
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-bonded assemblies of iron(II) spin crossover complexes†
Verónica
Jornet-Mollá
 ,
Marina I.
Rodríguez-Tarrazó
,
Miquel J.
Dolz-Lozano
,
Carlos
Giménez-Saiz
and
Francisco M.
Romero
*
,
Marina I.
Rodríguez-Tarrazó
,
Miquel J.
Dolz-Lozano
,
Carlos
Giménez-Saiz
and
Francisco M.
Romero
*
Departament de Química Inorgànica, Universitat de València, C/Dr. Moliner, 50, E-46100 Burjassot, Spain. E-mail: fmrm@uv.es
First published on 9th April 2024
Abstract
The paper reports on the synthesis, crystal structure, thermal and magnetic properties of spin crossover (SCO) salts containing the [Fe(bpp)2]2+ cation (bpp = 2,6-bis(pyrazol-3-yl)pyridine) and different rigid polycarboxylate anions, such as anthracene-9,10-dicarboxylate (ADC), benzene-1,3,5-tricarboxylate (BTC) and biphenyl-4,4′-dicarboxylate (BPDC). Compound [Fe(bpp)2](ADC)·9H2O (1) shows a porous hydrogen-bonded structure with water molecules sitting in the channels. It contains low-spin (LS) Fe2+ cations that undergo crossover to the high-spin (HS) state upon dehydration. Anhydrous 1 remains HS on cooling at low temperatures. A similar magnetic behaviour is obtained for the partially protonated BTC salt [Fe(bpp)2](HBTC)·5H2O (2), showing a spin change concomitant with dehydration to a HS phase that undergoes gradual and partial SCO on cooling, affecting 25% of the Fe2+ cations. Instead, the BPDC salt [Fe(bpp)2](BPDC)·5H2O (3) has a ground HS state in its fully hydrated form.
Introduction
Iron(II) complexes that undergo spin crossover (SCO) between their low-spin (LS, S = 0) and high-spin (HS, S = 2) states are certainly the most studied switchable molecular materials.1 As a response to external physical stimuli (light irradiation, temperature, pressure, and magnetic or electric fields),2–4 these compounds are able to show drastic changes in many of their physical properties, such as colour, magnetic susceptibility, dielectric constant, vibrational properties and crystal structure. In addition, solvent molecules strongly influence SCO properties.5 These features make SCO materials promising candidates for a wide variety of applications such as molecular switches, memory devices, thermometry, sensors and spintronics.6Among the different compounds exhibiting SCO behaviour, we turned our attention to [Fe(bpp)2]2+ complexes (bpp = 2,6-bis(pyrazol-3-yl)pyridine, Chart 1).7–9 The main reason is that these iron(II) complexes have four pyrazolyl N–H moieties that can act as hydrogen-bond donors towards solvent and/or anionic hydrogen-bond acceptors, yielding two interesting features: (i) a marked dependence of SCO properties on the presence of solvent molecules in the crystal lattice; (ii) the possibility of using the high directionality of hydrogen bonds in the design of crystalline assemblies. For instance, we have shown that it is possible to use the principles of hydrogen bonding in the preparation of SCO salts with non-centrosymmetric structures.10 Inversely, the structural changes that accompany the SCO phenomenon can have an impact on the hydrogen-bonding connectivity inside the crystal, leading to exciting effects, such as supramolecular H-bond isomerization11 or proton migration along short-strong H-bonds.12 Years ago, we started a systematic study of [Fe(bpp)2]X SCO salts containing polycarboxylate anions, since the latter have been extensively used in the design of metal–organic13 and hydrogen-bonded14 nanoporous frameworks with interesting sorption properties.15 The aim was the obtention of porous H-bonded structures where the SCO properties are affected by the nature of the encapsulated solvent/guest,16 as an alternative to more complex SCO nanoporous materials based on coordination chemistry, paving the way to applications like solvent/small molecule sensing or humidity control. We have now focused on hydrogen-bonded [Fe(bpp)2]2+ salts containing different rigid polycarboxylate anions, such as anthracene-9,10-dicarboxylate (ADC), benzene-1,3,5-tricarboxylate (BTC) or biphenyl-4,4′-dicarboxylate (BPDC). In this paper, we report on the structural, thermal and magnetic properties of salts [Fe(bpp)2](ADC)·9H2O (1), [Fe(bpp)2](HBTC)·5H2O (2) and [Fe(bpp)2](BPDC)·5H2O (3).
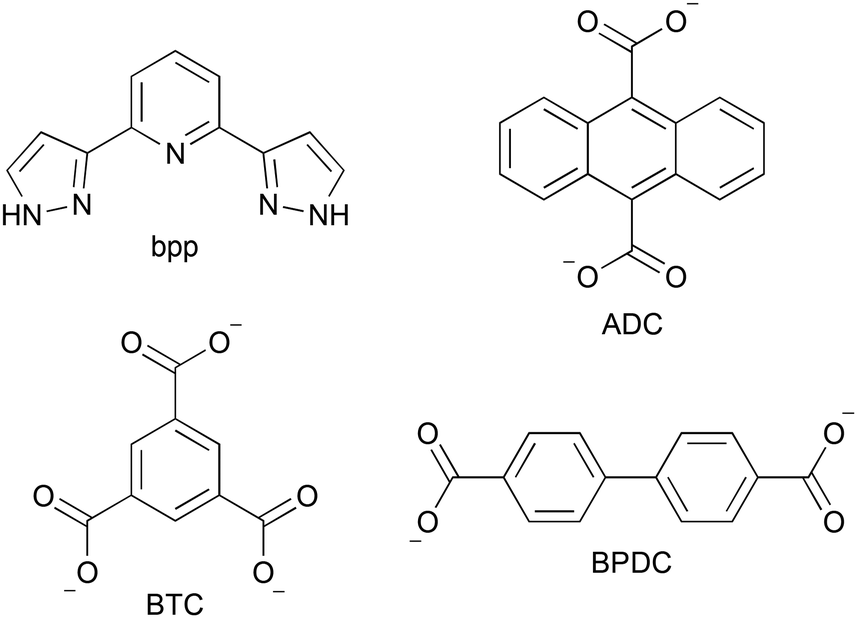 | ||
| Chart 1 Structures of the bpp ligand and several rigid polycarboxylate anions. | ||
Experimental section
Synthesis
Ligand bpp was prepared as previously published.17 All other reagents and solvents were used as received.Characterization techniques
Magnetic susceptibility measurements were performed on polycrystalline samples using a magnetometer (Quantum Design MPMS-XL-5) equipped with a SQUID sensor. Variable-temperature measurements were carried out in the temperature range 2–400 K in a magnetic field of 0.1 T. The temperature sweeping rate for compound 1 in the cooling and heating modes was as follows: 0.5 K min−1 (2–10 K), 3 K min−1 (10–400 K). For compounds 2 and 3, the sweeping rate in the 10–400 K temperature range was 2 K min−1. In all cases, for the first heating, the temperature of the sample was held at 400 K for 2 h to ensure complete dehydration. Measurements were performed on perforated plastic capsules in order to favour solvent loss.Thermogravimetric measurements were carried out in a Setaram Setsys TGA-ATD16/18 apparatus in the 295–1277 K temperature range (296–967 K for 2) under a nitrogen atmosphere and at a scan rate of 10 K min−1.
Differential scanning calorimetry measurements under nitrogen atmosphere were performed in a Mettler Toledo DSC 821e apparatus with warming and cooling rates equal to 10 K min−1 between 300 K and 450 K. A correction from the sample holder was automatically applied.
IR transmission measurements of KBr pellets were recorded at room temperature with a Nicolet Avatar 320 FT-IR spectrophotometer in the range 4000–400 cm−1.
CHN elemental analyses were carried out in a CE instruments EA 1110 CHNS analyzer.
Powder X-ray diffraction (PXRD) measurements were collected using Cu Kα radiation (λ = 1.54056 Å) at room temperature in a 2θ range from 2 to 40°. Polycrystalline samples of 1–3 were lightly ground in an agate mortar and filled into a 0.5 mm borosilicate capillary prior to being mounted and aligned on an Empyrean PANalytical powder diffractometer. The simulated diffractograms were obtained from single crystal X-ray data using the CrystalDiffract software.
Crystal structure determination
X-ray diffraction data of compounds 1–3 were collected at 120 K with a Rigaku Oxford Supernova diffractometer using a graphite monochromated Mo Kα radiation source (λ = 0.71073 Å). The CrysAlisPro program18 was used for cell refinements and data reduction. The structures were solved by direct methods using the SIR201419 program with the WinGX20 graphical user interface. The structure refinements were carried out with SHELXL-2018/3.21 A multiscan absorption correction, based on equivalent reflections22 was applied to the data using the program CrysAlisPro.H atoms on water molecules and N atoms were found in difference maps and refined positionally with geometric restraints (O–H = 0.82 Å, N–H = 0.89 Å and H⋯H = 1.30 Å) and with Uiso(H) = 1.5Ueq(O) for the H atoms on water molecules. The remaining H atoms were placed in calculated positions and refined with a riding model, with C–H = 0.95 Å, and Uiso(H) = 1.2Ueq(C,N). Four individual antibumping restraints were applied in 1, and one in 3, to prevent some hydrogen atoms on water molecules approaching too close to one another. One of the water molecules in 1 (O6W) had a refined occupancy close to 0.5, therefore their occupancy was fixed to that value in the final refinement cycle. All non-hydrogen atoms were refined anisotropically in all the structures.
The X-ray crystallographic structures have been deposited at the Cambridge Crystallographic Data Centre (deposition numbers CCDC 2335499 (1), 2335500 (2) and 2335503 (3)).†
Results and discussion
Synthesis and thermal properties
The synthetic method that has been generally applied for the synthesis of hydrogen-bonded networks containing the [Fe(bpp)2]2+ SCO units consists in a metathesis reaction of barium carboxylates with [Fe(bpp)2]SO4.16 This eliminates the presence of any other hydrogen-bond acceptor that could be in competition with the polycarboxylate anions (with the exception of the solvents). In the present case, probably due to the insolubility of the desired salts, the metathesis reaction was not necessary for 1 and 2 and these compounds were obtained by direct reaction from the sodium carboxylates obtained in situ by deprotonation of the corresponding carboxylic acids.The reaction outcome depends strongly on pH conditions after deprotonation. In the case of BTC (pH = 7.15), these conditions resulted in 2, a compound containing the monoprotonated dianion HBTC. Instead, for ADC, the same conditions (pH = 7.11) yielded 1, containing fully deprotonated ADC. However, lowering the pH after deprotonation to a value of 6.52 resulted in monoprotonated [Fe(bpp)2](HADC)2·5H2O. This compound is still under study and will be reported separately.
Concerning 3, the direct reaction gave unsatisfactory results and metathesis involving [Fe(bpp)2]Cl2 and Ag2(BPDC) as starting materials was used.
Thermogravimetric analyses of 1–3 have been performed. For 1, loss of the water molecules of crystallization takes place gradually from room temperature until 385 K (Fig. S2†). A weight decrease of 17% is observed (expected value for 9 H2O molecules: 18%). The huge mass loss at relatively low temperatures is in agreement with the presence of water sites in channels, loosely bound in an equivalent manner, as we shall see later. The dehydrated compound is stable at temperatures below 550 K. Above this temperature, it decomposes in several steps. In the case of 2, the loss of water molecules occurs in two smooth steps between room temperature and 440 K (Fig. S3†). The weight decrease (about 13.9%) is slightly higher than expected for the loss of five H2O molecules (12.3%). Decomposition of the sample starts at lower temperatures (520 K) as compared to the ADC salt. Finally, for 3, dehydration takes place in two steps at temperatures in the 345–415 K range, with a mass decrease of 10%. The sample keeps its integrity up to 520 K and decomposition occurs on further heating (Fig. S4†). The results indicate a good agreement with the formulation deduced from elemental analysis and X-ray crystal structure determinations (see below).
The differential scanning calorimetry (DSC) measurement of 1 is consistent with the thermogravimetric data. In the first heating process (Fig. S5,† curve 1), a broad endothermic feature centred at 350 K is observed, superposed with another endothermic peak located at 383 K. By comparison with TGA data, it can be deduced that the first feature corresponds to the loss of 4 H2O molecules, while the latter can be attributed to the loss of five additional molecules. In the case of 2, a very intense and large peak located around 345 K was recorded. This peak contains a high-temperature tail, in agreement with the gradual character of this dehydration process (Fig. S6,† curve 1). At last, for 3, two clearly separated endothermic processes are registered as peaks located at 344 and 392 K (Fig. S7,† curve 1). They correspond to the loss of 3 and 2 H2O molecules, respectively. Upon further heating, the already dehydrated compound exhibits a sharp and less intense peak at 425 K. For the three compounds, the DSC measurements in the subsequent cooling and heating cycles (Fig. S5–S7,† curves 2–3) show an essentially featureless plot, indicating that all the endothermic processes are irreversible. This is what should be expected for dehydration processes recorded in open sample holders. It indicates also that the sharp peak observed for 3 at 425 K is probably due to an irreversible structural rearrangement from a metastable phase.
Crystal structure analyses
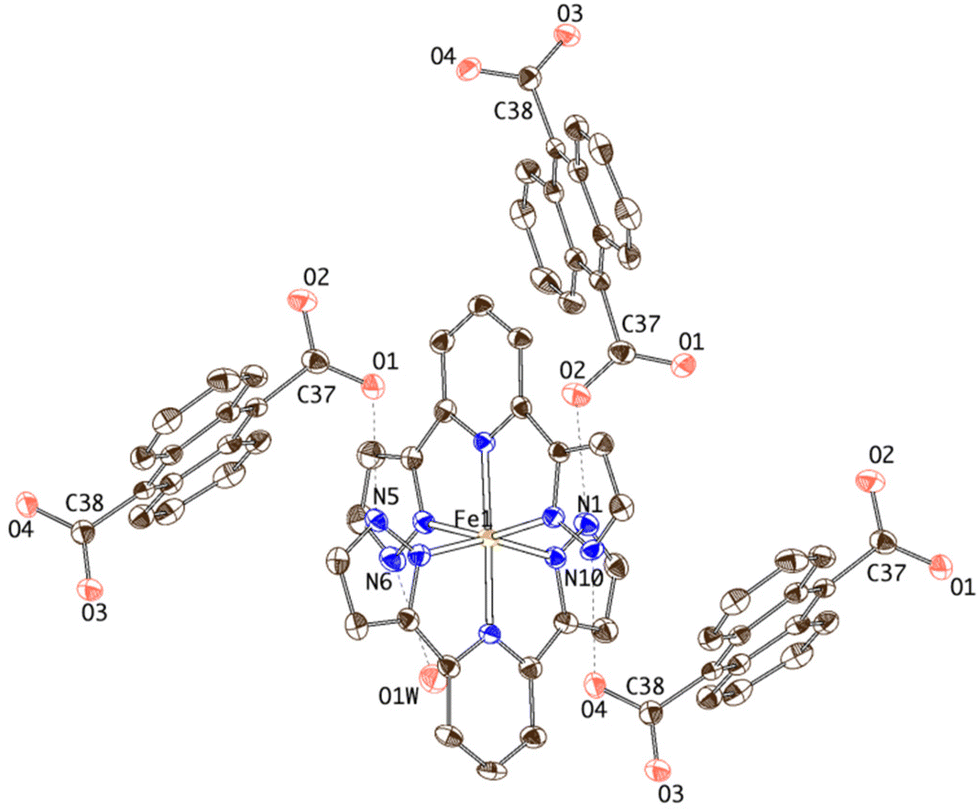 | ||
| Fig. 1 Thermal ellipsoid plot of the crystal structure of 1 showing the hydrogen-bonding connectivity of the [Fe(bpp)2]2+ complex. | ||
| Compound | 1 | 2 | 3 |
|---|---|---|---|
| a R 1 = Σ (Fo − Fc)/Σ(Fo); wR2 = [Σ[w(Fo2 – Fc2)2/Σ[w(Fo2)2]]1/2. | |||
| Formula | C38H44FeN10O13 | C31H32FeN10O11 | C36H36FeN10O9 |
| Formula weight | 904.68 | 776.51 | 808.60 |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | P212121 (no. 19) | P21/n (no. 14) | P21212 (no. 18) |
| a/Å | 11.9374(2) | 8.1212(2) | 28.8805(5) |
| b/Å | 14.5137(2) | 31.2063(8) | 15.3810(2) |
| c/Å | 23.8247(3) | 13.5547(4) | 8.49297(12) |
| α/° | 90 | 90 | 90 |
| β/° | 90 | 96.059(3) | 90 |
| γ/° | 90 | 90 | 90 |
| V/Å3 | 4127.77(10) | 3416.03(17) | 3772.67(10) |
| Z | 4 | 4 | 4 |
| T/K | 120.00(10) | 120.00(14) | 120.00(10) |
| D calcd/g cm−3 | 1.456 | 1.510 | 1.424 |
| λ/Å | 0.71073 | 0.71073 | 0.71073 |
| θ-range/° | 3.285–29.923 | 1.997–29.919 | 3.391–29.761 |
| No. of rflns collected | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 400 |
9255 | 10240 |
| No. of indep. rflns/Rint | 9136/0.1169 | 4236/0.2391 | 8885/0.0833 |
| Restraints/parameters | 35/629 | 20/528 | 22/560 |
| R 1/wR2 (I > 2σ(I))a | 0.0613/0.1145 | 0.0750/0.1474 | 0.0554/0.1011 |
| R 1/wR2 (all data)a | 0.0879/0.1238 | 0.1994/0.2055 | 0.0710/0.1061 |
| Δρmax and Δρmin/e Å−3 | 1.108/−0.422 | 0.599/−0.667 | 0.931/−0.895 |
Each ADC anion is in turn hydrogen-bonded to three [Fe(bpp)2]2+ cations. One of the carboxylate units (O1–C37–O2) acts as a bidentate syn, anti hydrogen-bond acceptor, bridging two [Fe(bpp)2]2+ complexes and organizing helical chains along the x axis (Fig. 2). The other carboxylate moiety (O3–C38–O4) is H-bonded to two water molecules (O3⋯O7W: 2.721(5) Å; O4⋯O1W: 2.667(5) Å) but it is also establishing an additional intrachain bifurcated hydrogen bond with a [Fe(bpp)2]2+ site. This dissimilar H-bonding interaction is translated in a less symmetric distribution of the electron density over the –CO2 fragment (C38–O3 = 1.246(5) Å and C38–O4 = 1.277(5) Å) with respect to the carboxylate bridging two [Fe(bpp)2]2+ complexes (C37–O1 = 1.253(6) Å and C37–O2 = 1.247(5) Å).
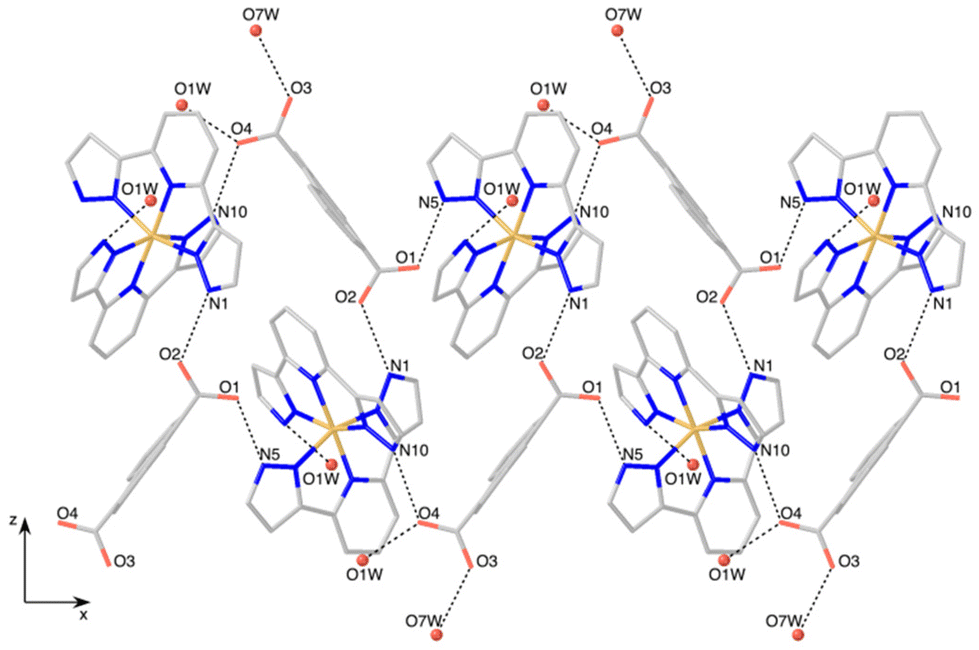 | ||
| Fig. 2 View of the crystal structure of 1 perpendicular to the xz plane, showing a double helical chain of [Fe(bpp)2]2+ cations and ADC anions alternating along the x axis. Only water molecules (O1W and O7W) directly connected to the chains are shown. Dashed lines refer to hydrogen bonds. | ||
The hydrogen-bonded helical chains are held together by N6⋯O1W⋯O4 bridges, forming hexagonal-shaped channels that run parallel to the x screw axis (Fig. 3). Water molecules fill these channels, establishing hydrogen bonds between them and with the chains. Indeed, the four carboxylate oxygen atoms are making additional H-bond interactions with the water molecules sitting in the cavities. Relevant hydrogen-bonding parameters are gathered in Table S2.†
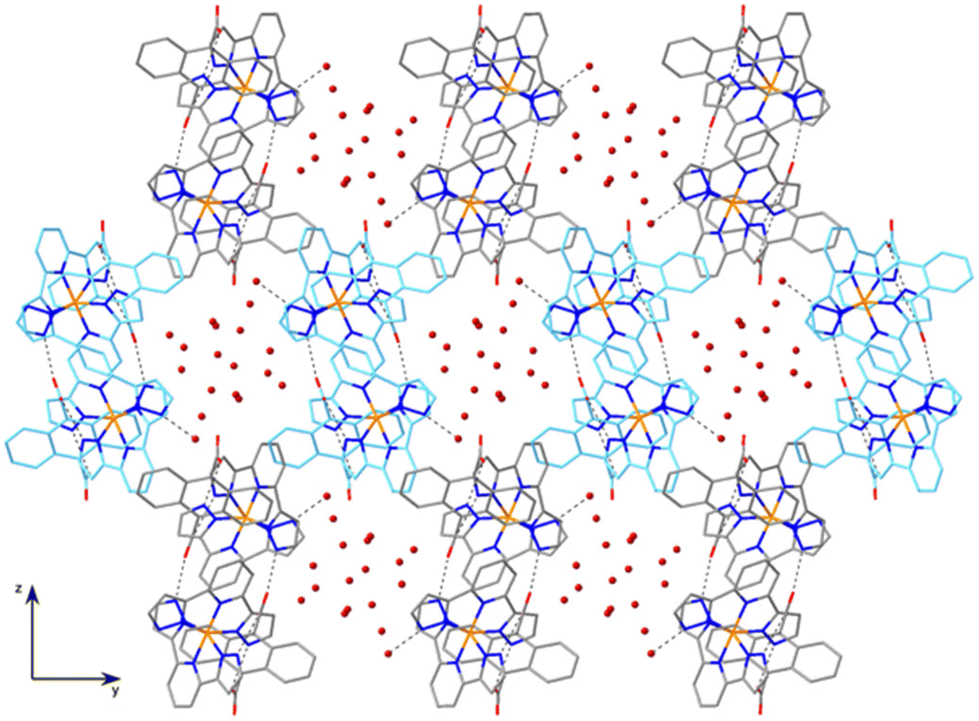 | ||
| Fig. 3 Projection of the crystal structure of 1 onto the bc plane showing the pseudohexagonal cavities filled with water molecules. Adjacent double helical chains are depicted in different colours. H atoms are omitted. Dashed lines refer to H bonds. | ||
A distinctive feature of the structure of 1 is the absence of π–π stacking interactions of any kind, despite the planar aromatic character of cations and anions. Thus, the typical terpyridine embrace interaction7 between [Fe(bpp)2]2+ complexes is prevented, the structure being governed mainly by H-bonding interactions. As a consequence, the iron(II) sites are well separated, the minimum Fe1–Fe1 distance being 9.0105(8) Å, much higher than the typical value commonly found in the terpyridine embrace packings (ca. 8 Å).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–OH moieties, respectively. Instead, the O1–C23–O2 carboxylate fragment is more symmetric (C23–O1: 1.265(5) Å; C23–O2: 1.254(5) Å), as expected for a deprotonated function. Further, the geometry around these two carboxylates corresponds to a syn, syn bridging mode. All these characteristics are similar to those previously found in [Fe(bpp)2](isonicNO)2·HisonicNO·5H2O (isonicNO = isonicotinate N-oxide anion), a compound showing an interplay between SCO and proton migration along SSHBs.12
O and C–OH moieties, respectively. Instead, the O1–C23–O2 carboxylate fragment is more symmetric (C23–O1: 1.265(5) Å; C23–O2: 1.254(5) Å), as expected for a deprotonated function. Further, the geometry around these two carboxylates corresponds to a syn, syn bridging mode. All these characteristics are similar to those previously found in [Fe(bpp)2](isonicNO)2·HisonicNO·5H2O (isonicNO = isonicotinate N-oxide anion), a compound showing an interplay between SCO and proton migration along SSHBs.12
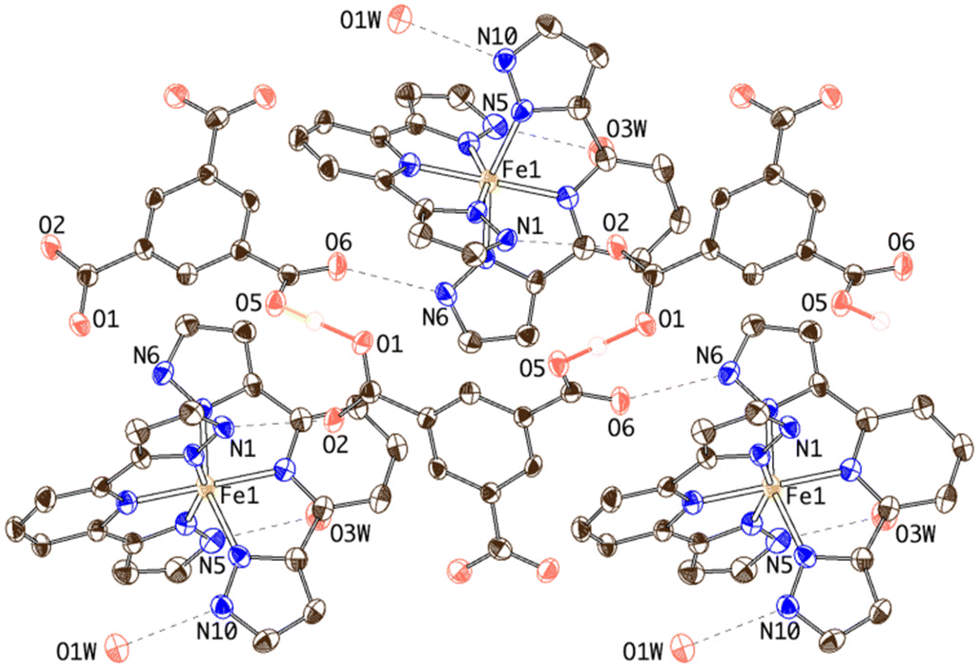 | ||
| Fig. 4 Thermal ellipsoid plot of the crystal structure of 2 showing the hydrogen-bonding connectivity of [Fe(bpp)2]2+ cations and HBTC anions. The O1⋯H1O⋯O5 SSHB interaction is depicted as red solid bonds. Dashed lines refer to N–H⋯O hydrogen bonds. | ||
The direct H-bonding interactions (Table S3†) between cations and anions organize alternating chains running along the [101] direction. These chains are further linked by the SSHBs to yield double helical chains (Fig. 5).
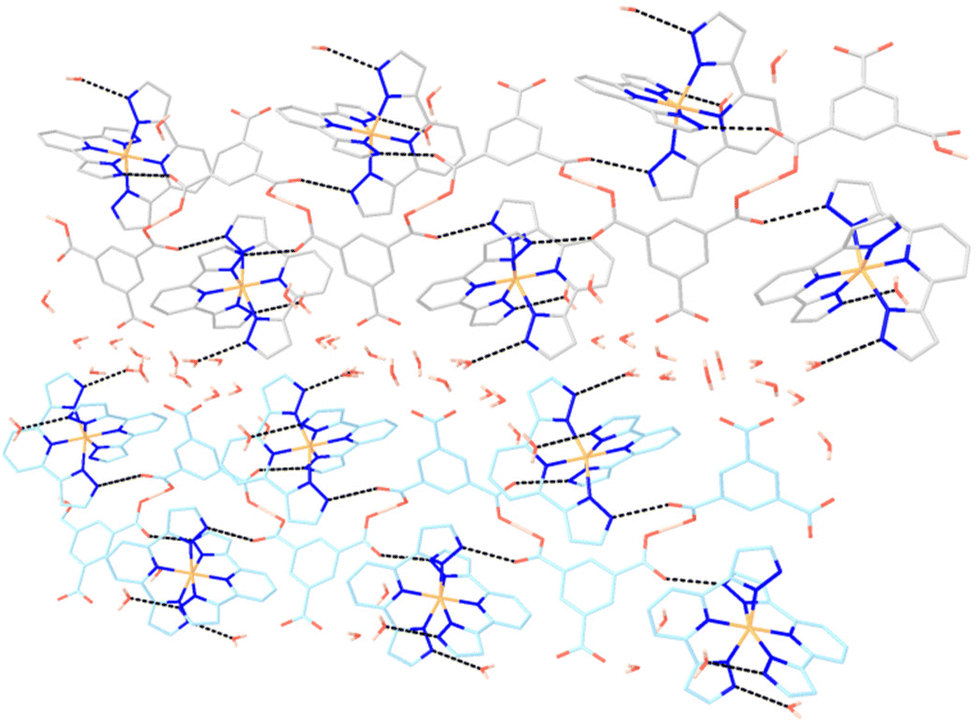 | ||
| Fig. 5 Perspective view of the crystal structure of 2 along the a axis, showing two double helical chains consisting of HBTC anions alternating with [Fe(bpp)2]2+ cations. Water molecules sit in the structure channels. Dashed lines refer to hydrogen bonds. | ||
The chains are assembled along the a axis to form double chains, owing to the existence of weak π–π stacking interactions between the pyrazolyl moieties of adjacent iron(II) complexes (Fig. S8†). Equivalent bpp ligands related by a translation along the x direction form stacks with a distance between adjacent bpp mean planes equal to 3.432(12) Å. The second interaction concerns bpp ligands related by a centre of symmetry (interplane distance: 3.897(10) Å). Since only three ligands are involved in π–π interactions, the formation of the classical square planar terpyridine embrace motif is prevented.
Two inequivalent BPDC anions, denoted BPDC(O1O2) and BPDC(O3O4) in reference to the numbering of the carboxylate O atoms, are found in the asymmetric unit. These two anions exhibit a nonplanar conformation, with torsion angles between phenyl rings of 22.73(7)° and 51.70(11)°, respectively. Both are located in special twofold positions, with the binary axis containing their geometric centres, thus intersecting the central C–C bond of the biphenyl units. BPDC(O1O2) connects two neighbouring [Fe(bpp)2]2+ cations in a syn, syn bridging mode, while BPDC(O3O4) is H-bonded to H2O molecules (Table S4†). As a result, the second coordination sphere of Fe1 is occupied (Fig. 6) by only two BPDC anions, with short H-bonding distances (N5⋯O1: 2.637(4) Å; N10⋯O2: 2.676(4) Å). These contacts propagate in the crystal to build layers consisting of chains of [Fe(bpp)2]2+ units that are pillared by the BPDC anion (Fig. 7). The layers lie parallel to the xy plane and are connected along the z direction by strong π–π stacking interactions (Fig. 8) between bpp ligands belonging to adjacent [Fe(bpp)2]2+ cations (interplane distance: 3.41(3) Å).
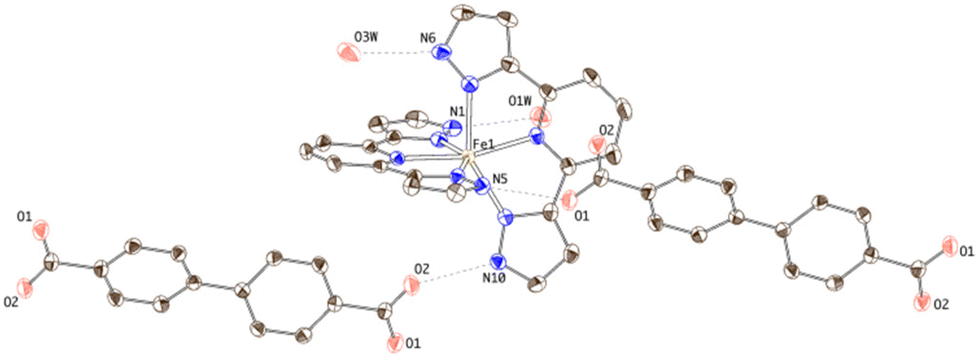 | ||
| Fig. 6 Thermal ellipsoid plot of the crystal structure of 3 showing the hydrogen-bonding connectivity of the [Fe(bpp)2]2+ complex. | ||
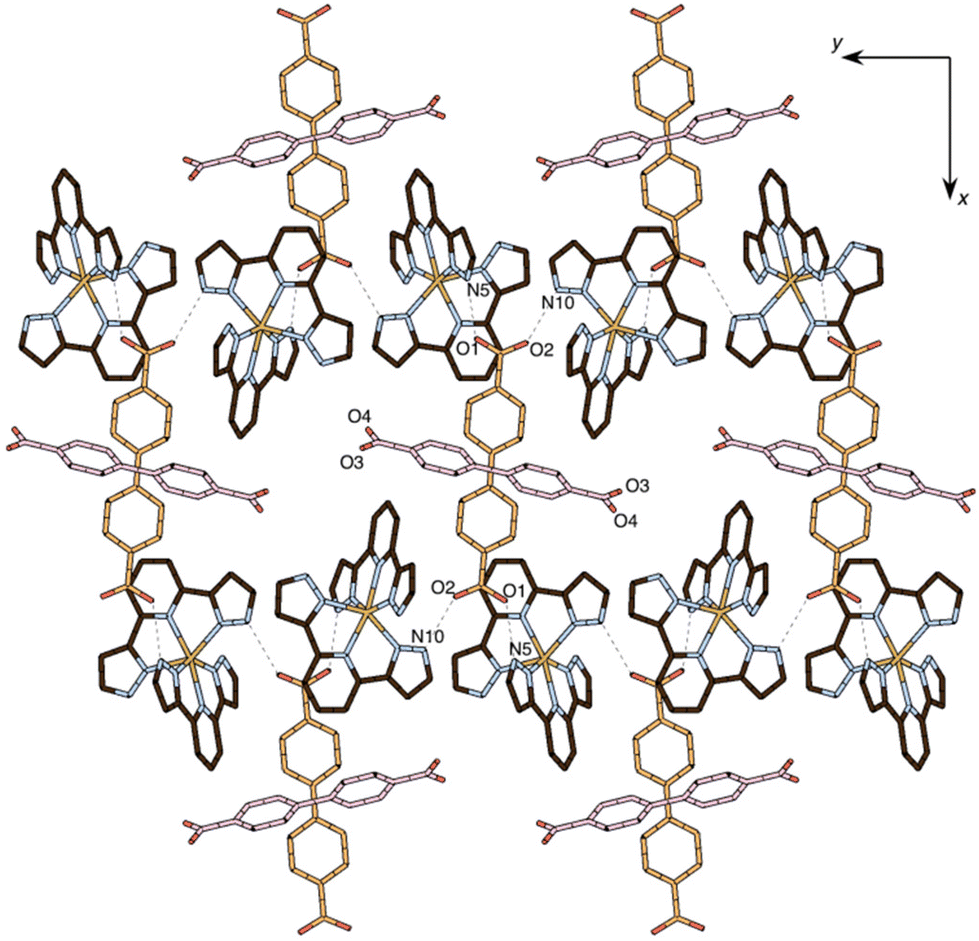 | ||
| Fig. 7 View of the crystal structure of 3 along the c axis, showing a single layer of high-spin Fe1 sites connected by BPDC anions (in orange). BPDC anions non-connected to the cationic sites are also shown (in pink). Water molecules are omitted. Dashed lines refer to hydrogen bonds. | ||
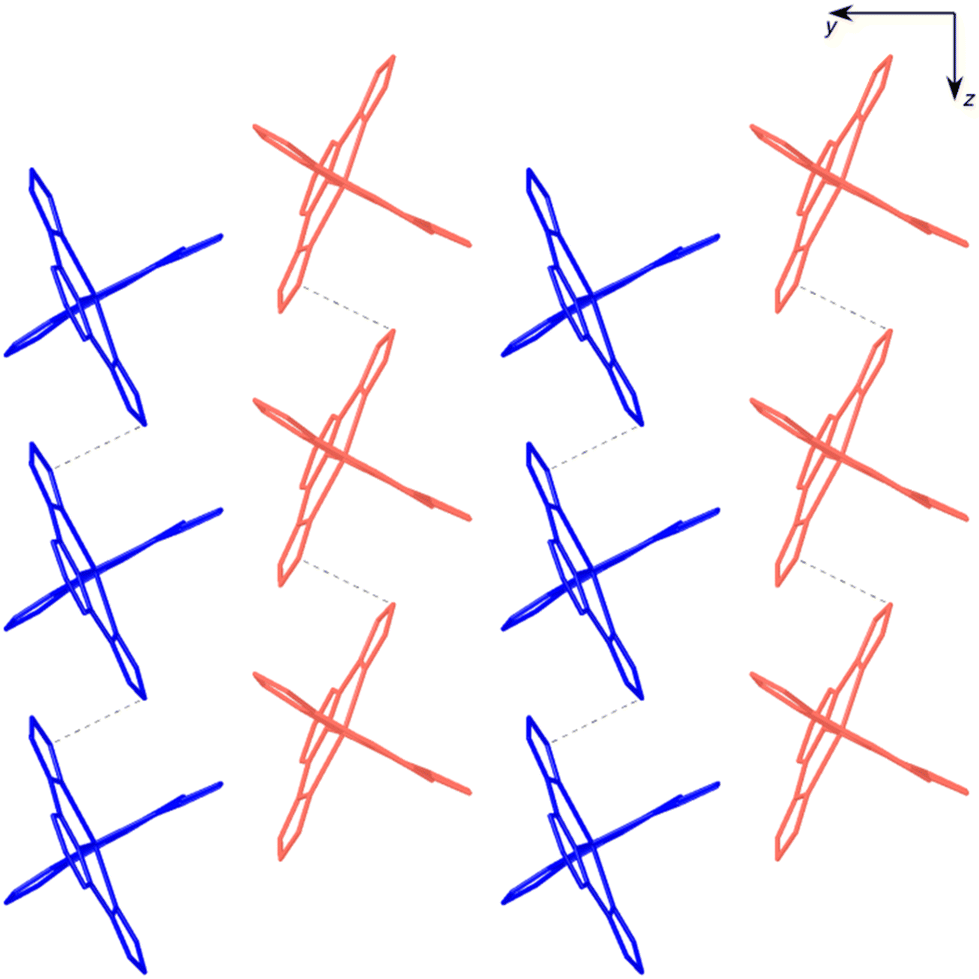 | ||
| Fig. 8 View of the crystal structure of 3 along the a axis, showing the stacking of three layers of [Fe(bpp)2]2+ cations along the z direction. Water molecules and BPDC anions are omitted. Dashed lines refer to π–π stacking interlayer interactions. | ||
Powder X-ray diffraction (PXRD) experiments have been performed for microcrystalline samples of 1–3. A comparison between the X-ray diffractograms measured at room temperature and the simulated diffractogram from the single crystal data recorded at 120 K (Fig. S9–S11†) shows that there is a good agreement between both patterns, excluding the existence of a phase transition between 120 K and room temperature, at least for compounds 1 and 3. In the case of the HBTC derivative (2), an important discrepancy between recorded and simulated data is observed, probably due to partial or complete dehydration of the sample (2 is the compound that has the lowest temperature of dehydration).
Magnetic properties
The temperature dependence of the χT product of 1 (χ = molar magnetic susceptibility; T = absolute temperature) is shown in Fig. 9. At low temperatures, the sample is diamagnetic, as expected for a low-spin ground state for this Fe(II) complex. Above 250 K (curve 1), χT increases continuously on heating up to 400 K, where it equals 3.03 emu K mol−1. This value is the one expected for one HS Fe2+ cation per formula unit (≈3.0 emu K mol−1), indicating a complete spin crossover that seems to initiate for the original nonahydrate sample but continues taking place simultaneously with dehydration. On cooling (curve 2), χT shows a nearly constant value around 3 emu K mol−1 down to 50 K, in agreement with a HS ground state for the anhydrous sample in the whole temperature range. Below 50 K, χT decreases sharply due to zero-field splitting (ZFS) of the S = 2 ground state. The second heating curve deviates slightly from the cooling plot in the low-temperature region but it is very similar in the region of interest. Clearly, the dehydrated sample is a HS phase lacking SCO behaviour, in agreement with DSC measurements.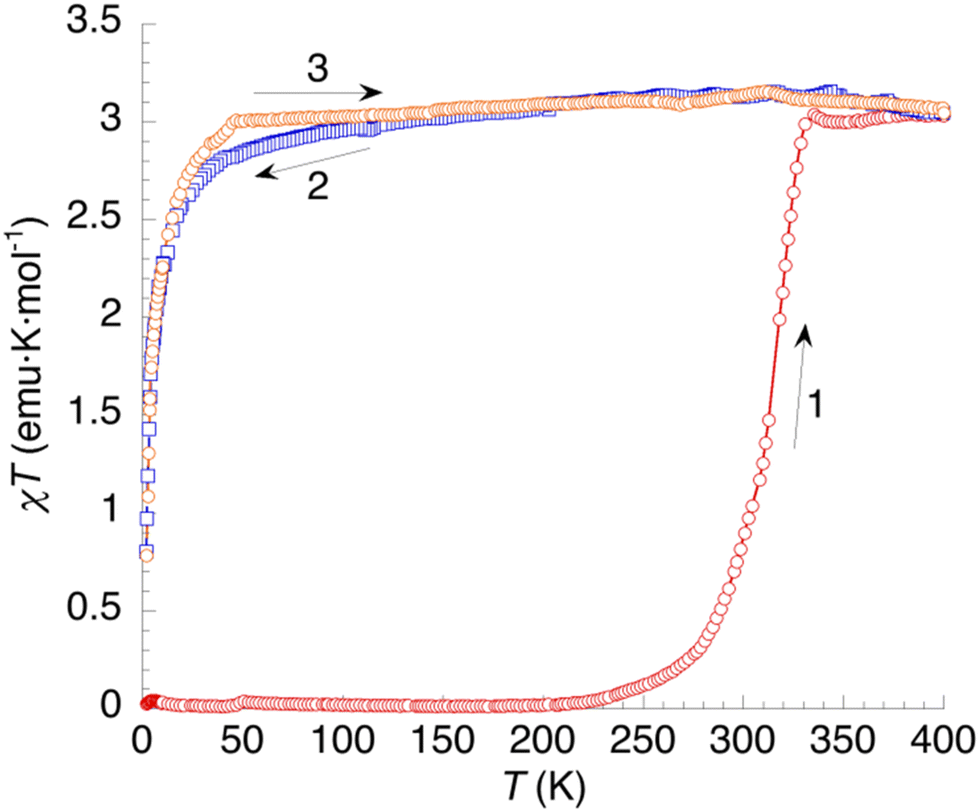 | ||
| Fig. 9 Thermal variation of the χT product of 1. Curve 1: first heating. Curves 2 and 3: subsequent temperature cycle (cooling and heating, respectively). | ||
Fig. 10 shows the thermal variation of χT of 2. The diamagnetic behaviour observed at low temperatures (curve 1) corresponds to a LS state (S = 0) for the Fe2+ cation. Above 200 K, χT starts to increase. First, with a small jump in the 200–250 K region; then more steadily until 330 K, and finally with a stepped behaviour at higher temperatures to reach a constant value of 3.12 emu K mol−1 at 400 K. Again, it seems that SCO starts prior to dehydration but accelerates as the sample is dehydrated. Cooling the compound (curve 2) results in a gradual decrease of χT to a value of 2.45 emu K mol−1 at 80 K, corresponding to a fraction of high-spin Fe2+ sites γHS = 0.75. Then, at lower temperatures, ZFS effects are clearly evidenced by a sharper decrease of χT. The second heating curve matches perfectly the behaviour upon cooling, indicating that the gradual SCO affecting ≈25% of the iron(II) sites is completely reversible.
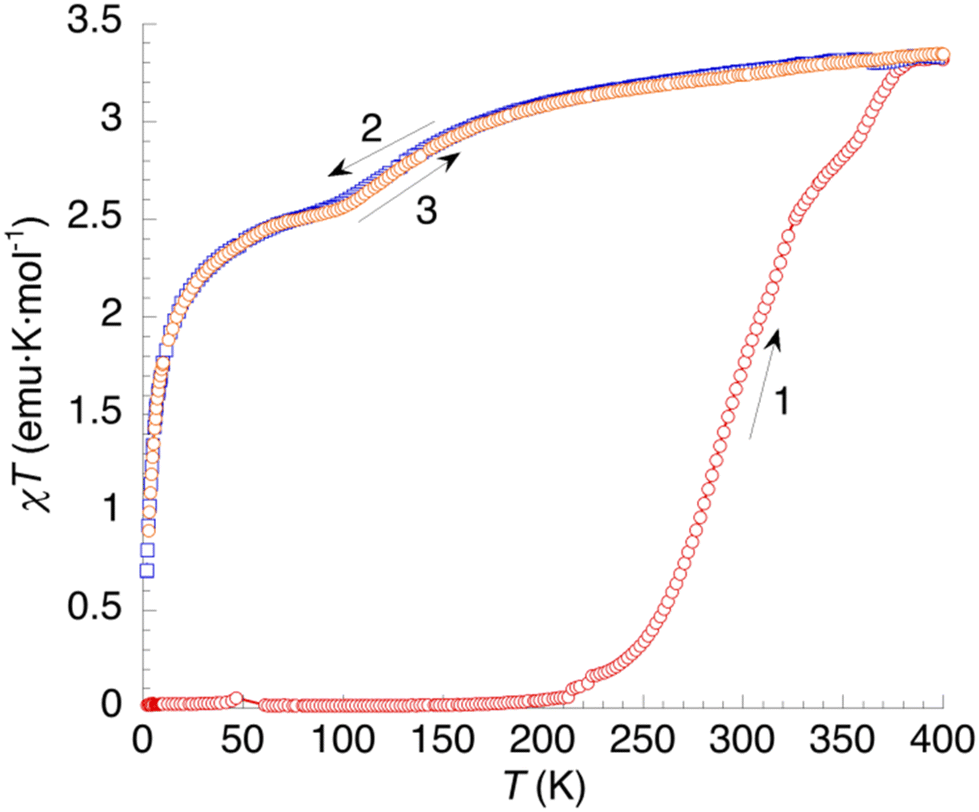 | ||
| Fig. 10 Thermal variation of the χT product of 2. Curve 1: first heating. Curves 2 and 3: subsequent temperature cycle (cooling and heating, respectively). | ||
The χT = f(T) plot for 3 (Fig. 11) shows an almost constant value of 3.75 emu K mol−1 in the 40–325 K range for the first heating measurement (curve 1). This corresponds to a HS state for the Fe2+ cations, a situation that is rather uncommon for hydrated [Fe(bpp)2]2+ salts. Between 2 K and 40 K, the magnetic moment increases very rapidly due to ZFS effects. At T > 325 K, dehydration is causing a small decrease of χT, reaching a minimum at 365 K and increasing again up to 400 K. The minimum can be attributed to a LS phase that is stable after the first dehydration process (see DSC). This phase is then destabilized after full dehydration. In the first cooling (curve 2) χT shows a gradual decrease to a value of 2.5 emu K mol−1 at 40 K. This means that one third of Fe2+ cations have undergone SCO at low temperatures (γHS = 0.66). The process is entirely reversible (curve 3).
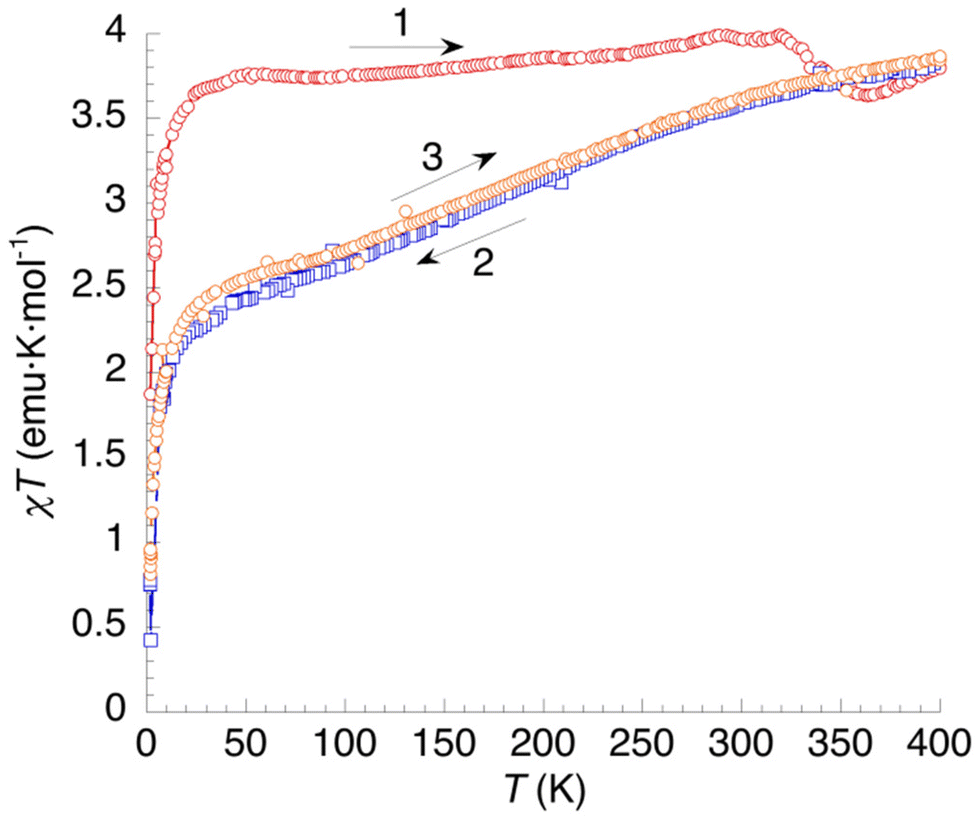 | ||
| Fig. 11 Thermal variation of the χT product of 3. Curve 1: first heating. Curves 2 and 3: subsequent temperature cycle (cooling and heating, respectively). | ||
Conclusions
Three hydrogen-bonded assemblies of [Fe(bpp)2]2+ cations and polycarboxylate anions have been obtained by direct combination or metathesis reactions. The crystal structures of the compounds have been elucidated. Two of them crystallize in noncentrosymmetric space groups and can be interesting for the study of cross effects between SCO and chirality.27,28 It is worth noting that the three crystal structures show a high degree of hydration, especially for 1, that results in the formation of channels. Preliminary assays have shown that it is possible to use a chemically-induced SCO phenomenon to detect selectively anionic species, such as nitrate salts. However, despite the rigidity of the anions, the thermal spin crossover of the reported materials is very gradual and concerns only a small fraction of metal sites, in contrast with other dehydrated compounds of this family.Author contributions
Verónica Jornet-Mollá: formal analysis, investigation, supervisión, visualization. Marina I. Rodríguez-Tarrazó: investigation. Miquel J. Dolz-Lozano: writing – review and editing. Carlos Giménez-Saiz: formal analysis, funding acquisition, supervision. Francisco M. Romero: conceptualization, formal analysis, funding acquisition, methodology, supervision, visualization, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank José M. Martínez-Agudo and Gloria Agustí for the magnetic measurements. We thank the grant CTQ2017-87201-P funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe” for financial support. This study forms part of the Advanced Materials programme and was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and by Generalitat Valenciana (CIPROM/2022/60 project).References
- Spin Crossover in Transition Metal Compounds, Topics in Current Chemistry, ed. P. Gütlich and H. A. Goodwin, Springer Verlag, Berlin-Heidelberg-New York, 2004, vol. 233–235 CrossRef CAS; Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, UK, 2013 CrossRef CAS; K. S. Kumar and M. Ruben, Coord. Chem. Rev., 2017, 346, 176 CrossRef CAS; R. W. Hogue, S. Singh and S. Brooker, Chem. Soc. Rev., 2018, 47, 7303 RSC.
- T. Delgado, A. Tissot, L. Guénée, A. Hauser, F. J. Valverde-Muñoz, M. Seredyuk, J. A. Real, S. Pillet, E.-E. Bendeif and C. Besnard, J. Am. Chem. Soc., 2018, 140, 12870 CrossRef CAS PubMed; J. Y. Ge, Z. Chen, L. Zhang, X. Liang, J. Su, M. Kurmoo and J.-L. Zuo, Angew. Chem., Int. Ed., 2019, 58, 8789 CrossRef PubMed; M. Darawsheh, L. A. Barrios, O. Roubeau, S. J. Teat and G. Aromí, Angew. Chem., Int. Ed., 2018, 57, 13509 CrossRef PubMed.
- T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 1 Search PubMed; D. Pinkowicz, M. Rams, M. Misek, K. V. Kamenev, H. Tomkowiak, A. Katrusiak and B. Sieklucka, J. Am. Chem. Soc., 2015, 137, 8795 CrossRef CAS PubMed; S. Ohkoshi, K. Imoto, Y. Tsunobuchi, S. Takano and H. Tokoro, Nat. Chem., 2011, 3, 564 CrossRef PubMed.
- H. Zheng, Y.-S. Meng, G. L. Zhou, C.-Y. Duan, O. Sato, S. Hayami, Y. Luo and T. Liu, Angew. Chem., Int. Ed., 2018, 57, 8468 CrossRef CAS PubMed.
- K. Dankhoff, C. Lochenie, F. Puchtler and B. Weber, Eur. J. Inorg. Chem., 2016, 2136 CrossRef CAS; A. Djemel, O. Stefanczyk, M. Marchivie, E. Trzop, E. Collet, C. Desplanches, R. Delimi and G. Chastanet, Chem. – Eur. J., 2018, 24, 14760 CrossRef PubMed; J. E. Clements, P. R. Airey, F. Ragon, V. Shang, C. J. Kepert and S. M. Neville, Inorg. Chem., 2018, 57, 14930 CrossRef; S. Rodríguez-Jiménez and S. Brooker, Inorg. Chem., 2019, 58, 8188 CrossRef PubMed; I. C. Berdiell, R. Kulmacezwski, N. Shahid, O. Cespedes and M. A. Halcrow, Chem. Commun., 2021, 57, 6566 RSC; D. J. Mondal, A. Mondal, A. Paul and S. Konar, Inorg. Chem., 2022, 61, 4572 CrossRef PubMed.
- T. Shiga, Y. Sato, M. Tachibana, H. Sato, T. Matsumoto, H. Sagayama, R. Kumai, Y. Murakami, G. N. Newton and H. Oshio, Inorg. Chem., 2018, 57, 14013 CrossRef CAS PubMed; D. Gao, Y. Liu, B. Miao, C. Wei, J.-G. Ma, P. Cheng and G.-M. Yang, Inorg. Chem., 2018, 57, 12475 CrossRef PubMed; W.-K. Han, H.-X. Zhang, Y. Wang, W. Liu, X. Yan, T. Li and Z.-G. Gu, Chem. Commun., 2018, 54, 12646 RSC; I.-R. Jeon, J. G. Park, C. R. Haney and T. D. Harris, Chem. Sci., 2014, 5, 2461 RSC; A. Tissot, X. Kesse, S. Giannopoulou, I. Stenger, L. Binet, E. Rivière and C. Serre, Chem. Commun., 2019, 55, 194 RSC; C. J. Kepert, Chem. Commun., 2006, 7, 695 RSC; E. Resines-Urien, E. Fernandez-Bartolome, A. Martinez-Martinez, A. Gamonal, L. Piñeiro-López and J. Sanchez Costa, Chem. Soc. Rev., 2023, 52, 705 RSC; M. Gavara-Edo, R. Córdoba, F. J. Valverde-Muñoz, J. Herrero-Martín, J. A. Real and E. Coronado, Adv. Mater., 2022, 34, 2202551 CrossRef PubMed; K. S. Kumar and M. Ruben, Angew. Chem., Int. Ed., 2021, 60, 7502 CrossRef PubMed.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493 CrossRef CAS; G. A. Craig, O. Roubeau and G. Aromí, Coord. Chem. Rev., 2014, 269, 13 CrossRef.
- K. H. Sugiyarto, W.-A. McHale, D. C. Craig, D. Rae, M. L. Scudder and H. A. Goodwin, Dalton Trans., 2003, 2443 RSC; A. Bhattacharjee, V. Ksenofontov, K. H. Sugiyarto, H. A. Goodwin and P. Gütlich, Adv. Funct. Mater., 2003, 13, 877 CrossRef CAS.
- C. Carbonera, J. Sánchez Costa, V. A. Money, J. Elhaïk, J. A. K. Howard, M. A. Halcrow and J.-F. Létard, Dalton Trans., 2006, 3058 RSC; D. Fedaoui, Y. Bouhadja, A. Kaiba, P. Guionneau, J.-F. Létard and P. Rosa, Eur. J. Inorg. Chem., 2008, 1022 CrossRef CAS; A. Kaiba, H. J. Shepherd, D. Fedaoui, P. Rosa, A. E. Goeta, N. Rebbani, J.-F. Létard and P. Guionneau, Dalton Trans., 2010, 39, 2910 RSC.
- V. Jornet-Mollá, Y. Duan, C. Giménez-Saiz, Y.-Y. Tang, P.-F. Li, F. M. Romero and R.-G. Xiong, Angew. Chem., 2017, 129, 14240 CrossRef.
- V. Jornet-Mollá, C. Giménez-Saiz, D. S. Yufit, J. A. K. Howard and F. M. Romero, Chem. – Eur. J., 2021, 27, 740 CrossRef PubMed.
- V. Jornet-Mollá, C. Giménez-Saiz, L. Cañadillas-Delgado, D. S. Yufit, J. A. K. Howard and F. M. Romero, Chem. Sci., 2021, 12, 1038 RSC.
- J. Kim, B. Chen, T. M. Reineke, H. Li, M. Eddaoudi, D. B. Moler, M. O′Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2001, 123, 8239 CrossRef CAS PubMed; Z. Ni, A. Yassar, T. Antoun and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 12752 CrossRef PubMed; H. Kumagai, M. Akita-Tanaka, K. Inoue, K. Takahashi, H. Kobayashi, S. Vilminot and M. Kurmoo, Inorg. Chem., 2007, 46, 5949 CrossRef PubMed.
- B. R. Bhogala and A. Nangia, Cryst. Growth Des., 2003, 3, 547 CrossRef CAS; B. R. Bhogala, S. Basavoju and A. Nangia, CrystEngComm, 2005, 7, 551 RSC.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastré, J. Mater. Chem., 2006, 16, 626 RSC.
- E. Coronado, M. C. Giménez-López, C. Giménez-Saiz and F. M. Romero, CrystEngComm, 2009, 11, 2198 RSC.
- Y. Lin and S. A. Lang, J. Heterocycl. Chem., 1977, 14, 345 CrossRef CAS.
- CrysAlisPro Software System, Agilent Technologies, Yarnton, Oxfordshire, UK, 2012 Search PubMed.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, J. Appl. Crystallogr., 2015, 48, 306 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849 CrossRef CAS.
- G. M. Sheldrick, SHELXL-2018, Universität Göttingen, Göttingen, Germany, 2018 Search PubMed.
- R. H. Blessing, J. Appl. Crystallogr., 1997, 30, 421 CrossRef CAS.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97 CrossRef CAS.
- K. H. Sugiyarto and H. A. Goodwin, Aust. J. Chem., 1988, 41, 1645 CrossRef CAS.
- L. K. Saunders, H. Nowell, L. E. Hatcher, H. J. Shepherd, S. J. Teat, D. R. Allan, P. R. Raithby and C. C. Wilson, CrystEngComm, 2019, 21, 5249 RSC.
- M. L. Scudder, D. C. Craig and H. A. Goodwin, CrystEngComm, 2005, 7, 642 RSC; P. King, J. J. Henkelis, C. A. Kilner and M. A. Halcrow, Polyhedron, 2013, 52, 1449 CrossRef CAS.
- I. A. Kühne, A. Ozarowski, A. Sultan, K. Esien, A. B. Carter, P. Wix, A. Casey, M. Heerah-Booluck, T. D. Keene, H. Müller-Bunz, S. Felton, S. Hill and G. G. Morgan, Inorg. Chem., 2022, 61, 3458 CrossRef PubMed.
- C. T. Kelly, R. Jordan, S. Felton, H. Müller-Bunz and G. G. Morgan, Chem. – Eur. J., 2023, 29, e202300275 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2335499, 2335500 and 2335503. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4dt00579a |
| This journal is © The Royal Society of Chemistry 2024 |