
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTemperature induced single-crystal to single-crystal transformation of uranium azide complexes†
Kai
Li
a,
Thayalan
Rajeshkumar
b,
Yue
Zhao
 a,
Tianwei
Wang
a,
Laurent
Maron
*b and
Congqing
Zhu
*a
a,
Tianwei
Wang
a,
Laurent
Maron
*b and
Congqing
Zhu
*a
aState Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: zcq@nju.edu.cn
bLPCNO, CNRS & INSA, Université Paul Sabatier, 135 Avenue de Rangueil, Toulouse 31077, France. E-mail: laurent.maron@irsamc.ups-tlse.fr
First published on 16th February 2024
Abstract
The monomeric and dimeric uranium azide complexes {[(CH3)2NCH2CH2NPiPr2]2U(N3)2} (2) and {[(CH3)2NCH2CH2NPiPr2]2U(N3)2}2 (3) were synthesized by treating complex 1 with NaN3 at 60 and −20 °C, respectively. A temperature-induced single-crystal to single-crystal transformation of 3 to 2 was observed. The reduction of either 2 or 3 with KC8 yields a uranium nitride complex {[(CH3)2NCH2CH2NPiPr2]4U2(μ-N)2} (4).
Single-crystal to single-crystal (SCSC) transformation is accompanied by the breaking of old chemical bonds and the formation of new bonds in the crystalline state, which can affect the dimensionality of complexes.1 However, maintaining crystallinity during SCSC transformations is often challenging, which may contribute to their rarity and unpredictable nature.2,3 Heating and light irradiation are two important external stimuli used to mediate SCSC transformations.4,5 Whilst there are abundant examples of SCSC transformations reported for main group elements,6 transition metals,7 and rare-earth metals,8 instances of such transformations for actinide elements are few.9 Based on our ongoing interest in uranium chemistry supported by double layer N–P ligands,10 we hereby report the isolation of monomeric and dimeric uranium azides 2 and 3 through salt metathesis between 1 and NaN3 at different temperatures. A temperature-induced irreversible SCSC transformation from 3 to 2 was observed. In addition, the reactivity of 2 and 3 with KC8 was investigated, leading to the isolation of uranium nitride 4.
Treatment of complex 1 with two equivalents of NaN3 in THF at room temperature (RT) for 10 h afforded a mixture containing both yellow crystalline complex 2 and green crystalline complex 3 after recrystallization at RT (Scheme 1). Interestingly, upon storing the filtrate of this reaction at 60 °C, we observed that the color of the filtrate changed from green to yellow, and the crystals of 2 were isolated with a 78% yield. Conversely, when the filtrate of this reaction was stored at −20 °C overnight, the crystals of 3 were formed with a 76% isolated yield (Scheme 1).
 | ||
| Scheme 1 Synthesis of complexes 2–4. | ||
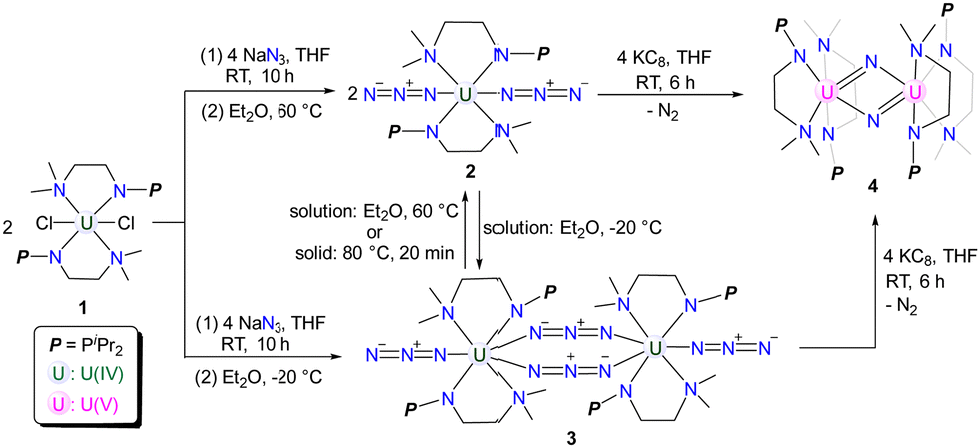
The structures of complexes 2 and 3 have been confirmed by single-crystal X-ray diffraction. Complex 2 is a monomer containing a single U center, which is coordinated by two monoanionic N–P ligands and two N3− units (Fig. 1). The bond lengths of U1–N1/N1′ (2.6990(18) Å) are longer than those of U1–N2/N2′ (2.2665(18) Å), reflecting the dative bond between U1 and N1/N1′. The bond lengths of U1–N3, N3–N4, and N4–N5 are 2.340(2) Å, 1.157(3) Å, and 1.152(3) Å, respectively. These bond lengths are consistent with the structure of U–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N+N− for the terminal azide unit. The bond angles of U1–N3–N4 and N3–N4–N5 are 159.0(2)° and 179.7(3)°, respectively.
N+N− for the terminal azide unit. The bond angles of U1–N3–N4 and N3–N4–N5 are 159.0(2)° and 179.7(3)°, respectively.
 | ||
| Fig. 1 Molecular structure of 2 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms were simplified into lines. | ||
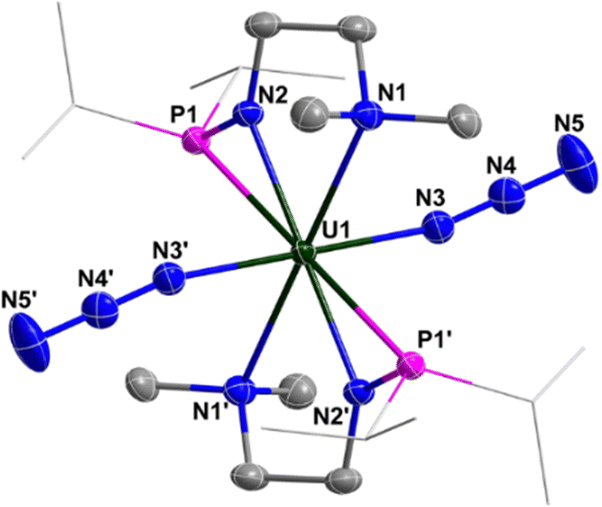
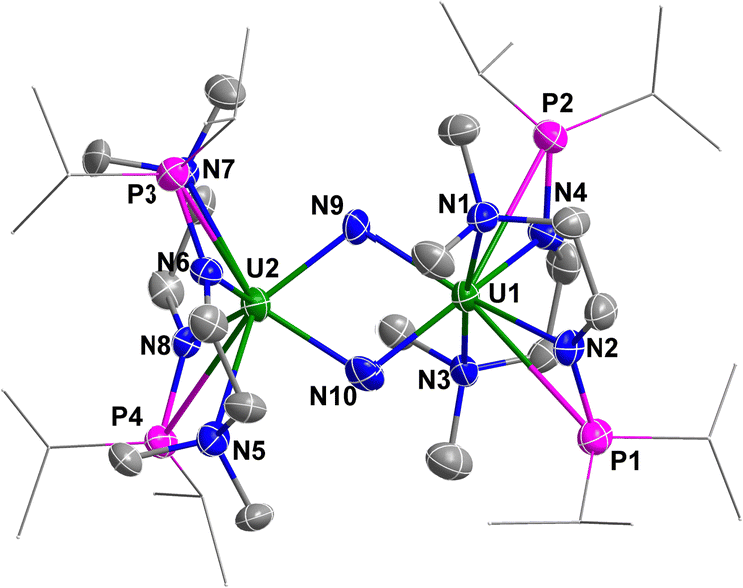
Complex 3 is a dimer containing two U centers connected by two N3− units (Fig. 2). The bond angles of U1–N8–N9 (140.0(10)°) and U1–N10–N11 (142.0(9)°) are smaller than the U1–N3–N4 angle (159.0(2)°) in complex 2. The bond lengths of U1–N8 (2.546(11) Å) and U1–N10 (2.484(10) Å) are longer than the bond length of U1–N5 (2.331(8) Å) due to the bridging nature of N8 and N10. Correspondingly, the bond lengths of N8–N9 (1.164(11) Å) and N10–N11 (1.180(10) Å) in the bridged N3− units are longer than those in the terminal N3− units, such as N6–N7 (1.151(14) Å).
 | ||
| Fig. 2 Molecular structure of 3 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms were simplified into lines. | ||
In solution, complexes 2 and 3 can be interconverted at −20 °C and 60 °C. Consistently, the 1H NMR spectra of complexes 2 or 3 recorded at RT demonstrated a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio between the monomer and dimer forms (Fig. S5 and S6, ESI†). Therefore, variable-temperature 1H NMR spectra of complex 3 were measured in d8-THF from 213 K to 333 K (Fig. 3). At 213 K, a range of signals between +82.1 and −76.3 ppm was attributed to resonances for complex 3. As the temperature increased, the signals for complex 3 gradually disappeared, and a set of broad signals for complex 2 emerged, indicating the conversion from 3 to 2. From these variable-temperature 1H NMR spectral data, we calculated ΔH to be 3.2 ± 0.4 kcal mol−1 (Fig. S14, ESI†), which is in close proximity to the value obtained from DFT calculations (5.0 kcal mol−1, Fig. S18, ESI†). Upon lowering the temperature from 333 K to 213 K, the signals for 2 disappeared and those for complex 3 reappeared (Fig. S1, ESI†). These results suggest that temperature-induced invertible transformation between complexes 2 and 3 is feasible in solution. The Fourier-transform infrared (FT-IR) spectra of complexes 2 and 3 were measured in the solid state at room temperature (Fig. S10 and S11, ESI†). Complex 3 exhibits two strong azide stretching vibrations at 2079 and 2134 cm−1, characteristic of U-bound terminal and bridged azide ligands, whereas complex 2 shows a single strong vibration at 2075 cm−1, indicative of U-bound terminal azide ligands.10a,11
:1 ratio between the monomer and dimer forms (Fig. S5 and S6, ESI†). Therefore, variable-temperature 1H NMR spectra of complex 3 were measured in d8-THF from 213 K to 333 K (Fig. 3). At 213 K, a range of signals between +82.1 and −76.3 ppm was attributed to resonances for complex 3. As the temperature increased, the signals for complex 3 gradually disappeared, and a set of broad signals for complex 2 emerged, indicating the conversion from 3 to 2. From these variable-temperature 1H NMR spectral data, we calculated ΔH to be 3.2 ± 0.4 kcal mol−1 (Fig. S14, ESI†), which is in close proximity to the value obtained from DFT calculations (5.0 kcal mol−1, Fig. S18, ESI†). Upon lowering the temperature from 333 K to 213 K, the signals for 2 disappeared and those for complex 3 reappeared (Fig. S1, ESI†). These results suggest that temperature-induced invertible transformation between complexes 2 and 3 is feasible in solution. The Fourier-transform infrared (FT-IR) spectra of complexes 2 and 3 were measured in the solid state at room temperature (Fig. S10 and S11, ESI†). Complex 3 exhibits two strong azide stretching vibrations at 2079 and 2134 cm−1, characteristic of U-bound terminal and bridged azide ligands, whereas complex 2 shows a single strong vibration at 2075 cm−1, indicative of U-bound terminal azide ligands.10a,11
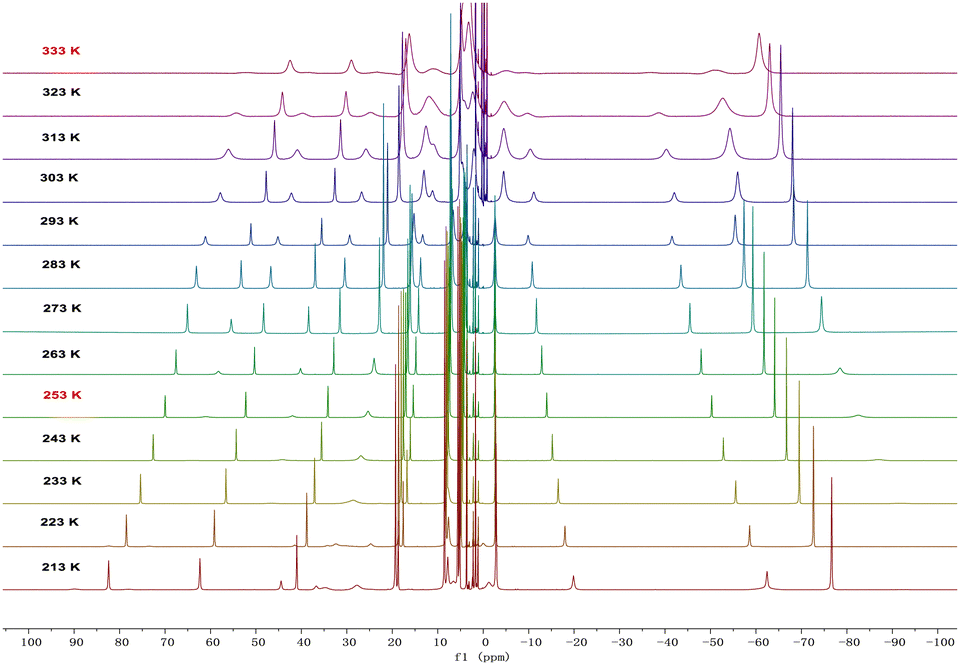 | ||
| Fig. 3 Variable-temperature 1H NMR spectra (from 213 K to 333 K) of complex 3 in d8-THF. | ||
The UV-Vis-NIR electronic absorption spectra of complexes 2 and 3 were measured in THF at RT (Fig. S7, ESI†). Complexes 2 and 3 display an intense absorption peak at 227 nm and 238 nm, respectively. In addition, a weak absorption peak at 452 nm for complex 2 and at 456 nm for complex 3 is also observed in the visible region. Furthermore, a series of low-intensity absorptions are observed in the NIR region. For example, peaks at 821, 893, 1129, 1339, 1409, and 1510 nm were observed for complex 2, while peaks at 819, 1129, 1335, 1427, and 1525 nm were observed for complex 3. These low-intensity absorptions (ε < 200 M−1 cm−1) in the NIR region for complexes 2 and 3 are attributed to the 5f–5f transitions, which are typical characteristics of U(IV) species.
Variable-temperature magnetic data for complexes 2 and 3 in the solid state were collected using a superconducting quantum interference device (SQUID) magnetometer (Fig. 4). The magnetic moment of complex 2 at 370 K is 3.10μB, which declines slowly to 2.49μB at 50 K, and then drops sharply to 0.55μB at 1.8 K with a trend toward zero. The effective moment for complex 3 at 1.8 K is 0.87μB, which increases sharply to 3.02μB at 50 K. With an increase in temperature from 50 K to 340 K, the magnetic moment of complex 3 reaches 3.31μB. When the temperature rises from 340 K to 370 K, the magnetic moments of complex 3 drop noticeably from 3.31 to 3.16μB, which is comparable to the moment of complex 2 at 370 K (3.10μB). With a reduction in temperature from 370 K to 1.8 K, the magnetic moment of complex 3 exhibits a similar trend and values to those of complex 2. The magnitudes of μeff and the temperature dependence for complexes 2 and 3 are comparable to those observed for previously reported U(IV) complexes.12 These results demonstrate that the dimeric complex 3 converts to the monomeric complex 2 at around 350 K in the solid state, suggesting a probable SCSC process.
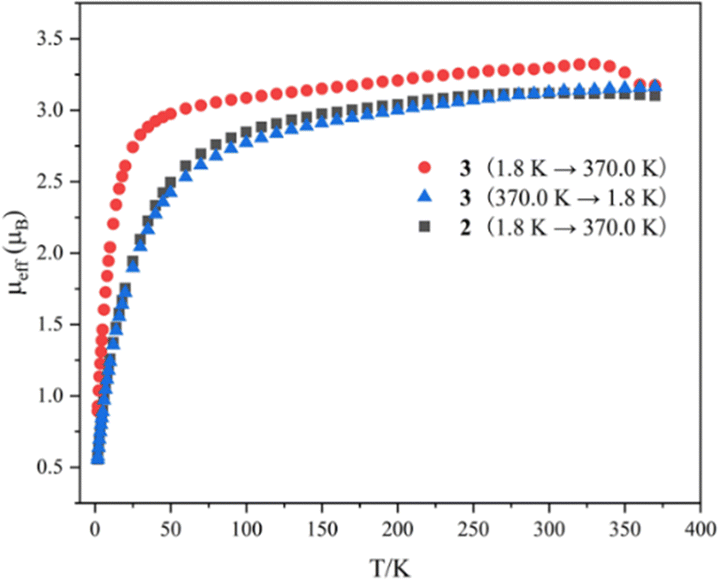 | ||
| Fig. 4 Variable-temperature effective magnetic moment data. Magnetic moment per uranium ion for 2 and 3. | ||
To corroborate the process of SCSC transformation, two approaches were employed. Initially, a crystal of complex 3 was placed on a diffractometer and X-ray diffraction data were collected at different temperatures (313 K, 323 K, 333 K, 343 K, and 353 K). Analysis of these data revealed that complex 3 underwent a transformation to complex 2 at 353 K (Table S2, ESI†), which is further corroborated by a color change of the crystal from green to yellow at this temperature (Fig. S15, ESI†). The second method involved heating a crystal of complex 3 at incremental temperatures (40 °C, 50 °C, 60 °C, 70 °C, and 80 °C) within a glovebox for 20 minutes, followed by X-ray diffraction data collection at 193 K. The analysis of these data also confirmed that complex 3 converted to complex 2 after heating at 80 °C (Table S3, ESI†). Photographic evidence of the crystal additionally supports this transformation (Fig. S16, ESI†). The persistence of crystallinity during heating suggests an authentic SCSC transformation process.
Calculations were performed on 2 and 3 at the DFT level using the B3PW91 functional and including solvent effects (see ESI† for details). The formation of complex 2 from precursor 1 is exothermic by 16.5 kcal mol−1. The dimerization of complex 2 to yield complex 3 is found to be exothermic by 5.0 kcal mol−1, in line with the thermal equilibrium between complexes 2 and 3 reported experimentally. The optimized geometry is agreement with the experimental one, particularly the unsymmetrical coordination of the monoanionic N–P ligand (with U–N1 and U–N2 distances of 2.27 Å and 2.71 Å, respectively). Similarly, the azide coordination to the uranium is well reproduced, with the U–N3 distance being 2.35 Å. Natural Bonding Orbital (NBO) analysis indicates the presence of a polarized U–N3 bond (90% character on N) with a Wiberg Bond Index (WBI) of 0.78. The N3–N4 and N4–N5 WBIs are 1.71 and 2.14, respectively, consistent with the presence of two relatively covalent NN double bonds interacting with a U(IV) center.
Geometry optimization was also performed on complex 3, and different spin states were considered, namely the quintet, triplet, and open-shell singlet. The quintet and open-shell singlet states were found to be degenerate, while the triplet state is 10.5 kcal mol−1 higher in energy. The unpaired spin density plots (Fig. S20, ESI†) show that both uranium centers are in a +IV oxidation state. The distinction between the quintet and the open-shell singlet states lies in the spin coupling between the two U(IV) centers—ferromagnetic for the quintet and antiferromagnetic for the open-shell singlet—with the small energy difference indicating that there is no strong coupling between them. The associated UV-visible spectrum (Fig. S22, ESI†), computed in THF solution, displays intense absorption in the UV region with an absorption feature in the visible region (around 400 nm), corroborating the experimental observations.
Uranium azides are well-known precursors for the synthesis of uranium nitride via photolysis or reduction. However, the photolysis of complex 2 under a 40 W UV lamp for 10 hours only generates intractable results (Fig. S9, ESI†). Fortunately, treating complex 2 with two equivalents of KC8 in THF affords a brownish solid, complex 4, in a 67% yield after recrystallization from Et2O at RT (Scheme 1). Complex 4 can also be synthesized by reducing complex 3 with four equivalents of KC8 under the same conditions. The 1H NMR spectrum of 4 exhibits five signals ranging from +7.6 to −30.1 ppm, with the signal of the N–CH3 group located at −30.1 ppm.
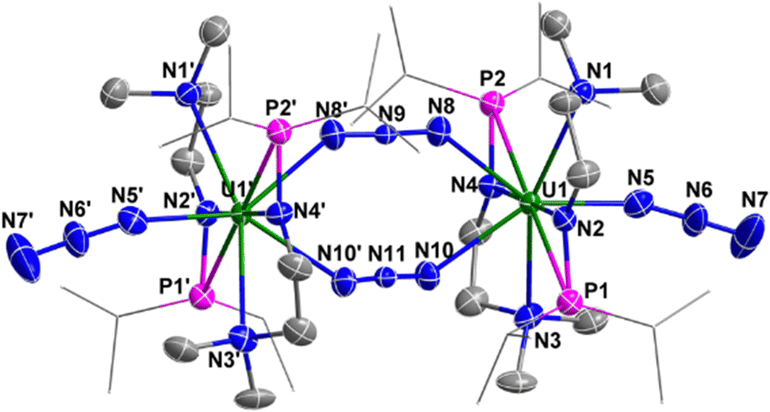
The solid-state structure of complex 4 was determined by single-crystal X-ray diffraction (Fig. 5). The diamond-shaped U2N2 core is supported by four monoanionic N–P ligands. The U1–N9 (2.090(9) Å), U1–N10 (2.113(7) Å), U2–N9 (2.087(7) Å) and U2–N10 (2.068(8) Å) bond lengths are comparable to the U–N bond lengths in [{K(DME)(calix[4]tetrapyrrole)UV}2(μ-NK)2][K(DME)4] (2.076(6) and 2.099(5) Å),13 [KUV(μ-N)(OSi(OtBu)3)3]2 (2.022(5) and 2.101(6) Å),14 [Cs3{UV(OSi(OtBu)3)3}(μ-N)2{UV(OSi(OtBu)3)2(κ-O)}][CsOSi(OtBu)3] (1.85(1), 2.09(1), 2.13(1) and 2.34(1) Å),15 [(μ-N)(UV(N[tBu]Ar)3)2][B(ArF)4] (Ar = 3,5-Me2C6H3, ArF = 3,5-(CF3)2C6H3) (2.0470(3) and 2.0511(3) Å),16 and in U(IV) nitride containing U2N2 core (2.153(2) Å).17 However, these U–N bond lengths are slightly longer than terminal uranium(V) nitrides supported by TrenTIPS ligand (1.80–1.89 Å).18
 | ||
| Fig. 5 Molecular structure of 4 with thermal ellipsoids at the 50% probability level. Hydrogen atoms are omitted for clarity. The iPr groups on P atoms were simplified into lines. | ||
The UV-visible-NIR electronic absorption spectrum of complex 4 was measured in THF at room temperature (Fig. S12, ESI†), displaying an intense absorption peak at 225 nm and a series of low-intensity absorptions in the NIR region. These broad features in the NIR region are comparable to those reported for U(V) nitride.11c,14,19 The effective magnetic moment at 300 K is 2.94μB (Fig. S13, ESI†), which is close to the theoretical value of 2.54μB for a U(V) ion.10f,12 With decreasing temperature, the moment decreases to 0.83μB at 1.8 K per uranium. The variation in μeff and the temperature dependence of complex 4 are comparable to those of reported U(V) complexes.20
Finally, calculations on complex 4 considered three different spin states. The triplet and open-shell singlet were found to be degenerate, whereas the closed-shell singlet is higher in energy by 30.7 kcal mol−1. The weak coupling between the two U centers is indicated by the small energy difference between the ferro- and antiferromagnetic states. The UN Wiberg Bond Indices (WBIs) are 1.43 and 1.37, consistent with the presence of delocalized UN double bonds. The U–U WBI is 0.5, highlighting electron delocalization within the U2N2 moiety.
In summary, monomeric and dimeric uranium azides, complexes 2 and 3, were isolated by reacting complex 1 with NaN3 at different temperatures. A temperature-induced irreversible SCSC transformation was observed from complex 3 to complex 2. The uranium nitride complex 4, featuring a U2N2 core, was isolated by reducing complexes 2 or 3 with KC8.
This research was supported by the National Key R&D Program of China (2021YFA1502500), the National Natural Science Foundation of China (22271138), the Natural Science Foundation of Jiangsu Province (BK20220065). High Performance Computing Center of Nanjing University is also acknowledged. L. M. is a senior member of the Institut Universitaire de France. CalMip is acknowledged for a generous grant of computing time.
Conflicts of interest
The authors declare no competing financial interests.Notes and references
- (a) G. Chakraborty, I.-H. Park, R. Medishetty and J. J. Vittal, Chem. Rev., 2021, 121, 3751 CrossRef CAS PubMed; (b) H. Wang, W. Meng, J. Wu, J. Ding, H. Hou and Y. Fan, Coord. Chem. Rev., 2016, 307, 130 CrossRef CAS; (c) S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2005, 34, 109 RSC; (d) Y. Chen, Q. Chen and Z. Zhang, Chin. J. Org. Chem., 2021, 41, 3826 CrossRef CAS.
- (a) J. J. Vittal, Coord. Chem. Rev., 2007, 251, 1781 CrossRef CAS; (b) O. V. Zenkina, E. C. Keske, R. Wang and C. M. Crudden, Angew. Chem., Int. Ed., 2011, 50, 8100 CrossRef CAS PubMed; (c) S. Rodríguez-Jiménez, H. L. C. Feltham and S. Brooker, Angew. Chem., Int. Ed., 2016, 55, 15067 CrossRef PubMed.
- (a) C. G. Bezzu, M. Helliwell, J. E. Warren, D. R. Allan and N. B. Mckeown, Science, 2010, 327, 1627 CrossRef CAS PubMed; (b) M. Kawano and M. Fujita, Coord. Chem. Rev., 2007, 251, 2592 CrossRef CAS; (c) Q. Liu, M. Lu, L. Yu, Y. Song and J. Lang, Chin. J. Chem., 2021, 39, 647 CrossRef CAS; (d) Y. Cui and D. Xia, Chin. J. Org. Chem., 2021, 41, 907 CrossRef CAS.
- (a) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755 RSC; (b) E. Fernandez-Bartolome, A. Martinez-Martinez, E. Resines-Urien, L. Pineiro-Lopez and J. S. Costa, Coord. Chem. Rev., 2022, 452, 214281 CrossRef CAS; (c) W. He, S. Li and Y. Lan, Inorg. Chem. Front., 2018, 5, 279 RSC.
- (a) S. Huang, T. Andy Hor and G. Jin, Coord. Chem. Rev., 2017, 346, 112 CrossRef CAS; (b) B. B. Rath and J. J. Vittal, Acc. Chem. Res., 2022, 55, 1445 CrossRef CAS PubMed; (c) Akhtaruzzaman, S. Khan, B. Dutta, T. S. Kannan, G. K. Kole and M. H. Mir, Coord. Chem. Rev., 2023, 483, 215095 CrossRef CAS; (d) M. Zhang, X. Yang, Z. Xu, J. Yin and S. Liu, Chin. J. Chem., 2022, 40, 2853 CrossRef CAS.
- (a) A. J. Thompson, A. I. C. Orué, A. J. Nair, J. R. Piece, J. McMurtrie and J. K. Clegg, Chem. Soc. Rev., 2021, 50, 11725 RSC; (b) A. K. Saini, K. Natarajan and S. M. Mobin, Chem. Commun., 2017, 53, 9870 RSC.
- (a) H. Li, S. Zhao, S. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364 RSC; (b) E. A. Patrick, M. E. Bowden, J. D. Erickson, R. M. Bullock and B. L. Tran, Angew. Chem., Int. Ed., 2023, e202304648 CAS.
- (a) Y. So and W. Leung, Coord. Chem. Rev., 2017, 340, 172 CrossRef CAS; (b) W. Chen, Y. Chen, G. Huang, J. Liu, J. Jia and M. Tong, Chem. Commun., 2018, 54, 10886 RSC; (c) Y. Xin, J. Wang, M. Zychowicz, J. J. Zakrzewski, K. Nakabayashi, B. Sieklucka, S. Chorazy and S.-I. Ohkoshi, J. Am. Chem. Soc., 2019, 141, 18211 CrossRef CAS PubMed.
- (a) K. Lv, S. Fichter, M. Gu, J. Marz and M. Schmidt, Coord. Chem. Rev., 2021, 446, 214011 CrossRef CAS; (b) T. Zheng, Y. Gao, D. Gui, L. Chen, D. Sheng, J. Diwu, Z. Chai, T. Albrecht-Schmitt and S. Wang, Dalton Trans., 2016, 45, 9031 RSC; (c) Z. Xie, L. Mei, Q. Wu, K. Hu, L. Xia, Z. Chai and W. Shi, Dalton Trans., 2017, 46, 7392 RSC; (d) L. Chen, Y. Zhang, Z. Weng, Z. Liu, J. Zhang, Y. Wang and S. Wang, Chin. J. Chem., 2021, 39, 597 CrossRef CAS; (e) L. Mei, L. Wang, L. Yuan, S. An, Y. Zhao, Z. Chai, P. C. Burns and W. Shi, Chem. Commun., 2015, 51, 11990 RSC.
- (a) X. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Zhu, Natl. Sci. Rev., 2023, 10, nwac144 CrossRef CAS PubMed; (b) X. Xin, I. Douair, T. Rajeshkumar, Y. Zhao, S. Wang, L. Maron and C. Zhu, Nat. Commun., 2022, 13, 3809 CrossRef CAS PubMed; (c) X. Xin, I. Douair, Y. Zhao, S. Wang, L. Maron and C. Zhu, J. Am. Chem. Soc., 2020, 142, 15004 CrossRef CAS PubMed; (d) P. Wang, Y. Zhao and C. Zhu, Organometallics, 2022, 41, 2448 CrossRef CAS; (e) P. Wang, I. Douair, Y. Zhao, S. Wang, J. Zhu, L. Maron and C. Zhu, Angew. Chem., Int. Ed., 2021, 60, 473 CrossRef CAS PubMed; (f) J. Shen, T. Rajeshkumar, G. Feng, Y. Zhao, S. Wang, L. Maron and C. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202303379 CrossRef CAS PubMed; (g) G. Feng, M. Zhang, D. Shao, X. Wang, S. Wang, L. Maron and C. Zhu, Nat. Chem., 2019, 11, 248 CrossRef CAS PubMed; (h) G. Feng, M. Zhang, P. Wang, S. Wang, L. Maron and C. Zhu, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17654 CrossRef CAS PubMed; (i) P. Wang, I. Douair, Y. Zhao, R. Ge, J. Wang, S. Wang, L. Maron and C. Zhu, Chemistry, 2022, 8, 1361 CrossRef CAS; (j) G. Feng, K. N. McCabe, S. Wang, L. Maron and C. Zhu, Chem. Sci., 2020, 11, 7585 RSC; (k) W. Fang, S. Pan, W. Su, S. Wang, L. Zhao, G. Frenking and C. Zhu, CCS Chem., 2022, 4, 1921 CrossRef CAS; (l) Q. Zhu, W. Fang, L. Maron and C. Zhu, Acc. Chem. Res., 2022, 55, 1718 CrossRef CAS PubMed; (m) W. Fang, Q. Zhu and C. Zhu, Chem. Soc. Rev., 2022, 51, 8434 RSC; (n) X. Sun, X. Gong, Z. Xie and C. Zhu, Chin. J. Chem., 2022, 40, 2047 CrossRef CAS; (o) W. Fang, I. Douair, A. Hauser, K. Li, Y. Zhao, P. W. Roesky, S. Wang, L. Maron and C. Zhu, CCS Chem., 2022, 4, 2630 CrossRef CAS; (p) K. Li, J. He, Y. Zhao and C. Zhu, Inorg. Chem. Front., 2023, 10, 5622 RSC.
- (a) R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723 CrossRef CAS PubMed; (b) R. Haiges, M. Vasiliu, D. A. Dixon and K. O. Christe, Chem. – Eur. J., 2017, 23, 652 CrossRef CAS PubMed; (c) J. Du, D. M. King, L. Chatelain, E. Lu, F. Tuna, E. J. L. Mclnnes, A. J. Wooles, L. Maron and S. T. Liddle, Chem. Sci., 2019, 10, 3738 RSC.
- D. R. Kindra and W. J. Evans, Chem. Rev., 2014, 114, 8865 CrossRef CAS PubMed.
- I. Korobkov, S. Gambarotta and G. Yap, Angew. Chem., Int. Ed., 2002, 41, 3433 CrossRef CAS PubMed.
- (a) C. Camp, J. Pécaut and M. Mazzanti, J. Am. Chem. Soc., 2013, 135, 12101 CrossRef CAS PubMed; (b) L. Barluzzi, L. Chatelain, F. Fadaei-Tirani, I. Zivkovic and M. Mazzanti, Chem. Sci., 2019, 10, 3543 RSC; (c) L. Barluzzi, F.-C. Hsueh, R. Scopelliti, B. E. Atkinson, N. Kaltsoyannis and M. Mazzanti, Chem. Sci., 2021, 12, 8096 RSC.
- N. Jori, T. Rajeshkumar, R. Scopelliti, I. Zivkovic, A. Sienkiewicz, L. Maron and M. Mazzanti, Chem. Sci., 2022, 13, 9232 RSC.
- A. R. Fox, P. L. Arnold and C. C. Cummins, J. Am. Chem. Soc., 2010, 132, 3250 CrossRef CAS PubMed.
- D. M. King, B. E. Atkinson, L. Chatelain, M. Gregson, J. A. Seed, A. J. Wooles, N. Kaltsoyannis and S. T. Liddle, Dalton Trans., 2022, 51, 8855 RSC.
- (a) D. M. King, F. Tuna, E. J. L. Mclnnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482 CrossRef CAS PubMed; (b) D. M. King, P. A. Cleaves, A. J. Wooles, B. M. Gardner, N. F. Chilton, F. Tuna, W. Lewis, E. J. L. Mclnnes and S. T. Liddle, Nat. Commun., 2016, 7, 13773 CrossRef CAS PubMed.
- P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412 CrossRef CAS PubMed.
- (a) P. L. Arnold, A.-F. Pecharman, E. Hollis, A. Yahia, L. Maron, S. Parsons and J. B. Love, Nat. Chem., 2010, 2, 1056 CrossRef CAS PubMed; (b) O. P. Lam, F. W. Heinemann and K. Meyer, C. R. Chim., 2010, 13, 803 CAS; (c) D. Patel, F. Moro, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 10388 CrossRef CAS PubMed; (d) O. P. Lam, F. W. Heinemann and K. Meyer, Chem. Sci., 2011, 2, 1538 RSC; (e) D. M. King, F. Tuna, E. J. L. Mclnnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717 CrossRef CAS PubMed; (f) D. Patel, F. Tuna, E. J. L. Mclnnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2013, 42, 5224 RSC; (g) C. T. Palumbo, L. Barluzzi, R. Scopelliti, I. Zivkovic, A. Fabrizio, C. Corminboeuf and M. Mazzanti, Chem. Sci., 2019, 10, 8840 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2310068–2310070 and 2329971–2329980. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc00546e |
| This journal is © The Royal Society of Chemistry 2024 |