
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A solar desalination charger for water treatment and value-added chemical production†
Seonghun
Kim
 a,
Dong Suk
Han
b and
Hyunwoong
Park
*a
a,
Dong Suk
Han
b and
Hyunwoong
Park
*a
aSchool of Energy Engineering, Kyungpook National University, Daegu 41566, Korea. E-mail: hwp@knu.ac.kr; Tel: +82-53-950-8973
bCenter for Advanced Materials, Qatar University, Doha 2713, Qatar
First published on 10th April 2024
Abstract
This study presents a photoelectrocatalytic desalination charger for the remediation of aquatic pollutants and the production of value-added chemicals. Under 1 sun irradiation, a Co-WBVO (BiVO4 doped with W and deposited with CoOOH) photoelectrode and aqueous NaxC electrode (Na on carbon felt, NaxC) pair efficiently desalinates brackish water (0.171 M NaCl) through ion-exchange membranes at an ion transport efficiency of ∼100%. The desalted chloride is partially oxidized by photogenerated holes into reactive chlorine species (RCSs) at a faradaic efficiency (FE) of >90%. The in situ generated RCSs are actively involved in the sequential oxidation of As(III) and NH4+. Meanwhile, the desalted Na+ is rapidly inserted into NaxC without any accumulation. Upon coupling with the charged NaxC, the electrocatalytic production of H2O2via O2 reduction with carbon nanotubes, H2via H2O reduction with NiMoS, and HCOOH via CO2 reduction with porous Bi are achieved at FEs of >80%. The as-designed PEC hybrid of the proof-of-concept can be applied to various purposes, including desalination, seawater electrolysis, production of value-added chemicals, and energy storage.
Broader contextProducing carbon-neutral chemicals and securing clean water are considered the most critical issues facing humanity over the next five decades. Among the proposed technical solutions to address these challenges, photoelectrocatalytic (PEC) systems working with inexhaustible carbon-free solar energy have been proven to be environmentally benign and technically feasible. Considering the maritime transport of green chemicals (e.g., H2), PEC systems must be operated near coastal areas with abundant saline water for cheap production.This study presents a PEC desalination charger for hybrid water reuse and solar chemical production. The primary feature of the PEC system is that it simultaneously drives many valuable reactions in a single device. During the desalination of brackish water, the desalted Cl− is oxidized to reactive chlorine species (RCSs; represented by HOCl/OCl−), which effectively mediate the oxidation of aquatic contaminants in the photoanode compartment. The desalted Na+ is concurrently accumulated in an aqueous Na metal electrode (Na on carbon felt, NaxC). We designed a PEC desalination cell comprising visible light–active photoanodes (W-doped BiVO4 deposited with CoOOH) with selective chloride oxidation reaction and a NaxC cathode with saline water (NaCl at 10 g L−1). The photoanodes were particularly tailored to partially oxidize the desalted chloride to RCSs while minimizing hyperoxidation into less reactive species (e.g., ClO3−) under simulated sunlight. These in situ generated RCSs effectively mediated the oxidation of mixed aquatic contaminants (As3+ and NH3). NaxC was photocharged simultaneously with the desalted Na+. After termination of irradiation, the charged NaxC was used to produce H2O2via O2 reduction reaction (E° = 0.695 V), H2via HER (E° = 0 V), and formic acid via CO2RR (E° = −0.2 V) with the as-designed carbon nanotubes (CNTs), NiMoS, and Bi, respectively. To the best of our knowledge, this is the first attempt to demonstrate a solar desalination charger. This approach should address an intrinsic challenge facing PEC systems whose operation is limited to the intermittent nature of sunlight with daily fluctuations and unavailability during nighttime. |
1. Introduction
Producing carbon-neutral chemicals and securing clean water are considered the most critical issues facing humanity over the next five decades.1–5 Among the proposed technical solutions to address these challenges, photoelectrocatalytic (PEC) systems working with inexhaustible, carbon-free solar energy have been proven to be environmentally benign and technically feasible.6–8 For example, a PEC device with 10% efficiency can produce molecular hydrogen at ∼0.7 g s−1via splitting clean water under the standard terrestrial 1 sun with an average daily power of ∼250 W m−2.7,9–11 Considering the maritime transport of H2 and its liquid-phase carriers (e.g., ammonia), the PEC systems must be operated near coastal areas with abundant saline water for cheap production. However, if obtaining the clean water from saline water is energy-intensive and significantly carbon-footprinted, the technoeconomic merits of the PEC system would weaken.Recently, PEC desalination systems have been proposed for hybrid water reuse and solar chemical production.12 In contrast to the conventional PEC reactions, the PEC desalination mimics electrodialysis in terms of system construction. The primary feature of the PEC desalination process is to simultaneously drive many valuable reactions in a single device (Scheme 1). Specifically, desalted Cl− is oxidized to reactive chlorine species (RCSs, represented by HOCl/OCl−) which effectively mediate the oxidation of aquatic contaminants in the photoanode compartment.12 The desalted Na+ is concurrently accumulated in the cathode compartment and increases the catholyte conductivity, assisting the H2 evolution reaction (HER) and CO2 reduction reaction (CO2RR). This proof-of-concept was further demonstrated using an expandable multi-stack PEC desalination device for the chloride oxidation reaction (ClOR), and metal composite cathodes for the HER and CO2RR.12–14 During the decomposition of aqueous substrates in the photoanode compartment, either HER or CO2RR occurred at a faradaic efficiency of >90%. Concurrent desalination of saline water (salinity 5–36 g L−1) proceeded with low specific energy consumption (1.1 kW h m−3). The as-produced H2 energy was estimated to further contribute to saving energy by 25–30%.13
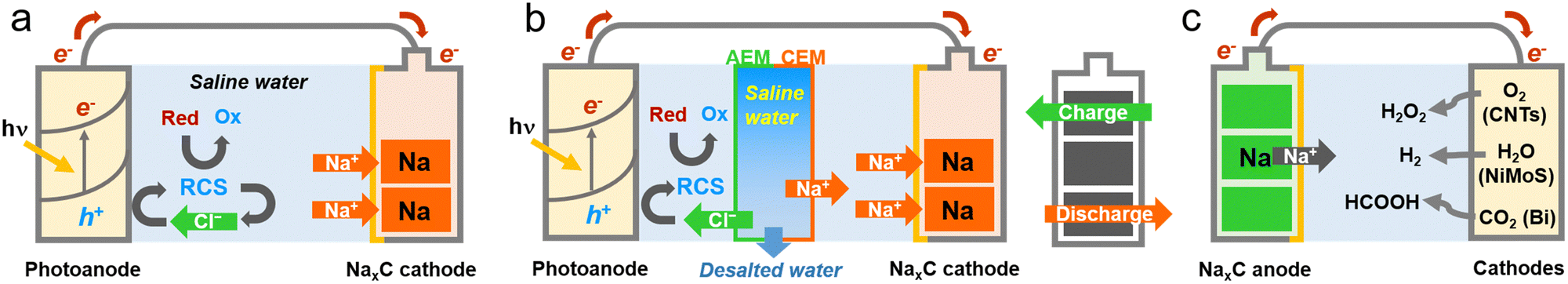 | ||
| Scheme 1 Schematic illustrations of (a) solar charge with water treatment, (b) solar water treatment and charge with desalination, and (c) the discharge process with value-added chemical production. (a) A photoelectrode (bare and modified BiVO4) and an Na metal array (NaxC) electrode are directly coupled in saline water with As(III) and NH3. Upon irradiation, the photoelectrode oxidizes Cl− to reactive chlorine species (RCSs) mediating the oxidation of As(III) and NH3. Simultaneously, Na+ ions are inserted into the NaxC electrode. (b) Both electrodes are separated by anion- and cation-exchange membranes (AEM and CEM, respectively), and the photoelectrode is placed in non-saline water with As(III) and NH3. The middle compartment between the two membranes (desalination cell) is filled with saline water. Upon irradiation, desalination proceeds, and the desalted Cl− and Na+ undergo the same reactions as those in (a). (c) The charged NaxC electrode is coupled to three different electrodes (CNTs, NiMoS, and porous Bi) to produce H2O2via O2 reduction, H2via H2O reduction, and HCOOH via CO2 reduction, respectively. | ||
We noted the potential use of the accumulated Na+ in charging an aqueous Na metal electrode. This approach should address an intrinsic challenge facing PEC systems whose operation is limited to an intermittent nature of sunlight with daily fluctuation and unavailability during nighttime. The aqueous Na metal electrode is characterized by its theoretical specific energy (1165 mA h g−1) and high cell voltage (−2.71 V).15,16 In this study, we designed a PEC desalination cell comprising visible light-active photoanodes (W-doped BiVO4 deposited with CoOOH) with a selective ClOR and aqueous Na metal cathode (Na on carbon felt, NaxC) with saline water (NaCl at 10 g L−1). The photoanode was particularly tailored to partially oxidize the desalted chloride to RCSs while minimizing hyper-oxidation into less reactive species (e.g., ClO3−) under simulated sunlight. These in situ generated RCSs effectively mediated the oxidation of mixed aquatic contaminants (As3+ and NH3). Simultaneously, the NaxC was photocharged with desalted Na+. After termination of irradiation, the charged NaxC was utilized for the production of H2O2via the O2 reduction reaction (ORR) (E° = 0.695 V), H2via HER (E° = 0 V), and formic acid via the CO2RR (E° = −0.2 V) with the as-designed CNTs, NiMoS, and Bi, respectively. To the best of our knowledge, this is the first attempt to demonstrate a solar desalination charger.
2. Experimental section
2.1. Synthesis of materials
Bare and W-doped BiVO4 (BVO and WBVO, respectively) were synthesized by electrodeposition and calcination.17,18 Fluorine-doped SnO2 (FTO) substrates (∼50 nm-thick FTO layer coated onto soda lime glass with resistivity of 6–9 Ω sq−1, Pilkington) were cleansed with ethanol (99.9%, Duksan) under ultrasonication and dried with an N2 stream. For Bi deposition, BiNO3·5H2O (75 mM, 10 mL, Junsei) was dissolved in aqueous KI solution (0.4 M, Sigma-Aldrich) adjusted at pH 1.7 using HNO3 (60%, Daejung). Then, p-benzoquinone (0.23 M, dissolved in 99.9% ethanol, Sigma-Aldrich) was added to the Bi solution and stirred for 5 min. The as-prepared FTO substrate (working electrode) with a Pt rod (counter electrode) and saturated calomel electrode (SCE, reference electrode) was immersed in the Bi solution and held at −0.2 V vs. SCE for 15 min using a potentiostat (Ivium). The Bi-deposited electrode was rinsed with deionized water (18 MΩ cm) and dried under ambient conditions. Then, vanadyl acetylacetonate (VO(C5H7O2)2, 0.2 M, 0.26 mL, Sigma-Aldrich) dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich) was dropped onto the Bi-deposited electrode and annealed at 450 °C for 2 h. The sample electrodes were immersed in KOH (1 M) solutions for 30 min and rinsed with deionized water to remove residual V2O5. For the synthesis of WBVO, tungsten chloride (WCl6, Sigma-Aldrich) was added to the vanadyl acetylacetonate solution at various concentrations (0.55–2.77 mM, corresponding to doping levels of 0.3–1.5 at%). When necessary, the as-synthesized WBVO samples were immersed in aqueous Co(II) solution (Co(NO3)2·6H2O, 0.1 M, Sigma-Aldrich) and biased at −0.9 V vs. SCE with deposition charges of 5–30 mC (denoted as Co-WBVO). Unless otherwise specified, the W doping level was 0.3 at% and the Co deposition charge was 10 mC. Prior to photoelectrolysis, the Co-WBVO electrode underwent six times potential sweeping from −0.4 to 1.5 V vs. SCE.For photocharging processes, an arrayed aqueous Na metal electrode (Na on carbon felt, NaxC, 4 TO ONE Co., Ulsan, Korea) with four-unit (coin-type) cells was coupled to the photoelectrodes. Each array was composed of a top cap, NASICON (Na3Si2Zr2PO12, diameter of 16 mm, ion conductivity of ∼2.5 × 10−3 S cm−1), 1 M NaCF3SO3 (Sigma-Aldrich) dissolved in tetraethylene glycol dimethyl ether (Sigma-Aldrich), sodium metal on carbon felt, a spacer, a spring, and a bottom cap. All fabrication processes were conducted in glove boxes.19 For H2 production, NiMoS (mixed Ni2S3 and MoS2) was electrochemically fabricated onto porous Ni substrates.13 For H2O2 production, multi-walled carbon nanotubes (CNTs) were loaded onto a commercial carbon paper (315 μm-thick, Sigracet 39 BB).20 For CO2 reduction to formic acid, a porous, dendrite-structured Bi electrode was fabricated on Cu substrates via galvanostatic electrodeposition (5 A cm−2) for 5 s.21 Detailed synthetic procedures of the electrocatalysts were described elsewhere.13,20,21
2.2. Photoelectrocatalytic and electrocatalytic activity tests
Linear sweep voltammograms of the as-synthesized bare and modified BVO electrodes were obtained at a scan rate of 20 mV s−1 under simulated sunlight (AM 1.5G, 100 mW cm−2, ABET Tech) in a single compartment cell containing 0.171 M NaCl (Daejung, 99%). An SCE and Pt foil were used as reference and counter electrodes, respectively. Electrochemical impedance spectra and Mott–Schottky plots were obtained in the frequency range of 0.01–100 kHz at 0 V and 1 kHz at 0.73 V vs. SCE, respectively. The incident photon-to-current efficiency (IPCE) was estimated using the following equation:| IPCE (%) = 1239.8 (V nm) × Jph × 100%/Plight × λ | (1) |
For oxidation of aquatic substrates with simultaneous charging with Na ions, the as-synthesized photoelectrodes (working electrode) were coupled to the NaxC electrode (counter electrode) and SCE in a single compartment cell with 0.171 M NaCl containing 1 mM NH3 (NH4Cl, Aldrich) and/or 1 mM arsenite (As(III), NaAsO2, Aldrich) at pH ∼ 6.5. (Scheme 1a). While applying a constant Jph (1 mA cm−2), a photopotential (Eph) of the working electrode (vs. SCE) and a potential difference (Ecell) between the photoelectrode and NaxC electrode were simultaneously recorded using a potentiostat (Ivium) and multimeter (Keysight, 34461A), respectively. For simultaneous ternary reactions (desalination of saline water, oxidation of ammonia and/or As(III), and charging with Na ions), a cell comprising a photoanode compartment with 0.171 M NaCl (20 mL) containing 1 mM As(III) and/or NH3, desalination compartment with saline water (0.171 M NaCl, 5 mL), and cathode compartment with 5 mM NaCl (20 mL) was designed with an anion exchange membrane (AEM, AMI-7001S, Membranes International) and a cation exchange membrane (CEM, CMI-7000S, Membranes International) (Scheme 1b). The desalination compartment was circulated at a flow rate of 10 mL min−1 using a peristaltic pump (Ismatec, Reglo ICC). While applying a constant Jph of 1 mA cm−2, Cl− and Na+ in the electrolyte and saline water were quantified to examine the inter-compartment ion transport.
After the PEC processes, the charged NaxC electrode was wired to (i) a CNT electrode in 0.1 M K2SO4 purged with O2 (pH 6.5) for hydrogen peroxide (H2O2) production via O2 reduction, (ii) an NiMoS electrode in 0.1 M KOH purged with N2 (pH ∼ 13) for H2 evolution via H2O reduction, or (iii) a Bi electrode in 0.1 M KHCO3 purged with CO2 (pH 6.8) for formate (HCOO−) production via CO2 reduction (Scheme 1c). Prior to bulk electrolysis, linear sweep voltammograms (vs. SCE) of each electrocatalyst (working electrode) were obtained with the charged NaxC (counter electrode) in a single compartment cell. While applying J of −1 and −3 mA cm−2 to the working electrodes, the potentials of the working electrodes (vs. SCE) and Ecells (potentials difference between the working and counter electrode) were simultaneously recorded using the potentiostat and multimeter, respectively. In addition, the CNTs and charged NaxC were galvanically coupled via direct wiring or through an LED bulb (λ = 566 nm, size of 3ϕ, forward voltage of 1.8–2.2 V, max current of 20 mA, DFRobot Co.).
During the PEC desalination and discharging processes, aliquots were intermittently sampled and analyzed. The anions (Cl−, ClO3−, NO2−, and NO3−) and cations (Na+ and NH4+) were analyzed using ion chromatographs (Thermo Scientific, DIONEX ICS-1100) equipped with a conductivity detector, IonPac As-11HC (4 × 250 mm) column for anions, and IonPac CS-12A (4 × 250 mm) column for cations, respectively.22 RCSs (HClO/ClO−) were quantified using the N,N-diethyl-p-phenylene-diamine (DPD) reagent (Hach method).22 As(V) and H2O2 were quantified using the molybdenum blue method23 and DMP method,23,24 respectively. Formate was quantified using high-performance liquid chromatography (Waters 2695 Separation Module) equipped with a dual absorbance detector (Waters 2487, 210 nm) and a column (BIO-RAD, Aminex HPX-87H, 300 mm × 7.8 mm).21,25 H2 was quantified using a gas chromatograph (GC, Agilent 7820) equipped with a thermal conductivity detector (TCD) and carboxen 1000 column. Detailed analytical methods for the as-mentioned chemicals were described in the cited references.
The Faradic efficiency (FE), specific energy consumption (SEC, for 50% desalination), and ion transport efficiency (ITE) were estimated by the following equations:
| FE (%) = (Amount of product × 2F) × 100%/(J × A × t) | (2) |
| SEC (kW h m−3) = Ecell × J × A × t/(saline water volume) | (3) |
| ITE (%) = (Amount of transported monovalent ion)/(J × A × t) | (4) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), operating cell voltage (V), operating current density (mA cm−2), electrode area (cm2), and time (s), respectively. In eqn (2), the product refers to chemicals produced with 2 electron-transfers (i.e., HClO, H2O2, H2, and HCOOH). To reflect pH differences, when necessary, “V vs. SCE” was converted to “VRHE” by the following: VRHE = V vs. SCE + 0.241 + 0.059 × pH.
485 C mol−1), operating cell voltage (V), operating current density (mA cm−2), electrode area (cm2), and time (s), respectively. In eqn (2), the product refers to chemicals produced with 2 electron-transfers (i.e., HClO, H2O2, H2, and HCOOH). To reflect pH differences, when necessary, “V vs. SCE” was converted to “VRHE” by the following: VRHE = V vs. SCE + 0.241 + 0.059 × pH.
2.3. Surface characterization
The top and cross-sectional morphologies of the as-synthesized bare and modified BVO samples were examined using a field emission scanning electron microscope (FE-SEM, Hitachi SU8200; accelerating voltage of 5 kV; emission current of 9.8 μA; working distance 10.8 mm). X-ray diffraction (XRD, PANalytical EMPUREAN) with Cu-Ka radiation (40 kV and 30 mA) and X-ray photoelectron spectroscopy (XPS, NEXSA, Thermo Fisher Scientific) with Al-Kα (hν = 1486.6 eV) were employed to examine the crystallite structures of the samples and binding states of the component elements, respectively. A UV-visible absorption spectrometer (Shimadzu, UV-2450) was used to examine the transmittance of the samples. The time-resolved photoluminescence (TRPL) study was carried out using a confocal microscope (MicroTime-200, Picoquant, Germany). The lifetime measurements were performed at the Korea Basic Science Institute (KBSI), Daegu Center, Korea. A single-mode pulsed diode laser (375 nm with 30 ps pulse width and average power of ∼10 μW operating in 2 MHz repetition rate) was used as an excitation source. The steady-state PL spectrum was obtained by dividing and guiding emission photons through an optical fiber to the external spectrometer (F-7000, Hitachi). A detailed analytical procedure of TRPL was described elsewhere.263. Results and discussion
3.1. Photoelectrocatalytic activity of the as-synthesized electrodes
The as-synthesized BVO sample exhibited the typical monoclinic crystal structure (ICDD# 04-010-5713) (Fig. 1a). W-doping did not influence the crystallinity of BVO. Neither W- nor Co-oriented XRD peaks (e.g., WO3 and Co2O3, respectively) were observed with WBVO and Co-WBVO, because of trace amounts of doped W and deposited Co. The bare and modified BVO samples showed the morphologies grown via interparticle connection, with an overall film thickness of ∼1.8 μm (Fig. S1, ESI†). Compared to the bare BVO, the morphology of WBVO was more distinct with small particles, while Co-WBVO had a rougher surface and less agglomeration. Meanwhile, XPS analysis with Co-WBVO obviously revealed the presence of W and Co. The atomic level of W was estimated to be 0.25% with respect to Bi (Fig. 1b). However, the W doping did not induce spectral difference of the Bi 4f and V 2p bands (Fig. S2, ESI†). Notably, the oxidation states of the deposited Co were mixed with Co2+ (781.6 eV) and Co3+ (780.5 eV) at a Co3+ fraction of 13.4% (Fig. 1c). After pre-anodization, the fraction of Co3+ increased to ∼34% (Fig. 1d). A concurrent decrease in the metal-OH intensity of the O 1s band was also observed. This change should indicate that the as-deposited Co species was primarily Co(OH)227,28 and oxidized to CoOOH upon repetitive anodic scans.29 Co deposition onto WBVO did not induce a significant change in the film thickness. This further indicates that the thin Co layer did not interfere with light absorption.30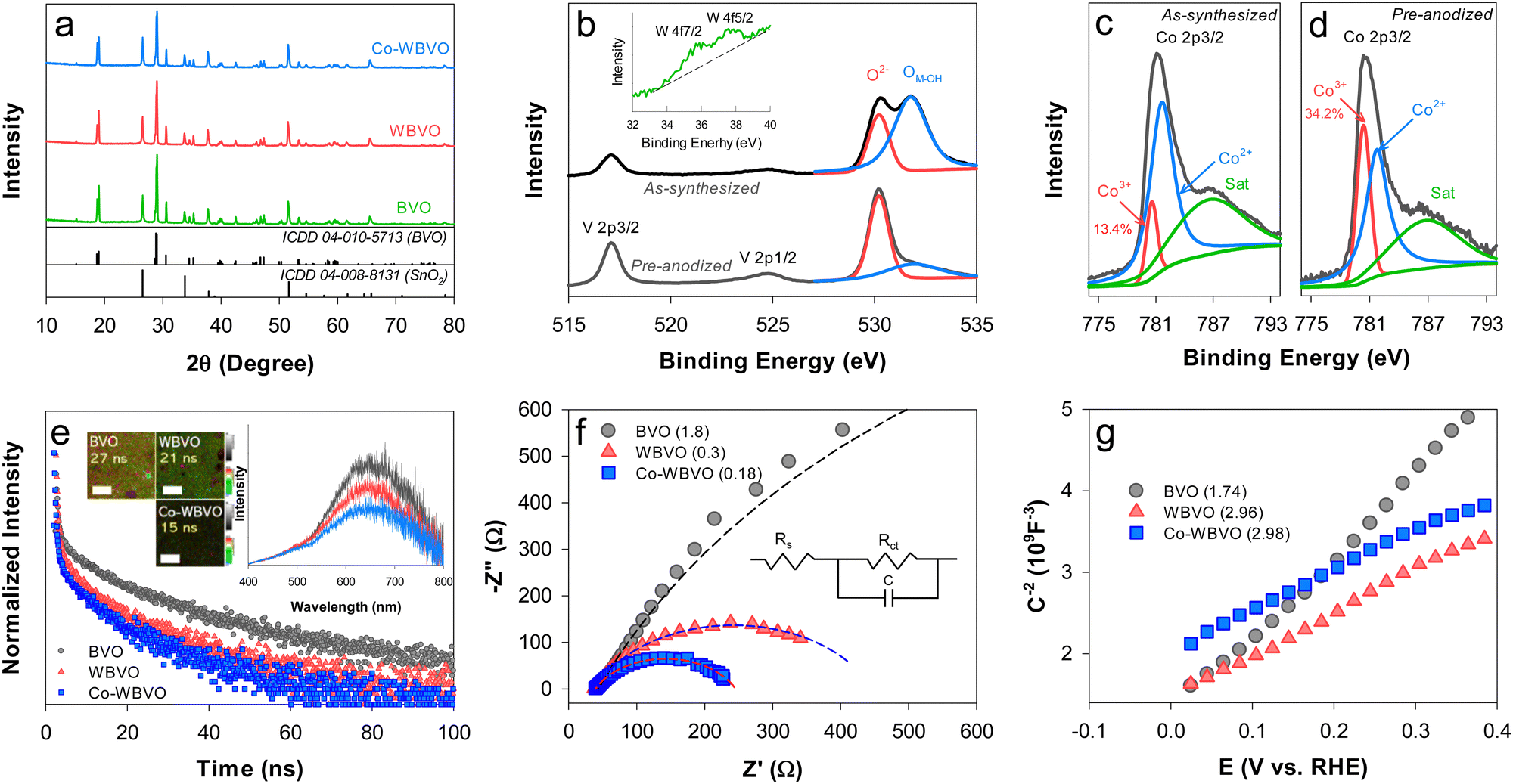 | ||
| Fig. 1 (a) XRD patterns of the FTO substrate, BVO, WBVO, and Co-WBVO. XPS spectra of (b) the V 2p and O 1s bands (inset: W 4f bands) and (c) and (d) the Co 2p3/2 bands for Co-WBVO (as-synthesized and pre-anodized). The numbers in (c) and (d) refer to the areal fractions of the deconvoluted Co3+ peaks with respect to the Co peaks. (e)–(g) Optical and electrochemical characterization of BVO, WBVO, and Co-WBVO: (e) TRPL spectra. The inset shows 2D emission lifetime images and PL spectra. The white bars represent 20 μm, and the numbers are average emission decay lifetimes (τ). (f) EIS spectra in 0.171 M NaCl. The numbers in parentheses refer to the charge transfer resistance (Rct) values (kΩ) estimated by fitting the spectra to the Randles circuit model. (g) Mott–Schottky plots in 0.171 M NaCl. The numbers in parentheses refer to donor density (ND, ×1022 cm−3). | ||
TRPL was used to examine the charge transfer dynamics of the bare and modified BVO films (Fig. 1e). Upon excitation at λ = 375 nm, the PL spectra showed broad emission bands in the λ range of 400–800 nm, with the maximum at λ ∼ 650 nm (ca. 1.9 eV) (Fig. 1e inset). These emission bands were attributed to the radiative recombination of photogenerated electron and hole pairs via intermediate trap states within the bandgap.31,32 However, the maximum emission intensity was lowered by W-doping and Co-deposition, indicating inhibited charge recombination. The normalized TRPL emission intensity with bare BVO exponentially decayed with an average lifetime (τ) of 27 ns. WBVO exhibited the same decay profile yet with a relatively small τ value (21 ns). Co-deposition further decreased the τ value to 15 ns. The 2D-emission lifetime images showed that the brightness was not uniform throughout the detected regions with BVO and WBVO (Fig. 1e inset). However, Co-WBVO was entirely dark due to the fast, uniform charge transfer kinetics. EIS analysis was also performed to gain insight into the interfacial charge transfer resistance (Rct) (Fig. 1f). The Nyquist plots showed a large semicircle with Rct of ∼1.8 kΩ for BVO, whereas WBVO and Co-WBVO exhibited significantly reduced semicircles with Rct values of ∼0.3 and 0.18 kΩ, respectively. The predominant effect of W(VI)-doping on the reduction of Rct was attributed to an increase in donor density (ND ∼ 3 × 1022 cm−3), enhancing the electrical conductivity of BVO (ND ∼ 1.7 × 1022 cm−3) (Fig. 1g). Co-deposition also contributed to reduction in Rct but marginally, which was attributed to a similar ND value to that of WBVO.
The linear sweep voltammograms with bare and modified BVO photoanodes were obtained in aqueous NaCl solutions (0.171 M, pH ∼6.5) (Fig. 2a). The bare BVO showed an onset potential (Eon) of ∼0.8 VRHE with Jph ∼ 1.45 mA cm−2 for ClOR (Jph,ClOR, estimated at E°(Cl2/Cl−) = 1.36 VRHE). This Jph,ClOR value is quite comparable to those in the literature (Table S1, ESI†). W-doping significantly influenced the voltammograms, leading to the lowest Eon (∼0.6 VRHE) and the highest Jph,ClOR (2.4 mA cm−2) at doping levels of 0.3–0.6 at% (Fig. S3, ESI†). Although the primary role of the W dopant is to enhance charge separation (see Fig. 1e), its heavy doping induces a structural change from the monoclinic scheelite to tetragonal scheelite.33,34 This should decrease the PEC activity at W-doping levels over 0.6 at%. Notably, the Co-WBVO showed different voltammograms with repetitive potential sweeps. In the first sweep, Eon (∼1.7 VRHE) was ∼0.9 V more positive than those without Co deposition (i.e., BVO and WBVO) and Jph,ClOR increased dramatically at E > Eon. From the second to the fifth sweep, Eon gradually shifted to low potential and reached to 0.4 VRHE with Jph,ClOR of 3.55 mA cm−2 (Fig. S4, ESI†). These changes in the voltammogram were attributed to gradual oxidation of the as-deposited Co(OH)2 to CoOOH during the oxidative potential sweeps (see Fig. 1c and d).30 This Co oxidation shifted the flatband potential of WBVO from 0 to −0.1 VRHE (see Fig. 1g), enhancing band-bending and charge separation.28,35 It was also found that the optimal charge amount for Co deposition was 10 mC (Fig. S5, ESI†). A further deposition at 30 mC degenerated Co-WBVO. In agreement with the voltammogram profiles, the IPCE values of Co-WBVO were greater than those of BVO and WBVO (Fig. 2a inset). However, the onset wavelengths for Jph generation and IPCE were the same at λ ∼ 505 nm for bare and modified BVOs, owing to their similar bandgaps of 2.45 eV (Fig. S6, ESI†). This indicates that the employed surface modification enhanced the charge transfer efficiency,36 not affecting the intrinsic electronic structure of BVO.
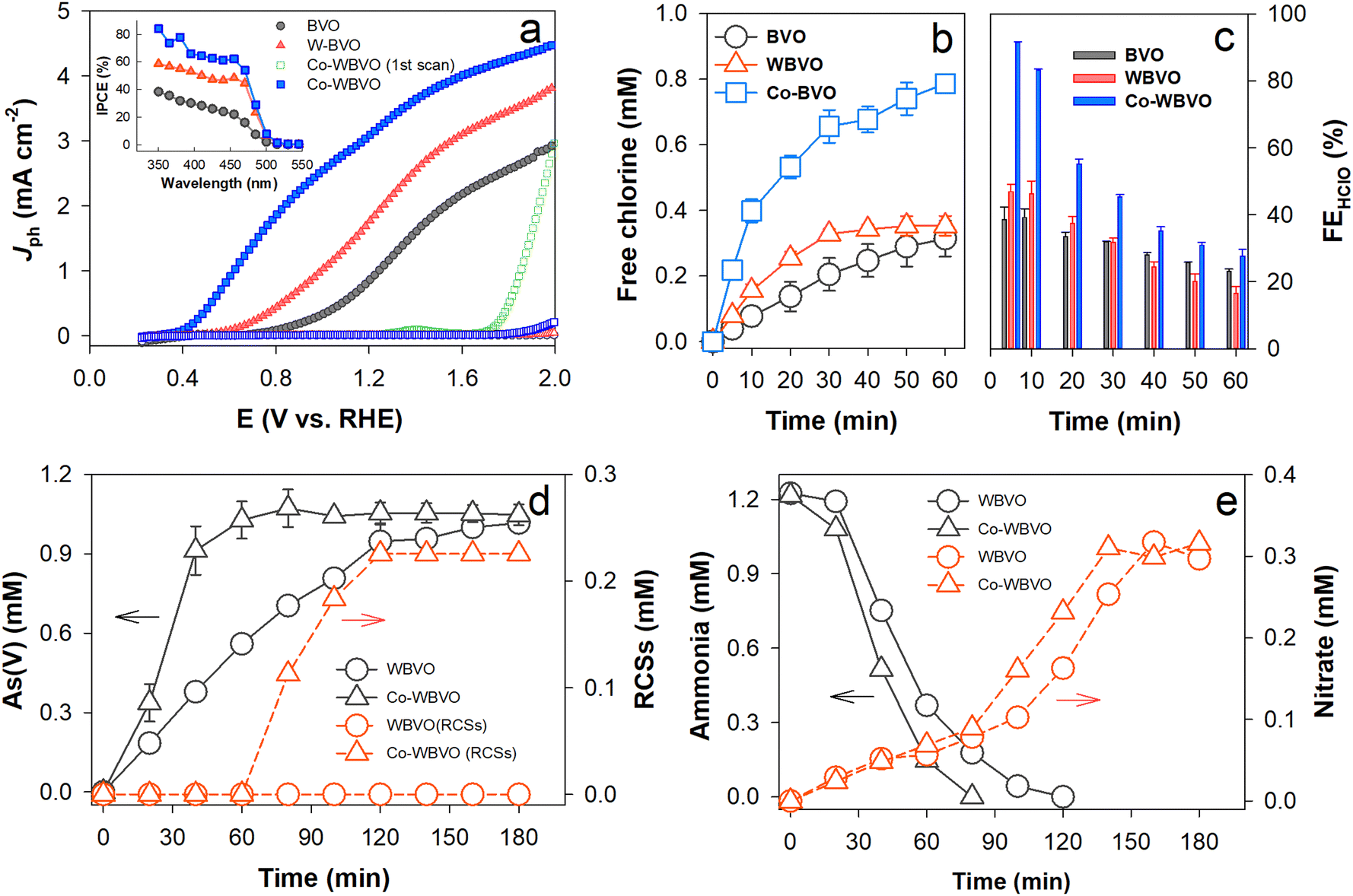 | ||
| Fig. 2 PEC activities of the bare and modified BVO samples in 0.171 M NaCl. (a) Linear sweep voltammograms. The inset shows IPCE profiles at E = 1.36 VRHE. (b) and (c) RSC (represented by HClO) production and FERCSs. (d) Oxidation of As(III) (1 mM) to As(V) and production of RCSs with Co-WBVO. (e) Oxidation of NH3 (1 mM) and concurrent production of nitrate (NO3−). The bulk PEC reactions (b)–(e) were performed in a two-compartment cell at E = 0.73 VSCE (corresponding to E = 1.36 VRHE: note that the solution pH changed with time, and hence, the SCE scale was used). | ||
The ClOR was further examined at E = 1.36 VRHE in a two-compartment cell divided by a proton-exchange membrane (Fig. 2b). With bare BVO, a stable Jph,ClOR of ∼1.5 mA cm−2 flowed over 60 min (Fig. S7, ESI†) and RCSs (represented by HClO) were produced via ClOR with photogenerated holes (h+) (reactions (R1) and (R2)).
| 2Cl− + 2h+ → Cl2 | (R1) |
| Cl2 + H2O → HClO + Cl− + H+ | (R2) |
The as-produced RCSs were active for in situ oxidation of As(III) to As(V) (reaction (R3)). With WBVO at E = 1.36 VRHE in NaCl solution, As(V) was produced with an apparent rate constant (kapp) of 0.45 min−1 (Fig. 2d). With Co-WBVO, the As(V) production was significantly enhanced with kapp of 0.78 min−1. RCSs were produced right after the complete oxidation of As(III) (∼60 min), confirming that the RCSs were the primary reactive species.37 No further increase in the amount of RCSs in ∼120 min was attributed to concurrent oxidation of HClO to ClO3−. The decomposition of ammonia was also examined (Fig. 2e). Similar to the case of As(III) oxidation, Co-WBVO exhibited a faster decomposition kinetics than WBVO. For both photoelectrodes, the ammonia decomposition accompanied the gradual production of nitrate. Even after a complete decomposition of ammonia, the nitrate production continued because of the sequential oxidation of N-chloroamine intermediates (e.g., NH3−xClx) by HClO (reactions (R4) and (R5)).38 Compared to As(III), the slower oxidation of ammonia was attributed to the eight-electron transfer reaction, requiring four HClO molecules per 1 ammonia molecule.
| AsIII(OH)3 + HClO → H2AsVO− + Cl− + 2H+ | (R3) |
| NH3 + xHClO → NH3−xClx + xH2O | (R4) |
| NCl3 + HClO + 2H2O → NO3− + 4Cl− + 5H+ | (R5) |
3.2. PEC charging and electrocatalytic discharging reactions
To drive PEC ClOR and charging with sodium ions, WBVO and Co-WBVO photoelectrodes were coupled to arrayed NaxC electrodes (four units) in aqueous NaCl solutions containing both As(III) and NH3. Fig. 3a shows the linear sweep voltammograms with the photoelectrodes and simultaneous changes in the cell voltages (Ecells) of the photoelectrode and NaxC array electrode pairs. The overall voltammogram profiles were similar to conventional ones with the Pt counter electrode (see Fig. 2a). However, Jph with NaxC was greater than that with Pt. Increasing a number of NaxC enhanced the Jph values (Fig. S9a, ESI†). It appears that the Na+ insertion (charging) kinetics with NaxC was comparable to the H+ reduction with Pt and partially influenced the behavior of the photoelectrodes.39 With increasing potential bias, Ecell increased linearly because of the PEC charging of NaxC, yet less significantly with more unit NaxC electrodes (Fig. S9b, ESI†). When Jph of 1 mA cm−2 was applied to WBVO and Co-WBVO, photopotentials (Eph) of ∼0.7 VSCE and ∼0.5 VSCE (1.06 and 0.86 VRHE, respectively) were maintained over 8 h, with Ecells of ∼4.0 and ∼3.7 V, respectively (Fig. S10, ESI†). During the PEC ClOR with Co-WBVO and simultaneous charging of NaxC arrays, As(III) was continuously oxidized to As(V) for 4 h (Fig. 3b). Soon after completion of the As(III) oxidation, the ammonia decomposition was initiated and completed in ∼4 h (hence a total ∼8 h for completion of both As(III) oxidation and ammonia decomposition). A similar behavior was observed for WBVO yet with retarded kinetics. Compared to WBVO, the faster decomposition rates with Co-WBVO should be attributed to greater FERCSs values (i.e., greater amounts of RCSs; see Fig. 2b).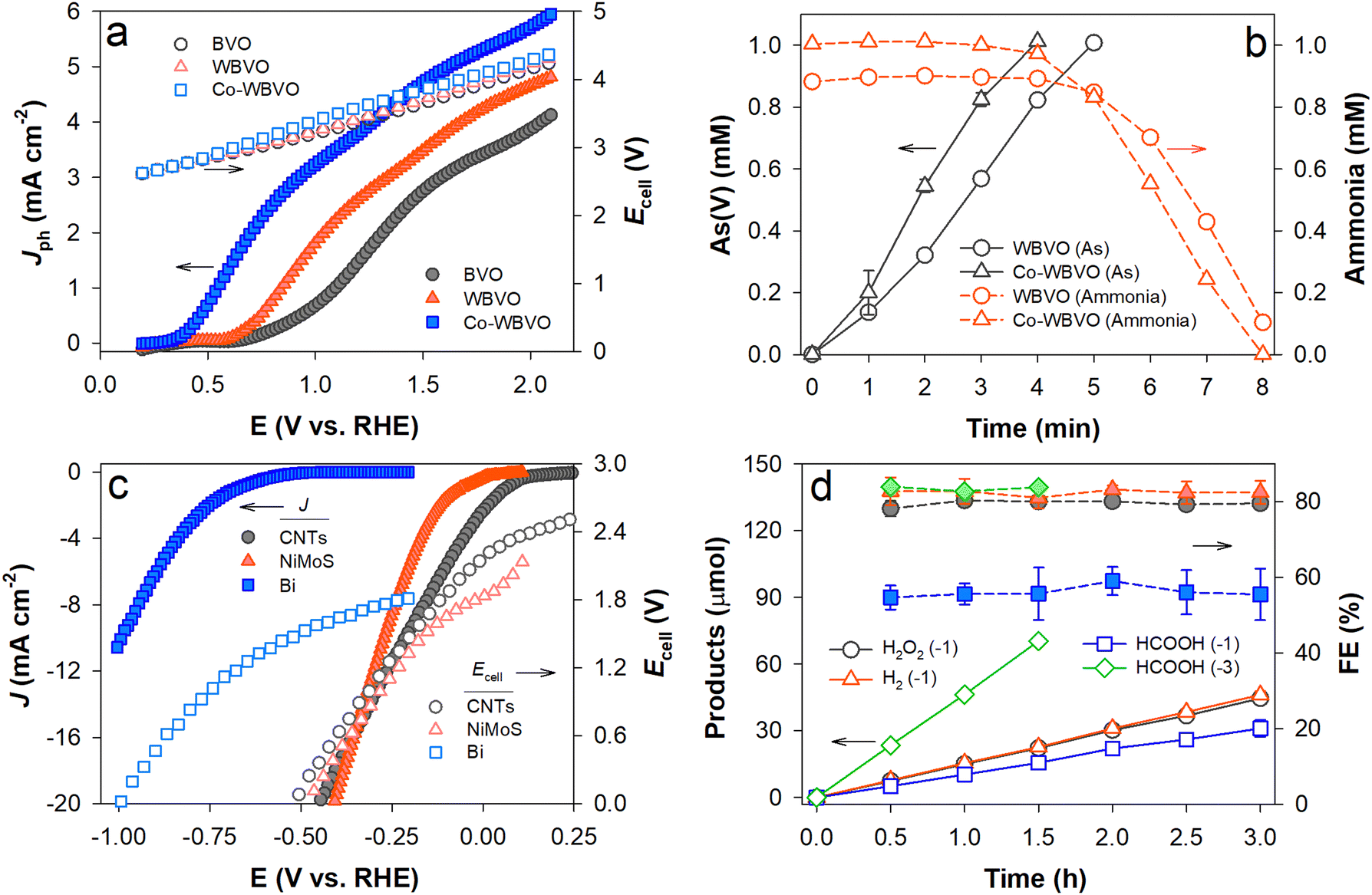 | ||
| Fig. 3 (a) and (b) PEC charge and (c) and (d) electrocatalytic discharge reactions. In the photocharge stage, the photoanodes were coupled to the NaxC electrode in a 0.171 M NaCl solution with mixed As(III) and NH3. Scheme 1a shows the experimental setup. In the discharging stage, the charged NaxC electrode was disconnected from the photoanodes. It was then connected to each of three different electrocatalysts (CNTs for O2 reduction in 0.1 M K2SO4 purged with O2 at pH 6.5; NiMoS for H2 evolution in 0.1 M KOH purged with N2 at pH 13; Bi for CO2 reduction in 0.1 M KHCO3 purged with CO2 at pH 6.8). Scheme 1c shows the experimental setup. (a) Linear sweep voltammograms of the photoelectrodes and concurrent changes in the cell voltages (Ecell) of the photoelectrode–NaxC pairs. (b) Simultaneous oxidation of As(III) (1 mM) and NH3 (1 mM) at J = 1 mA cm−2 during the photocharge stage. (c) Linear sweep voltammograms of the electrocatalysts and concurrent changes in the Ecell values of the electrocatalyst and charged NaxC pairs. (d) Production and FEs of H2O2via O2 reduction with CNTs, H2via water reduction with NiMoS, and HCOOH via CO2 reduction with Bi during the discharge stage. The numbers in parentheses are the applied J values (unit: mA cm−2). | ||
After the PEC processes, the charged NaxC electrode with 8 mA h (= 1 mA cm−2 × 1 cm2 × 8 h) was coupled to CNTs,20 NiMoS,13,40 and porous Bi electrodes21,41 specifically designed for the production of H2O2, H2, and HCOOH (representing formic acid and formate) via reductions of O2, H2O, and CO2, respectively, under various conditions (reactions (R6)–(R8)).
| O2 + 2H+ + 2e− → H2O2 | (R6) |
| 2H+ + 2e− → H2 | (R7) |
| CO2 + 2H+ + 2e− → HCOOH | (R8) |
The voltammogram with the as-synthesized CNT electrode showed Eon of 0.2 VRHE, corresponding to the open-circuit Ecell of 2.5 V for the CNTs and NaxC pair (Fig. 3c). Cathodic JCNTs increased linearly with increasing negative ECNTs, whereas Ecell decreased because of the discharging process with NaxC. Upon a constant JCNTs of −1 mA cm−2, ECNTs of 0.035 VRHE and Ecell of ∼2.25 V were maintained over 3 h (Fig. S11, ESI†). Simultaneously, H2O2 was linearly produced with time with an FE of >80% (Fig. 3d). The obtained FE values were quite similar to those in the literature,20 indicating that the PEC charging and post-discharging process successfully worked.
The voltammograms with the NiMoS and Bi electrodes showed that the HER and CO2RR need ∼0.2 and ∼0.7 V-greater Eon values, respectively, than that for the H2O2 production reaction. These led to decreases in the open-circuit Ecell values to 1.85 and 1.48 V, respectively. At a constant J of −1 mA cm−2, H2 and HCOOH were produced linearly over 3 h, while constant potentials (ENiMoS = −0.085 VRHE; EBi = −0.7 VRHE) and Ecell values (1.8 V and 1.27 V, respectively) were maintained (Fig. S11a, ESI†). The FE of H2 production was ∼80%, which was quite similar to that in the literature.13 In contrast, the FE of HCOOH production was as low as ∼60%. Our previous study showed that the optimal potential with the Bi electrode for CO2RR was found to be approximately −0.85 VRHE, whereas FE decreased to ∼55% at EBi of −0.7 VRHE.21 Hence, when JBi increased to −3 mA cm−2, a stable EBi of −0.8 VRHE was obtained and the FE of HCOOH production enhanced to >80%. The productions of H2O2 and H2 were also enhanced at J of −3 mA cm−2 with FEs of >80% (Fig. S12, ESI†). However, the NaxC electrode was rapidly discharged at J of −3 mA cm−2 and Ecells dropped in ∼1 h (Fig. S11b, ESI†).
3.3. PEC desalination-coupled charging of NaxC
For coupling PEC charging and desalination processes, a membrane device composed of a photoelectrode compartment with As(III) and NH3, desalination compartment with saline water, and cathode compartment with NaxC was designed (Scheme 1b). With Co-WBVO at a constant Jph of 1 mA cm−2, Eph rapidly increased to ∼0.53 VSCE in 1 h and was stabilized at ∼0.6 VSCE over 10 h (Fig. 4a). This behavior was quite similar to that without the desalination process (Fig. S10, ESI†). The device Ecell followed the same trend as that of Eph, reaching ∼4.1 V in 10 h. During the PEC charging process, the amount of chloride in the saline water decreased linearly with time and the amount of chloride in the anolyte simultaneously increased (Fig. 4b). The total ΔCl− in the saline water and anolyte in 10 h were the same as ∼0.45 mmol, indicating a unidirectional Cl− transport from the saline water to the anolyte. Accordingly, the ITE for Cl− was maintained to be ∼100% over 10 h (Fig. 4b inset). The SEC for 50% desalination was estimated to be 7.7 kW h m−3, which was higher than a previous study by 1.48 times.13 Meanwhile, the oxidation of As(III) (1 mM) to As(V) was completed in 2 h; subsequently, the ammonia decomposition proceeded for the following 4 h (Fig. 4c). Similar behaviors were also found in the case without the desalination compartment (see Fig. 3b).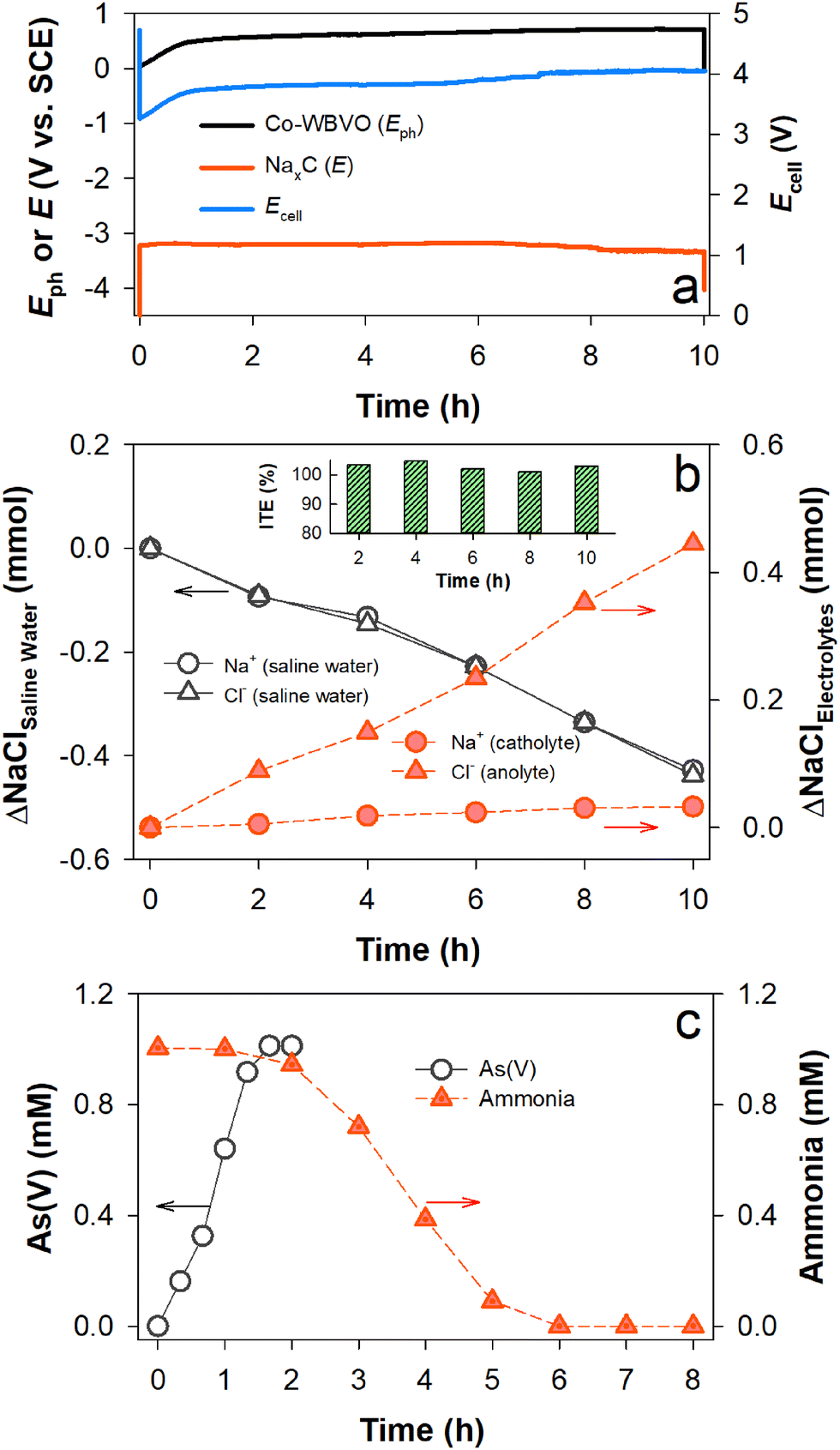 | ||
| Fig. 4 Solar charge process with desalination of saline water (0.171 M NaCl) and oxidation of mixed As(III) and NH3 (each 1 mM). A Co-WBVO photoelectrode (with the SCE) and an NaxC electrode were immersed in 0.171 M NaCl solutions. Scheme 1b shows the experimental setup. (a) Changes in the Eph of Co-WBVO and the Ecell of the Co-WBVO–NaxC pair at Jph of 1 mA cm−2. (b) Changes in the amounts of Na+ and Cl− in saline water, Na+ in the catholyte, and Cl− in the anolyte. The inset shows the ITEs. (c) Changes in As(V) and NH3 concentrations. For the WBVO–NaxC pair, see Fig. S13 (ESI†). | ||
It should be noted that the amount of Na+ in the catholyte insignificantly changed, although the amount of Na+ in the desalination compartment continuously decreased (ITE of ∼100% for Na+). The different behavior of Na+ from Cl− was attributed to the insertion of Na+ into the NaxC electrode fast enough to keep the amount of Na+ unchanged over 10 h. Based on the photocharges of 10 mA h, the amount of inserted Na+ was estimated to be ∼0.37 mmol. Considering that the actual amount of inserted Na+ for 10 h was ∼0.35 mmol, the charge storage efficiency was estimated to be ∼95%. All the observed behaviors with Co-WBVO were the same as those with WBVO (Fig. S13, ESI†), except for the faster oxidation kinetics of As(III) and ammonia due to higher FERCSs with Co-WBVO.
The as-photocharged NaxC electrode was directly wired to a CNT electrode in a single compartment cell containing K2SO4 (pH 6.5) purged with O2, and Ecell of 0 V was applied to the NaxC and CNT electrode pair. JCNTs of −24 mA cm−2 flowed in the initial stage and abruptly decreased in 20 min (Fig. 5a), while the initial ECNTs (−0.4 VRHE) increased to ∼0.3 VRHE in the later stage (Fig. 5b). During the discharging period, H2O2 production reached a plateau of ∼155 μmol in 1 h with FE of ∼80% (Fig. 5c and d). When the photocharged NaxC was coupled to the NiMoS electrode for H2 production, JNiMoS decayed more rapidly than JCNTs at FE of ∼90% for H2 production, while ENiMoS gradually decreased. With the Bi electrode, the initial value of JBi (∼5 mA cm−2) and the amount of HCOOH were much lower than those with the other electrodes despite the same two-electron transfer process. This behavior with Bi was attributed to a large potential required for the CO2RR. Nevertheless, the FE of HCOOH production was ∼80%. Finally, the NaxC and CNT electrodes were wired through an LED bulb at Ecell of 0 V. A stable discharging JCNTs of −2 mA cm−2 was obtained over 2 h with the generation of LED light (Fig. 5e). H2O2 was produced linearly with time at an FE of >80% (Fig. 5f).
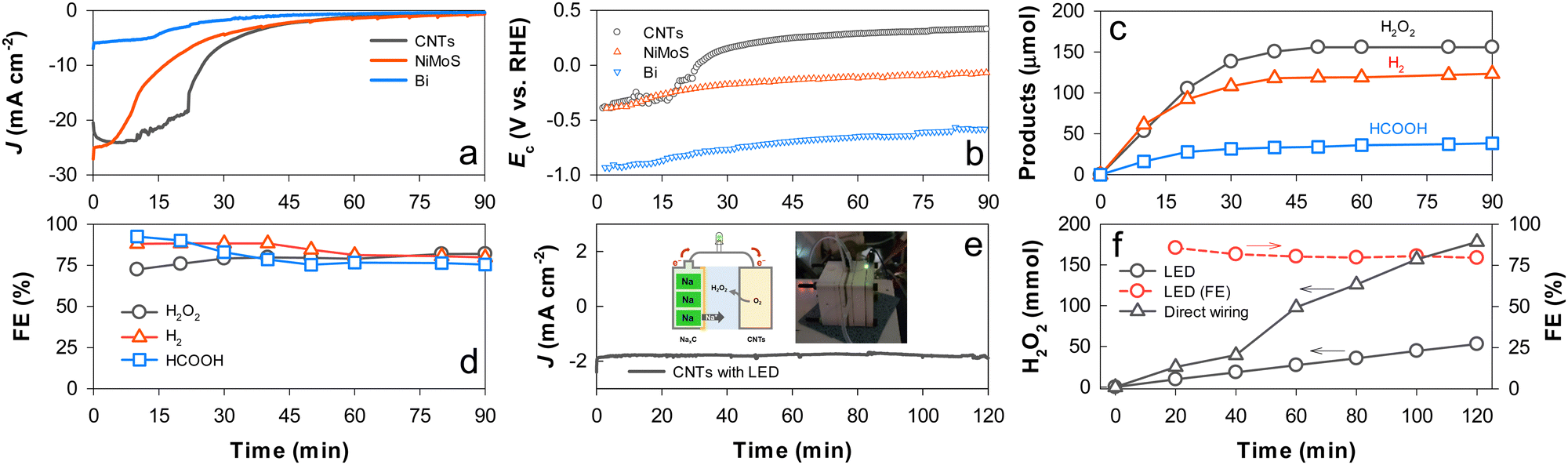 | ||
| Fig. 5 (a)–(d) Electrocatalytic production of H2O2 with CNTs, H2 with NiMoS, and HCOOH with Bi coupled to a PEC charged NaxC electrode at an applied Ecell of 0 V (see Scheme 1c): (a) changes in the discharge J values of the electrocatalysts; (b) changes in the potentials (Ec) of the electrocatalysts; (c) production of H2O2, H2, and HCOOH; and (d) FEs for chemical production. (e) and (f) Electrocatalytic production of H2O2 with CNTs coupled to the charged NaxC electrode via an LED bulb: (e) changes in the discharge J value of CNTs and (f) H2O2 production and FE values with time. For comparison, H2O2 production without the LED bulb is shown. | ||
4. Conclusions
We demonstrated a desalination-coupled PEC hybrid system capable of remediating aquatic pollutants via redox reactions of desalted Cl− and charging an aqueous sodium metal battery with photogenerated electrons and desalted Na+. Significant efforts have been made to synthesize high efficiency BVO-based photoelectrodes via W-doping and Co-deposition. The optimized Co-WBVO oxidized chlorides into RCSs at an FE > 90%, with minimized production of non-reactive chlorine species. These in situ generated but storable RCSs effectively oxidized As(III) to As(V) and sequentially NH4+ to nitrate in mixed As(III) and NH4+ solution. During the anodic reaction with chloride, Na+ was successfully inserted into a wired NaxC electrode. The photocharged NaxC was further demonstrated to drive electrocatalytic conversions of O2 to H2O2 with CNTs, H2O to H2 with NiMoS, and CO2 to HCOOH with Bi, at FEs of >80%. This PEC charging–discharging reaction with Na+ and Cl− was successfully coupled to a desalination process with brackish water in a three-compartment cell with ion-exchange membranes. While desalination proceeded at an SEC of 7.7 kW h m−3, Na+ and Cl− were unidirectionally transported to the NaxC electrode compartment and photoelectrode compartment, respectively, at an ITE of ∼100%. Despite a proof-of-concept, the as-designed PEC hybrid can be applied for various purposes, including desalination, seawater electrolysis, production of value-added chemicals, and energy storage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the National Research Foundation of Korea (RS-2023-00254645 and 2022R1C1C2011712). This publication was made possible by a grant from the Qatar National Research Fund under its National Priorities Research Program (NPRP 13S-0202-200228).References
- D. T. Pio, A. C. M. Vilas-Boas, N. F. C. Rodrigues and A. Mendes, Green Chem., 2022, 24, 5403–5428 RSC.
- A. González-Garay, N. Mac Dowell and N. Shah, Discover. Chem. Eng., 2021, 1, 2 CrossRef.
- UNESCO World Water Assessment Programme, The United Nations World Water Development Report 2020: Water and Climate change, https://www.unwater.org/publications/un-world-water-development-report-2020, (accessed Jan 26, 2023).
- M. Ahmed, M. O. Mavukkandy, A. Giwa, M. Elektorowicz, E. Katsou, O. Khelifi, V. Naddeo and S. W. Hasan, npj Clean Water, 2022, 5, 12 CrossRef CAS.
- S. Guo and S. C. Tan, Joule, 2024, 8, 291–294 CrossRef.
- A. Landman, H. Dotan, G. E. Shter, M. Wullenkord, A. Houaijia, A. Maljusch, G. S. Grader and A. Rothschild, Nat. Mater., 2017, 16, 646–651 CrossRef CAS PubMed.
- Z. P. Ifkovits, J. M. Evans, M. C. Meier, K. M. Papadantonakis and N. S. Lewis, Energy Environ. Sci., 2021, 14, 4740–4759 RSC.
- L. Yang, L. Loh, D. K. Nandakumar, W. Lu, M. Gao, X. L. C. Wee, K. Zeng, M. Bosman and S. C. Tan, Adv. Mater., 2020, 32, 2000971 CrossRef CAS PubMed.
- B. Koo, D. Kim, P. Boonmongkolras, S. R. Pae, S. Byun, J. Kim, J. H. Lee, D. H. Kim, S. Kim and B. T. Ahn, ACS Appl. Energy Mater., 2020, 3, 2296–2303 CrossRef CAS.
- P. Varadhan, H. C. Fu, Y. C. Kao, R. H. Horng and J. H. He, Nat. Commun., 2019, 10, 5282 CrossRef PubMed.
- C. S. Tan, K. W. Kemp, M. R. Braun, A. C. Meng, W. Tan, C. E. Chidsey, W. Ma, F. Moghadam and P. C. McIntyre, Sustainable Energy Fuels, 2019, 3, 1490–1500 RSC.
- S. Kim, G. Piao, D. S. Han, H. K. Shon and H. Park, Energy Environ. Sci., 2018, 11, 344–353 RSC.
- S. Kim, D. S. Han and H. Park, Appl. Catal., B, 2021, 284, 119745 CrossRef CAS.
- B. J. Kim, G. Piao, S. Kim, S. Y. Yang, Y. Park, D. S. Han, H. K. Shon, M. R. Hoffmann and H. Park, ACS Sustainable Chem. Eng., 2019, 7, 15320–15328 CrossRef CAS.
- B. Sun, P. Li, J. Zhang, D. Wang, P. Munroe, C. Wang, P. H. L. Notten and G. Wang, Adv. Mater., 2018, 30, e1801334 CrossRef PubMed.
- M. Q. Zhu, S. M. Li, B. Li, Y. J. Gong, Z. G. Du and S. B. Yang, Sci. Adv., 2019, 5, eaau6264 CrossRef CAS PubMed.
- T. W. Kim and K.-S. Choi, Science, 2014, 343, 990–994 CrossRef CAS PubMed.
- K. J. McDonald and K.-S. Choi, Energy Environ. Sci., 2012, 5, 8553–8557 RSC.
- S. Park, B. SenthilKumar, K. Kim, S. M. Hwang and Y. Kim, J. Mater. Chem. A, 2016, 4, 7207–7213 RSC.
- S. Y. Choi, S. Kim, K. J. Lee, J. Y. Kim, D. S. Han and H. Park, Appl. Catal., B, 2019, 252, 55–61 CrossRef CAS.
- G. Piao, S. H. Yoon, D. S. Han and H. Park, ChemSusChem, 2020, 13, 698–706 CrossRef CAS PubMed.
- S. Kim, G. Piao, D. S. Han, H. K. Shon and H. Park, Energy Environ. Sci., 2018, 11, 344–353 RSC.
- W. Choi, M. Kim, B.-j Kim, Y. Park, D. S. Han, M. R. Hoffmann and H. Park, Appl. Catal., B, 2020, 265, 118607 CrossRef CAS.
- S. Y. Choi, S. Kim, K. J. Lee, J. Y. Kim, D. S. Han and H. Park, Appl. Catal., B, 2019, 252, 55–61 CrossRef CAS.
- U. Kang and H. Park, J. Mater. Chem. A, 2017, 5, 2123–2131 RSC.
- H. W. Jeong, W.-S. Chae, B. Song, C.-H. Cho, S.-H. Baek, Y. Park and H. Park, Energy Environ. Sci., 2016, 9, 3143–3150 RSC.
- R. T. Gao, D. He, L. Wu, K. Hu, X. Liu, Y. Su and L. Wang, Angew. Chem., Int. Ed., 2020, 132, 6272–6277 CrossRef.
- F. Tang, W. Cheng, H. Su, X. Zhao and Q. Liu, ACS Appl. Mater. Interfaces, 2018, 10, 6228–6234 CrossRef CAS PubMed.
- B. B. Zhang, X. J. Huang, H. Y. Hu, L. J. Chou and Y. P. Bi, J. Mater. Chem. A, 2019, 7, 4415–4419 RSC.
- B. J. Taitt, D.-H. Nam and K.-S. Choi, ACS Catal., 2018, 9, 660–670 CrossRef.
- I. Abdellaoui, M. M. Islam, M. Remeika, Y. Higuchi, T. Kawaguchi, T. Harada, C. Budich, T. Maeda, T. Wada, S. Ikeda and T. Sakurai, J. Phys. Chem. C, 2020, 124, 3962–3972 CrossRef CAS.
- T. Tachikawa, T. Ochi and Y. Kobori, ACS Catal., 2016, 6, 2250–2256 CrossRef CAS.
- M. Rohloff, B. Anke, S. Zhang, U. Gernert, C. Scheu, M. Lerch and A. Fischer, Sustainable Energy Fuels, 2017, 1, 1830–1846 RSC.
- S. Tokunaga, H. Kato and A. Kudo, Chem. Mater., 2001, 13, 4624–4628 CrossRef CAS.
- B. Zhang, X. Huang, H. Hu, L. Chou and Y. Bi, J. Mater. Chem. A, 2019, 7, 4415–4419 RSC.
- H. W. Jeong, T. H. Jeon, J. S. Jang, W. Choi and H. Park, J. Phys. Chem. C, 2013, 117, 9104–9112 CrossRef CAS.
- W. Choi, D. S. Han and H. Park, J. Phys. Chem. A, 2022, 126, 8459–8467 CrossRef CAS PubMed.
- B.-j Kim, G. Piao, S. Kim, S. Y. Yang, Y. Park, D. S. Han, H. K. Shon, M. R. Hoffmann and H. Park, ACS Sustainable Chem. Eng., 2019, 7, 15320–15328 CrossRef CAS.
- G. Hodes, J. Phys. Chem. Lett., 2012, 3, 1208–1213 CrossRef CAS PubMed.
- B. J. Kim, H. K. Shon, D. S. Han and H. Park, Desalination, 2023, 551, 116431 CrossRef CAS.
- G. Piao, S. H. Yoon, H. G. Cha, D. S. Han and H. Park, J. Mater. Chem. A, 2022, 10, 24006 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ee00782d |
| This journal is © The Royal Society of Chemistry 2024 |