. Au and L. Jiang, Inorg. Chem., 2018, 57, 10081–10089 CrossRef CAS PubMed.
- J. Kim, S.-N. Kim, H.-G. Jang, G. Seo and W.-S. Ahn, Appl. Catal., A, 2013, 453, 175–180 CrossRef CAS.
- Z. Shi, M. Pan, X. Wei and D. Wu, Int. J. Energy Res., 2021, 46, 1285–1298 CrossRef.
- N. Rui, Z. Wang, K. Sun, J. Ye, Q. Ge and C. Liu, Appl. Catal., B, 2017, 218, 488–497 CrossRef CAS.
- C. Luo, J. Zhao, Y. Li, W. Zhao, Y. Zeng and C. Wang, Appl. Surf. Sci., 2018, 447, 627–635 CrossRef CAS.
- W. C. Wilfong, C. S. Srikanth and S. S. C. Chuang, ACS Appl. Mater. Interfaces, 2014, 6, 13617–13626 CrossRef CAS PubMed.
- M. B. Ansari, R. S. Shukla, Y.-H. Mo and S.-E. Park, Adv. Chem. Eng., 2021, 6, 100102 CrossRef CAS.
- J. Baltrusaitis, J. H. Jensen and V. H. Grassian, J. Phys. Chem. B, 2006, 110, 12005–12016 CrossRef CAS PubMed.
- S. Eckle, H.-G. Anfang and R. J. R. Behm, J. Phys. Chem. C, 2011, 115, 1361–1367 CrossRef CAS.
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- Y. Wei, F. Liu, J. Ma, C. Yang, X. Wang and J. Cao, Mol. Catal., 2022, 525, 112354 CrossRef CAS.
- Z. Cai, J. Dai, W. Li, K. B. Tan, Z. Huang, G. Zhan, J. Huang and Q. Li, ACS Catal., 2020, 10, 13275–13289 CrossRef CAS.
- S. Zhu, N. Li, D. Zhang and T. Yan, J. CO2 Util., 2022, 64, 102177 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Professor Geoffrey Ozin on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ey00261f |
| This journal is © The Royal Society of Chemistry 2024 |